

Significance
Malaria remains a major global health threat. In the face of increasing resistance to available chemotherapeutics, new antimalarial drugs with new modes of action are urgently needed. The causative agent of malaria is a single-celled parasite that invades and replicates within red blood cells. Escape from the red cell, a process called egress, involves a proteolytic pathway triggered by an essential parasite subtilisin-like serine protease called SUB1. Here, we describe the development and rational optimization of a potent, membrane-permeable substrate-based boronic acid compounds that block egress and parasite proliferation by direct inhibition of SUB1 activity. The compounds could form the basis of a new type of antimalarial medicine that would both protect against infection and treat disease.
Keywords: serine protease, boronic acid, egress, Plasmodium falciparum, malaria
Abstract
Malaria is a devastating infectious disease, which causes over 400,000 deaths per annum and impacts the lives of nearly half the world’s population. The causative agent, a protozoan parasite, replicates within red blood cells (RBCs), eventually destroying the cells in a lytic process called egress to release a new generation of parasites. These invade fresh RBCs to repeat the cycle. Egress is regulated by an essential parasite subtilisin-like serine protease called SUB1. Here, we describe the development and optimization of substrate-based peptidic boronic acids that inhibit Plasmodium falciparum SUB1 with low nanomolar potency. Structural optimization generated membrane-permeable, slow off-rate inhibitors that prevent P. falciparum egress through direct inhibition of SUB1 activity and block parasite replication in vitro at submicromolar concentrations. Our results validate SUB1 as a potential target for a new class of antimalarial drugs designed to prevent parasite replication and disease progression.
Malaria, a disease caused by obligate intracellular parasites of the genus Plasmodium, is a global health problem threatening more than half the earth’s population (1). Recent decades have seen a considerable reduction in the incidence of clinical malaria and malaria-related mortality, largely due to the availability of efficacious chemotherapies and control of the mosquito vector (2). However, efforts toward malaria eradication are impeded by the alarming spread of drug-resistant parasites, rendering existing drugs ineffective in many regions (3, 4). Of particular concern, resistance has now been reported to nearly all clinically used antimalarial drugs including artemisinins, the current front line drug class (5). There is therefore an urgent need to bolster the antimalarial drug arsenal with new chemotherapeutics, particularly those with as yet unexploited mechanisms of action.
Clinical malaria results from repeated rounds of replication of the parasite in circulating red blood cells (RBCs). Merozoites invade the cells and divide asexually within a membrane-bound parasitophorous vacuole (PV) to produce a mature multinucleated form called a schizont. This then undergoes segmentation to generate 16 or more daughter merozoites, which are eventually released through a lytic process called egress, in the process destroying the infected RBC. Shortly before egress, activation of a parasite cyclic GMP-dependent protein kinase called PKG induces the discharge of a subtilisin-like serine protease called SUB1 from specialized merozoite secretory organelles called exonemes (6, 7). Upon its release into the PV lumen, SUB1 rapidly cleaves and activates a number of PV-resident and merozoite surface proteins, leading within minutes to explosive rupture of the PV membrane (PVM) and RBC membrane to allow merozoite release (8–12). The free parasites immediately invade fresh RBCs to repeat the cycle.
All Plasmodium species, including the most important human malaria pathogens Plasmodium falciparum, Plasmodium vivax, and Plasmodium knowlesi, possess a single ortholog of SUB1 with similar (though not identical) substrate specificity (13). Genetic experiments have shown that SUB1 is indispensable for parasite survival, with SUB1 gene disruption leading in asexual blood stages and the preceding liver stages of infection to a complete block in merozoite egress (12, 14, 15). This, together with the lack of structural resemblance of SUB1 to human serine proteases (16, 17), has focused interest on SUB1 as an attractive pharmacological target for antimalarial drug discovery. However, the identification of potent drug-like SUB1 inhibitors has proven to be a difficult task. Attempts to identify ligands of SUB1 by screening of synthetic or natural product libraries, and through in silico screening, met with limited success (6, 18, 19), probably due to the relatively shallow and elongated cavity of the enzyme active site (16, 17). We have previously reported the rational design of peptidic ketoamide inhibitors of P. falciparum SUB1 (PfSUB1) based on the substrate specificity of the enzyme (Fig. 1) (13, 20). Preliminary structure-activity relationships analysis of these inhibitors revealed a tetrapeptide mimic on the nonprime side and an oxycarbonylethyl group on the prime side as structural features required to attain submicromolar inhibitory potency. Given the capacity of boronic acids to form strong covalent but reversible bonds with the catalytic Ser residue of serine proteases, here we have investigated peptidic boronic acids as PfSUB1 inhibitors. These efforts have generated nanomolar PfSUB1 inhibitors that can access PfSUB1 in the intraerythrocytic parasite and prevent parasite replication through direct inhibition of egress.
Fig. 1.

Development of rationally designed peptidic PfSUB1 inhibitors.
Results
Discovery of Potent Substrate-Based Peptidyl Boronic Acid Inhibitors of PfSUB1.
We previously described the development of a fluorescence-based in vitro assay suitable for the evaluation of substrate-based PfSUB1 inhibitors, using recombinant PfSUB1 (rPfSUB1) and fluorogenic peptide substrates based on cleavage sites within endogenous protein substrates of PfSUB1 (13, 21). In our earlier work (13, 20), we used the assay to identify a substrate-based pentapeptidic α-ketoamide with a P4 Ile residue and P2 Gly residue as our most potent inhibitor 1 (IC50 ∼900 nM; Fig. 1). Unfortunately, this and related α-ketoamides showed no antiparasite activity in vitro. This was perhaps unsurprising due to the high molecular mass and polar nature of these compounds, including the presence of a carboxylic acid moiety that was designed to mimic endogenous PfSUB1 protein substrates by interacting with the basic S’ surface of the PfSUB1 active-site cleft (16). Collectively, these features likely rendered the compounds poorly membrane penetrant.
To build on that work, we first explored a range of P4 substituents of the N-acetyl peptidyl α-ketoamide scaffold, maintaining the P1 Ala, P2 Gly, and P3 Thr sidechains unaltered. Replacement of the P4 Ile side chain with a cyclopentane improved potency, resulting in an peptidic α-ketoamide 2a with an IC50 ∼370 nM (SI Appendix, Table S1). Reasoning that the exploration of alternative warheads with known activity against serine proteases might prove fruitful, we replaced the α-ketoamide functionality of this compound with a boronic acid warhead, in the process removing the prime side carboxylate. This resulted in compound 3a, which gratifyingly demonstrated an ∼sevenfold increase in potency over the best α-ketoamide (Table 1). Combining the features of the α-ketoamide and compound 3a by adding back the P4 cyclopentane improved potency by a further ∼13-fold, leading to the low nanomolar IC50 compound 3b (Table 1).
Table 1.
PfSUB1 enzyme inhibitory and parasite growth inhibitory potency of peptidic boronic acids
| Entry | Compound | Structure | IC50 (nM) (rPfSUB1)* | EC50 (μM) (parasite growth)† |
| 1 | 3a |  |
69.4 ± 1.2 | 1.8 ± 0.6 |
| 2 | 3b |  |
9.3 ± 0.5 | 2.3 ± 1.4 |
| 3 | 3c |  |
60.1 ± 2.1 | 18.4 ± 1.8 |
| 4 | 3d | 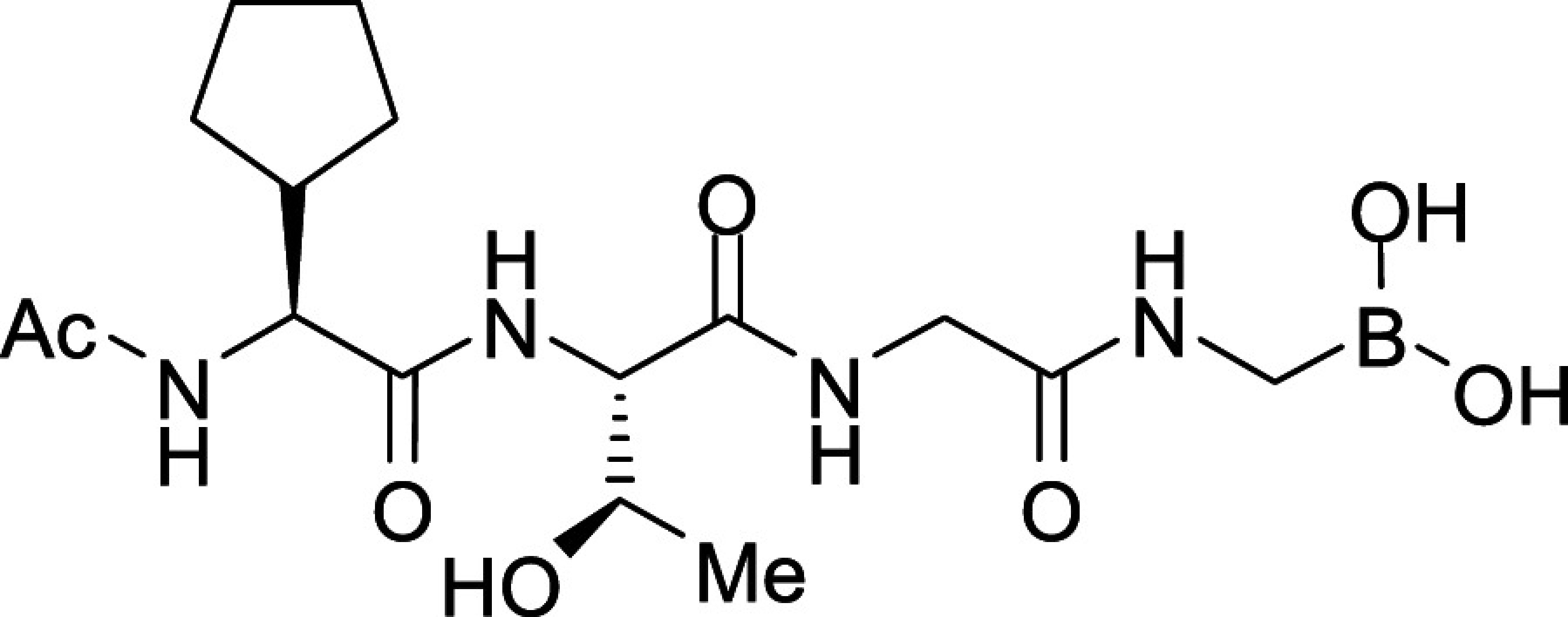 |
54.3 ± 1.1 | 1.9 ± 0.4 |
| 5 | 3e |  |
4.6 ± 0.1 | 15.0 ± 3.6 |
| 6 | 3f |  |
204.2 ± 7.5 | N.D. |
| 7 | 3g |  |
18.7 ± 1.3 | N.D. |
| 8 | 3h |  |
112.0 ± 2.3 | N.D. |
| 9 | 3i |  |
7.8 ± 0.3 | 0.34 ± 0.08 |
| 10 | 3j |  |
5.7 ± 0.1 | 0.26 ± 0.06 |
IC50 values were determined by quantifying inhibition of rPfSUB1-mediated proteolytic cleavage of a fluorogenic peptide substrate. Values are mean averages from at least three independent measurements ± SD.
EC50 values were obtained by quantifying inhibition of P. falciparum growth in vitro over a period of 96 h (two erythrocytic growth cycles) using the DNA-binding fluorescent dye SYBR Green I to measure parasite replication (22). Values are mean averages from at least three independent measurements ± SD.
N.D., not determined.
To examine the importance of the stereochemistry of the aminoboronic acid substructure at the P1 position, the PfSUB1 inhibitory potency of boronic acid epimer 3c was examined (Table 1). We found that 3c was significantly less potent than 3b (Table 1), indicating the requirement for a chiral center configuration matching that of the L-amino acid in native substrates of SUB1. We therefore maintained this stereochemistry in all subsequent boronic acid analogs.
Further work focused on enhancing the potency of the compound 3b structural template. Removal of the methyl side chain at the P1 subsite (compound 3d) reduced potency by eightfold. On the other hand, attempts to improve potency by exploring extended alkyl or phenyl substituents at the P1 subsite (compounds 3e, 3f, 3g, and 3h) met with only limited success, although compound 3e bearing a hydroxyethyl substituent displayed ∼twofold increased potency over compound 3b. This appears to contradict earlier substrate specificity studies, which indicated a preference for the S1 subpocket of PfSUB1 to accommodate polar sidechains (13). The observation may be explained by a preference of nucleophilic P1 side-chain residues to form cyclic boronic acids, preventing the polar hydroxyl group from engaging in interactions with the enzyme.
Conditional gene disruption experiments have shown that PfSUB1 is essential for asexual blood-stage parasite survival in vitro (12). To assess the capacity of the compounds to interfere with parasite replication, we used an in vitro growth assay, which exploits the DNA-binding fluorescent dye SYBR Green I to measure parasite proliferation in human RBCs (which do not possess a nucleus) (22). This showed that while all the compounds inhibited parasite replication, with EC50 values as low as 1.8 μM, there was a poor correlation between growth inhibition and the PfSUB1 enzyme-inhibitory potency of the compounds (Table 1). In particular, the most potent inhibitor of PfSUB1 enzymatic activity, compound 3e, was more than sixfold less growth inhibitory than compound 3b. We reasoned that the polar nature of 3e likely limits its membrane permeability. It was concluded that this set of compounds suffered from poor access to PfSUB1 within the intracellular parasite, probably due to low cellular permeability.
P3 Modification Results in Peptidic Boronic Acids with Submicromolar Parasite Growth Inhibitory Activity.
To seek insights into how the most potent PfSUB1-inhibitory compounds of this first boronic acid series might be accommodated into the PfSUB1 active-site groove, we took advantage of the X-ray crystal structure of PfSUB1 (16) to perform in silico molecular docking of compounds (3b and 3e; Fig. 2). We examined the bound molecules in a docked pose in which the boron atom was involved in a tetrahedrally coordinated intermediate involving the catalytic His428 (Nε2H) and the oxyanion hole partner N520 (Nδ) and engaged in a covalent bond with the Oγ of the catalytic Ser606 of PfSUB1. This showed conservation of the substrate-enzyme canonical H-bond pattern, with the inhibitor peptidic backbone interacting with PfSUB1 residues Gly467 (NH), Ser490 (NH), and Ser492 (NH). For both inhibitors, the P4 cyclopentane was nicely accommodated into the S4 pocket (shaded green for hydrophobicity and delimited by a thick solid line to indicate optimal steric filling in Fig. 2B). The inhibitor 3b P1 Ala side chain did not fill the S1 pocket entirely (indicated by the absence of a solid line at the bottom of the S1 pocket) but occupied the hydrophobic part of the pocket. In the case of inhibitor 3e, docked in the form of an acyclic boronic acid, the P1 hydroxyethyl extension filled the S1 pocket fully and was stabilized by hydrogen bonding with Ser490 Oγ and Ser492 Oγ at the bottom of the pocket. Despite this, little improvement in potency of inhibitor 3e over inhibitor 3a was observed, which as mentioned above we suspect is likely explained by compound 3e adopting the preferential cyclic form of the boronic acid.
Fig. 2.

Substrate-based development of peptidic boronic acid inhibitors of PfSUB1. (A) ICM-Pro best docking poses for the PfSUB1-inhibitory compounds 3b (ICM-Pro score −27), 3e (ICM-Pro score −32), and 3j (ICM-Pro score −34) in the active site of PfSUB1 (4LVN). The inhibitors are represented as colored balls and sticks. Hydrogen atoms are shown, while hydrogen bond interactions are indicated (dotted lines). The enzyme molecular surface is shown in transparent mode (green, hydrophobic; blue, hydrogen bond donor; and red, hydrogen bond acceptor). ICM docking score values below −32 (unitless) are considered good docking scores. (B) Corresponding two-dimensional interaction diagrams, with green shading for hydrophobic regions, blue shading for hydrogen bond acceptors, gray dashed arrows for hydrogen bonds (including length in Å), broken thick lines around ligand shape for accessible surfaces, and gray parabolas for large accessible surface areas. Interacting PfSUB1 residues are labeled and enclosed in oval shapes, the size of which varies depending on the degree of residue contribution. In all cases, the ligand boron atom is shown covalently bound to the active-site Ser606 Oγ through a boron ester bond, and positions of the S1 and S4 PfSUB1 active-site pockets are indicated. The P3 position is annotated in red in B.
Consistent with the X-ray crystal structure of PfSUB1, which includes its propeptide bound into the active-site groove of the catalytic domain in a substrate-like manner, the P3 Thr side chain of the docked compounds 3b and 3e was observed to extend into solvent, with no significant contacts with the molecular surface of the PfSUB1 catalytic domain. Interestingly, however, in both docking poses we noticed potential for modifying and/or extending the P3 side chain (openness depicted in gray in the two-dimensional diagram) in order to promote hydrophobic interactions with the side-chain carbon atoms of Lys465 and Leu466 that line the side of the S3 pocket. In silico replacement of the P3 Thr with Val supported this, revealing potential hydrophobic interactions between the Val P3 side chain and the side chains of Leu466 and Lys465 (Fig. 2B).
In accord with this, we prepared compounds 3i and 3j in which the P3 Thr of compound 3b was replaced, respectively, with an Ala and Val side chain (Table 1). The new compounds showed slightly improved (3i; IC50 ∼7.8 nM) or nearly twofold improved (3j; IC50 ∼5.7 nM) potency relative to compound 3b in the in vitro PfSUB1 enzyme assay. Significantly, compounds 3i and 3j displayed ∼10-fold improved growth inhibitory potency in the SYBR Green I parasite growth assay, likely due to their increased lipophilicity, which was expected to confer better membrane permeability.
Inhibition of P. falciparum Egress by Selective Peptidic Boronic Acids that Access PfSUB1 in Intracellular Parasites.
To determine their mode of action, the four most potent growth inhibitory compounds were next evaluated using very short-term cell-based assays focused on the narrow window within the asexual blood-stage lifecycle during which the parasite undergoes egress from host RBCs and invasion into fresh cells. For this, P. falciparum cultures containing synchronous, highly mature schizonts were supplemented with compounds 3b, 3e, 3i, and 3j at a range of dilutions, then allowed to undergo egress and invasion for just 4 h in the continued presence of the compounds, before assessing formation of newly invaded “ring” stage parasites by flow cytometry. This confirmed a dose-dependent inhibitory effect on the transition from schizont to ring stage, with the relatively lipophilic compounds 3i and 3j displaying similar EC50 values that were significantly lower than those of 3b and 3e (Fig. 3A). Microscopic examination of the cultures revealed schizonts arrested by compounds 3i and 3j, confirming inhibition of schizont rupture. Importantly, the arrested schizonts were morphologically indistinguishable from those arrested by the reversible PKG inhibitor (4-[7-[(dimethylamino)methyl]-2-(4-fluorphenyl)imidazo[1,2-α]pyridine-3-yl]pyrimidin-2-amine (C2)), appearing as segmented forms trapped within an apparently intact PVM and RBC membrane (Fig. 3B). This egress-arrest phenotype is similar to that obtained by genetic disruption of PfSUB1 and was clearly different from that following arrest by the cysteine protease inhibitor E64 (Fig. 3B), which does not inhibit PfSUB1 directly but which blocks a cysteine protease-dependent step in egress following SUB1 discharge and PVM rupture (12).
Fig. 3.

Peptidic boronic acid PfSUB1 inhibitors prevent P. falciparum egress. (A) Dose–response curves showing ring formation following incubation of highly mature 3D7 schizonts with RBCs for 4 h in the presence of the indicated compounds. Values are means of three independent experiments. Calculated EC50 values were as follows: compound 3b, 12.7 ± 0.8 μM; 3e, 15.7 ± 2.5 μM; 3i, 2.0 ± 0.1 μM; and 3j 2.5 ± 0.3 μM. Error bars, SD. (B) Light micrographs of Giemsa-stained thin films prepared from selected cultures similar to those described in A, sampled prior to start or following the 4 h incubation step. Extensive ring formation is evident in the control culture (examples indicated by arrows). In contrast, cultures containing compounds 3i or 3j (10 μM) show arrest of unruptured schizonts with no ring formation. Note that the phenotype of the 3i- or 3j-arrested schizonts is similar to that of C2-treated parasites but distinct from those arrested by the cysteine protease inhibitor E64, where PVM rupture occurs allowing release of the enclosed merozoites into the RBC cytosol. (Scale bar, 20 μm.)
Examination of the inhibitory activity of compound 3j against the mammalian trypsin-family serine proteases trypsin, chymotrypsin, and elastase revealed a high degree of selectivity for PfSUB1 (>>100-fold; SI Appendix, Figs. S1–S3), encouraging us to focus subsequent work on this compound. To directly visualize the inhibitory effects of compound 3j on parasite egress and to examine the reversibility of inhibition, we used live time-lapse video microscopy to observe the behavior of schizonts exposed to the compound for just 1 h immediately prior to egress. For this, we used a transgenic parasite line expressing a PVM protein (EXP2) fused with the green fluorescent protein mNeon Green, facilitating real-time visualization of PVM integrity as previously reported by Glushakova and colleagues (23). As shown in Fig. 4A and Movie S1, this clearly demonstrated significant inhibition of PVM rupture and egress in parasites treated with 3j, with no signs of egress even 30 min following washout of the compound. Importantly, 3j-treated parasites remained viable, as shown by their continued capacity to incorporate the vital mitochondrial dye MitoTracker Red CMXRos (24) (SI Appendix, Fig. S4), but showed no signs of the PVM rounding and other morphological changes that typically precede egress (23, 25), indicating a complete and selective block in the egress pathway. These egress-associated transitions were also absent from PfSUB1-null parasites (12), indicating that the effects of 3j closely mimic genetic disruption of PfSUB1. Further extended incubation of the treated, washed schizonts with fresh RBCs resulted in only very limited appearance of new ring stage parasites (Fig. 4B). This confirmed that even short-term treatment with compound 3j could dramatically impede parasite escape from the host RBC and that the egress inhibition over these timescales was effectively irreversible.
Fig. 4.

Washout experiments show that peptidic boronic acid 3j is a membrane-permeable inhibitor of PfSUB1 and P. falciparum egress. (A) Stills from time-lapse video microscopic monitoring of purified schizonts following washout of the indicated treatments. The parasites express an mNeonGreen fusion of the PVM protein EXP2. Washout of drug from schizonts arrested with the reversible PKG inhibitor C2 (1 μM) resulted in normal egress, initiating at ∼6.5 min following washout. In contrast, no egress occurred over the course of 30 min following washout of parasites treated with saturating amounts (10 μM) of compound 3j, and there was no discernible change in shape or integrity of the PVM (although slight time-dependent photobleaching of the fluorescence signal is evident). Identical results were obtained in four independent experiments. (Scale bar, 20 μm.) Also reference Movies S1 and S2. (B) Ring formation following incubation with fresh RBCs of schizonts pretreated with vehicle only (DMSO), C2 (1 μM), or compound 3j. Drugs were washed away before addition of RBCs. Ring production was assessed at 24 h. Parasitaemia was also assessed at 48 h to ensure that the rings detectable at 24 h were viable. No rings were produced by the 10 μM 3j pretreated schizonts, whereas schizonts pretreated with the reversible PKG inhibitor C2 produced rings efficiently. Results shown are from three independent experiments in different batches of blood. Error bars, ± SD. (C) Western blot analysis of mature schizonts and culture supernatants thereof following pretreatment for 4 h with compound 3j at the indicated concentrations, then washout before analysis of egress. The parasite PV protein SERA5, which is proteolytically converted to the P50 fragment through the action of PfSUB1, appeared in the supernatants of control schizonts (which underwent egress) but remained intracellular in its intact, full-length form at higher concentrations of 3j. As expected, SERA5 processing was also blocked by C2 (positive control). (D) Quantitation of hemoglobin release into culture supernatants (an indicator of the extent of egress) in the assay analyzed in C. Error bars, SD. Data shown are typical of four independent experiments.
These results suggested that compound 3j can access and inhibit PfSUB1 in an intracellular location (i.e., within the intraerythrocytic parasite, or in the PV, or both). To seek unambiguous confirmation that PfSUB1 is the intracellular target of compound 3j, we examined the effects of the compound on the PfSUB1-mediated proteolytic processing of the established endogenous PfSUB1 substrate SERA5, an abundant parasite PV protein that only becomes accessible to cleavage upon discharge of PfSUB1 into the PV in the minutes leading up to egress and is then released in a processed form into culture supernatants (6, 11, 12). As shown in Fig. 4C, treatment with compound 3j reproducibly prevented proteolytic processing and release of SERA5 into culture supernatants in a dose-dependent manner, even following compound washout. Crucially, at higher concentrations of the drug where egress and release of processed SERA5 was completely blocked, no intracellular processing of SERA5 was evident in the intact egress-arrested schizonts. Quantitation of egress in these same washout assays by measuring release of residual hemoglobin from the rupturing schizonts showed an EC50 of ∼2 μM (Fig. 4D), similar to the EC50 value for new ring generation previously determined by flow cytometry (Fig. 3A). It was concluded that compound 3j prevents egress and parasite proliferation through direct inhibition of intracellular PfSUB1.
An Optimized Membrane-Penetrant Peptidic Boronic Acid Displays Time-Dependent, Slowly Reversible Binding Kinetics to PfSUB1.
Boronic acids form reversible covalent bonds with serine and threonine proteases (26). Inhibition is generally time dependent, and the covalent nature of the binding can result in relatively long target occupancy times despite the reversibility of the bond. That this might be the case with compound 3j binding to PfSUB1 was initially suggested by our washout experiments (Fig. 4), which showed that egress inhibition by compound 3j following washout was much longer lasting than the rapidly reversible egress inhibition mediated by C2. To analyze the kinetic characteristics of the interaction between 3j and rPfSUB1, we used progress curve analysis to continuously monitor rPfSUB1-mediated cleavage of a fluorogenic substrate in the presence of a range of concentrations of 3j. As shown in SI Appendix, Fig. S5, under conditions where substrate cleavage in the absence of inhibitor (control reaction) displayed a linear relationship with time, indicating negligible substrate depletion, progress curves in the presence of compound 3j became progressively nonlinear, characteristic of slow-binding (time-dependent) inhibition. Under such conditions, fit of the progress curves by nonlinear regression to Eq. 1 (see Materials and Methods) allows determination of kobs, the pseudo first-order rate constant for onset of inhibition. The kobs is effectively a composite of the on and off rates, so least linear squares regression of the calculated kobs values against inhibitor concentration allows determination of values of the pseudo first-order dissociation rate constant koff and the second-order association rate constant kon for the inhibitor-rPfSUB1 interaction, based respectively on values from the y-intercept and slope. The y-intercept value corresponds to a koff of 3.7 × 10−4 s−1, which equates to a t1/2off (bound half-life) of ∼31 min. The calculated kon value was 3.6 × 105 M−1.s−1, allowing calculation of an apparent equilibrium inhibition constant KIkin (koff/kon) of 1.0 nM. It was concluded that compound 3j is a potent, slowly reversible inhibitor of PfSUB1, completely consistent with the washout data.
Discussion
Prior to parasite egress from the confines of its host RBC, SUB1 is stored in membrane-bound merozoite secretory organelles called exonemes before its discharge into the PV lumen minutes before egress to encounter its endogenous substrates. As a result, in order to gain access to the intracellular enzyme prior to substrate cleavage, exogenously applied inhibitory compounds likely need to cross at least two and as many as four distinct biological membranes: the RBC membrane, the PVM, the parasite plasma membrane, and the exoneme membrane (Fig. 5). This poses particular challenges for the design of substrate-based inhibitors. In the case of covalent modifying compounds, such as those described here, access to the exoneme-resident enzyme could potentially allow inactivation of the stored SUB1 long before its PKG-regulated discharge into the PV. In this work, we did not determine the intracellular site of PfSUB1 inhibition, so we cannot state whether inhibition took place within the PV, or the exonemes, or both. Regardless, by gradual optimization of the structure and lipophilicity of our compounds we have now successfully developed potent PfSUB1-inhibitory compounds that can functionally inactivate PfSUB1 within intact, parasite-infected RBCs and block egress.
Fig. 5.

Schematic indicating the requirement for inhibitors of SUB1 to cross at least two and up to four membranes to access and inactivate the enzyme in intraerythrocytic parasites. Inhibition likely occurs either in the PV (route A), or the exonemes (route B), or both.
Our conclusion that the intracellular inhibition of PfSUB1 mediated by compound 3j is directly and causally responsible for the observed block in egress is most clearly supported by the phenotype of the arrested schizonts, which was indistinguishable from that resulting from conditional genetic disruption of the PfSUB1 (12) or PKG gene (27), or following treatment with the PKG inhibitor C2, with no signs of the morphological changes that typically precede egress such as PVM rounding or PV rupture. We cannot rule out the possibility of effects on other parasite enzymes at the concentrations used to obtain complete egress inhibition, even in the short-term assays designed to focus on the short window of the parasite life cycle over which egress occurs. However, we consider off-target effects unlikely given that no other parasite serine protease has been implicated in egress; the only two other subtilisin-like enzymes expressed in the parasite, SUB2 and SUB3, are respectively dispensable for egress (28) or nonessential in blood stages (29). The ∼10-fold higher potency of 3j in the long-term SYBR Green–based parasite growth assay (EC50 0.26 μM; Table 1) may be a result of the fact that this assay captures the combined effects of egress inhibition over the course of two erythrocytic cycles in the continuous presence of the drug, although again off-target effects cannot be formally ruled out. We anticipate that further optimization of the PfSUB1-inhibitory potency and membrane permeability of 3j is highly feasible. Work is already underway to determine the atomic structure of the 3j-PfSUB1 complex to facilitate structure-based inhibitor improvement.
Peptidic boronic acids have long-established therapeutic potential, as best exemplified by the widespread clinical use for multiple myeloma of the proteasome inhibitors bortezomib (Velcade) and ixazomib, the latter of which is orally bioavailable in its citric acid form, Ninlaro. The clinical success of these compounds is in part due to the long drug target residence times that can be obtained with slowly reversible covalent inhibitors. Target binding by boronic acid protease inhibitors is generally time dependent, perhaps further explaining the differences in potency we observed between the long-term and short-term cellular assays with compounds 3i and 3j. However, our washout experiments with compound 3j suggest that, once bound to PfSUB1 in the exonemes and/or PV, it takes at least 30 min for the level of target engagement to fall below a threshold that allows successful egress. Examination of the capacity of schizonts treated with saturating levels of 3j to productively egress and form new rings following compound washout showed that the egress block under these conditions was effectively irreversible. An alternative explanation for this apparently irreversible inhibition of egress by 3j is that the inhibitor is not easily washed out due to its accumulation in the parasite (or infected RBC) at high concentrations. We cannot formally rule out this possibility. However, our ex vivo kinetic analysis of the inhibition of rPfSUB1 by compound 3j fully supports a slow off rate, with an estimated bound half-life (t1/2off) of ∼30 min, very similar to that of the interaction between bortezomib and the β5 chymotrypsin-like subunit of the human proteasome (30–32). Even if cellular accumulation does contribute to the prolonged egress inhibition exerted by 3j, this is only likely to favor efficacy; indeed, intracellular accumulation is an important component of the mode of action of the important antimalarial 4-aminoquinoline chloroquine (33). While peptide-based drug development can present challenges for in vivo applications due to metabolic instability, covalent compounds can be effective even with relatively short plasma half-lives, since target residence time can be longer than plasma half-life. Peptidyl boronic acids can anyway have excellent pharmacodynamic properties; for example, the terminal half-life of ixazomib is ∼9.5 d (a fact that allows weekly dosing of patients for treatment of multiple myeloma; ref. 34), which is nearly five times the duration of the P. falciparum asexual blood-stage lifecycle.
SUB1 has an unusual substrate specificity, which differs subtly between different Plasmodium species, suggesting that the enzyme and its multiple cognate parasite substrates have coevolved to ensure optimal cleavage efficiency (13). As a result, inhibitors of PfSUB1 are unlikely to show similar potency against SUB1 orthologs from rodent malaria parasite species such as Plasmodium berghei, making these parasite species unsuitable as model systems for assessing the in vivo efficacy of our compounds. Importantly, SUB1 also lacks structural resemblance to any known human serine protease (16), reducing the likelihood of substrate-based SUB1 inhibitors displaying toxicity due to off-target activity against host enzymes. In support of this, we found here that 3j is only poorly potent against the mammalian serine proteases examined. Toxicity can be especially problematic where long-term or life-long treatment regimens are required due to chronic infection (e.g., with HIV). However, malaria is an acute disease, and long-term therapeutic regimens are rare; indeed, current standard treatments for uncomplicated falciparum malaria are just 3 d long. Since SUB1 plays an essential role in the development and release of exoerythrocytic (liver-stage) merozoites that initiate blood-stage infection (14, 15), medicines based on SUB1 inhibitors have prophylactic as well as therapeutic potential. Optimized SUB1 inhibitors could also potentially be combined with inhibitors of other essential enzymes in the egress pathway, including PKG (35–37) and the SUB1 aspartic protease maturase plasmepsin X (38–41). Such combinations could yield additive or synergistic enhancement of potency and decrease opportunities to select for drug resistance.
In conclusion, we have produced substrate-based peptidic boronic acids that block asexual blood-stage P. falciparum proliferation through direct, effectively irreversible inhibition of intracellular PfSUB1. Further investigation of the pharmacokinetic properties and structure-based improvement of these compounds has the potential to generate compounds suitable for preclinical trials in animal models of malaria.
Materials and Methods
P. falciparum Maintenance and Manipulation.
Asexual blood stages of P. falciparum (clones 3D7 and B11) (42) were routinely maintained at 37 °C in human erythrocytes at 1 to 4% hematocrit in RPMI 1640 containing Albumax II (Thermo Scientific) supplemented with 2 mM L-glutamine in a low oxygen atmosphere using standard procedures (43). Human blood was obtained from anonymized donors through the UK National Blood and Transplant service and was used within 2 wk of receipt. No ethical approval is required for its use. For synchronization, mature schizont-stage parasites were isolated on cushions of 70% (volume[vol]/vol) Percoll (GE Healthcare) adjusted to isotonicity as described (43). Routine microscopic examination of parasite growth was performed by fixing air-dried thin blood films with 100% methanol before staining with 10% Giemsa stain (VWR international) in 6.7 mM phosphate buffer, pH 7.1.
Generation of the B11-EXP2-mNeonGreen line was achieved by fusing mNeonGreen to the endogenous C terminus of EXP2 using Cas9-mediated gene editing, following the methods of ref. 23. A pair of guide RNAs were designed targeting a region toward the 3′ end of the EXP2 locus (oligo 1: ATTGATATTATGTACAGTATCTGA, oligo 2: AAACTCAGATACTGTACATAATAT) (Sigma). This oligonucleotide pair was annealed with T4 PNK ligase (New England Biolabs) and ligated with T4 ligase (New England Biolabs) into the U6 cassette of the Cas9 vector (pDC2-Cas9_U6-hDHFR) previously digested with Bsb1-HF (New England Biolabs). The plasmid was propagated under ampicillin selection in Escherichia coli and sequenced to check for correct incorporation of the guide (Genewiz). The resulting plasmid was cotransfected into B11 schizonts along with the repair plasmid pyPM2GT-EXP2-mNG (a kind gift of Josh Beck, Iowa State University, Ames, IA), linearized with AflII (New England Biolabs). Drug selection for integration was carried out with 2.5 nM WR99210 (Sigma-Aldrich) from 24 h posttransfection, and clonal lines of the resulting B11-EXP2-mNeonGreen line were obtained by limiting dilution cloning and treatment with 1 μM Ancotil (5-fluorocytosine) before use.
Parasite Growth, Egress, and Invasion Assays.
The impact of the peptidic boronic acids on replication of asexual blood-stage P. falciparum (clone 3D7) was assessed using a SYBR Green I assay, essentially as described by Smilkstein et al. (22). Briefly, test compounds (dissolved in dimethyl sulfoxide [DMSO] at concentrations ranging from 4 mM to 5 μM) were added in triplicate to wells of flat bottomed, 96-well microtitre plates (1 μL per well). Wells were then supplemented with 100 μL per well of a P. falciparum parasite culture at 0.1% parasitaemia, 1% hematocrit. Each assay plate also included DMSO-only control wells (1% vol/vol), as well as additional control wells containing uninfected RBCs only. Plates were incubated in sealed, humidified gassed chambers at 37 °C for 96 h to allow the parasites to undergo two entire cycles of erythrocytic growth. Wells were then supplemented with 100 μL 1:5,000 dilution of stock SYBR Green I (Life Technologies, catalog no. S7563) diluted in 20 mM Tris HCl pH 7.5, 5 mM EDTA, 0.008% (weight/vol) saponin, 0.08% (vol/vol) Triton ×100. Plates were agitated to mix, incubated for a further 1 h in the dark at room temperature, then 150 μL samples from each well transferred to a fresh white microwell plate and fluorescence quantified using a SpectraMax M5e plate reader and SoftMax Pro-6.3 software (Ex 485 nm/Em 530 nm). IC50 values were determined from dose–response curves obtained after subtracting background fluorescence values (obtained from the RBC-only wells) from all experimental readings.
Short-term egress, invasion, and washout assays were performed essentially as described previously (27, 28, 42). Briefly, highly synchronous mature Percoll-enriched schizonts with or without added fresh RBCs (∼5% parasitaemia final) were incubated with compounds under test or vehicle only (DMSO, 1% vol/vol). For washout assays, schizonts were treated with C2 or various concentrations of inhibitor 3j for 1 to 4 h, then washed extensively (at least four times) prior to addition to fresh RBCs where required. After incubation at 37 °C for just 1 to 4 h (or overnight for invasion assays) to allow schizont rupture, cells were pelleted. Clarified culture supernatants were assessed for extent of hemoglobin release (a measure of schizont rupture) by absorption spectroscopy at 415 nm as described previously (28) or analyzed by Western blot using antibodies against SERA5 (7, 11). To quantify generation of new rings, samples of the cultures were fixed with 4% paraformaldehyde/0.02% glutaraldehyde and stained with SYBR Green I (Life Technologies) and then analyzed by flow cytometry on a BD FACSVerse using BD FACSuite software. Data were analyzed using FlowJo software. All cultures were also routinely analyzed by microscopic examination of Giemsa-stained thin films to visually assess parasite morphology.
Time-Lapse and Live Fluorescence Microscopy.
Viewing chambers for live parasite microscopic examination were constructed as previously described (7). All images were recorded on a Nikon Eclipse Ni light microscope fitted with a Hamamatsu C11440 digital camera and Nikon N Plan Apo λ 63×/1.45NA oil immersion objective. For time-lapse video microscopy, differential interference contrast (DIC) images were taken at 10 s intervals over 30 min while fluorescence (mNeon Green) images were taken every 2 min to prevent bleaching. Time-lapse videos were analyzed and annotated using Fiji (44). For viability staining using the vital mitochondrial dye MitoTracker Red CMXRos (ThermoFisher Scientific; stored as a 10 μM stock in DMSO), the dye was added (20 nM final concentration) to a suspension of schizonts pretreated for 1 h with either DMSO (control, 1% vol/vol) or compound 3j (10 μM). The schizonts were incubated with the dye for 15 min at 37 °C, then washed twice, transferred to a viewing chamber, and observed immediately by dual DIC/fluorescence microscopy.
Protease Inhibition Assays: IC50 Calculations and Progress Curve Kinetics.
Proteolytic activity of rPfSUB1 was quantified at room temperature by monitoring cleavage of the peptidic fluorogenic substrate SERA4st1F-6R12 (Ac-CKITAQDDEESC-OH possessing tetramethylrhodamine labeling of both cysteine residues) (13). Fluorescence of this peptide is quenched by rhodamine dimerization in the intact substrate but increases upon cleavage at the internal Q-D bond. Chymotrypsin-treated rPfSUB1 (expressed and purified as described previously (16) was stored at −80 °C as a 228 U/mL stock in 20 mM Tris–HCl pH 8.2, 150 mM NaCl, 10% glycerol, and diluted for use (1:500 or 1:600) in reaction buffer (20 mM Tris–HCl pH 8.2, 150 mM NaCl, 12 mM CaCl2, 25 mM CHAPS). Peptidic boronic acid inhibitors were dissolved in 100% DMSO at 10 or 20 mM, then further diluted in DMSO to generate stock solutions ranging from 500 to 0.01 μM and then used diluted 1:100 in the enzyme reactions. All reactions were performed in wells of white 96-well microwell plates (Nunc); 50 μL diluted rPfSUB1 was preincubated for 5 min with 1 μL each of the serially diluted boronic acid inhibitors, followed by addition of 50 μL substrate solution (0.1 μM final). Subsequent fluorescence increase was continuously monitored using a SpectraMax M5e plate reader and SoftMax Pro-6.3 software, with readings taken every 5 min for 60 min using excitation and emission values of 552 and 580 nm, respectively. Initial rates were calculated over the first 25 min of the assay, during which period progress curves were linear, and IC50 values were calculated with GraphPad Prism 8.0 using the nonlinear regression, [inhibitor] versus response, variable slope (four parameters). All experiments were performed in duplicate.
Details of the methodology used to evaluate the effects of compound 3j on the proteolytic activity of the mammalian serine proteases trypsin, chymotrypsin, and elastase are provided in SI Appendix.
Progress Curve Kinetic Analysis of Compound 3j.
Progress curves of SERA4st1F-6R12 cleavage by rPfSUB1 were acquired at seven concentrations of inhibitor compound 3j over a period of 35 min, during which fluorescence increases in the absence of inhibitor were linear. The obtained progress curves (four independent replicates) were fit using GraphPad Prism 8.0 software to the following time-dependent inhibition equation:
| [1] |
In the equation, Vi is the initial velocity, Vs is the final steady-state velocity, and kobs reflects the observed pseudo first-order rate of inactivation. The obtained kobs values were plotted against compound concentration using a linear least squares fit. All statistical analysis was carried out using GraphPad Prism 8.
Covalent Docking.
Flexible covalent docking of peptidyl boronic acid compounds into the active site of PfSUB1 (Protein Data Bank: 4LVN) was performed using the Internal Coordinate Mechanics software (ICM-Pro) package version 3.9-1c/MacOSX (Molsoft LLC). The inhibitors were drawn using the ICM chemistry molecular editor and compiled into an sdf docking table. After adding hydrogen atoms to the structure, the C-terminal region of the SUB1 propeptide (P4 to P1 positions that occupy the SUB1 active site) was used to define boundaries within the enzyme active site for the docking procedure and then removed from the active site along with all water molecules prior to docking. The catalytic histidine (His428, Nε2) was protonated as part of the catalytic process, resulting from covalent binding of the boron atom ligand to the active Ser (Ser606 Oγ). The boronic acid covalent mechanism was selected from the ICM program reactions list. Potential energy maps of the SUB1 receptor pocket and docking preferences were set up using the program default parameters. Energy terms were based on the all-atom vacuum force field ECEPP/3, and conformational sampling was based on the biased probability Monte Carlo procedure (45). Four independent docking runs were performed per compound, with a length of simulation (thoroughness) varied from three to four and the selection of two docking poses. Ligands were ranked according to their ICM energetics (ICM score, unitless), which weighs the internal force-field energy of the ligand combined with other ligand-receptor energy parameters.
Statistical Analysis.
All statistical analysis was carried out using GraphPad Prism 8.0.
Supplementary Material
Acknowledgments
We gratefully acknowledge Josh Beck (Iowa State University) and Joshua Zimmerberg (NIH) for the kind gift of construct pyPM2GT-EXP2-mNeonGreen and are indebted to Justin Molloy (The Francis Crick Institute) for invaluable discussions and advice on the kinetic analysis. This research was funded in part by the Wellcome Trust (Grant 106239/Z/14/A to M.J.B.). This work was supported by funding to M.J.B. from the Francis Crick Institute (https://www.crick.ac.uk/), which receives its core funding from Cancer Research United Kingdom (FC001043; https://www.cancerresearchuk.org), the UK Medical Research Council (FC001043; https://mrc.ukri.org/), and the Wellcome Trust (FC001043; https://wellcome.org/). The work was also supported by funding to A.J. from the European Regional Development Fund (Agreement No. 1.1.1.1/16/A/290) and by Wellcome ISSF2 funding to the London School of Hygiene & Tropical Medicine. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
The authors declare no competing interest.
This article is a PNAS Direct Submission.
See online for related content such as Commentaries.
This article contains supporting information online at https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.2022696118/-/DCSupplemental.
Data Availability
All study data are included in the article and/or supporting information.
References
- 1.WHO , World malar. Rep. 2018 World Health Organ. Geneva 2016. https://www.who.int/malaria/publications/world-malaria-report-2018/report/en. Accessed 29 October 2020.
- 2.Wells T. N., Hooft van Huijsduijnen R., Van Voorhis W. C., Malaria medicines: A glass half full? Nat. Rev. Drug Discov. 14, 424–442 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Wells T. N., Alonso P. L., Gutteridge W. E., New medicines to improve control and contribute to the eradication of malaria. Nat. Rev. Drug Discov. 8, 879–891 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Hyde J. E., Drug-resistant malaria–An insight. FEBS J. 274, 4688–4698 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dondorp A. M., et al., Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 361, 455–467 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeoh S., et al., Subcellular discharge of a serine protease mediates release of invasive malaria parasites from host erythrocytes. Cell 131, 1072–1083 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Collins C. R., et al., Malaria parasite cGMP-dependent protein kinase regulates blood stage merozoite secretory organelle discharge and egress. PLoS Pathog. 9, e1003344 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koussis K., et al., A multifunctional serine protease primes the malaria parasite for red blood cell invasion. EMBO J. 28, 725–735 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silmon de Monerri N. C., et al., Global identification of multiple substrates for Plasmodium falciparum SUB1, an essential malarial processing protease. Infect. Immun. 79, 1086–1097 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Das S., et al., Processing of Plasmodium falciparum merozoite surface protein MSP1 activates a spectrin-binding function enabling parasite egress from RBCs. Cell Host Microbe 18, 433–444 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins C. R., Hackett F., Atid J., Tan M. S. Y., Blackman M. J., The Plasmodium falciparum pseudoprotease SERA5 regulates the kinetics and efficiency of malaria parasite egress from host erythrocytes. PLoS Pathog. 13, e1006453 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas J. A., et al., A protease cascade regulates release of the human malaria parasite Plasmodium falciparum from host red blood cells. Nat. Microbiol. 3, 447–455 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Withers-Martinez C., et al., Plasmodium subtilisin-like protease 1 (SUB1): Insights into the active-site structure, specificity and function of a pan-malaria drug target. Int. J. Parasitol. 42, 597–612 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suarez C., Volkmann K., Gomes A. R., Billker O., Blackman M. J., The malarial serine protease SUB1 plays an essential role in parasite liver stage development. PLoS Pathog. 9, e1003811 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tawk L., et al., A key role for Plasmodium subtilisin-like SUB1 protease in egress of malaria parasites from host hepatocytes. J. Biol. Chem. 288, 33336–33346 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Withers-Martinez C., et al., The malaria parasite egress protease SUB1 is a calcium-dependent redox switch subtilisin. Nat. Commun. 5, 3726 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giganti D., et al., A novel Plasmodium-specific prodomain fold regulates the malaria drug target SUB1 subtilase. Nat. Commun. 5, 4833 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Bouillon A., et al., In Silico screening on the three-dimensional model of the Plasmodium vivax SUB1 protease leads to the validation of a novel anti-parasite compound. J. Biol. Chem. 288, 18561–18573 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arastu-Kapur S., et al., Identification of proteases that regulate erythrocyte rupture by the malaria parasite Plasmodium falciparum. Nat. Chem. Biol. 4, 203–213 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Kher S. S., et al., Substrate derived peptidic α-ketoamides as inhibitors of the malarial protease PfSUB1. Bioorg. Med. Chem. Lett. 24, 4486–4489 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Blackman M. J., et al., Structural and biochemical characterization of a fluorogenic rhodamine-labeled malarial protease substrate. Biochemistry 41, 12244–12252 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Smilkstein M., Sriwilaijaroen N., Kelly J. X., Wilairat P., Riscoe M., Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 48, 1803–1806 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glushakova S., et al., Rounding precedes rupture and breakdown of vacuolar membranes minutes before malaria parasite egress from erythrocytes. Cell. Microbiol. 20, e12868 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jogdand P. S., et al., Flow cytometric readout based on Mitotracker Red CMXRos staining of live asexual blood stage malarial parasites reliably assesses antibody dependent cellular inhibition. Malar. J. 11, 235 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glushakova S., et al., New stages in the program of malaria parasite egress imaged in normal and sickle erythrocytes. Curr. Biol. 20, 1117–1121 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smoum R., Rubinstein A., Dembitsky V. M., Srebnik M., Boron containing compounds as protease inhibitors. Chem. Rev. 112, 4156–4220 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Koussis K., Withers-Martinez C., Baker D. A., Blackman M. J., Simultaneous multiple allelic replacement in the malaria parasite enables dissection of PKG function. Life Sci. Alliance 3, e201900626 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collins C. R., et al., The malaria parasite sheddase SUB2 governs host red blood cell membrane sealing at invasion. eLife 9, e61121 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang M., et al., Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 360, eaap7847 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hasinoff B. B., Progress curve analysis of the kinetics of slow-binding anticancer drug inhibitors of the 20S proteasome. Arch. Biochem. Biophys. 639, 52–58 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Williamson M. J., et al., Comparison of biochemical and biological effects of ML858 (salinosporamide A) and bortezomib. Mol. Cancer Ther. 5, 3052–3061 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Kupperman E., et al., Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 70, 1970–1980 (2010). Corrected in: Cancer Res.70, 3853 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Ecker A., Lehane A. M., Clain J., Fidock D. A., PfCRT and its role in antimalarial drug resistance. Trends Parasitol. 28, 504–514 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gupta N., et al., Clinical pharmacology of ixazomib: The first oral proteasome inhibitor. Clin. Pharmacokinet. 58, 431–449 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matralis A. N., et al., Development of chemical entities endowed with potent fast-killing properties against Plasmodium falciparum malaria parasites. J. Med. Chem. 62, 9217–9235 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baker D. A., et al., A potent series targeting the malarial cGMP-dependent protein kinase clears infection and blocks transmission. Nat. Commun. 8, 430 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vanaerschot M., et al., Inhibition of resistance-refractory P. Falciparum kinase PKG delivers prophylactic, blood stage, and transmission-blocking antiplasmodial activity. Cell Chem. Biol. 27, 806–816.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pino P., et al., A multistage antimalarial targets the plasmepsins IX and X essential for invasion and egress. Science 358, 522–528 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nasamu A. S., et al., Plasmepsins IX and X are essential and druggable mediators of malaria parasite egress and invasion. Science 358, 518–522 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zogota R., et al., Peptidomimetic plasmepsin inhibitors with potent anti-malarial activity and selectivity against cathepsin D. Eur. J. Med. Chem. 163, 344–352 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Favuzza P., et al., Dual plasmepsin-targeting antimalarial agents disrupt multiple stages of the malaria parasite life cycle. Cell Host Microbe 27, 642–658.e12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perrin A. J., et al., The actinomyosin motor drives malaria parasite red blood cell invasion but not egress. MBio 9, e00905–e00918 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blackman M. J., Purification of Plasmodium falciparum merozoites for analysis of the processing of merozoite surface protein-1. Methods Cell Biol. 45, 213–220 (1994). [DOI] [PubMed] [Google Scholar]
- 44.Schindelin J., et al., Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abagyan R., Totrov M., Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 235, 983–1002 (1994). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All study data are included in the article and/or supporting information.