

SUMMARY
Despite remarkable successes in the clinic, cancer targeted therapy development remains challenging and the failure rate is disappointingly high. This problem is partly due to the misapplication of the targeted therapy paradigm to therapeutics targeting pan-essential genes, which can result in therapeutics whereby efficacy is attenuated by dose-limiting toxicity. Here we summarize the key features of successful chemotherapy and targeted therapy agents, and use case studies to outline recurrent challenges to drug development efforts targeting pan-essential genes. Finally, we suggest strategies to avoid previous pitfalls for ongoing and future development of pan-essential therapeutics.
INTRODUCTION
A key component of successful drug development is the assessment of the therapeutic index (TI), the ratio of the dose or exposure of a drug required to elicit the desired therapeutic effect compared with the dose or exposure at which toxicity becomes limiting (Figure 1). While drugs with a high TI effectively kill cancer cells with manageable toxicities, drugs with a low TI or even “inverted” TIs cause significant side effects at or below efficacious doses. Cytotoxic chemotherapies, which typically target proliferating cells, generally have low TIs and thus require dose and schedule optimization and “rescue” interventions to mitigate side effects. The development of targeted therapeutics has provided alternative routes to achieving high TIs by either targeting cancer dysregulated genes with limited requirements for homeostasis in adults (e.g., ABL, KIT, TRK, ALK) or by developing mutation-biased inhibitors (e.g., EGFR, BRAF, IDH1/2, KRASG12C). However, therapeutics targeting of pan-essential genes (e.g., those genes where inactivation leads to loss of fitness in multiple normal human tissues, see later section for details) are often aggregated within this “targeted” paradigm. Yet such therapeutics will often have low TIs and in many ways are more similar to chemotherapy. A lack of consideration for the specific problems of targeting pan-essential genes likely contributes to high clinical failure rates. Here, we focus on small-molecule targeted therapeutics; however, we believe that these principles would apply to antibody-based therapeutics such as antibody-drug conjugates and other newer modalities.
Figure 1. Therapeutic index of cancer therapeutic agents.
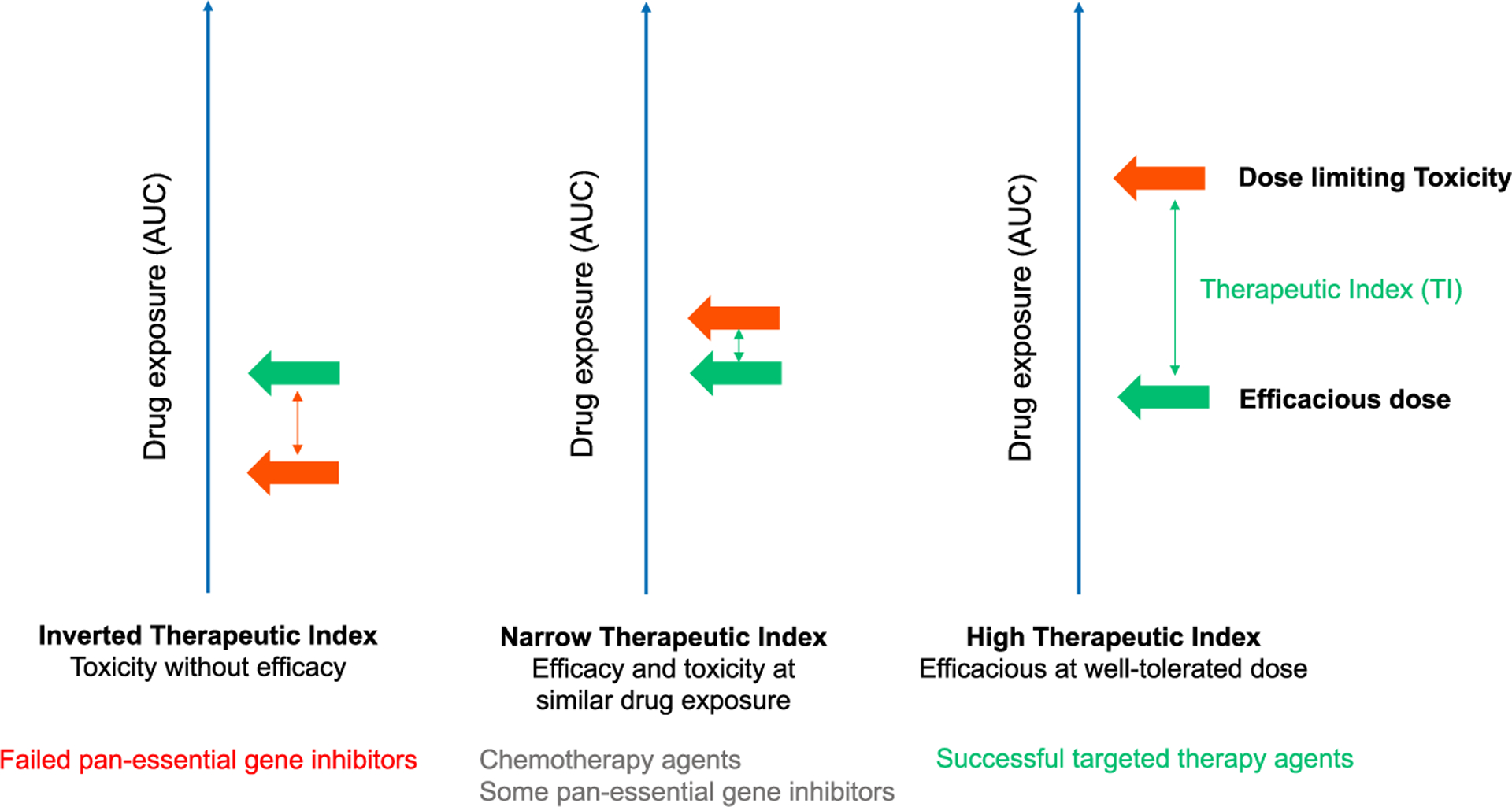
Therapeutic index (TI) is the ratio of the dose or exposure of a drug required to elicit the desired therapeutic effect (green arrow) compared with the dose or exposure at which toxicity becomes limiting (orange arrow). High-TI drugs (many successful targeted therapy drugs) are efficacious at well-tolerated doses; narrow-TI drugs (chemotherapies, some pan-essential gene inhibitors) often have high-efficacy doses slightly below doses leading to dose-limiting toxicities; the effective doses of inverted-TI drugs (some pan-essential gene inhibitors) are lower than doses that lead to severe toxicities, and these drugs often fail in clinical development.
STRATEGIES TO INCREASE THE THERAPEUTIC INDEX FOR CYTOTOXIC CHEMOTHERAPIES
Cytotoxic chemotherapeutics remain pillars of systemic cancer therapy. The majority broadly inhibit proliferating cells by disrupting key mechanisms involved in DNA replication and cell division. As a result, many cancers are sensitive to chemotherapy. Normal proliferating cells are also vulnerable to chemotherapy, thus side effects such as nausea, vomiting, mucositis, anemia, thrombocytopenia, and leukopenia are prevalent (Nurgali et al., 2018). To achieve TIs with chemotherapy, several strategies have been implemented.
Schedule optimization
Intermittent dosing of chemotherapy drugs, rather than the continuous administration, decreases side effects while retaining anti-tumor activity (Kirkwood et al., 1981). The on-off cycles enable sufficient total drug exposure to kill tumor cells while allowing normal cell recovery during “drug holidays” (Foote, 1998).
Side-effect mitigation
Supportive medications have been key to enabling advances in chemotherapy. “Leucovorin rescue” after methotrexate treatment restores folate levels and rescues normal cells (Papac et al., 1973). G-CSF (granulocyte colony-stimulating factor) and GM-CSF (granulocyte-macrophage colony-stimulating factor) accelerate white blood cell recovery (Neidhart et al., 1992; Sheridan et al., 1992) and similarly, bone marrow transplant rescues hematopoiesis from myeloablative doses of chemotherapy (Tallman et al., 1997). Finally, the development of 5-HT3 antagonists dramatically improved the tolerability of highly emetogenic agents such as cisplatin (Gralla et al., 2005).
Formulation optimization
Formulation strategies have been developed to try to shift drug distribution toward the tumor. For example, liposome-encapsulated or nanoparticle formulations are thought to lead to fewer toxicities than standard formulations of doxorubicin and paclitaxel (Leonard et al., 2009; Scripture et al., 2005), although few truly dose- and exposure-equivalent clinical studies of such formulations have been reported.
Personalized dosing
The dose of chemotherapy drugs is generally determined on the basis of each patient’s body surface area, weight, and renal function (Van Den Bongard et al., 2000), allowing individualized dosing in each patient to avoid under- or overdosing. This would result in an aggregate improvement of TI in the patient population.
KEY FEATURES OF HIGH-THERAPEUTIC-INDEX TARGETED THERAPIES
Compared with normal cells, cancer cells rely on specific oncogenic pathways for proliferation and survival (“oncogene addiction”) or have genetic or phenotypic features that confer vulnerabilities to specific perturbations (“non-oncogene addiction” and “synthetic lethality”), and thus can be specifically targeted by drugs without major adverse effects on normal tissues (Francies et al., 2020; Luo et al., 2009). Successful targeted therapies have achieved high TIs through distinct modalities including the development of the following agents.
Mutant-selective or mutant-biased inhibitors
EGFR, IDH1/2, BRAF, and KRASG12C inhibitors are mutant-biased or mutant-selective either by serendipity or by design (Canon et al., 2019; Chapman et al., 2011; DiNardo et al., 2018; Tsai et al., 2008). Most recently, clinical data from EGFR inhibitor osimertinib and KRASG12C inhibitors have all demonstrated impressive TIs in patients with the corresponding mutations (Hallin et al., 2020; Hong et al., 2020; Mok et al., 2017).
Lineage-restricted inhibitors
BTK and PI3Kδ inhibitors, anti-CD20 antibodies, and CD19-directed CAR-T cells, among others, all effectively treat B cell malignancies despite also killing normal B cells, an important but dispensable lineage (Burger and Wiestner, 2018; Hendriks et al., 2014). Similarly, hormonal therapies for breast and prostate cancers act on mechanisms operant in both tumor and normal tissues, yet the normal tissue side effects are modest at the organismal level.
Synthetic lethal gene inhibitors
PARP inhibitors selectively impair the viability of cells lacking functional BRCA1/2 (Huang et al., 2020), and in a recent study patients with mutations in ATM and CHK2 also conferred sensitivity (Mateo et al., 2015).
Widely differential surface antigen expression (e.g., HER2, EGFR)
Certain therapeutic antibodies take advantage of marked increases in target abundance in cancer by their ability to completely occupy every target and drive enhanced killing through antibody-dependent cellular cytotoxicity, often with threshold effects sparing lower-expressing normal tissues.
Predictive biomarkers
Predictive biomarkers (e.g., HER2 overexpression, EGFRmut, PI3Kmut, BRAFmut), restricting treatment to those with a high probability of response, substantially increases the population benefit (Hyman et al., 2017).
“Pan-ESSENTIAL” CANCER TARGETS: PROBLEMS REVEALED IN CLINICAL DEVELOPMENT
The success of targeted therapies led to a surge in cancer drug development, with more than 3,000 oncology phase 1 trials initiated between 2006 and 2015. Despite new genomic technologies for target validation and patient selection, only 5.1% of oncology drugs entering phase 1 progressed to Food and Drug Administration (FDA) approval—the lowest among 21 major diseases (Thomas et al., 2015). We posit that a contributing factor to this failure rate is the misapplication of the targeted therapy paradigm to the drugging of pan-essential genes. More specifically, the selective targeting of a pan-essential gene might on its surface appear to be “targeted” therapy, but is in fact closer to the chemotherapy paradigm. By not recognizing this distinction, the peril is to ignore the difficulties in empiric drug discovery and the lessons learned from chemotherapeutic development.
What is a pan-essential gene? The definition of pan-essential can vary in the context of cellular level and organism level, and here we refer to a gene as pan-essential if losing that gene leads to loss of fitness or cell death in multiple normal tissues or cell lineages in humans. Pan-essential genes in single-cell model organisms have been extensively characterized by transposon/chemical-induced mutagenesis and single-gene knockouts (Rancati et al., 2018). Technological advancements, in particular CRISPR knockout (Haley and Roudnicky, 2020), have enabled the robust identification of genes essential for cell growth and fitness in human cancer cell lines (Behan et al., 2019; Hart et al., 2015; Tsherniak et al., 2017; Wang et al., 2015), human haploid cell lines (Blomen et al., 2015), and human pluripotent stem cells (Mair et al., 2019; Yilmaz et al., 2018). The criteria for defining essential genes generally include genes where single guide RNAs (sgRNAs) are strongly depleted during cell propagation in a majority of cell lines (although cutoffs are slightly different across studies). For example, Hart et al. (2015) included genes whose depletion robustly impaired viability effect in at least three out of five screened cell lines, yielding a list of 1,580 genes (at a stringent 5% false discovery rate threshold). Pan-essential genes identified from these different datasets show high degrees of correlation (Chen et al., 2019b). Recent advances in dual CRISPR screens using multiplexed sgRNAs reveals additional pan-essential paralogous gene pairs. Although performed for limited numbers of genes and cell lines, such studies show that paralogous pairs such as CDK4/6, MEK1/2, and HDAC1/2 exhibit pan-essential profiles (DeWeirdt et al., 2020; Gonatopoulos-Pournatzis et al., 2020; Han et al., 2017). Additional pan-essential paralogous pairs can also be inferred from the genome-wide single-gene CRISPR and RNAi screens (Viswanathan et al., 2018).
Since the characterization of human pan-essential genes became available only recently, it is likely that many cancer targets initially posited to manifest context-specific dependence are in retrospect pan-essential. We compiled a list of molecular targets of clinical-stage oncology drug candidates from canSAR and the Therapeutic Target Database (Tym et al., 2016; Wang et al., 2020) and compared this with a list of human pan-essential genes in at least two published CRISPR single-gene datasets (Dempster et al., 2019; Hart et al., 2015; Yilmaz et al., 2018) or in one paralog dataset. We identified therapeutics that target ~20 pan-essential genes, including regulators of the cell cycle (PLK1, CDK1, CDK7, CDK9, AURKA, AURKB, CDK4/6), epigenetic regulators (DNMT1, BRD4, HDAC3), protein homeostasis regulators (NEDD8, 20S proteasome subunits), and DNA-damage response modulators (ATR, WEE1, CHK1) (Table 1). Targeting such pan-essential genes could be associated with limiting on-target toxicities and difficulties in patient stratification. Indeed, such drug candidates have suffered numerous phase 2/3 trial failures. Examination of the cell-line sgRNA knockout vulnerability distributions of genes targeted by inhibitors such as BRAF and ALK (Tsherniak et al., 2017) shows impaired viability in only a small fraction of cell lines. In contrast, sgRNAs against chemotherapy target genes such as TOP1, TOP2, and DHFR show broad viability effects with median CERES score close to −1 (Figures 2A and 2B). Similarly, the knockout phenotype of recent pan-essential targets also shows broadly lethal patterns (Figure 2C). Likewise, many inhibitors of pan-essential targets show broad cytotoxic patterns whereas targeted therapeutics show selective sensitivity in small subsets of cell lines (Figures 2E–2G). Thus, large-scale genetic or compound profiling can unveil broadly cytotoxic profiles of compounds that might have initially appeared to have contextual specificities. For example, BRD4 was initially thought of as a therapeutic target for MYC-amplified cancers, and indeed both genetic knockdown and small-molecule inhibition of BRD4 lead to anti-tumor effect in various relevant preclinical models (Delmore et al., 2011; Zuber et al., 2011). However, in the drug-sensitivity profiles across >700 cell lines for two clinical-stage BRD4 inhibitors, neither MYC copy numbers nor BRD4 knockout effects correlate with the sensitivity of either BRD4 inhibitors. In contrast is the high correlations between the sensitivity to a BRAF or ABL inhibitor (dabrafenib or nilotinib, respectively), the viability effects of BRAF or ABL1 knockouts, and the presence of a BRAF mutation or ABL fusion, respectively (Figure 3).
Table 1.
Representative pan-essential genes as oncology drug targets
| Pan-essential genes | Cell function | Representative drug and most recent progress |
|---|---|---|
| AURKA | cell-cycle regulation | alisertib (failed in a phase 3 trial in PTCL) |
| AURKB | cell-cycle regulation | barasertib (failed in a phase 2 trial in AML) |
| PLK1 | cell-cycle regulation | volasertib (failed in a phase 3 trial in AML) |
| CDC7 | cell-cycle regulation | TAK-931 (currently in a phase 2 trial in advanced solid tumors) |
| CDK4/6 | cell-cycle regulation | palbociclib (approved in combination with fulvestrant for treating HR + HER- breast cancer) |
| CDK1 | cell-cycle regulation | dinaciclib (failed in a phase 3 trial in CLL) |
| CDK9 | cell-cycle regulation | dinaciclib (failed in a phase 3 trial in CLL) |
| BRD4 | epigenetic regulation | CPI-0610 (currently in a phase 3 trial in myelofibrosis) |
| HDAC3 | epigenetic regulation | vorinostat (approved for CTCL, failed in 100 + clinical trials for various oncology indications) |
| DNMT1 | epigenetic regulation | azacitidine (approved for treating MDS) |
| PRMT5 | epigenetic regulation | GSK-3326595 (currently in phase 2 trials for various oncology indications) |
| SF3B1 | splicing regulation | H-3B8800 (currently in phase 2 trial in MDS) |
| NEDD8 | protein homeostasis | MLN4924 (currently in a phase 3 trial in AML and MDS) |
| 20S proteasome subunits | protein homeostasis | bortezomib (approved for treating multiple myeloma and MCL) |
| ATM | DNA-damage response | AZD0156 (currently in a phase 1 clinical trial in solid tumors) |
| ATR | DNA-damage response | VX-970 (currently in phase 2 trials in multiple solid tumors) |
| WEE1 | DNA-damage response | AZD1775 (multiple phase 2 trials were terminated because of safety concerns) |
| CHK1 | DNA-damage response | MK8776 (failed in a phase 2 trial in AML) |
| KIF11 | microtubule stability | ARRY-520 (development halted after a phase 2 trial in multiple myeloma) |
| MEK1/2 | proliferation | trametinib (approved for treating BRAF-mutant advanced melanoma) |
| XPO1 | nuclear export | selinexor (approved for treating advanced diffuse large B cell lymphoma, and multiple myeloma) |
Figure 2. Genome-wide CRISPR knockout and compound profiling in cancer cell lines revealed the pan-essential nature of many cancer drug targets.
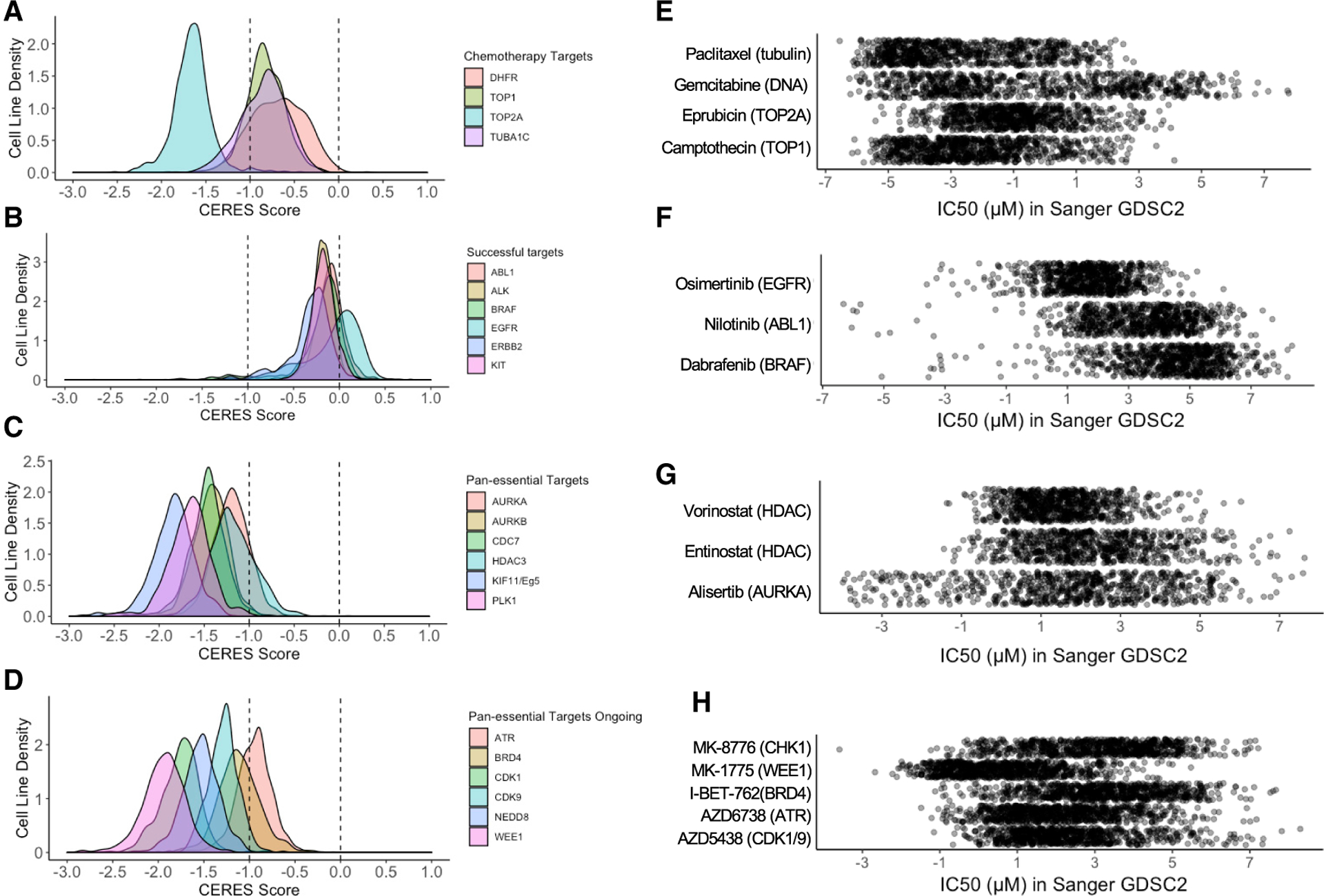
(A–D) Density plots representing the CERES score distribution of cancer cell lines after the indicated gene knockout. A CERES score of −1 represents the median effect of knocking out essential genes, and a CERES score of 0 represents no growth disadvantage. CRISPR (Avana) Public 20Q2 dataset from Broad Institute was used in this analysis.
(E–H) Scatterplots representing the IC50 distributions of the indicated drugs and their molecular targets in ~800 cancer cell lines. GDSC2 dataset from Sanger Institute was used in this analysis.
Figure 3. Biomarker correlations with BRD inhibitor, BRAF inhibitor, and ABL inhibitor drug sensitivities.
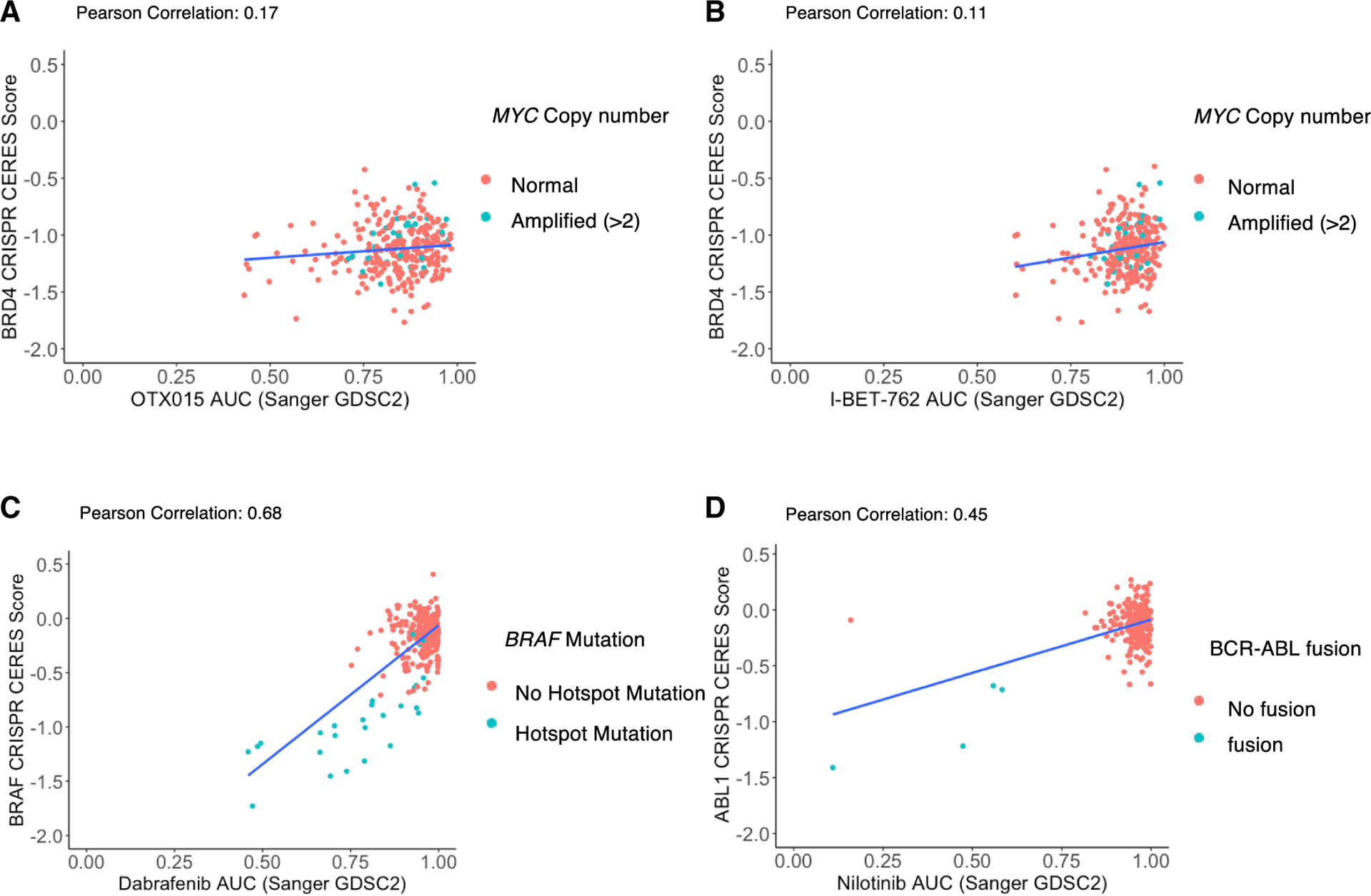
The AUC values of two BET inhibitors, OTX015 (A) and I-BET-762 (B), are plotted against the CERES score of BRD4 knockouts in the same cell line. The AUC values of the BRAF inhibitor dabrafenib (C) and ABL inhibitor nilotinib (D) are plotted against the CERES score of BRAF knockout in the same cell line. Each dot represents a cancer cell line and red colors represent MYC amplification (A and B), BRAF hotspot mutation (C), or BCR-ABL fusion (D) in the cell line. Genomic and CRISPR data were obtained from Broad Institute Depmap portal. BET inhibitor AUC data were obtained from Genomics of Drug Sensitivity in Cancer Portal.
Since targeting pan-essential genes will lead to broad cytotoxic effects, “apparent” validation of a specific anti-cancer effect of interest is essentially guaranteed when performing experiments in only a few cell lines or xenografts. This bias will therefore prioritize pan-essential mechanisms or inhibitors in cancer drug discovery (see below for examples). However, inhibitors of pan-essential targets are likely to manifest difficulty in clinical development with respect to both the identification of responding patients and an enriched responding patient population, and with respect to tolerability. This divergence has led to costly failures in late clinical development. Here we take a deeper look into inhibitors of several pan-essential cancer targets that have attracted significant interest and investment in drug discovery, but failed in multiple phase 2 and phase 3 clinical trials.
CASE STUDIES OF INHIBITORS TARGETING PAN-ESSENTIAL GENES IN CANCER
Histone deacetylase inhibitors
Hyperpolar compounds such as hexamethylene bisacetamide (HMBA) were observed to induce leukemic cancer cell differentiation. Modifications led to SAHA (vorinostat), which also had pro-differentiation and anti-proliferative effects, and was found to inhibit histone deacetylation (Dokmanovic et al., 2007; Marks et al., 2004). Intriguingly, the differentiation phenotype was also induced by HMBA, which lacked histone deacetylase (HDAC) inhibitory activity, hence the link between the differentiation phenotype and either HDAC inhibition or epigenetic regulation was not established. Nonetheless, the empiric anti-proliferative activity and the relative ease of targeting HDACs led to the widespread pursuit of pharmaceutical HDAC inhibitors.
In this pursuit three challenges emerged. First, HDAC inhibition leads to broad cytotoxic effects in a large selection of cancer preclinical models. Thus, if one experimentally “looks under the lamppost” at a specific cancer lineage or molecular class in isolation, one will by default generate positive supportive data. Second, the majority of HDAC inhibitors in clinical development are broad-spectrum inhibitors targeting multiple isoforms of HDACs (Falkenberg and Johnstone, 2014). Thus, it is challenging to attribute the anti-tumor effects of inhibitors to the specific function of a single HDAC isoform and allow optimization. Third, nearly all HDACs are ubiquitously expressed in essential tissues of humans, and the individual knockouts of Hdac1, Hdac2, Hdac3, Hdac4, Hdac5, Hdac7, and Hdac8 in mice are either embryonic/perinatal lethal or lead to major organ defects after birth (Falkenberg and Johnstone, 2014). Thus, the broad requirement for HDAC activity in normal human tissues along with inhibitor polypharmacology made it likely that side effects would be limiting.
Four HDAC inhibitors are FDA approved: vorinostat in cutaneous T cell lymphoma (CTCL), belinostat in peripheral T cell lymphoma (PTCL), romidepsin in PTCL and CTCL, and panobinostat in multiple myeloma. While this is a measure of success, the efficacy is modest and the overall clinical benefit is marginal due to the severity of adverse events. In the phase 3 trial of panobinostat in refractory multiple myeloma, patients in the panobinostat treatment group showed a longer median progression-free survival (mPFS) than the control group (11.99 versus 8.08 months) (San-Miguel et al., 2014). However, 96% of patients in the panobinostat-combination group experienced grade 3/4 severe adverse events. The FDA initially rejected the new drug application in refractory multiple myeloma because “the drug’s benefits did not outweigh its risks,” but later approved it in a narrower indication in myeloma (Jalloh, 2015). A later analysis showed that the overall survival (OS) of patients was not improved (San-Miguel et al., 2016).
The broad cellular activity of HDAC inhibitors and the description of such inhibitors as “epigenetic regulators” concomitantly led to numerous new hypotheses. Again, the pan-lethal nature of the inhibitors guaranteed that every proposed cell-line-based indication when tested in isolation would appear positive (absent robust controls). Such hypotheses lacking more robust validation led to multiple phase 2/3 failures of HDAC inhibitors in non-small cell lung cancer (NSCLC), acute myeloid leukemia (AML), mesothelioma, ovarian cancer, and high-grade gliomas among others. These trials showed low response rates and high grade-3/4 adverse event rates consistently raising the red flag of low TI. Vorinostat itself was studied in 156 phase 1, 139 phase 2, and 9 phase 3 trials with only the phase 2 trial in CTCL leading to FDA approval (Figure 4), thus invalidating most, if not all preclinical HDAC therapeutic hypotheses. Despite these data, HDAC-inhibitor hypotheses continue to emerge with investigators testing entinostat + endocrine therapy in a failed phase 3 trial (Yeruva et al., 2018), entinostat + anti-PD1 antibody in a failed phase 2 trial (O’Shaughnessy et al., 2020), and recent proposals to test vorinostat in pediatric glioma (Hashizume, 2017), despite cellular IC50 data in that indication being indistinguishable from the IC50 distribution of vorinostat in large cell-line panels.
Figure 4. Clinical development trajectories of a selection of pan-essential and highly selective targeted therapy drug candidates.
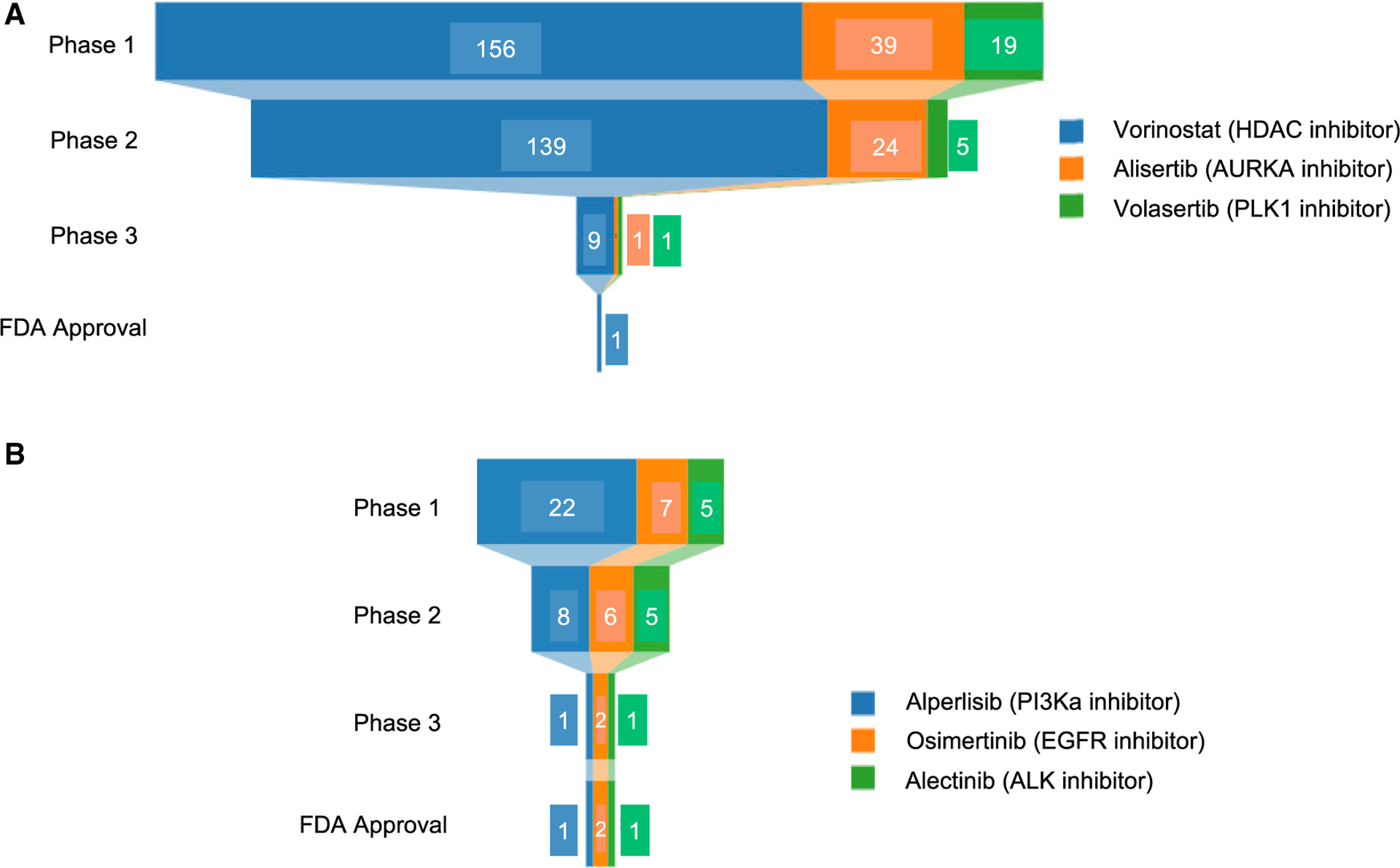
Clinical trial information for three pan-essential drug candidates discussed in case studies (A) and three successful targeted therapy agents (B) were obtained from clinicaltrials.gov. Clinical trials of corresponding drug candidates are included for this analysis if they have the status suspended, terminated, completed, or withdrawn, or have published trial results.
Aurora kinase inhibitors
Aurora kinases regulate entry into mitosis, spindle assembly, and cytokinesis. Aurora kinase A (AURKA) localizes to centrosomes and drives centrosome maturation, separation, and spindle assembly while aurora kinase B (AURKB) is a chromosomal passenger protein localizing along the chromosome and centromere kinetochore to facilitate mitosis. AURKA and AURKB are amplified in several types of cancers, leading to the notion that they might make attractive cancer targets (Lens et al., 2010; Mehra et al., 2013). Paradoxically, AURKA and AURKB function as both oncogenes and tumor suppressors. In mice, Aurka transgenic overexpression increases mammary and skin carcinomas while Aurka heterozygous deletion leads to lymphoma. Similarly, Aurkb overexpression increases lymphomagenesis while heterozygous deletion gives rise to multiple cancers (Otto and Sicinski, 2017). While AURKA was thought to be selectively lethal in NMYC-driven neuroblastoma, in CRISPR screening datasets, AURKA and AURKB are both pan-essential genes (Figure 1C), and inhibition of AURKB impairs cell viability in actively proliferating tumor and normal cells (Mehra et al., 2013). Therefore, we can anticipate a low TI.
Since 2010, more than ten aurora kinase inhibitors have entered clinical development (Otto and Sicinski, 2017), but none have achieved FDA approval. Alisertib is an AURKA inhibitor with >200-fold selectivity over AURKB that was studied in 24 phase 2 trials in breast, bladder, and prostate cancers, SCLC, NSCLC, multiple myeloma, neuroblastoma, melanoma, and PTCL. From these trials alisertib progressed to phase 3 testing only in PTCL (Figure 4), where alisertib was not superior to standard of care (33% versus 45% overall response rate [ORR]) and had a median PFS of 115 versus 104 days (O’Connor et al., 2019).
Similarly, AURKB inhibitors have not achieved clinical success. To date, only barasertib/AZD1152 has reached phase 2 testing. In a phase 2 trial comparing barasertib with low-dose cytarabine (LDAC) in elderly AML patients, while treatment with barasertib led to higher response rates, serious adverse events were significantly higher in patients treated with barasertib compared with LDAC (stomatitis 29% versus 0%, febrile neutropenia 50% versus 19%, pneumonia 23% versus 8%), leading to its discontinuation (Kantarjian et al., 2013). Similarly, the development of AURKB inhibitor BI811283 was discontinued after a phase 1 trial showed 0% ORR (Mross et al., 2016).
PLK inhibitors
Polo-like kinases (PLKs) regulate cell cycle and mitosis. PLK1 has an essential role in regulating G2/M transition, centrosome maturation, sister chromatid separation, mitosis exit, and cytokinesis initiation (Strebhardt and Ullrich, 2006). PLK1 is overexpressed in a series of solid and hematologic malignancies, and is associated with poor prognosis (Strebhardt, 2010). PLK1 is also a negative regulator of tumor suppressor p53 (Ando et al., 2004). Thus, PLK1 was considered an interesting cancer target.
Plk1 knockout or knockdown leads to viability effects in various cancer models; however, whether such effects are cancer selective remains questionable. While small interfering RNA silencing data in transgenic mice and primary mammalian cell lines shows that toxicity of targeting PLK1 might be tolerable (Raab et al., 2011), Plk1 homozygous knockouts are embryonic lethal at the 4/8-cells stage (Lu et al., 2008) and PLK1 is essential in human pluripotent stem cells. Interestingly, Plk1 heterozygous knockout mice show aneuploidy and increased tumor development at advanced ages (Strebhardt, 2010), and PLK1 overexpression prevents the development of KRAS- and HER2-induced mammary gland tumors in transgenic mouse models (de Cárcer et al., 2018). PLK1 knockdown results in anti-proliferative effects in many cancer cell lines without robust biomarkers for distinguishing sensitive cell lines (Strebhardt and Ullrich, 2006) and in CRISPR screening datasets PLK1 is a pan-essential gene (Figure 2C).
To date, no PLK1 inhibitors have been approved by the FDA. Two inhibitors have progressed to phase 3 testing although both have suffered multiple clinical failures. Volasertib is a selective PLK1 inhibitor with >1,000-fold selectivity against other kinases (Otto and Sicinski, 2017). In a phase 3 trial in previously untreated AML patients, volasertib + LDAC failed to improve either ORR or OS compared with LDAC alone, but produced higher rates of grade 3/4/5 adverse events (AEs), especially higher grade 5 serious AEs and treatment-related deaths (27.9% versus 15.2%) (Döhner et al., 2016). In NSCLC, volasertib alone or in combination with pemetrexed shortened PFS and increased toxicity compared with pemetrexed (Ellis et al., 2015). In platinum-resistant ovarian cancer, volasertib shortened PFS (13.1 versus 20.6 weeks) and increased grade 3/4 AEs compared with chemotherapy (61.1% versus 30.9%) (Pujade-Lauraine et al., 2016). Finally, in a single-arm phase 2 trial in bladder cancer, volasertib failed to meet the prespecified criteria for activity while 62% of patients experienced grade 3/4 AEs (Stadler et al., 2014).
Rigosertib is a multi-kinase inhibitor with modest PLK1 selectivity (Otto and Sicinski, 2017). In a phase 3 trial in high-risk myelodysplastic syndrome (MDS), rigosertib failed to improve OS and increased grade 3/4 AEs, including anemia, thrombocytopenia, and neutropenia (Garcia-Manero et al., 2016). In a phase 2/3 metastatic pancreas cancer trial, adding rigosertib to gemcitabine failed to improve PFS and increased grade 3/4 AEs (O’Neil et al., 2015). Currently, rigosertib is in phase 2 and 3 trials in second-line higher-risk MDS patients, and alone or in combination with azacitidine.
CDC7 inhibitors
CDC7 is a serine-threonine kinase that phosphorylates and activates MCM2 and regulates initiation of DNA synthesis, chromosomal segregation in mitosis, DNA-damage response, response to DNA-replication stress, and entry into mitosis (Montagnoli et al., 2004; Yamada et al., 2014). Although CDC7 is upregulated and associated with poor prognosis in several types of cancer (Kulkarni et al., 2009), there are no data supporting an oncogenic role for CDC7 nor that the overexpression confers sensitivity to CDC7 inhibition. Indeed, CDC7 expression might simply correlate with the portion of cells in S phase (Montagnoli et al., 2010). Cdc7 knockouts are embryonic lethal in mice between embryonic day 3.5 (E3.5) and E6.5 (Kim et al., 2002), and CRISPR screening data show that CDC7 is pan-lethal in human cell lines (Figure 2C). Thus, CDC7 inhibitors are likely to behave similarly to low-TI chemotherapies.
To date, no CDC7 inhibitors have reached phase 3, and several have failed or were terminated in phase 1/2. BMS-863233/XL413 is a potent and selective oral CDC7 inhibitor with good preclinical pharmacokinetic (PK) properties and in vivo anti-tumor activity in a colorectal cancer xenograft study (Koltun et al., 2012). Two phase 1/2 trials in advanced hematologic and solid cancers (NCT00838890, NCT00886782) were terminated prior to completion due to issues with drug metabolism and lack of efficacy (EU Final Clinical Study Report CA198002). In further characterizing this inhibitor in 64 cancer cell lines neither mutational status, doubling time, nor CDC7, DBF4, or MCM2 mRNA expression was associated with sensitivity. NMS-1116354 is a potent and selective CDC7 inhibitor with in vitro and in vivo anti-tumor activity in colon cancer models (Montagnoli et al., 2008). NMS-1116354 was terminated early after two phase 1 trials in advanced solid tumors (NCT01016327 and NCT01092052). TAK-931 is a potent (IC50 < 0.3 nM) and selective oral CDC7 inhibitor still in clinical development. In large-panel cell-line data, TAK-931 showed anti-tumor activities in KRAS-mutant cells than KRAS wild-type cells but with questionable statistical significance (Iwai et al., 2019). In a dose-escalation phase 1 trial in advanced solid tumors, the majority of patients experience grade ≥3 AEs, including neutropenia, decreased white blood cell count, and leukopenia, although 5 out of 25 patients had partial remission or stable disease (Shimizu et al., 2018). TAK-931 is currently being evaluated in a phase 2 clinical trial of patients with advanced solid tumors.
LESSONS LEARNED
We can observe several recurrent themes worth considering for future development of therapeutics in this space.
Misidentifying pan-essential genes as selective-essential targets based on limited preclinical modelling
Before large-scale characterized cancer cell lines and patient-derived xenografts were available (Barretina et al., 2012; Gao et al., 2015; McDonald et al., 2017), the evaluation of most cancer drug targets was limited to small sets of preclinical models. Even today, academic labs lack the resources required to conduct large-scale cell-line testing and hence preclinical hypothesis validation remains underpowered. In addition, the publication pressure to equate novel cancer biology with a potential therapeutic intervention is also a contributing factor. For example, HDAC and bromodomain inhibitors are frequently used to provide evidence in support of a selective epigenetic hypothesis when in fact these are broadly cytotoxic agents. More importantly, the mistaken identification of a pan-essential target as selectively essential will lead to the clinical testing of erroneous hypotheses, lack of efficacy, and the exposure of patients to unnecessary toxicity.
The inability to stratify and enrich the clinical trial population
When the pan-lethal activity of a therapeutic is recognized, it is not clear that cell-line testing, even when there is a broader range of effect (Figure 2C), will provide clear translatable predictive biomarkers. It is possible that the ability to define increased tumor sensitivity for cytotoxic targets is impaired by the relative normalized growth rates of most cell lines. In the absence of a predictive genetic feature, overexpression of the target is often invoked as a sensitizing feature. However, as pertains to small-molecule inhibitors, from a kinetic view increased levels of a pan-essential target will create resistance to a therapeutic, not sensitivity. Indeed, DHFR amplification results in resistance, not sensitivity, to methotrexate (Trent et al., 1984). Ultimately, clinical development then progresses based on idiosyncratic responses in the clinic and a haphazard empirical development approach.
Inadequate attention to therapeutic pharmacology
Chemotherapeutics were typically developed using intermittent schedules, drugs having shorter half-lives, individualized patient dosing, and often intravenous formulations. This type of pharmacology might be ideal for tuning therapeutics having a narrow range of tolerated doses (narrow TI). Fixed daily oral dosing and longer half-lived therapies emerging from the Gleevec paradigm provide far less flexibility for narrow-TI drugs. It is notable that the CDC7 inhibitors in clinical developments are all oral despite the knowledge that neutropenia and other cytopenia would be the dose-limiting toxicity.
Lack of a therapeutic window due to on-target toxicity
We expect that therapeutics targeting pan-essential genes will exhibit low-TI profiles; however, the inability to predict whether there is an adequate TI or overestimating the TI from preclinical studies remains problematic. First, there are obvious differences between human and rodent and dog toxicology. Second, new pan-essential targets may have poorly understood cytotoxic effects in non-proliferating cells, creating new classes of AEs. For example, patients treated with HDAC inhibitors consistently experienced profound fatigue or asthenia when mitigation strategies were not known (Krug et al., 2015; O’Shaughnessy et al., 2020; San-Miguel et al., 2014). Third, overestimation of efficacy is common. Slowed tumor progression in preclinical models, rather than regression, is often interpreted as efficacy, whereas in humans this constitutes progression. This leads to lower estimated efficacious drug concentrations and therefore an overestimation of the TI (Figure 1). Finally, because cancer drugs are “expected” to have a narrow TI, untoward toxicity in dose-range finding and good-lab-practice toxicology studies is seen as acceptable.
Lack of a therapeutic window due to inhibitor polypharmacology
The problem of polypharmacology is not limited to targeting pan-essential genes; however, as pan-essential targets can have highly related family members (e.g., 18 human HDACs, 9 human cyclin-dependent kinases [CDKs]), fewer selective inhibitors may target multiple pan-essential genes. For example, several CDK inhibitors broadly target CDK1, 2, 7, and 9, which are all pan-essential (Otto and Sicinski, 2017). While selective polypharmacology can enhance the anti-tumor effect of certain drugs (e.g., CDK4/6 inhibitors, MEK1/2 inhibitors), balancing on-target synergies and toxicities of inhibiting multiple pan-essential targets is often challenging.
Therapeutic failure occurs late in drug development rather than during therapeutic optimization
Several factors contribute to this issue: first, pan-lethal targets are often selected because of their “druggable” enzymatic function. Thus, the odds of making a potent drug are high. Second, because the target is pan-lethal, many if not most preclinical models will appear to be responsive. Third, as even the viability responses to pan-lethal inhibitors still follow a distribution (Figure 2), in vitro responses to a therapeutic can appear to be differential in small-cell-line sets. Fourth, the broad cellular activity will result in many positive experiments in the academic preclinical setting, generating enthusiasm for the target. In aggregate, these features facilitate and pave the way for preclinical drug development eliminating many typical points of preclinical failure. Thus, HDAC inhibitors were “easy” to make in the preclinical setting, yet were disastrous in clinical development.
FUTURE STRATEGIES TO IMPROVE THE DEVELOPMENT OF “NEXT-GENERATION” PAN-ESSENTIAL TARGET INHIBITORS
The success of chemotherapy and of pan-essential inhibitors such as CDK4/6 and MEK inhibitors suggests that targeting pan-essential genes will remain an important strategy for cancer therapeutics development. It is also clear that many of the above challenges are likely to apply to a “next generation” of inhibitors of pan-essential targets including BRD4, ATR, WEE1, NEDD8, CHK1, and CDK9 inhibitors, among others. Akin to prior pan-essential targets, these genes control key cellular processes such as cell-cycle regulation, DNA-damage response, proteolysis homeostasis, and transcriptional control, and fit the criteria of pan-essential genes (Figure 2D). Indeed, the distribution of cellular sensitivity to many of these next-generation therapeutics including inhibitors of CDK1/2/9 (AZD5438), ATR (AZD6738), WEE1 (MK-1775), CHK1 (MK-8776), and BRD4 (I-BET-762) are similar to the distributions of chemotherapeutics (Figure 2H).
Since it is likely that the development of newer pan-essential therapeutics will face hurdles similar to those outlined above, strategies are needed to prevent an ongoing “death row” of clinical development failures. It is essential that we improve our ability to validate and prosecute pan-essential targets, as well as to separate and optimize those likely to provide patient benefits while deprioritizing and discontinuing those likely to engender toxicity over efficacy.
Target evaluation and validation in multi-omic and functional datasets
Large-scale projects have enabled the characterization of mRNA and protein expression levels in many normal human cell lines and tissues (Ardlie et al., 2015; Consortium et al., 2019; Regev et al., 2017; Uhlén et al., 2015). Simultaneously, profiling of tumors and tumor models has been greatly expanded. The Cancer Cell Line Encyclopedia (CCLE) has generated comprehensive “omic” data in more than 1,000 cancer cells (Barretina et al., 2012; Ghandi et al., 2019). While The Cancer Genome Atlas and related projects have made genetic, mRNA expression, and proteomic data in thousands of patients readily available (Chen et al., 2019a; Gillette et al., 2020), the availability of these data, must be coupled with far greater sophistication in their use beyond the monocular assumption that overexpression should predict drug sensitivity. While overexpression of cell-surface proteins indeed enhances tumor killing by antibodies through increased antibody recruitment to the tumor, the overexpression of low-molecular-weight inhibitor targets is just as likely to confer resistance by raising the enzymatic target concentration. Moreover, there is now clear evidence that heterozygous loss of a pan-essential gene, or homozygous loss of one gene from a pan-essential paralog pair, confers selective lethality (Nichols et al., 2020; Viswanathan et al., 2018). Therefore, it would be wise to equally consider the possibility that reduced concentrations of pan-essential enzymes could confer increased tumor sensitivity. In addition, mRNA expression analyses often compare a tumor with its “matched” normal. However, for dispensable lineages (e.g., B cells, breast and prostate epithelium) targeting both the tumor and the normal tissue is acceptable. Thus, we should instead consider defining “critical normal” tissues as key comparators for data mining (e.g., heart, brain, liver, gastrointestinal tract, bone marrow, stem cells).
Unbiased target discovery is increasingly enabled through CRISPR knockout screens. Notably, when comparing large-scale short hairpin RNA (shRNA) screens and CRISPR screens, certain pan-essential genes (e.g., PRMT5) are selectively lethal when inactivated by shRNA. This raises the question as to whether genome-wide partial-loss-of-function studies focused on the pan-lethal genes might provide clearer evidence for differential effects across cancer (see further discussion below). Additionally, genome-wide CRISPR screens have so far been performed only in a small number of “normal” cell lines including fibroblast and pluripotent stem cells, thus limiting the ability to discern cancer-specific effects of pan-lethal genes. Future efforts to characterize a panel of normal cell lines from diverse “critical normal” lineages might serve as robust controls for this purpose.
Small-molecule perturbation projects have generated sensitivities of more than 300 cancer cell lines to more than 400 small molecules (Iorio et al., 2016; Seashore-Ludlow et al., 2015). This approach has been extended to a “drug repurposing hub,” where more than 4,500 compounds have been tested in more than 500 cancer cell lines (Corsello et al., 2020). These large-scale drug-sensitivity datasets might allow the robust identification of response or resistance biomarkers to certain compounds, and compound mechanism of action (MOA) determination by correlation with CRISPR loss-of-function datasets. Thus, therapeutic hypotheses initially derived from the profiling of small cell-line sets should be immediately assessed in such datasets. Lastly, in vitro profiles are nearly always assessed using continuous compound exposure often over 48 to 72 h. In contrast, in vitro 10-h pulse dosing of HDM2 inhibitors revealed induction of apoptosis not seen in continuously exposed cells (Jeay et al., 2018). Such data raise the possibility that in vitro pulse profiling might result in differential cytotoxicity missed by continuous exposure.
New technologies to enable partial/temporal target inhibition in preclinical models
Since the complete and permanent knockout of pan-essential genes can lead to broad cytotoxicity, partial and temporal loss-of-function perturbations in preclinical models will be pivotal in defining the therapeutic indices of pan-essential inhibitors. Previously, such perturbations required the generation of tool compounds or even optimized drug candidates. Rapid generation of highly effective tool compound remains an indispensable aspect for target validation. However, the degree of optimization required for an informative tool compound can be challenging for many targets. For example, the phenotypic differences mentioned above elicited by pulsatile versus continuous HDM2 inhibition was not recognizable until a potent clinical candidate was developed (Jeay et al., 2018). The discovery of ligand-mediated degrons, including the auxin-inducible degron (Zhang et al., 2015), Small-Molecule-Assisted Shutoff (SMASh) tag, CRBN-recruiting dTAG (Nabet et al., 2018), and VHL-recruiting dTAG (Nabet et al., 2020), has enabled conditional and temporal control of protein degradation and to some degree might bypass the need for tool compounds. Here, the fusion of degrons to an endogenous pan-essential protein of interest by CRISPR-mediated locus-specific knockin will allow conditional controlled perturbation of the target protein in cell lines, xenografts (Nabet et al., 2018, 2020; Natsume et al., 2016), and genetically engineered mice (Banaszynski et al., 2008), and could enable direct assessment of efficacy and toxicity much earlier in the drug-discovery process.
Similarly, using CRISPR technology to induce partial rather than complete loss of function may be advantageous. To this end, engineered panels of mismatch “attenuated” sgRNAs were shown to establish differentially graded gene expression outputs through a dCAS9-based CRISPRi system (Jost et al., 2020). These and other technologies are likely to be of enormous benefit in validating specific hypotheses pertaining to pan-lethal targets.
Biomarker identification through expanded preclinical models
Biomarkers that enable patient stratification can maximize the therapeutic benefit by reducing the non-responder population. For MEK1/2 inhibitors, BRAF mutant cancer cell lines are more sensitive than BRAF wild-type cell lines. For CDK4/6 inhibitors, hormone receptor (HR)-positive breast cancers show increased sensitivity and often have frequent cyclin D1 amplification or CDKN2 inactivation (sensitive biomarkers) but infrequent RB (resistance biomarker) inactivation. These features, at least in part, account for the clinical success of these therapeutics despite their relatively broad requirement for cell viability (Álvarez-Fernández and Malumbres, 2020). For the more potent pan-essential therapeutics, it has been nearly impossible to discover such markers prior to clinical development. The application of novel preclinical models may help with this challenge.
PRISM is a pooled mixture of more than 750 barcoded CCLE cell lines (Yu et al., 2016) enabling efficient compound testing across many preclinical models simultaneously. This approach can be used to identify the anti-tumor MOA of previously reported compounds and confirm the MOA of lead compounds (Corsello et al., 2020; Li et al., 2019; You et al., 2020). Thus, PRISM might help overcome the throughput and cost limitations of large-scale cell-line profiling.
As noted earlier, cell-line models might have a limited ability to discern a range of sensitivities for pan-lethal target inhibitors. Thus, in addition to cancer cell lines, scalable patient-derived xenografts (PDXs) and organoids might expand the preclinical oncology model repertoire for responder versus non-responder stratifications by enabling testing at tolerated doses (in the PDX setting) and enabling the profiling of tumor types that are under-represented in cell-line space. Large-scale efforts such as the PDX encyclopedia (PDXE) and ProXe, as well as several organoid biobanks, provide both resources and the conceptual framework for future scaling-up and compound screens (Dijkstra et al., 2018; Gao et al., 2015; Townsend et al., 2016). For example, in the PDXE dataset chemotherapy drugs such as paclitaxel and abraxane show differential and limited responses across PDXs of different lineages. Notably, this is substantially different from the pan-lethal profiles of the same drug in in vitro cell-line screens. Future expansions of patient-derived cancer models might provide new opportunities for biomarker identification of drug candidates with pan-essential properties.
Dosing and schedule optimization
It is likely that most therapies directed at pan-essential genes will be administered intermittently. Hence, we should not predicate drug-discovery efforts on the default assumptions of continuous and/or oral administration. Instead, exploring different dosing strategies based on PK, pharmacodynamic, and toxicology considerations should precede the commitment to a given PK profile. While rodents and dogs are unlikely to provide an exact guide for the human PK and schedule, the pharmacologic parameters most closely associated with efficacy and toxicity can be elucidated—for example, determining whether efficacy or toxicity is driven by Cmax, area under the curve (AUC), or the duration of exposure over a Cmin value.
For certain targeted therapies whereby toxicity was dose limiting, alternative dosing strategies or PK profiles have also improved the TIs. Palbociclib and ribociclib adopted 3-week-on/1-week-off dosing to mitigate myelotoxicity (Klein et al., 2018) while the flat PK profile of trametinib with a peak-to-trough ratio of 1.8 resulted in exposures falling within a narrow tolerated range of MEK inhibition (Infante et al., 2012). Unfortunately, insufficient attention is paid to the preclinical testing of these parameters, and instead trial and error typically takes place during clinical development, ultimately delaying the discovery of an optimal regimen or leading to clinical failure due to intolerability.
Formulation optimization or enhanced tumor delivery
Nanoparticle formulations for chemotherapy agents have been actively explored to increase the TI of chemotherapeutic agents, and several have gained FDA approval (Shi et al., 2017). In general, the goal is to enhance tumor exposure over normal tissue. However, it should be noted that simply modulating tissue exposure away from the most sensitive normal tissue might be sufficient to substantially improve the TI of a drug (e.g., bone marrow sparing). Interestingly, a nanoparticle formulation of AURKB inhibitor barasertib (AZD1152) is being tested in phase 1 (NCT03217838) based on preclinical data showing improved anti-tumor activity and reduced bone marrow toxicities in preclinical AML models (Ashton et al., 2016). Despite the opportunities to improve TI, many chemotherapy nanoparticle formulations have failed to improve clinical outcomes (Shi et al., 2017). The preclinical testing of such formulations remains largely fixated on bulky tumor xenografts that are unlikely to model the human tumor microenvironment. In this setting, formulations with low volumes of distribution (a characteristic of formulations confined to the vascular space) are likely to simply leak into the xenograft and appear to have dramatically improved TIs, which then fail to translate to the clinical setting. Therefore, investigators should consider formulation testing in lower volume dispersed or orthotopic models. While tail vein and portal vein injections are not true models for lung or liver metastasis, they nonetheless result in multiple small-volume tumors and are likely better for detecting meaningful improvements in drug distribution.
Recent developments in novel therapeutic modalities have also enabled the selective delivery of low-TI therapeutics (cytotoxic drugs or immune cells) to tumor cells. Antibody-drug conjugates (ADCs) combine antibodies targeting antigens abundant on tumor cell surfaces with highly potent cytotoxic drugs, allowing the tumor-specific delivery of cytotoxic drug and minimizing toxicities to normal cells. Four ADCs have obtained FDA approval, followed by more than 60 ongoing clinical trials of ADCs in many different tumor types (Khongorzul et al., 2020). In the ADC space, dose-limiting toxicities are still driven by off-target side effects, thus antibody-based delivery of cytotoxic therapeutics requires further optimization. In the immune oncology space, the tumor-specific delivery of cytotoxic T cells and natural killer cells by engineered chimeric antigen receptors (CARs) or bispecific antibodies similarly exploits surface antigens that are selectively abundant on tumor cells, sometimes at the expense of dispensable lineage normal cells (e.g., CD19 and CD20 on B cell acute lymphoblastic leukemia cells and normal B cells, B cell maturation antigen on multiple myeloma cells and normal plasma cells). These new modality therapeutics provide exciting future directions to increase the TIs of therapeutics often considered too toxic to administer systematically.
Combinations with highly selective therapeutics
Combination strategies might ultimately provide a path to significantly improving the TI of therapeutics targeting pan-essential genes. A common pitfall in the development of HDAC and PLK1 inhibitors was the combination with other low-TI drugs leading to marked increases in serious AEs (e.g., HDAC inhibitors with bortezimib). An alternative approach for low-TI therapies would be to prioritize combinations with high-TI drugs. Three CDK4/6 inhibitors are approved in metastatic ER+/HER2− breast cancer in combination with endocrine therapy whereby the pivotal trials consistently showed >60% improvement in the median PFS (Klein et al., 2018). Similarly, three MEK1/2 inhibitors (trametinib, cobimetinib, and binimetinib) are approved in combination with BRAF inhibitors to treat BRAF-mutant melanoma, again delivering significant clinical benefits (Yaeger and Corcoran, 2019). In these examples, although CDK4/6 and MEK1/2 double knockouts show a pan-lethal effect and CDK4/6 and MEK1/2 inhibitors are broadly acting, when the inhibitors are combined with either tissue-specific therapeutics (e.g., fulvestrant) or mutant-selective agents (BRAF inhibitors) (Im et al., 2019; Long et al., 2015; Robert et al., 2019) the additive or synergistic activity is confined to the tumor, thus resulting in a better than expected TI for the combination. This strategy could also be beneficial in creating barriers to the development of acquired resistance. For example, AURKA and AURKB can be resistance nodes to third-generation EGFR inhibitors in NSCLC models (Bertran-Alamillo et al., 2019; Shah et al., 2019), and AURKA appears to be a node for KRASG12C-specific inhibitor resistance (Xue et al., 2020). Thus, the combination of aurora kinase inhibitors with EGFR or KRASG12C inhibitors in patients with such mutations could provide clinical benefit in the future. Lastly, in indications such as microsatellite instability cancers where PD-1/PD-L1 antagonists have biomarker-driven anti-cancer activity, combinations with pan-essential inhibitors again might shift the TI in a more favorable direction.
CONCLUSION
The high failure rate in oncology drug development has impeded our efforts to efficiently bring new therapeutics to cancer patients. The 5.1% success rate from phase 1 to FDA approval is far from optimal, yet we can learn from these prior experiences. From the case studies in this review, it is apparent that not all cancer targets are created equal. Developing therapeutics that target pan-essential genes requires careful target prioritization and validation, biomarker identification, pharmacokinetic optimization, and combination strategies to increase the TI. While it is obvious that testing another pan-HDAC inhibitor monotherapy in another non-stratified solid tumor indication should be avoided, implementing new strategies to improve our current drug development steps is not trivial. Hence, in addition to the mindset shift, future research innovations including extending genomic and functional datasets to normal lineages, new genetic technologies for partial loss-of-function experiments, advances in cancer preclinical models, and comprehensive drug combination assessment in preclinical settings will be pivotal to further optimizing drug-discovery efforts to benefit cancer patients. Finally, future basic science efforts to thoroughly understand the fundamental biology of pan-essential genes, and their specific involvements in different cancer types, will also benefit the discovery of the next generation of cancer therapeutics.
ACKNOWLEDGMENTS
We thank Kornelia Polyak, Keith Flaherty, and members of the Sellers lab for the critical reading of the manuscript and helpful discussions.
DECLARATION OF INTERESTS
W.R.S. is a board or SAB member and holds equity in Ideaya Biosciences, Civetta Therapeutics, and Bluebird bio and has consulted for Array, Astex, Dynamo Therapeutics, Epidarex Capital, Ipsen, PearlRiver Therapeutics, Sanofi, Servier and Syndax Pharmaceuticals, and receives research funding from Pfizer Pharmaceuticals, Merck Pharmaceuticals, Ideaya Biosciences and Ridgeline Discovery. W.R.S. is a co-patent holder on EGFR mutation diagnostic patents. T.I. is an employee and shareholder of Scorpion Therapeutics.
REFERENCES
- Álvarez-Fernández M, and Malumbres M (2020). Mechanisms of sensitivity and resistance to CDK4/6 inhibition. Cancer Cell 37, 514–529. [DOI] [PubMed] [Google Scholar]
- Ando K, Ozaki T, Yamamoto H, Furuya K, Hosoda M, Hayashi S, Fukuzawa M, and Nakagawara A (2004). Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J. Biol. Chem 279, 25549–25561. [DOI] [PubMed] [Google Scholar]
- Ardlie KG, DeLuca DS, Segrè AV, Sullivan TJ, Young TR, Gelfand ET, Trowbridge CA, Maller JB, Tukiainen T, Lek M, et al. (2015). The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton S, Song YH, Nolan J, Cadogan E, Murray J, Odedra R, Foster J, Hall PA, Low S, Taylor P, et al. (2016). Aurora kinase inhibitor nanoparticles target tumors with favorable therapeutic index in vivo. Sci. Transl. Med 8, 325ra17. [DOI] [PubMed] [Google Scholar]
- Banaszynski LA, Sellmyer MA, Contag CH, Wandless TJ, and Thorne SH (2008). Chemical control of protein stability and function in living mice. Nat. Med 14, 1123–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behan FM, Iorio F, Picco G, Gonçalves E, Beaver CM, Migliardi G, Santos R, Rao Y, Sassi F, Pinnelli M, et al. (2019). Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 568, 511–516. [DOI] [PubMed] [Google Scholar]
- Bertran-Alamillo J, Cattan V, Schoumacher M, Codony-Servat J, Giménez-Capitán A, Cantero F, Burbridge M, Rodríguez S, Teixidó C, Roman R, et al. (2019). AURKB as a target in non-small cell lung cancer with acquired resistance to anti-EGFR therapy. Nat. Commun 10, 10.1038/s41467-019-09734-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomen VA, Májek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, Sacco R, Van Diemen FR, Olk N, Stukalov A, et al. (2015). Gene essentiality and synthetic lethality in haploid human cells. Science 350, 1092–1096. [DOI] [PubMed] [Google Scholar]
- Burger JA, and Wiestner A (2018). Targeting B cell receptor signalling in cancer: preclinical and clinical advances. Nat. Rev. Cancer 18, 148–167. [DOI] [PubMed] [Google Scholar]
- Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, Gaida K, Holt T, Knutson CG, Koppada N, et al. (2019). The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223. [DOI] [PubMed] [Google Scholar]
- de Cárcer G, Venkateswaran SV, Salgueiro L, El Bakkali A, Somogyi K, Rowald K, Montañés P, Sanclemente M, Escobar B, de Martino A, et al. (2018). Plk1 overexpression induces chromosomal instability and suppresses tumor development. Nat. Commun 9, 3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et al. (2011). Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med 364, 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Chandrashekar DS, Varambally S, and Creighton CJ (2019a). Pan-cancer molecular subtypes revealed by mass-spectrometry-based proteomic characterization of more than 500 human cancers. Nat. Commun 10, 5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Zhang Z, Jiang S, Li R, Li W, Zhao C, Hong H, Huang X, Li H, and Bo X (2019b). New insights on human essential genes based on integrated analysis and the construction of the HEGIAP web-based platform. Brief. Bioinform 2019, 1397–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium H, Human NIH, and Atlas B (2019). The human body at cellular resolution: the NIH Human Biomolecular Atlas Program. Nature 574, 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsello SM, Nagari RT, Spangler RD, Rossen J, Kocak M, Bryan JG, Humeidi R, Peck D, Wu X, Tang AA, et al. (2020). Discovering the anticancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 1, 235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. (2011). BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster JM, Pacini C, Pantel S, Behan FM, Green T, Krill-Burger J, Beaver CM, Younger ST, Zhivich V, Najgebauer H, et al. (2019). Agreement between two large pan-cancer CRISPR-Cas9 gene dependency data sets. Nat. Commun 10, 10.1038/s41467-019-13805-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Bongard HJGD, Mathôt RAA, Beijnen JH, and Schellens JHM (2000). Pharmacokinetically guided administration of chemotherapeutic agents. Clin. Pharmacokinet 39, 345–367. [DOI] [PubMed] [Google Scholar]
- DeWeirdt PC, Sanson KR, Sangree AK, Hegde M, Hanna RE, Feeley MN, Griffith AL, Teng T, Borys SM, Strand C, et al. (2020). Optimization of AsCas12a for combinatorial genetic screens in human cells. Nat. Biotechnol 10.1038/s41587-020-0600-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra KK, Cattaneo CM, Weeber F, Chalabi M, van de Haar J, Fanchi LF, Slagter M, van der Velden DL, Kaing S, Kelderman S, et al. (2018). Generation of tumor-reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell 174, 1586–1598.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNardo CD, Stein EM, De Botton S, Roboz GJ, Altman JK, Mims AS, Swords R, Collins RH, Mannis GN, Pollyea DA, et al. (2018). Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N. Engl. J. Med 378, 2386–2398. [DOI] [PubMed] [Google Scholar]
- Döhner H, Symeonidis A, Sanz MA, Deeren D, Demeter J, Anagnostopoulos A, Esteve J, Fiedler W, Porkka K, Kim HJ, et al. (2016). Phase III randomised trial of volasertib plus low-dose cytarabine (LDAC) versus placebo plus LDAC in patients aged ≥65 years with previously untreated AML, ineligible for intensive therapy21st Congress of the European Hematology Association. Haematologica 101 (s1), Abstract S501. [Google Scholar]
- Dokmanovic M, Clarke C, and Marks PA (2007). Histone deacetylase inhibitors: overview and perspectives. Mol. Cancer Res 5, 981–989. [DOI] [PubMed] [Google Scholar]
- Ellis PM, Leighl NB, Hirsh V, Reaume MN, Blais N, Wierzbicki R, Sadrolhefazi B, Gu Y, Liu D, Pilz K, et al. (2015). A randomized, open-label phase II trial of volasertib as monotherapy and in combination with standard-dose pemetrexed compared with pemetrexed monotherapy in second-line treatment for non-small-cell lung cancer. Clin. Lung Cancer 16, 457–465. [DOI] [PubMed] [Google Scholar]
- Falkenberg KJ, and Johnstone RW (2014). Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov 13, 673–691. [DOI] [PubMed] [Google Scholar]
- Foote M (1998). The importance of planned dose of chemotherapy on time: do we need to change our clinical practice? Oncologist 3, 365–368. [PubMed] [Google Scholar]
- Francies HE, McDermott U, and Garnett MJ (2020). Genomics-guided pre-clinical development of cancer therapies. Nat. Cancer 1, 482–492. [DOI] [PubMed] [Google Scholar]
- Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, Zhang C, Schnell C, Yang G, Zhang Y, et al. (2015). High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med 21, 1318–1325. [DOI] [PubMed] [Google Scholar]
- Garcia-Manero G, Fenaux P, Al-Kali A, Baer MR, Sekeres MA, Roboz GJ, Gaidano G, Scott BL, Greenberg P, Platzbecker U, et al. (2016). Rigosertib versus best supportive care for patients with high-risk myelodysplastic syndromes after failure of hypomethylating drugs (ONTIME): a randomised, controlled, phase 3 trial. Lancet Oncol. 17, 496–508. [DOI] [PubMed] [Google Scholar]
- Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, Barretina J, Gelfand ET, Bielski CM, Li H, et al. (2019). Next-generation characterization of the cancer cell line encyclopedia. Nature 569, 503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillette MA, Satpathy S, Cao S, Dhanasekaran SM, Vasaikar SV, Krug K, Petralia F, Li Y, Liang WW, Reva B, et al. (2020). Proteogenomic characterization reveals therapeutic vulnerabilities in lung adenocarcinoma. Cell 182, 200–225.e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonatopoulos-Pournatzis T, Aregger M, Brown KR, Farhangmehr S, Braunschweig U, Ward HN, Ha KCH, Weiss A, Billmann M, Durbic T, et al. (2020). Genetic interaction mapping and exon-resolution functional genomics with a hybrid Cas9-Cas12a platform. Nat. Biotechnol 38, 638–648. [DOI] [PubMed] [Google Scholar]
- Gralla RJ, de Wit R, Herrstedt J, Carides AD, Ianus J, Guoguang-Ma J, Evans JK, and Horgan KJ (2005). Antiemetic efficacy of the neurokinin-1 antagonist, aprepitant, plus a 5HT3 antagonist and a corticosteroid in patients receiving anthracyclines or cyclophosphamide in addition to high-dose cisplatin. Cancer 104, 864–868. [DOI] [PubMed] [Google Scholar]
- Haley B, and Roudnicky F (2020). Functional genomics for cancer drug target discovery. Cancer Cell 38, 31–43. [DOI] [PubMed] [Google Scholar]
- Hallin J, Engstrom LD, Hargi L, Calinisan A, Aranda R, Briere DM, Sudhakar N, Bowcut V, Baer BR, Ballard JA, et al. (2020). The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 10, 54–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K, Jeng EE, Hess GT, Morgens DW, Li A, and Bassik MC (2017). Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat. Biotechnol 35, 463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, et al. (2015). High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 163, 1515–1526. [DOI] [PubMed] [Google Scholar]
- Hashizume R (2017). Epigenetic targeted therapy for diffuse intrinsic pontine glioma. Neurol. Med. Chir. (Tokyo) 57, 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks RW, Yuvaraj S, and Kil LP (2014). Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat. Rev. Cancer 14, 219–232. [DOI] [PubMed] [Google Scholar]
- Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, Falchook GS, Price TJ, Sacher A, Denlinger CS, et al. (2020). KRAS G12C inhibition with sotorasib in advanced solid tumors. N. Engl. J. Med 383, 1207–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A, Garraway LA, Ashworth A, and Weber B (2020). Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov 19, 23–38. [DOI] [PubMed] [Google Scholar]
- Hyman DM, Taylor BS, and Baselga J (2017). Implementing genome-driven oncology. Cell 168, 584–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SA, Lu YS, Bardia A, Harbeck N, Colleoni M, Franke F, Chow L, Sohn J, Lee KS, Campos-Gomez S, et al. (2019). Overall survival with ribociclib plus endocrine therapy in breast cancer. N. Engl. J. Med 381, 307–316. [DOI] [PubMed] [Google Scholar]
- Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, DeMarini DJ, Cox DS, Xu Y, Morris SR, et al. (2012). Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 13, 773–781. [DOI] [PubMed] [Google Scholar]
- Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Gonçalves E, Barthorpe S, Lightfoot H, et al. (2016). A landscape of pharmacogenomic interactions in cancer. Cell 166, 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai K, Nambu T, Dairiki R, Ohori M, Yu J, Burke K, Gotou M, Yamamoto Y, Ebara S, Shibata S, et al. (2019). Molecular mechanism and potential target indication of TAK-931, a novel CDC7-selective inhibitor. Sci. Adv 5, eaav3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalloh M (2015). Panobinostat: a surprising approval for the treatment of multiple myeloma. Pharm. Today 21, 38. [Google Scholar]
- Jeay S, Ferretti S, Holzer P, Fuchs J, Chapeau EA, Wartmann M, Sterker D, Romanet V, Murakami M, Kerr G, et al. (2018). Dose and schedule determine distinct molecular mechanisms underlying the efficacy of the p53-MDM2 inhibitor HDM201. Cancer Res. 78, 6257–6267. [DOI] [PubMed] [Google Scholar]
- Jost M, Santos DA, Saunders RA, Horlbeck MA, Hawkins JS, Scaria SM, Norman TM, Hussmann JA, Liem CR, Gross CA, et al. (2020). Titrating gene expression using libraries of systematically attenuated CRISPR guide RNAs. Nat. Biotechnol 38, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarjian HM, Martinelli G, Jabbour EJ, Quintás-Cardama A, Ando K, Bay JO, Wei A, Gröpper S, Papayannidis C, Owen K, et al. (2013). Stage I of a phase 2 study assessing the efficacy, safety, and tolerability of barasertib (AZD1152) versus low-dose cytosine arabinoside in elderly patients with acute myeloid leukemia. Cancer 119, 2611–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khongorzul P, Ling CJ, Khan FU, Ihsan AU, and Zhang J (2020). Antibody-drug conjugates: a comprehensive review. Mol. Cancer Res 18, 3–19. [DOI] [PubMed] [Google Scholar]
- Kim JM, Nakao K, Nakamura K, Saito I, Katsuki M, Arai K, and Masai H (2002). Inactivation of Cdc7 kinase in mouse ES cells results in S-phase arrest and p53-dependent cell death. EMBO J. 21, 2168–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood JM, Canellos GP, Ervin TJ, Pitman SW, Weichselbaum R, and Miller D (1981). Increased therapeutic index using moderate dose methotrexate and leucovorin twice weekly versus weekly high dose methotrexate-leucovorin in patients with advanced squamous carcinoma of the head and neck: a safe new effective regimen. Cancer 47, 2414–2421. [DOI] [PubMed] [Google Scholar]
- Klein ME, Kovatcheva M, Davis LE, Tap WD, and Koff A (2018). CDK4/6 Inhibitors: the mechanism of action may not be as simple as once thought. Cancer Cell 34, 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltun ES, Tsuhako AL, Brown DS, Aay N, Arcalas A, Chan V, Du H, Engst S, Ferguson K, Franzini M, et al. (2012). Discovery of XL413, a potent and selective CDC7 inhibitor. Bioorg. Med. Chem. Lett 22, 3727–3731. [DOI] [PubMed] [Google Scholar]
- Krug LM, Kindler HL, Calvert H, Manegold C, Tsao AS, Fennell D,Öhman R, Plummer R, Eberhardt WEE, Fukuoka K, et al. (2015). Vorinostat in patients with advanced malignant pleural mesothelioma who have progressed on previous chemotherapy (VANTAGE-014): a phase 3, double-blind, randomised, placebo-controlled trial. Lancet Oncol. 16, 447–456. [DOI] [PubMed] [Google Scholar]
- Kulkarni AA, Kingsbury SR, Tudzarova S, Hong HK, Loddo M, Rashid M, Rodriguez-Acebes S, Prevost AT, Ledermann JA, Stoeber K, et al. (2009). Cdc7 kinase is a predictor of survival and a novel therapeutic target in epithelial ovarian carcinoma. Clin. Cancer Res 15, 2417–2425. [DOI] [PubMed] [Google Scholar]
- Lens SMA, Voest EE, and Medema RH (2010). Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat. Rev. Cancer 10, 825–841. [DOI] [PubMed] [Google Scholar]
- Leonard RCF, Williams S, Tulpule A, Levine AM, and Oliveros S (2009). Improving the therapeutic index of anthracycline chemotherapy: focus on liposomal doxorubicin (Myocet™). Breast 18, 218–224. [DOI] [PubMed] [Google Scholar]
- Li H, Ning S, Ghandi M, Kryukov GV, Gopal S, Deik A, Souza A, Pierce K, Keskula P, Hernandez D, et al. (2019). The landscape of cancer cell line metabolism. Nat. Med 25, 850–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long GV, Stroyakovskiy D, Gogas H, Levchenko E, De Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, et al. (2015). Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet 386, 444–451. [DOI] [PubMed] [Google Scholar]
- Lu L-Y, Wood JL, Minter-Dykhouse K, Ye L, Saunders TL, Yu X, and Chen J (2008). Polo-like kinase 1 is essential for early embryonic development and tumor suppression. Mol. Cell. Biol 28, 6870–6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Solimini NL, and Elledge SJ (2009). Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136, 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair B, Tomic J, Masud SN, Cohen B, Keller G, and Correspondence JM (2019). Essential gene profiles for human pluripotent stem cells identify uncharacterized genes and substrate dependencies. Cell Reports 27, 599–615.e12. [DOI] [PubMed] [Google Scholar]
- Marks PA, Richon VM, Miller T, and Kelly WK (2004). Histone deacetylase inhibitors. Adv. Cancer Res 91, 137–168. [DOI] [PubMed] [Google Scholar]
- Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, et al. (2015). DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med 373, 1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald ER, de Weck A, Schlabach MR, Billy E, Mavrakis KJ, Hoffman GR, Belur D, Castelletti D, Frias E, Gampa K, et al. (2017). Project DRIVE: a compendium of cancer dependencies and synthetic lethal relationships uncovered by large-scale, deep RNAi screening. Cell 170, 577–592.e10. [DOI] [PubMed] [Google Scholar]
- Mehra R, Serebriiskii IG, Burtness B, Astsaturov I, and Golemis EA (2013). Aurora kinases in head and neck cancer. Lancet Oncol. 14, e425–e435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok TS, Wu Y-L, Ahn M-J, Garassino MC, Kim HR, Ramalingam SS, Shepherd FA, He Y, Akamatsu H, Theelen WSME, et al. (2017). Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N. Engl. J. Med 376, 629–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagnoli A, Tenca P, Sola F, Carpani D, Brotherton D, Albanese C, and Santocanale C (2004). Cdc7 inhibition reveals a p53-dependent replication checkpoint that is defective in cancer cells. Cancer Res. 64, 7110–7116. [DOI] [PubMed] [Google Scholar]
- Montagnoli A, Valsasina B, Croci V, Menichincheri M, Rainoldi S, Marchesi V, Tibolla M, Tenca P, Brotherton D, Albanese C, et al. (2008). A Cdc7 kinase inhibitor restricts initiation of DNA replication and has antitumor activity. Nat. Chem. Biol 4, 357–365. [DOI] [PubMed] [Google Scholar]
- Montagnoli A, Moll J, and Colotta F (2010). Targeting cell division cycle 7 kinase: a new approach for cancer therapy. Clin. Cancer Res 16, 4503–4511. [DOI] [PubMed] [Google Scholar]
- Mross K, Richly H, Frost A, Scharr D, Nokay B, Graeser R, Lee C, Hilbert J, Goeldner R-G, Fietz O, et al. (2016). A phase I study of BI 811283, an Aurora B kinase inhibitor, in patients with advanced solid tumors. Cancer Chemother. Pharmacol 78, 405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet B, Roberts JM, Buckley DL, Paulk J, Dastjerdi S, Yang A, Leggett AL, Erb MA, Lawlor MA, Souza A, et al. (2018). The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol 14, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet B, Ferguson FM, Seong BKA, Kuljanin M, Leggett AL, Mohardt ML, Robichaud A, Conway AS, Buckley DL, Mancias JD, et al. (2020). Rapid and direct control of target protein levels with VHL-recruiting dTAG molecules. BioRxiv. 10.1101/2020.03.13.980946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume T, Kiyomitsu T, Saga Y, and Kanemaki MT (2016). Rapid protein depletion in human cells by auxin-inducible degron tagging with short homology donors. Cell Rep. 15, 210–218. [DOI] [PubMed] [Google Scholar]
- Neidhart JA, Mangalik A, Stidley CA, Tebich SL, Sarmiento LE, Pfile JE, Oette DH, and Oldham FB (1992). Dosing regimen of granulocyte-macrophage colony-stimulating factor to support dose-intensive chemotherapy. J. Clin. Oncol 10, 1460–1469. [DOI] [PubMed] [Google Scholar]
- Nichols CA, Gibson WJ, Brown MS, Kosmicki JA, Busanovich JP, Wei H, Urbanski LM, Curimjee N, Berger AC, Gao GF, et al. (2020). Loss of heterozygosity of essential genes represents a widespread class of potential cancer vulnerabilities. Nat. Commun 11, 2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurgali K, Jagoe RT, and Abalo R (2018). Editorial: adverse effects of cancer chemotherapy: anything new to improve tolerance and reduce sequelae? Front. Pharmacol 9, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto T, and Sicinski P (2017). Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 17, 93–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor OA, Ozcan M, Jacobsen ED, Roncero JM, Trotman J, Demeter J, Masszi T, Pereira J, Ramchandren R, Beaven A, et al. (2019). Randomized phase III study of alisertib or investigator’s choice (selected single agent) in patients with relapsed or refractory peripheral T-cell lymphoma. J. Clin. Oncol 37, 613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neil BH, Scott AJ, Ma WW, Cohen SJ, Aisner DL, Menter AR, Tejani MA, Cho JK, Granfortuna J, Coveler L, et al. (2015). A phase II/III randomized study to compare the efficacy and safety of rigosertib plus gemcitabine versus gemcitabine alone in patients with previously untreated metastatic pancreatic cancer. Ann. Oncol 26, 1923–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shaughnessy J, Moroose RL, Babu S, Baramidze K, Chan D, Leitner SP, Nemsadze G, Ordentlich P, Quaranto C, Meyers ML, et al. (2020). Results of ENCORE 602 (TRIO025), a phase II, randomized, placebo-controlled, double-blinded, multicenter study of atezolizumab with or without entinostat in patients with advanced triple-negative breast cancer (aTNBC). J. Clin. Oncol 38, 1014. [Google Scholar]
- Papac RJ, Donnall T, Skeel RT, Marsh JC, and Mitchell MS (1973). Improved therapeutic index of methotrexate with “leucovorin rescue”. Cancer Res. 33, 1729–1734. [PubMed] [Google Scholar]
- Pujade-Lauraine E, Selle F, Weber B, Ray-Coquard I-L, Vergote I, Sufliarsky J, Del Campo JM, Lortholary A, Lesoin A, Follana P, et al. (2016). Volasertib versus chemotherapy in platinum-resistant or -refractory ovarian cancer: a randomized phase II Groupe des Investigateurs Nationaux pour l’Etude des Cancers de l’Ovaire study. J. Clin. Oncol 34, 706–713. [DOI] [PubMed] [Google Scholar]
- Raab M, Kappel S, Krämer A, Sanhaji M, Matthess Y, Kurunci-Csacsko E, Calzada-Wack J, Rathkolb B, Rozman J, Adler T, et al. (2011). Toxicity modelling of Plk1-targeted therapies in genetically engineered mice and cultured primary mammalian cells. Nat. Commun 2, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancati G, Moffat J, Typas A, and Pavelka N (2018). Emerging and evolving concepts in gene essentiality. Nat. Rev. Genet 19, 34–49. [DOI] [PubMed] [Google Scholar]
- Regev A, Teichmann S, Lander E, Amit I, Benoist C, Birney E, Bodenmiller B, Campbell P, Carninci P, and Enard W (2017). Science forum: the human cell atlas. eLife 6, e27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Grob JJ, Stroyakovskiy D, Karaszewska B, Hauschild A, Levchenko E, Chiarion Sileni V, Schachter J, Garbe C, Bondarenko I, et al. (2019). Five-year outcomes with dabrafenib plus trametinib in metastatic melanoma. N. Engl. J. Med 381, 626–636. [DOI] [PubMed] [Google Scholar]
- San-Miguel JF, Hungria VTM, Yoon SS, Beksac M, Dimopoulos MA, Elghandour A, Jedrzejczak WW, Günther A, Nakorn TN, Siritanaratkul N, et al. (2014). Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 15, 1195–1206. [DOI] [PubMed] [Google Scholar]
- San-Miguel JF, Hungria VTM, Yoon SS, Beksac M, Dimopoulos MA, Elghandour A, Jedrzejczak WW, Günther A, Nakorn TN, Siritanaratkul N, et al. (2016). Overall survival of patients with relapsed multiple myeloma treated with panobinostat or placebo plus bortezomib and dexamethasone (the PANORAMA 1 trial): a randomised, placebo-controlled, phase 3 trial. Lancet Haematol. 3, e506–e515. [DOI] [PubMed] [Google Scholar]
- Scripture CD, Figg WD, and Sparreboom A (2005). Paclitaxel chemotherapy: from empiricism to a mechanism-based formulation strategy. Ther. Clin. Risk Manag 1, 107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seashore-Ludlow B, Rees MG, Cheah JH, Coko M, Price EV, Coletti ME, Jones V, Bodycombe NE, Soule CK, Gould J, et al. (2015). Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov. 5, 1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah KN, Bhatt R, Rotow J, Rohrberg J, Olivas V, Wang VE, Hemmati G, Martins MM, Maynard A, Kuhn J, et al. (2019). Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat. Med 25, 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan WP, Fox RM, Begley CG, Maher D, McGrath KM, Begley CG, Juttner CA, To LB, Szer J, and Mostyn G (1992). Effect of peripheral-blood progenitor cells mobilised by filgrastim (G-CSF) on platelet recovery after high-dose chemotherapy. Lancet 339, 640–644. [DOI] [PubMed] [Google Scholar]
- Shi J, Kantoff PW, Wooster R, and Farokhzad OC (2017). Cancer nano-medicine: progress, challenges and opportunities. Nat. Rev. Cancer 17, 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Doi T, Kondo S, Takahashi H, Yamamoto N, Sheldon-Waniga E, Zhou X, Bahamon B, Li H, and Kuboki Y (2018). First-in-human phase 1 study of TAK-931, an oral cell division cycle 7 (CDC7) inhibitor, in patients (pts) with advanced solid tumors. J. Clin. Oncol 36, 2506. [Google Scholar]
- Stadler WM, Vaughn DJ, Sonpavde G, Vogelzang NJ, Tagawa ST, Petrylak DP, Rosen P, Lin CC, Mahoney J, Modi S, et al. (2014). An open-label, single-arm, phase 2 trial of the polo-like kinase inhibitor volasertib (BI 6727) in patients with locally advanced or metastatic urothelial cancer. Cancer 120, 976–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strebhardt K (2010). Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat. Rev. Drug Discov 9, 643–660. [DOI] [PubMed] [Google Scholar]
- Strebhardt K, and Ullrich A (2006). Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer 6, 321–330. [DOI] [PubMed] [Google Scholar]
- Tallman MS, Rademaker AW, Jahnke L, Brown SG, Bauman A, Mangan C, Kelly C, Rubin H, Kies MS, Shaw J, et al. (1997). High-dose chemotherapy, autologous bone marrow or stem cell transplantation and post-transplant consolidation chemotherapy in patients with advanced breast cancer. Bone Marrow Transpl. 20, 721–729. [DOI] [PubMed] [Google Scholar]
- Thomas DW, Burns J, Audette J, Carroll A, Dow-Hygelund C, and Hay M (2015). Clinical Development Success Rates 2006–2015 (BIO/Informa/Amplion). https://www.bio.org/sites/default/files/legacy/bioorg/docs/Clinical%20Development%20Success%20Rates%202006-2015%20-%20BIO,%20Biomedtracker,%20Amplion%202016.pdf.
- Townsend EC, Murakami MA, Christodoulou A, Christie AL, Köster J, DeSouza TA, Morgan EA, Kallgren SP, Liu H, Wu SC, et al. (2016). The public repository of xenografts enables discovery and randomized phase II-like trials in mice. Cancer Cell 29, 574–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trent JM, Buick RN, Olson S, Horns RC, and Schimke RT (1984). Cytologic evidence for gene amplification in methotrexate-resistant cells obtained from a patient with ovarian adenocarcinoma. J. Clin. Oncol 2, 8–15. [DOI] [PubMed] [Google Scholar]
- Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, Bremer R, Gillette S, Kong J, Haass NK, et al. (2008). Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. U S A 105, 3041–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a cancer dependency map. Cell 170, 564–576.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tym JE, Mitsopoulos C, Coker EA, Razaz P, Schierz AC, Antolin AA, and Al-Lazikani B (2016). canSAR: an updated cancer research and drug discovery knowledgebase. Nucleic Acids Res. 44, D938–D943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. (2015). Tissue-based map of the human proteome. Science 80, 347. [DOI] [PubMed] [Google Scholar]
- Viswanathan SR, Nogueira MF, Buss CG, Krill-Burger JM, Wawer MJ, Malolepsza E, Berger AC, Choi PS, Shih J, Taylor AM, et al. (2018). Genome-scale analysis identifies paralog lethality as a vulnerability of chromosome 1p loss in cancer. Nat. Genet 50, 937–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, and Sabatini DM (2015). Identification and characterization of essential genes in the human genome. Science 350, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang S, Li F, Zhou Y, Zhang Y, Wang Z, Zhang R, Zhu J, Ren Y, Tan Y, et al. (2020). Therapeutic target database 2020: enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res. 48, D1031–D1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B, Kim D, Li C, Stanchina E. De, Mazutis L, et al. (2020). Rapid non-uniform adaptation to conformation-specific KRAS (G12C) inhibition. Nature 577, 421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaeger R, and Corcoran RB (2019). Targeting alterations in the RAF-MEK pathway. Cancer Discov. 9, 329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Masai H, and Bartek J (2014). Regulation and roles of Cdc7 kinase under replication stress. Cell Cycle 13, 1859–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeruva SLH, Zhao F, Miller KD, Tevaarwerk AJ, Wagner LI, Gray RJ, Sparano JA, and Connolly RM (2018). E2112: randomized phase III trial of endocrine therapy plus entinostat/placebo in patients with hormone receptor-positive advanced breast cancer. Npj Breast Cancer 4, 10.1038/s41523-017-0053-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz A, Peretz M, Aharony A, Sagi I, and Benvenisty N (2018). Defining essential genes for human pluripotent stem cells by CRISPR-Cas9 screening in haploid cells. Nat. Cell Biol 20, 610–619. [DOI] [PubMed] [Google Scholar]
- You I, Erickson EC, Donovan KA, Eleuteri NA, Fischer ES, Gray NS, and Toker A (2020). Discovery of an AKT degrader with prolonged inhibition of downstream signaling. Cell Chem. Biol 27, 66–73.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Mannan AM, Yvone GM, Ross KN, Zhang YL, Marton MA, Taylor BR, Crenshaw A, Gould JZ, Tamayo P, et al. (2016). High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat. Biotechnol 34, 419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Ward JD, Cheng Z, and Dernburg AF (2015). The auxin-inducible degradation (AID) system enables versatile conditional protein depletion in C. elegans. Dev 142, 4374–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. (2011). RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]