

Abstract
Background & Aims:
Primary sclerosing cholangitis (PSC) is a rare, cholestatic liver disease with no currently approved therapies. Obeticholic acid (OCA) is a potent farnesoid X receptor (FXR) agonist approved for the treatment of primary biliary cholangitis. We investigated the efficacy and safety of OCA in patients with PSC.
Methods:
AESOP was a phase II, randomized, double-blind, placebo-controlled, dose-finding study. Eligible patients were 18 to 75 years of age with a diagnosis of PSC and serum alkaline phosphatase (ALP) ≥2× the upper limit of normal (ULN) and total bilirubin <2.5× ULN. Patients were randomized 1:1:1 to receive placebo, OCA 1.5–3.0 mg, or OCA 5–10 mg once daily for a 24-week, double-blind phase followed by a 2-year, long-term safety extension (LTSE). Primary endpoints were change in ALP from baseline to week 24, and safety.
Results:
The intent-to-treat population comprised 76 patients randomized to placebo (n = 25), OCA 1.5–3.0 mg (n = 25), and OCA 5–10 mg (n = 26). At week 24, serum ALP was significantly reduced with OCA 5–10 mg vs. placebo (least-square [LS] mean difference = −83.4 [SE = 40.3] U/L; 95% CI −164.28 to −2.57; p = 0.043). Serum ALP was not significantly reduced with OCA 1.5–3.0 mg vs. placebo at week 24 (LS mean [SE] difference = −78.29 [41.81] U/L; 95% CI −162.08 to 5.50; p = 0.067). Total bilirubin remained comparable to baseline in all groups. The most common treatment-emergent adverse event was dose-related pruritus (placebo 46%; OCA 1.5–3.0 mg 60%; OCA 5–10 mg 67%). Reductions in ALP were maintained during the LTSE, and no new safety signals emerged.
Conclusions:
Treatment with OCA 5–10 mg reduced serum ALP in patients with PSC. Mild to moderate dose-related pruritus was the most common adverse event.
Registration:
ClinicalTrials.gov: NCT02177136; EudraCT: 2014-002205-38.
Keywords: Cholestasis, Farnesoid X receptor, Ursodeoxycholic acid
Lay summary
Primary sclerosing cholangitis (PSC) is a long-term disease that damages the bile ducts in the liver over time. In the AESOP clinical study in patients with PSC, obeticholic acid reduced serum alkaline phosphatase (a potential marker of disease severity) during an initial 24-week treatment period. The result was sustained during the 2-year, long-term extension of the study. The most common side effect of obeticholic acid in the study was itchy skin, which is consistent with earlier clinical studies.
Graphical Abstract
Introduction
Primary sclerosing cholangitis (PSC) is a rare, life-threatening, progressive, cholestatic liver disease in which inflammation and fibrosis lead to the destruction of intrahepatic and extrahepatic bile ducts with eventual onset of cirrhosis and end-stage liver disease.1 Diagnosis of PSC is based on a combination of biochemical and radiological criteria.2,3 Serum alkaline phosphatase (ALP) is elevated in most patients, consistent with cholestasis.1 Compared with ALP levels in primary biliary cholangitis (PBC), ALP levels in PSC are highly variable and fluctuate on the individual level.1 Using data from 1,001 patients with PSC, the UK-PSC consortium reported that serum ALP <2.4× upper limit of normal (ULN) at 1 year after diagnosis was associated with improved transplant-free survival,4 and other studies have reported associations between reduction in serum ALP and longer survival.5–7
Currently, there are no approved pharmacological therapies for PSC, and liver transplantation is the only therapeutic alternative demonstrated to improve clinical outcomes.3 Ursodeoxycholic acid (UDCA) has been used to treat PSC, but the results are mixed:3 treatment with UDCA has led to improved serum liver biochemistry, and similar results have recently been reported with the side-chain shortened derivative norUDCA;8 however, there is currently no evidence that UDCA alters the clinical course of PSC or prolongs survival.9–11 Therefore, effective systemic therapies for PSC represent a substantial unmet medical need.
The farnesoid X receptor (FXR) is an essential regulator of bile acid homeostasis; FXR activation is associated with release of fibroblast growth factor 19 (FGF19), decreased bile acid synthesis, and improved choleresis.12–14 In recent phase II studies in patients with PSC, the nonsteroidal FXR agonist cilofexor improved markers of cholestasis and liver biochemistry,15 and NGM282 (an FGF19 analog) inhibited bile acid synthesis and decreased markers of fibrosis.14
Obeticholic acid (OCA) is a selective and potent FXR agonist. In phase II and III clinical studies in patients with PBC, treatment with OCA resulted in significant reductions in ALP, a marker of hepatic damage and inflammation which is associated with long-term clinical outcomes in PBC.13,16,17 Currently, OCA is approved for the treatment of PBC in combination with UDCA in adults with an inadequate response to UDCA, or as monotherapy in patients unable to tolerate UDCA.18
Recent phase II studies have used serum ALP as an efficacy endpoint for the assessment of novel therapies in PSC, and a study of UDCA to improve ALP has been proposed by the American College of Gastroenterology guidance on PSC.3,8,15 Based on the known biological effects of FXR activation12,14 and the efficacy of OCA for treating PBC,13 OCA has been selected to be studied in patients with PSC. The objectives of this phase II study were to evaluate the effect of OCA on cholestatic markers and safety in patients with PSC.
Patients and methods
Study design and treatment
AESOP (Assessment of Efficacy and Safety of OCA in PSC) was a randomized, double-blind (DB), placebo-controlled, dose-finding study evaluating the efficacy and safety of OCA in patients with PSC, followed by an open-label, long-term safety extension (LTSE) (Fig. 1). Eligible patients were randomly assigned 1:1:1 to 3 treatment groups using an interactive web response system (IWRS): OCA 1.5 mg titrating to 3.0 mg (hereafter referred to as OCA 1.5–3.0 mg); OCA 5 mg titrating to 10 mg (hereafter referred to as OCA 5–10 mg); or placebo. Allocation was concealed by the use of matching tablets. Randomization was stratified by concomitant UDCA use (yes or no) and total bilirubin value (≤1.5× ULN, or >1.5× ULN). Study drug and placebo were administered orally, once daily. For the first 12 weeks, patients received OCA 1.5 mg, OCA 5 mg, or placebo. After 12 weeks, providing there were no limiting safety or tolerability concerns, the dose was up titrated from 1.5 to 3.0 mg (OCA 1.5–3.0 mg group), 5 to 10 mg (OCA 5–10 mg group), and the placebo group remained on placebo. The DB treatment was continued for a further 12 weeks at the titrated dose, for a total of 24 weeks treatment.
Fig. 1. Study design.
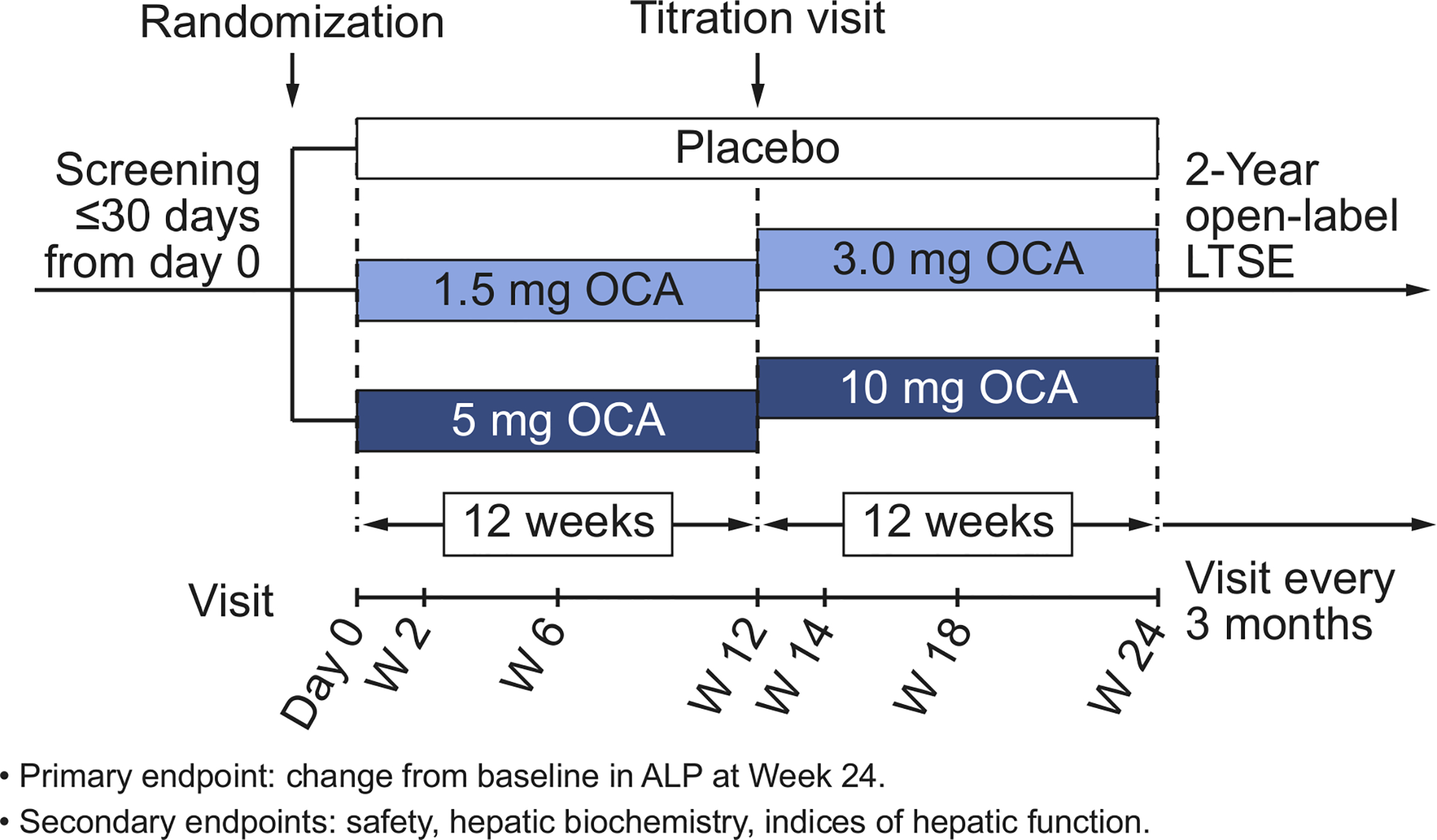
ALP, alkaline phosphatase; LTSE, long-term safety extension; OCA, obeticholic acid.
Following completion of the double-blind (DB) phase, patients reconsented to the LTSE, with visits scheduled every 3 months for up to an additional 24 months. The end of treatment visit (week 24) in the DB phase was used as the day 1 visit for the LTSE. The study blind was broken to assign the starting OCA dose for the LTSE. All patients, including those who had received placebo during the DB phase, were planned to start OCA treatment at 5 mg, except for those who completed treatment in the DB phase with OCA 10 mg; these patients continued with the same dose unless safety and tolerability warranted a reduction to 5 mg. Patients could titrate to higher doses of OCA, at a frequency not greater than every 3 months, up to a maximum dose of 10 mg daily. The study was terminated early for administrative reasons before the majority of patients completed the LTSE.
Patient population
Eligible patients were males or females 18 to 75 years of age with a diagnosis of PSC based on cholangiography. At screening, all patients had serum ALP ≥2× ULN and total bilirubin <2.5× ULN. Concomitant therapy with UDCA was permitted in a maximum of 50% of the randomized population. For patients receiving UDCA, the dose needed to be stable for ≥3 months prior to and including day 0 and must not have exceeded 20 mg/kg/day. Key exclusion criteria were evidence of a secondary cause of sclerosing cholangitis (e.g., immunoglobulin G4 [IgG4] >4× ULN or evidence of IgG4-assoriated cholangitis), clinical complications of chronic liver disease or clinically significant hepatic decompensation (e.g., Child-Pugh classification B or C), clinically relevant dominant strictures, or history of other chronic liver diseases. Patients were stratified by UDCA use and total bilirubin.
The study protocol was approved by regulatory authorities and institutional review boards or ethics committees. All patients provided written informed consent prior to study entry. The study was conducted in accordance with the European Union Clinical Trials Directive (2001/20/EC and subsequent amendments), 21CFR Part 312, Good Clinical Practice (Committee for Proprietary Medicinal Products/International Conference on Harmonisation/135/95), and the Declaration of Helsinki ethical principles.
Endpoints
The primary efficacy endpoint (DB phase) was change from baseline in serum ALP at week 24 in the intent-to-treat (ITT) population, which was made up of all randomized patients who received investigational product in the DB phase. Secondary endpoints included: hepatic function and biochemistry, such as alanine aminotransferase (ALT), aspartate aminotransferase (AST), gamma-glutamyl transferase (GGT), direct bilirubin and total bilirubin; fibrosis as assessed by measuring the enhanced liver fibrosis (ELF™) score; FXR activity as assessed by changes in FGF19 and 7α-hydroxy-4-cholesten-3-one (C4); and disease-specific symptoms. Pruritus was evaluated at baseline and at each study visit by a 100 mm visual analog scale (pruritus VAS scale) ranging from “no itching” to “worst possible itching”; VAS scores are reported as mean (SD). The primary endpoint of the LTSE was safety; changes in ALP, hepatic biochemistry, and other features of hepatic function and damage were secondary endpoints.
Samples for laboratory assessment of serum chemistry parameters were obtained at screening and weeks 2, 6, 12, 14, 18, and 24 (end of study visit). Safety was monitored and assessed by adverse events (AEs) and serious AEs (SAEs), medical history, physical examination, vital signs measurements, electrocardiogram results, and clinical laboratory assessments. AEs were evaluated at every study visit and were graded for severity (mild, moderate, or severe). During the LTSE, laboratory sampling and AE assessments were conducted at day 1 (week 24 DB visit) and months 3, 6, 9, 12, 15, 18, 21, and 24.
Statistics
A sample size of 25 patients per treatment group was calculated to provide at least 90% power (based on anticipated percentage change from baseline) to detect a mean difference in ALP of 15 units between treatment groups, assuming a 20% dropout rate and a mean absolute change in ALP of −20 and −5 units for OCA and placebo groups, respectively, with a pooled SD of 16, based on a 2-sided independent 2-group t test at an alpha level of 0.05. The primary efficacy analysis compared the change from baseline in ALP at week 24 between the OCA groups and placebo using an analysis of covariance (ANCOVA) model with treatment group and randomization strata as fixed effects, and baseline ALP as a covariate. Both ANCOVA and repeated-measures ANCOVA using a mixed model were used to provide estimates of ALP change from baseline and corresponding 95% CI at each visit. For statistical testing, changes from baseline in biochemical parameters obtained from the model are reported as least-square (LS) mean with SE; tests were performed at a significance level of 0.05. All analyses were based on available data.
A hierarchical approach was used for multiplicity adjustments. If the primary efficacy analysis was statistically significant (p <0.05), the following order was used in the testing procedure to compare the change from baseline in ALP between OCA and placebo: i) OCA 5–10 mg group, week 12; ii) OCA 1.5–3.0 mg group, week 24; iii) OCA 1.5–3.0 mg group, week 12. If at any step the comparison was not statistically significant, then all subsequent comparisons were exploratory rather than confirmatory. Multiplicity adjustments were not applied for any other statistical testing. The study was registered with ClinicalTrials.gov (NCT02177136) and EudraCT (2014–002205-38).
Results
Patient population
A total of 77 patients were enrolled and randomized to 1 of 3 treatment groups: placebo (n = 25), OCA 1.5–3.0 mg (n = 26), and OCA 5–10 mg (n = 26) (Fig. S1). One patient randomized to the OCA 1.5–3.0 mg group withdrew consent prior to dosing, and 1 patient randomized to the placebo group received OCA 5 mg titrated to 10 mg; therefore, 24 patients actually received placebo, 25 received OCA 1.5 mg titrated to 3 mg, and 27 received OCA 5 mg titrated to 10 mg. The patient who was randomly assigned to placebo but was treated with OCA was included in the placebo group for the ITT analyses and in the OCA 5–10 mg group for the safety analyses to provide conservative estimates of efficacy and safety; a post hoc assessment of ALP in the safety population was also performed, and this population was used for assessments of efficacy in subpopulations. A total of 65 patients (86%) completed the first 12 weeks of the DB phase and had a week 12 ALP assessment without any major protocol deviations, and 61 patients (80%) completed the entire 24-week DB phase. The first patient entered the DB study in February 2015, and the last patient completed the study in March 2017.
Among the 61 patients who completed the DB phase, 59 patients (97%) entered the LTSE and received OCA (Fig. S1). The median weighted average daily dose was 5 mg. Among the patients who entered the LTSE, 31 patients (53%) discontinued due to close of study by the sponsor, and 4 patients (7%) had 2 years or more in the LTSE; the last patient completed the LTSE in March 2018.
Demographics and baseline characteristics were generally consistent with those of patients with PSC (Table 1). Mean baseline ALP values were markedly elevated among all groups. The majority of patients had baseline ALP >3× ULN (placebo 68%; OCA 1.5–3.0 mg 60%; OCA 5–10 mg 54%). A substantial proportion of patients reported pruritus at baseline (placebo 64%; OCA 1.5–3.0 mg 40%; OCA 5–10 mg 35%), and 46 to 48% of patients were receiving UDCA at study entry (Table 1). Mean (SD) baseline pruritus VAS scores were higher in the placebo group than in the OCA groups (18.2 [21.9] in the placebo group, 15.7 [19.0] in the OCA 1.5–3.0 mg group, and 11.6 [21.3] in the OCA 5–10 mg group). More than 50% of patients had inflammatory bowel disease (IBD) at study entry (ulcerative colitis [UC] in 42% and Crohn’s disease in 14%). At baseline, UC was mild in 40% of patients in the placebo group, 48% of patients in the OCA 1.5–3.0 mg group, and 38% of patients in the OCA 5–10 mg group; mean (SD) partial Mayo scores were 1.1 (1.2) in the placebo group, 0.3 (0.5) in the OCA 1.5–3.0 mg group, and 0.9 (1.0) in the OCA 5–10 mg group. The most common concomitant treatment for IBD at study entry was mesalamine (placebo 16%; OCA 1.5–3.0 mg 40%; OCA 5–10 mg 35%).
Table 1.
Baseline demographics and disease characteristics (ITT population).
| Placebo (n = 25) | OCA 1.5–3.0 mg (n = 25) | OCA 5–10 mg (n = 26) | |
|---|---|---|---|
| Age, year; mean (SD) | 43.7 (13.1) | 41.6 (12.6) | 44.9 (14.3) |
| >65 y, n (%) | 1 (4) | 1 (4) | 3 (12) |
| Sex, n (%) | |||
| Male | 14 (56) | 15 (60) | 12 (46) |
| Female | 11 (44) | 10 (40) | 14 (54) |
| Race, n (%) | |||
| White | 22 (88) | 21 (84) | 22 (85) |
| Black | 3 (12) | 3 (12) | 4 (15) |
| Asian | 0 | 1 (4) | 0 |
| Ethnicity, n (%) | |||
| Hispanic or Latino | 1 (4) | 2 (8) | 2 (8) |
| Age at diagnosis, y; mean (SD) | 36.2 (14.5) | 35.3 (11.8) | 36.0 (14.5) |
| UDCA | |||
| Current use, n (%) | 12 (48) | 12 (48) | 12 (46) |
| Highest previous daily dose, mg/kg; mean (SD) | 17 (4) | 13 (3) | 13 (4) |
| ALP, U/L; mean (SD) | 562.8 (300.2) | 422.5 (123.1) | 428.5 (178.2) |
| Total bilirubin, mg/dl; mean (SD) | 1.2 (0.7) | 1.0 (0.5) | 1.1 (0.6) |
| IBD, n (%) | 13 (52) | 15 (60) | 15 (58) |
| UC | 10 (40) | 12 (48) | 10 (38) |
| Crohn’s disease | 3 (12) | 3 (12) | 5 (19) |
| Partial Mayo score; mean (SD) | 1.1 (1.2) | 0.3 (0.5) | 0.9 (1.0) |
| Intrahepatic disease, n(%) | 16 (64) | 14 (56) | 14 (54) |
| Intra- and extrahepatic disease, n (%) | 9 (36) | 11 (44) | 10 (38) |
| Pruritus, n (%) | |||
| Baseline | 16 (64) | 10 (40) | 9 (35) |
| Previous | 2 (8) | 5 (20) | 7 (27) |
| Moderate severity of most recent pruritus event | 4 (16) | 4 (16) | 8 (31) |
| Pruritus VAS, mean (SD) | 18.2 (21.9) | 15.7 (19.0) | 11.6 (21.3) |
ALP, alkaline phosphatase; IBD, inflammatory bowel disease; ITT, intent-to-treat; OCA, obeticholic acid; UC, ulcerative colitis; UDCA, ursodeoxycholic acid; VAS, visual analog scale.
Efficacy
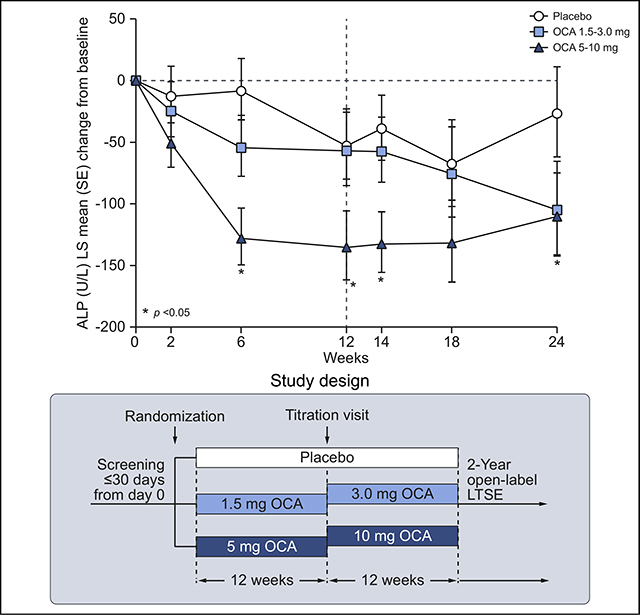
The primary efficacy analysis (ITT population, DB phase) demonstrated a statistically significant decrease from baseline in serum ALP in the OCA 5–10 mg group compared with placebo at week 24, with an LS mean (SE) difference of –83.42 (40.34) U/L (95% CI −164.28 to −2.57; p = 0.043) (Fig. 2A). At week 12, the decrease in ALP in the OCA 5–10 mg group also was significantly different from placebo (LS mean [SE] difference of −82.35 [33.39] U/L) (95% CI −149.11 to −15.59; p = 0.017). A reduction in ALP in the OCA 5–10 mg group vs. placebo was observed as early as week 6, when all patients were receiving the OCA 5 mg dose. The ALP response was maintained throughout the 24-week treatment period; however, titration of OCA from 5 to 10 mg did not lead to further reductions in ALP. At week 24 in the OCA 1.5–3.0 mg group, there was no significant reduction in ALP compared with placebo (LS mean [SE] difference = −78.29 [41.81] U/L, 95% CI −162.08 to 5.50; p = 0.067). Similarly, there was no significant reduction in ALP with OCA 1.5–3.0 mg compared with placebo at week 12.
Fig. 2. Mean change from baseline in ALP over time by treatment group.
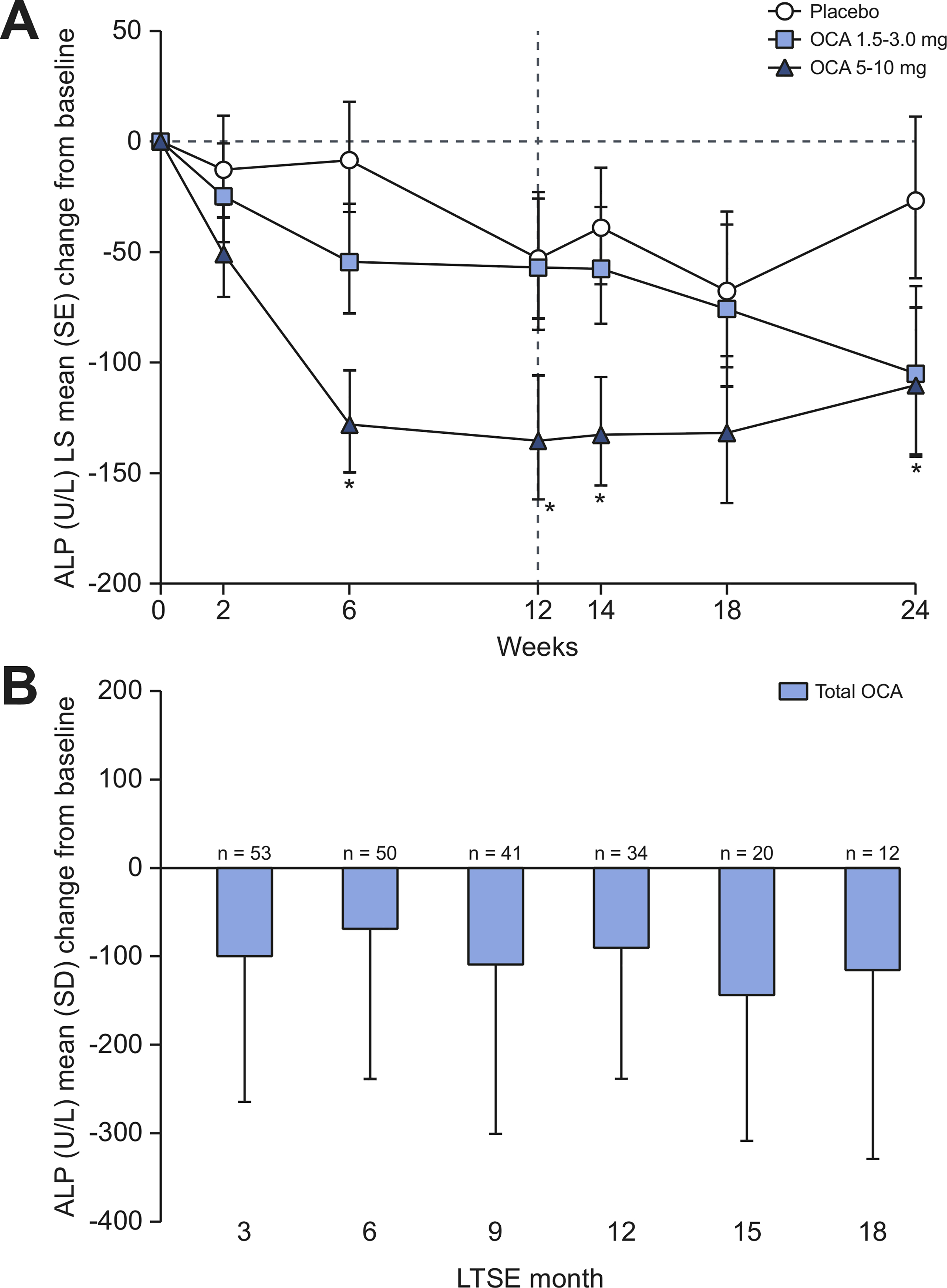
(A) LS mean (SE) change from baseline in serum ALP (U/L) in the DB phase (ITT population). Line at week 12 indicates dose titration. Mean baseline ALP (U/L) (SEM): 562.8 (60.0) (placebo), 422.5 (24.6) (OCA 1.5–3.0 mg), 428.5 (35.0) (OCA 5–10 mg). (B) Mean (SD) change from baseline in serum ALP in the LTSE (pooled data). Mean baseline ALP (U/L) (SD): 456.6 (224.6). ANCOVA was used for these analyses. *p <0.05; OCA dose vs. placebo. ALP, alkaline phosphatase: ANCOVA, analysis of covariance; DB, double-blind; ITT, intent-to-treat; LS, least-square; LTSE, long-term safety extension; OCA, obeticholic acid.
The reductions in ALP observed with OCA treatment during the DB phase persisted during the LTSE in patients who continued OCA treatment (n = 39), and reductions in ALP were observed starting at the month 6 visit among patients who crossed over from placebo (n = 20); pooled data are presented in Fig. 2B. Serum ALP levels were generally stable throughout the DB phase in the 20 patients receiving placebo who entered the LTSE (DB baseline value of 546 U/L, with a −23 U/L reduction at LTSE day 1 [i.e., prior to initiating OCA treatment]) (Fig. S2). In contrast, these patients’ serum ALP values were reduced −87 U/L at LTSE month 6 compared to at DB baseline. In patients who received OCA during the DB phase, ALP values were essentially unchanged from baseline to LTSE month 12 (nominal p >0.05) but significant reductions relative to baseline were observed in patients who crossed over from placebo (nominal p = 0.013).
To evaluate the efficacy of OCA when administered both with and without UDCA, a post hoc analysis of ALP values in the safety population was performed according to baseline UDCA use. During the DB phase, changes in ALP from baseline to week 24 in the safety population demonstrated a better response with OCA vs. placebo regardless of baseline UDCA use. The magnitude of ALP reduction with OCA vs. placebo was substantially greater (25% to 30% reductions in the OCA 5–10 mg group) for patients without baseline UDCA treatment compared with those who were receiving UDCA at baseline (14% to 16% reductions in the OCA 5–10 mg group) (Fig. 3). During the LTSE, with the exception of month 6, ALP levels were reduced with OCA treatment regardless of UDCA status, but as in the DB phase, the magnitude of ALP reduction was greater in patients not using UDCA at baseline (Fig. 3).
Fig. 3. Mean change from baseline in ALP over time by UDCA baseline use.
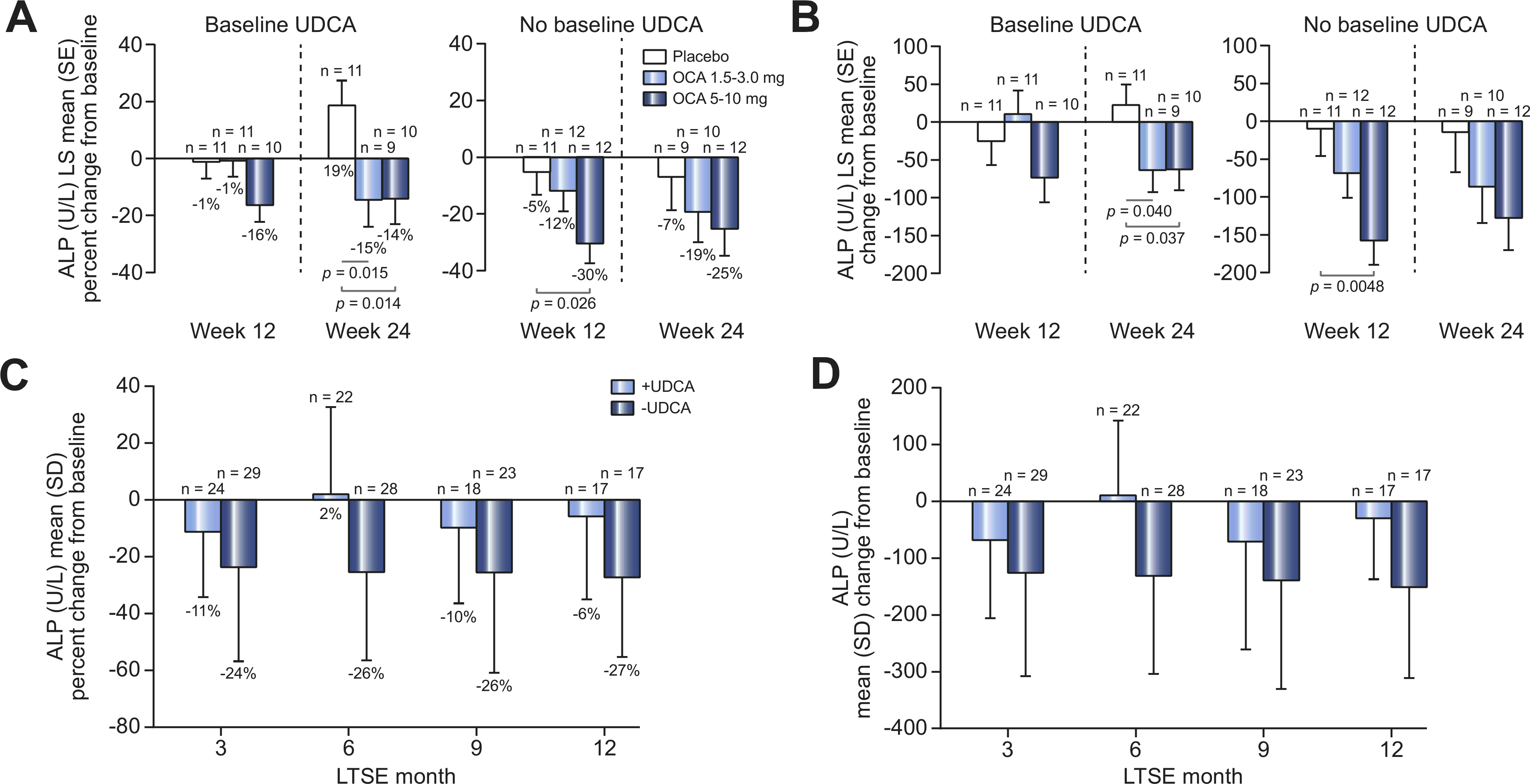
(A) Percentage or (B) absolute LS mean (SE) change from baseline in serum ALP (U/L) by UDCA baseline use in the DB phase (safety population). Mean baseline ALP (U/L) (SEM) UDCA use: 485.8 (104.0) (placebo), 432.4 (36.5) (OCA 1.5–3.0 mg), 387.1 (33.3) (OCA 5–10 mg); no UDCA use: 642.2 (66.8) (placebo), 413.4 (34.5) (OCA 15–3.0 mg), 468.6 (54.1) (OCA 5–10 mg). (C) Percentage or (D) absolute mean (SD) change from baseline in serum ALP by UDCA baseline use for all patients in the LTSE (pooled data). Mean baseline ALP (U/L) (SD) UDCA use, 427.5 (249.6); no UDCA use, 482.9 (199.8). ANCOVA was used for these analyses. ALP, alkaline phosphatase; ANCOVA, analysis of covariance; DB, double-blind; LS, least-square; LTSE, long-term safety extension; OCA, obeticholic acid; UDCA, ursodeoxycholic acid.
Secondary endpoints
Liver biochemistry was comprehensively evaluated in the safety population throughout the study, and serum values were studied relative to DB baseline during the LTSE. Median values of total and direct bilirubin were within the normal reference range in all treatment groups at DB baseline and generally fluctuated around the respective baseline values throughout the DB phase and the LTSE (Fig. S3 and S4). Median AST and ALT values generally decreased during the DB phase, with no significant differences observed among treatment groups (Fig. S5 and S6). Median GGT values decreased throughout the DB phase, and greater reductions were observed in the OCA 5–10 mg group vs. the placebo and OCA 1.5–3.0 mg groups (Fig. S7). Median AST and ALT values were generally lower at LTSE day 1 vs. baseline and AST reductions remained stable throughout the LTSE. Levels of ALT decreased slightly; however, the number of patients on study was small after month 15 (Fig. S5 and S6). Median GGT values were lower at LTSE day 1 vs. baseline and generally remained stable during the LTSE (Fig. S7). Baseline ELF scores (median [IQR]) were similar in the placebo (9.95 [2.00]), OCA 1.5–3.0 mg (9.70 [1.40]) and OCA 5–10 mg (10.10 [2.10]) groups, and no significant changes were observed over time in the DB phase or LTSE (Fig. S8).
Consistent with its FXR activation, OCA treatment was associated with changes from baseline in FGF19 and C4 over time (Fig. 4 and 5). Baseline values of these parameters were generally similar across groups. At week 24, a mean (SD) decrease in FGF19 of −81 (328) pg/ml was observed in the placebo group compared with increases of 66 (144) pg/ml in the OCA 1.5–3.0 mg group and 476 (775) pg/ml in die OCA 5–10 mg group. An increase in C4 in was observed in the placebo group at week 24 (mean [SD] = 2.2 [18.0] ng/ml), compared with decreases of-5.6 (17.5) ng/ml and −7.2 (10.8) ng/ml in the OCA 1.5–3.0 mg and 5–10 mg groups, respectively. These patterns of OCA-related increases in FGF19 and decreases in C4 persisted in the USE, although the number of observations was limited at month 24 (Fig. 4 and 5).
Fig. 4. Mean change from baseline in FGF19 over time by treatment group.
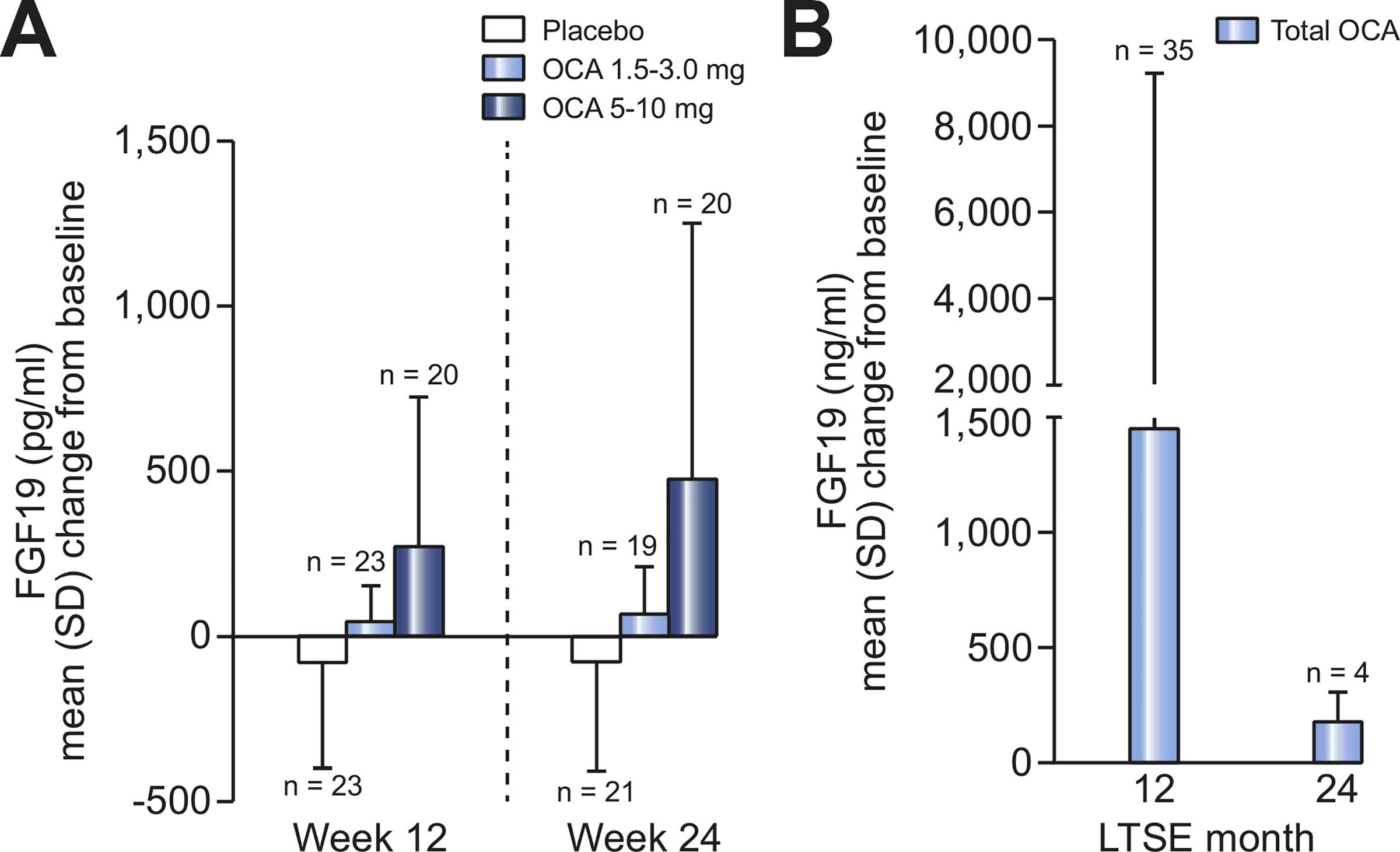
(A) Mean (SD) change from DB baseline in serum FGF19 (pg/ml) in DB phase (ITT population). Une indicates dose titration. Mean baseline FGF19 (pg/ml) (SD): 241.8 (339.1) (placebo), 143.2 (88.7) (OCA 1.5–3.0 mg), 187.7 (171.6) (OCA 5–10 mg). (B) Mean (SD) change from baseline in serum FGF19 in LTSE (pooled data). Mean baseline FGF19 (pg/ml) (SD): 182.0 (225.0). DB, double-blind; FGF19, fibroblast growth factor 19; ITT, intent-to-treat; LTSE, long-term safety extension; OCA, obeticholic add.
Fig. 5. Mean change from baseline in C4 over time by treatment group.
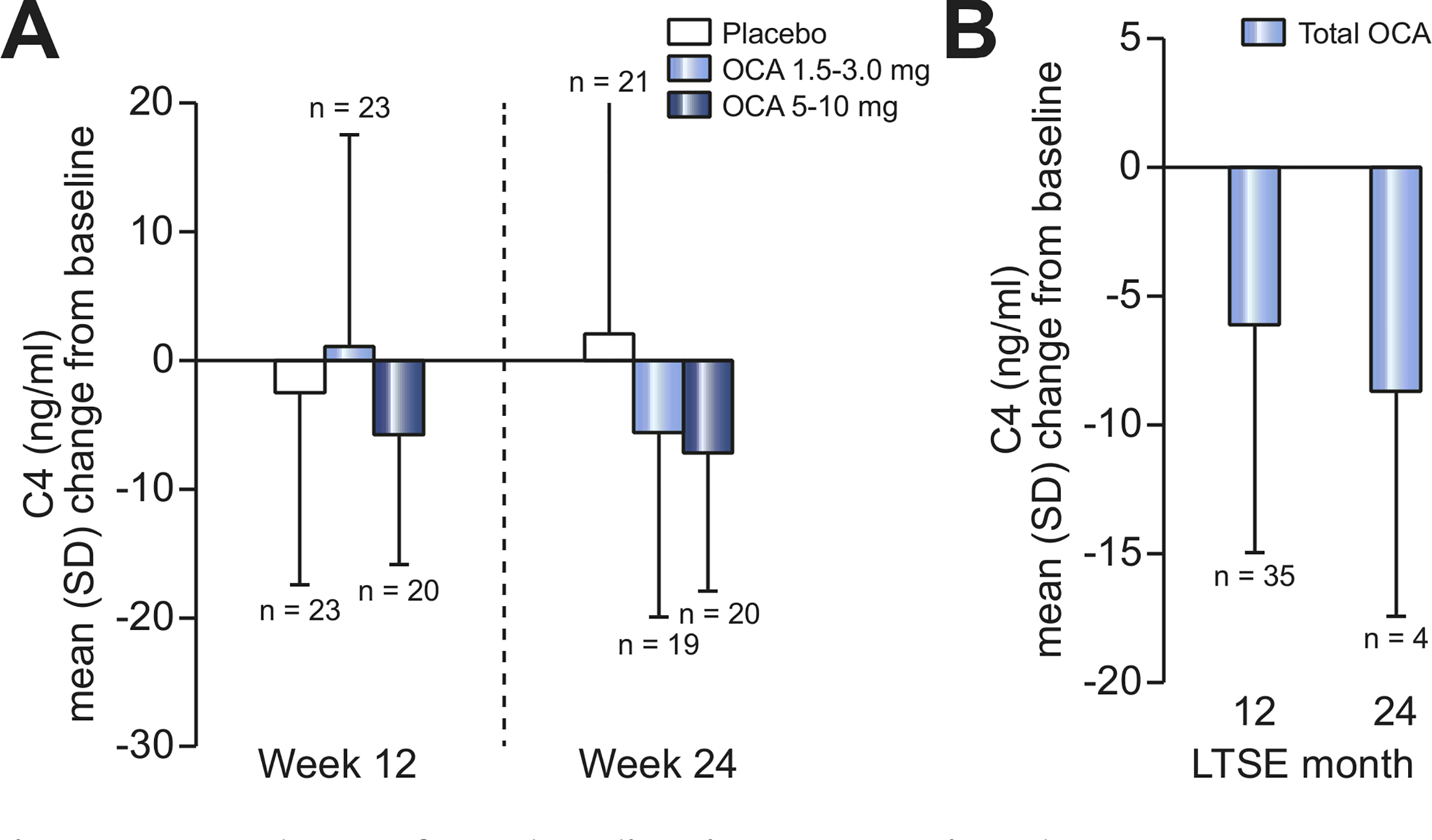
(A) Mean (SD) change from DB baseline in serum C4 (ng/ml) in DB phase (ITT population). Line indicates dose titration. Mean baseline C4 (ng/ml) (SD): 11.9 (15.8) (placebo), 13.8 (24.3) (OCA 1.5–3.0 mg), 11.1 (12.6) (OCA 5–10 mg). (B) Mean (SD) change from baseline in serum C4 in LTSE (pooled data). Mean baseline FGF19 (pg/ml) (SD): 13.5 (19.5). C4, 7α-hydroxy-4-cholesten-3-one; DB, double-blind; FGF19, fibroblast growth factor 19; ITT, intent-to-treat; LTSE, long-term safety extension; OCA, obeticholic add.
Treatment- and dose-related increases from baseline in pruritus VAS scores were observed over time, indicating worsening pruritus in the OCA 5–10 mg group. At week 12, mean (SD) pruritus VAS scores were 23.0 (25.3) in the placebo group, 15.9 (19.4) in the OCA 1.5–3.0 mg group, and 26.4 (23.4) in the OCA 5–10 mg group. By week 24, the mean (SD) VAS score remained relatively stable in the placebo and OCA 1.5–3.0 mg groups (19.1 [21.7] and 18.6 [21.1], respectively), but had increased to 33.1 (33.3) in the OCA 5–10 mg group.
Safety
In the DB phase, a majority of patients in each treatment group (88% to 96%) reported at least 1 treatment-emergent AE (TEAE) (Table 2). Most TEAEs were mild to moderate in severity but a higher proportion of patients in the OCA groups experienced severe TEAEs than in the placebo group (OCA 5–10 mg 52%; OCA 1.5–3.0 mg 28%; placebo 17%). Ten patients experienced SAEs: 2 patients in the placebo group (ascites, cholecystitis acute, and esophageal varices hemorrhage), 4 patients in the OCA 1.5–3.0 mg group (peripheral swelling [n = 2], bile duct stenosis, cholangitis infective, hyperbilirubinemia, pancreatitis, and Pseudomonas sepsis), and 4 patients in the OCA 5–10 mg group (cholangitis [n = 2], ascites, pharyngitis, and bacterial pneumonia). No deaths were reported during the study.
Table 2.
Treatment-emergent adverse events (all grades) occurring in ≥10% of patients during the DB phase (safety population).
| System organ class preferred term, n (%) | Placebo (n = 24) | OCA 1.5–3.0 mg (n = 25) | OCA 5–10 mg (n = 27) |
|---|---|---|---|
| All TEAEs | 21 (88) | 23 (92) | 26 (96) |
| Skin and subcutaneous tissue disorders | |||
| Pruritusa | 11 (46) | 15 (60) | 18 (67) |
| Gastrointestinal disorders | |||
| Nausea | 3 (13) | 2 (8) | 6 (22) |
| Diarrhea | 2 (8) | 1 (4) | 3 (11) |
| Abdominal pain | 4 (17) | 2 (8) | 2 (7) |
| Abdominal pain upperb | 4 (17) | 1 (4) | 2 (7) |
| Vomiting | 4 (17) | 1 (4) | 1 (4) |
| General disorders and administration site conditions | |||
| Pyrexia | 1 (4) | 3 (12) | 4 (15) |
| Fatigue | 2 (8) | 1 (4) | 3 (11) |
| Respiratory, thoracic, and mediastinal disorders | |||
| Oropharyngeal pain | 1 (4) | 2 (8) | 3 (11) |
| Cough | 3 (13) | 1 (4) | 1 (4) |
| Investigations | |||
| Blood bilirubin increased | 3 (13) | 1 (4) | 3 (11) |
| Infections and infestations | |||
| Sinusitis | 0 | 1 (4) | 3 (11) |
| Nasopharyngitis | 3 (13) | 1 (4) | 0 |
| Metabolism and nutrition disorders | |||
| Decreased appetite | 3 (13) | 0 | 0 |
| Psychiatric disorders | |||
| Insomnia | 1 (4) | 0 | 3 (11) |
DB, double-blind; OCA, obeticholic acid; TEAE, treatment-emergent adverse event.
Severe pruritus: placebo, n = 2 (8%); OCA 1.5–3.0 mg, n = 4 (16%); OCA 5–10 mg, n = 11 (41%). One patient in the OCA 5–10 mg group had pruritus generalized (moderate severity).
Severe abdominal pain upper: placebo, n = 0: OCA 1.5–3.0 mg, n = 0; OCA 5–10 mg, n = 1 (4%).
A total of 45 patients (57%) experienced pruritus or pruritus generalized during the DB phase, and 4 patients discontinued treatment due to pruritus (1 patient in the OCA 1.5–3.0 mg group and 3 patients in the OCA 5–10 mg group). The overall incidence of treatment-emergent pruritus was greater with OCA 5–10 mg (67%) and OCA 1.5–3.0 mg (60%) compared with placebo (46%).
Between 38% and 48% of patients had a history of UC at baseline, with mean partial Mayo scores between 0.3 and 1.1. Mean partial Mayo scores fluctuated during the DB phase, but no clear treatment or dose-related differences were observed over time. At week 24, a majority of patients with UC had partial Mayo scores of ≤2, indicative of UC remission.
The safety profile in the LTSE was consistent with that observed during the DB phase, and the majority of TEAEs were mild to moderate in intensity (Table 3). A total of 13 patients discontinued during the LTSE because of a TEAE: pruritus (n = 3), blood bilirubin increased (n = 3), ALP increased (n = 1), hepatic enzymes increased (n = 1), liver function test abnormal (n = 1), transaminases increased (n = 1), ascites (n = 1), jaundice (n = 1), and hyperbilirabinemia (n = 1). A total of 19 patients (32%) experienced SAEs during the LTSE; gastrointestinal disorders (17%) and infections/infestations (12%) were the most common. Aside from abdominal pain, which occurred in 3 patients, no SAE occurred in more than 1 patient, and no events were considered related or likely related to OCA. No SAEs led to treatment discontinuation during the LTSE.
Table 3.
Treatment-emergent adverse events occurring in ≥5% of patients during the LTSE.
| System organ class preferred term | All patients, n (%) (N = 59) |
Exposure-adjusted incidence (all grades)a | |
|---|---|---|---|
| All grades | Severe | ||
| All TEAEs | 57 (97) | 26 (44) | 367.6 |
| Gastrointestinal disorders | |||
| Vomiting | 8 (14) | 1 (2) | 13.0 |
| Diarrhea | 8 (14) | 0 | 12.9 |
| Nausea | 8 (14) | 0 | 12.8 |
| Abdominal pain | 7 (12) | 1 (2) | 11.4 |
| Abdominal pain upper | 7 (12) | 2 (3) | 11.2 |
| Ascites | 4 (7) | 0 | 6.1 |
| Constipation | 4 (7) | 0 | 6.3 |
| Ulcerative colitis | 4 (7) | 0 | 6.2 |
| Hemorrhoids | 4 (7) | 0 | 6.2 |
| Abdominal pain lower | 3 (5) | 0 | 4.7 |
| Esophageal varices | 3 (5) | 0 | 4.6 |
| Crohn’s disease | 3 (5) | 1 (2) | 4.5 |
| Skin and subcutaneous tissue disorders | |||
| Pruritus | 33 (56) | 9 (15) | 77.8 |
| Rash | 4 (7) | 0 | 6.0 |
| Infections and infestations | |||
| Urinary tract infection | 4 (7) | 0 | 6.3 |
| Bronchitis | 4 (7) | 0 | 6.2 |
| Nasopharyngitis | 4 (7) | 0 | 6.0 |
| Influenza | 3 (5) | 0 | 4.7 |
| Investigations | |||
| Blood bilirubin increased | 4 (7) | 0 | 6.0 |
| ALT increased | 3 (5) | 2 (3) | 4.5 |
| LFT abnormal | 3 (5) | 1 (2) | 4.5 |
| Hepatobiliary disorders | |||
| Cholangitis | 5 (8) | 1 (2) | 7.6 |
| Jaundice | 5 (8) | 0 | 7.5 |
| Bile duct stenosis | 3 (5) | 0 | 4.5 |
| Hyperbilirubinemia | 3 (5) | 1 (2) | 4.5 |
| Portal hypertension | 3 (5) | 0 | 4.5 |
| General disorders | |||
| Pyrexia | 4 (7) | 0 | 6.4 |
| Fatigue | 4 (7) | 1 (2) | 6.2 |
| Peripheral edema | 3 (5) | 0 | 4.5 |
| Other | |||
| Hypokalemia | 3 (5) | 0 | 4.5 |
| Insomnia | 3 (5) | 0 | 4.5 |
ALT, alanine aminotransferase; LFT, liver function test; LTSE, long-term safety extension; TEAE, treatment-emergent adverse event.
Number of patients with an event per 100 patient exposure years.
Discussion
This phase II, DB, placebo-controlled study demonstrated that treatment with OCA 5–10 mg led to significant reductions in ALP, a marker of cholestasis, in patients with PSC after 24 weeks. Improvements in ALP were apparent as early as 2 weeks after OCA initiation and were sustained throughout treatment. A subgroup analysis demonstrated reductions in ALP regardless of UDCA use, although reductions were more robust in patients not receiving UDCA at baseline.
The doses selected for the higher dosing regimen in this study (5 and 10 mg daily) are known to be therapeutic for PBC,17 but lower doses (1.5 and 3.0 mg) were also evaluated in this study. The 1.5 mg dose administered in the first 12 weeks of the study showed no meaningful biochemical response. However, while the 3.0 mg dose appeared to show additional improvement over 1.5 mg, this was not statistically different from placebo, although the patient numbers were relatively small and the variability in the biochemical data was larger than seen in the phase III PBC study with OCA.17 Initial doses of 1.5 and 3.0 mg were incorporated to allow identification of the minimally safe and effective dose while further characterizing the exposure-response relationship. The nested, dose-escalation approach allowed for characterization of the value of titration to address potential concerns about tolerability and intra-subject variability. The relationship between ALP response and OCA treatment was dose-dependent. Reductions in ALP following titration from 1.5 to 3.0 mg OCA suggest that 3.0 mg may represent the minimally effective dose of OCA in patients with PSC. The reductions in ALP relative to placebo observed at week 24 with OCA 10 mg were similar to those observed at week 12 with OCA 5 mg, suggesting that the 5 mg dose level may be adequately efficacious. Based on improvements in the primary endpoint, doses in the range of 3 to 5 mg may be beneficial in the treatment of patients with PSC. However, although reductions in ALP have been associated with improved prognosis,4 PSC is a complex disease and no single surrogate endpoint is currently established as a strong predictor of clinical benefit.19
The overall reduction in serum ALP observed in this study is lower than that observed with UDCA monotherapy in PSC; Lindor and colleagues reported reductions of approximately 41–46%.9 The observed efficacy of OCA with respect to ALP in the present study appears similar to that of recent studies of norUDCA (n = 161) (26% reduction in serum ALP after 12 weeks of treatment with 1,500 mg/day [p <0.0001 vs. placebo]),8 and cilofexor (n = 52) (21% reduction in serum ALP after 12 weeks of treatment with 100 mg/day [p = 0.029 vs. placebo]).15 However, comparisons across different studies must be made with caution due to different populations and study conduct.
Obeticholic acid at doses up to 10 mg was generally well tolerated during both phases of the study. Pruritus was the most common TEAE and occurred more often in the OCA groups during the DB phase. Most TEAEs were mild to moderate in severity; however, compared to the placebo group, a higher proportion of patients in the OCA groups experienced severe TEAEs. The disparity between treatment groups was attributable to the higher incidence of severe pruritus (8% of patients in the placebo group, 16% in the OCA 1.5–3.0 mg group, and 41% in the OCA 5–10 mg group). Severe pruritus was also the most common severe TEAE during the LTSE (reported in 15% of patients, with 3 patients discontinuing due to pruritus that began during the LTSE). Pruritus has been shown to be manageable in other diseases18 and the apparent dose-dependent relationship between OCA, pruritus, and clinical response in PSC should be further evaluated.
Design and conduct of clinical studies in PSC are confounded by a number of clinical factors. In addition to the rarity of PSC, the natural history of the disease is long, with duration of transplant-free survival ranging from 13 to 21 years.20 Serum ALP has been used as a surrogate proof-of-concept endpoint, but levels of ALP are highly variable and may be normal in a substantial number of patients.1,20 International study groups working in conjunction with regulatory authorities have suggested a combination of ALP reduction and histologic improvement of fibrosis may be a useful surrogate endpoint.19,20 However, due to the lack of a single, validated surrogate endpoint that regulators acknowledge as reasonably likely to predict patient outcomes, the optimal endpoints for clinical studies in PSC have yet to be established.19,20
One of the study limitations was the early termination (due to administrative reasons) of the LTSE. Because the majority (93%) of patients in the LTSE discontinued OCA before the month 24 visit, the number of patients evaluable for all liver biochemistry outcomes declined after month 12 and the results must be interpreted with caution.
In summary, this phase II study demonstrated that OCA treatment in patients with PSC resulted in sustained reductions in serum ALP levels in the presence or absence of UDCA, consistent with a potential for clinical benefit of OCA in PSC. OCA was generally safe and well tolerated with no evidence of worsening total bilirubin or other liver chemistries. Further clinical investigations of potential treatments for PSC, including OCA, are needed along with validation and acceptance of surrogate endpoints.
Supplementary Material
Highlights.
Novel treatments for PSC are an urgent unmet need.
This phase II study evaluated OCA in patients with PSC.
OCA 5–10 mg significantly reduced serum ALP levels at 24 weeks.
The safety profile of OCA was consistent with previous studies.
Acknowledgements
Financial support for medical editorial assistance was provided by Intercept Pharmaceuticals, Inc. We thank William Sinkins, PhD, ProEd Communications, Inc., for his medical editorial assistance with this manuscript.
Financial support
Intercept Pharmaceuticals, Inc.
Conflict of interest
Dr. Kowdley reports grants from Intercept during the conduct of the study; grants and personal fees from Gilead, Genfit, HighTide, Intercept, NGM Biopharma and CymaBay; personal fees from Assembly, Blade, Conatus, Abbvie and Roche and royalties from Up-To-Date (educational website), outside the submitted work. Dr. Vuppalanchi reports financial support to his institution from Gilead Sciences, Zydus Cadilla, Lajolla Pharmaceuticals for conduct of clinical trials during the conduct of the current study. He received financial support for consulting on the matters of drug safety from LabCorp/Covance. Dr. Levy reports grants from Intercept, grants from Gilead, grants from CymaBay, grants from Novartis, grants from Enanta, grants from Genkyotex, grants from NGM, grants from Lilly, grants and personal fees from GSK, personal fees from Pliant, grants from Genfit, personal fees from TARGET PharmaSolutions, personal fees from Shire, personal fees from Cara Therapeutics, outside the submitted work. Dr. Floreani reports no conflict of interest. Dr. Andreone reports personal fees from Intercept Pharmaceuticals, Inc., outside the submitted work. Dr. LaRusso has nothing to disclose. Dr. Shrestha has nothing to disclose. Dr. Trotter reports no conflict of interest. Dr. Goldberg reports grants from Merck, grants from Gilead, grants from Zydus, outside the submitted work. Dr. Rushbrook reports no conflict of interest. Dr. Hirschfield reports personal fees from Intercept Pharmaceuticals, Inc., personal fees from Cymabay, personal fees from GSK, grants from Gilead, grants from Falk Pharma, outside the submitted work. Dr. Schiano reports being a consultant for Alexion; and his institution received a grant for research from Progenity. Dr. Jin reports no conflict of interest. Dr. Pencek reports personal fees and is a shareholder and former employee of Intercept Pharmaceuticals, Inc., outside the submitted work. Dr. MacConell is an employee and shareholder of Intercept Pharmaceuticals, Inc., outside the submitted work. Dr. Shapiro is an employee and shareholder of Intercept Pharmaceuticals, Inc., outside the submitted work. Dr. Bowlus reports grants from Intercept, during the conduct of the study; grants from Gilead, grants and personal fees from Cymabay, grants and personal fees from Intercept, personal fees from Pliant, personal fees from Parvus, personal fees from BiomX, outside the submitted work.
Please refer to the accompanying ICMJE disclosure forms for further details.
Abbreviations
- AEs
adverse events
- ALP
alkaline phosphatase
- ANCOVA
analysis of covariance
- AST
aspartate aminotransferase
- C4
7α-hydroxy- 4-cholesten-3-one
- DB
double-blind
- ELF
enhanced liver fibrosis
- FGF19
fibroblast growth factor 19
- FXR
famesoid X receptor
- GGT
gamma-glutamyltransferase
- IBD
inflammatory bowel disease
- ITT
intent-to-treat
- LFT
liver function test
- LS
least-square
- LTSE
long-term safety extension
- PBC
primary biliary cholangitis
- PSC
primary sclerosing cholangitis
- OCA
obeticholic acid
- SAEs
serious adverse events
- TEAE
treatment-emergent adverse event
- UC
ulcerative colitis
- UDCA
ursodeoxycholic acid
- ULN
upper limit of normal
- VAS
visual analog scale.
Footnotes
Data sharing
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jhep.2020.02.033.
References
- [1].Karlsen TH, Folseraas T, Thorbum D, Vesterhus M. Primary sclerosing cholangitis - a comprehensive review. J Hepatol 2017;67(6): 1298–1323. [DOI] [PubMed] [Google Scholar]
- [2].Lazaridis KN, LaRusso NF. Primary sclerosing cholangitis. N Engl J Med 2016;375(12):1161–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lindor KD, Kowdley KV, Harrison ME, American College of Gastroenterology. ACG clinical guideline: primary sclerosing cholangitis. Am J Gastroenterol 2015;110(5):646–659. quiz 660. [DOI] [PubMed] [Google Scholar]
- [4].Goode EC, Clark AB, Mells GF, Srivastava B, Spiess K, Gelson WTH, et al. Factors associated with outcomes of patients with primary sclerosing cholangitis and development and validation of a risk scoring system. Hepatology 2019;69(5):2120–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Al Mamari S, Djordjevic J, Halliday JS, Chapman RW. Improvement of serum alkaline phosphatase to <1.5 upper limit of normal predicts better outcome and reduced risk of cholangiocarcinoma in primary sclerosing cholangitis. J Hepatol 2013;58(2):329–334. [DOI] [PubMed] [Google Scholar]
- [6].Lindstrom L, Hultcrantz R, Boberg KM, Friis-Uby I, Bergquist A. Association between reduced levels of alkaline phosphatase and survival times of patients with primary sclerosing cholangitis. Clin Gastroenterol Hepatol 2013;11(7):841–846. [DOI] [PubMed] [Google Scholar]
- [7].Rupp C Rossler A, Halibasic E, Sauer P, Weiss KH, Friedrich K, et al. Reduction in alkaline phosphatase is associated with longer survival in primary sclerosing cholangitis, independent of dominant stenosis. Aliment Pharmacol Ther 2014;40(11–12):1292–1301. [DOI] [PubMed] [Google Scholar]
- [8].Fickert P, Hirschfield GM, Denk G, Marschall HU, Altorjay I, Farkkila M, et al. norUrsodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J Hepatol 2017;67(3):549–558. [DOI] [PubMed] [Google Scholar]
- [9].Lindor KD. Ursodiol for primary sclerosing cholangitis. Mayo primary sclerosing cholangitis-ursodeoxycholic add study group. N Engl J Med 1997;336(10):691–695. [DOI] [PubMed] [Google Scholar]
- [10].Olsson R, Boberg KM, de Muckadell OS, Lindgren S, Hultcrantz R, Folvik G, et al. High-dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5-year multicenter, randomized, controlled study. Gastroenterology 2005;129(5):1464–1472. [DOI] [PubMed] [Google Scholar]
- [11].Lindor KD, Kowdley KV, Luketic VA, Harrison ME, McCashland T, Befeler AS, et al. High-dose ursodeoxycholic add for the treatment of primary sclerosing cholangitis. Hepatology 2009;50(3):808–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Papazyan R, Liu X, Liu J, Dong B, Plummer EM, Lewis RD 2nd, et al. FXR activation by obeticholic acid or nonsteroidal agonists induces a humanlike lipoprotein cholesterol change in mice with humanized chimeric liver. J Lipid Res 2018;59(6):982–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kowdley KV, Luketic V, Chapman R, Hirschfield GM, Poupon R, Schramm C, et al. A randomized trial of obeticholic acid monotherapy in patients with primary biliary cholangitis. Hepatology 2018;67(5):1890–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hirschfield GM, Chazouilleres O, Drenth JP, Thorburn D, Harrison SA, Landis CS, et al. Effect of NGM282, an FGF19 analogue, in primary sclerosing cholangitis: a multicenter, randomized, double-blind, placebo-controlled phase II trial. J Hepatol 2019;70(3):483–493. [DOI] [PubMed] [Google Scholar]
- [15].Trauner M, Gulamhusein A, Hameed B, Caldwell S, Shiffman ML, Landis C, et al. The nonsteroidal farnesoid X receptor agonist cilofexor (GS-9674) improves markers of cholestasis and liver Injury in patients with primary sclerosing cholangitis. Hepatology 2019;70(3):788–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hirschfield GM, Mason A Luketic V, Lindor K, Gordon SC, Mayo M, et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology 2015;148(4):751–761.e8. [DOI] [PubMed] [Google Scholar]
- [17].Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med 2016;375(7):631–643. [DOI] [PubMed] [Google Scholar]
- [18].Pate J, Gutierrez JA, Frenette CT, Goel A, Kumar S, Manch RA et al. Practical strategies for pruritus management in the obeticholic acid-treated patient with PBC: proceedings from the 2018 expert panel. BMJ Open Gastroenterol 2019;6(1):e000256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ponsioen CY, lindor KD, Mehta R, Dimick-Santos L Design and endpoints for clinical trials in primary sclerosing cholangitis. Hepatology 2018;68(3):1174–1188. [DOI] [PubMed] [Google Scholar]
- [20].Ponsioen CY, Chapman RW, Chazouilleres O, Hirschfield GM, Karlsen TH, Lohse AW, et al. Surrogate endpoints for clinical trials in primary sclerosing cholangitis: review and results from an International PSC Study Group consensus process. Hepatology 2016;63(4):1357–1367. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.