

Abstract
Moving from macroscale preparative systems in proteomics to micro- and nanotechnologies offers researchers the ability to deeply profile smaller numbers of cells that are more likely to be encountered in clinical settings. Herein a recently developed microscale proteomic method, microdroplet processing in one pot for trace samples (microPOTS), was employed to identify proteomic changes in ∼200 Barrett’s esophageal cells following physiologic and radiation stress exposure. From this small population of cells, microPOTS confidently identified >1500 protein groups, and achieved a high reproducibility with a Pearson’s correlation coefficient value of R > 0.9 and over 50% protein overlap from replicates. A Barrett’s cell line model treated with either lithocholic acid (LCA) or X-ray had 21 (e.g., ASNS, RALY, FAM120A, UBE2M, IDH1, ESD) and 32 (e.g., GLUL, CALU, SH3BGRL3, S100A9, FKBP3, AGR2) overexpressed proteins, respectively, compared to the untreated set. These results demonstrate the ability of microPOTS to routinely identify and quantify differentially expressed proteins from limited numbers of cells.
Keywords: proteomics, microPOTS, lithocholic acid, Barrett’s esophagus, X-ray
1. Introduction
Mass spectrometry (MS) has emerged as the most powerful technology for analysis and discovery of proteins.1 Since the term proteome was coined in 1994,2 researchers have used MS to comprehensively define the molecular mechanisms that underpin cellular functions. To achieve this goal, there are two main approaches to proteomics: top-down and bottom-up, with the latter being more often applied today.3,4 Significant gains have been made by applying these approaches in large-scale studies5 to fully profile protein expression and their post-translational modifications. However, standard proteomic analysis demands substantial amounts of starting material to exhaustively characterize a proteome. For instance, about 105 to millions of cells have typically been used to achieve a high proteome coverage.6 Historically, utilization of such large amounts of starting material often precluded the ability for proteomics to compete with genomics in the analysis of small numbers of cells. This is because genomics allows sample material to be amplified through polymerase chain reaction (PCR).7 Proteomics on the other hand has had to pay special attention to sample preparation of small numbers of cells in order to avoid adsorptive losses of low abundance proteins. Therefore, this limitation has hampered the application of proteomics to study samples of limited availability such as human tissues from, e.g., biopsies.
Against the backdrop of the shortcomings of traditional macroscale sample preparation, mostly inherited from the field of protein chemistry, methods for working with limited numbers of cells have recently been reported, including laser capture microdissection, immobilized enzyme reactors, fluorescence-activated cell sorting (FACS), and microfluidics formats.8−14 For example, using laser capture microdissection, Clair et al.10 identified >3400 proteins from 4000 cells. However, none of these recent developments allowing characterization of proteomes from fewer than 1000 cells by MS-based proteomics can yet be said to be the method of choice for exploring proteomes. Thus, there is burgeoning interest in the community to develop and optimize highly sensitive and specific microscale proteomic workflows to interrogate protein changes in both health and disease. Consequently, reports of micro- and nanoscale MS-based proteomics have dramatically increased in number lately because they facilitate new opportunities to explore trace levels of samples previously out of reach to researchers.15−18 For instance, an ultrasensitive nanoscale method, which used gold nanoparticles, identified 650 proteins from a proteomic analysis of 80 cells with a detection limit of proteins reaching 50 zmol.19 Additionally, many integrated proteome methods optimized for single-cell analysis are increasingly becoming commonplace.20,21 Some newly assembled proteome analysis devices have reported high numbers of identified proteins, e.g., 328 proteins identified from analysis of 10 single HeLa cells, and with a detection limit approximated to be between 1.7–170 zmol.22
One new microfluidics-based platform termed nanodroplet processing in one pot for trace samples (nanoPOTS) that was developed recently has demonstrated remarkable results from the proteomic analysis of small samples.23 By applying a bottom-up proteomic approach, nanoPOTS proved to be capable of processing samples in nanowells with volumes of less than 200 nL.23 This method was applied in the analysis of about 10 to 140 cells, and over 1500 proteins were confidently identified. Recently, nanoPOTS was also integrated with a top-down proteomic workflow, and ∼170 to ∼620 proteoforms from ∼70 to ∼770 HeLa cells were quantitatively identified with high confidence.24 An adaptation of nanoPOTS that utilizes conventional micropipettes and operates in low-microliter range called microdroplet processing in one pot for trace samples (microPOTS) has also been developed to address a few bottlenecks such as the demands for nanoliter pipetting platform and highly skilled personnel to run nanoPOTS. Initially, microPOTS was applied to ∼25 cultured HeLa cells and 50 μm square mouse liver tissue thin sections, and about 1800 and 1200 unique proteins were generated from HeLa cells and mouse liver, respectively.25 Additionally, high reproducibility was reported based on pairwise Pearson’s correlation coefficient values of 0.96–0.98, and with median CVs of ≤12.4% from the results of the previously mentioned analysis.
In this study, we applied the microPOTS to characterize proteomes of ∼200 cells used as a Barrett’s esophagus cell model following various perturbations. Barrett’s esophagus is a premalignant condition thought to arise in the lower esophagus due to chronic reflux of gastric acid and bile leading to genotoxic stress and mutation of the gatekeeper genes TP53 and SMAD4.26 Barrett’s confers an approximately 100-fold greater risk of development of esophageal adenocarcinoma and understanding the molecular changes during carcinogenesis may be useful to guide preventative therapy.27 It is known that gut bacteria modify bile acids derived from cholic acid and chenodeoxycholic acid to deoxycholic acid and lithocholic acid (LCA).28 In turn, deoxycholic acid and lithocholic acid are conjugated to yield a variety of conjugated bile salts that can exist in wide-ranging concentrations in patients being monitored for the acid-reflux disease. It is not known whether there is a specific role for bile acids in the selection for specific genetic mutations in esophageal adenocarcinoma progression. Recent work has demonstrated a novel sponge-device (Cytosponge) can sample small numbers of surface cell populations from the esophagus without endoscopy and can determine the presence of Barrett’s esophagus.29 This device can also be used to triage patients with Barrett’s for more intensive endoscopic surveillance according to the presence of markers of progression to esophageal adenocarcinoma.30 As these devices become integrated into clinical practice the molecular changes during the progression from Barrett’s to esophageal adenocarcinoma need to be identified from the small numbers of cells retrieved during sampling. Identifying protein markers of progression that can be tested by immunohistochemistry will aid in improving the sensitivity and translation of this technology. The findings of this study reveal that microPOTS allowed for the identification of >1500 proteins from fewer than 200 cells, and radiologic and physiologic stress induce proteomic changes in cell models.
2. Experimental Section
2.1. Materials
MicroPOTS chips were fabricated in-house as described previously.25 The microwell chips were designed with a diameter of 2.2 mm and a well-to-well spacing of 4.5 mm. LC-MS grade water and acetonitrile, formic acid (FA), iodoacetamide (IAA), and dithiothreitol (DTT) were purchased from Thermo Fisher Scientific (Waltham, MA). N-Dodecyl β-d-maltose (DDM) was a product of Sigma-Aldrich (St. Louis MO). Both Lys-C and trypsin were purchased from Promega (Madison, WI).
2.2. Cell Culture
CP-A cells were cultured in keratinocyte media (ThermoFisher) supplemented with human recombinant epidermal growth factor (rEGF), bovine pituitary extract (BPE), 1% penicillin/streptomycin (Invitrogen) and incubated at 37 °C with 5% CO2. All other chemicals and reagents were obtained from Sigma unless otherwise mentioned. The guide RNAs targeting the p53 and smad4 genes to generate isogenic gene knockout cells are described in a separate manuscript. Briefly, the guide RNAs were either cloned into lentiCRISPRv2 transfer plasmid or procured as custom synthetic crRNAs from Integrated DNA Technologies (IDT), USA; tracrRNA was also manufactured by IDT. Cells were either transfected using attractene transfection reagent (Qiagen) or electroporated using Nucleofector Kit V (Amaxa, Lonza). After the electroporation, cells were transferred into a 6-well plate and allowed to recover for 3–5 days. The bulk population were single-cell isolated using flow cytometry and individual cells were deposited into 96-well plates using BD FACSJazz cell sorter. Individual colonies obtained were later replicated into 96-well plates and screened for successful gene deletion using immunocytochemistry against the p53 or Smad4. The clonal lines that stained negative for corresponding proteins were further expanded, and the loss of functional p53 and Smad4 protein was confirmed by immunoblotting against respective antibodies and Sanger sequencing for selected knockout clones using the primers flanking the gRNA cleavage site confirmed the genetic editing.
2.3. Cells Treatment with LCA and X-ray
CPA wildtype, p53 null, and p53 Smad4 double null cell lines were cultured in triplicate and treated with 10 μM lithocholic acid (LCA) or DMSO as a control for 24 h or irradiated with 2 Gy X-ray and cultured for further 24 h. Following the treatment, the cells were trypsinized, washed with PBS, counted and samples were prepared with 2 × 105 cells in 50 μL PBS for each condition.
2.4. Proteomic Sample Preparation in Microwells
To prepare samples for LC-MS analysis, 5 μL of 1% DDM and 0.5 μL of 500 mM DTT were added to 50 μL of sample, followed by incubation at 65 °C and 600 rpm for 1 h to lyse the cells and denature proteins. The cell lysates were diluted to 200 cells/500 nL with 50 mM ABC (pH 8.5) and 500 nL of cell suspension was pipetted into the microwells. Next, 500 nL of 10 mM IAA was added, and the samples were allowed to incubate in the dark at room temperature for 45 min. Two-step enzymatic digestion was applied by sequentially adding 500 nL of 10 ng/μL Lys-C and 500 nL of 20 ng/ μL trypsin in Ammonium bicarbonate buffer, followed by incubation at 37 °C for 3 and 10 h, respectively. Thereafter, 500 nL of 5% FA was added, followed by incubation at RT for 1 h. The chips were stored in the humidified box sealed in a Ziploc bag at 4 °C until analysis.
2.5. LC-MS/MS Analysis
A nanoPOTS autosampler was employed to introduce samples in microwells into LC-MS.31 The samples in microwell chips were extracted and loaded into a solid-phase extraction (SPE) column (4 cm long, 150 μm i.d., packed with 3 μm, 300 Å C18 particles, Phenomenex, Torrance, CA, USA) using 100% buffer A (0.1% formic acid) delivered by a Dionex UltiMate NCP-3200RS pump. After sample loading, the concentrated peptides were separated using a 50 cm long, 50 μm i.d. nanoLC column with an integrated electrospray emitter (PicoFrit column, New Objective, Woburn, MA, USA). The LC column was packed in house with the same C18 particles used for the SPE column described above. The LC flow rate was 150 nL/min. A 50 min linear gradient from 8% to 22% buffer B (0.1% formic acid in ACN) was used for peptide elution, followed by raising the gradient to 35% buffer B in 10 min to elute hydrophobic peptides. The column was then washed by flushing the column with 80% buffer B for 5 min. Finally, the column was equilibrated using 2% buffer B for 15 min before the next injection.
An Orbitrap Fusion Lumos Tribrid MS (ThermoFisher, San Jose, USA) operating in data dependent acquisition mode was employed for peptide signal collection. To trigger electrospray, a high voltage of 2200 V was applied at the metal union (Valco, Houston, USA) between the SPE column and LC column. The ion transfer tube was set at 200 °C for desolvation and the radio frequency of the ion funnel was set at 30% for optimal peptide transmission. For MS1 acquisition, an Orbitrap resolution of 120 000, a MS scan range from 375 to 1600, an AGC level of 1 × 106, and a maximum injection time of 100 ms were used. Precursor ions with charges between +2 and +7, and intensity values over 1 × 104 were selected for HCD fragmentation and MS2 scanning. Precursors were isolated with an m/z window of 2 and fragmented by high energy dissociation (HCD) set at 30%. The fragment ions were transferred to Orbitrap for MS2 acquisition at a scan resolution of 60 000 and a maximum injection time of 118 ms. To reduce repeated sampling, an exclusion duration of 30 s and m/z tolerance of ±10 ppm were applied.
2.6. Data and Statistical Analysis
The .raw files from LC-MS/MS were loaded in the MaxQuant software (v1.6.7.0) for analysis. Identification of peptides was performed using the built-in Andromeda to search against the reviewed UniProt human proteome database (2019 release with a total of 42 427 entries, where 20 350 were reviewed and 22 077 were unreviewed). All of the search parameters were used in their default setting. The enzyme was set to trypsin, and maximum missed cleavages set to 2, while fixed modification was set to carbamidomethylation of cysteines and a false discovery rate (FDR) at peptide-to-spectrum matches (PSM) and protein levels set to 0.01. The resulting .txt output files from MaxQuant were loaded into R statistical environment (v4.0.2) and preprocessed before analyzing with DEP (differentially enrichment analysis of proteomic data) package.32,33 The DEP package offers a robust and reproducible analysis workflow for MS-based proteomics data when determining differentially enriched/expressed proteins. DEP filters out contaminant and reverse protein sequences, logarithmically transforms the data, and then normalizes the data by variance stabilizing normalization (vsn) method. Subsequently, it imputes missing values and runs statistical tests to determine proteins with significantly altered expression levels. The latter step is made feasible by the test_diff function, which performs a differential enrichment test based on protein-wise linear models and empirical Bayes statistics using limma.34,35 Data visualization was carried out using the BPG (v6.0.1)36 and BioVenn (v1.0.2)37 packages. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE38 partner repository with the data set identifier PXD020741.
2.7. Protein Annotation and Assessment of Physicochemical Aspects
The sequences of the identified proteins and gene ontology (GO) for protein annotation regarding cellular components were accessed and retrieved from UniProtKB.39 To calculate the grand average of hydropathy (GRAVY) value for the protein sequences, an online GRAVY calculator was used.40
3. Results and Discussion
3.1. Analyzing Barrett’s Esophageal Cell Samples by MicroPOTS
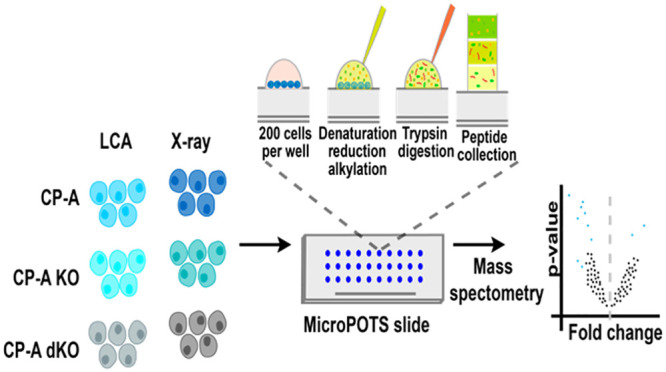
The field of proteomics is rapidly changing with new technologies and methods advancing the capacity to examine a larger proportion of the proteome from small numbers of cells even down to the single-cell41 level with a high degree of certainty. Over the past five years, the quest to design and develop highly sensitive and specific proteomic methodologies for this purpose has become intense, and many such methods have now been made available.42−44 Moving away from macroscale preparative techniques in proteomics to micro- and nanotechnologies offers researchers the ability to characterize smaller numbers of cells that are more likely to be encountered in clinical settings. The recently unveiled microPOTS system is one of the methodologies that are promising and poised to widen the window of possibilities in proteomics research.25 This study aimed at applying the microPOTS separation system to identify proteome signatures of either physiological stress or radiation in 200 Barrett’s esophageal cells. Initially, CP-A p53 single null (CP-A KO) and CP-A p53-SMAD4 double null cells (CP-A dKO) were generated, and together with the parental CP-A wild-type cells (CP-A WT), were subsequently subjected to either LCA or X-ray treatments. The microPOTS system was then applied for protein extraction and a nano-LC-MS workflow followed as depicted in Scheme 1.
Scheme 1. Overview of the Workflow of MicroPOTS for the Identification of Proteomic Changes in ∼200 CP-A Cells after Treatment with Either LCA or X-ray.

First, cells were grown in keratinocyte media, and the generated CP-A null cells and wild-type were subjected to different stressors as previously described. (a) Samples were then processed for protein extraction and further digested into peptides using the microPOTS system. (b) The collected peptides were subsequently subjected to LC-MS/MS analysis. (c) A spectrum showing the relative intensity and mass to charge ratio (m/z) of the ions being analyzed. (d) The resulting files were loaded into MaxQuant for peptide identification, after which the output files of this step were next imported into R environment and analyzed using the DEP package. (e) The results of the analysis were then visualized using the previously mentioned R packages.
3.2. Protein Identification from Different CP-A Genotypes and Stress Conditions
All the samples reported a high number of identified protein groups, with each replicate having over 1500 protein groups from only 200 cells. Replicates from each sample type were averaged and the mean value of protein groups plotted in a bar graph (Figure 1a). The average number of confidently identified protein groups for each sample type was as follows with average number of proteins identified in parentheses: CP-A WT (2273), CP-A KO (1673), CP-A dKO (2008), CP-A LCA-WT (1685), CP-A LCA-KO (2004), CP-A LCA-dKO (1821), CP-A X-ray-WT (2300), CP-A X-ray-dKO (2345), and CP-A X-ray-KO (2367). The total number of proteins obtained from three replicates increased to over 2000 for all sample types when results were pooled from three wells for each sample type (Figure 1b). Next, an intersection analysis was performed to assess the number of shared and uniquely identified proteins. From this comparison, it was shown that over 1800 (60%) proteins could be identified in all sample types. Comparison of the total proteins from three replicates of CP-A LCA-WT, CP-A LCA-KO, and CP-A LCA-dKO produced a high protein overlap of 67% with 1867 proteins (Figure 1c). Out of these 1867 proteins, 93 (3.4%), 185 (6.7%), and 195 (7%) were unique to LCA-WT, LCA-KO, and LCA-dKO, respectively. The X-ray treatment equally reported a high overlap of 2354 (77%) proteins between the groups (Figure 1d). Also, the WT, KO, and dKO samples were overlapped with their corresponding treatment groups (LCA and X-ray) to determine the number of shared proteins and proteins that were unique to each sample. There was a high overlap of greater than 60% for the number of identified proteins for each sample type (Figure S1). The number of identified proteins in this work is comparable to a previous study that performed proteomic analysis on small numbers of cells (∼100) on an LTQ-Orbitrap system, and identified ∼1500 proteins.45
Figure 1.

Barplots and Venn diagram plots respectively showing the number of identified protein groups and overlap for CP-A WT, CP-A KO, and CP-A dKO samples of ∼200 cells. (a) Average numbers of identified protein groups with each bar representing the mean and standard deviation (error bars) of replicates from individual sample type. (b) Total numbers of identified protein groups from three replicates for each sample type. (c) A 67% overlap of the identified proteins for samples treated with LCA. (d) Samples treated with X-ray shows a 77% overlap for the identified protein groups.
3.3. Evaluation of Protein Extraction Efficiency and Reproducibility
The results show that over 1500 proteins were confidently identified from 200 cells in all sample types. This high number of identified proteins from only 200 cells indicates the high extraction efficiency of the microPOTS system for microscale proteomic analysis. A look at the mean and standard deviations across the sample types reveals consistency in the number of identified proteins. As illustrated in Figure S2, almost all replicates had over 75% fully tryptic cleavage sites with fewer than 25% missed cleavages, indicating good tryptic digestion, which also translates into a high extraction efficiency.
The evaluation of microPOTS reproducibility in this study was predicated on two approaches—qualitative and quantitative analysis. Just like in the case of other method comparison studies46−48 that often use this kind of approach, we decided to use the strategy to assess the performance of microPOTS. The reproducibility of the measurements is important in evaluation of the results and correct identification of proteins is critical to discovering new proteomic signatures with high certainty and specificity.49−52 Qualitative reproducibility was achieved by comparing the overlap of the identified proteins between sample types and illustrating the shared proteins in an area-proportional Venn diagram. In Figure 2a, results for LCA treated CP-A dKO cells showed over 52% protein overlap between the replicates. Whereas for the X-ray treated CP-A dKO cells, an overlap of 73% was reported between replicates. Additionally, protein overlap between replicates for the rest of samples was assessed, and high protein overlap of over 50% was reported for almost all comparisons (Figure S3). Further, quantitative reproducibility between the replicates was assessed using the LFQ values of replicates to perform a pairwise Pearson’s correlation coefficient analysis. As shown in Figure 2b, the quantitative assessment of reproducibility demonstrated a high Pearson’s correlation coefficient value (R > 0.9). A high correlation coefficient (R > 0.9) was observed for almost all the pairwise comparisons that were conducted (Figure S4). CP-A KO replicates reported a lower correlation coefficient (R = 0.83) relative to the rest of the samples. Reproducibility of the MicroPOTS system was further assessed by computing the coefficient of variation (CV) for individual protein intensities in each sample condition (Figure 2c). All samples except CP-A KO showed little variation with a median CV that is less than 50% (Table S1).
Figure 2.

Qualitative and quantitative comparison of the identified proteins between replicates. (a) The area-proportional Venn diagrams illustrate the proportion of shared and unique proteins between technical replicates. (b) Scatter plots showing Pearson’s correlation coefficient between logarithmically (log10) transformed quantile normalized LFQ values of the corresponding replicates. (c) A boxplot representing the distribution of coefficient of variation for quantile normalized protein intensities across different experimental conditions. Each dot represents the CV of an individual protein intensity within each sample type.
3.4. Comparison of MicroPOTS Data to Bulk Proteomics Data Set
The microPOTS data were compared to an existing data set of the same cell line (CP-A). The samples that generated the bulk data were prepared and analyzed according to standard proteomic workflows as described in Box S1. The resulting data were processed and analyzed using bioinformatics tools as stated in Box S2. Next, we correlated all the overlapping proteins that were confidently identified between the two data sets using Pearson correlation method. As illustrated in the scatter plot in Figure S5, there was a high positive correlation of R = 0.625.
3.5. Comparison of Physicochemical Aspects of Identified Proteins
It has been established that high molecular weight (MW) and basic proteins are often challenging to extract due to their propensity to undergo intra- and intermolecular interactions.53,54 As such, we were interested in exploring the MW, and GRAVY distribution pattern of the identified proteins. The GRAVY distribution scores for LCA treated CP-A dKO cells indicated that a high number of hydrophobic proteins were detected with microPOTS (Figure 3a). A similar distribution trend of GRAVY was noticed from all the sample types with the majority of identified protein IDs within 0.4 to −0.2 range (Figure S6). LCA treated CP-A WT cells showed that the most abundant proteins had a MW between 20 and 30 kDa in all sample types (Figure 3b). Additionally, a similar MW distribution pattern was observed across all sample types, which demonstrates that the proteins show no major difference regarding their physicochemical characteristics (Figure S7). Next, the distribution of identified proteins according to their subcellular localization was explored, which showed that over 50% of the identified proteins resided within the cytosolic region, and this was observed across all sample types. Nearly 20% of confidently identified proteins for all the sample types were located within the cytosol (Figure 3c). All samples revealed that few ribosomal proteins could be detected by the microPOTS system, and this finding is consistent with Zhu et al.23 reported in their study. Ribosomal and cytoskeleton-derived proteins were the least likely to be identified with a percentage of less than 5%. The distribution pattern for subcellular proteins was almost the same across all sample types (Figure S8).
Figure 3.

Assessment of the physicochemical features and subcellular localization of the identified proteins. Distribution of (A) GRAVY in CP-A LCA-dKO, (B) molecular weight in CP-A LCA-WT, and (C) subcellular localization of identified proteins in CP-A X-ray-KO.
3.6. Effect of Stress on Protein Expression
Differential expression levels of the identified proteins were determined between CP-A WT, CP-A KO, and CP-A dKO as well as between their corresponding treatment set (LCA and X-ray treated groups). A pairwise comparison for all sample types was carried out to evaluate differentially expressed proteins between cells treated with different stresses. The findings from the present study indicate that LCA and X-ray induced changes in the proteome of CP-A cells, which is also consistent with the findings of Proungvitaya et al. that reported bile acids-induced alteration of protein expression in model cells system.55 Significant alterations were observed in CPA-dKO cells following LCA and X-ray treatment. Specifically, a pairwise comparison between CP-A LCA-dKO and CP-A dKo revealed that 21 proteins were upregulated, and 13 proteins were downregulated, some of which include ASNS, RALY, CSRP1, CTSD, FAM120A, ESTD, and GSR (Figure 4a). Also, CP-A X-ray-dKO and CP-A dKO comparison reported 32 upregulated proteins with 14 downregulated proteins, including NONO, SAR1A, HNRL2, PLEC.1, FARSA, S100P, FKBP3, and AGR2 (Figure 4b). Table S2 and Table S3 represent the complete list of proteins that were significantly differentially expressed for LCA and X-ray treated CP-A dKO cells, respectively. Anterior Gradient 2 (AGR2) is a member of the protein disulfide isomerase family, and its overexpression has been associated with many human cancers including neoplasia of esophagus.56,57 This evidence is consistent with the present study, which has shown that AGR2 is overexpressed in Barrett’s esophageal cells (CP-A X-ray-dKO). Having detected ∼1500 proteins from fewer than 200 cells, and capturing differential expression, signify a potential use of the microPOTS-LC-MS method to explore subcellular populations within a tissue/tumor microenvironment including T-cells, fibroblasts and macrophages.
Figure 4.

Volcano plots illustrating the fold difference in the expression levels of proteins in (a) CP-A LCA-dKO vs CP-A dKO and (b) CP-A X-ray-dKO vs CP-A dKO. The horizontal coordinate represents the log2 fold change difference, while the vertical coordinate represents the −log10p-value. Black color indicates proteins with nonsignificant changes in abundance, blue color represents significantly downregulated proteins, and red color represents significantly upregulated proteins.
Conclusion
In this study, a recently developed proteomic method called microPOTS was applied to identify proteins and determine the changes in the proteome of ∼200 cells (including an isogenic cell panel being used for the Barrett’s esophageal studies) following radiation and physiological stress treatment. The results show that the microPOTS method is applicable for use in qualitative and quantitative proteomic studies where only low cell numbers are available. Ionizing radiation was one stress used since it is DNA damaging and can activate p53 function. LCA was used since it is a component of bile acids that can impact on acid reflux disease and cancer progression in this tissue. The results were highly reproducible (R > 0.9) between replicates, allowing us to investigate confidently the effect of stress on the cells important in biological applications. With the microPOTS method ∼1500 unique proteins were quantified in all the samples. Moreover, results for the cells treated with LCA revealed differential expression analysis of 21 upregulated proteins and 13 downregulated proteins (CP-A LCA-dKO vs CP-A dKO), some of which include RALY, CSRP1, ASNS, ESTD, and FAM120A. Also, a comparison set between CP-A X-ray-dKO and CP-A dKO reported 33 significantly overexpressed proteins and 15 underexpressed proteins, including NONO, SKP1, HNRL2, PLEC.1, S100P, FKBP3, and AGR2. The results of the present study offer a basis for further studies to deeply interrogate in the future the molecular mechanisms that underpin LCA induction of proteome changes using clinical biopsies, which could aid in uncoupling the distinct role of bile acids in the selection for specific genetic mutations in esophageal adenocarcinoma progression. Importantly, the use of microPOTS, while not as sensitive as its companion technique nanoPOTS, was implemented and will be used in future studies.
Acknowledgments
This work was funded by the International Research Agenda’s Program of the Foundation for Polish Science (MAB/2017/03). The “International Centre for Cancer Vaccine Science” project is carried out within the International Research Agendas programme of the Foundation for Polish Science cofinanced by the European Union under the European Regional Development Fund. The authors would also like to thank the CI-TASK, Gdansk and the PL-Grid Infrastructure, Poland for providing their hardware and software resources. The University of Victoria-Genome BC Proteomics Centre is grateful to Genome Canada and Genome British Columbia for financial support for Genomics Technology Platforms (GTP) funding for operations and technology development (264PRO). The experiment was performed using EMSL (grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research and located at PNNL. The Brno CP-A study was supported by the European Regional Development Fund – Project ENOCH (No. CZ.02.1.01/0.0/0.0/16_019/0000868) and by the Ministry of Health Development of Research Organization, MH CZ – DRO (MMCI, 00209805).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.0c00629.
Figure S1: Venn diagram illustrating the shared number of identified proteins among different sample types, as well as proteins that are unique to each sample type; Figure S2: Number of missed cleavages are shown for all replicates and are expressed in percentage; Figure S3: Qualitative assessment of reproducibility of the microPOTS system; Figure S4: Quantitative assessment of reproducibility of the microPOTS system; Table S1: Median coefficient of variation (CV) for quantile normalized protein LFQ values for each sample type; Box S1: Sample preparation and LC-MS analysis for bulk proteomics data set; Box S2: Bulk data analysis and comparison to microPOTS data; Figure S5: Scatter plot with associated Pearson’s correlation coefficient (R = 0.625) between microPOTS and bulk proteomics for all overlapping 1066 proteins that were identified; Figure S6: Assessment of physicochemical characteristics; Figure S7: Assessment of physicochemical characteristics; Figure S8: Subcellular localization; Table S2: List of differentially expressed proteins between CP-A dKO cells with or without LCA treatment; Table S3: List of differentially expressed proteins between CP-A dKO cells with or without X-ray treatment (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Mann M.; Hendrickson R. C.; Pandey A. Analysis of Proteins and Proteomes by Mass Spectrometry. Annu. Rev. Biochem. 2001, 70, 437–473. 10.1146/annurev.biochem.70.1.437. [DOI] [PubMed] [Google Scholar]
- Wasinger V. C.; Cordwell S. J.; Cerpa-Poljak A.; Yan J. X.; Gooley A. A.; Wilkins M. R.; Duncan M. W.; Harris R.; Williams K. L.; Humphery-Smith I. Progress with Gene-Product Mapping of the Mollicutes: Mycoplasma Genitalium. Electrophoresis 1995, 16 (7), 1090–1094. 10.1002/elps.11501601185. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Fonslow B. R.; Shan B.; Baek M.-C.; Yates J. R. Protein Analysis by Shotgun/Bottom-up Proteomics. Chem. Rev. 2013, 113 (4), 2343–2394. 10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayne J.; Ning Z.; Zhang X.; Starr A. E.; Chen R.; Deeke S.; Chiang C.-K.; Xu B.; Wen M.; Cheng K.; Seebun D.; Star A.; Moore J. I.; Figeys D. Bottom-Up Proteomics (2013–2015): Keeping up in the Era of Systems Biology. Anal. Chem. 2016, 88 (1), 95–121. 10.1021/acs.analchem.5b04230. [DOI] [PubMed] [Google Scholar]
- Fagerberg L.; Strömberg S.; El-Obeid A.; Gry M.; Nilsson K.; Uhlen M.; Ponten F.; Asplund A. Large-Scale Protein Profiling in Human Cell Lines Using Antibody-Based Proteomics. J. Proteome Res. 2011, 10 (9), 4066–4075. 10.1021/pr200259v. [DOI] [PubMed] [Google Scholar]
- Branca R. M. M.; Orre L. M.; Johansson H. J.; Granholm V.; Huss M.; Pérez-Bercoff Å.; Forshed J.; Käll L.; Lehtiö J. HiRIEF LC-MS Enables Deep Proteome Coverage and Unbiased Proteogenomics. Nat. Methods 2014, 11 (1), 59–62. 10.1038/nmeth.2732. [DOI] [PubMed] [Google Scholar]
- Wang G.; Brennan C.; Rook M.; Wolfe J. L.; Leo C.; Chin L.; Pan H.; Liu W.-H.; Price B.; Makrigiorgos G. M. Balanced-PCR Amplification Allows Unbiased Identification of Genomic Copy Changes in Minute Cell and Tissue Samples. Nucleic Acids Res. 2004, 32 (9), e76. 10.1093/nar/gnh070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N.; Xu M.; Wang P.; Li L. Development of Mass Spectrometry-Based Shotgun Method for Proteome Analysis of 500 to 5000 Cancer Cells. Anal. Chem. 2010, 82 (6), 2262–2271. 10.1021/ac9023022. [DOI] [PubMed] [Google Scholar]
- Parapatics K.; Huber M. L.; Lehmann D.; Knoll C.; Superti-Furga G.; Bennett K. L.; Rudashevskaya E. L. Proteomic Analysis of Low Quantities of Cellular Material in the Range Obtainable from Scarce Patient Samples. J. Integr. OMICS 2015, 5 (1), 30–43–43. 10.5584/jiomics.v5i1.172. [DOI] [Google Scholar]
- Clair G.; Piehowski P. D.; Nicola T.; Kitzmiller J. A.; Huang E. L.; Zink E. M.; Sontag R. L.; Orton D. J.; Moore R. J.; Carson J. P.; Smith R. D.; Whitsett J. A.; Corley R. A.; Ambalavanan N.; Ansong C. Spatially-Resolved Proteomics: Rapid Quantitative Analysis of Laser Capture Microdissected Alveolar Tissue Samples. Sci. Rep. 2016, 6, 6. 10.1038/srep39223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coscia F.; Doll S.; Bech J. M.; Mund A.; Lengyel E.; Lindebjerg J.; Madsen G. I.; Moreira J. M. A.; Mann M.. A Streamlined Mass Spectrometry-Based Proteomics Workflow for Large Scale FFPE Tissue Analysis. bioRxiv, Sep 23, 2019, 779009. 10.1101/779009. [DOI] [PubMed]
- Martin J. G.; Rejtar T.; Martin S. A. Integrated Microscale Analysis System for Targeted Liquid Chromatography Mass Spectrometry Proteomics on Limited Amounts of Enriched Cell Populations. Anal. Chem. 2013, 85 (22), 10680–10685. 10.1021/ac401937c. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H.; Miyazaki M.; Honda T.; Briones-Nagata M. P.; Arima K.; Maeda H. Rapid and Efficient Proteolysis for Proteomic Analysis by Protease-Immobilized Microreactor. Electrophoresis 2009, 30 (18), 3257–3264. 10.1002/elps.200900134. [DOI] [PubMed] [Google Scholar]
- Myers S. A.; Rhoads A.; Cocco A. R.; Peckner R.; Haber A. L.; Schweitzer L. D.; Krug K.; Mani D. R.; Clauser K. R.; Rozenblatt-Rosen O.; Hacohen N.; Regev A.; Carr S. A. Streamlined Protocol for Deep Proteomic Profiling of FAC-Sorted Cells and Its Application to Freshly Isolated Murine Immune Cells. Mol. Cell. Proteomics 2019, 18 (5), 995–1009. 10.1074/mcp.RA118.001259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W.; Li H.; Liu L.; Huang P.; Wang Z.; Chen W.; Ye M.; Yu X.; Tian R. An Integrated Strategy for High-Sensitive and Multi-Level Glycoproteome Analysis from Low Micrograms of Protein Samples. J. Chromatogr. A 2019, 1600, 46–54. 10.1016/j.chroma.2019.04.041. [DOI] [PubMed] [Google Scholar]
- Chen W.; Wang S.; Adhikari S.; Deng Z.; Wang L.; Chen L.; Ke M.; Yang P.; Tian R. Simple and Integrated Spintip-Based Technology Applied for Deep Proteome Profiling. Anal. Chem. 2016, 88 (9), 4864–4871. 10.1021/acs.analchem.6b00631. [DOI] [PubMed] [Google Scholar]
- Dou M.; Zhu Y.; Liyu A.; Liang Y.; Chen J.; Piehowski P. D.; Xu K.; Zhao R.; Moore R. J.; Atkinson M. A.; Mathews C. E.; Qian W.-J.; Kelly R. T. Nanowell-Mediated Two-Dimensional Liquid Chromatography Enables Deep Proteome Profiling of < 1000 Mammalian Cells. Chem. Sci. 2018, 9 (34), 6944–6951. 10.1039/C8SC02680G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Scheibinger M.; Ellwanger D. C.; Krey J. F.; Choi D.; Kelly R. T.; Heller S.; Barr-Gillespie P. G. Single-Cell Proteomics Reveals Changes in Expression during Hair-Cell Development. eLife 2019, 8, e50777. 10.7554/eLife.50777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao X.; Zhang X. Design of Five-Layer Gold Nanoparticles Self-Assembled in a Liquid Open Tubular Column for Ultrasensitive Nano-LC-MS/MS Proteomic Analysis of 80 Living Cells. Proteomics 2017, 17 (8), 1600463. 10.1002/pmic.201600463. [DOI] [PubMed] [Google Scholar]
- Chen Q.; Yan G.; Gao M.; Zhang X. Ultrasensitive Proteome Profiling for 100 Living Cells by Direct Cell Injection, Online Digestion and Nano-LC-MS/MS Analysis. Anal. Chem. 2015, 87 (13), 6674–6680. 10.1021/acs.analchem.5b00808. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Clair G.; Chrisler W. B.; Shen Y.; Zhao R.; Shukla A. K.; Moore R. J.; Misra R. S.; Pryhuber G. S.; Smith R. D.; Ansong C.; Kelly R. T. Proteomic Analysis of Single Mammalian Cells Enabled by Microfluidic Nanodroplet Sample Preparation and Ultrasensitive NanoLC-MS. Angew. Chem., Int. Ed. 2018, 57 (38), 12370–12374. 10.1002/anie.201802843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao X.; Wang X.; Guan S.; Lin H.; Yan G.; Gao M.; Deng C.; Zhang X. Integrated Proteome Analysis Device for Fast Single-Cell Protein Profiling. Anal. Chem. 2018, 90 (23), 14003–14010. 10.1021/acs.analchem.8b03692. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Piehowski P. D.; Zhao R.; Chen J.; Shen Y.; Moore R. J.; Shukla A. K.; Petyuk V. A.; Campbell-Thompson M.; Mathews C. E.; Smith R. D.; Qian W.-J.; Kelly R. T. Nanodroplet Processing Platform for Deep and Quantitative Proteome Profiling of 10–100 Mammalian Cells. Nat. Commun. 2018, 9 (1), 882. 10.1038/s41467-018-03367-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Uwugiaren N.; Williams S. M.; Moore R. J.; Zhao R.; Goodlett D.; Dapic I.; Paša-Tolić L.; Zhu Y. Sensitive Top-Down Proteomics Analysis of a Low Number of Mammalian Cells Using a Nanodroplet Sample Processing Platform. Anal. Chem. 2020, 92 (10), 7087–7095. 10.1021/acs.analchem.0c00467. [DOI] [PubMed] [Google Scholar]
- Xu K.; Liang Y.; Piehowski P. D.; Dou M.; Schwarz K. C.; Zhao R.; Sontag R. L.; Moore R. J.; Zhu Y.; Kelly R. T. Benchtop-Compatible Sample Processing Workflow for Proteome Profiling of < 100 Mammalian Cells. Anal. Bioanal. Chem. 2019, 411 (19), 4587–4596. 10.1007/s00216-018-1493-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes C. S.; Becq J.; Warren A.; Cheetham R. K.; Northen H.; O’Donovan M.; Malhotra S.; di Pietro M.; Ivakhno S.; He M.; Weaver J. M. J.; Lynch A. G.; Kingsbury Z.; Ross M.; Humphray S.; Bentley D.; Fitzgerald R. C. Whole-Genome Sequencing Provides New Insights into the Clonal Architecture of Barrett’s Esophagus and Esophageal Adenocarcinoma. Nat. Genet. 2015, 47 (9), 1038–1046. 10.1038/ng.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters Y.; Al-Kaabi A.; Shaheen N. J.; Chak A.; Blum A.; Souza R. F.; Di Pietro M.; Iyer P. G.; Pech O.; Fitzgerald R. C.; Siersema P. D. Barrett Oesophagus. Nat. Rev. Dis. Primer 2019, 5 (1), 35. 10.1038/s41572-019-0086-z. [DOI] [PubMed] [Google Scholar]
- Hofmann A. F. The Continuing Importance of Bile Acids in Liver and Intestinal Disease. Arch. Intern. Med. 1999, 159 (22), 2647–2658. 10.1001/archinte.159.22.2647. [DOI] [PubMed] [Google Scholar]
- Fitzgerald R. C.; di Pietro M.; O’Donovan M.; Maroni R.; Muldrew B.; Debiram-Beecham I.; Gehrung M.; Offman J.; Tripathi M.; Smith S. G.; Aigret B.; Walter F. M.; Rubin G.; Bagewadi A.; Patrick A.; Shenoy A.; Redmond A.; Muddu A.; Northrop A.; Groves A.; Shiner A.; Heer A.; Takhar A.; Bowles A.; Jarman A.; Wong A.; Lucas A.; Gibbons A.; Dhar A.; Curry A.; Lalonde A.; Swinburn A.; Turner A.; Lydon A.-M.; Gunstone A.; Lee A.; Nambi A.; Ariyarathenam A.; Elden A.; Wilson A.; Donepudi B.; Campbell B.; Uszycka B.; Bowers B.; Coghill B.; de Quadros B.; Cheah C.; Bratten C.; Brown C.; Moorbey C.; Clisby C.; Gordon C.; Schramm C.; Castle C.; Newark C.; Norris C.; A’Court C.; Graham C.; Fletcher C.; Grocott C.; Rees C.; Bakker C.; Paschalides C.; Vickery C.; Schembri D.; Morris D.; Hagan D.; Cronk D.; Goddard D.; Graham D.; Phillips D.; Prabhu D.; Kejariwal D.; Garg D.; Lonsdale D.; Butterworth D.; Clements D.; Bradman D.; Blake D.; Mather E.; O’Farrell E.; Markowetz F.; Adams F.; Pesola F.; Forbes G.; Taylor G.; Collins G.; Irvine G.; Fourie G.; Doyle H.; Barnes H.; Bowyer H.; Whiting H.; Beales I.; Binnian I.; Bremner I.; Jennings I.; Troiceanu I.; Modelell I.; Emmerson I.; Ortiz J.; Lilley J.; Harvey J.; Vicars J.; Takhar J.; Larcombe J.; Bornschein J.; Aldegather J.; Johnson J.; Ducker J.; Skinner J.; Dash J.; Walsh J.; Miralles J.; Ridgway J.; Ince J.; Kennedy J.; Hampson K.; Milne K.; Ellerby K.; Priddis K.; Rainsbury K.; Powell K.; Gunner K.; Ragunath K.; Knox K.; Baseley L.; White L.; Lovat L.; Berney L.; Crockett L.; Murray L.; Westwood L.; Chalkley L.; Leggett L.; Dale L.; Scovell L.; Brooks L.; Saunders L.; Owen L.; Dilwershah M.; Baldry M.; Corcoran M.; Roy M.; Macedo M.; Attah M.; Anson M.-J.; Rutter M.; Wallard M.; Gaw M.; Hunt M.; Lea-Hagerty M.; Penacerrada M.; Bianchi M.; Baker-Moffatt M.; Czajkowski M.; Sleeth M.; Brewer N.; Wooding N.; Todd N.; Millen N.; Zolle O.; Whitehead O.; Ojechi P.; Moore P.; Banim P.; Spellar P.; Bhandari P.; Kant P.; Nixon R.; Russell R.; Roberts R.; Skule R.; West R.; Fox R.; Beesley R.; Gibbins R.; Osborne R.; Thiagarajan S.; Bastiman S.; Warburton S.; Pai S.; Leith-Russell S.; Utting S.; Watson S.; Wytrykowski S.; Singh S.; Malhotra S.; Woods S.; Conway S.; Mateer S.; Macrae S.; Singh S.; Fourie S.; Campbell S.; Parslow-Williams S.; Goel S.; Dellar S.; Jones S.; Knight S.; Mackay-Thomas S.; Mukherjee S.; Allen S.; Henry S.; Evans T.; Leighton T.; Bray T.; Shackleton T.; Santosh V.; Glover V.; Chandraraj V.; Elson W.; Briggs W.; Barron Z.; Khan Z.; Sasieni P. Cytosponge-Trefoil Factor 3 versus Usual Care to Identify Barrett’s Oesophagus in a Primary Care Setting: A Multicentre, Pragmatic, Randomised Controlled Trial. Lancet 2020, 396 (10247), 333–344. 10.1016/S0140-6736(20)31099-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes C. S.; Chettouh H.; Achilleos A.; Galeano-Dalmau N.; Debiram-Beecham I.; MacRae S.; Fessas P.; Walker E.; Varghese S.; Evan T.; Lao-Sirieix P. S.; O’Donovan M.; Malhotra S.; Novelli M.; Disep B.; Kaye P. V.; Lovat L. B.; Haidry R.; Griffin M.; Ragunath K.; Bhandari P.; Haycock A.; Morris D.; Attwood S.; Dhar A.; Rees C.; Rutter M. D.; Ostler R.; Aigret B.; Sasieni P. D.; Fitzgerald R. C. Risk Stratification of Barrett’s Oesophagus Using a Non-Endoscopic Sampling Method Coupled with a Biomarker Panel: A Cohort Study. Lancet Gastroenterol. Hepatol. 2017, 2 (1), 23–31. 10.1016/S2468-1253(16)30118-2. [DOI] [PubMed] [Google Scholar]
- Williams S. M.; Liyu A. V.; Tsai C.-F.; Moore R. J.; Orton D. J.; Chrisler W. B.; Gaffrey M. J.; Liu T.; Smith R. D.; Kelly R. T.; Pasa-Tolic L.; Zhu Y. Automated Coupling of Nanodroplet Sample Preparation with Liquid Chromatography–Mass Spectrometry for High-Throughput Single-Cell Proteomics. Anal. Chem. 2020, 92 (15), 10588–10596. 10.1021/acs.analchem.0c01551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEP: Differential Enrichment analysis of Proteomics data version 1.10.0 from Bioconductor, https://rdrr.io/bioc/DEP/ (accessed Jul 4, 2020).
- Zhang X.; Smits A. H.; van Tilburg G. B.; Ovaa H.; Huber W.; Vermeulen M. Proteome-Wide Identification of Ubiquitin Interactions Using UbIA-MS. Nat. Protoc. 2018, 13 (3), 530–550. 10.1038/nprot.2017.147. [DOI] [PubMed] [Google Scholar]
- test_diff: Differential enrichment test in DEP: Differential Enrichment analysis of Proteomics data, https://rdrr.io/bioc/DEP/man/test_diff.html (accessed Jul 5, 2020).
- Ritchie M. E.; Phipson B.; Wu D.; Hu Y.; Law C. W.; Shi W.; Smyth G. K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43 (7), e47–e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- P’ng C.; Green J.; Chong L. C.; Waggott D.; Prokopec S. D.; Shamsi M.; Nguyen F.; Mak D. Y. F.; Lam F.; Albuquerque M. A.; Wu Y.; Jung E. H.; Starmans M. H. W.; Chan-Seng-Yue M. A.; Yao C. Q.; Liang B.; Lalonde E.; Haider S.; Simone N. A.; Sendorek D.; Chu K. C.; Moon N. C.; Fox N. S.; Grzadkowski M. R.; Harding N. J.; Fung C.; Murdoch A. R.; Houlahan K. E.; Wang J.; Garcia D. R.; de Borja R.; Sun R. X.; Lin X.; Chen G. M.; Lu A.; Shiah Y.-J.; Zia A.; Kearns R.; Boutros P. C. BPG: Seamless, Automated and Interactive Visualization of Scientific Data. BMC Bioinf. 2019, 20 (1), 42. 10.1186/s12859-019-2610-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsen T.; de Vlieg J.; Alkema W. BioVenn – a Web Application for the Comparison and Visualization of Biological Lists Using Area-Proportional Venn Diagrams. BMC Genomics 2008, 9 (1), 488. 10.1186/1471-2164-9-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y.; Csordas A.; Bai J.; Bernal-Llinares M.; Hewapathirana S.; Kundu D. J.; Inuganti A.; Griss J.; Mayer G.; Eisenacher M.; Pérez E.; Uszkoreit J.; Pfeuffer J.; Sachsenberg T.; Yılmaz Ş.; Tiwary S.; Cox J.; Audain E.; Walzer M.; Jarnuczak A. F.; Ternent T.; Brazma A.; Vizcaíno J. A. The PRIDE Database and Related Tools and Resources in 2019: Improving Support for Quantification Data. Nucleic Acids Res. 2019, 47 (D1), D442–D450. 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt , https://www.uniprot.org/ (accessed Jun 25, 2020).
- Protein GRAVY, https://www.bioinformatics.org/sms2/protein_gravy.html (accessed Jun 25, 2020).
- Budnik B.; Levy E.; Harmange G.; Slavov N. SCoPE-MS: Mass Spectrometry of Single Mammalian Cells Quantifies Proteome Heterogeneity during Cell Differentiation. Genome Biol. 2018, 19 (1), 161. 10.1186/s13059-018-1547-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou M.; Clair G.; Tsai C.-F.; Xu K.; Chrisler W. B.; Sontag R. L.; Zhao R.; Moore R. J.; Liu T.; Pasa-Tolic L.; Smith R. D.; Shi T.; Adkins J. N.; Qian W.-J.; Kelly R. T.; Ansong C.; Zhu Y. High-Throughput Single Cell Proteomics Enabled by Multiplex Isobaric Labelling in a Nanodroplet Sample Preparation Platform. Anal. Chem. 2019, 91 (20), 13119–13127. 10.1021/acs.analchem.9b03349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng S. S. H.; Demir F.; Ergin E. K.; Dirnberger S.; Uzozie A.; Tuscher D.; Nierves L.; Tsui J.; Huesgen P. F.; Lange P. F. Sensitive Determination of Proteolytic Proteoforms in Limited Microscale Proteome Samples. Mol. Cell. Proteomics 2019, 18 (11), 2335–2347. 10.1074/mcp.TIR119.001560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasuga K.; Katoh Y.; Nagase K.; Igarashi K. Microproteomics with Microfluidic-Based Cell Sorting: Application to 1000 and 100 Immune Cells. Proteomics 2017, 17 (13–14), 1600420. 10.1002/pmic.201600420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J.; Zhang G. L.; Li S.; Ivanov A. R.; Fenyo D.; Lisacek F.; Murthy S. K.; Karger B. L.; Brusic V. Pathway Analysis and Transcriptomics Improve Protein Identification by Shotgun Proteomics from Samples Comprising Small Number of Cells - a Benchmarking Study. BMC Genomics 2014, 15 (Suppl 9), S1. 10.1186/1471-2164-15-S9-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanca A.; Abbondio M.; Pisanu S.; Pagnozzi D.; Uzzau S.; Addis M. F. Critical Comparison of Sample Preparation Strategies for Shotgun Proteomic Analysis of Formalin-Fixed, Paraffin-Embedded Samples: Insights from Liver Tissue. Clin. Proteomics 2014, 11 (1), 28. 10.1186/1559-0275-11-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Föll M. C.; Fahrner M.; Oria V. O.; Kühs M.; Biniossek M. L.; Werner M.; Bronsert P.; Schilling O. Reproducible Proteomics Sample Preparation for Single FFPE Tissue Slices Using Acid-Labile Surfactant and Direct Trypsinization. Clin. Proteomics 2018, 15 (1), 11. 10.1186/s12014-018-9188-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broeckx V.; Boonen K.; Pringels L.; Sagaert X.; Prenen H.; Landuyt B.; Schoofs L.; Maes E. Comparison of Multiple Protein Extraction Buffers for GeLC-MS/MS Proteomic Analysis of Liver and Colon Formalin-Fixed, Paraffin-Embedded Tissues. Mol. BioSyst. 2016, 12 (2), 553–565. 10.1039/C5MB00670H. [DOI] [PubMed] [Google Scholar]
- Tabb D. L.; Vega-Montoto L.; Rudnick P. A.; Variyath A. M.; Ham A.-J. L.; Bunk D. M.; Kilpatrick L. E.; Billheimer D. D.; Blackman R. K.; Cardasis H. L.; Carr S. A.; Clauser K. R.; Jaffe J. D.; Kowalski K. A.; Neubert T. A.; Regnier F. E.; Schilling B.; Tegeler T. J.; Wang M.; Wang P.; Whiteaker J. R.; Zimmerman L. J.; Fisher S. J.; Gibson B. W.; Kinsinger C. R.; Mesri M.; Rodriguez H.; Stein S. E.; Tempst P.; Paulovich A. G.; Liebler D. C.; Spiegelman C. Repeatability and Reproducibility in Proteomic Identifications by Liquid Chromatography–Tandem Mass Spectrometry. J. Proteome Res. 2010, 9 (2), 761–776. 10.1021/pr9006365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmotte N.; Lasaosa M.; Tholey A.; Heinzle E.; van Dorsselaer A.; Huber C. G. Repeatability of Peptide Identifications in Shotgun Proteome Analysis Employing Off-Line Two-Dimensional Chromatographic Separations and Ion-Trap MS. J. Sep. Sci. 2009, 32 (8), 1156–1164. 10.1002/jssc.200800615. [DOI] [PubMed] [Google Scholar]
- Resing K. A.; Meyer-Arendt K.; Mendoza A. M.; Aveline-Wolf L. D.; Jonscher K. R.; Pierce K. G.; Old W. M.; Cheung H. T.; Russell S.; Wattawa J. L.; Goehle G. R.; Knight R. D.; Ahn N. G. Improving Reproducibility and Sensitivity in Identifying Human Proteins by Shotgun Proteomics. Anal. Chem. 2004, 76 (13), 3556–3568. 10.1021/ac035229m. [DOI] [PubMed] [Google Scholar]
- Washburn M. P.; Ulaszek R. R.; Yates J. R. Reproducibility of Quantitative Proteomic Analyses of Complex Biological Mixtures by Multidimensional Protein Identification Technology. Anal. Chem. 2003, 75 (19), 5054–5061. 10.1021/ac034120b. [DOI] [PubMed] [Google Scholar]
- Magdeldin S.; Yamamoto T. Toward Deciphering Proteomes of Formalin-Fixed Paraffin-Embedded (FFPE) Tissues. Proteomics 2012, 12 (7), 1045–1058. 10.1002/pmic.201100550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes E.; Broeckx V.; Mertens I.; Sagaert X.; Prenen H.; Landuyt B.; Schoofs L. Analysis of the Formalin-Fixed Paraffin-Embedded Tissue Proteome: Pitfalls, Challenges, and Future Prospectives. Amino Acids 2013, 45 (2), 205–218. 10.1007/s00726-013-1494-0. [DOI] [PubMed] [Google Scholar]
- Proungvitaya S.; Klinthong W.; Proungvitaya T.; Limpaiboon T.; Jearanaikoon P.; Roytrakul S.; Wongkham C.; Nimboriboonporn A.; Wongkham S. High Expression of CCDC25 in Cholangiocarcinoma Tissue Samples. Oncol. Lett. 2017, 14 (2), 2566–2572. 10.3892/ol.2017.6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brychtova V.; Vojtesek B.; Hrstka R. Anterior Gradient 2: A Novel Player in Tumor Cell Biology. Cancer Lett. 2011, 304 (1), 1–7. 10.1016/j.canlet.2010.12.023. [DOI] [PubMed] [Google Scholar]
- Suwanmanee G.; Yosudjai J.; Phimsen S.; Wongkham S.; Jirawatnotai S.; Kaewkong W. Upregulation of AGR2vH Facilitates Cholangiocarcinoma Cell Survival under Endoplasmic Reticulum Stress via the Activation of the Unfolded Protein Response Pathway. Int. J. Mol. Med. 2019, 45 (2), 669–677. 10.3892/ijmm.2019.4432. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.