

ABSTRACT
The rise of antibodies as a promising and rapidly growing class of biotherapeutic proteins has motivated numerous studies to characterize and understand antibody structures. In the past decades, the number of antibody crystal structures increased substantially, which revolutionized the atomistic understanding of antibody functions. Even though numerous static structures are known, various biophysical properties of antibodies (i.e., specificity, hydrophobicity and stability) are governed by their dynamic character. Additionally, the importance of high-quality structures in structure–function relationship studies has substantially increased. These structure–function relationship studies have also created a demand for precise homology models of antibody structures, which allow rational antibody design and engineering when no crystal structure is available. Here, we discuss various aspects and challenges in antibody design and extend the paradigm of describing antibodies with only a single static structure to characterizing them as dynamic ensembles in solution.
KEYWORDS: Antibody structure, antibody structure prediction, antibody design, ensembles in solution
Introduction
Antibodies are protective agents used by the adaptive immune system to recognize and neutralize foreign objects through interactions with the target antigen. Long half-life, specificity to their respective antigen, and efficacy are beneficial attributes of antibodies.1 Because of their ability to recognize targets, they offer an innovative and efficient way to control pathogens by binding to their surfaces and thereby inactivating them. The immunoglobulin repertoire contains enormous diversity, which facilitates the recognition of a wide variety of different antigens. Antibodies have become one of the fastest growing fields in terms of academic and industrial research.1 Three of the top 5 selling drugs in 2019, 2020 and 2021 are in fact antibodies.2–4 This substantial interest has led to a vast amount of experimental data, including affinity and stability measurements as well as structural information.
The antigen binding fragment
The ability of an antibody to recognize a broad variety of different pathogens, such as viruses and bacteria, is determined by the antigen-binding fragment (Fab). This region consists of a heavy and a light chain that can each be subdivided into a constant (CH1, CL) and a variable domain (VH, VL).5 The variable region, also known as Fv, is the focal point of recombination and somatic hypermutation events.6–8 The diversity of an antibody in sequence and structure is concentrated within six hypervariable loops, the so-called complementarity-determining regions (CDRs), forming the antigen-binding site of an antibody.9–12 The heavy and light chains contain three loops each, known as CDR-H1, CDR-H2, CDR-H3 and CDR-L1, CDR-L2, CDR-L3, respectively.
Although there is great variation in the sequence and size of the CDRs, five of the six loops (CDR-H3 is the exception) have been classified into so-called canonical structures, assuming that they can only adopt a limited number of backbone conformations.11,13–18 Furthermore, the different amino acids at position H71 (Kabat nomenclature)10,12 are thought to influence both the position and the canonical cluster assignment of the CDR-H2 loop, and thus potentially affect antigen binding.17,19,20 Generally, the major determinants of specificity and affinity of these five CDR loops for an antigen are the size, shape and biophysical complementarity of their surface residues and their relative positions to each other.11 The CDR-H3 loop reveals the highest diversity in length, sequence and structure and has the ability to adopt various different conformations during the V(D)J recombination and somatic hypermutation. Thus, the accurate prediction of CDR-H3 loop structure remains challenging.21–23 The CDR-H3 loop is also known to play a central role in antigen-binding and recognition as it has on average the highest counts of contacts with the antigen.21 Additionally, the length and structure of the CDR-H3 loop can directly influence the antigen-binding patterns, and thereby have an effect on the specificity of the paratope.21,22
Recent studies that investigated the conformational diversity of the CDR-H3 loop in solution have shown that, in particular, CDR-H3 loop conformations in unbound antibody X-ray structures can be distorted by crystal packing effects and that the actual dominant CDR-H3 loop conformation in solution is optimized to bind the antigen. Thus, special care has to be taken when characterizing antibody CDR-H3 loops based on “unbound” Fab X-ray structures.24
Furthermore, it was shown that one single static structure is not enough to capture the high flexibility of any of the CDR loops. All CDR loops, not just CDR-H3, should thus be described as conformational ensembles in solution. Conformational rearrangements of the individual CDR loops and transitions between different canonical clusters were observed in the micro-to-millisecond timescale. Some canonical clusters even belong to the same kinetic minimum in solution, and hence might be combined.25,26
The regions of the variable domains apart from these loops are known as framework and are highly conserved in both sequence and main-chain conformations.10–12 This variability in the antigen-binding site is achieved by V(D)J recombination,27 somatic hypermutation,6 class switching7, and the combinatorial diversity via heavy and light-chain pairing.9 Apart from the length and sequence composition of the CDR loops, the relative orientation of VH and VL codetermines the shape of the antigen-binding site. Reorientations in the relative VH-VL orientation directly change the binding site geometry, and thereby have an effect on the specificity and affinity of the paratope. Especially in the field of antibody engineering, the preservation of the VH-VL orientation is essential to retain the original antibody properties.28–30 The VH-VL interface also strongly influences the stability of the Fv region. Because numerous residues in the VH-VL binding interface are highly retained, the role of conserved residues on the Fab function and consequently binding has been studied. Mutations that are distant from the CDR loops, however, also have effects on binding, which indicates that they indirectly affect antigen binding by favoring different VH-VL interface orientations.31–33 In addition, the influence of amino acids at position H23 (Kabat nomenclature)34 have been shown to have an effect on antigen-binding.33 Changes in the VH-VL interdomain orientations of up to 5° have also been reported upon antigen-binding and have been interpreted to follow the induced-fit mechanism of antigen recognition through rigid-body rotations of the VH and VL domains.35,36
Molecular dynamics simulations of whole Fvs and Fabs reveal fluctuations in these relative VH-VL interdomain orientations.37 The observed variability between these domains has been confirmed by nuclear magnetic resonance (NMR) experiments and is in line with the idea that these relative interdomain orientations can be interpreted as an additional structural feature of antibodies that increases the antibody repertoire and enlarges the number of possible binding partners. By applying fast Fourier transformation to the interface angles, timescales of 0.1 to 10 GHz could be assigned to the fastest collective interdomain movements, while the slower components of the observed dynamics are governed by conformational changes in the CDR loops that occur in the micro-to-millisecond timescale.37,38
In contrast to the prevalent static view of the binding interface, it was shown that antibodies exist as ensembles of paratope states.39 These paratope states are defined by a characteristic combination of CDR loop conformations and interdomain orientations. They interconvert into each other in the micro-to-millisecond timescale by correlated loop and interdomain rearrangements. Several studies have shown that crystal packing effects in unbound crystal structures can distort the paratope and thus result in misleading X-ray structures.24,40 For the first time, a complete description of conformations, thermodynamics and kinetics of the whole-binding paratope in solution can be achieved, which provides a new paradigm in the understanding of CDR binding loop states, antibody-antigen recognition, relative VH-VL interface and elbow angle distributions and their respective dynamics (Figures 1 and 2). In addition, it has been shown that these conformational ensembles also determine the hydrophobicity of antibodies, which makes them particularly relevant for tackling antibody developability issues.41,42
Figure 1.
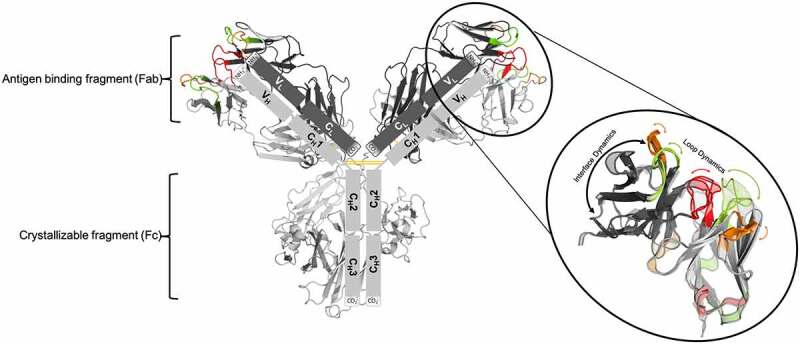
Structure of an IgG1 antibody and schematic illustration of the unique modular anatomy. the arms of the Y-shaped structure allow the antibody to carry out two functions, on the one hand antigen-binding and on the other hand biological activity mediation. the arms of the antibody are known as antigen-binding fragments (Fabs). The Fab is composed of a constant and a variable domain of each of the heavy and the light chain. the variable domains shape the antigen binding site (paratope) at the amino-terminal end of the antibody. the variable fragment (Fv) is highlighted in the picture. the CDR 1 loops are colored in green, the CDR 2 loops are depicted in orange and the CDR 3 loops are shown in red. The close up to the Fv also indicates the high flexibility of the CDR loops and the relative VH-VL interface and shows that the antibody binding site exists as ensembles of paratope states. the tail region of the antibody, also known as Fc region, is responsible for the communication with the immune system and interacts with the cell surface receptors, called Fc receptors
Figure 2.

Antibodies exist as ensembles in solution. summary of antibody Fab dynamics and their respective timescales. bond vibrations and sidechain rotations can already be captured in the femto-to-picosecond timescale. Interface and elbow angle dynamics occur in the low nanosecond timescale, while conformational transitions between CDR loops can be sampled in the microsecond timescale. the combination of interface angles and different CDR loop conformations have been described as ensembles of paratope states in solution, which interconvert between each other in the micro-to-millisecond timescale
The overall stability of a Fab is governed by the high degree of cooperation between the elbow angle and the VH/VL and CH1/CL interface, while the direct interactions of the VL and CL/VH and CH1 domains do not influence the stability of either domain.43 Similar to the relative VH-VL interface, the CH1-CL interdomain orientations also reveal high variability and can be captured in the low nanosecond timescale. However, even though the captured dynamics are similar between the Fab interfaces, the nature and number of interface interactions can differ. The constant domains of the Fab show hydrophobic interactions at the center of the interface surrounded by a small number of salt-bridges, while the Fv interface is strongly dominated by framework interactions and conformations of the CDR loops.34,43 Structurally, the CH1-CL domains resemble the CH3-CH3 domains. Apart from the VH/VL and CH1/CL interface, the elbow angle is also influenced by the shape of the paratope and might contribute to antigen specificity. The elbow angle is defined as the angle between the pseudo-2-fold axes relating to VH-VL and CH1-CL, and has been shown to increase Fab flexibility and allow the same antibody to recognize different antigens.44,45 Mutations in the Fab elbow region have been reported to influence conformational flexibility and paratope plasticity.19,45–49
The crystallizable fragment region
The tail region of the antibody, known as the crystallizable fragment (Fc), is responsible for interactions with the cell surface, immune system activation and extension of the molecular half-life.5 Antibody Fabs and Fc domains are linked together via a flexible unstructured hinge region. The Fc can be divided into a CH2-CH2 and a CH3-CH3 dimer. The CH2-CH2 dimer is mainly responsible for interacting with type I or type II Fc receptors (FcRs), which can be located on effector cells or on B cells, and thereby modulate both the adaptive and innate immune response. The interface between the two CH2-CH2 domains contains conserved glycosylation sites at Asn297, which are conjugated to a core heptasaccharide, forming a biantennary Fc glycan. The glycans modulate the functions, affinities and Fc conformations.50–53 The hydrogen bonding in the CH2-CH2 interface can be observed either directly between the two carbohydrate chains, or through a dynamic water network.54 Detailed structural and dynamic analysis of the CH2-CH2 interface in IgG1 and IgG2 has revealed that movements of the CH2 domains originate from pivoting around a highly conserved ball- and socket-like joint, formed by the CH2 L251 sidechain (ball) with the CH3 residues M428, H429, E430 and H435 (socket).54
The CH3 domains bind tightly with each other by both hydrophobic interactions at the center, surrounded by salt bridges, thereby forming the foundation for the heavy-chain dimer association.54 Mutations in the CH3-CH3 interface have been shown to not only strongly influence the stability and the association of the two domains, but also alter glycosylation and result in structural changes of the CH2 domain.55 By mutating residues in the interface, the energetic contributions of single amino acids could be quantified. Thereby, three contacts within the interface were found to highly stabilize the interface, with the hydrogen bond between T366 and Y407 in the center of the interface described as the most important interaction. Similarly, the charge–charge interaction between K409 and D399 was shown to have a high energetic contribution, as well as the hydrophobic interactions of L368 and F405.56,57
Heterodimeric Fc variants have been engineered primarily through the replacement of homodimer-favoring interactions at the interface with heterodimer-favoring interactions by asymmetric mutations in both heavy chains. These rational approaches can be classified into different strategies, with some of the strategies relying on steric complementarity (also known as the Knobs-into-Holes approach), and others involving the introduction of asymmetric charged interactions.58,59
Various studies have investigated the influence of the Fab, the Fc and the glycans on the activity of an antibody.52,60 It was recently shown that antigen binding induces conformational changes in the Fc domain, followed by Fc receptor activation. Thus, antigen binding also allosterically promotes Fc receptor binding and recognition.61 Consequently, conformational rearrangements in the Fc directly modulate the activity and binding affinity toward binding and recognizing Fc receptors.52
Antibody specificity – antibody affinity maturation
The most striking aspect of antibodies, and at the same time a fundamental requirement of the immune system, is the specific nature of their interaction with an antigen.62 The specificity of an antibody evolves through various rounds of somatic hypermutations, followed by selection in the germinal centers.6,63 Repeated exposure of the same antigen results in a selection of antibodies with higher affinities and specificities. Studies investigating various different aspects of humoral and cellular immunity have contributed to the present view of specificity as part of the complexity of molecular recognition.7,64–68
Antibodies were first identified at the end of the 1800s, yet the process by which can a limited repertoire of antibodies recognize an effectively limitless number of antigens is still not fully understood.69 Sufficient evidence showing that antibodies are not infinitely specific has accumulated. Numerous studies have in fact demonstrated that antibodies can recognize more than one antigen and thus can be described as functionally promiscuous or multi-specific.66,69–72 This was already discussed in the 1940s, when Pauling and Landsteiner suggested that antibodies follow the concept of conformational diversity.73,74
Following Landsteiner’s idea that there are ‘different ways of folding the same polypeptide chain’, Pauling proposed the idea of having an ensemble of preexisting conformations out of which the functional ones are selected.73 This view was also supported by the conformational selection or population shift model originating from the Monod-Wyman-Changeux model.75–77 In the early 1990s, Milstein and Foote revived this idea,78,79 which was subsequently also demonstrated Wedemayer.80 The concept of conformational selection suggests that, within this preexisting ensemble of conformations, the binding competent state is selected, accompanied by a population shift.76,81 The probability of the conformation chosen by the antigen determines the binding mechanism, which can be either “lock and key”,36,82 “conformational selection”,75,76,83 or “induced fit”.36,83,84 Historically, protein–protein interactions such as antibody-antigen binding were assumed to follow the “lock and key” mechanism. This “lock and key” binding mechanism can especially be observed for matured antibodies, where the apo conformation is selected as the binding competent conformation.80,85 Studies investigating the consequences of affinity maturation have observed a substantial rigidification of the antigen-binding site as a consequence of the increase in specificity.38,39,80,85–87 Even though rigidification might only be one of the various consequences of affinity maturation, it still represents a fundamental mechanism resulting in an increase in specificity (Figure 3).
Figure 3.
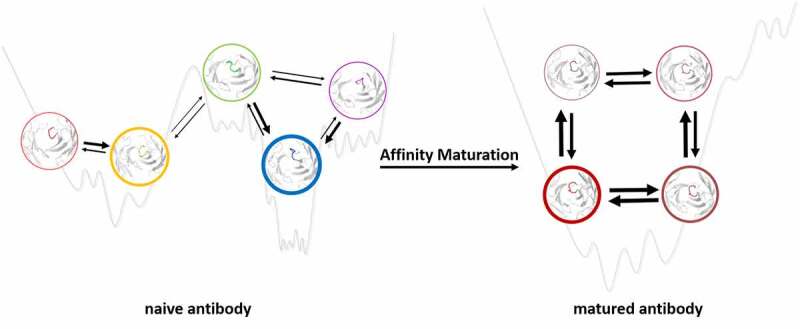
Effect of maturation on the free energy landscape. the potential energy hypersurface of the naive and the matured antibody are represented as 1D basins, showing accessible conformational substates. the wide basins of the naive antibody illustrate the possibility of binding a diverse set of antigens. The increased depth and at the same time decreased number of basins upon affinity maturation indicate the enhanced enthalpic interactions that formed, which are accompanied by a decrease in conformational entropy
If the binding occurs to a rare conformation in solution, which cannot be detected before binding, the process can be interpreted as induced fit binding.88 Both induced fit and conformational selection have been discussed in current literature to elucidate the binding preferences of multi-specific antibodies, which can recognize various structurally unrelated antigens with low affinity due to their inherently more flexible-binding site.65,72,83,84 Thus, promiscuity might arise from a multitude of weakly populated conformations, each of which is able to bind different binding partners. Rigidification upon affinity maturation shifts the probabilities toward a smaller number of states, and thereby reduces the number of potential-binding partners.86
Future perspective and recommendations to the community
As the functions and properties of antibodies are strongly governed by their dynamic nature, both Fab as well as Fcs should be considered as ensembles in solution. Especially, the Fab, which is responsible for antigen binding and recognition, should be described as having interconverting states in solution. The probabilities of these states determine the specificity, promiscuity and affinity. These different conformations of the antigen-binding site are characterized by different paratope states in solution and CDR loop state-dependent interdomain orientations. In-depth understanding of these states and their dynamic interconversion is a paradigm change for rational antibody design and engineering. Furthermore, allosteric effects resulting in signal transduction from the antigen-binding site, reaching as far as to the Fc receptor-binding site, have to be expected. This signal is surmised to be transmitted by interdomain rearrangements of the VH-VL, CH1-CL, CH2-CH2 and CH3-CH3 interfaces.
Thus, what could be done differently in practice? First of all, the one single structure characterizing an antibody the best is the dominant conformation in solution, which does not necessarily coincide with the (apo) X-ray structure. The community should strive to predict this dominant structure in solution instead of trying to predict X-ray structures potentially distorted by crystal packing effects. Obviously, developing such predictions is a time- and resource-consuming effort, as it is necessary to systematically characterize and, if possible, experimentally verify (e.g., by NMR), a large number of dominant conformations in solution. For a deeper understanding of binding properties (e.g., finetuning of specificity) and eventually also other biophysical properties (e.g., developability liabilities), only looking at the dominant structure in solution is not sufficient. These properties can only be understood quantitatively by considering all important structures in solution weighted by their probabilities. In particular, docking might profit from such an approach.
Funding Statement
This work was supported by the Austrian Science Fund (FWF) via the grant P30565 and P30737 and DOC 30.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed. G.G. is a Roche employee; Roche has an interest in developing antibody-based therapeutics.
Author contributions
The manuscript was discussed and written through the contributions of all authors. All
authors have given approval to the final version of the manuscript.
List of Abbreviations
CDRs – complementarity-determining regions
Fab – antigen-binding fragment
Fc – crystallizable fragment
Fv – variable region
NMR – nuclear magnetic resonance
References
- 1.Chames P, Van Regenmortel M, Weiss E, Baty D.. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157(2):220–7. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaplon H, Reichert JM. Antibodies to watch in 2019. mAbs. 2019;11(2):219–38. doi: 10.1080/19420862.2018.1556465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaplon H, Muralidharan M, Schneider Z, Reichert JM. Antibodies to watch in 2020. mAbs. 2020;12(1):1703531. doi: 10.1080/19420862.2019.1703531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaplon H, Reichert JM. Antibodies to watch in 2021. MAbs. 2021;13(1):1860476. doi: 10.1080/19420862.2020.1860476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schroeder HW, Cavacini L. Structure and function of immunoglobulins. J Allergy Clin Immunol. 2010;125(2):S41–52. doi: 10.1016/j.jaci.2009.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teng G, Papavasiliou FN. Immunoglobulin somatic hypermutation. Annu Rev Genet. 2007;41(1):107–20. doi: 10.1146/annurev.genet.41.110306.130340. [DOI] [PubMed] [Google Scholar]
- 7.Wabl M, Charles. S. Affinity maturation and class switching. Current Opinion in Immunology 1996:8(1);89-92. [DOI] [PubMed] [Google Scholar]
- 8.Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302(5909):575–81. doi: 10.1038/302575a0. [DOI] [PubMed] [Google Scholar]
- 9.Chailyan A, Marcatili P, Tramontano A. The association of heavy and light chain variable domains in antibodies: implications for antigen specificity. FEBS Journal. 2011;278(16):2858–66. doi: 10.1111/j.1742-4658.2011.08207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kabat EA. National Institutes of Health (U.S.), Columbia University. sequences of proteins of immunological interest. Bethesda (MD): U.S. Dept. of Health and Human Services, Public Health Service, National Institutes of Health; 1991. [Google Scholar]
- 11.Al-Lazikani B, Lesk AM, Chothia C. Standard conformations for the canonical structures of immunoglobulins1. J Mol Biol. 1997;273(4):927–48. doi: 10.1006/jmbi.1997.1354. [DOI] [PubMed] [Google Scholar]
- 12.Wu TT, Kabat EA. An analysis of the sequences of the variable regions of Bence Jones proteins and myeloma light chains and their implications for antibody complementarity. J Exp Med. 1970;132(2):211–50. doi: 10.1084/jem.132.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol. 1987;196(4):901–17. doi: 10.1016/0022-2836(87)90412-8. [DOI] [PubMed] [Google Scholar]
- 14.Chothia C, Lesk AM, Tramontano A, Levitt M, Smith-Gill SJ, Air G, Sheriff S, Padlan EA, Davies D, Tulip WR, et al. Conformations of immunoglobulin hypervariable regions. Nature. 1989;342(6252):877–83. doi: 10.1038/342877a0. [DOI] [PubMed] [Google Scholar]
- 15.North B, Lehmann A, Dunbrack Jr RL. A new clustering of antibody CDR loop conformations. J Mol Biol. 2011;406(2):228–56. doi: 10.1016/j.jmb.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martin ACR, Thornton JM. Structural families in loops of homologous proteins: automatic classification, modelling and application to antibodies. J Mol Biol. 1996;263(5):800–15. doi: 10.1006/jmbi.1996.0617. [DOI] [PubMed] [Google Scholar]
- 17.Tramontano A, Chothia C, Lesk AM. Framework residue 71 is a major determinant of the position and conformation of the second hypervariable region in the VH domains of immunoglobulins. J Mol Biol. 1990;215(1):175–82. doi: 10.1016/S0022-2836(05)80102-0. [DOI] [PubMed] [Google Scholar]
- 18.Tomlinson IM, Cox JP, Gherardi E, Lesk AM, Chothia C. The structural repertoire of the human V kappa domain. EMBO J. 1995;14(18):4628–38. doi: 10.1002/j.1460-2075.1995.tb00142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davenport TM, Gorman J, Joyce MG, Zhou T, Soto C, Guttman M, Moquin S, Yang Y, Zhang B, Doria-Rose NA, et al. Somatic hypermutation-induced changes in the structure and dynamics of HIV-1 broadly neutralizing antibodies. Structure. 2016;24(8):1346–57. doi: 10.1016/j.str.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernández-Quintero ML, Kroell KB, Hofer F, Riccabona JR, Liedl KR. Mutation of framework residue H71 results in different antibody Paratope States in solution. Front Immunol. 2021;12:243. doi: 10.3389/fimmu.2021.630034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Regep C, Georges G, Shi J, Popovic B, Deane CM. The H3 loop of antibodies shows unique structural characteristics. Proteins. 2017;85(7):1311–18. doi: 10.1002/prot.25291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishigami H, Kamiya N, Nakamura H. Revisiting antibody modeling assessment for CDR-H3 loop. Protein Eng Des Sel. 2016;29(11):477–84. doi: 10.1093/protein/gzw028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stein A, Kortemme T, Zhang Y. Improvements to robotics-inspired conformational sampling in Rosetta. PLoS ONE. 2013;8(5):e63090. doi: 10.1371/journal.pone.0063090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fernández-Quintero ML, Kraml J, Georges G, Liedl KR. CDR-H3 loop ensemble in solution – conformational selection upon antibody binding. MAbs. 2019;11(6):1077–88. doi: 10.1080/19420862.2019.1618676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fernández-Quintero ML, Math BF, Loeffler JR, Liedl KR. Transitions of CDR-L3 loop canonical cluster conformations on the micro-to-millisecond timescale. Front Immunol. 2019;10:2652. doi: 10.3389/fimmu.2019.02652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fernández-Quintero ML, Heiss MC, Pomarici ND, Math BA, Liedl KR. Antibody CDR loops as ensembles in solution vs. Canonical Clusters from X-ray Structures mAbs. 2020;12:1744328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Market E, Papavasiliou FN. V(D)J recombination and the evolution of the adaptive immune system. PLoS Biol. 2003;1(1):e16. doi: 10.1371/journal.pbio.0000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bujotzek A, Dunbar J, Lipsmeier F, Schäfer W, Antes I, Deane CM, Georges G. Prediction of VH–VL domain orientation for antibody variable domain modeling. Proteins. 2015;83(4):681–95. doi: 10.1002/prot.24756. [DOI] [PubMed] [Google Scholar]
- 29.Bujotzek A, Lipsmeier F, Harris SF, Benz J, Kuglstatter A, Georges G. VH-VL orientation prediction for antibody humanization candidate selection: a case study. mAbs. 2016;8(2):288–305. doi: 10.1080/19420862.2015.1117720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dunbar J, Fuchs A, Shi J, Deane CM. ABangle: characterising the VH–VL orientation in antibodies. Protein Engineering, Design and Selection. 2013;26(10):611–20. doi: 10.1093/protein/gzt020. [DOI] [PubMed] [Google Scholar]
- 31.Chatellier J, Van Regenmortel MHV, Vernet T, Altschuh D. Functional mapping of conserved residues located at the VL and VH domain interface of a Fab. J Mol Biol. 1996;264(1):1–6. doi: 10.1006/jmbi.1996.0618. [DOI] [PubMed] [Google Scholar]
- 32.Roguska MA, Pedersen JT, Keddy CA, Henry AH, Searle SJ, Lambert JM, Goldmacher VS, Blättler WA, Rees AR, Guild BC. Humanization of murine monoclonal antibodies through variable domain resurfacing. Proc Natl Acad Sci USA. 1994;91(3):969. doi: 10.1073/pnas.91.3.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adair JR, Athwal DS, Emtage JS. Humanised antibodies. 1999. [Google Scholar]
- 34.Honegger A, Malebranche AD, Röthlisberger D, Plückthun A. The influence of the framework core residues on the biophysical properties of immunoglobulin heavy chain variable domains. Protein Engineering, Design and Selection. 2009;22(3):121–34. doi: 10.1093/protein/gzn077. [DOI] [PubMed] [Google Scholar]
- 35.Teplyakov A, Obmolova G, Malia T, Gilliland G. Antigen recognition by antibody C836 through adjustment of VL/VH packing. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2011;67(10):1165–67. doi: 10.1107/S1744309111027746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koshland Daniel E. The key–lock theory and the induced fit theory. Angewandte Chemie International Edition in English. 1995;33(2324):2375–78. doi: 10.1002/anie.199423751. [DOI] [Google Scholar]
- 37.Fernández-Quintero ML, Hoerschinger VJ, Lamp LM, Bujotzek A, Georges G, Liedl KR. VH-VL interdomain dynamics observed by computer simulations and NMR. Proteins 88, 830–839. doi: 10.1002/prot.25872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fernández-Quintero ML, Kroell KK, Heiss MC, Loeffler JR, Quoika PK, Waibl F, Bujotzek A, Moessner E, Guy Georges K, Liedl R. Surprisingly fast interface and elbow angle dynamics of antigen-binding fragments. Front Mol Biosci (2020:7;1–10. doi: 10.3389/fmolb.2020.609088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fernández-Quintero ML, Pomarici ND, Math BA, Kroell KK, Waibl F, Bujotzek A, Guy Georges K, Liedl. R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fernández-Quintero ML, Pomarici ND, Math BA, Kroell KB, Waibl F, Bujotzek A, Georges G, Liedl KR. Antibodies exhibit multiple paratope states influencing VH–VL domain orientations. Communications Biology. 2020;3(1):589. doi: 10.1038/s42003-020-01319-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raybould MIJ, Marks C, Krawczyk K, Taddese B, Nowak J, Lewis AP, Bujotzek A, Shi J, Deane CM. Five computational developability guidelines for therapeutic antibody profiling. Proc Natl Acad Sci USA. 2019;116(10):4025. doi: 10.1073/pnas.1810576116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Waibl F, Fernández-Quintero ML, Kamenik AS, Kraml J, Hofer F, Kettenberger H, Georges G, Liedl KR. Conformational ensembles of antibodies determine their hydrophobicity. Biophys J. 2021;120(1):143–57. doi: 10.1016/j.bpj.2020.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Röthlisberger D, Honegger A, Plückthun A. Domain interactions in the Fab fragment: a comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. J Mol Biol. 2005;347(4):773–89. doi: 10.1016/j.jmb.2005.01.053. [DOI] [PubMed] [Google Scholar]
- 44.Stanfield RL, Zemla A, Wilson IA, Rupp B. Antibody Elbow angles are influenced by their light chain class. J Mol Biol. 2006;357(5):1566–74. doi: 10.1016/j.jmb.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 45.Sotriffer CA, Rode BM, Varga JM, Liedl KR. Elbow flexibility and Ligand-induced domain rearrangements in antibody Fab NC6.8: large effects of a small Hapten. Biophys J. 2000;79(2):614–28. doi: 10.1016/S0006-3495(00)76320-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sotriffer CA, Liedl KR, Linthicum DS, Rode BM, Varga JM. Ligand-induced domain movement in an antibody fab: molecular dynamics studies confirm the unique domain movement observed experimentally for fab NC6.8 upon complexation and reveal its segmental flexibility11Edited Wilson I. J Mol Biol. 1998;278(2):301–06. doi: 10.1006/jmbi.1998.1684 [DOI] [PubMed] [Google Scholar]
- 47.Niederfellner G, Lammens A, Mundigl O, Georges GJ, Schaefer W, Schwaiger M, Franke A, Wiechmann K, Jenewein S, Slootstra JW, et al. Epitope characterization and crystal structure of GA101 provide insights into the molecular basis for type I/II distinction of CD20 antibodies. Blood. 2011;118(2):358–67. doi: 10.1182/blood-2010-09-305847. [DOI] [PubMed] [Google Scholar]
- 48.Mössner E, Brünker P, Moser S, Püntener U, Schmidt C, Herter S, Grau R, Gerdes C, Nopora A, Van Puijenbroek E, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell–mediated B-cell cytotoxicity. Blood. 2010;115(22):4393–402. doi: 10.1182/blood-2009-06-225979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meyer S, Evers M, Jansen JHM, Buijs J, Broek B, Reitsma SE, Moerer P, Amini M, Kretschmer A, Ten Broeke T; Meyer S, Evers M, Jhm Jansen, Buijs J, Broek B, Se Reitsma, Moerer P, Amini M, Kretschmer A, Ten Broeke T, et al. New insights in type I and II CD20 antibody mechanisms-of-action with a panel of novel CD20 antibodies. Br J Haematol. 2018;180(6):808–20. doi: 10.1111/bjh.15132. [DOI] [PubMed] [Google Scholar]
- 50.Krapp S, Mimura Y, Jefferis R, Huber R, Sondermann P. Structural analysis of human IgG-Fc Glycoforms reveals a correlation between Glycosylation and structural integrity. J Mol Biol. 2003;325(5):979–89. doi: 10.1016/S0022-2836(02)01250-0. [DOI] [PubMed] [Google Scholar]
- 51.Matsumiya S, Yamaguchi Y, Saito J, Nagano M, Sasakawa H, Otaki S, Satoh M, Shitara K, Kato K. Structural comparison of Fucosylated and Nonfucosylated Fc fragments of human Immunoglobulin G1. J Mol Biol. 2007;368(3):767–79. doi: 10.1016/j.jmb.2007.02.034. [DOI] [PubMed] [Google Scholar]
- 52.Frank M, Walker RC, Lanzilotta WN, Prestegard JH, Barb AW. Immunoglobulin G1 Fc domain motions: implications for Fc engineering. J Mol Biol. 2014;426(8):1799–811. doi: 10.1016/j.jmb.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sazinsky SL, Ott RG, Silver NW, Tidor B, Ravetch JV, Wittrup KD. Aglycosylated immunoglobulin G1 variants productively engage activating Fc receptors. Proc Natl Acad Sci USA. 2008;105(51):20167. doi: 10.1073/pnas.0809257105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Teplyakov A, Zhao Y, Malia TJ, Obmolova G, Gilliland GL. IgG2 Fc structure and the dynamic features of the IgG CH2–CH3 interface. Mol Immunol. 2013;56(1–2):131–39. doi: 10.1016/j.molimm.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 55.Rose RJ, Van Berkel PHC, Van Den Bremer ETJ, Labrijn AF, Vink T, Schuurman J, Heck AJR, Parren PWHI. Mutation of Y407 in the CH3 domain dramatically alters glycosylation and structure of human IgG. MAbs. 2013;5(2):219–28. doi: 10.4161/mabs.23532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ha J-H, Kim J-E, Kim Y-S. Immunoglobulin Fc Heterodimer platform technology: from design to applications in therapeutic antibodies and proteins. Front Immunol. 2016;7:394. doi: 10.3389/fimmu.2016.00394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dall’Acqua W, Simon AL, Mulkerrin MG, Carter P. Contribution of domain interface residues to the stability of antibody CH 3 domain Homodimers. Biochemistry. 1998;37(26):9266–73. doi: 10.1021/bi980270i. [DOI] [PubMed] [Google Scholar]
- 58.Leaver-Fay A, Froning KJ, Atwell S, Aldaz H, Pustilnik A, Lu F, Huang F, Yuan R, Hassanali S, Chamberlain AK, et al. Computationally designed bispecific antibodies using negative state Repertoires. Structure. 2016;24(4):641–51. doi: 10.1016/j.str.2016.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ridgway JBB, Presta LG, Carter P. ‘Knobs-into-holes’ engineering of antibody CH 3 domains for heavy chain heterodimerization. Protein Engineering, Design and Selection. 1996;9(7):617–21. doi: 10.1093/protein/9.7.617. [DOI] [PubMed] [Google Scholar]
- 60.Janda A, Bowen A, Greenspan NS, Casadevall A. Ig constant region effects on variable region structure and function. Front Microbiol. 2016;7:22. doi: 10.3389/fmicb.2016.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao J, Nussinov R, Ma B. Antigen binding allosterically promotes Fc receptor recognition. MAbs. 2019;11(1):58–74. doi: 10.1080/19420862.2018.1522178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Richards F, Konigsberg W, Rosenstein R, Varga J. On the specificity of antibodies. Science. 1975;187(4172):130. doi: 10.1126/science.46122. [DOI] [PubMed] [Google Scholar]
- 63.De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol. 2015;15(3):137–48. doi: 10.1038/nri3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. 2012;30(1):429–57. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- 65.Van Regenmortel MHV. Specificity, polyspecificity, and heterospecificity of antibody-antigen recognition. Journal of Molecular Recognition. 2014;27(11):627–39. doi: 10.1002/jmr.2394. [DOI] [PubMed] [Google Scholar]
- 66.Jain D, Salunke DM. Antibody specificity and promiscuity. Biochemical Journal. 2019;476(3):433–47. doi: 10.1042/BCJ20180670. [DOI] [PubMed] [Google Scholar]
- 67.Krishnan L, Sahni G, Kaur KJ, Salunke DM. Role of antibody paratope conformational flexibility in the manifestation of molecular Mimicry. Biophys J. 2008;94(4):1367–76. doi: 10.1529/biophysj.107.108654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guthmiller JJ, Wilson PC. It’s hard to teach an old B cell new tricks. Cell. 2020;180(1):18–20. doi: 10.1016/j.cell.2019.12.019. [DOI] [PubMed] [Google Scholar]
- 69.James LC, Roversi P, Tawfik DS. Antibody multispecificity mediated by conformational diversity. Science. 2003;299(5611):1362–67. doi: 10.1126/science.1079731. [DOI] [PubMed] [Google Scholar]
- 70.Inman JK. Multispecificity of the antibody combining region and antibody diversity [Internet]. Sercarz EE, Williamson AR, Fox CF, editors. The Immune System. Academic Press; 1974. page 37–52.Available from: . http://www.sciencedirect.com/science/article/pii/B9780126371505500110 [Google Scholar]
- 71.James LC, Tawfik DS. Conformational diversity and protein evolution – a 60-year-old hypothesis revisited. Trends Biochem Sci. 2003;28(7):361–68. doi: 10.1016/S0968-0004(03)00135-X. [DOI] [PubMed] [Google Scholar]
- 72.Guthmiller JJ, Lan -LY-L, Fernández-Quintero ML, Han J, Utset HA, Bitar DJ, Hamel NJ, Stovicek O, Li L, Tepora M, et al. Polyreactive broadly neutralizing B cells are selected to provide defense against pandemic threat influenza viruses. Immunity. 2020;53(6):1230–1244. doi: 10.1016/j.immuni.2020.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pauling LA. A theory of the structure and process of formation of antibodies. J Am Chem Soc. 1940;62(10):2643–57. doi: 10.1021/ja01867a018. [DOI] [Google Scholar]
- 74.Landsteiner K. The specificity of serological reactions. New York 14 (N. Y.): Dover Publications, Inc; 1962. 180 Varick St. [Google Scholar]
- 75.Ma B, Kumar S, Tsai C-J, Nussinov R. Folding funnels and binding mechanisms. Protein Eng. 1999;12(9):713–20. doi: 10.1093/protein/12.9.713. [DOI] [PubMed] [Google Scholar]
- 76.Tsai C-J, Kumar S, Ma B, Nussinov R. Folding funnels, binding funnels, and protein function. Protein Science. 1999;8(6):1181–90. doi: 10.1110/ps.8.6.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Monod J, Wyman J, Changeux J-P. On the nature of allosteric transitions: a plausible model. J Mol Biol. 1965;12(1):88–118. doi: 10.1016/S0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 78.Foote J, Milstein C.. Conformational isomerism and the diversity of antibodies. Proceedings of the National Academy of Sciences of the United States of America 1994; 91:10370–74. doi: 10.1073/pnas.91.22.10370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Foote J, Milstein C. Kinetic maturation of an immune response. Nature. 1991;352(6335):530–32. doi: 10.1038/352530a0. [DOI] [PubMed] [Google Scholar]
- 80.Wedemayer GJ, Patten PA, Wang LH, Schultz PG, Stevens RC. Structural insights into the evolution of an antibody combining site. Science. 1997;276(5319):1665. doi: 10.1126/science.276.5319.1665. [DOI] [PubMed] [Google Scholar]
- 81.Nussinov R, Ma B. Protein dynamics and conformational selection in bidirectional signal transduction. BMC Biol. 2012;10(1):2. doi: 10.1186/1741-7007-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Braden BC, Dall’Acqua W, Eisenstein E, Fields BA, Goldbaum FA, Malchiodi EL, Mariuzza RA, Schwarz FP, Ysern X, Poljak RJ. Protein motion and lock and key complementarity in antigen-antibody reactions. Pharm Acta Helv. 1995;69(4):225–30. doi: 10.1016/0031-6865(94)00046-X. [DOI] [PubMed] [Google Scholar]
- 83.Wang W, Ye W, Yu Q, Jiang C, Zhang J, Luo R, Chen H-F; Wang W, Ye W, Yu Q, Jiang C, Zhang J, Luo R, Chen H-F. Conformational Selection and . Induced fit in specific antibody and antigen recognition: SPE7 as a case study. J Phys Chem B. 2013;117(17):4912–23. doi: 10.1021/jp4010967. [DOI] [PubMed] [Google Scholar]
- 84.Csermely P, Palotai R, Nussinov R. Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem Sci. 2010;35(10):539–46. doi: 10.1016/j.tibs.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fernández-Quintero ML, Loeffler JR, Bacher LM, Waibl F, Clarissa A, Seidler K, Liedl. R. Local and global rigidification upon antibody affinity maturation. Front Mol Biosci. 2020;7:182. doi: 10.3389/fmolb.2020.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fernández-Quintero ML, Loeffler JR, Kraml J, Kahler U, Kamenik AS, Liedl KR. Characterizing the diversity of the CDR-H3 loop conformational ensembles in relationship to antibody binding properties. Front Immunol. 2019;9:3065. doi: 10.3389/fimmu.2018.03065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Thorpe IF, Brooks CL. Molecular evolution of affinity and flexibility in the immune system. Proc Natl Acad Sci USA. 2007;104(21):8821. doi: 10.1073/pnas.0610064104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fernández-Quintero ML, Pomarici ND, Loeffler JR, Seidler CA, Liedl KR. T-cell receptor CDR3 loop conformations in solution shift the relative Vα-Vβ domain distributions. Front Immunol. 2020;11:1440. doi: 10.3389/fimmu.2020.01440. [DOI] [PMC free article] [PubMed] [Google Scholar]