

Abstract
Cell-cell interactions control the physiology and pathology of the central nervous system (CNS). To study astrocyte cell interactions in vivo, we developed rabies barcode interaction detection followed by sequencing (RABID-seq), which combines barcoded viral tracing and single-cell RNA sequencing (scRNA-seq). Using RABID-seq, we identified axon guidance molecules as candidate mediators of microglia-astrocyte interactions that promote CNS pathology in experimental autoimmune encephalomyelitis (EAE) and, potentially, multiple sclerosis (MS). In vivo cell-specific genetic perturbation EAE studies, in vitro systems, and the analysis of MS scRNA-seq datasets and CNS tissue established that Sema4D and Ephrin-B3 expressed in microglia control astrocyte responses via PlexinB2 and EphB3, respectively. Furthermore, a CNS-penetrant EphB3 inhibitor suppressed astrocyte and microglia proinflammatory responses and ameliorated EAE. In summary, RABID-seq identified microglia-astrocyte interactions and candidate therapeutic targets.
INTRODUCTION:
Glial cells of the central nervous system (CNS), including astrocytes and microglia, play critical roles in development, tissue repair, and homeostasis. However, dysregulated astrocyte and microglia responses contribute to the pathogenesis of neurologic diseases. Indeed, environmental chemicals, microbial metabolites, and cell-cell interactions have been shown to modulate disease-promoting responses in astrocytes and microglia in the context of multiple sclerosis (MS) and its model, experimental autoimmune encephalomyelitis (EAE). In particular, although astrocyte interactions with microglia are known to play important roles in the pathology of MS and other neurologic diseases, the pathways that facilitate astrocyte-microglia cross-talk are poorly understood, and consequently, few therapeutic inventions are available to target them.
RATIONALE:
Understanding the complexity of astrocyte-microglia cross-talk in CNS inflammation requires the study of precise neuroimmune interactions in vivo, but methodologies for defining the specific cell types, pathways, and molecules that mediate these interactions are limited. We developed a virus-based barcoding method for the identification of thousands of CNS cell interactions in vivo and the simultaneous analysis of the transcriptome of interacting cells with single-cell resolution. We applied this technique, named rabies barcode interaction detection followed by sequencing (RABID-seq), to the study of microglia-astrocyte communication in the context of CNS inflammation in EAE and MS.
RESULTS:
To develop RABID-seq, we engineered an mRNA-barcoded library in glycoprotein G–deficient pseudorabies virus (RabΔG-BC), which spreads between interacting cells but can only replicate in cells that transgenically express viral glycoprotein G. We pseudotyped the RabΔG-BC plasmid library using envelope protein of subgroup A (EnvA) packaging. Thus, the pseudotyped virus only infects cells that transgenically express the EnvA receptor, TVA After its replication in cells that express TVA and viral glycoprotein G, RabΔG-BC infects interacting cells, labelling them with the virus-encoded barcode. To study RABID-seq astrocyte interactions in vivo during CNS inflammation in the EAE model of MS, we used transgenic mice expressing glycoprotein G and TVA in astrocytes under the control of the Gfap promoter. These studies identified several axon guidance molecules as critical mediators of microglia-astrocyte interactions in the context of inflammation. By combining RABID-seq with genetic perturbation studies in vivo, validation with primary mouse and human cells in vitro, and the analysis of MS patient samples by immunostaining and single-cell RNA-seq, we established that microglia-astrocyte interactions mediated by Sema4D-PlexinB1, Sema4D-PlexinB2, and Ephrin-B3–EphB3 promote CNS pathology in EAE—and potentially MS. Notably, Ephrin-B3–EphB3 participated in forward and reverse signaling, which boosted both microglia and astrocyte pathogenic activities via the regulation of nuclear factor κB and mammalian target of rapamycin, respectively. Finally, we demonstrated that a CNS-penetrant small-molecule inhibitor of the kinase activity of the EphB3 intracellular domain ameliorates EAE in both acute and chronic progressive models.
CONCLUSION:
We developed RABID-seq, a novel approach for the simultaneous investigation of cell interactions and the transcriptome of interacting cells in vivo with single-cell resolution. RABID-seq identified signaling pathways controlled by the axon guidance molecules Sema4D-PlexinB1, Sema4D-PlexinB2, and Ephrin-B3/EphB3 as mediators of microglia-astrocyte interactions that promote CNS pathogenesis and also as candidate targets for therapeutic intervention in neurologic disorders.
Graphical Abstract
Elucidation of microglia-astrocyte interactions by rabies barcode interaction detection followed by sequencing (RABID-seq).
Pseudotyped rabies virus expressing barcoded mRNA targets Gfap+ astrocytes, where it replicates before infecting neighboring cells, leaving a barcoded trace. Single-cell RNA sequencing reads both cellular mRNAs and viral barcodes, allowing for the reconstruction of in vivo cell interactions and the transcriptional analysis of interacting cells with single-cell resolution.
Astrocytes are central nervous system (CNS)–resident glial cells with important roles in health and disease. Astrocyte functions in development, homeostasis, and disease are controlled by cell interactions (1–10). For example, astrocyte interactions with microglia regulate synaptic pruning (11), neurodegeneration (2), and CNS inflammation (12). In the context of autoimmune CNS disorders such as multiple sclerosis (MS) and its preclinical model, experimental autoimmune encephalomyelitis (EAE), astrocyte activation is modulated by T cells and other peripheral immune cells recruited to the inflamed CNS (3, 6, 10, 12–16). However, the full extent of cell interactions that control astrocyte responses and the molecular mechanisms involved are poorly understood. The investigation of those interactions is further complicated by the heterogeneity of astrocytes and other cell types, as well as the need to define the specific cell subsets participating in interactions of interest.
High-throughput genomic approaches such as single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics can profile thousands of individual cells, but challenges remain in applying these approaches to study cell interactions. Moreover, although some techniques can profile immune cell interactions (17, 18) and cell networks based on the sequencing of microdissected units (19) or the use of photoactivatable markers (20), these approaches cannot easily profile cell interactions in the CNS and may fail to detect interactions involving only a small subset of cells.
We developed rabies barcode interaction detection followed by sequencing (RABID-seq) to identify astrocyte cell interactions and the molecular phenotypes of interacting cells in vivo. RABID-seq uses glycoprotein G–deficient pseudorabies virus (RabAG) engineered to express a fluorescent mRNA-encoded barcode as it spreads between interacting cells, allowing the reconstruction of cellular cross-talk in vivo via scRNA-seq. By encoding spatial relationships directly into the transcriptome, RABID-seq detects cell interactions that otherwise would not be detected by single-cell profiling alone. Using RABID-seq, we identified the axon guidance molecules Sema4D-PlexinB2 and Ephrin-B3–EphB3 as mediators of microglia-astrocyte interactions that promoted CNS pathology in EAE. Moreover, we identified a CNS-penetrant small-molecule inhibitor of EphB3 signaling that ameliorates acute and chronic progressive EAE. In summary, RABID-seq provides an approach for the comprehensive investigation of cell interactions in the CNS with single-cell resolution, identifying microglia-astrocyte interactions and candidate targets for therapeutic intervention in neurologic disorders.
RABID-seq overview
RabΔG is a powerful tool for studying cell interactions because it can be targeted to specific cell types, including astrocytes and other glia (21–25) (Fig. 1A). To study astrocyte cell interactions, we engineered the RabΔG virus to express barcoded mCherry (RabΔG-mCherry-BC). Because barcode sequences are inserted before the transcriptional stop of the polyadenylated mCherry transcript, the transcribed mRNA barcode can be analyzed by scRNA-seq (Fig. 1B). In addition, mCherry allows the isolation by flow cytometry of fluorescently labeled barcoded cells in a RabΔG-transduced cell network (Fig. 1C), enabling the simultaneous analysis of cell transcriptomes and RabΔG barcodes by high-throughput droplet-based scRNA-seq (Fig. 1D).
Fig. 1. Reconstruction of single-cell transcriptomes and connectomes using RABID-seq.
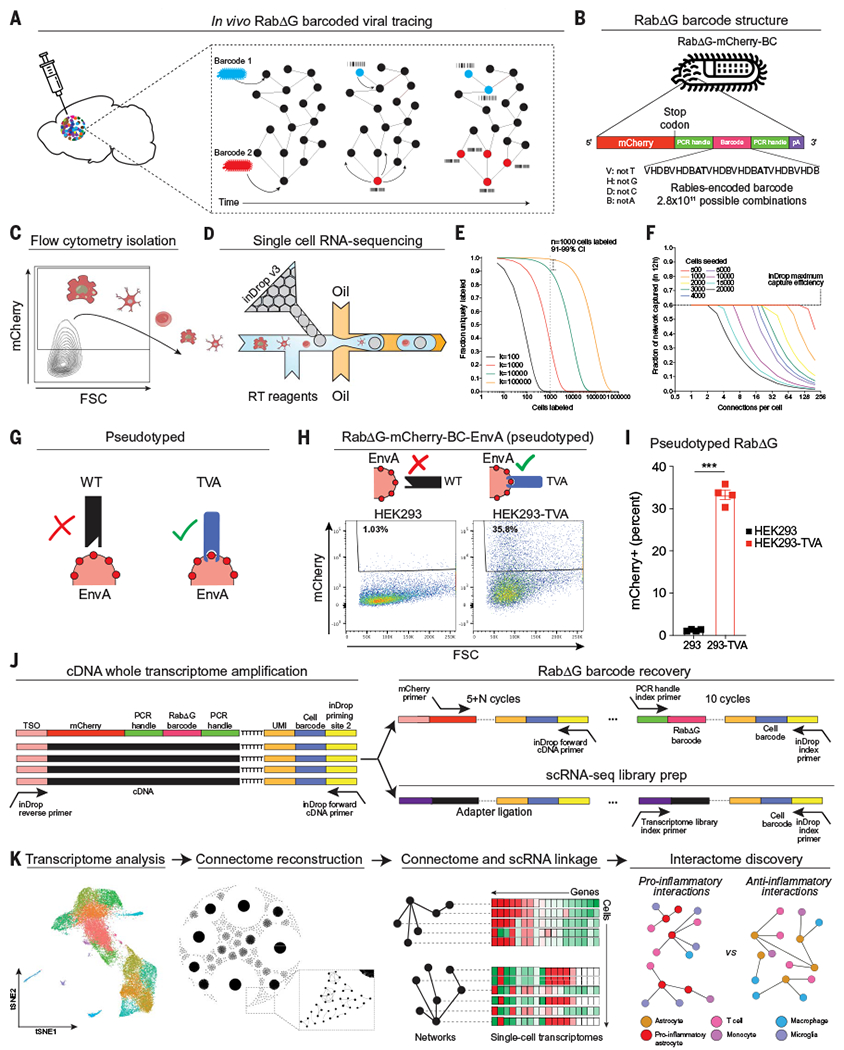
(A) Barcoded RabΔG virus is delivered via intracranial injection, and barcodes transfer to neighboring cells as RabΔG virus spreads throughout interacting cells. (B) The RabΔG genome expresses mCherry, which enables the recovery and sequencing of virus-infected cells. The mCherry transcript harbors a unique barcode with semirandom structure, flanked by constant regions to facilitate amplification. Base pair lengths are not to scale. (C) Flow cytometry recovery of mCherry+ cells from the CNS. FSC, forward scatter. (D) Single-cell RNA-sequencing of mCherry+ cells. RT, reverse transcription. (E) Fraction of uniquely labeled cells as a function of RabDG barcode library diversity and number of cells transduced. (F) Fraction of the in vivo network captured using in Drop (maximum 60% cell capture rate over a maximum period of 12 hours of encapsulation) as a function of the number of connections that each cell makes for different numbers of transduced cells. (G to I) RabΔG pseudotyping for cell targeting. (G) Schematic of RabΔG pseudotyping workflow and cell infectability. WT, wild type. (H) Fluorescence-activated cell sorting (FACS) analysis showing that pseudotyped virus only infects HEK293-TVA cells in vitro. HEK293, human embryonic kidney-293 cells. (I) Percent of HEK293 or HEK293-TVA cells infected with pseudotyped RabΔG virus. n = 4 samples per group. Unpaired two-tailed t test. (J) Generation of scRNA-seq libraries from inDrop using a SMART-seq approach with template switching and whole-transcriptome amplification (WTA). (Top right) WTA material is further amplified using a two-step approach with mCherry-specific primers followed by PCR primers targeting the constant region flanking the barcode. (Bottom right) Sequencing libraries are prepared from WTA product to produce scRNA-seq libraries. TSO, template-switching oligonucleotide. (K) Linkage of transcriptome and connectome data enables reconstruction of genome-wide transcriptional signatures of interacting cells in vivo. Data shown as means ± SEM. ***P < 0.001.
After amplification and sequencing, we detected ~1.5 million unique sequences in the barcoded RabΔG-mCherry-BC plasmid library (fig. S1, A to C). We pseudotyped the rabies virus from the barcoded RabΔG-mCherry-BC plasmid library using envelope protein of subgroup A (EnvA) packaging, which only infects cells expressing the EnvA receptor TVA and thereby allows the genetic targeting of cells of interest in vivo (21, 25, 26). Because the resulting pseudotyped rabies virus library was estimated to contain 104 to 105 unique barcodes (fig. S1D), we predicted that 91 to 99% of infected cells will be uniquely barcoded if 1000 cells were initially infected with the pseudotyped RabΔG-mCherry-BC virus library (Fig. 1, E and F).
We used an in vitro system to confirm that infection with pseudotyped RabΔG-mCherry-BC virus is restricted to TVA-expressing cells (Fig. 1, G to I) and developed a polymerase chain reaction (PCR)-based strategy for amplifying rabies connection barcodes from cDNA generated by the inDrop workflow (Fig. 1J and fig. S1, E and F). Notably, RabΔG-mCherry-BC sequencing libraries retain three crucial pieces of information: (i) a scRNA-seq cell barcode to assign RabΔG rabies barcodes to single-cell transcriptome data, (ii) a unique rabies barcode structure that allows efficient error correction, and (iii) a unique molecular identifier (UMI) to count RabΔG barcode transcripts (Fig. 1J). After sequencing, barcodes were identified, counted, and associated with individual cells captured by scRNA-seq. Interactions between cells were determined by the presence of shared barcodes (fig. S1, G to H), allowing for the reconstruction of cellular networks with genome-wide transcriptional information in vivo at single-cell resolution (Fig. 1K). Thus, RABID-seq allows the high-throughput analysis of single-cell interactions in vivo.
RABID-seq detects astrocyte cell interactions in naïve and EAE mice
We used transgenic mice expressing the rabies glycoprotein G and the EnvA receptor (TVA) under the control of the Gfap promoter (GfapTVA/G mice) (27) to target the initial infection of pseudotyped RabΔG-mCherry-BC virus to astrocytes and limit the subsequent transfer of barcodes (Fig. 2A and fig. S2A). Specifically, the RabΔG-mCherry-BC virus can only initially infect Gfap+ astrocytes expressing TVA, which also express the rabies glycoprotein G. New viral particles produced by Gfap+ astrocytes incorporate the rabies glycoprotein G into their envelopes, thereby acquiring the ability to infect and barcode neighboring cells. However, because Gfap− neighboring cells do not express the rabies glycoprotein G, they cannot further disseminate the virus (28, 29). This approach genetically targets Gfap+ astrocytes for initial barcoding, revealing astrocyte-centric cell interaction networks. Using this system, we detected the spread of RabΔG-mCherry-BC by flow cytometry, which peaked 7 days after transduction (fig. S2B). In validation studies, we confirmed the expression of TVA/G in astrocytes in GfapTVA/G mice (fig. S2C), titered the RabΔG-mCherry-BC virus to seed 1000 cells in GfapTVA/G mice upon infection (fig. S2, D and E), and detected RabΔG-mCherry-BC spread to neighboring cells, such as microglia (fig. S2F). Next, we transduced the forebrain of naïve GfapTVA/G mice or EAE GfapTVA/G mice with RabΔG-mCherry-BC virus 7 or 12 days after immunization with MOG35-55 to target priming and peak EAE, respectively (Fig. 2B). At 7.5 days after transduction, we sorted mCherry+ cells by flow cytometry (fig. S3A) and analyzed them by scRNA-seq (figs. S3, B to F; S4; and S5). Cell types were evenly distributed across each sample analyzed by RABID-seq in naïve and EAE mice (figs. S3 to S5). We detected RabΔG-mCherry–encoded barcodes in all samples analyzed and confirmed sufficient sequencing depth for all samples (fig. S6 and S7, A to C). Specifically, we detected an average of 1000 barcodes per mouse across the eight mice analyzed (fig. S6, D to F), consistent with our target seeding rate (fig. S2E), previously set to minimize barcode collisions (Fig. 1, E and F). In vitro studies suggested that early- versus late-seeded cells could be distinguished on the basis of barcode UMI abundance (figs. S7D).
Fig. 2. RABID-seq analysis of astrocyte cell interactions in naïve and EAE mice.
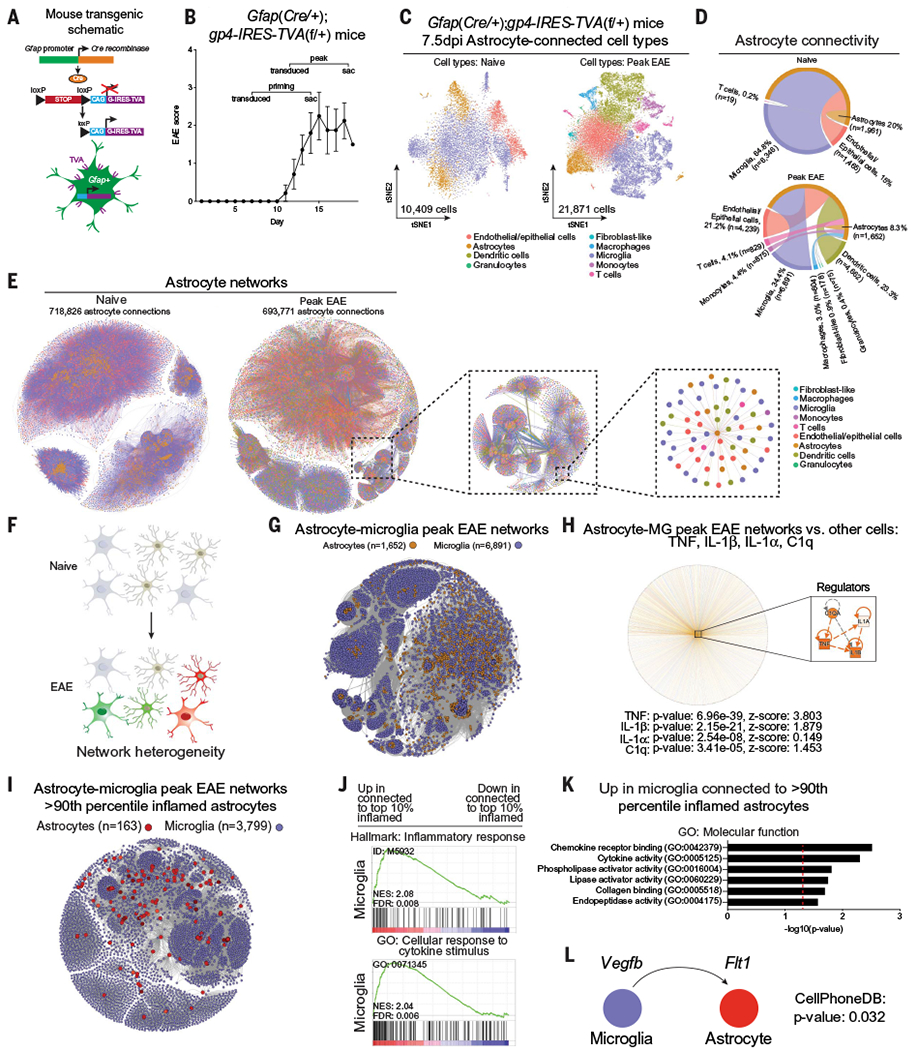
(A) Transgenic mouse line generated to target Gfap-expressing cells with the EnvA-TVA system. (B) EAE disease course. Mice were transduced with barcoded rabies virus, and brains were harvested 7.5 days after infection for scRNA-seq.Error bars indicate mean ± SEM. (C) t-distributed stochastic neighbor embedding (tSNE) plots of single-cell RABID-seq data from naïve and peak-EAE mice. The number of cells that passed bioinformatic filters is displayed near the origin. (D) Circos plots of astrocyte cell interactions in naïve and peak-EAE mice. Percentages are shown relative to the total number of connections. n is the number of cells of each cell type. (E) Network representation of astrocyte cell interactions. To provide a sense of scale, increasingly smaller portions of the network are selected and enlarged. Cells are colored by cell type, as determined using scRNA-seq data. (F to K) Analysis of astrocyte-microglia interactions during peak EAE by RABID-seq. (F) Schematic of heterogeneous interactions between astrocytes and microglia during EAE. (G) Network representation of astrocyte-microglia interactions detected by RABID-seq. (H) IPA (ingenuity pathway analysis) network analysis of single-cell RABID-seq data showing predicted upstream regulators in astrocytes connected to microglia (MG) versus astrocytes connected to other cells. Statistical analysis: right-tailed Fisher’s exact test. (I) Visualization of >90th percentile proinflammatory astrocyte-microglia subnetworks. (J) GSEA (gene set enrichment analysis) preranked analysis of scRNA-seq data comparing microglia connected to >90th percentile proinflammatory astrocytes versus microglia connected to <10th percentile proinflammatory astrocytes. (K) Analysis by gene ontology: molecular function of microglia connected to >90th percentile proinflammatory astrocytes. (L) CellPhoneDB identification of VEGFB-FLT1 signaling between microglia and >90th percentile proinflammatory astrocytes.
We used scRNA-seq to analyze 32,280 RabΔG barcoded cells, including astrocytes, microglia, monocytes, and T cells, from naïve or peak-EAE mice (Fig. 2C). Our cell isolation method removed oligodendrocytes and neurons to enable us to focus on astrocytes, microglia, and infiltrating immune cells. We built astrocyte-centric cell interaction networks by including only rabies barcodes that were detected in at least one astrocyte, limiting potential confounding effects that could result from Cre leakiness. In naïve mice, we detected astrocyte-astrocyte interactions, as well as interactions with microglia and other cells. In EAE mice at the peak of disease, astrocyte interaction networks were more diverse (Fig. 2, D and E) and included interactions with peripheral cells, such as T cells, that were recruited to the inflamed CNS (3, 13, 16, 30).
We developed an inflammation score based on the activation of the inflammatory response defined by the Gene Ontology initiative (fig. S8, A to C, and data S1). We then selected astrocytes with the highest (>90th percentile) and lowest (<10th percentile) proinflammatory transcriptional phenotypes in EAE that displayed interactions with T cells (fig. S8D). Astrocytes with the highest proinflammatory scores were connected to T cells that exhibited proinflammatory phenotypes and high tumor necrosis factor–α (TNFα) signaling via nuclear factor κB (NF-κB) (136 astrocytes, 506 T cells, 3796 connections) (fig. S8, E and F, and data S2), in agreement with the reported increase in proinflammatory astrocyte responses by proinflammatory T cells (3). Conversely, T cells connected to astrocytes displaying the lowest proinflammatory phenotype (132 astrocytes, 684 T cells, 3847 connections) showed higher expression of molecules associated with the suppression of inflammation (e.g., Ctla4, Ikzf4, Il2ra, and Il10) (fig. S8G). Indeed, when we analyzed subnetworks of Il10ra+ astrocytes and Il10+ T cells (fig. S8H), we detected interleukin-2 (IL-2)–signal transducer and activator of transcription 5 (STAT5) signaling pathways that have been associated with regulatory T cells (fig. S8I) (31), recapitulating IL-10–driven anti-inflammatory effects of T cells on astrocytes (32, 33). Thus, RABID-seq can be used to simultaneously identify astrocyte cell interactions and the transcriptional features of interacting cells at the single-cell level.
Identification of microglia-astrocyte signaling via axon guidance molecules by RABID-seq
Microglia-astrocyte interactions play important roles during CNS development, homeostasis, and disease (2, 11, 12). However, a comprehensive understanding of these interactions and how they shift during inflammation is still missing (Fig. 2F). RABID-seq detected microglial control of astrocytes mediated by IL-1, TNF, and C1q in EAE mice relative to naüve mice, consistent with previous reports (2) (Fig. 2, G and H, and data S3 to S5). Moreover, we detected the activation of proinflammatory signatures and chemokine-mediated signaling in microglia connected to astrocytes displaying a high proinflammatory phenotype (>90th percentile) (Fig. 2, I and J). Indeed, the analysis of ligand-receptor interactions (34) between these high-proinflammatory phenotype astrocytes (>90th percentile) and microglia in peak EAE recapitulated previous reports (12) of increased proinflammatory FLT1 signaling and decreased aryl hydrocarbon receptor–driven anti-inflammatory responses in astrocytes triggered by microglia-produced VEGF-B (vascular endothelial growth factor B) (Fig. 2, K and L) (12). The microglial phagocytosis of cell debris containing mCherry protein did not generate a fluorescent signal strong enough to mask the signal generated by the RabΔG-based tracing of cell interactions (35) (fig. S2D).
We next used RABID-seq to identify mechanisms of microglia-astrocyte communication in EAE mice at the peak of disease, detecting the activation of pathways associated with axon guidance molecules (Fig. 3A and fig. S9A). Axon guidance molecules play important roles during development but are co-opted by tumors and inflammatory processes in the periphery (36). Thus, we examined axon guidance pathways associated with semaphorin-plexin, ephrin-EPH, netrin, and Slit/Robo signaling in the microglia-astrocyte cell networks that we identified in naüve and peak-EAE mice. The analysis of the single-cell RABID-seq dataset detected the activation of Sema4D/PlexinB during peak EAE, driven by Sema4d expression in microglia and Plxnb2 expression in astrocytes (Fig. 3, B to D, and fig. S9B). We made similar observations during the priming phase of EAE (fig. S9, C and D) and when studying Plxnb2+ astrocyte interactions with Sema4d+ monocytes during peak EAE (fig. S9, F to H). We also detected significant activation of Ephrin-B–mediated signaling (Fig. 3, B and C).
Fig. 3. RABID-seq identifies a role for Sema4D-PlexinB2 signaling in microglia-astrocyte communication.
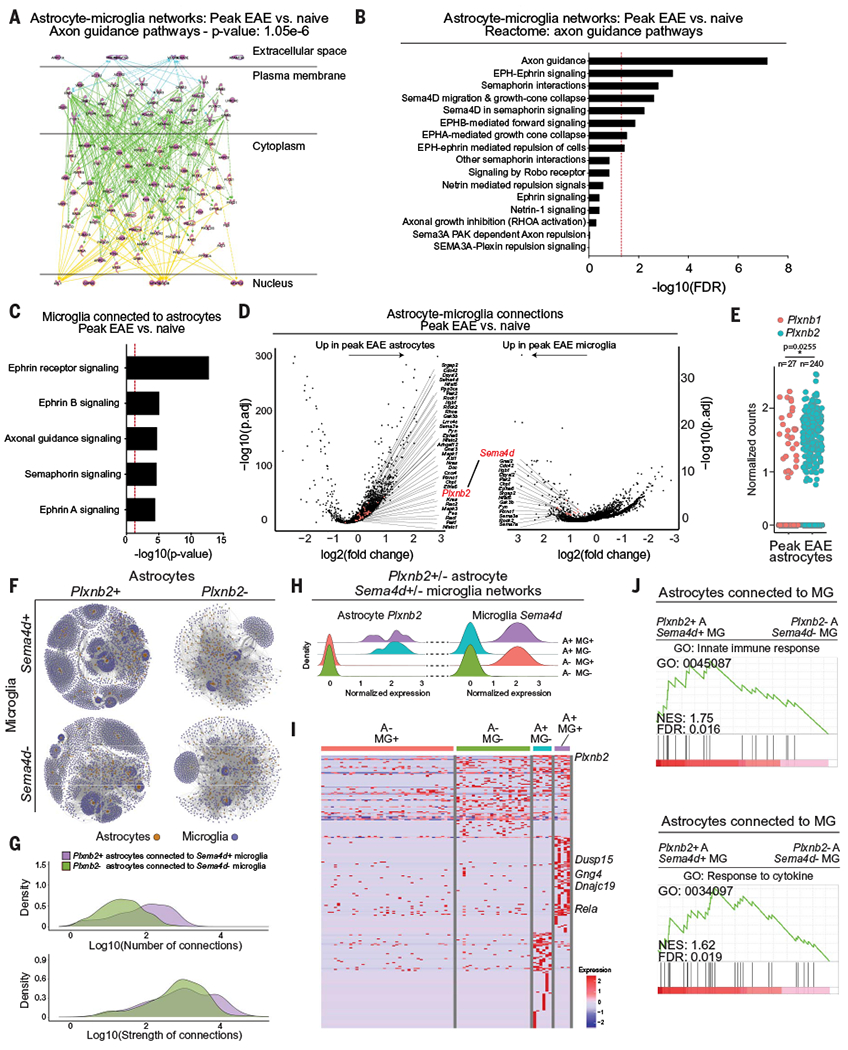
(A) IPA analysis of axon guidance pathway genes activated in astrocyte-microglia networks in peak-EAE versus naïve mice. (B) Differentially regulated axon guidance pathways in astrocyte-microglia networks in peak-EAE versus naïve mice. FDR, false discovery rate. (C) Differentially regulated axon guidance pathways in microglia connected to astrocytes in peak-EAE versus naíve mice. (D) Differential gene expression analysis of astrocytes connected to microglia in peak-EAE versus naíve mice. Differentially expressed genes [adjusted P value (P.adj) < 0.05] from axon guidance pathways in astrocytes (left) and microglia (right) are colored and labeled by gene name on volcano plots of −log10(P.adj) versus fold change. (E) Expression of Plxnbl and Plxnb2 in peak-EAE astrocytes. Two-tailed paired t test on percent per mouse. (F) Subnetworks of Plxnb2+/− astrocytes connected to Sema4d+/− microglia. (G) Density plots of the number of interactions between Plxnb2+ astrocytes connected to Sema4d+ microglia and Plxnb2− astrocytes connected to Sema4d− microglia. (H) Normalized single-cell expression of Plxnb2 in astrocytes and Sema4d in microglia within the subnetworks shown in (F). A+: Plxnb2+ astrocytes; A−: Plxnb2− astrocytes; MG+: Sema4d+ microglia; MG−: Sema4d− microglia. (I) Differential gene expression between astrocytes in the Plxnb2-Sema4d subnetworks determined by RABID-seq. (J) GSEA preranked analysis of single-cell RABID-seq data comparing Plxnb2+ astrocytes connected to Sema4d+ microglia (A+ MG+) to Plxnb2− astrocytes connected to Sema4d− microglia (A− MG−). NES, normalized enrichment score.
Microglia-astrocyte Sema4D-PlexinB2 signaling promotes CNS inflammation in EAE
Sema4D signals through the PlexinB1 and PlexinB2 receptors (36, 37), but the analysis of the RABID-seq dataset detected higher Plxnb2 expression in astrocytes during EAE (Fig. 3, D and E, and fig. S9B). Thus, to investigate the role of Sema4D-PlexinB2 signaling during EAE, we analyzed the single-cell transcriptional signatures of interacting Sema4d+ microglia and Plxnb2+ astrocytes detected by RABID-seq. We subdivided our RABID-seq data into nonoverlapping networks of Plxnb2+ or Plxnb2− astrocytes, connected to Sema4d+ or Sema4d− microglia (Fig. 3, F to H). Plxnb2+ astrocytes interacted preferentially with Sema4d+ microglia (Fig. 3, F and G), exhibiting increased activation of semaphorin-plexin signaling concomitant with increased activation of proinflammatory responses (Fig. 3, H to J, and data S6). In addition, the analysis of a published scRNA-seq dataset of 48 MS patients and matched controls (3) detected increased interactions between microglial SEMA4D and astrocytic PLXNB2 in MS patients (fig. S10, A to C), similar to observations made in astrocytes and microglia when we merged our RABID-seq data with previously published scRNA-seq data from MS patients (3) (fig. S10, D and E). Thus, microglia-astrocyte interactions mediated by Sema4D-PlexinB2 promote CNS inflammation during EAE—and potentially MS.
We next investigated the role of microglia-astrocyte interactions mediated by Sema4D-PlexinB2 signaling in EAE pathogenesis. By immunostaining, we detected increased Sema4D in microglia and PlexinB2 in astrocytes during EAE (Fig. 4A) and in MS patient samples (Fig. 4B), validating our RABID-seq findings. Moreover, the activation of primary mouse microglia in culture with TNFα/IL-1β proinflammatory cytokines known to contribute to the pathogenesis of EAE and MS (3) increased Sema4d expression (Fig. 4C). In addition, the treatment of primary mouse or human astrocytes with a recombinant Sema4D fragment with plexin agonist activity (38–40) increased the expression of the proinflammatory genes Nos2 and Il1b (Fig. 4, D and E, fig. S11, A and B), suggesting that Sema4D-triggered Plexin signaling boosts astrocyte proinflammatory responses.
Fig. 4. Microglia-astrocyte Sema4D-PlexinB2 signaling promotes CNS inflammation in EAE.
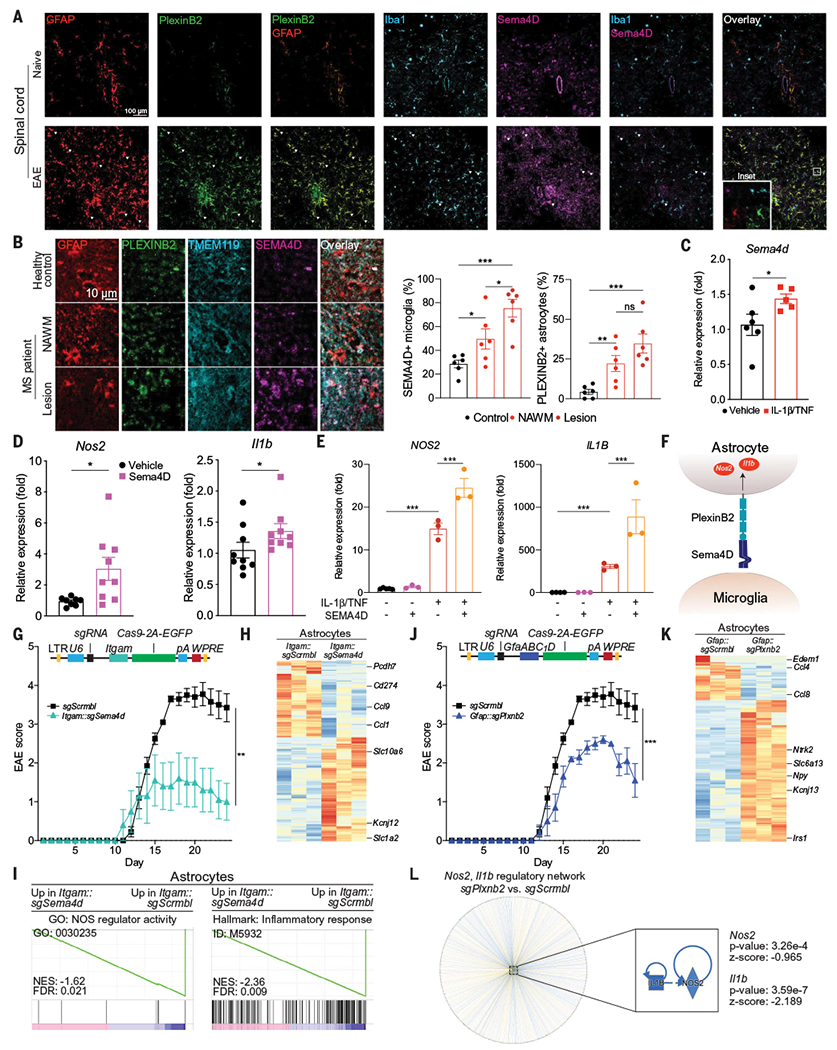
(A) Immunostaining analysis of PlexinB2, GFAP, Ibal, and Sema4D in the spinal cords of naïve and peak-EAE mice. Images are representative of n = 3 mice per group. (B) Immunostaining of MS patient and healthy control CNS tissue. n = 6 images from N = 3 patients per region. Statistical analysis: unpaired two-tailed t test. NAWM, normally appearing white matter. (C) Sema4d expression determined by quantitative PCR (qPCR) in primary mouse microglia treated with IL-1β/TNF versus vehicle. n = 6 vehicle, n = 5 IL-1β/TNF. Statistical analysis: Kolmogorov-Smirnov t test. (D) Nos2 and Il1b expression determined by qPCR in primary mouse astrocytes treated with a recombinant Sema4D fragment with agonistic activity. n = 9 samples per group, n = 8 samples for Nos2 vehicle. Statistical analysis: Kolmogorov-Smirnov t test per group. (E) Nos2 and Il1b expression determined by qPCR in primary human fetal astrocytes treated with the indicated compounds. n = 5 vehicle Nos2, n = 4 vehicle Il1b, n = 3 otherwise. Statistical analysis: unpaired two-tailed t test. (F) Schematic depicting microglial Sema4D binding PlexinB2 expressed in astrocytes. (G) EAE disease course in mice transduced with Itgam::Cas9 coexpressing sgSema4d- or sgScrmbl-targeting lentiviruses. n = 10 sgScrmbl, n = 5 sgSema4d mice. Statistical analysis: two-way repeated measures analysis of variance (ANOVA). (Top) Schematic of lentiviral vector. Lentiviral transduction occurred 7 days before EAE induction to avoid targeting recruited myeloid cells. (H and I) RNA-seq analysis of gene expression (H) and GSEA preranked (I) of astrocytes isolated from mice transduced with Itgam::sgSema4d or Itgam::sgScrmbl. (J) EAE disease course in mice transduced with Gfap::Cas9 coexpressing sgPlxnb2 or sgScrmbl. n = 10 sgScrmbl, n = 5 sgPlxnb2 mice. Statistical analysis: two-way repeated measures ANOVA. (Top) Schematic of lentiviral vector. (K) Differential gene expression determined by RNA-seq in astrocytes from mice transduced with Gfap::sgPlxnb2 versus Gfap::sgScrmbl. (L) Upstream regulator analysis by IPA of Gfap::sgPlxnb2 astrocytes relative to Gfap::sgScrmbl shows down-regulation of Nos2- and Il1b-driven proinflammatory pathways. Data shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. ns, not significant.
To determine whether microglia-astrocyte interactions mediated by Sema4D-PlexinB2 promote CNS inflammation during EAE (Fig. 4F), we developed CRISPR-Cas9 lentiviruses to inactivate Sema4d in microglia and Plxnb2 in astrocytes using Itgam- or Gfap-driven Cas9 and targeting single guide RNA, respectively (fig. S11, C to F). The inactivation of Sema4d in microglia ameliorated EAE (Fig. 4G). Moreover, astrocytes isolated from sgSema4d-targeting knockdown mice coexpressing Itgam-driven Cas9 showed decreased activation of Nos2 and proinflammatory signaling, supporting a role for microglia-derived Sema4D in promoting astrocyte pathogenic activities (Fig. 4, H and I). Indeed, Plxnb2 inactivation in astrocytes also resulted in EAE amelioration (Fig. 4J), concomitant with the down-regulation of proinflammatory pathways in astrocytes associated with NOS2 and IL-1β (Fig. 4, K and L, and fig. S11, C and D).
PlexinB1 and PlexinB2 show redundancy in some biological systems (41, 42). The analysis of our RABID-seq dataset suggested that Plxnb1 and Plxnb2 are expressed by different astrocyte subpopulations and that Plxnb2+ astrocytes are more abundant than Plxnb1+ astrocytes (Fig. 3E and fig. S11G). Thus, we investigated whether Sema4D signaling via PlexinB1 might also contribute to EAE pathogenesis. Knockdown via Gfap-driven Cas9 coexpressing sgPlxnb1 in astrocytes also ameliorated EAE via IL-1β and NOS2, although to a lesser extent than Plxnb2 (fig. S11, H to K). Indeed, we also detected increased interactions between microglial SEMA4D and PLXNB1 expressed in astrocytes in MS patients (3), although the detected increase in this interaction in MS was lower than the increase detected for SEMA4D-PLXNB2 (fig. S10, A and C). Thus, microglia-astrocyte Sema4D-PlexinB2 (and, to a lesser extent, Sema4D-PlexinB1) interactions promote CNS inflammation during EAE.
EphB3 receptor signaling boosts astrocyte proinflammatory activities
Our RABID-seq studies of astrocyte interactions in EAE also detected microglia-astrocyte signaling mediated by erythropoietin-producing human hepatocellular B (EphB) receptors (Fig. 3, A and B), which belong to a family of transmembrane receptor tyrosine kinases that have important roles in axon guidance, among other biological processes (43). EphB receptors are activated by interactions with their membrane-bound ligands Ephrin-B1, Ephrin-B2, and Ephrin-B3, encoded by Efnb1, Efnb2, and Efnb3, respectively (43). Efnb3 is mostly expressed in the CNS, whereas Efnb1 and Efnb2 show a broader expression pattern (44). We detected increased expression of Efnb3, but not Efnb1 or Efnb2, in microglia during EAE (fig. S12A). Efnb3 expression levels in monocytes recruited to the CNS during EAE were similar to the expression levels detected in microglia in naüve mice (fig. S12A), suggesting that the Ephrin-B3 in monocytes does not play a major role in the control of EphB3 receptor-dependent astrocyte proinflammatory activities during EAE. Moreover, flow cytometry analysis of neonatal mouse microglia-astrocyte cocultures detected higher EphB3 expression in astrocytes than in microglia (fig. S12B). Finally, the analysis of CNS human samples detected an increase in EPHRINB3+ microglia and EPHB3+ astrocytes in MS lesions (Fig. 5, A to C). Thus, signaling between microglial Ephrin-B3 and EphB3 receptors in astrocytes may play a role in CNS inflammation in EAE and, potentially, MS.
Fig. 5. EphB3 receptor signaling boosts astrocyte proinflammatory responses.
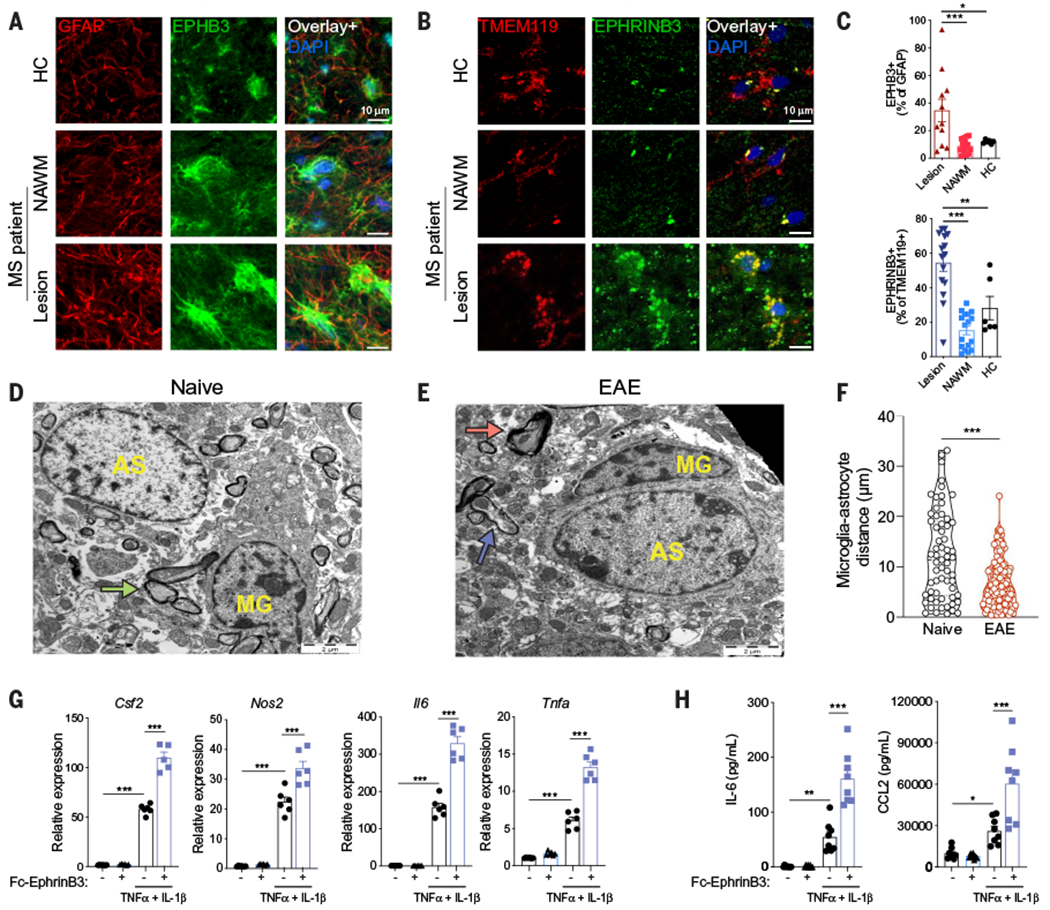
(A to C) Immunostaining analysis of MS and healthy control (HC) CNS tissue samples for the colocalization of EPHB3 and GFAP in astrocytes (A) or EPHRINB3 and TMEM119 in microglia (B). (C) Quantification of immunostaining data. n = 6 or 16 images from N = 3 patients per region. Statistical analysis: one-way ANOVA and Dunnett post-test. (D and E) Representative electron micrographs of naïve (D) and EAE (E) spinal cords. Microglia (MG) cells exhibit elongated and dark nuclei with clumped chromatin and dark cytoplasm, and astrocytic cells (AS) are characterized by pale nuclei that are usually regular in shape with a thin rim of heterochromatin beneath the nuclear membrane.Green arrow, intact myelin; red arrow, myelin destruction; blue arrow, remyelination. The black space in the top right corner of (E) indicates the edge of the tissue section. (F) Quantification of microglia-astrocyte distance in electron microscopy images. n = 63 naïve mice, n = 167 EAE mice. Statistical analysis: unpaired two-tailed t test. (G) Csf2, Nos2, Il6, and Tnfa expression determined by qPCR in neonatal mouse astrocytes stimulated with TNFα and IL-1β in the presence of plate-bound Ephrin-B3–Fc chimera. n = 6 samples per group. Statistical analysis: one-way ANOVA and Dunnett post-test. Data are representative of two independent experiments. (H) IL-6 and CCL2 concentration in supernatants of neonatal mouse astrocytes stimulated with TNFα and IL-1β in the presence of plate-bound Ephrin-B3–Fc chimera. n = 6 samples per group. Statistical analysis: one-way ANOVA and Dunnett post-test. Data are representative oftwo independent experiments. Data shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Both EphB3 and its ligand Ephrin-B3 are plasma membrane proteins (43). We used transmission electron microscopy to detect increased microglia-astrocyte contacts during EAE (Fig. 5, D to F), suggesting a potential reason for increased Eph signaling mediated by microglia Ephrin-B3 and astrocyte EphB3 interactions. To study the effects of EphB3 signaling in astrocytes, we treated neonatal mouse astrocytes in culture with plate-bound Ephrin-B3–Fc chimera and activated them with TNFα and IL-1β. EphB3 activation boosted the expression of genes associated with astrocyte proinflammatory activities such as Il6, Nos2, Csf2, and Tnfa (Fig. 5G); EphB3 activation also boosted IL-6 and CCL2 production (Fig. 5H). We obtained similar results when we analyzed the effects of EphB3 activation with plate-bound Ephrin-B3–Fc chimera on primary human astrocytes in culture (fig. S12C). Thus, EphB3 signaling may boost astrocyte proinflammatory activities.
Microglial Ephrin-B3 and astrocyte EphB3 promote CNS pathology in EAE
To study the role of Ephrin-B3 and EphB3 in the regulation of microglial and astrocyte responses in the context of CNS inflammation, we knocked down Ephb3 in astrocytes and Efnb3 in microglia during EAE using lentivirus-delivered short hairpin RNAs (shRNAs) expressed under the control of Gfap or Itgam promoters, respectively (12) (fig. S13, A and B). EAE mice were injected intracerebroventricularly at day 7 after immunization, before disease onset, to target CNS resident cells as previously described (3, 9, 12, 13). We did not detect reduced Efnb3 expression in CNS-infiltrating monocytes, confirming that these cells were not affected by the knockdown (fig. S13B). The lentiviruses reached the spinal cord, and the knockdown of Ephb3 in astrocytes or Efnb3 in microglia resulted in a comparable amelioration of EAE (Fig. 6A and fig. S13, C to G) but did not affect T cell responses (fig. S13, H to K). Ephb3 and Efnb3 knockdown led to a reduction in proinflammatory Ly6CHi monocytes recruited to the CNS during EAE (45) (Fig. 6B). The simultaneous knockdown of Ephb3 in astrocytes and Efnb3 in microglia did not ameliorate the disease more than single knockdowns did (fig. S13L). Moreover, no EAE amelioration was detected after Efnb3 knockdown in astrocytes (fig. S13M).
Fig. 6. Microglia-astrocyte Ephrin-B3–EphB3 signaling promotes CNS pathology in EAE.
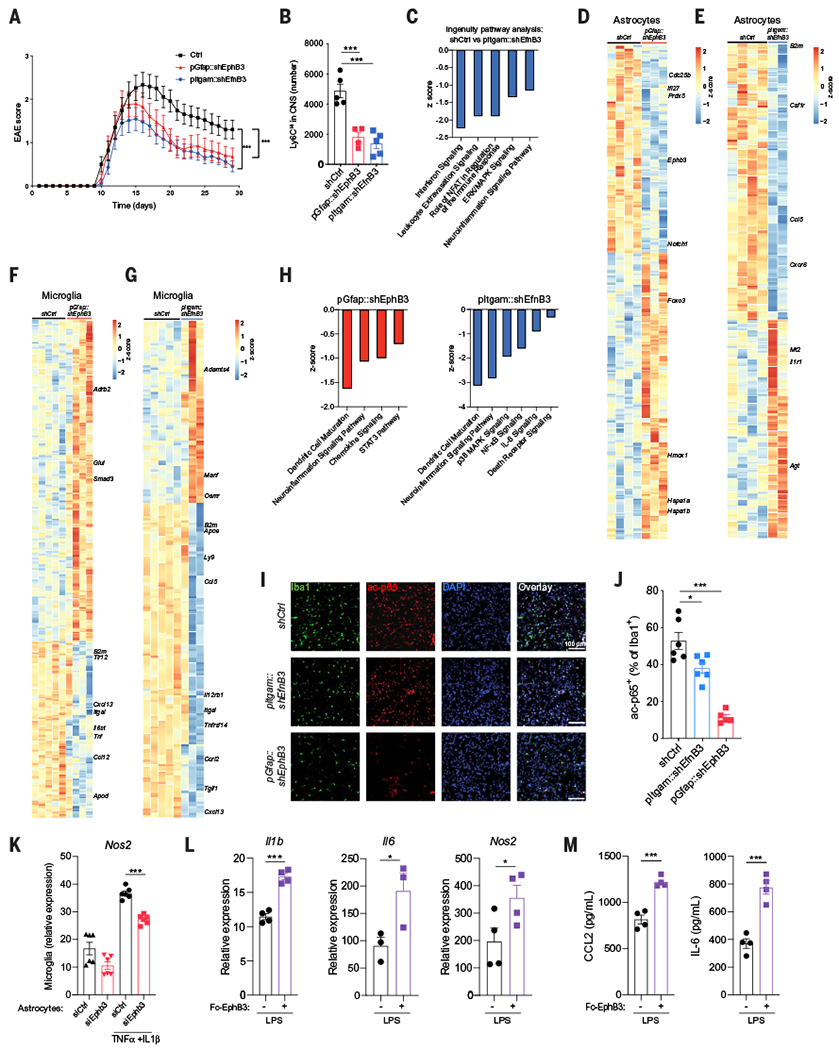
(A) EAE in knockdown mice transduced with lentiviral constructs targeting Ephb3 in astrocytes (red), Efnb3 in microglia (blue), or nontargeting control (black) Ctrl (n = 24 mice), pGfap::shEphb3 (n = 17 mice), pItgam::shEfnb3 (n = 22 mice). Statistical analysis: two-way ANOVA. (B) Quantification of proinflammatory monocytes in the CNS of EAE mice after the knockdown of Ephb3 in astrocytes (red), Efnb3 in microglia (blue), or nontargeting control (black). n = 5 mice per group. Statistical analysis: one-way ANOVA and Dunnett post-test. (C to E) RNA-seq analysis of astrocytes. (C) Differentially regulated pathways in astrocytes after Efnb3 knockdown in microglia analyzed by ingenuity pathways analysis. (D and E) Heatmap of differentially expressed genes in astrocytes after the knockdown of Ephb3 in astrocytes (D) or Efnb3 in microglia (E). (F to H) RNA-seq analysis of microglia isolated from EAE mice transduced with lentiviral vectors targeting Ephb3 in astrocytes, Efnb3 in microglia, or non-targeting control. (F and G) Heatmap of differentially expressed genes in microglia after knockdown of Ephb3 in astrocytes (F) or Efnb3 in microglia (G). (H) Relevant pathways selected from ingenuity pathway analysis of the genes differentially expressed in microglia after Ephb3 knockdown in astrocytes (left) and Efnb3 knockdown in microglia (right). (I) Immunostaining analysis of acetylated p65 (ac-p65) and Iba1 in the CNS of EAE knockdown mice. (J) Quantification of ac-p65+ Iba1+ cells. n = 6 mice per group. Statistical analysis: one-way ANOVA and Tukey post-test. (K) Astrocytes treated with control or Ephb3-targeting small interfering RNA and pretreated with TNFα and IL-1β were cocultured overnight with microglia, and microglial Nos2 expression was determined by qPCR. n = 6 samples per group. Statistical analysis: one-way ANOVA and Tukey post-test. (L and M) Neonatal mouse microglia cultured in plates precoated with EphB3-FcChimera and stimulated with LPS. (L) Illb, Il6, and Nos2 expression determined by qPCR and (M) CCL2 and IL-6 production quantified by enzyme-linked immunosorbent assay (ELISA) in supernatants. n = 4 samples per group. Statistical analysis: unpaired two-tailed t test. Data are representative of two independent experiments. Data shown as mean ± SEM. *P < 0.05, ***P < 0.001.
Analysis of the transcriptional response of astrocytes from EAE mice after the knockdown of Ephb3 in astrocytes or Efnb3 in microglia revealed decreased expression of genes associated with inflammation and neurodegeneration (Fig. 6, C to E). Thus, microglial Ephrin-B3 and astrocyte EphB3 receptors participate in the control of astrocyte proinflammatory activities during EAE.
Reverse Ephrin-B3 signaling boosts NF-κB driven responses in microglia during EAE
In addition to forward signaling in Ephb-expressing cells, the interaction between EphB receptors and their membrane-bound Ephrin-B ligands triggers reverse signaling in Efnb-expressing cells (46–50). In support of a role of reverse Ephrin-B3 signaling in the control of microglial responses during EAE, the knockdown of Efnb3 in microglia or Ephb3 in astrocytes decreased proinflammatory gene expression in microglia (Fig. 6, F to H). Moreover, in agreement with decreased microglial proinflammatory transcriptional responses detected by RNA-seq, we detected decreased NF-κB activation after the knockdown of Efnb3 in microglia or Ephb3 in astrocytes, suggesting that astrocyte Ephrin-B3 signaling boosts NF-κB driven proinflammatory transcriptional programs in microglia (Fig. 6, I and J).
Ephrin-B3–EphB3 interactions may modulate microglial responses via reverse signaling through Ephrin-B3 in microglia and also indirectly via EphB3-controlled astrocyte secreted factors. To study the role of reverse Ephrin-B3 signaling in the control of microglial responses, we cocultured mouse neonatal microglia in vitro with mouse neonatal astrocytes prestimulated with TNFα and IL-1β . Coculture with prestimulated astrocytes increased microglial Nos2 expression, and this increase was diminished by Ephb3 knockdown in astrocytes (Fig. 6K), suggesting that EphB3-induced reverse signaling contributes to the control of microglia activation by astrocytes. Indeed, plate-bound EphB3–Fc chimera boosted Il1b, Il6, and Nos2 expression as well as CCL2 and IL-6 secretion by mouse primary microglia activated in culture with lipopolysaccharide (LPS) (Fig. 6, L and M). Thus, reverse Ephrin-B3 signaling boosts microglial proinflammatory activities during EAE.
Pharmacologic inhibition of EphB3 receptor kinase ameliorates EAE
Multiple signaling events are triggered by EphB3 receptor activation; one of these signaling mechanisms is EphB3 kinase activity (43). A38 is a CNS-penetrant small molecule that inhibits EphB3 kinase activity (Fig. 7A) (51). We detected a dose-dependent inhibition of EphB3 kinase activity by A38 in a cell-free assay (fig. S14A). A38 did not induce cytotoxicity or apoptosis (fig. S14, B and C). To study the effects of EphB3 signaling in astrocytes, we pretreated neonatal mouse astrocytes in culture with A38 and activated them with TNFα and IL-1β. A38 reduced I16 expression in astrocytes in a dose-dependent manner (Fig. 7B); it also decreased the production of IL-6, CCL2, and TNFα (Fig. 7C). Similar results were obtained when we used C9, an additional inhibitor of EphB3 kinase activity (52) (fig. S14, D and E), or when A38 was tested on adult rodent astrocytes (fig. S14, F and G) or primary human astrocytes cultured in the presence of serum or in serum-free medium (fig. S14, H to J). Consistent with the higher levels of EphB3 detected in astrocytes compared with microglia (fig. S12B), A38 showed no effect on the microglial expression of the proinflammatory genes Nos2 and Tnfa induced by LPS stimulation (Fig. 7D), confirming that EphB3 intracellular signaling is important for the regulation of astrocyte responses but not microglial responses.
Fig. 7. Pharmacologic inhibition of EphB3 receptor kinase ameliorates EAE.
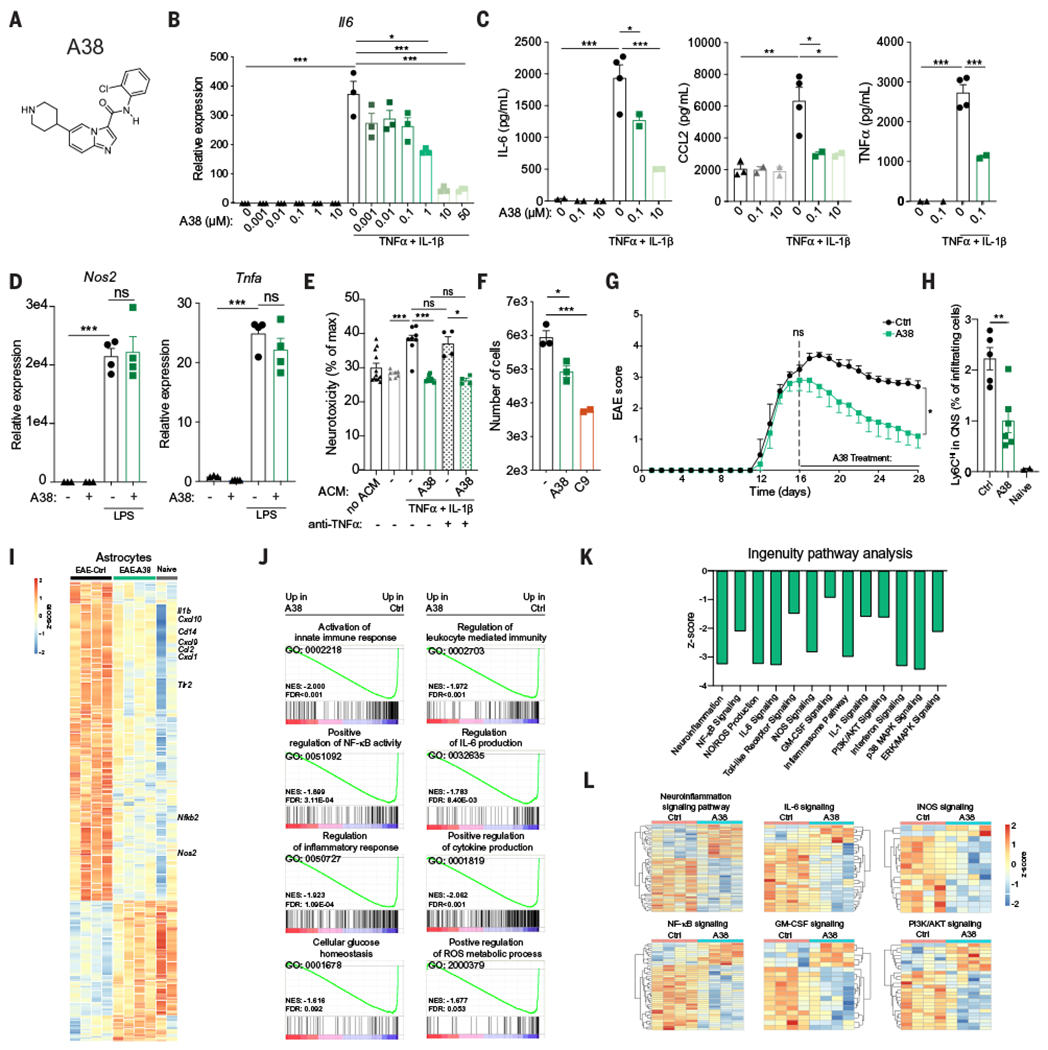
(A) A38 structure. (B) Il6 mRNA expression determined by qPCR in neonatal mouse astrocytes stimulated with TNFα and IL-1β in the presence of the indicated concentrations of A38. n = 3 samples per group. Statistical analysis: one-way ANOVA and Sidak post-test. (C) IL-6, CCL2, and TNFα concentration measured by ELISA in supernatants of neonatal mouse astrocytes stimulated as in (B) with the indicated concentrations of A38. n = 4 and 2 samples (0.1 and 10 groups). Statistical analysis: one-way ANOVA and Dunnett post-test. Data are representative of three independent experiments. (D) Nos2 and Tnfa mRNA expression determined by qPCR in neonatal mouse microglia stimulated with LPS in the presence of A38. n = 4 samples. Statistical analysis: one-way ANOVA and Tukey post-test. Data are representative of two independent experiments. (E and F) Primary mouse astrocytes were activated with TNFα and IL-1β, and treated with A38 or C9. Media was replaced, cells were extensively washed, and new medium was added 24 hours later; ACM was collected 48 hours later. (E) ACM was added to the mouse neuron cell line N2A preactivated with IFNγ, and cytotoxicity was determined by quantifying lactate dehydrogenase release after 24 hours; TNF blocking antibody was added where indicated. n = 8 samples per group but n = 4 samples for anti-TNFα groups. Statistical analysis: one-way ANOVA and Tukey post-test. Data are representative of two independent experiments. (F) Migration assay of splenic CD11b+ monocytes performed using ACM. n = 3 samples (- and A38), n = 3 samples (C9). Statistical analysis: one way ANOVA and Dunnett post-test. Data are representative of two independent experiments. (G) EAE in C57Bl/6J mice treated twice a day with vehicle or 20 mg/kg A38 injected intraperitoneally, starting at the peak of the disease. n = 5 mice per group. Statistical analysis: two-way ANOVA. Data are representative of two independent experiments. (H) Quantification of monocytes in the CNS of C57Bl/6J mice treated as in (A). n = 5 Ctrl mice, n = 6 A38 mice, and n = 2 naïve mice. Statistical analysis: one-way ANOVA and Tukey post-test. (I to L) RNA-seq analysis of astrocytes from naïve or EAE mice treated with A38 or vehicle. (I) Heatmap of differentially expressed genes in astrocytes. (J) GSEA of astrocytes. (K and L) Ingenuity pathway analysis of genes differentially expressed in astrocytes from A38-treated mice compared with vehicle-treated mice. Data shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. ns, not significant.
TNFα (53, 54) and nitric oxide (13, 55) have been linked to astrocyte pathogenic activities. Moreover, CCL2 produced by astrocytes promotes monocyte recruitment to the CNS (5, 56). Thus, based on the effects of A38 and C9 on TNFα, Nos2, and CCL2 expression, we evaluated neurotoxic and chemotactic activities of astrocyte-conditioned medium (ACM) collected from astrocytes pretreated with A38 or C9. The pharmacological inhibition of EphB3 in astrocytes reduced ACM neurotoxic and chemoattractant activity (Fig. 7, E and F). The neurotoxic activity was TNF independent (Fig. 7E and fig. S14K), suggesting that its inhibition by A38 involved the regulation of additional neurotoxicity mechanisms.
To evaluate the potential of the EphB3 kinase as a therapeutic target during CNS inflammation, we induced EAE in B6 wild-type mice by immunization with MOG35–55 and initiated treatment with A38 (20 mg/kg body weight twice a day by intraperitoneal injection) at the peak of the disease, using vehicle as a control. A38 administration ameliorated EAE, as indicated by the reduction in EAE scores (Fig. 7G) and the reduced demyelination and axonal loss detected in histopathological analyses (figs. S13, D to G). A38 also reduced the recruitment of Ly6CHi proinflammatory monocytes to the CNS (Fig. 7H) but did not affect T cell responses (fig. S15, A to C). A38 did not affect the glia limitans when compared with the vehicle-treated group (fig. S15D), as determined by the analysis of CLDN5 as described (57). Moreover, A38 administration decreased astrocyte and microglial expression of transcriptional modules associated with CNS inflammation and neurodegeneration, as determined by RNA-seq (Fig. 7, I to L, and fig. S15, E to H). A38 administration concomitant with Ephb3 knockdown in astrocytes did not further increase the therapeutic effects achieved by these interventions alone (figs. S14, D to G, and S15I), suggesting that the amelioration of EAE by A38 involves the inhibition of EphB3 kinase activity in astrocytes.
To further validate the potential of EphB3 kinase inhibition for the therapeutic modulation of CNS inflammation and neurodegeneration, we used the nonobese diabetic (NOD) mouse model of chronic progressive EAE induced by immunization with MOG35–55, which recapitulates aspects of secondary progressive MS, including the progressive and irreversible accumulation of neurologic disability (5, 9, 58, 59). Specifically, we evaluated the effects of A38 administration or Ephb3 knockdown in astrocytes during the progressive phase of NOD EAE (fig. S16A). Both the pharmacological inactivation of EphB3 kinase activity with A38 and the knockdown of Ephb3 in astrocytes by lentivirus-delivered shRNAs ameliorated NOD EAE, as indicated by the reduced clinical scores and the decreased recruitment of proinflammatory monocytes to the CNS; we did not detect changes in the peripheral T cell response (fig. S16, A to C). Additionally, A38 administration and Ephb3 knockdown in astrocytes suppressed the astrocyte and microglial expression of transcriptional modules associated with CNS inflammation and neurodegeneration (fig. S16, D to G). Thus, EphB3 signaling in astrocytes promotes CNS inflammation and is a candidate target for therapeutic intervention.
EphB3 kinase activates mTOR and boosts mitochondrial ROS production in astrocytes
Finally, we investigated the mechanisms involved in the control of astrocyte proinflammatory activities by EphB3 signaling. We established a signature score for the activation of the Ephrin pathway in astrocytes and analyzed microglia-astrocyte subnetworks that contained astrocytes with high (>90th percentile) and low (<10th percentile) scores (Fig. 8A). Differential expression analysis of astrocytes in these subnetworks identified PI3K-AKT-MTOR signaling as a potential pathway involved in Ephrin receptor signaling (Fig. 8B). Further analysis of protein interaction networks modulated by A38 treatment identified Pik3r1 as a candidate mediator of the effects of EphB3 signaling (Fig. 8C). PIK3R1 encodes the regulatory subunits (p85α, p55α, and p50α) of class I PI3Kα, which is associated with the control of innate immunity (60). Supporting a role for PIK3R1 in EphB3 signaling in astrocytes, A38 suppressed the phosphorylation of p85α, p55α, and their downstream signaling molecule AKT in primary mouse astrocytes in culture (Fig. 8D). Similarly, the pharmacological inhibition of class I PI3Kα by ZSTK474 suppressed the phosphorylation of its target AKT (Fig. 8D).
Fig. 8. EphB3 kinase activates mTOR and boosts mitochondrial ROS production in astrocytes.
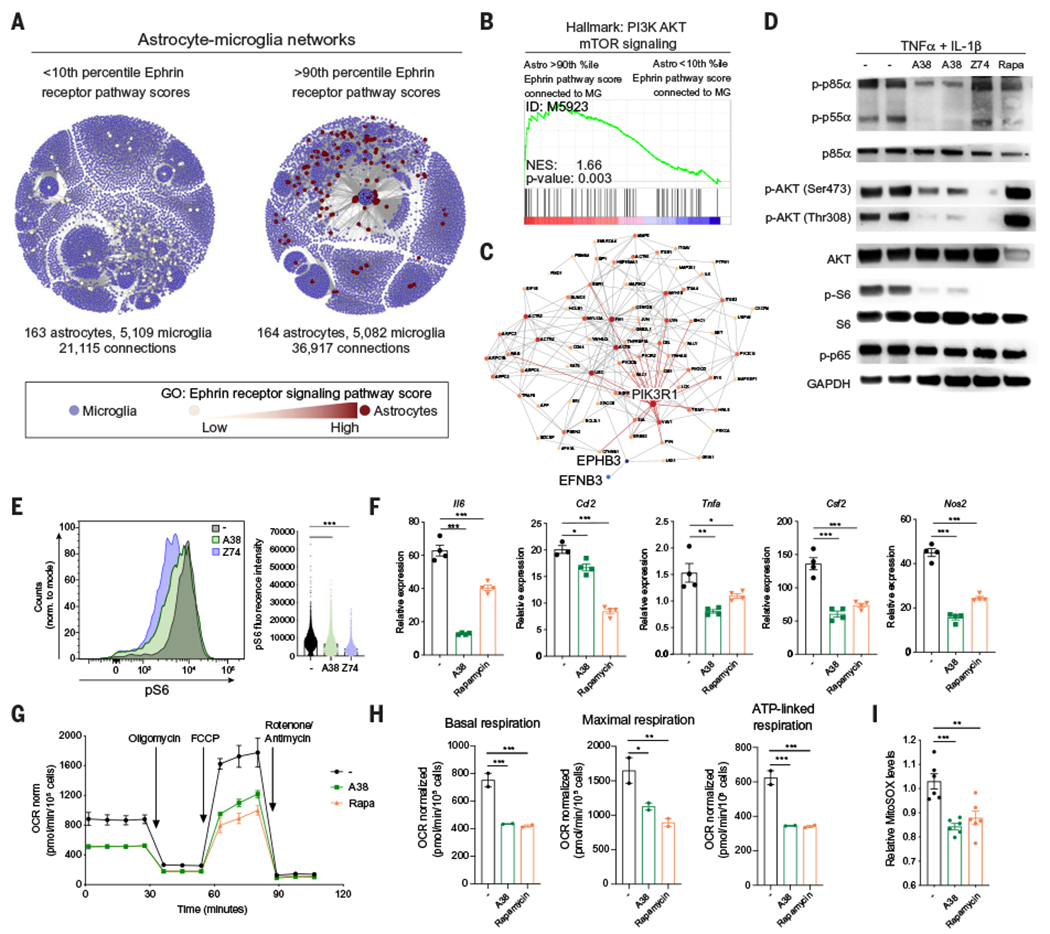
(A) Subnetworks of astrocytes interacting with microglia, as determined by RABID-seq and binned (<10th versus >90th percentile) based on their expression of Ephrin receptor pathway genes (MSigDB ID: M5923). (B) GSEA preranked analysis of RABID-seq data comparing >90th percentile astrocytes to <10th percentile astrocytes. (C) Protein-protein interaction analysis of the effects of A38 on astrocytes. (D) Western blot analysis of phosphorylated (p-) or total protein for p85α PIK3R1, p55α PIK3R1, AKT, S6, p65, and GAPDH in primary neonatal mouse astrocytes activated for 30 min with TNFα and IL-1β in the presence of the indicated compounds. Z74, ZSTK74 (class I PI3K isoforms inhibitor); Rapa, rapamycin. Blots are representative of three independent experiments. (E) Analysis of S6 phosphorylation determined by intracellular staining and flow cytometry of astrocytes stimulated as in (D). Statistical analysis: one way ANOVA and Tukey post-test. (F) Il6, Ccl2, Tnfa, Csf2, and Nos2 expression determined by qPCR in neonatal mouse astrocytes stimulated with TNFα and IL-1β in the presence of A38 or rapamycin. n = 4 samples per group. Statistical analysis: one way ANOVA and Dunnett post-test. Data are representative of three independent experiments. (G) Seahorse mitochondrial stress test performed on astrocytes pretreated overnight with A38 or rapamycin. n = 2 samples. OCR, oxygen consumption rate; FCCP, trifluoromethoxy carbonylcyanide phenylhydrazone. (H) Quantification of basal mitochondrial respiration, maximal mitochondrial respiration, and ATP-linked respiration calculated from mitochondrial stress assay from (E). n = 2 samples per group. Statistical analysis: one-way ANOVA and Tukey post-test. Data are representative of three independent experiments. (I) Mitochondrial ROS measured by MitoSOX staining after overnight treatment with IL-1β/TNFα and A38 or rapamycin. n = 6 samples per group. Statistical analysis: one-way ANOVA and Tukey post-test. Data are representative of three independent experiments. Data shown as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
AKT is reported to activate the transcription factor NF-κB and the mammalian target of rapamycin (mTOR) (61). The inhibition of AKT phosphorylation by A38 did not suppress NF-κB activation, as determined by the analysis of its phosphorylation and nuclear translocation in primary astrocytes stimulated with TNFα and IL-1β (Fig. 8D and fig. S17A). However, A38 suppressed S6 phosphorylation downstream of mTORC1, suggesting that A38 interferes with mTOR activation (Fig. 8D). Indeed, we detected reduced S6 phosphorylation when we used flow cytometry to analyze the effects of the pharmacological inhibition of EphB3 kinase or PI3K in primary astrocytes activated in vitro with TNFα and IL-1β (Fig. 8E). Thus, through its effects on the PI3K-AKT axis, EphB3 kinase activity promotes mTOR activation in astrocytes.
To investigate the role of mTOR on astrocyte responses, we used the mTOR inhibitor rapamycin (62). Rapamycin treatment suppressed the expression of proinflammatory genes in astrocytes stimulated in vitro with TNFα and IL-1β (Fig. 8F), recapitulating our previous observations on the effects of A38 on astrocytes. Consistent with its inhibitory effects on mTOR, rapamycin suppressed S6 phosphorylation but did not suppress the phosphorylation of AKT, p85, or NF-κB subunit p65 (Fig. 8D).
mTOR controls mitochondrial function (63), which has been linked to pathogenic activities of astrocytes and microglia in neurologic disorders (9, 64). Indeed, mTOR-driven mitochondrial respiration produces reactive oxygen species (ROS), which promote proinflammatory gene expression and contribute to neurodegeneration (65–67). Thus, we investigated the effects of A38 on mitochondrial function and ROS production. The inhibition of EphB3 kinase activity by A38 decreased basal and maximal mitochondrial respiration, as well as adenosine triphosphate (ATP)–linked respiration in primary astrocytes (Fig. 8, G and H); similar results were obtained with rapamycin. Consistent with its suppressive effects on mitochondrial respiration, A38 and rapamycin also decreased the mitochondrial production of ROS induced in astrocytes by stimulation with TNFα and IL-1β (Fig. 8I). Accordingly, we detected a decrease in the expression of genes related to ROS metabolic processes in astrocytes isolated from A38-treated EAE mice (Fig. 7J). Moreover, treatment with rotenone and antimycin A, which increase mitochondrial ROS levels (68–70) (fig. S17B), boosted the expression of proinflammatory genes in astrocytes (fig. S17C). Thus, the production of mitochondrial ROS driven by PI3K-AKT-mTOR signaling contributes to the proinflammatory effects of EphB3 kinase activation in astrocytes.
Discussion
Several techniques have been developed to infer cell interactions on the basis of spatial transcriptomics (71–77), physical interactions (17, 18) or colocalization in tissue (19, 20). However, these approaches remain difficult to apply to the study of cells in the CNS, owing to technical complexity, throughput limitations, or a lack of single-cell resolution with respect to transcriptional or interaction information. To overcome these limitations, we developed RABID-seq, an unbiased, high-throughput, and accessible technology for the study of cell interactions. RABID-seq uses glycoprotein G–deficient pseudotyped rabies virus engineered to express a unique mCherry mRNA barcode to capture interactions directly from transcriptomic data, thereby exploiting the maturity and ubiquity of next-generation sequencing (78) and lowering the barrier to technology adoption.
Microglia-astrocyte communication plays a central role in CNS physiology (79). Using RABID-seq, we identified axon guidance molecules as participants in microglia-astrocyte communication in the context of CNS inflammation. Axon guidance molecules have been shown to be co-opted in the context of cancer and inflammation (80, 81), but their participation in microglia-astrocyte interactions was unknown. We identified a role for Sema4D-PlexinB1 and Sema4D-PlexinB2 interactions in the microglial control of astrocytes during EAE and MS. Sema4D is described to participate in neurodevelopment (82, 83), T cell activation (36, 84), and T cell–driven microglial activation in EAE (36, 85). Our findings suggest that developing MS therapies that target SEMA4D could be improved if the therapeutic agent reaches the CNS (86).
Most microglia-astrocyte interactions known to participate in the control of CNS inflammation are mediated by soluble factors (2, 5, 6, 11, 12, 58, 87). We identified Ephrin-B3–EphB3 signaling as a participant in microglia-astrocyte contact–dependent cross-talk. Eph receptor signaling plays important roles in development (43); immune regulation (88); maintenance of epithelial architecture (89); and control of neural progenitor proliferation, axonal guidance, and synapse formation (90–92). Eph receptors participate in astrocyte-neuron communication (93–95), which suggests that, in the context of inflammation, Ephrin-B3–EphB3 signaling may mediate interactions of astrocytes with cell types other than microglia. Notably, EphA receptor up-regulation in astrocytes has been reported in MS patients (96), and EphB2 (1) and EphB3 (97) are increased in astrocytes after spinal cord injury. Eph receptor signaling has also been linked to the pathology of Alzheimer’s disease (98), Parkinson’s disease (99), amyotrophic lateral sclerosis (100), and schizophrenia (101). In this sense, Eph receptor signaling resembles IL-33 (11, 102) and complement (103–106), which participate in CNS development but are also reactivated in neurologic diseases. However, a distinguishing feature of Ephrin-Eph receptor interactions is the induction of reverse signaling in Ephrin expressing cells (46–50), which amplifies NF-κB–driven proinflammatory responses in microglia.
We found that EphB3 kinase activity in astrocytes modulates mTOR activation and mitochondrial ROS production. In myeloid cells in the CNS, the genetic inhibition of ROS production decreases proinflammatory gene expression (67). ROS triggers the production of proinflammatory cytokines via the regulation of NLRP3 and MAPK (65, 66). EphB3 and EphA4 signaling in astrocytes induces the production of d-serine (107), which promotes synaptic damage by acting on N-methyl-d-aspartate receptors (108). Thus, EphB3 signaling provides a mechanism for the microglial control of astrocyte metabolism and its multiple effects on CNS inflammation. It is conceivable that microglial control of astrocyte metabolism via EphB3 not only affects proinflammatory and neurotoxic responses but also interferes with the metabolic support of neurons by astrocytes in the context of inflammation, as recently reported (5, 9).
Our studies identify Ephrin-B3–EphB3 signaling as a candidate target for the therapeutic modulation of astrocyte and microglial pathogenic activities in MS. The therapeutic blockade of Ephrin-B3–EphB3 signaling may interfere with disease-promoting responses in astrocytes and microglia, as well as additional mechanisms associated with MS pathology and linked to this pathway, including the disruption of the blood-brain barrier (109) and the inhibition of remyelination (110). In this context, CNS-penetrant small molecules, such as A38 described in this work, provide better therapeutic approaches than other Eph targeting therapies (111) that have limited access to the CNS. However, potential off-target effects are possible as a result of the multiple cell- and context-specific signaling mechanisms that have been linked to Eph receptors (43, 112, 113).
In summary, we developed RABID-seq, an approach for the high-throughput identification of cell interactions and the molecular phenotype of interacting cells with single-cell resolution. RABID-seq enabled the identification of microglia-astrocyte interactions mediated by the axon guidance molecules Sema4D-PlexinB1, Sema4D-PlexinB2, and Ephrin-B3–EphB3, which represent candidate targets for therapeutic intervention in MS and other neurologic disorders.
Materials and methods summary
The supplementary materials provide a detailed description of our materials and methods. Adult C57BL/6J (no. 000664) and NOD/ShiLtJ (NOD mice) (no. 001976) were obtained from The Jackson Laboratory. B6.Cg-Tg(Gfap-cre) 73.12Mvs/J hemizygous mice (The Jackson Laboratory, no. 012886) were crossed to homozygous B6;129P2-Gt(ROSA)26Sortm1(CAG-RABVgp4,-TVA)Arenk/J mice (The Jackson Laboratory, no. 024708). Tg(CAG-Kaede)15Kgwa mice (114) on a C57Bl/6J background were obtained from RIKEN BRC. EAE was induced as previously described (3, 9, 13).
Barcoded rabies virus was created by replacing green fluorescent protein (GFP) with mCherry in pSADΔG-GFP-F2 (Addgene, no. 32635), followed by insertion of a 28–base pair semirandom anchored barcode downstream of the mCherry translational stop codon using Gibson assembly. Pseudotyped G-deficient rabies virus was produced as previously described (115). Intracranial delivery of RabΔG was performed largely as described previously (3, 13). The forebrain was targeted unilaterally using the following coordinates: +1.25 (lateral), +1.0 (rostral), −3.0 (ventral) relative to Bregma. Cells were isolated by flow cytometry on a FACS Aria IIu cell sorter (BD Biosciences). Sorting of mCherry+ cells at low flow rates through a 100-μm nozzle was judged in the PE-Texas Red channel using a yellow-green laser. After sorting, cells were scRNA-sequenced using the inDrops workflow (116) (v3 beads, Harvard Single Cell Core) with modifications to the molecular biology to enable reverse transcription with template switching in drops. Illumina sequencing libraries for transcriptome sequencing were prepared from purified whole-transcriptome–amplified product using an adapter ligation approach with the NEBNext Ultra II FS Kit (NEB, no. E7805). Illumina sequencing libraries for rabies barcode sequencing were prepared from the same material using a two-step nested PCR protocol. InDrops scRNA-seq data were processed using the publicly available bioinformatics pipeline (https://github.com/indrops/indrops) (117). Connectome data were analyzed using scripts developed for this work that are publicly available on GitHub at https://github.com/Zha0rong/RABID-seq.
Supplementary Material
ACKNOWLEDGMENTS
We thank all members of the Quintana laboratory for helpful advice and discussions; G. Lipof for technical assistance; D. Kozoriz, R. Krishnan, and A. Chicoine for technical assistance with flow cytometry studies; the Harvard Medical School Rodent Histopathology Core, which provided histopathology service; the Harvard Medical School Microfluidics Facility for access to soft lithography equipment; the NeuroTechnology Studio at Brigham and Women’s Hospital for providing NextSeq550 instrument access; and S. Boswell, A. Ratner, and the Harvard Medical School Single Cell Core for assistance with the inDrop workflow and discussions and protocols regarding the SMART inDrop workflow.
Funding: This work was supported by grants NS102807, ES02530, ES029136, and AI126880 from the NIH; RG4111A1 and JF2161-A-5 from the National Multiple Sclerosis Society; RSG-14-198-01-LIB from the American Cancer Society; and PA-160408459 from the International Progressive MS Alliance (to F.J.Q.). F.J.Q. and M.P. were supported by the DFG-funded CRC/TRR167 “NeuroMac.” C.G.-V. was supported by an Alfonso Martin Escudero Foundation postdoctoral fellowship and by a postdoctoral fellowship (ALTF 610-2017) from the European Molecular Biology Organization. M.A.W. was supported by NIH (1K99NS114111 and F32NS101790), a training grant from the NIH and Dana-Farber Cancer Institute (T32CA207201), and a traveling neuroscience fellowship from the Program in Interdisciplinary Neuroscience at Brigham and Women’s Hospital. I.C.C. and M.A.W. received support from the Women’s Brain Initiative at Brigham and Women’s Hospital. V.R. received support from an educational grant from Mallinkrodt Pharmaceuticals (A219074) and a fellowship from the German Research Foundation (DFG RO4866 1/1). S.E.J.Z. was supported by a joint FRQS-EndMS postdoctoral fellowship. C.-C.C. received support from a postdoctoral research abroad program (104-2917-I-564-024) from the Ministry of Science and Technology, Taiwan K.V.B. was supported by F30AG063463 from the NIH. P.L. was supported by 5T32AG000222 from the NIH. C.M.P. was supported by a fellowship from FAPESP BEPE (2019/13731-0). D.L.R. was supported by R21AG061678 from the NIH. A.Pr. holds a senior Canada Research Chair in Multiple Sclerosis. Sanger sequencing was carried out at the DNA Resource Core of Dana-Farber/Harvard Cancer Center (funded in part by NCI Cancer Center support grant 2P30CA006516-48).
Footnotes
Competing interests: I.C.C., M.A.W., C.G.-V., K.J.H., and F.J.Q. have filed a provisional patent application for the barcoding strategy and the therapeutic targeting of EphB signaling outlined in this paper.
Data and materials availability: Sequencing data have been deposited into GEO under the SuperSeries accession number GSE150858. The code that supports the findings of this study has been deposited in GitHub at https://github.com/Zha0rong/RABID-seq. All other materials will be made available upon reasonable request.
SUPPLEMENTARY MATERIALS
science.sciencemag.org/content/372/6540/eabf1230/suppl/DC1
MDAR Reproducibility Checklist
SUPPLEMENTARY MATERIALS
http://science.sciencemag.org/content/suppl/2021/04/21/372.6540.eabf1230.DC1
REFERENCES AND NOTES
- 1.Anderson MA et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200 (2016). doi: 10.1038/nature17623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liddelow SA et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017). doi: 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wheeler MA et al. MAFG-driven astrocytes promote CNS inflammation. Nature 578, 593–599 (2020). doi: 10.1038/s41586-020-1999-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenhalgh AD, David S, Bennett FC, Immune cell regulation of glia during CNS injury and disease. Nat. Rev. Neurosci 21, 139–152 (2020). doi: 10.1038/s41583-020-0263-9 [DOI] [PubMed] [Google Scholar]
- 5.Mayo L. et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat. Med 20, 1147–1156 (2014). doi: 10.1038/nm.3681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rothhammer V. et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med 22, 586–597 (2016). doi: 10.1038/nm.4106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sofroniew MV, Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci 16, 249–263 (2015). doi: 10.1038/nrn3898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wheeler MA, Quintana FJ, Regulation of Astrocyte Functions in Multiple Sclerosis. Cold Spring Harb. Perspect. Med 9, a029009 (2019). doi: 10.1101/cshperspect.a029009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chao CC et al. Metabolic Control of Astrocyte Pathogenic Activity via cPLA2-MAVS. Cell 179, 1483–1498.e22 (2019). doi: 10.1016/j.cell.2019.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanmarco LM et al. Gut-licensed IFNα+ NK cells drive LAMP1+TRAIL+ anti-inflammatory astrocytes. Nature 590, 473–479 (2021). doi: 10.1038/s41586-020-03116-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vainchtein ID et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 359, 1269–1273 (2018). doi: 10.1126/science.aal3589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rothhammer V. et al. Microglial control of astrocytes in response to microbial metabolites. Nature 557, 724–728 (2018). doi: 10.1038/s41586-018-0119-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wheeler MA et al. Environmental Control of Astrocyte Pathogenic Activities in CNS Inflammation. Cell 176, 581–596.e18 (2019). doi: 10.1016/j.cell.2018.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fontana A, Fierz W, Wekerle H, Astrocytes present myelin basic protein to encephalitogenic T-cell lines. Nature 307, 273–276 (1984). doi: 10.1038/307273a0 [DOI] [PubMed] [Google Scholar]
- 15.Sun D, Wekerle H, Ia-restricted encephalitogenic T lymphocytes mediating EAE lyse autoantigen-presenting astrocytes. Nature 320, 70–72 (1986). doi: 10.1038/320070a0 [DOI] [PubMed] [Google Scholar]
- 16.Locatelli G. et al. Mononuclear phagocytes locally specify and adapt their phenotype in a multiple sclerosis model. Nat. Neurosci 21, 1196–1208 (2018). doi: 10.1038/s41593-018-0212-3 [DOI] [PubMed] [Google Scholar]
- 17.Giladi A. et al. Dissecting cellular crosstalk by sequencing physically interacting cells. Nat. Biotechnol 38, 629–637 (2020). doi: 10.1038/s41587-020-0442-2 [DOI] [PubMed] [Google Scholar]
- 18.Pasqual G. et al. Monitoring T cell-dendritic cell interactions in vivo by intercellular enzymatic labelling. Nature 553, 496–500 (2018). doi: 10.1038/nature25442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turczyk BM et al. Spatial Sequencing: A Perspective. J. Biomol. Tech 31, 44–46 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stoeckius M. et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017). doi: 10.1038/nmeth.4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hagendorf N, Conzelmann KK, Pseudotyping of G-Gene-Deficient Rabies Virus. Cold Spring Harb. Protoc 2015, prot089417 (2015). doi: 10.1101/pdb.prot089417 [DOI] [PubMed] [Google Scholar]
- 22.Mount CW, Yalçin B, Cunliffe-Koehler K, Sundaresh S, Monje M, Monosynaptic tracing maps brain-wide afferent oligodendrocyte precursor cell connectivity. eLife 8, e49291 (2019). doi: 10.7554/eLife.49291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Motori E et al. Inflammation-induced alteration of astrocyte mitochondrial dynamics requires autophagy for mitochondrial network maintenance. Cell Metab. 18, 844–859 (2013). doi: 10.1016/j.cmet.2013.11.005 [DOI] [PubMed] [Google Scholar]
- 24.Card JP et al. , Pseudorabies virus infection of the rat central nervous system: Ultrastructural characterization of viral replication, transport, and pathogenesis. J. Neurosci 13, 2515–2539 (1993). doi: 10.1523/JNEUROSCI.13-06-02515.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wickersham IR et al. Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron 53, 639–647 (2007). doi: 10.1016/j.neuron.2007.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wall NR, Wickersham IR, Cetin A, De La Parra M, Callaway EM, Monosynaptic circuit tracing in vivo through Cre-dependent targeting and complementation of modified rabies virus. Proc. Natl. Acad. Sci. U.S.A 107, 21848–21853 (2010). doi: 10.1073/pnas.1011756107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia AD, Doan NB, Imura T, Bush TG, Sofroniew MV, GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat. Neurosci 7, 1233–1241 (2004). doi: 10.1038/nn1340 [DOI] [PubMed] [Google Scholar]
- 28.Callaway EM, Luo L, Monosynaptic Circuit Tracing with Glycoprotein-Deleted Rabies Viruses. J. Neurosci 35, 8979–8985 (2015). doi: 10.1523/JNEUROSCI.0409-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reardon TR et al. Rabies Virus CVS-N2c(ΔG) Strain Enhances Retrograde Synaptic Transfer and NeuronalViability. Neuron 89, 711–724 (2016). doi: 10.1016/j.neuron.2016.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang Z. et al. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity 32, 414–425 (2010). doi: 10.1016/j.immuni.2010.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie L, Choudhury GR, Winters A, Yang SH, Jin K, Cerebral regulatory T cells restrain microglia/macrophage-mediated inflammatory responses via IL-10. Eur. J. Immunol 45, 180–191 (2015). doi: 10.1002/eji.201444823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mayo L. et al. IL-10-dependent Tr1 cells attenuate astrocyte activation and ameliorate chronic central nervous system inflammation. Brain 139, 1939–1957 (2016). doi: 10.1093/brain/aww113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ito M. et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 565, 246–250 (2019). doi: 10.1038/s41586-018-0824-5 [DOI] [PubMed] [Google Scholar]
- 34.Vento-Tormo R. et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 563, 347–353 (2018). doi: 10.1038/s41586-018-0698-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang KW, Sabatini BL, Single-Cell Analysis of Neuroinflammatory Responses Following Intracranial Injection of G-Deleted Rabies Viruses. Front. Cell. Neurosci 14, 65 (2020). doi: 10.3389/fncel.2020.00065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumanogoh A, Kikutani H., Immunological functions of the neuropilins and plexins as receptors for semaphorins.Nat. Rev. Immunol 13, 802–814 (2013). doi: 10.1038/nri3545 [DOI] [PubMed] [Google Scholar]
- 37.Tamagnone L. et al. Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell 99, 71–80 (1999). doi: 10.1016/S0092-8674(00)80063-X [DOI] [PubMed] [Google Scholar]
- 38.Elhabazi A, Delaire S, Bensussan A, Boumsell L, Bismuth G., Biological activity of soluble CD100. I. The extracellular region of CD100 is released from the surface of T lymphocytes by regulated proteolysis. J. Immunol 166, 4341–4347 (2001). doi: 10.4049/jimmunol.166.7.4341 [DOI] [PubMed] [Google Scholar]
- 39.Janssen BJC et al. , Structural basis of semaphorin-plexin signalling. Nature 467, 1118–1122 (2010). doi: 10.1038/nature09468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu L. et al. , Regulated surface expression and shedding support a dual role for semaphorin 4D in platelet responses to vascular injury. Proc. Natl. Acad. Sci. U.S.A 104, 1621–1626 (2007). doi: 10.1073/pnas.0606344104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fazzari P. et al. Plexin-B1 plays a redundant role during mouse development and in tumour angiogenesis. BMC Dev. Biol 7, 55 (2007). doi: 10.1186/1471-213X-7-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paldy E. et al. Semaphorin 4C Plexin-B2 signaling in peripheral sensory neurons is pronociceptive in a model of inflammatory pain. Nat. Commun 8, 176 (2017). doi: 10.1038/s41467-017-00341-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kania A, Klein R, Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat. Rev. Mol. Cell Biol 17, 240–256 (2016). doi: 10.1038/nrm.2015.16 [DOI] [PubMed] [Google Scholar]
- 44.Imondi R, Wideman C, Kaprielian Z, Complementary expression of transmembrane ephrins and their receptors in the mouse spinal cord: A possible role in constraining the orientation of longitudinally projecting axons. Development 127, 1397–1410 (2000). [DOI] [PubMed] [Google Scholar]
- 45.Mildner A. et al. CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain 132, 2487–2500 (2009). doi: 10.1093/brain/awp144 [DOI] [PubMed] [Google Scholar]
- 46.Brückner K, Pasquale EB, Klein R, Tyrosine phosphorylation of transmembrane ligands for Eph receptors. Science 275, 1640–1643 (1997). doi: 10.1126/science.275.5306.16407 [DOI] [PubMed] [Google Scholar]
- 47.Cowan CA, Henkemeyer M, The SH2/SH3 adaptor Grb4 transduces B-ephrin reverse signals. Nature 413, 174–179 (2001). doi: 10.1038/35093123 [DOI] [PubMed] [Google Scholar]
- 48.Henkemeyer M. et al. Nuk controls pathfinding of commissural axons in the mammalian central nervoussystem. Cell 86, 35–46 (1996). doi: 10.1016/S0092-8674(00)80075-6 [DOI] [PubMed] [Google Scholar]
- 49.Holland SJ et al. Bidirectional signalling through the EPH-family receptor Nuk and its transmembrane ligands. Nature 383, 722–725 (1996). doi: 10.1038/383722a0 [DOI] [PubMed] [Google Scholar]
- 50.Klein R, Bidirectional modulation of synaptic functions by Eph/ephrin signaling. Nat. Neurosci 12, 15–20 (2009). doi: 10.1038/nn.2231 [DOI] [PubMed] [Google Scholar]
- 51.Qiao L. et al. Structure-activity relationship study of EphB3 receptor tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett 19, 6122–6126 (2009). doi: 10.1016/j.bmcl.2009.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kung A. et al. Development of Specific, Irreversible Inhibitors for a Receptor Tyrosine Kinase EphB3. J. Am. Chem. Soc 138, 10554–10560 (2016). doi: 10.1021/jacs.6b05483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lock C. et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat. Med 8, 500–508 (2002). doi: 10.1038/nm0502-500 [DOI] [PubMed] [Google Scholar]
- 54.Ye L. et al. IL-1β and TNF-α induce neurotoxicity through glutamate production: A potential role for neuronal glutaminase. J. Neurochem 125, 897–908 (2013). doi: 10.1111/jnc.12263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hewett SJ, Csernansky CA, Choi DW, Selective potentiation of NMDA-induced neuronal injury following induction of astrocytic iNOS. Neuron 13, 487–494 (1994). doi: 10.1016/0896-6273(94)90362-X [DOI] [PubMed] [Google Scholar]
- 56.Weiss JM, Berman JW, Astrocyte expression of monocyte chemoattractant protein-1 is differentially regulated by transforming growth factor beta. J. Neuroimmunol 91, 190–197 (1998). doi: 10.1016/S0165-5728(98)00183-0 [DOI] [PubMed] [Google Scholar]
- 57.Alvarez JI et al. , The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 334, 1727–1731 (2011). doi: 10.1126/science.1206936 [DOI] [PubMed] [Google Scholar]
- 58.Rothhammer V. et al. Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc. Natl. Acad. Sci. U.S.A 114, 2012–2017 (2017). doi: 10.1073/pnas.1615413114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Simmons SB, Pierson ER, Lee SY, Goverman JM, Modeling the heterogeneity of multiple sclerosis in animals. Trends Immunol. 34, 410–422 (2013). doi: 10.1016/j.it.2013.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fruman DA et al. The PI3K Pathway in Human Disease. Cell 170, 605–635 (2017). doi: 10.1016/j.cell.2017.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Inoki K, Li Y, Zhu T, Wu J, Guan KL, TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol 4, 648–657 (2002). doi: 10.1038/ncb839 [DOI] [PubMed] [Google Scholar]
- 62.Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH, RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 78, 35–43 (1994). doi: 10.1016/0092-8674(94)90570-3 [DOI] [PubMed] [Google Scholar]
- 63.Cunningham JT et al. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 450, 736–740 (2007). doi: 10.1038/nature06322 [DOI] [PubMed] [Google Scholar]
- 64.Ulland TK et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 170, 649–663.e13 (2017). doi: 10.1016/j.cell.2017.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bulua AC et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med 208, 519–533 (2011). doi: 10.1084/jem.20102049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ip WKE, Hoshi N, Shouval DS, Snap per S, Medzhitov R, Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 356, 513–519 (2017). doi: 10.1126/science.aal3535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mendiola AS et al. Transcriptional profiling and therapeutic targeting of oxidative stress in neuroinflammation. Nat. Immunol 21, 513–524 (2020). doi: 10.1038/s41590-020-0654-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li N. et al. Mitochon drial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem 278, 8516–8525 (2003). doi: 10.1074/jbc.M210432200 [DOI] [PubMed] [Google Scholar]
- 69.Sarafian TA et al. Disruption of astrocyte STAT3 signaling decreases mitochondrial function and increases oxidative stress in vitro. PLOS ONE 5, e9532 (2010). doi: 10.1371/journal.pone.0009532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang L. et al. Mitochondrial Respiratory Chain Inhibitors Involved in ROS Production Induced by Acute High Concentrations of Iodide and the Effects of SOD as a Protective Factor. Oxid. Med. Cell. Longev 2015, 217670 (2015). doi: 10.1155/2015/217670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang X. et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 361, eaat5691 (2018). doi: 10.1126/science.aat5691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moffitt JR et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362, eaau5324 (2018). doi: 10.1126/science.aau5324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee JH et al. Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat. Protoc 10, 442–458 (2015). doi: 10.1038/nprot.2014.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rodriques SG et al. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019). doi: 10.1126/science.aaw1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ståhl PL et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016). doi: 10.1126/science.aaf2403 [DOI] [PubMed] [Google Scholar]
- 76.Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X, RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015). doi: 10.1126/science.aaa6090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee JH et al. Highly multiplexed subcellular RNA sequencing in situ. Science 343, 1360–1363 (2014). doi: 10.1126/science.1250212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zador AM et al. Sequencing the connectome. PLOS Biol. 10, e1001411 (2012). doi: 10.1371/journal.pbio.1001411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vainchtein ID, Molofsky AV, Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci. 43, 144–154 (2020). doi: 10.1016/j.tins.2020.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nishide M, Kumanogoh A, The role of semaphorins in immune responses and autoimmune rheumatic diseases. Nat. Rev. Rheumatol 14, 19–31 (2018). doi: 10.1038/nrrheum.2017.201 [DOI] [PubMed] [Google Scholar]
- 81.Neufeld G, Kessler O, The semaphorins: Versatile regulators of tumour progression and tumour angiogenesis. Nat. Rev. Cancer 8, 632–645 (2008). doi: 10.1038/nrc2404 [DOI] [PubMed] [Google Scholar]
- 82.Tran TS, Kolodkin AL, Bharadwaj R, Semaphorin regulation of cellular morphology. Annu. Rev. Cell Dev. Biol 23, 263–292 (2007). doi: 10.1146/annurev.cellbio.22.010605.093554 [DOI] [PubMed] [Google Scholar]
- 83.Pasterkamp RJ, Getting neural circuits into shape with semaphorins. Nat. Rev. Neurosci 13, 605–618 (2012). doi: 10.1038/nrn3302 [DOI] [PubMed] [Google Scholar]
- 84.Kumanogoh A. et al. Requirement for the lymphocyte semaphorin, CD100, in the induction of antigen-specific T cells and the maturation of dendritic cells. J. Immunol 169, 1175–1181 (2002). doi: 10.4049/jimmunol.169.3.1175 [DOI] [PubMed] [Google Scholar]
- 85.Okuno T. et al. Roles of Sema4D-plexin-B1 interactions in the central nervous system for pathogenesis of experimental autoimmune encephalomyelitis. J. Immunol 184, 1499–1506 (2010). doi: 10.4049/jimmunol.0903302 [DOI] [PubMed] [Google Scholar]
- 86.Worzfeld T, Offermanns S, Semaphorins and plexins as therapeutic targets. Nat. Rev. Drug Discov 13, 603–621 (2014). doi: 10.1038/nrd4337 [DOI] [PubMed] [Google Scholar]
- 87.Linnerbauer M, Wheeler MA, Quintana FJ, Astrocyte Crosstalk in CNS Inflammation. Neuron 108, 608–622 (2020). doi: 10.1016/j.neuron.2020.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lu P, Shih C, Qi H, Ephrin B1-mediated repulsion and signaling control germinal center T cell territoriality and function. Science 356, eaai9264 (2017). doi: 10.1126/science.aai9264 [DOI] [PubMed] [Google Scholar]
- 89.Batlle E. et al. EphB receptor activity suppresses colorectal cancer progression. Nature 435, 1126–1130 (2005). doi: 10.1038/nature03626 [DOI] [PubMed] [Google Scholar]
- 90.Contractor A. et al. Trans-synaptic Eph receptor-ephrin signaling in hippocampal mossy fiber LTP. Science 296, 1864–1869 (2002). doi: 10.1126/science.1069081 [DOI] [PubMed] [Google Scholar]
- 91.Lim BK, Matsuda N, Poo MM, Ephrin-B reverse signaling promotes structural and functional synaptic maturation in vivo. Nat. Neurosci 11, 160–169 (2008). doi: 10.1038/nn2033 [DOI] [PubMed] [Google Scholar]
- 92.McClelland AC, Sheffler-Collins SI, Kayser MS, Dalva MB, Ephrin-B1 and ephrin-B2 mediate EphB-dependent presynaptic development via syntenin-1. Proc. Natl. Acad. Sci. U.S.A 106, 20487–20492 (2009). doi: 10.1073/pnas.0811862106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Carmona MA, Murai KK, Wang L , Roberts AJ, Pasquale EB, Glial ephrin-A3 regulates hippocampal dendritic spine morphology and glutamate transport. Proc. Natl. Acad. Sci. U.S.A 106, 12524–12529 (2009). doi: 10.1073/pnas.0903328106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Filosa A. et al. Neuron-glia communication via EphA4/ephrin-A3 modulates LTP through glial glutamate transport. Nat. Neurosci 12, 1285–1292 (2009). doi: 10.1038/nn.2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Murai KK, Nguyen LN, Irie F, Yamaguchi Y, Pasquale EB, Control of hippocampal dendritic spine morphology through ephrin-A3/EphA4 signaling. Nat. Neurosci 6, 153–160 (2003). doi: 10.1038/nn994 [DOI] [PubMed] [Google Scholar]
- 96.Sobel RA, Ephrin A receptors and ligands in lesions and normal-appearing white matter in multiple sclerosis. Brain Pathol. 15, 35–45 (2005). doi: 10.1111/j.1750-3639.2005.tb00098.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Miranda JD et al. Induction of Eph B3 after spinal cord injury. Exp. Neurol 156, 218–222 (1999). doi: 10.1006/exnr.1998.7012 [DOI] [PubMed] [Google Scholar]
- 98.Cissé M. et al. Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature 469, 47–52 (2011). doi: 10.1038/nature09635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nkiliza A. et al. RNA-binding disturbances as a continuum from spinocerebellar ataxia type 2 to Parkinson disease. Neurobiol. Dis 96, 312–322 (2016). doi: 10.1016/j.nbd.2016.09.014 [DOI] [PubMed] [Google Scholar]
- 100.Van Hoecke A. et al. EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat. Med 18, 1418–1422 (2012). doi: 10.1038/nm.2901 [DOI] [PubMed] [Google Scholar]
- 101.Parrinello S. et al. EphB signaling directs peripheral nerve regeneration through Sox2-dependent Schwann cell sorting. Cell 143, 145–155 (2010). doi: 10.1016/j.cell.2010.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gadani SP, Walsh JT, Smirnov I, Zheng J, Kipnis J, The glia-derived alarmin IL-33 orchestrates the immune response and promotes recovery following CNS injury. Neuron 85, 703–709 (2015). doi: 10.1016/j.neuron.2015.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hong S. et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716 (2016). doi: 10.1126/science.aad8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sekar A. et al. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183 (2016). doi: 10.1038/nature16549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Stevens B. et al. The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178(2007). doi: 10.1016/j.cell.2007.10.036 [DOI] [PubMed] [Google Scholar]
- 106.Werneburg S. et al. Targeted Complement Inhibition at Synapses Prevents Microglial Synaptic Engulfment and Synapse Loss in Demyelinating Disease. Immunity 52, 167–182.e7 (2020). doi: 10.1016/j.immuni.2019.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhuang Z. et al. EphrinBs regulate D-serine synthesis and release in astrocytes. J. Neurosci 30, 16015–16024 (2010). doi: 10.1523/JNEUROSCI.0481-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Perez EJ et al. Enhanced astrocytic d-serine underlies synaptic damage after traumatic brain injury. J. Clin. Invest 127, 3114–3125 (2017). doi: 10.1172/JCI92300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Assis-Nascimento P, Tsenkina Y, Liebl DJ, EphB3 signaling induces cortical endothelial cell death and disrupts the blood-brain barrier after traumatic brain injury. Cell Death Dis. 9, 7 (2018). doi: 10.1038/s41419-017-0016-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Syed YA et al. Antibody-mediated neutralization of myelinassociated EphrinB3 accelerates CNS remyelination. Acta Neuropathol. 131, 281–298 (2016). doi: 10.1007/s00401-015-1521-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Boyd AW, Bartlett PF, Lackmann M, Therapeutic targeting of EPH receptors and their ligands. Nat. Rev. Drug Discov 13, 39–62 (2014). doi: 10.1038/nrd4175 [DOI] [PubMed] [Google Scholar]
- 112.Dalva MB et al. EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell 103, 945–956 (2000). doi: 10.1016/S0092-8674(00)00197-5 [DOI] [PubMed] [Google Scholar]
- 113.Takasu MA, Dalva MB, Zigmond RE, Greenberg ME, Modulation of NMDA receptor-dependent calcium influx and gene expression through EphB receptors. Science 295, 491–495 (2002). doi: 10.1126/science.1065983 [DOI] [PubMed] [Google Scholar]
- 114.Tomura M. et al. Monitoring cellular movement in vivo with photoconvertible fluorescence protein “Kaede” transgenic mice. Proc. Natl. Acad. Sci. U.S.A 105, 10871–10876 (2008). doi: 10.1073/pnas.0802278105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Osakada F, Callaway EM, Design and generation of recombinant rabies virus vectors. Nat. Protoc 8, 1583–1601 (2013). doi: 10.1038/nprot.2013.094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zilionis R. et al. Single-cell barcoding and sequencing using droplet microfluidics. Nat. Protoc 12, 44–73 (2017). doi: 10.1038/nprot.2016.154 [DOI] [PubMed] [Google Scholar]
- 117.Klein AM et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015). doi: 10.1016/j.cell.2015.04.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.