

ABSTRACT
Adoptive cell therapy using TCR-engineered T cells (TCR-T cells) represents a promising strategy for treating relapsed and metastatic cancers. We previously established methods to identify neoantigen-specific TCRs based on patients’ PBMCs. However, in clinical practice isolation of PBMCs from advanced-stage cancer patients proves to be difficult. In this study, we substituted blood-derived T cells for tumor-infiltrating lymphocytes (TILs) and used an HLA-matched cell line of antigen-presenting cells (APCs) to replace autologous dendritic cells. Somatic mutations were determined in head and neck squamous cell carcinoma resected from two patients. HLA-A*02:01-restricted neoantigen libraries were constructed and transferred into HLA-matched APCs for stimulation of patient TILs. TCRs were isolated from reactive TIL cultures and functionality was tested using TCR- T cells in vitro and in vivo. To exemplify the screening approach, we identified the targeted neoantigen leading to recognition of the minigene construct that stimulated the strongest TIL response. Neoantigen peptides were used to load MHC-tetramers for T cell isolation and a TCR was identified targeting the KIAA1429D1358E mutation. TCR-T cells were activated, exhibited cytotoxicity, and secreted cytokines in a dose-dependent manner, and only when stimulated with the mutant peptide. Furthermore, comparable to a neoantigen-specific TCR that was isolated from the patient’s PBMCs, KIAA1429D1358E-specific TCR T cells destroyed human tumors in mice. The established protocol provides the required flexibility to methods striving to identify neoantigen-specific TCRs. By using an MHC-matched APC cell line and neoantigen-encoding minigene libraries, autologous TILs can be stimulated and screened when patient PBMCs and/or tumor material are not available anymore.
Abbreviations:
Head and neck squamous cell carcinoma (HNSCC); adoptive T cell therapy (ACT); T cell receptor (TCR); tumor-infiltrating lymphocytes (TIL); cytotoxic T lymphocyte (CTL); peripheral blood mononuclear cell (PBMC); dendritic cell (DC); antigen-presenting cells (APC)
KEYWORDS: Head and neck squamous cell carcinoma, adoptive T cell therapy, neoantigen, tumor-infiltrating lymphocytes, T cell receptor engineered T cells
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer type and has usually a poor prognosis.1–3 HNSCC is often highly immunosuppressive, which is characterized by low numbers of T cells in the tumor microenvironment.4–7 Adoptive cell therapy (ACT) using tumor-specific TCR-engineered T cells, which recognize cancer-specific neoantigens, is a promising option to overcome the limitation of presently available treatments. In previous work, our group established effective and rapid methods to identify neoantigen-specific TCRs and to generate TCR-engineered T cells (TCR-T cells) in a short time. We identified neoantigen-specific T cells from PBMCs derived from either patients or healthy donors and characterized several TCRs targeting neoantigens in HNSCC and ovarian cancer.8–10 However, given the intense treatment regimens and the severe myelosuppression that affect many late-stage cancer patients,11–13 obtaining a sufficient number of T cells and dendritic cells (DCs) to screen the autologous repertoire for neoantigen-specific TCRs is often problematic. Although HLA-matched healthy donors can be an alternative to obtaining tumor-specific T cells from patient material, there are limitations for patients having rare MHC alleles.
Instead of using blood samples from healthy donors or advanced-stage cancer patients, tumor-infiltrating lymphocytes (TILs) could be another source of neoantigen-specific T cells. Accumulated evidence has shown that tumor-specific T cells are present in the tumor microenvironment.14–18 ACT using TILs achieved complete and durable regression in a small subset of melanoma patients whose tumors had a large number of somatic mutations.19–21 However, clinical benefit by ACT using TILs was very limited for other types of solid tumors, which may correlate with the lower number of somatic mutations, resulting in a lower number of neoantigen-specific T cells in the TIL populations.22–24 In addition, only 27% of patients, who expected to have the ACT, could finally receive the treatment,25,26 mainly because both T cells in the tumor microenvironment were exhausted and could not well proliferate to obtain a sufficient number of TILs for ACT.27–29 In contrast, TCR T-cell therapy utilizes peripheral blood T cells, which process explosive proliferative potentials and more resistance to exhausting tumor microenvironment than TILs, representing another advantage over TIL therapy. Hence, the rapid identification of functional TCR from TILs and their subsequent use to generate neoantigen-specific TCR-T cells could overcome these problems.
In the present study, we established a new strategy by using in vitro expanded TILs and HLA-matched allogeneic C1R cells as APCs to screen for neoantigen-specific TCRs. HLA-matched allogeneic C1R cells derived from the B cells could efficiently present the antigen for the recognization of T cells and were widely used to be the substitute of the autologous DCs to evaluate the function of the T cells.8,10,30,31 The use of minigene-encoded neoantigen libraries for APC loading allowed to quickly narrow the number of neoantigen targets. The function of neoantigen-specific TCRs was tested in vitro and their ability to regress tumors was shown in mice bearing human tumors expressing the targeted mutation. The established strategy could be a primary option for patients from which isolation of PBMC material is not possible in sufficient quantity. Combined with our strategy to screen for neoantigen-specific TCRs in the pool of patient PBMCs, ACT using TCR-T cells can be tailored for each patient based on the availability of PBMCs or TILs.
Materials and methods
Patients
Eight patients with HNSCC who were treated at the University of Chicago Medical Center were enrolled in this study after obtaining written informed consent as we described previously.32 The clinicopathological features of these patients are summarized in SupplementaryTable 1. Fresh tumors from these patients (B1-B8) were collected from November 2016 to April 2017. Blood samples or normal adjacent tissue (pathology reviewed, e.g. uninvolved lymph nodes, muscle, etc.) were collected as normal controls. The study protocol was approved by the Institutional Review Board of the University of Chicago (approval number 8980, 13–0797, and 13–0526) and conducted in compliance with the declaration of Helsinki.
Table 1.
List of peptides expressed by minigenes
| mutated Peptide |
Wild-type peptide |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Minigene | gene | Amino acid substitution | Sequence | Affinity to HLA-A (IC50 nM) | Sequence | Affinity to HLA-A (IC50 nM) | tumor_var (RNA) |
mutation ratio |
HLA Alleles |
| B2-1 | AASDH | S589F | FLNSGGDFL | 21 | FLNSGGDSL | 48 | 4 | 27% | A*02: 01 |
| ABCA2 | S715F | MVIFWVYSV | 3 | MVISWVYSV | 3 | 16 | 24% | A*02: 01 | |
| ACOT8 | M252L | FLVSLDHSM | 25 | FMVSLDHSM | 19 | 27 | 25% | A*02: 01 | |
| ACP2 | F393L | LLLIVLLLTV | 23 | FLLIVLLLTV | 10 | 49 | 26% | A*02: 01 | |
| ADCY9 | S938F | YVFLCPDSSV | 39 | YVSLCPDSSV | 182 | 7 | 19% | A*02: 01 | |
| AKR1C3 | P119S | YLIHSSMSL | 4 | YLIHSPMSL | 6 | 23 | 13% | A*02: 01 | |
| AKT2 | S34F | FLLKSDGFFI | 9 | FLLKSDGSFI | 28 | 46 | 10% | A*02: 01 | |
| ALKBH1 | P354S | VLATDQNFSL | 42 | VLATDQNFPL | 25 | 12 | 18% | A*02: 01 | |
| ALMS1 | R4120W | WIYEQLPEV | 4 | RIYEQLPEV | 5 | 6 | 15% | A*02: 01 | |
| ARID1B | S2118 F | FMALLSNLA | 42 | SMALLSNLA | 341 | 34 | 26% | A*02: 01 | |
| ATG2B | E1609K | FLMKIQLSKV | 6 | FLMEIQLSKV | 4 | 7 | 12% | A*02: 01 | |
| ATP6V0A2 | R755K | KLWALSLAHA | 30 | RLWALSLAHA | 47 | 22 | 20% | A*02: 01 | |
| ATXN10 | L159F | FLIITDFFL | 8 | FLIITDLFL | 25 | 54 | 15% | A*02: 01 | |
| BCAR1 | R255Q | ALAQLQQGV | 47 | ALARLQQGV | 74 | 119 | 28% | A*02: 01 | |
| C11orf74 | P63S | SISSCIPFV | 5 | SIPSCIPFV | 8 | 16 | 30% | A*02: 01 | |
| CDCA2 | P815L | KLMESSSVV | 5 | KPMESSSVV | 7200 | 6 | 20% | A*02: 01 | |
| CEPT1 | Q199R | FMFYCAHWRT | 13 | FMFYCAHWQT | 13 | 13 | 15% | A*02: 01 | |
| CHMP2B | E132K | VLDEIGIKI | 47 | VLDEIGIEI | 17 | 35 | 17% | A*02: 01 | |
| B2-2 | CHST2 | P150L | GMAGVAAPL | 21 | GMAGVAAPP | 10959 | 29 | 32% | A*02: 01 |
| CLMP | S2F | FLLLLLLLV | 9 | SLLLLLLLV | 30 | 1 | 30% | A*02: 01 | |
| COL12A1 | T1055R | YQIGWDRFCV | 16 | YQIGWDTFCV | 6 | 17 | 21% | A*02: 01 | |
| COPZ1 | G97R | ALLENMERL | 21 | ALLENMEGL | 15 | 102 | 14% | A*02: 01 | |
| CRYBG3 | A2862T | SLADTRTTSV | 28 | SLADTRATSV | 49 | 1 | 27% | A*02: 01 | |
| CTNS | P73S | TILELSDEV | 24 | TILELPDEV | 36 | 2 | 25% | A*02: 01 | |
| CYP2R1 | E212K | KLFSENVEL | 8 | ELFSENVEL | 277 | 7 | 23% | A*02: 01 | |
| DLST | P25S | SLGRRSLPGV | 49 | PLGRRSLPGV | 4021 | 71 | 28% | A*02: 01 | |
| DNPH1 | E159K | ALLDRYFKA | 10 | ALLDRYFEA | 5 | 39 | 20% | A*02: 01 | |
| DOHH | E208K | ALFRHKVGYV | 38 | ALFRHEVGYV | 22 | 26 | 33% | A*02: 01 | |
| DPY19L3 | V424I | FILSITVIV | 7 | FVLSITVIV | 10 | 7 | 11% | A*02: 01 | |
| DSP | S2077F | SLQDAVFQGV | 18 | SLQDAVSQGV | 37 | 1349 | 14% | A*02: 01 | |
| ECE1 | R524Q | MQFFNFSWRV | 15 | MRFFNFSWRV | 960 | 39 | 15% | A*02: 01 | |
| EPB41L1 | P115L | FAFTVKFYL | 26 | FAFTVKFYP | 8426 | 21 | 16% | A*02: 01 | |
| FAM171B | P645S | VMTSFSSEL | 28 | VMTPFSSEL | 33 | 1 | 23% | A*02: 01 | |
| FANCG | P243L | GLCPRLVLV | 48 | GLCPRPVLV | 119 | 4 | 34% | A*02: 01 | |
| FARS2 | S368L | VINDILFWL | 12 | VINDISFWL | 19 | 61 | 16% | A*02: 01 | |
| FAT1 | P2085L | LLYYAVVKV | 13 | LPYYAVVKV | 11303 | 52 | 12% | A*02: 01 | |
| FURIN | M434I | GLLDAGAIV | 21 | GLLDAGAMV | 13 | 85 | 19% | A*02: 01 | |
| B2-3 | FZD6 | S238F | IIYYFVCYSI | 28 | IIYYSVCYSI | 47 | 9 | 12% | A*02: 01 |
| GAK | R578K | ILVKAVVMTPV | 24 | ILVRAVVMTPV | 33 | 10 | 35% | A*02: 01 | |
| HSPH1 | S393F | FVTDAVPFPI | 19 | SVTDAVPFPI | 140 | 123 | 20% | A*02: 01 | |
| KLF4 | P195L | SLSGGFVAEL | 31 | SPSGGFVAEL | 16501 | 97 | 20% | A*02: 01 | |
| LTN1 | K1279N | FLNYCSSPL | 6 | FLKYCSSPL | 35 | 14 | 18% | A*02: 01 | |
| MAP4K1 | S108F | FLSELQISYV | 4 | SLSELQISYV | 8 | 2 | 18% | A*02: 01 | |
| MED13 | P1679L | FLEMVQTLPL | 29 | FLEMVQTLPP | 4077 | 23 | 23% | A*02: 01 | |
| MED14 | P1344L | VQFCLTIPL | 19 | VQFCLTIPP | 7241 | 25 | 39% | A*02: 01 | |
| MFSD10 | P199L | LLLEMAPWFA | 6 | LPLEMAPWFA | 5755 | 85 | 21% | A*02: 01 | |
| MLXIP | P270L | LLLDTDMLM | 34 | PLLDTDMLM | 3213 | 5 | 19% | A*02: 01 | |
| MRGBP | I111N | VLPEENIQEV | 44 | VLPEEIIQEV | 24 | 72 | 18% | A*02: 01 | |
| MTOR | P896S | ALDSYKHKV | 28 | ALDPYKHKV | 33 | 4 | 18% | A*02: 01 | |
| MYCBP2 | S4018F | YCFELLFMV | 8 | YCFELLSMV | 34 | 24 | 13% | A*02: 01 | |
| MYCBPAP | E606K | VLQKLLMGV | 20 | VLQELLMGV | 8 | 1 | 15% | A*02: 01 | |
| MYO1B | M14I | SLLDNIIGV | 3 | SLLDNMIGV | 3 | 66 | 22% | A*02: 01 | |
| NCOA6 | P657S | LMSQGQMMV | 27 | LMPQGQMMV | 43 | 2 | 21% | A*02: 01 | |
| NRBP2 | F365L | SLMELDKFL | 13 | SFMELDKFL | 6739 | 35 | 15% | A*02: 01 | |
| NUMB | P40L | FLVKYLGHV | 10 | FPVKYLGHV | 6584 | 15 | 15% | A*02: 01 | |
| NVL | S19F | HMNSSLLFL | 28 | HMNSSLLSL | 57 | 8 | 27% | A*02: 01 | |
| B8-1 | AMPD2 | E494K | KLHLFLEHV | 39 | ELHLFLEHV | 1271 | 5 | 25% | A*02: 01 |
| ATN1 | P356S | TLAPSSHSL | 36 | TLAPSPHSL | 50 | 16 | 33% | A*02: 01 | |
| BEND3 | P583L | LLVHLFLEL | 28 | LLVHLFPEL | 11 | 2 | 16% | A*02: 01 | |
| BRPF1 | F292Y | ILYCDMCNL | 21 | ILFCDMCNL | 16 | 8 | 22% | A*02: 01 | |
| C14orf93 | L303F | FVLSKLVHNV | 27 | LVLSKLVHNV | 137 | 8 | 28% | A*02: 01 | |
| C17orf58 | P8L | TLDGFFFRV | 4 | TPDGFFFRV | 8749 | 13 | 21% | A*02: 01 | |
| CAD | S1928Y | SLVGQHILYV | 14 | SLVGQHILSV | 28 | 9 | 19% | A*02: 01 | |
| CCDC28A | P19S | SLGAWRLYLL | 23 | PLGAWRLYLL | 1285 | 4 | 48% | A*02: 01 | |
| CDK5R1 | S96F | FTFAQPPPA | 33 | STFAQPPPA | 551 | 19 | 17% | A*02: 01 | |
| CHST15 | F468C | YLLDWLSVC | 29 | YLLDWLSVF | 23 | 9 | 37% | A*02: 01 | |
| DALRD3 | P427S | GLYSTFPPV | 3 | GLYPTFPPV | 3 | 17 | 24% | A*02: 01 | |
| DDX19B | S129F | LLFSATFEDFV | 42 | SVWKFAQKV | 130 | 19 | 18% | A*02: 01 | |
| ELMSAN1 | S625F | FIAPPVYFNI | 32 | FIAPPVYSNI | 178 | 46 | 31% | A*02: 01 | |
| ESRP2 | P655L | ALASALTSV | 9 | ALASAPTSV | 22 | 36 | 14% | A*02: 01 | |
| FADS2 | S429F | ALLDIIRFL | 8 | ALLDIIRSL | 13 | 1 | 18% | A*02: 01 | |
| FKBP15 | H413Y | SLYPAHPAL | 20 | SLHPAHPAL | 107 | 16 | 20% | A*02: 01 | |
| FLII | A42V | YLPEELVAL | 7 | YLPEELAAL | 8 | 83 | 36% | A*02: 01 | |
| FMO4 | G519R | YLKAWRAPV | 24 | YLKAWGAPV | 12 | 4 | 33% | A*02: 01 | |
| IQGAP3 | P560S | GLDDVSLSV | 6 | GLDDVSLPV | 4 | 10 | 23% | A*02: 01 | |
| ITGB1 | S277F | FTDAGFHFA | 39 | STDAGFHFA | 726 | 260 | 18% | A*02: 01 | |
| B8-2 | RAPGEF2 | S806L | LMFAIISGL | 9 | SMFAIISGL | 10 | 3 | 25% | A*02: 01 |
| RBBP5 | I313F | IIASFSSGV | 37 | FPSPILKV | 11180 | 15 | 17% | A*02: 01 | |
| SEC23IP | P436L | ILDGEMPQV | 9 | IPDGEMPQV | 13477 | 19 | 22% | A*02: 01 | |
| SLC25A30 | P274S | GSWNIIFFV | 20 | GPWNIIFFV | 344 | 11 | 16% | A*02: 01 | |
| SMTN | R665W | KLIWAALREL | 41 | KLIRAALREL | 143 | 23 | 21% | A*02: 01 | |
| SNX19 | L387F | IMFMTPGSFL | 51 | IMLMTPGSFL | 71 | 19 | 29% | A*02: 01 | |
| STRN3 | P406S | ALAFHSVEPV | 19 | ALAFHPVEPV | 23 | 20 | 20% | A*02: 01 | |
| TBL2 | R162C | WLANGDTLCV | 31 | WLANGDTLRV | 33 | 16 | 19% | A*02: 01 | |
| TMCO1 | I65V | FADTLLIVFV | 48 | TLLIVFISV | 24 | 151 | 20% | A*02: 01 | |
| TMEM125 | V126I | VLLSGLVLLI | 24 | VLLSGLVLLV | 10 | 1 | 14% | A*02: 01 | |
| TRAPPC9 | E639K | SLPAKSGLYPV | 44 | SLPAESGLYPV | 41 | 3 | 30% | A*02: 01 | |
| TRIAP1 | P58 L | ILIEGLEFM | 11 | IPIEGLEFM | 11912 | 18 | 13% | A*02: 01 | |
| TRRAP | S702 F | RLPEMGFNV | 16 | RLPEMGSNV | 161 | 1 | 28% | A*02: 01 | |
| UBE3C | P991 L | HLVIKVFWRV | 44 | PVIKVFWRV | 1236 | 20 | 11% | A*02: 01 | |
| URGCP | P310L | GLVEISWFFL | 20 | GLVEISWFFP | 1333 | 11 | 14% | A*02: 01 | |
| USP15 | P800S | KLDLWSLPSV | 5 | KLDLWSLPPV | 4 | 24 | 14% | A*02: 01 | |
| WSB2 | S194L | VLLGHLQWV | 5 | VLSGHLQWV | 10 | 20 | 35% | A*02: 01 | |
| ZNF33A | S288F | FTLSKPHGV | 16 | STLSKPHGV | 183 | 5 | 11% | A*02: 01 | |
| B8-3 | KIAA1429 | D1358E | FLAEHEYGL | 3 | FLAEHDYGL | 3 | 16 | 25% | A*02: 01 |
| KIAA1958 | P400S | KLNKFSVFNI | 37 | KLNKFPVFNI | 39 | 1 | 21% | A*02: 01 | |
| KIF23 | S334F | ITISQLFLV | 11 | ITISQLSLV | 106 | 32 | 27% | A*02: 01 | |
| LIG3 | G76R | YLVFLPRLHV | 35 | YLVFLPGLHV | 29 | 11 | 15% | A*02: 01 | |
| LNPEP | S349F | VQDEFFESV | 21 | VQDEFSESV | 31 | 5 | 20% | A*02: 01 | |
| LPHN3 | H1049Y | KMFHYTAIL | 8 | KMFHHTAIL | 12 | 1 | 27% | A*02: 01 | |
| MAPK13 | G137V | YLVYQMLKV | 9 | YLVYQMLKG | 5484 | 50 | 28% | A*02: 01 | |
| MROH7 | S214F | SLDLDFNPLL | 48 | SLDLDSNPLL | 81 | 1 | 23% | A*02: 01 | |
| MTHFD1L | V418I | VLIAGITPT | 31 | VLVAGITPT | 139 | 31 | 67% | A*02: 01 | |
| MYADM | S68F | FMFTWCFCF | 15 | SMFTWCFCF | 135 | 22 | 15% | A*02: 01 | |
| NID1 | G495E | GIIEWMFAV | 3 | GIIGWMFAV | 4 | 17 | 37% | A*02: 01 | |
| NLRP1 | P614L | ILQEHLIPL | 5 | ILQEHPIPL | 9 | 3 | 10% | A*02: 01 | |
| NPHP4 | P159L | RLYHGTLRAL | 49 | RLYHGTPRAL | 101 | 5 | 18% | A*02: 01 | |
| NUP214 | P1463S | SLPTSFPTL | 41 | PLPTSFPTL | 4229 | 53 | 24% | A*02: 01 | |
| PADI1 | S118F | YLTGVDIFL | 8 | YLTGVDISL | 11 | 1 | 16% | A*02: 01 | |
| PAQR7 | S119F | YLFFSALAHL | 8 | YLSFSALAHL | 15 | 15 | 17% | A*02: 01 | |
| PKP2 | F491 L | TLTENIIIPL | 25 | TLTENIIIPF | 1221 | 5 | 33% | A*02: 01 | |
| PLEKHG4 | S635F | ALPQAFPTV | 19 | ALPQASPTV | 29 | 2 | 25% | A*02: 01 | |
| PTK7 | D463N | MLISENSRFEV | 31 | MLISEDSRFEV | 44 | 103 | 29% | A*02: 01 | |
Whole-exome sequencing and transcriptome analysis
The DNA/RNA extraction and Whole-exome sequencing and transcriptome analysis were performed as described previously.32 Briefly, genomic DNAs and total RNAs of the tumor and adjacent normal tissue were extracted using the AllPrep DNA/RNA mini kit (Qiagen, Catalog number 80207). Genomic DNAs of peripheral blood were extracted using QIAamp DNA Blood Midi Kit (Qiagen, Catalog number 51183).
Whole-exome libraries were built up as previously described33 and sequenced by 100-bp paired-end reads on HiSeq2500 Sequencer (Illumina, San Diego, CA, USA). Somatic variants (single nucleotide variations (SNVs) and indels) were called using the following parameters, (i) base quality ≥ 15, (ii) sequence depth ≥ 10, (iii) variant depth ≥ 4, (iv) variant frequency in tumor ≥10%, (v) variant frequency in normal < 2%, and (vi) Fisher P-value < 0.05.34 SNVs and indels were annotated based on RefGene using ANNOVAR.35 Transcriptome analysis was performed with TruSeq RNA Library Prep Kit v2 (Illumina, Catalog number 15026495) on HiSeq2500 Sequencer (Illumina, San Diego, CA, USA) according to the manufacturer’s instruction.
Identification of potential neoantigens
HLA class I genotypes of these patients were determined by the OptiType algorithm36 using whole-exome data of normal samples. The binding affinities of all possible 8- to 11-mer peptides harboring each amino acid substitution to HLA class I molecules was examined using NetMHC v4.0 software37–39 and the peptides of which predicted binding affinity to HLA-A lower than 500 nM were filtered out. Besides, non-synonymously mutant peptides with a defined level of gene expressions (at least 10 reads among ~20,000,000 sequence reads according to the transcriptome data) in tumor cells were chosen. The neoantigen candidate peptides were synthesized by Innopep Inc. (San Diego, CA, USA).
Cell line
HLA-homozygous immortalized cell line C1R A02:01 was created based on human B-cell lymphoblastoid cell line C1R40 (originally lacking HLA-A and HLA-B) and was kindly provided by OncoTherapy Science, Inc (Kawasaki, Japan). C1R A02:01 cells were maintained in RPMI-1640 medium (Hyclone) supplied with 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin (Hyclone). The expression of HLA-A02:01 was routinely examined with an HLA-A02 specific antibody (MBL International) by FACS.
Minigene construction
Tandem minigenes were constructed for each non-synonymous variant identified and consisted of the mutant amino acid flanked by 12 amino acids of the wild-type protein sequence. Eighteen or twenty minigenes were strung together and separated by proteasomal cleavage site AAY and linked to the GFP reporter gene by a 2A self-cleaving peptide at the C-terminus, which allows monitoring transfection efficiency. These minigene constructs were codon-optimized and cloned into a pMS-T vector (GeneArt, Thermo Fisher).
Preparation of peptide-MHC I-tetramer complex
The APC conjugated peptide-specific tetramer was generated with QuickSwitchTM Quant Tetramer Kit (MBL International, Catalog number TB-7300-K2) according to the manufacturer’s instruction. Briefly, 50 µL of the Tetramer at 50 µg/mL was mixed with 1 µL of 10 mM peptide solution and 1 µL of the proprietary Peptide Exchange Factor and incubated for 4 hours at room temperature in dark. The exchanging efficiency was evaluated by FACS as described in the manual and qualified peptide-MHC I-tetramers were stored at 4°C until use.
Isolation and expansion of TILs
TILs isolation and culture were performed following a modified traditional TILs culture protocol as described in our previous paper.32 Briefly, fresh tumors were chopped into small chunks less than 1 × 1 mm, then digested with collagenase mixture in 37°C shaking incubator for 1–4 hours until the single-cell suspension was obtained. The cells were cultured with 1000 IU/mL IL-2 (R&D Systems, Catalog number 202-IL-050) in a RetroNectin-coated flask (Takara Bio, Catalog number T100A) for 2 weeks. CD8+ subgroups of expanded TILs were isolated with Dynabeads CD8 positive isolation kit (Thermo Fisher Scientific, Catalog numbers 11333D).
Induction of neoantigen-specific cytotoxic T lymphocytes (CTLs) with expanded TILs and APC cells pulsed with in vitro transcribed RNA or peptides
Induction of neoantigen-specific T cells was performed by the protocol we previously developed 10. CMV pp65 antigen peptide (restricted to HLA-A02:01) was used as a positive control. C1R A02:01 cells were used as APC cells in replacement of autologous DCs. mRNA of neoantigens were transduced into C1R A02:01 cells by electroporation. Briefly, messenger RNA coding tandem neoantigen minigene was produced by in vitro transcription (IVT) using mMACHINE T7 kit (Ambion, AM1344) and Poly(A) Tailing Kit (Ambion, AM1350) as instructed by the manufacturer. Later, RNA was introduced into C1R A02:01 cells via electroporation. C1R A02:01 cells were resuspended in Opti-MEM (Life Technologies) at 8 × 106 cells/ml. 10 µg of IVT RNA was aliquoted into the bottom of an electroporation cuvette with a 4-mm gap, and 250 µl of C1R A02:01 cells were added. C1R A02:01 cells were electroporated at 350 V and 250µF for one pulse with Gene Pulser Xcell electroporation system (Bio-Rad). Cells were gently resuspended into the medium and transferred to the cell incubator. Transfection efficiency was routinely assessed with GFP expression by FACS 16-hour post-transfection. Transfected C1R A02:01 cells were used as APC and co-cultured with TILs for CTL induction. On the first day of CTLs induction, 2 × 105 pulsed or non-pulsed APC cells were co-cultured with 5 × 105 T cells. On the tenth day after co-culture, neoantigen-specific T cells were stained with peptide-HLA tetramers for respective neoantigen peptides and analyzed by flow cytometry. CD8+/Tetramer+ T cells were sorted out and used for the following TCR sequencing analysis.
TCR sequencing analysis of sorted neoantigen-specific T cells
The libraries for TCR sequencing were prepared by following the protocol as previously described.38,41,42 Briefly, total RNAs of sorted CD8+/Tetramer+ T cells were extracted by PicoPure RNA Isolation kit (Life Technologies, Catalog number KIT0204). The quality of RNA was evaluated with TapeStation 2200 (Agilent Technologies). 5ʹ rapid amplification of cDNA end adapter was added during the cDNA synthesis using SMART cDNA library construction kit (Clontech Laboratories, Inc., Japan). TCRA and TCRB sequences were amplified using a forward primer for the SMART adapter and a reverse primer specific to the TCR constant region. Then Illumina sequence adapter with barcode sequences was added using the Nextera XT Index kit (Illumina, Catalog numbers FC-131-2001, FC-131-2002, FC-131-2003). The final prepared libraries were sequenced by 300-bp paired-end reads on the Illumina MiSeq platform using missed Reagent v3 600-cycles kit (Illumina, Catalog number MS-102-3001). Sequencing data analysis was performed using Tcrip as described previously.41 Briefly, sequencing reads were mapped to the TCR reference sequences obtained from IMGT/GENE-DB (http://www.imgt.org) using Bowtie2 aligner (version 2.1.0), and after decomposition of sequencing reads into V, (D) and J segments, CDR3 were searched.
Construction of TCR-engineered T cells
TCR sequences were codon-optimized and cloned into the lentiviral vector pLVX (Clontech). Mouse TCR constant regions were used in replacement of human TCR constant regions to reduce endogenous TCR mismatch and improve TCR surface expression.43 Lentiviruses were generated with HEK293T cells, and PBMCs from healthy donors were transduced as described previously.44 TCR-engineered T cells were maintained in GT551-H3 medium (Takara) supplied with 5% human serum (GemCell), 1% penicillin/streptomycin (Hyclone), and 400 IU/mL IL-2. The expression of TCR gene was routinely examined with a mouse TCRβ specific antibody (BD Biosciences) by FACS.
Enzyme-linked immunospot (ELISPOT) and enzyme-linked immunosorbent assay (ELISA) assay
IFNγ secretion of TILs was measured by ELISPOT using Human IFNγ ELISpotPRO kit (MABTECH, Catalog number 3420–2APW-10) following the manufacturer’s instruction. Briefly, 5 × 104 resting TILs were co-cultured with 2 × 104 peptide-pulsed C1R A02:01 cells at 37°C for 20 hours in a 96-well plate. Spots were captured and analyzed by an automated ELISPOT reader, ImmunoSPOT S4 (Cellular Technology Ltd, Shaker Heights, OH), and analyzed with the ImmunoSpot Professional Software package, Version 5.1 (Cellular Technology Ltd).
The amount of secreted IFNγ, IL-2, and TNFα in the supernatant of T cells were quantified with ELISA set (BD Biosciences, Catalog number 555142, 555190, 555212). Briefly, 5 × 104 resting KIAA1429D1358E TCR-T cells were co-cultured with 2 × 104 peptide-pulsed C1R A02:01 cells at 37°C for 20 hours in a 96-well plate. The supernatant was collected, and the concentration of each protein was measured according to the manufacturer’s instruction.
Cytotoxic assay
CytoTox 96 Non-Radioactive Cytotoxicity Assay kit (Promega, Catalog number G1780) was used to determine the cytotoxic activities of T cells, following the manufacturer’s instruction. Briefly, effector cells (TCR-engineered T cells) and target cells (C1R A02:01 cells) were co-cultured at different ratios (2:1, 5:1, 10:1, and 20:1). After 4-hour incubation, the supernatant was collected, and the amount of lactate dehydrogenase (LDH) was measured according to the manufacturer’s instruction. The maximum LDH release of target cells was measured by lysing cells with 0.9% Triton X-100. The spontaneous LDH release of effector and target cells was measured by separate incubation of the respective population. The percentage of cytotoxic activity was calculated according to the following formula: % Cytotoxicity = [(Experimental − Effector Spontaneous − Target Spontaneous)/(Target Maximum − Target Spontaneous)] × 100. Experiments were performed in quadruplicate.
Generation of the neoantigen-expressing tumor cell lines
The minigenes containing the KIAA1429D1358E (sequence: FLAEHEYGL) or MAGOHBG17A (sequence: RYYVGHKAKF) mutation8 were codon-optimized, synthesized and cloned into a lentiviral vector pLVX-GFP which contains a GFP reporter gene and a puromycin resistance gene. Lentiviral supernatants were generated by using HEK293T cells, then, human tumor cell line SW480 cells (HLA A*02:01, HLA A*24:02) was infected with the lentivirus to generate the neoantigen-stably expressing cell lines (SW480- KIAA1429D1358E, SW480- MAGOHBG17A). Cells were selected with 1 μg/mL puromycin for 3 days. Transfection efficiency was routinely assessed with GFP expression by FACS.
Treatment of established tumors in NCG mouse model
The animal experiment proceeded under the approval of the ethics committee of Shenzhen people’s hospital, China (approval number LL-KY-2020033). NCG mice (NOD/ShiltJGpt-Prkdcem26Cd52Il2rgem26Cd22/Gpt), which are created by sequential CRISPR/Cas9 knockout of the Prkdc and Il2rg loci in the NOD/Nju mouse and are lack of functional/mature T, B, and NK cells, were used in this study. Five weeks-old NCG mice (GemPharmatech Co., Ltd., China) were inoculated subcutaneously with 2 × 106 SW480-KIAA1429D1358E tumor cells or SW480-MAGOHBG17A tumor cells. Mice were randomly divided into three groups, TCR-engineered T cells treated mice, control T cells treated mice, and tumor control group (n = 7 or n = 5 for each group). Control T cells were derived from the same donor’s PBMCs as TCR-T cells and were maintained under the same culture condition. T cells were cultured in vitro for 14 days and then used for in vivo infusion. T cell treatments were given on day 8 following tumor inoculation, which consisted of an intravenous injection of 3 × 107 T cells for one injection. Mice received an intraperitoneal injection of adjuvant IL-2 (Kingsley Pharmaceutical Co., Ltd., China) 400,000 IU daily 6 times on day 0–2, day 8–10 after T cell injection. Tumor size was determined by caliper measurement.
Statistical analysis
The student’s t-test was performed for the functional assays of TCR-engineered T cells. Statistical analyses were done using GraphPad Prism version 8.0 (GraphPad Software, La Jolla, CA). P-value <0.05 was considered to be statistically significant.
Results
Whole-exome sequencing and neoantigen prediction
We previously reported that whole-exome sequencing of genomic DNA from freshly resected tumor tissues and corresponding normal cells of 8 HNSCC patients identified a total of 7207 non-synonymous mutations (11–4290 non-synonymous mutations and that three of the eight patients, B2, B5, and B8 showed extremely high tumor mutation burden).32 In the present study, we additionally analyzed the HLA A alleles of each patient, as the information is summarized in Supplementary Table 1.
Using WES and RNA transcriptome data, we predicted neoantigens for these patients, as shown in Supplementary Table 2. Considering the present availability of HLA tetramers, we had chosen patients B2 and B8 who have an HLA A02:01 allele for further analysis. We predicted 251 and 273 neoantigen candidates for HLA A02:01 in the patient B2 and B8, respectively (binding affinity less than 500 nM), or 125 and 75 neoantigens when the binding affinity was less than 50 nM. To screen the neoantigen-specific TCRs, we designed 6 minigenes, each of which is designed to express 18 to 20 neoantigen peptides, for patient B2 and B8, respectively (3 minigenes for each patient), as shown in Table 1.
After in vitro transcription, mRNAs of corresponding minigenes were generated and used to pulse the antigen-presenting C1R A02:01 cells by electroporation. The transfection efficiency was calculated as between 50% and 90%, as shown in Supplementary Figure 1. Then, pulsed C1R A02:01 cells were co-cultured with the expanded CD8+ T cells isolated from the expanded TILs of patient B2 and B8, and the IFNγ secretion of CD8+ T cells was measured by ELISPOT analysis. We found that most of the minigenes could activate T cells to some extent. Among them, the B8-3 minigene showed stronger stimulation as shown in Figure 1. Based on the expression levels of the mutated peptides, we finally chose 10 peptides from the B8-3 minigene and synthesized each of them, which are summarized in Table 2.
Figure 1.
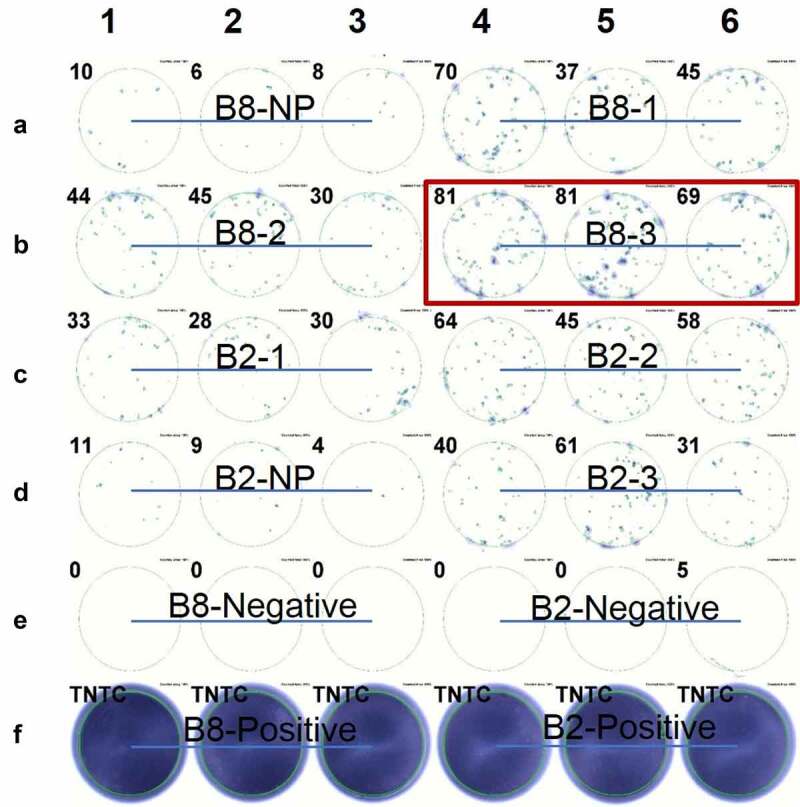
ELISPOT assay showed the IFNγ secretion of expanded TILs induced by CIR A02:01 cells loaded with in vitro transcribed RNA. TILs derived from patient B2 or B8 were co-cultured with neoantigens-pulsed CIR A02:01 for ten days, and IFNγ secretions of which were detected by ELISPOT. Non-pulsed (NP) control result of B8 was shown as A1-A3; the result of minigene B8-1 pulsed C1R A02:01 cells as A4-A6; B8-2 as B1-B3; B8-3 as B4-B6; B2-1 as C1-C3; B2-2 as C4-C6; non-pulsed of B2 as D1-D3; B2-3 as D4-D6; positive controls of B8 and B2, which were treated with PMA and Ionomycin, were presented as F1-F3 and F4-F6, respectively. Experiments were conducted in triplicate. TNTC represents too numerous to count
Table 2.
List of peptides checked for CTL induction
| mutated Peptide |
Wild-type peptide |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Minigene | gene | Amino acid substitution |
Sequence | Affinity to HLA-A (IC50 nM) | Sequence | Affinity to HLA-A (IC50 nM) | tumor_var (RNA) |
mutation ratio |
HLA Alleles |
| B8-3 | PTK7 | D463N | MLISENSRFEV | 31 | MLISEDSRFEV | 44 | 103 | 29% | A*02: 01 |
| NUP214 | P1463S | SLPTSFPTL | 41 | PLPTSFPTL | 4229 | 53 | 24% | A*02: 01 | |
| MAPK13 | G137V | YLVYQMLKV | 9 | YLVYQMLKG | 5484 | 50 | 28% | A*02: 01 | |
| KIF23 | S334 F | ITISQLFLV | 11 | ITISQLSLV | 106 | 32 | 27% | A*02: 01 | |
| MTHFD1L | V418I | VLIAGITPT | 31 | VLVAGITPT | 139 | 31 | 67% | A*02: 01 | |
| MYADM | S68F | FMFTWCFCF | 15 | SMFTWCFCF | 135 | 22 | 15% | A*02: 01 | |
| NID1 | G495E | GIIEWMFAV | 3 | GIIGWMFAV | 4 | 17 | 37% | A*02: 01 | |
| KIAA1429 | D1358E | FLAEHEYGL | 3 | FLAEHDYGL | 3 | 16 | 25% | A*02: 01 | |
| PAQR7 | S119 F | YLFFSALAHL | 8 | YLSFSALAHL | 15 | 15 | 17% | A*02: 01 | |
| LIG3 | G76R | YLVFLPRLHV | 35 | YLVFLPGLHV | 29 | 11 | 15% | A*02: 01 | |
Isolation of neoantigen-specific T cells from TILs and TCR sequencing of sorted neoantigen-reactive T cells
Ten peptides with higher RNA expression levels (TOP10 of tumor_var (RNA), shown in Table 2) were further tested for CTL induction. After 11 days of co-culturing TILs with C1R A02:01 cells pulsed or not pulsed with each of these 10 mutated peptides, neoantigen-reactive CD8+ T cells were sorted with each of respective peptide-HLA-tetramer complexes by flow cytometry. CD8+/tetramer+ T cells for two neoantigens, KIAA1429D1358E and MAPK13G137V were recognized by this method: the proportions of positive cells for KIAA1429D1358E and MAPK13G137V were 0.14% (412 cells) and 0.014% (112 cells), respectively (Figure 2).
Figure 2.
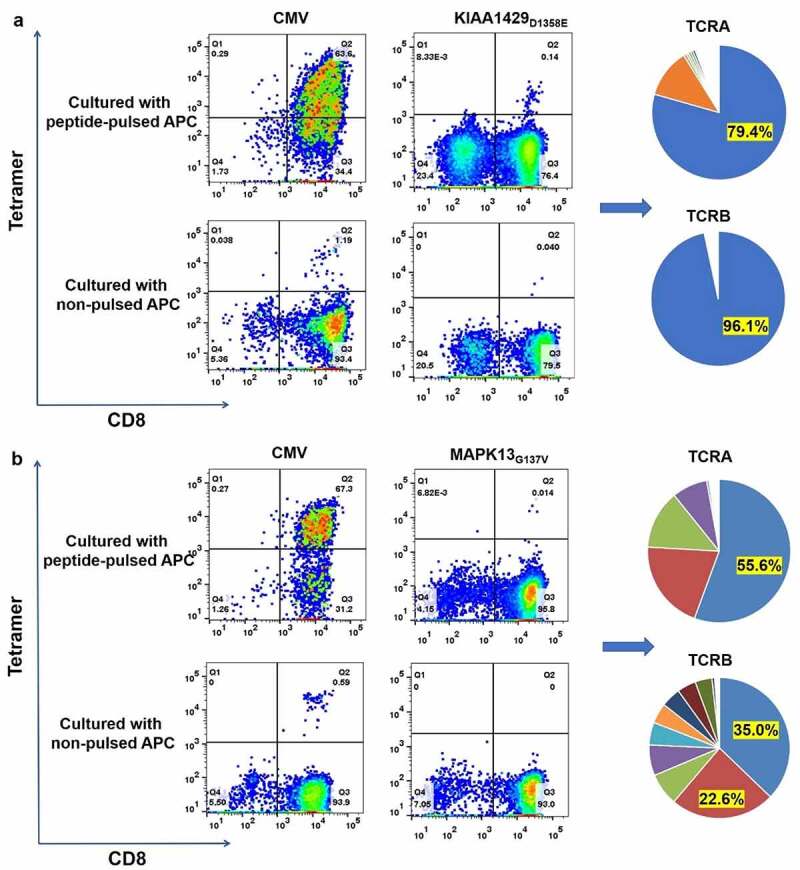
Induction of neoantigen-specific CTLs and identification of TCRA and TCRB sequences of sorted CD8+/tetramer+ T cells. (a) Peptide-HLA Tetramer assay for TILs co-cultured with C1R A02:01 pulsed with or without KIAA1429D1358E peptide (left); the pie-chart showed the frequencies of TCRA, TCRB and CDR3 sequences of sorted CD8+/tetramer+ T cells (right). Antigen peptide of CMV pp65 for HLA-A02:01 was used as a positive control. (b) Peptide-HLA Tetramer assay for TILs co-cultured with C1R A02:01 pulsed with or without MAPK13G137V peptide (left); the pie-chart showed the frequencies of TCRA, TCRB and CDR3 sequences of sorted CD8+/tetramer+ T cells (right). Antigen peptide of CMV pp65 for HLA-A02:01 was used as a positive control
Subsequently, we performed TCRA and TCRB sequencing of sorted CD8+/tetramer+ T cells for KIAA1429D1358E and MAPK13G137V using a method reported previously.45 The data are shown in Figure 2. A set of single dominant TCRA (79.4%) and TCRB (96.1%) sequences were detected for KIAA1429D1358E. On the other hand, multiple TCRA and TCRB clonotypes were observed for MAPK13G137V. The detailed information about TCRA/B and CDR3 sequences were provided in Supplementary Table 3 and 4. Besides, the dominant TCRA and TCRB sequences for KIAA1429D1358E were also found in both expanded TILs and original primary tumors (data not shown), while none of TCRA or TCRB clones for MAPK13G137V were found in either expanded TILs or the original tumor microenvironment of patient B8.
KIAA1429D1358E-TCR-engineered T cells recognized the KIAA1429D1358E peptide
Using the dominant TCRA/TCRB pair sequences for the KIAA1429D1358E mutant peptide, we generated the KIAA1429D1358E-TCR-engineered T cells. We constructed the vector designed to express TCRA/TCRB including mouse TCR constant regions and introduced it into PBMC isolated from a human healthy donor (since endogenous human TCRs are expressed in T cells, the replacement of human constant regions with mouse constant regions makes the efficiency to produce a pair of introduced TCRA and TCRB to be high). FACS analysis with mouse TCRB antibody showed that the proportion of CD8+ T cells expressing neoantigen-specific engineered TCR was 37.8%, as shown in Figure 3a. The peptide-tetramer staining assay showed that exogenous-TCR expressing cells recognized the mutated KIAA1429D1358E peptide but not the wild-type KIAA1429 peptide (Figure 3b).
Figure 3.
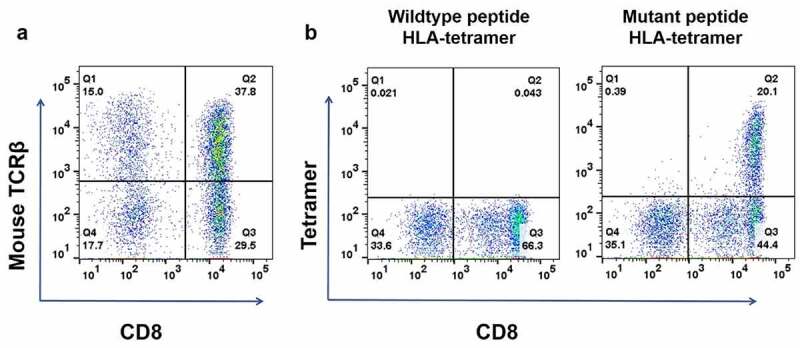
Construction of KIAA1429D1358E- TCR-engineered T cells. (a) The proportion of T cells expressing KIAA1429D1358E specific TCR were stained with human CD8 antibody and an antibody against the mouse constant region of TCRβ. (b) KIAA1429D1358E- TCR-engineered T cells were stained with an HLA-Tetramer loaded with the corresponding wild-type or mutated peptide
We then performed functional analysis of the TCR-engineered T cells using T cells co-cultured with C1R A02:01 cells as APCs. IFNγ ELISPOT assay showed mutated peptide-specific and peptide-dose-dependent activities of KIAA1429D1358E-TCR-engineered T cells when they were co-cultured with mutated peptide-pulsed C1R A02:01 cells, while no IFNγ secretion was observed in C1R A02:01 cells loaded with the corresponding wild-type peptide (Figure 4a).
Figure 4.
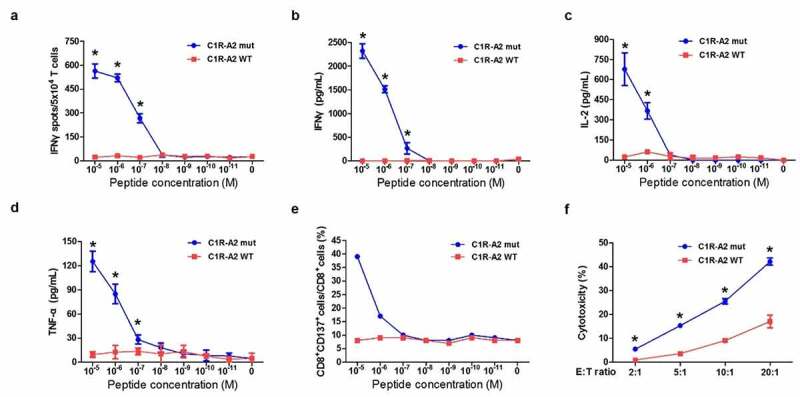
Functional analysis of KIAA1429D1358E- TCR-engineered T cells. (a) IFNγ ELISPOT assay of KIAA1429D1358E- TCR-engineered T cells stimulated by C1R A02:01 cells pulsed with different concentrations of mutated or wild-type peptide. (b) IFNγ ELISA assay of KIAA1429D1358E- TCR-engineered T cells stimulated by C1R A02:01 cells loaded with different concentrations of the mutated or wild-type peptide. (c) IL-2 ELISA assay of KIAA1429D1358E- TCR-engineered T cells stimulated by C1R A02:01 cells loaded with different concentrations of the mutated or wild-type peptide. (d) TNF-α ELISA assay of KIAA1429D1358E- TCR-engineered T cells stimulated by C1R A02:01 cells loaded with different concentrations of the mutated or wild-type peptide. (e) CD137 staining of KIAA1429D1358E- TCR-engineered T cells stimulated by C1R A02:01 cells loaded with different concentrations of the mutated or wild-type peptide. (f) Cytotoxic activity of KIAA1429D1358E- TCR-engineered T cells under different effector cell/target cell ratios. Four different ratios (2:1, 5:1, 10:1 and 20:1) were examined. The asterisks indicate the statistically significant difference (p < .05) between two groups
The levels of several cytokines (IFNγ, TNFα, and IL-2) were also quantified with an ELISA assay and were comparable with the IFNγ ELISPOT results (Figure 4b-d). In consist with cytokine secretion, CD137 upregulation was observed in KIAA1429D1358E-TCR-engineered T cells upon the co-culture with C1R A02:01 cells pulsed with the mutated peptide (Figure 4e). To further validate whether these TCR-engineered T cells have cytotoxic activities, a cell-mediated cytotoxic assay was performed and the lactate dehydrogenase, which would be released from damaged target cells, was measured. The data showed that significant cytotoxic activities were detected on KIAA1429D1358E-TCR-engineered T cells against mutant-peptide-pulsed C1R A02:01 cells in the effector/target cell ratio-dependent manner (Figure 4f). Modest cytotoxicity against wildtype-peptide-loaded C1R A02:01 cells was detected when the effector/target cell ratios were very high (20:1).
Both TCRs screened from patient’s PBMCs and TILs could effectively recognize the corresponding mutated peptides and caused regression of tumors in murine models
To examine the in vivo activity of neoantigen specific-TCR obtained by different approaches, NCG mice, in which human tumors carrying either KIAA1429D1358E or MAGOHBG17A mutation were subcutaneously established, were treated with respective TCR-engineered T cells (TCR-T group, n = 7, and n = 5, respectively) or cytokine-induced nonspecific T cells (control T cell group, n = 7, and n = 5), or were remained to be untreated (untreated group, n = 7, and n = 5) as the control groups. The treatment schedule is shown in Figure 5a. The KIAA1429D1358E carrying minigene-GFP-puromycin resistance gene or MAGOHBG17A carrying minigene-GFP-puromycin resistance gene was transduced into human SW480 cells by lentivirus (transduction efficiencies were 95.8% and 98.1% after puromycin selection, as shown in Supplementary Figure 2a and 2b respectively). After the puromycin selection, the percentage of GFP-positive SW480 cells reached almost 100% and were used to form tumors in NCG mice.
Figure 5.
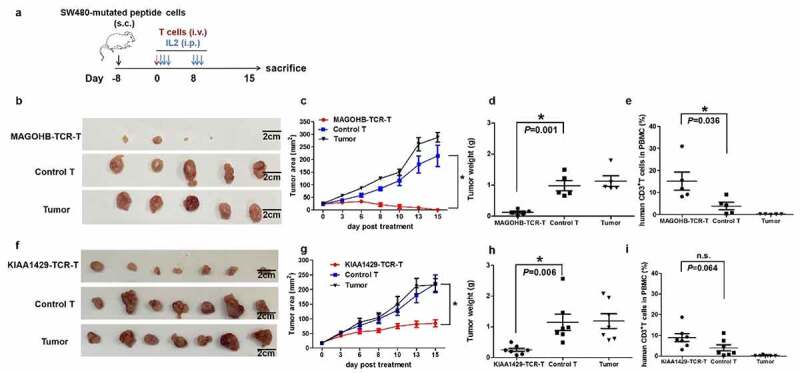
Evaluation of the in vivo activities of MAGOHBG17A- TCR-engineered T cells and KIAA1429D1358E- TCR-engineered T cells. (a) NCG mice were burdened with subcutaneous tumors carrying MAGOHBG17A mutation or KIAA1429D1358E mutation for eight days, and treated with intravenous injections of corresponding TCR-engineered T cells, respectively. 400,000 IU systemic IL-2 was given daily by intraperitoneal injection for 3 days. For mice treated with MAGOHBG17A- TCR-engineered T cells: (b) tumor sizes, (c) tumor area of tumor growth over time, (d) tumor weights and (e) proportions of human CD3+ T cell in the peripheral blood at the end of this experiment were recorded. For mice treated with KIAA1429D1358E- TCR-engineered T cells: (f) tumor sizes, (g) tumor area of tumor growth over time, (h) tumor weights and (i) proportions of human CD3+ T cell in the peripheral blood at the end of this experiment were recorded
The same dose of MAGOHBG17A-TCR-engineered T cells or KIAA1429D1358E-TCR-engineered T cells (3 × 107 T cells per mouse) was administered intravenously once on day 8 after the tumor-cell inoculation to treat tumors in mouse models. At the beginning of the MAGOHBG17A-TCR-engineered T cells treatment, the tumor sizes among the three groups were similar (22.96 ± 4.40 mm2 for the TCR-T group, 23.60 ± 4.23 mm2 for the control T cell group, and 25.70 ± 6.03 mm2 for the untreated group). Administration of MAGOHBG17A-TCR-engineered T cells significantly regressed tumor growth after 3 days of the treatment and kept tumors progression-free including a tumor in one mouse which showed complete eradication at day 8. Tumors in the remaining four mice also showed significant regression. On the other hand, no regression was observed in the control T cell group or the untreated group. The dissection of tumor tissues confirmed the significant differences in the tumor size among the three groups (Figure 5b-d). Furthermore, we checked the number of exogenous T cells in peripheral blood of mice by FACS, and found an increase in the number of MAGOHBG17A-TCR-engineered T cells compared to those in the control T cell group, indicating proliferation of MAGOHBG17A-TCR-engineered T cells possibly after the exposure to neoantigens (Figure 5e).
An intravenous injection of KIAA1429D1358E-TCR-engineered T cells or nonspecific cytokine-induced T cells was also administered on day 8 after the tumor-cell inoculation. At the starting point of the KIAA1429D1358E-TCR-engineered T cells treatment, there were no significant differences in the tumor sizes among the three groups (18.19 ± 13.74 mm2 for the TCR-T group, 17.36 ± 13.31 mm2 for the control T cell group, and 17.49 ± 12.66 mm2 for the untreated group). Administration of KIAA1429D1358E-TCR-engineered T cells showed significant inhibition on the tumor growth after 3 days of the treatment and kept tumors progression-free, while the control T cell-treated group and the untreated group showed rapid tumor progression. The dissection of tumor tissues confirmed the significant differences in the tumor size among the three groups (Figure 5f-h). Furthermore, we investigated the number of exogenous T cells in peripheral blood of mice by FACS and found an increased number of KIAA1429D1358E-TCR-engineered T cells in comparison with the control T cells, indicating proliferation of KIAA1429D1358E-TCR-engineered T cells after the exposure to neoantigens (Figure 5i). These results revealed that both MAGOHBG17A-TCR-engineered T cells and KIAA1429D1358E-TCR-engineered T cells mediated anti-tumor activity against neoantigen-expressing tumors.
Discussion
HNSCC, which is classified as a highly immunosuppressive cancer type, shows a low level of lymphocyte infiltration (including neoantigen-specific T cells),46–48 suggesting HNSCC patients are unlikely to have sufficient numbers of tumor-reacting TILs for adoptive cell transfer. Hence, the ex vivo generation of TCR-engineered T cells targeting tumor-specific neoantigens would be one of the promising options to overcome the immunosuppressive status in the tumor microenvironment.
To facilitate the screen of tumor-reactive TCRs, previous studies have raised an ingenious TCR screening technique by using neoantigen peptide immunized HLA-A02:01/11:01 transgenic mice, and several TCRs targeting prevalent neoantigens KRASG12V/G12D,49 p53264–272,50 CEA691-69951 and MAGE-A352 were successfully found by this method. However, these studies depended on human HLA transgenic mice-based protocol, and identified TCRs were of murine species, which might be an obstacle for the clinical translation of their techniques. In contrast, the technique raised by our group, consisting of 7 steps: (1) whole-exome sequencing, (2) prediction of neoantigen epitopes based on HLA alleles, (3) isolation of neoantigen-specific T cells, (4) identification of neoantigen-specific TCR sequences, (5) production of viral vectors for the transfer of neoantigen-specific TCR genes, (6) transfection of T cells with neoantigen-specific TCRs, (7) functional evaluation of TCR-T cells,9,10 can be conducted within to 2–3 months from cancer tissues to TCR-engineered T cells prepared. This means there is a possibility that even advanced-stage cancer patients have an opportunity to receive TCR-T cell therapy. However, since our previous protocols were performed by the use of PBMCs of patients or healthy donors, there are some limitations when patients have rare MHC alleles or have a severe myelosuppressive condition.
Our group previously compared the TCR repertoires between expanded TILs and original ones in primary cancer tissues in HNSSC patients.32 The data showed that TILs derived from three HNSCC patients with very high mutation burden showed high levels of in vitro proliferation. We also found that the TCR composition (repertoires) of expanded TILs was very similar to original TILs in the tumor microenvironment. This might partly explain that TIL treatment or immune checkpoint inhibitors showed good clinical responses in some patients carrying tumors with a higher mutation burden.
Since it was uncertain whether expanded TILs (or maybe lymphocytes in draining lymph nodes53) could replace PBMCs for induction of neoantigen-specific CTLs, we tested this hypothesis in this study. The data showed that almost all minigenes designed from patients B2 and B8 with tumors having the high mutation burden could induce CTLs successfully. We chose the B8-3 minigene, which caused a slightly higher response to APCs expressing antigens from the minigene, to further screen the neoantigen-specific TCRs and their target. By an approach using expanded B8 TILs as the source of CD8+ T cells and C1R A02:01 cells as the antigen-presenting cells, we finally obtained neoantigen-specific TCR sequences and constructed the KIAA1429D1358E-TCR-engineered T cells which could cause regression of the tumors carrying KIAA1429D1358E mutation in the murine model.
By ten peptides of one minigene, we confirmed the feasibility of our new strategy based on expanded TILs. Given that it seemed almost all minigenes could activate the corresponding expanded TILs, we think it is possible to screen more TCRs using other minigenes. However, in the present study, we did not test more peptides considering the cost and the availability of Tetramers.
Notably, though the treatment of the KIAA1429D1358E-TCR-engineered T cells could suppress the growth of the tumor, it did not completely eradicate the tumors while the MAGOHBG17A-TCR-engineered T cells, which we constructed in our previous study,8 could cause the almost complete regression of the tumors carrying MAGOHBG17A mutation under the same treatment design. The factors that could affect the TCR-T treatment efficacy include: (1) the proportion of tumor cells expressing an antigen (some cells might have lost the protein expression); (2) the affinity of the specific TCR against the MHC-neoantigen complex; (3) the expression levels of neoantigens and their affinity to MHC; (4) the expression levels and turnover rates of the mutated protein.54–56 Among these factors, our analysis indicated that almost all tumor cells carry the targeted mutated peptide since we constructed the selection marker (puromycin-resistant gene) together with the mutant peptide gene. Interestingly, during the in vitro evaluation of TCR-T cells in which we used the C1R cells pulsed with an extremely high concentration of the peptide (20 μM) to stimulate TCR-T cells, we noticed that with a similar antigen-presenting system, the IFNγ secretion level of KIAA1429D1358E-TCR-engineered T cells is about 30–40% of that of the MAGOHBG17A-TCR-engineered T cells (Supplementary Figure 3a-b), suggesting that the interaction between TCR and neoantigen-MHC complex may affect the cytotoxic activity of TCR-engineered T cells. We certainly need to accumulate a large amount of information to conclude this hypothesis. However, since we repeatedly constructed KIAA1429D1358E-TCR-engineered T cells using T cells from several healthy donors, the tumor regressive effects using the different donors were similar, further implying that the lower tumor-regressive effect of KIAA1429D1358E-TCR-engineered T cells was likely to be independent of sources of donor T cells.
Our present work proves the feasibility of isolating neoantigen-specific T cells from TILs, identifying their TCR sequences, and constructing TCR-engineered T cells. This modified approach could be especially useful for patients with a high TMB and particularly for those having rare HLA alleles, further broadening the application of our approach in the clinic.
Supplementary Material
Funding Statement
This work was supported by the National Natural Science Foundation of China [82002956]; Guangdong Basic and Applied Basic Research Foundation [2019A1515110149]; China Postdoctoral Science Foundation [2019M663390]; Shenzhen Basic Research Program (JCYJ20190807150615224); Shenzhen International Collaborative Innovation Program (GJHZ20190821162003794).
Disclosure of potential conflicts of interest
Y. N. is a stockholder and a scientific advisor of OncoTherapy Science, Inc. No potential conflicts of interest were disclosed by the other authors.
Authors’ contributions
L.R. and Y.N. designed, supervised the project, and edited the manuscript; L.R. performed CTL induction; T.W. built the engineered T cells, performed evaluation of TCR-engineered T cells and wrote the manuscript; M.L. directed the construction of engineered T cells; M.X., N.L., C.Z. and J.J. assisted experiments; K.K. analyzed data and interpreted data; M.H. performed the TCR-sequencing of sorted cells; L.L., H.Q., and C.D. directed and supervised the techniques involved; N.A. provided the samples, clinical information, and advice to the project.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Global Burden of Disease Cancer, C , et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol. 2017;3:524–13. doi: 10.1001/jamaoncol.2016.5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Razzaghi H, Saraiya M, Thompson TD, Henley SJ, Viens L, Wilson R. Five-year relative survival for human papillomavirus-associated cancer sites. Cancer. 2018;124:203–211. doi: 10.1002/cncr.30947 [DOI] [PMC free article] [PubMed]
- 3.Chaturvedi AK, Anderson WF, Lortet-Tieulent J, Curado MP, Ferlay J, Franceschi S, Rosenberg PS, Bray F, Gillison ML. Worldwide trends in incidence rates for oral cavity and oropharyngeal cancers. J Clin Oncol. 2013;31(36):4550–4559. doi: 10.1200/JCO.2013.50.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moskovitz JM, Ferris RL, Immunology T.. Immunotherapy for head and neck squamous cell carcinoma. J Dent Res. 2018;97:622–626. doi: 10.1177/0022034518759464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferris RL. Immunology and immunotherapy of head and neck cancer. J Clin Oncol. 2015;33:3293–3304. doi: 10.1200/JCO.2015.61.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Economopoulou P, Agelaki S, Perisanidis C, Giotakis EI, Psyrri A. The promise of immunotherapy in head and neck squamous cell carcinoma. Ann Oncol. 2016;27(9):1675–1685. doi: 10.1093/annonc/mdw226. [DOI] [PubMed] [Google Scholar]
- 7.Solomon B, Young RJ, Rischin D. Head and neck squamous cell carcinoma: genomics and emerging biomarkers for immunomodulatory cancer treatments. Semin Cancer Biol. 2018;52:228–240. doi: 10.1016/j.semcancer.2018.01.008. [DOI] [PubMed] [Google Scholar]
- 8.Ren L, Leisegang M, Deng B, Matsuda T, Kiyotani K, Kato T, Harada M, Park JH, Saloura V, Seiwert T, Vokes E, Agrawal N, Nakamura Y. Identification of neoantigen-specific T cells and their targets: implications for immunotherapy of head and neck squamous cell carcinoma. Oncoimmunology. 2019;8:e1568813. doi: 10.1080/2162402X.2019.1568813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kato T, Matsuda T, Ikeda Y, Park JH, Leisegang M, Yoshimura S, Hikichi T, Harada M, Zewde M, Sato S, Hasegawa K, Kiyotani K, Nakamura Y. Effective screening of T cells recognizing neoantigens and construction of T-cell receptor-engineered T cells. Oncotarget. 2018;9:11009–11019. doi: 10.18632/oncotarget.24232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsuda T, Leisegang M, Park JH, Ren L, Kato T, Ikeda Y, Harada M, Kiyotani K, Lengyel E, Fleming GF, Nakamura Y. Induction of neoantigen-specific cytotoxic T cells and construction of T-cell receptor-engineered T cells for ovarian cancer. Clin Cancer Res. 2018;24:5357–5367. doi: 10.1158/1078-0432.CCR-18-0142. [DOI] [PubMed] [Google Scholar]
- 11.Madeddu C, Gramignano G, Astara G, Demontis R, Sanna E, Atzeni V, Maccio A. Pathogenesis and treatment options of cancer related anemia: perspective for a targeted mechanism-based approach. Front Physiol. 2018;9:1294. doi: 10.3389/fphys.2018.01294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaspar BL, Sharma P, Das R. Anemia in malignancies: pathogenetic and diagnostic considerations. Hematology. 2015;20(1):18–25. doi: 10.1179/1607845414Y.0000000161. [DOI] [PubMed] [Google Scholar]
- 13.Epstein RS, Aapro MS, Basu Roy UK, Salimi T, Krenitsky J, Leone-Perkins ML, Girman C, Schlusser C, Crawford J. Patient burden and real-world management of chemotherapy-induced myelosuppression: results from an online survey of patients with solid tumors. Adv Ther. 2020;37(8):3606–3618. doi: 10.1007/s12325-020-01419-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Groot R, Van Loenen MM, Guislain A, Nicolet BP, Freen-Van Heeren JJ, Verhagen O, Van Den Heuvel MM, De Jong J, Burger P, Van Der Schoot CE, et al. Polyfunctional tumor-reactive T cells are effectively expanded from non-small cell lung cancers, and correlate with an immune-engaged T cell profile. Oncoimmunology. 2019;8(11):e1648170. doi: 10.1080/2162402X.2019.1648170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Westergaard MCW, Andersen R, Chong C, Kjeldsen JW, Pedersen M, Friese C, Hasselager T, Lajer H, Coukos G, Bassani-Sternberg M, et al.Tumour-reactive T cell subsets in the microenvironment of ovarian cancer. Br J Cancer. 2019;120(4):424–434. doi: 10.1038/s41416-019-0384-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan Q, Zhang C, Yang W, Liu Y, Heyilimu P, Feng D, Xing L, Ke Y, Lu Z. Isolation of T cell receptor specifically reactive with autologous tumour cells from tumour-infiltrating lymphocytes and construction of T cell receptor engineered T cells for esophageal squamous cell carcinoma. J Immunother Cancer. 2019;7(1):232. doi: 10.1186/s40425-019-0709-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, Parkhurst MR, Ankri C, Prickett TD, Crystal JS, et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest. 2015;125(10):3981–3991. doi: 10.1172/JCI82416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dijkstra KK, Cattaneo CM, Weeber F, Chalabi M, van de Haar J, Fanchi LF, Slagter M, van der Velden DL, Kaing S, Kelderman S, et al. Generation of tumor-reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell. 2018;174(6):1586–1598, e1512. doi: 10.1016/j.cell.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roncati L, Palmieri B. Adoptive cell transfer (ACT) of autologous tumor-infiltrating lymphocytes (TILs) to treat malignant melanoma: the dawn of a chimeric antigen receptor T (CAR-T) cell therapy from autologous donor. Int J Dermatol. 2020;59(7):763–769. doi: 10.1111/ijd.14945. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen LT, Saibil SD, Sotov V, Le MX, Khoja L, Ghazarian D, Bonilla L, Majeed H, Hogg D, Joshua AM, et al. Phase II clinical trial of adoptive cell therapy for patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and low-dose interleukin-2. Cancer Immunol Immunother. 2019;68(5):773–785. doi: 10.1007/s00262-019-02307-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saint-Jean M, Knol AC, Volteau C, Quereux G, Peuvrel L, Brocard A, Pandolfino MC, Saiagh S, Nguyen JM, Bedane C, et al. Adoptive cell therapy with tumor-infiltrating lymphocytes in advanced melanoma patients. J Immunol Res. 2018;2018:3530148. doi: 10.1155/2018/3530148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pedersen M, Westergaard MCW, Milne K, Nielsen M, Borch TH, Poulsen LG, Hendel HW, Kennedy M, Briggs G, Ledoux S, et al. Adoptive cell therapy with tumor-infiltrating lymphocytes in patients with metastatic ovarian cancer: a pilot study. Oncoimmunology. 2018;7:e1502905. doi: 10.1080/2162402X.2018.1502905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Butt SU, Malik L. Role of immunotherapy in bladder cancer: past, present and future. Cancer Chemother Pharmacol. 2018;81(4):629–645. doi: 10.1007/s00280-018-3518-7. [DOI] [PubMed] [Google Scholar]
- 24.Inada Y, Mizukoshi E, Seike T, Tamai T, Iida N, Kitahara M, Yamashita T, Arai K, Terashima T, Fushimi K, et al. Characteristics of immune response to tumor-associated antigens and immune cell profile in patients with hepatocellular carcinoma. Hepatology. 2019;69(2):653–665. doi: 10.1002/hep.30212. [DOI] [PubMed] [Google Scholar]
- 25.Goff SL, Smith FO, Klapper JA, Sherry R, Wunderlich JR, Steinberg SM, White D, Rosenberg SA, Dudley ME, Yang JC. Tumor infiltrating lymphocyte therapy for metastatic melanoma: analysis of tumors resected for TIL. J Immunother. 2010;33:840–847. doi: 10.1097/CJI.0b013e3181f05b91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348(6230):62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geukes Foppen MH, Donia M, Svane IM, Haanen JB. Tumor-infiltrating lymphocytes for the treatment of metastatic cancer. Mol Oncol. 2015;9(10):1918–1935. doi: 10.1016/j.molonc.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papaioannou NE, Beniata OV, Vitsos P, Tsitsilonis O, Samara P. Harnessing the immune system to improve cancer therapy. Ann Transl Med. 2016;4(14):261. doi: 10.21037/atm.2016.04.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee S, Margolin K. Tumor-infiltrating lymphocytes in melanoma. Curr Oncol Rep. 2012;14(5):468–474. doi: 10.1007/s11912-012-0257-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watanabe K, Tsukahara T, Toji S, Saitoh S, Hirohashi Y, Nakatsugawa M, Kubo T, Kanaseki T, Kameshima H, Terui T, et al. Development of a T-cell receptor multimer with high avidity for detecting a naturally presented tumor-associated antigen on osteosarcoma cells. Cancer Sci. 2019;110:40–51. doi: 10.1111/cas.13854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Waddington JC, Meng X, Illing PT, Tailor A, Adair K, Whitaker P, Hamlett J, Jenkins RE, Farrell J, Berry N, et al. Identification of flucloxacillin-haptenated HLA-B*57:01 ligands: evidence of antigen processing and presentation. Toxicol Sci. 2020;177:454–465. doi: 10.1093/toxsci/kfaa124. [DOI] [PubMed] [Google Scholar]
- 32.Ren L, Matsuda T, Deng B, Kiyotani K, Kato T, Park JH, Seiwert TY, Vokes EE, Agrawal N, Nakamura Y. Similarity and difference in tumor-infiltrating lymphocytes in original tumor tissues and those of in vitro expanded populations in head and neck cancer. Oncotarget. 2018;9:3805–3814. doi: 10.18632/oncotarget.23454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 35.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, Kohlbacher O. OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics. 2014;30:3310–3316. doi: 10.1093/bioinformatics/btu548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kiyotani K, Park JH, Inoue H, Husain A, Olugbile S, Zewde M, Nakamura Y, Vigneswaran WT. Integrated analysis of somatic mutations and immune microenvironment in malignant pleural mesothelioma. Oncoimmunology. 2017;6(2):e1278330. doi: 10.1080/2162402X.2016.1278330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choudhury NJ, Kiyotani K, Yap KL, Campanile A, Antic T, Yew PY, Steinberg G, Park JH, Nakamura Y, O'Donnell PH. Low T-cell receptor diversity, high somatic mutation burden, and high neoantigen load as predictors of clinical outcome in muscle-invasive bladder cancer. Eur Urol Focus. 2016;2:445–452. doi: 10.1016/j.euf.2015.09.007. [DOI] [PubMed] [Google Scholar]
- 39.Lundegaard C, Lund O, Nielsen M. Accurate approximation method for prediction of class I MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics. 2008;24:1397–1398. doi: 10.1093/bioinformatics/btn128. [DOI] [PubMed] [Google Scholar]
- 40.Zemmour J, Little AM, Schendel DJ, Parham P. The HLA-A,B “negative” mutant cell line C1R expresses a novel HLA-B35 allele, which also has a point mutation in the translation initiation codon. J Immunol. 1992;148:1941–1948. [PubMed] [Google Scholar]
- 41.Fang H, Yamaguchi R, Liu X, Daigo Y, Yew PY, Tanikawa C, Matsuda K, Imoto S, Miyano S, Nakamura Y. Quantitative T cell repertoire analysis by deep cDNA sequencing of T cell receptor alpha and beta chains using next-generation sequencing (NGS). Oncoimmunology. 2014;3:e968467. doi: 10.4161/21624011.2014.968467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kato T, Iwasaki T, Uemura M, Nagahara A, Higashihara H, Osuga K, Ikeda Y, Kiyotani K, Park JH, Nonomura N, Nakamura Y. Characterization of the cryoablation-induced immune response in kidney cancer patients. Oncoimmunology. 2017;6:e1326441. doi: 10.1080/2162402X.2017.1326441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006;66:8878–8886. doi: 10.1158/0008-5472.CAN-06-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leisegang M, Engels B, Meyerhuber P, Kieback E, Sommermeyer D, Xue SA, Reuss S, Stauss H, Uckert W. Enhanced functionality of T cell receptor-redirected T cells is defined by the transgene cassette. J Mol Med (Berl). 2008;86:573–583. doi: 10.1007/s00109-008-0317-3. [DOI] [PubMed] [Google Scholar]
- 45.Leisegang M, Turqueti-Neves A, Engels B, Blankenstein T, Schendel DJ, Uckert W, Noessner E. T-cell receptor gene-modified T cells with shared renal cell carcinoma specificity for adoptive T-cell therapy. Clin Cancer Res. 2010;16:2333–2343. doi: 10.1158/1078-0432.CCR-09-2897. [DOI] [PubMed] [Google Scholar]
- 46.Watermann C, Pasternack H, Idel C, Ribbat-Idel J, Bragelmann J, Kuppler P, Offermann A, Jonigk D, Kuhnel MP, Schrock A, et al. Recurrent HNSCC harbor an immunosuppressive tumor immune microenvironment suggesting successful tumor immune evasion. Clin Cancer Res. 2021;27:632–644. doi: 10.1158/1078-0432.CCR-20-0197. [DOI] [PubMed] [Google Scholar]
- 47.Canning M, Guo G, Yu M, Myint C, Groves MW, Byrd JK, Cui Y. Heterogeneity of the head and neck squamous cell carcinoma immune landscape and its impact on immunotherapy. Front Cell Dev Biol. 2019;7:52. doi: 10.3389/fcell.2019.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peltanova B, Raudenska M, Masarik M. Effect of tumor microenvironment on pathogenesis of the head and neck squamous cell carcinoma: a systematic review. Mol Cancer. 2019;18:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang QJ, Yu Z, Griffith K, Hanada K, Restifo NP, Yang JC. Identification of T-cell Receptors Targeting KRAS-Mutated Human Tumors. Cancer Immunol Res. 2016;4:204–214. doi: 10.1158/2326-6066.CIR-15-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cohen CJ, Zheng Z, Bray R, Zhao Y, Sherman LA, Rosenberg SA, Morgan RA. Recognition of fresh human tumor by human peripheral blood lymphocytes transduced with a bicistronic retroviral vector encoding a murine anti-p53 TCR. J Immunol. 2005;175:5799–5808. doi: 10.4049/jimmunol.175.9.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parkhurst MR, Joo J, Riley JP, Yu Z, Li Y, Robbins PF, Rosenberg SA. Characterization of genetically modified T-cell receptors that recognize the CEA:691-699 peptide in the context of HLA-A2.1 on human colorectal cancer cells. Clin Cancer Res. 2009;15:169–180. doi: 10.1158/1078-0432.CCR-08-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chinnasamy N, Wargo JA, Yu Z, Rao M, Frankel TL, Riley JP, Hong JJ, Parkhurst MR, Feldman SA, Schrump DS, et al. A TCR targeting the HLA-A*0201-restricted epitope of MAGE-A3 recognizes multiple epitopes of the MAGE-A antigen superfamily in several types of cancer. J Immunol. 2011;186(685–696). doi: 10.4049/jimmunol.1001775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matsuda T, Miyauchi E, Hsu YW, Nagayama S, Kiyotani K, Zewde M, Park JH, Kato T, Harada M, Matsui S, Ueno M, Fukuda K, Suzuki N, Hazama S, Nagano H, Takeuchi H, Vigneswaran WT, Kitagawa Y, Nakamura Y. TCR sequencing analysis of cancer tissues and tumor draining lymph nodes in colorectal cancer patients. Oncoimmunology. 2019;8:e1588085. doi: 10.1080/2162402X.2019.1588085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stone JD, Chervin AS, Kranz DM. T-cell receptor binding affinities and kinetics: impact on T-cell activity and specificity. Immunology. 2009;126(2):165–176. doi: 10.1111/j.1365-2567.2008.03015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Campillo-Davo D, Flumens D, Lion E. The quest for the best: how tcr affinity, avidity, and functional avidity affect TCR-Engineered T-Cell Antitumor Responses. Cells. 2020;9(7):1720. doi: 10.3390/cells9071720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Knapp B, Van Der Merwe PA, Dushek O, Deane CM. MHC binding affects the dynamics of different T-cell receptors in different ways. PLoS Comput Biol. 2019;15:e1007338. doi: 10.1371/journal.pcbi.1007338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.