

Abstract
Overactive bladder (OAB) is a pervasive clinical problem involving alterations in both neurogenic and myogenic activity. While there has been some progress in understanding neurogenic inputs to OAB, the mechanisms controlling myogenic bladder activity are unclear. We report the involvement of myocardin (MYOCD) and microRNA-1 (miR-1) in the regulation of connexin 43 (GJA1), a major gap junction in bladder smooth muscle, and the collective role of these molecules during post-natal bladder development. Wild-type (WT) mouse bladders showed normal development from early post-natal to adult including increases in bladder capacity and maintenance of normal sensitivity to cholinergic agents concurrent with down-regulation of MYOCD and several smooth muscle cell (SMC) contractile genes. Myocardin heterozygous-knockout mice exhibited reduced expression of Myocd mRNA and several SMC contractile genes concurrent with bladder SMC hypersensitivity that was mediated by gap junctions. In both cultured rat bladder SMC and in vivo bladders, MYOCD down-regulated GJA1 expression through miR-1 up-regulation. Interestingly, adult myocardin heterozygous-knockout mice showed normal increases in bladder and body weight but lower bladder capacity compared to WT mice. These results suggest that MYOCD down-regulates GJA1 expression via miR-1 up-regulation, thereby contributing to maintenance of normal sensitivity and development of bladder capacity.
The human bladder is composed of urothelial cells (UC) and bladder smooth muscle cells (BSMC) that coordinate the homeostatic control of storage and voiding of urine. Pathological changes in UC and/or BSMC mediate several clinically important bladder diseases, most notably urinary incontinence or overactive bladder (OAB), which is characterized by urinary urgency and frequent micturition. The pathophysiology of OAB relates mainly to neurogenic alterations in the innervation or myogenic activity of detrusor muscle (Brading, 1997; Li et al., 2007; Yoshimura, 2007). Recent studies have revealed the mechanisms underlying neurogenic changes associated with nerve growth factor, neurotransmitters and c-fiber activity (de Groat, 1997; Yoshimura et al., 2006; Yoshimura, 2007). In contrast, however, there is a paucity of molecular insight into the underlying mechanisms for myogenic changes that may directly impact OAB. Therefore, research for therapeutic targets of myogenic components of bladder overactivity in animal models is of clinical importance.
The mechanisms underlying myogenic changes have been evaluated mainly in the field of bladder outlet obstruction. Recent studies in that field demonstrated that up-regulation of connexin 43 (GJA1), a major gap junction protein in BSMC, induced bladder hypersensitivity through gap junction generation (Christ et al., 2003; Imamura et al., 2009). Gap junctions consist of channels between neighboring cells, mediating transfer of chemical molecules or electrical stimulation (Fry et al., 2004; Li et al., 2007). In some bladder or vascular smooth muscle diseases, GJA1 up-regulation and coupling of smooth muscle occurred, thereby inducing change in contractile property through up-regulation of cell–cell communication (Liao et al., 2007; Rocha et al., 2008; Imamura et al., 2009). Collectively, these findings suggest that alterations in the regulation of GJA1 expression could underlie BSMC dysfunction.
Recent clinical studies have demonstrated that OAB in adulthood is associated with lower urinary tract symptoms in childhood (Bower et al., 2005; Fitzgerald et al., 2006; Minassian et al., 2006). These results suggest that OAB may have a hereditary background. One of the possible hereditary aspects in OAB is low bladder capacity. Bladder capacity increases with age in childhood and reaches to the average level in adulthood (Koff, 1983; Neveus et al., 2006). Therefore, impairment to this maturation process for bladder capacity could result in low urinary capacitance. Previous studies demonstrated that myogenic changes with increased gap junction generation in BSMC caused low bladder capacity as part of OAB (Imamura et al., 2009). Interestingly, the promoter region of the GJA1 gene was shown to have an activator protein-1 binding site (Negoro et al., 2011). Thus, there is evidence for transcriptional regulation of the GJA1 gene in context of OAB.
The program of SMC differentiation is determined at the genome level by the presence of one or more 10 bp sequence elements known as CArG boxes located in the immediate proximal promoter or intronic region of most SMC-restricted genes (Miano, 2003; Owens et al., 2004). The CArG box binds a widely expressed transcription factor known as serum response factor (SRF; Norman et al., 1988). SRF possesses intrinsically weak transcriptional activity, but recruits a host of coactivators that mediate gene transcription (Miano et al., 2007). Among SRF cofactors, Myocardin (MYOCD) stands as one of nature’s most powerful effectors of gene expression (Wang et al., 2001). We first proposed that MYOCD was a vital switch for the SMC differentiation program (Chen et al., 2002). Indeed, MYOCD is both necessary (Li et al., 2003; Huang et al., 2008) and sufficient (Long et al., 2008) for a functionally active SMC differentiation program. In recent years, the SRF-MYOCD transcriptional switch has been shown to regulate several microRNAs, including the miR143/145 bicistronic gene (Cordes et al., 2009). Thus, the program for SMC differentiation is governed through both transcriptional (SRF-MYOCD) and post-transcriptional (microRNAs) processes.
The overall goal of this study was to evaluate the role of transcriptional and post-transcriptional events associated with post-natal BSMC function. We studied expression of MYOCD and several SMC contractile genes in 3- and 15-week-old bladders and correlated such molecular changes to bladder function. In general, 15-week-old bladders exhibited less MYOCD and contractile proteins that correlated with increases in bladder volume and reduced micturition. Interestingly, 3-week-old mice with only 1 Myocd allele phenocopied the down-regulated condition of SMC markers and miR-1 in adult wild-type (WT) mice. Finally, we showed that the maintenance of normal responsiveness in adult BSMC was in part related to an attenuation of MYOCD-induced miR-1 and the latter’s repression of GJA1 protein.
Materials and Methods
Cells and animals
For cell culture, rat bladder smooth muscle cells (RBSMC) were isolated from 1-week-old male Sprague-Dawley rats using a procedure described previously (Kanematsu et al., 2005; Imamura et al., 2010). For animal studies, 3- or 15-week-old male C57BL6 WT and myocardin heterozygous-knockout (Myocd+/−) mice were used. Myocd+/− mice were kindly provided by Prof. Eric Olson of the Department of Molecular Biology, University of Texas Southwestern Medical Center. Animals were treated in accordance with NIH animal care guidelines and all animal experiments were approved by the University Committee on Animal Resources at the University of Rochester.
RNA extraction from cells and tissues
Total RNA was extracted from cultured RBSMC, mouse tissues, rat bladder urothelium, or rat bladder muscle layers with the Trizol reagent (Invitrogen, Carlsbad, CA) as described previously (Long et al., 2009).
PCR analysis
cDNA was synthesized with First-strand cDNA synthesis kit (GE Healthcare, Buckinghamshire, UK). Expression levels of myocardin (Myocd), alpha smooth muscle actin (Acta2), calponin (Cnn1), smooth muscle myosin heavy chain (Myh11), cytokeratin 20 (Krt20), and connexin 43 (Gja1) were examined with conventional PCR or qPCR. For these experiments, expression levels were normalized against an internal (Gapd) control. Primers used in the experiments are listed in the Table 1. qPCR was performed in 20 μl reactions containing SYBR Green using a My-IQ real-time PCR machine (Bio-Rad Laboratories, Hercules, CA). Reaction mixtures were incubated with 50 cycles of following two steps: 95°C for 10 sec and 64°C for 30 sec.
TABLE 1.
List of primers used in this study
| No. | Gene | Primer sequence | Application | Size (bp) | |
|---|---|---|---|---|---|
| 1 | Myocd (mouse) | For | AAGGTCCATTCCAACTGCTC | qPCR | 216 |
| Rev | CCATCTCTACTGCTGTCATCC | ||||
| 2 | Acta2 (mouse) | For | GCTTCGCTGGTGATGATGCTC | qPCR | 177 |
| Rev | AGTTGGTGATGATGCCGTGTTC | ||||
| 3 | Cnn1 (mouse) | For | GGACCAGGCGACCATCAG | qPCR | 297 |
| Rev | TAGGCAGAGTTGTAGTAGTTGTG | ||||
| 4 | Myh 11 (mouse) | For | TGCCGACACAGCCTACAGAAG | qPCR | 130 |
| Rev | GGACGCCACCACAGCCAAG | ||||
| 5 | Gja1 (mouse) | For | AACAGCAGCAGACTTTGAAAC | qPCR | 234 |
| Rev | AATGAAGAGCACCGACAGC | ||||
| 6 | Gapd (mouse) | For | CGTGCCGCCTGGAGAAAC | RT-PCR and qPCR | 136 |
| Rev | TGGGAGTTGCTGTTGAAGTCG | ||||
| 7 | Myocd (mouse) | For | TTCTGCCGATGGATTCTTCCGTGA | RT-PCR | 208 |
| Rev | AGATCCTGGATTGTGTCCAGAGGA | ||||
| 8 | Cnn1 (rat) | For | ACACTTTAACCGAGGTCCTGCCTA | qPCR | 154 |
| Rev | CTTGAGGCCATCCATGAAGTTGCT | ||||
| 9 | Acta2 (rat) | For | GCTGTGCTATGTCGCTCTGG | qPCR | 157 |
| Rev | AATGAAAGATGGCTGGAAGAGG | ||||
| 10 | Krt20 (rat) | For | ATGCGGATAACTGTGGAAGC | qPCR | 187 |
| Rev | CCTCCACGTTGACATTGTTG | ||||
| 11 | Myocd (rat) | For | CAGAAAGTGACAAGAACGATACAG | qPCR | 265 |
| Rev | TGAAGCAGCCGAGCATAGG | ||||
| 12 | Gja1 (rat) | For | GAAAGAGAGGTGCCCAGAC | qPCR | 199 |
| Rev | GCCAGGTTGTTGAGTGTTAC | ||||
| 13 | Gapd (rat) | For | GCAAGTTCAACGGCACAGTCAAG | qPCR | 124 |
| Rev | ACATACTCAGCACCAGCATCACC |
Each number (No.) indicates the pair of primers used to amplify the gene listed.
MicroRNA expression analysis
The specific primers for miR-1, miR-145, and endogenous control snoRNA202 were purchased from Ambion (Austin, TX). qPCR for miR expression was performed in 20 μl of master mix containing 2× TaqMan Universal PCR Master Mix, 20× TaqMan MicroRNA Assay mix and sample in a My-IQ (Bio-Rad Laboratories). Reaction mixtures were incubated with 40 cycles of following two steps: 95°C for 15 sec and 60°C for 60 sec.
Western blotting
Extracted protein from tissues and cells was resolved by sodium dodecylsulfate polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. The membranes were incubated with antibodies against smooth muscle myosin heavy (MYH11, 1:2000, Biomedical Technologies, Stoughton, MA), calponin (CNN1, 1:5000, Sigma, St Louis, MO), alpha smooth muscle actin (ACTA2, 1:5000, Sigma), connexin 43 (GJA1, 1:2000, Sigma), cytokeratin18 (KRT18, 1:4000, Sigma), and myocardin (MYOCD, 1:1000, Santa Cruz Biotechnology, Santa Cruz, CA). α-tubulin (TUBA, 1:2000, Sigma) or Glyceraldehyde-3-phosphate dehydrogenase (GAPD, 1:200, Millipore, Bedford, MA) was used as an internal control. Each band was quantified by densitometry using Image J 1.33 software.
Immunohistochemistry
Tissues were obtained from 10% neutral buffered formalin perfusion fixed mice. Tissues were then processed, embedded, and cut (6 μm) followed by immunohistochemistry with the indicated antibodies using established protocols we have described in a previous report (Nanda and Miano, 2012).
VSOP method for bladder overactivity
For voiding behavior analysis, including bladder capacity and urinary frequency, the voided stain on paper (VSOP) method for functional bladder overactivity was performed according to a procedure described previously (Sugino et al., 2008). Mice were treated with 50 μl/g body weight distilled water by oral gavage before each experiment and then placed on a wire net tray covered with a transparent plastic cage. Filter paper was set 20 cm under the tray to record stain of the voided urine. Voiding behavior was analyzed for 2 h and the time on voiding and stain area were recorded during the experiment. Immediately after the last voiding, mice were sacrificed. Bladders were carefully harvested with ligation of bladder neck and then opened to check post void residual urine. Stain was scanned and its area was measured with Image J 1.33 software. Voided volume was calculated from each stain area following the prepared calibration curve which was created with analysis of stain area when fixed volume of distilled water was dropped 20 cm above filter paper. Bladder capacity was defined as mean voided volume in each mouse. Urinary frequency was defined as total times of voiding during recorded 2 h.
Muscle strip test for bladder contraction
Force measurement was performed as described previously (Kanematsu et al., 2003; Imamura et al., 2009). Mouse bladder samples (n = 6) were trimmed in strips 2 mm wide with urothelium. The strips were fixed to a strip holder and placed in an organ bath filled with Krebs’ solution at 37°C. After equilibration for 1 h with a passive force of 0.5 g, strips from each group were subjected to carbachol stimulation. Another set of strips was prepared for stimulation in the presence of α-glycyrrhetinic acid (GA) to inhibit gap junction activity. These strips were washed three times with Krebs’ solution after carbachol stimulation, placed in Krebs’ solution containing 30 μM α-GA, and then subjected to carbachol stimulation. The initial dose–response curve (10−8–3 × 10−5 M) revealed that maximum tension occurred at 3 × 10−6 or 10−5 M carbachol. The muscle contractile force at each strip was expressed as percentage of its maximum response. The pEC50 (−logEC50) values for each group were calculated using GraphPad Prism 5.0 for Windows (GraphPad Software, Inc., San Diego, CA).
Treatment of RBSMC with Ad-MYOCD and a miR-1 inhibitor
RBSMC were transduced with Ad-MYOCD (MOI 100) or Ad-LacZ, control vector. Twenty-four hours after transduction, total RNA was extracted from transduced cells and expression of miR-1 and GJA1 analyzed. For loss-of-function study of miR-1, RBSMC were transfected with an antisense miR-1 inhibitor (anti-miR1 30 nM, Ambion) or a negative control inhibitor (30 nM, Ambion) using siPORT NeoFX (Ambion) according to the manufacture’s protocol. Twenty-four hours after transfection, cells were transduced with Ad-MYOCD or Ad-LacZ and then cultured for 24 h in DMEM and 10% FBS. Lysates were extracted from cultured cells and expression of MYOCD, CNN1, GJA1, and TUBA were analyzed by Western blotting.
Statistical analysis
Data were analyzed with unpaired Student’s t-test or Tukey’s multiple comparison test using Graph Pad Prism 5.0 for Windows. P < 0.05 was accepted as significant.
Results
Bladder function during post-natal development
To evaluate changes in function during bladder development, 15-week-old adult mice were compared to 3-week-old neonatal mice. First, two different types of voiding function analysis were performed. VSOP results revealed that functional bladder capacity increased, but urinary frequency decreased significantly in 15-week-old mice when compared to those in 3-week-old mice (Fig. 1A–B). A muscle strip test revealed no difference in bladder sensitivity over a wide range of carbachol doses (Fig. 1C). We next analyzed the expression of several SRF-MYOCD dependent genes in bladder, including the myocardin coactivator itself. qPCR results revealed that mRNA expression of Myocd, Acta2, and Myh11 were lower in 15-week-old mice compared to 3-week-old mice (Fig. 1D). Further, there was a general trend toward similar changes in these markers at the protein level (Fig. 1E, Supplement 1). Immunohistochemistry also revealed qualitatively lower expression of ACTA2 in 15-week-old mouse bladders when compared to 3-week-old mouse bladders (Fig. 1F). These results establish an inverse correlative change in urinary bladder volume with molecular markers of BSMC activity with increasing age.
Fig. 1.
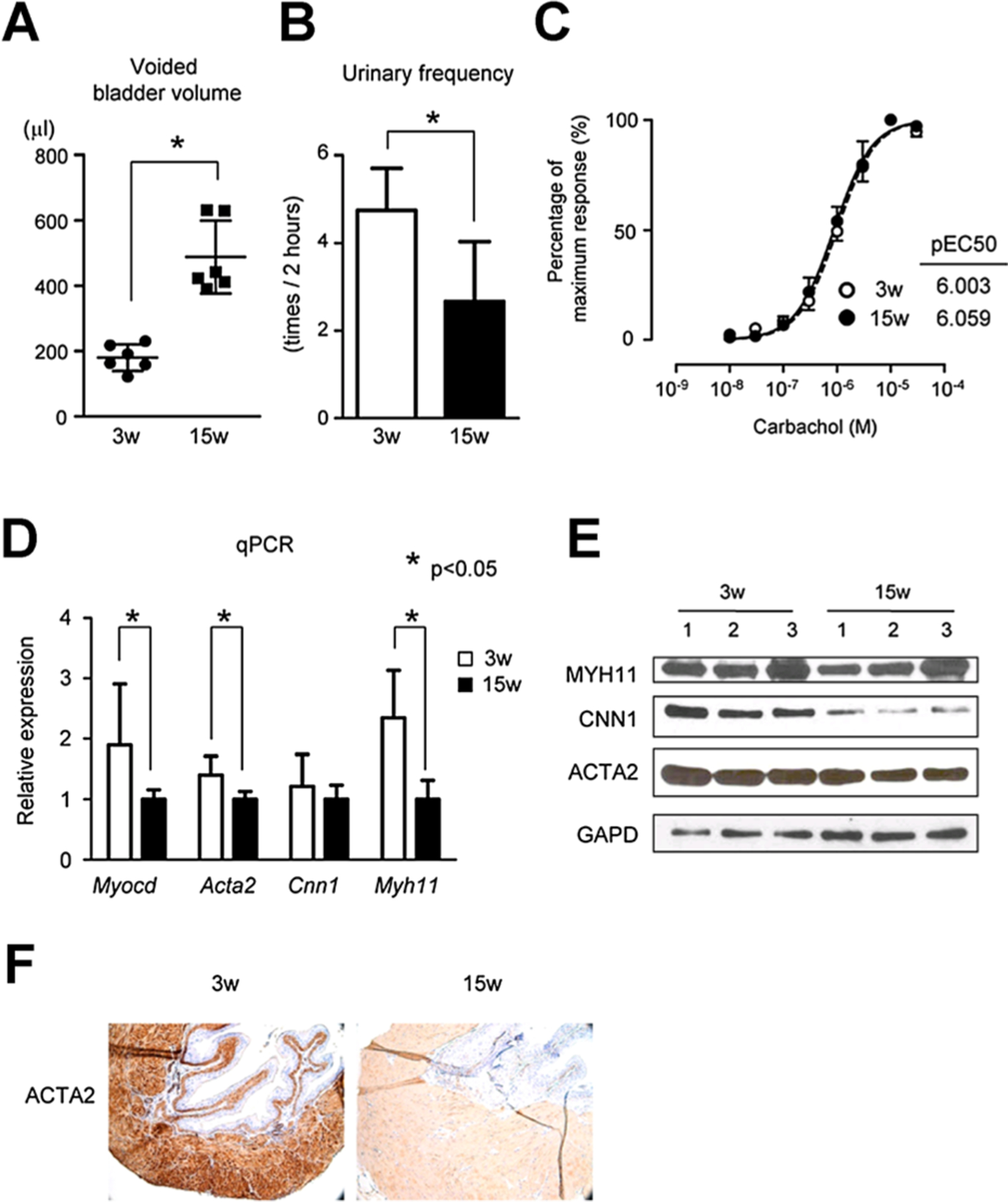
A, B: Voiding behavior analysis using the VSOP (Voided Stain On the Paper) method showed increase in bladder capacity (A) and decrease in urinary frequency (B) in 15-week-old mice compared to 3-week-old mice. C: Bladder contractility examination with bladder muscle strips showed same normal sensitivity to cholinergic agents in both 3-week-old mice and 15-week-old mice. D: qPCR data showed myocardin (MYOCD) and SM markers were down-regulated in 15-week-old mice compared to 3-week-old mice. E: Western blotting also revealed that SM markers were down-regulated in 15-week-old mice compared to 3-week-old mice. F: Immunohsitochemistry showed that ACTA2 was down-regulated in bladders of 15-week-old mice compared to those of 3-week-old mice. *Statistically significant difference (P < 0.05).
Developmental changes in BSMC marker expression in Myocd+/− mice
Conventional RT-PCR results revealed lower Myocd mRNA expression in 3-week-old Myocd+/− mouse bladders when compared to WT mouse bladders (Fig. 2A). qPCR results revealed reduced levels of Myocd mRNA expression in Myocd+/− mouse bladders at both 3- and 15-week-old when compared to WT mouse bladders (Fig. 2B–C). Western blotting revealed that expression of SM markers (CNN1 particularly) at both 3- and 15-week-old was lower in Myocd+/− mouse bladders versus WT mouse bladders (Fig. 2D, Supplement 2). Immunohistochemistry revealed lower expression of ACTA2 in 3-week-old Myocd+/− mouse bladders when compared to WT mouse bladders (Fig. 2E). These results establish reduced levels of Myocd and some of its target genes in Myocd+/− mouse bladders thus providing a molecular model to assess MYOCD-dependent functional assays in the bladder.
Fig. 2.
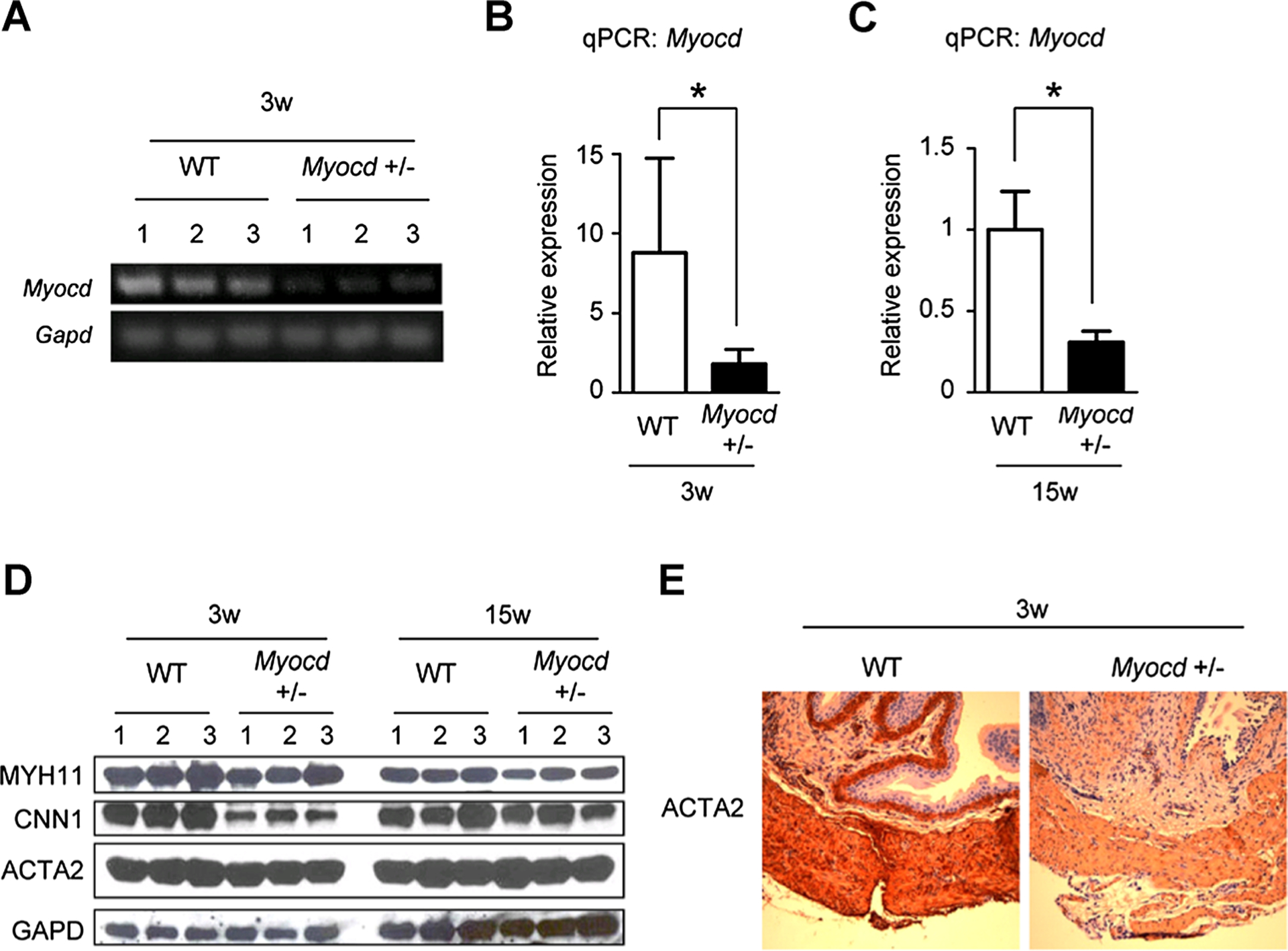
A:RT-PCRdata showed that MYOCD was down-regulatedin bladders of3-week-old Myocd+/− mice compared to those of wild-type(WT) mice. B, C: qPCR data also showed that MYOCD was down-regulated in bladders of 3(B) and 15-week-old (C) Myocd+/− mice compared to those of WT mice. D: Western blotting revealed that SM markers were down-regulated in bladders of 3 and 15-week-old Myocd+/− mice compared to those of WT mice. E: Immunohsitochemistry showed that ACTA2 was down-regulated in bladders of 3-week-old Myocd+/− mice compared to those of WT mice. *Statistically significant difference (P < 0.05).
Reduced myocardin induces hypersensitivity via gap junction generation
Previous studies demonstrated leftward shifts of the dose–response curve and increased pEC50 values in carbachol-induced contractions as evidence for myogenic hypersensitivity (Speakman et al., 1987; Brading, 1997). In 3-week-old mice, Myocd+/− mouse bladders revealed a leftward shift and significantly increased pEC50 values when compared to WT mouse bladders (Fig. 3A). Inhibition of gap junction activity with α-GA altered these effects of hypersensitivity in Myocd+/− mouse bladders, but did not induce changes in the dose–response curve and pEC50 values in WT mouse bladders (Fig. 3B–C). Similarly, in 15-week-old Myocd+/− mice, a leftward shift and significantly increased pEC50 values was observed over WT mouse bladders (Fig. 3D). These effects were altered upon addition of a gap junction inhibitor, α-GA, which did not induce changes in sensitivity of WT mouse bladders (Fig. 3E–F).
Fig. 3.
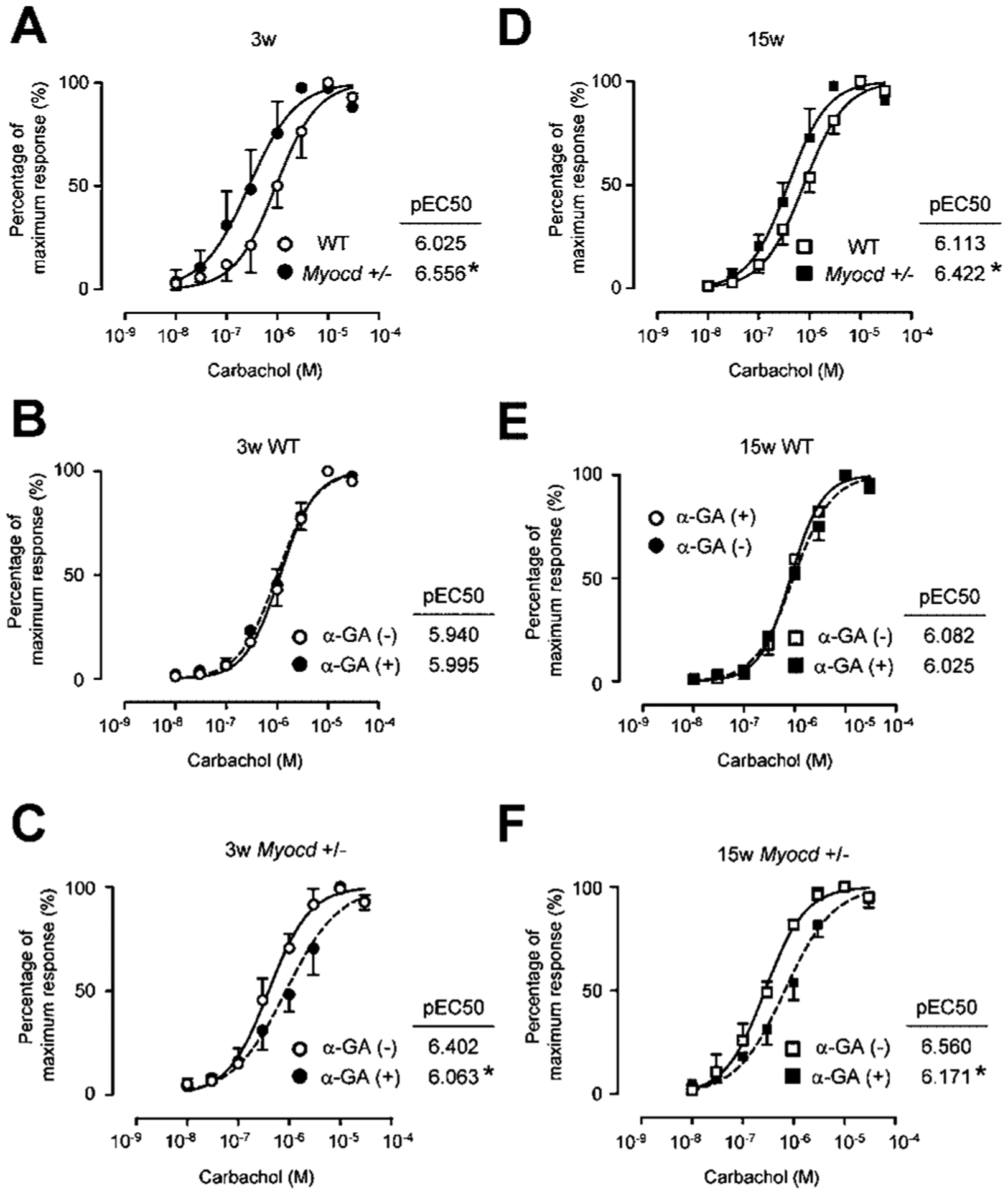
A–C: In 3-week-old mice, Myocd+/− mice showed left-shift and significantly elevated pEC50 values as an evidence of bladder hypersensitivity to cholinergic agents (A). α-glycyrrhetinic acid (GA), a gap junction inhibitor did not affect bladder sensitivity of WT mice (B) but reversed these changes in Myocd+/− mice (C). D–F: In 15-week-old mice, Myocd+/− mice also showed left-shift and significantly elevated pEC50 values as an evidence of hypersensitivity to cholinergic agents (D). α-GA did not affect bladder sensitivity of WT mice (E) but reversed these changes in Myocd+/− mice (F). *Statistically significant difference (P < 0.05).
Myocd+/− bladders show elevated GJA1 protein and reduced miR-1 levels
To evaluate expression site of MYOCD, GJA1, and miR-1, these expression in smooth muscle layers or urothelium from rat bladders was compared. qPCR results revealed that miR-1 was abundantly expressed in smooth muscle layers but not in urothelium, concurrently with mRNA expression of Myocd. Meanwhile, Gja1 was similarly expressed in urothelium and smooth muscle layer (Fig. 4A). However, Western blotting revealed that GJA1 protein was abundantly expressed in smooth muscle layers (Fig. 4B). Western blotting revealed that GJA1 protein expression was higher in Myocd+/− mouse bladders at 3 and 15 weeks of age (Fig. 4C). However, qPCR results demonstrated little change in Gja1 mRNA level in Myocd+/− mouse bladders versus WT mouse bladders at 3 and 15 weeks of age (Fig. 4D). As expected from previous work showing MYOCD-dependent miR-1 expression (Jiang et al., 2010), qPCR analysis of bladders from Myocd+/− mice revealed lower miR-1 at 3 weeks of age compared to WT mouse bladders. There was a similar trend at 15 weeks as well (Fig. 4E). Interestingly, there were only minor differences in the expression of miR-145 (Fig. 4F), a well-defined SMC miR that controls vascular SMC plasticity (Cordes et al., 2009).
Fig. 4.
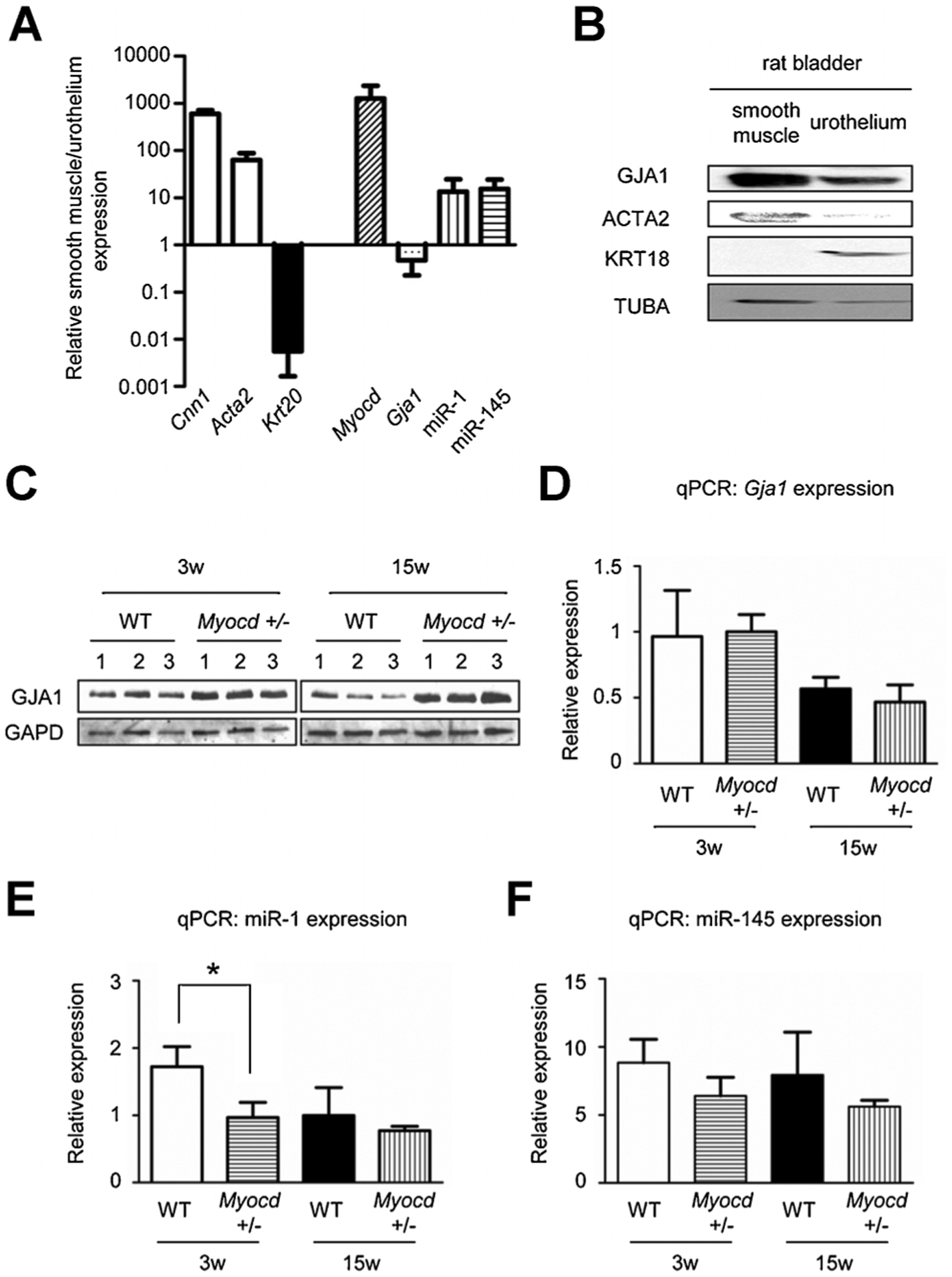
A: qPCR data showed that Myocd and miR-1 was predominantly expressed in the smooth muscle layers which was positive for smooth muscle markers (Cnn1, Acta2) but not in urothelium which was positive for an urothelial marker (Krt20). Gja1 was expressed in both. B: Western blotting showed that GJA1 protein was predominantly expressed in the smooth muscle layers positive for a smooth muscle marker (ACTA2) but not in urothelium positive for urothelial marker (KRT18). C: Western blotting revealed up-regulated protein level of GJA1 in bladders of 3 and 15-week-old Myocd+/− mice compared to those of WT mice. D: In contrast, qPCR data showed no difference in RNA level of Gja1 in bladders of WT and Myocd+/− mice. E: qPCR data showed miR-1 in bladders of Myocd+/− mice was down-regulated compared to those of WT mice. F: qPCR data also showed miR-145 in bladders of Myocd+/− mice was down-regulated compared to those of WT mice. *Statistically significant difference (P < 0.05).
GJA1 protein levels are regulated through a myocardin/miR-1 axis
qPCR results revealed high miR-1 expression in bladder and aorta as compared to liver although expression of miR-1 in bladder was about tenfold less than that seen in heart and skeletal muscle where miR-1 levels are abundantly expressed (Townley-Tilson et al., 2010) (Fig. 5A). For gain-of-function study of MYOCD, in vitro transduction of MYOCD into RBSMC was performed. qPCR results revealed that MYOCD overexpression in RBSMC induced expression of miR-1 with little change in Gja1 mRNA (Fig. 5B–C). A loss-of-function study of miR-1 was then performed in ectopic MYOCD-expressing RBSMC. Western blotting revealed that MYOCD down-regulated GJA1 protein level in RBSMC and a miR-1 inhibitor partially blocked this effect (Fig. 5D). These results suggest that MYOCD represses GJA1 expression at a post-transcriptional level via miR-1 up-regulation.
Fig. 5.
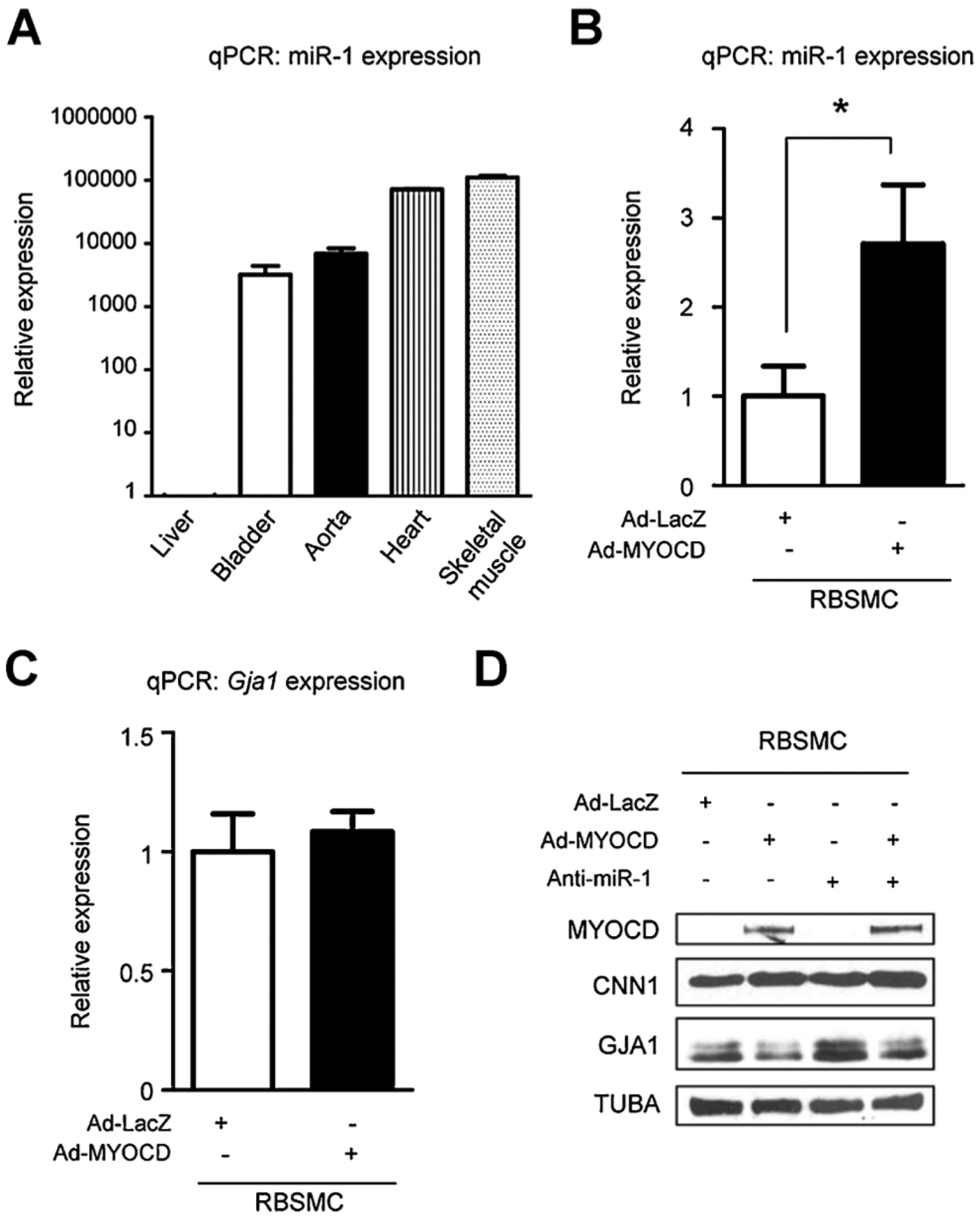
A: In vivo qPCR data showed that miR-1 expression level was high in murine bladder, aorta, heart and skeletal muscle compared to liver. B: In vitro qPCR data showed that MYOCD induced miR-1 up-regulation in rat bladder smooth muscle cells (RBSMC). C: MYOCD did not affect Gja1 RNA level in RBSMC. D: Western blotting showed that MYOCD down-regulated GJA1 protein level in RBSMC and a miR-1 inhibitor blocked this effect of GJA1 protein up-regulation induced by MYOCD. *Statistically significant difference (P < 0.05).
Down-regulation of myocardin impairs functional bladder capacity during bladder development
To investigate the effect of MYOCD for bladder overactivity during post-natal development, VSOP analysis of WT or Myocd+/− mouse bladders was performed at both 3 and 15 weeks of age. Analysis of whole body and bladder weights revealed no difference between WT and Myocd+/− mice at both 3 and 15 weeks of age (Fig. 6A–B). VSOP results showed little change in functional bladder capacity of Myocd+/− mice as compared to WT mice at 3 weeks of age; however, there was a significant decrease in voided bladder volume of Myocd+/− mice versus WT mice at 15 weeks of age (Fig. 6C). Interestingly, urinary frequency in Myocd+/− mice was significantly increased at 15 weeks of age when compared to WT mice (Fig. 6D). Post void residual urine was negligible in each mouse strain at both time points (data not shown).
Fig. 6.
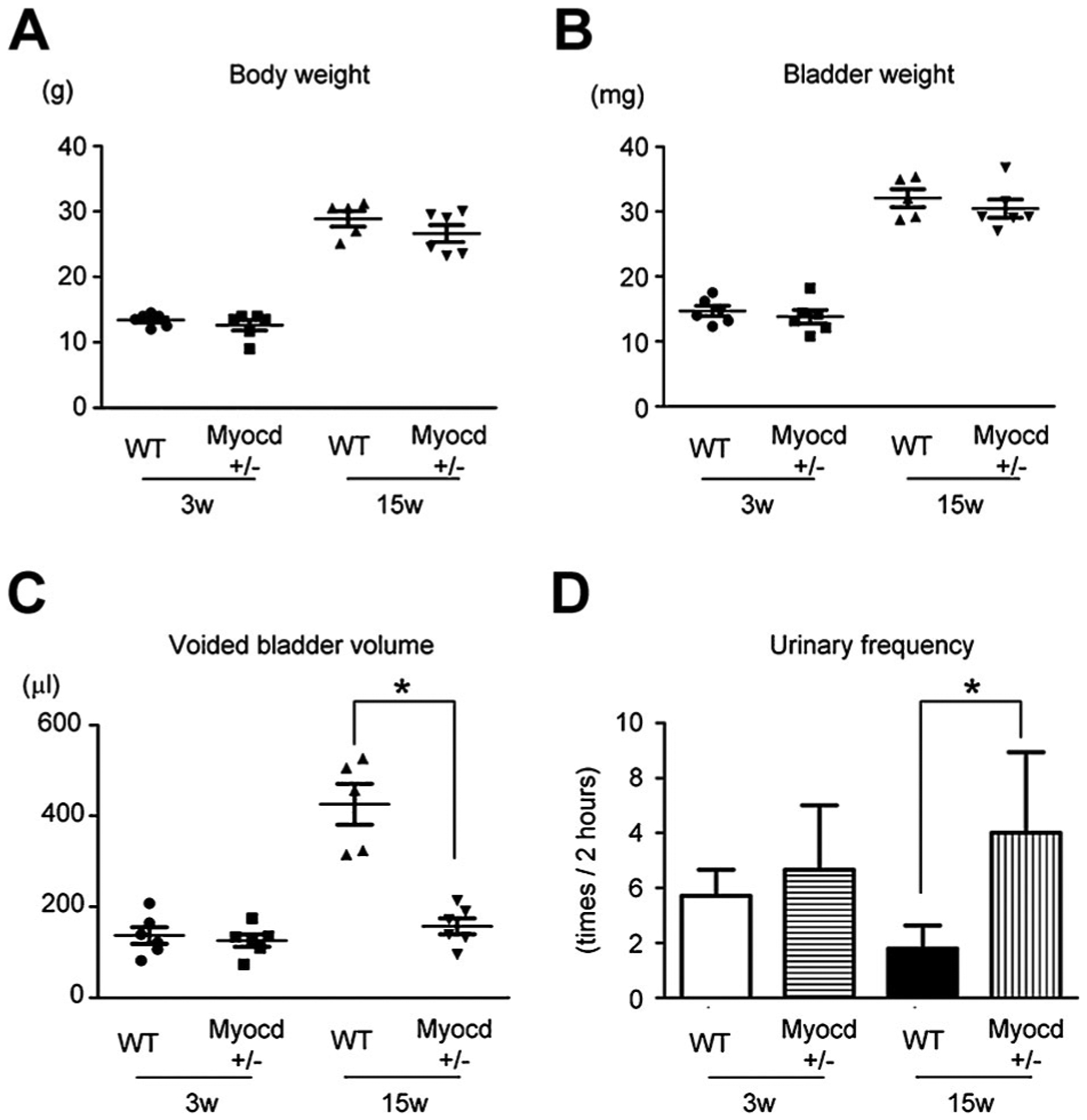
A, B: Both WT and Myocd+/− mice showed normal increase in body (A) and bladder weight (B) in 15-week-old mice compared to 3-week-old mice. C: Voiding behavior analysis showed normal bladder volume increase in 15-week-old WT mice. But in contrast, the bladder volume of 15-week-old Myocd+/− mice remained low as well as that of 3-week-old Myocd+/− mice. D: Voiding behavior analysis also showed normal reduction in urinary frequency. But in contrast, urinary frequency of 15-week-old Myocd+/− mice remained high as well as that of 3-week-old Myocd+/− mice. *Statistically significant difference (P < 0.05).
Discussion
In this study, we have demonstrated that Myocardin (MYOCD) regulates bladder capacity and sensitivity through connexin 43 (GJA1) and miR-1 expression during post-natal development. These results are supported by the following evidence: (1) bladder capacity in 15-week-old mice increased with normal sensitivity to carbachol and reduced MYOCD expression when compared to that in 3-week-old mice (Fig. 1); (2) MYOCD and BSMC markers were down-regulated in Myocd+/− mice (Fig. 2); (3) Myocd+/− mice showed bladder hypersensitivity dependent on functional gap junctions (Fig. 3); (4) MYOCD down-regulated GJA1 protein expression through miR-1 down-regulation (Figs. 4–5); (5) Myocd+/− mice exhibit an OAB-like phenotype as shown by significantly decreased bladder capacity and increased urinary frequency at 15 weeks of age (Fig. 6).
A major finding of this study is a novel physiological role for MYOCD in maintaining bladder homeostasis through its regulated expression of GJA1 and subsequent bladder capacity and urinary frequency during post-natal development. Previous studies demonstrated MYOCD as a crucial molecular component for the expression of contractile gene expression in SMCs and cardiac muscle cells (Wang et al., 2001; Chen et al., 2002; Li et al., 2003). However, it has been unclear whether these regulatory roles of MYOCD affect physiological properties in SMCs in a vivo setting. There are a couple of reports showing that MYOCD elicits functional activity in vascular SMC through contractile measurements (Jiang et al., 2010). However, essentially nothing has been reported about the role of MYOCD in bladder physiology. Here, we have provided a foundational study for the analysis of MYOCD in bladder during a time when dramatic changes occur in bladder function.
This study revealed the difference of post-natal bladder development in the normal condition or the condition of genetically down-regulated MYOCD. In the normal condition, MYOCD and SMC markers were down-regulated during bladder development. It is well known that SMC proliferation occurs upon down-regulation of SMC markers (Owens et al., 2004). Thereby, SRF-MYOCD pathways may contribute to normal post-natal bladder development due to down-regulation of SMC-related molecules. Meanwhile, in the condition of genetically down-regulated MYOCD, SMC markers and miR-1 were down-regulated, with an attending increase in GJA1 expression and bladder hypersensitivity. Our hypothesis for these findings is there exists a threshold level of MYOCD inducing miR-1 down-regulation and the following GJA1 up-regulation. In natural development, MYOCD expression level was maintained over threshold level; thus, MYOCD would not induce significant changes in miR-1 and GJA1 expression. In the condition of hypersensitivity induced by GJA1, bladder capacity did not increase. These results indicated that hypersensitivity overcame the effect of down-regulation in SMC markers for bladder capacity.
Of particular interest in this study is the molecular mechanism of MYOCD-miR-1-GJA1 axis in functional bladder activity. Our results reveal two new findings about this molecular mechanism. One is the role of miR-1 in smooth muscle. The effect of microRNAs for smooth muscle has been increasingly appreciated since the reporting that miR-143 and miR-145 promote MYOCD expression and the regulation of smooth muscle plasticity (Cordes et al., 2009). miR-1 is highly expressed in skeletal and cardiac muscle and regulates activity of these two muscle types through the expression of GJA1, which is a direct target of miR-1 (Yang et al., 2007; Townley-Tilson et al., 2010). Here, we show that miR-1 also has a functional role in bladder SMC. Interestingly, there are conflicting data over the role of miR-1 in vascular SMC with one report showing an effect on MYOCD-dependent growth (Chen et al., 2011) and another report claiming little to no role for miR-1 in vascular SMC (Torella et al., 2011). The expression data reported here would suggest that within SMC populations, miR-1 is likely to be dominant in bladder SMC as well as vascular SMC.
The other novel finding here is a new role for MYOCD as a repressor of GJA1 in smooth muscle. MYOCD has been known as a key regulator for SMC and cardiac contractile genes but its regulation of non-contractile genes is less clear. One recent in vitro study demonstrated that MYOCD induced the expression of an SMC-restricted ion channel gene with a coincident increase in electrical current (Long et al., 2009). Data in this study demonstrate that MYOCD indirectly down-regulates GJA1 through its repressive action on miR-1. GJA1 is a gap junction protein that mediates cell–cell communication and its up-regulation was shown to induce hypersensitivity in smooth muscle tissues (Imamura et al., 2009; Billaud et al., 2011). Smooth muscle tissues have the important function of phasic contraction; therefore, SMC have intrinsic regulatory mechanisms controlling relaxation and contraction (McHale et al., 2006). Given the role of GJA1 in mediating SMC coupling of transported molecules that likely impinge upon SMC contractile activity, we surmise that MYOCD may down-regulate the expression of GJA1 to fine-tune the contractile property of smooth muscle tissues such as in bladder.
Interestingly, our results showed that down-regulation of MYOCD induced hypersensitivity but did not affect bladder capacity in 3-week-old mice. Why was this apparent discrepancy observed? Our speculation is that other mechanisms independent of hypersensitivity caused minimum bladder capacity in 3-week-old WT mice. A salient characteristic of the neonatal (3-week) bladder is low capacity induced with premature contraction (Sillen, 2001). In fact, bladder smooth muscle in a hypersensitive status showed premature contraction which occurred under lower pressure than in normal status (Brading, 1997; Imamura et al., 2009). However, our data showed that 3-week-old WT mice did not show hypersensitivity (Fig. 1C), suggesting that hypersensitivity did not contribute to low capacity with premature contraction in neonatal bladder. Based on other studies demonstrating that neural mechanisms such as ion channel activity or acethylcholine release contributed to spontaneous contraction in neonatal bladders (Kanai et al., 2007; Ng et al., 2007), those neural mechanisms might regulate low bladder capacity.
It is notable that a MYOCD-miR-1 pathway, which can regulate GJA1 protein expression, could be involved in pathogenesis of bladder dysfunction while an SRF-MYOCD pathway, which can modulate expression of SMC-related molecules such as MYH11, CNN1, and ACTA2, could contribute to normal post-natal development of bladder function. One study recently reported that loss-of-function of uroplakin in urothelium caused sustained contraction (Aboushwareb et al., 2009). However, there has been no report showing a regulatory effect of the specific gene in smooth muscle for bladder function. Our data reveal that loss-of-function in smooth muscle with deletion of one Myocd allele caused bladder overactivity. The findings reported here support a MYOCD-miR-1 pathway as potentially new therapeutic targets for OAB. Currently, the main drugs for OAB are anticholinergics, which have side effects of reduction in bladder contractility. Regulation of a MYOCD-miR-1 pathway could improve symptoms related to OAB without changes in bladder contractility since this pathway regulates GJA1 expression, which has been reported to exert little influence on the contractile force in bladders (Imamura et al., 2009).
In conclusion, genetic down-regulation of MYOCD induced GJA1 expression and related hypersensitivity through miR-1 down-regulation, thereby inducing bladder overactivity during post-natal development. These data suggest that a MYOCD-miR-1 pathway could be pivotal for post-natal bladder development.
Supplementary Material
Acknowledgments
This work was supported by grants National Institutes of Health grants HL62572 and HL091168 and from SUMITOMO Life Social Welfare Services Foundation to MI.
Contract grant sponsor:
NIH;
Contract grant numbers:
HL62572, HL091168.
Contract grant sponsor:
SUMITOMO Life Social Welfare Services Foundation.
Footnotes
Additional supporting information may be found in the online version of this article.
References
- Aboushwareb T, Zhou G, Deng F-M, Turner C, Andersson K-E, Tar M, Zhao W, Melman A, D’Agostino R, Sun T-T, Christ GJ. 2009. Alterations in bladder function associated with urothelial defects in uroplakin II and IIIa knockout mice. Neurourol Urodyn 28:1028–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billaud M, Dahan D, Marthan R, Savineau J-P, Guibert C. 2011. Role of the gap junctions in the contractile response to agonists in pulmonary artery from two rat models of pulmonary hypertension. Respir Res 12:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bower WF, Yip SK, Yeung CK. 2005. Dysfunctional elimination symptoms in childhood and adulthood. J Urol 174:1623–1628. [DOI] [PubMed] [Google Scholar]
- Brading AF. 1997. A myogenic basis for the overactive bladder. Urology 50:57–67, discussion 68–73. [DOI] [PubMed] [Google Scholar]
- Chen J, Kitchen CM, Streb JW, Miano JM. 2002. Myocardin: A component of a molecular switch for smooth muscle differentiation. J Mol Cell Cardiol 34:1345–1356. [DOI] [PubMed] [Google Scholar]
- Chen J, Yin H, Jiang YL, Radhakrishnan SK, Huang ZP, Li JJ, Shi Z, Kilsdonk EPC, Gui Y, Wang DZ, Zheng XL. 2011. Induction of microRNA-1 by myocardin in smooth muscle cells inhibits cell proliferation. Arterioscler Thromb Vasc Biol 31:368–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ GJ, Day NS, Day M, Zhao W, Persson K, Pandita RK, Andersson KE. 2003. Increased connexin43-mediated intercellular communication in a rat model of bladder overactivity in vivo. Am J Physiol Regul Integr Comp Physiol 284:R1241–R1248. [DOI] [PubMed] [Google Scholar]
- Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee T-H, Miano JM, Ivey KN, Srivastava D. 2009. MiR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 460:705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groat WC. 1997. A neurologic basis for the overactive bladder. Urology 50:36–52, discussion 53–36. [DOI] [PubMed] [Google Scholar]
- Fitzgerald MP, Thom DH, Wassel-Fyr C, Subak L, Brubaker L, Van Den Eeden SK, Brown JS. 2006. Childhood urinary symptoms predict adult overactive bladder symptoms. J Urol 175:989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry CH, Sui GP, Severs NJ, Wu C. 2004. Spontaneous activity and electrical coupling in human detrusor smooth muscle: Implications for detrusor overactivity? Urology 63:3–10. [DOI] [PubMed] [Google Scholar]
- Huang J, Cheng L, Li J, Chen M, Zhou D, Lu MM, Proweller A, Epstein JA, Parmacek MS. 2008. Myocardin regulates expression of contractile genes in smooth muscle cells and is required for closure of the ductus arteriosus in mice. J Clin Invest 118:515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura M, Negoro H, Kanematsu A, Yamamoto S, Kimura Y, Nagane K, Yamasaki T, Kanatani I, Ito N, Tabata Y, Ogawa O. 2009. Basic fibroblast growth factor causes urinary bladder overactivity through gap junction generation in the smooth muscle. Am J Physiol Renal Physiol 297:F46–F54. [DOI] [PubMed] [Google Scholar]
- Imamura M, Long XC, Nanda V, Miano JM. 2010. Expression and functional activity of four myocardin isoforms. Gene 464:1–10. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Yin H, Zheng XL. 2010. MicroRNA-1 inhibits myocardin-induced contractility of human vascular smooth muscle cells. J Cell Physiol 225:506–511. [DOI] [PubMed] [Google Scholar]
- Kanai A, Roppolo J, Ikeda Y, Zabbarova I, Tai C, Birder L, Griffiths D, de Groat W, Fry C. 2007. Origin of spontaneous activity in neonatal and adult rat bladders and its enhancement by stretch and muscarinic agonists. Am J Physiol Renal Physiol 292:F1065–F1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanematsu A, Yamamoto S, Noguchi T, Ozeki M, Tabata Y, Ogawa O. 2003. Bladder regeneration by bladder a cellular matrix combined with sustained release of exogenous growth factor. J Urol 170:1633–1638. [DOI] [PubMed] [Google Scholar]
- Kanematsu A, Yamamoto S, Iwai-Kanai E, Kanatani I, Imamura M, Adam RM, Tabata Y, Ogawa O. 2005. Induction of smooth muscle cell-like phenotype in marrow-derived cells among regenerating urinary bladder smooth muscle cells. Am J Pathol 166:565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koff SA. 1983. Estimating bladder capacity in children. Urology 21:248. [DOI] [PubMed] [Google Scholar]
- Li S, Wang DZ, Wang Z, Richardson JA, Olson EN. 2003. The serum response factor coactivator myocardin is required for vascular smooth muscle development. Proc Natl Acad Sci USA 100:9366–9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Jiang C, Hao P, Li W, Song C, Song B. 2007. Changes of gap functional cell–cell communication in overactive detrusor in rats. Am J Physiol Cell Physiol 293:C1627–C1635. [DOI] [PubMed] [Google Scholar]
- Liao Y, Regan CP, Manabe I, Owens GK, Day KH, Damon DN, Duling BR. 2007. Smooth muscle-targeted knockout of connexin43 enhances neointimal formation in response to vascular injury. Arterioscler Thromb Vasc Biol 27:1037–1042. [DOI] [PubMed] [Google Scholar]
- Long X, Bell RD, Gerthoffer WT, Zlokovic BV, Miano JM. 2008. Myocardin is sufficient for a smooth muscle-like contractile phenotype. Arterioscler Thromb Vasc Biol 28:1505–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long X, Tharp DL, Georger MA, Slivano OJ, Lee MY, Wamhoff BR, Bowles DK, Miano JM. 2009. The smooth muscle cell-restricted KCNMB1 ion channel subunit is a direct transcriptional target of serum response factor and myocardin. J Biol Chem 284:33671–33682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHale N, Hollywood M, Sergeant G, Thornbury K. 2006. Origin of spontaneous rhythmicity in smooth muscle. J Physiol 570:23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miano JM. 2003. Serum response factor: Toggling between disparate programs of gene expression. J Mol Cell Cardiol 35:577–593. [DOI] [PubMed] [Google Scholar]
- Miano JM, Long X, Fujiwara K. 2007. Serum response factor: Master regulator of the actin cytoskeleton and contractile apparatus. Am J Physiol Cell Physiol 292:C70–C81. [DOI] [PubMed] [Google Scholar]
- Minassian VA, Lovatsis D, Pascali D, Alarab M, Drutz HP. 2006. Effect of childhood dysfunctional voiding on urinary incontinence in adult women. Obstet Gynecol 107: 1247–1251. [DOI] [PubMed] [Google Scholar]
- Nanda V, Miano JM. 2012. Leiomodin 1, a new serum response factor-dependent target gene expressed preferentially in differentiated smooth muscle cells. J Biol Chem 287:2459–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negoro H, Kanematsu A, Imamura M, Kimura Y, Matsuoka R, Tanaka M, Tabata Y, Ogawa O. 2011. Regulation of connexin 43 by basic fibroblast growth factor in the bladder: Transcriptional and behavioral implications. J Urol 185:2398–2404. [DOI] [PubMed] [Google Scholar]
- Neveus T, von Gontard A, Hoebeke P, Hjalmas K, Bauer S, Bower W, Jorgensen TM, Rittig S, Walle JV, Yeung C-K, Djurhuus JC. 2006. The standardization of terminology of lower urinary tract function in children and adolescents: Report from the Standardisation Committee of the International Children’s Continence Society. J Urol 176:314–324. [DOI] [PubMed] [Google Scholar]
- Ng Y-K, de Groat WC, Wu H-Y. 2007. Smooth muscle and neural mechanisms contributing to the downregulation of neonatal rat spontaneous bladder contractions during postnatal development. Am J Physiol Regul Integr Comp Physiol 292:R2100–R2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman C, Runswick M, Pollock R, Treisman R. 1988. Isolation and properties of cDNA clones encoding SRF, a transcription factor that binds to the c-fos serum response element. Cell 55:989–1003. [DOI] [PubMed] [Google Scholar]
- Owens GK, Kumar MS, Wamhoff BR. 2004. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84:767–801. [DOI] [PubMed] [Google Scholar]
- Rocha ML, Kihara AH, Davel AP, Britto LRG, Rossoni LV, Bendhack LM. 2008. Blood pressure variability increases connexin expression in the vascular smooth muscle of rats. Cardiovasc Res 80:123–130. [DOI] [PubMed] [Google Scholar]
- Sillen U 2001. Bladder function in healthy neonates and its development during infancy. J Urol 166:2376–2381. [DOI] [PubMed] [Google Scholar]
- Speakman MJ, Brading AF, Gilpin CJ, Dixon JS, Gilpin SA, Gosling JA. 1987. Bladder outflow obstruction—A cause of denervation supersensitivity. J Urol 138:1461–1466. [DOI] [PubMed] [Google Scholar]
- Sugino Y, Kanematsu A, Hayashi Y, Haga H, Yoshimura N, Yoshimura K, Ogawa O. 2008. Voided stain on paper method for analysis of mouse urination. Neurourol Urodyn 27: 548–552. [DOI] [PubMed] [Google Scholar]
- Torella D, Iaconetti C, Catalucci D, Ellison GM, Leone A, Waring CD, Bochicchio A, Vicinanza C, Aquila I, Curcio A, Condorelli G, Indolfi C. 2011. MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ Res 109:880–893. [DOI] [PubMed] [Google Scholar]
- Townley-Tilson WHD, Callis TE, Wang D. 2010. MicroRNAs 1, 133 and 206: Critical factors of skeletal and cardiac muscle development, function, and disease. Int J Biochem Cell Biol 42:1252–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, Krieg PA, Olson EN. 2001. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 105:851–862. [DOI] [PubMed] [Google Scholar]
- Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B, Zhang Y, Xu C, Bai Y, Wang H, Chen G, Wang Z. 2007. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med 13:486–491. [DOI] [PubMed] [Google Scholar]
- Yoshimura N 2007. Lower urinary tract symptoms (LUTS) and bladder afferent activity. Neurourol Urodyn 26:908–913. [DOI] [PubMed] [Google Scholar]
- Yoshimura N, Bennett NE, Hayashi Y, Ogawa T, Nishizawa O, Chancellor MB, de Groat WC, Seki S. 2006. Bladder overactivity and hyperexcitability of bladder afferent neurons after intrathecal delivery of nerve growth factor in rats. J Neurosci 26:10847–10855. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.