

Abstract
Introduction
Subtle cognitive impairment (SCI) may appear before pathological changes surpass thresholds for abnormality. We aimed to investigate whether SCI could predict Alzheimer's pathologies and advancement.
Methods
A total of 816 cognitively normal individuals were enrolled to assess the longitudinal neuropathological and clinical correlates of baseline SCI, via linear mixed‐effects and Cox proportional‐hazard models. Cross‐lagged panel models were used in specific time waves.
Results
SCI individuals had a faster increase in brain amyloid burden and a higher risk of conversion. They also showed greater rates of cerebrospinal fluid (CSF) phosphorylated tau (p‐tau)181 increase and glucose metabolism decrease. In addition, baseline SCI predicted worse clinical progression, whereas multi‐domain SCI advanced faster compared to the single domain group.
Discussion
Baseline SCI could be an imperative prediction indicator of clinical and pathological progression. It enables cognitive measures to be informative at a very early stage and provided objective criteria for high‐risk population screening.
Keywords: CSF, PET, SCI, amyloid, neurodegeneration, tau
1. INTRODUCTION
A prolonged asymptomatic phase exists in the course of the Alzheimer's continuum with the appearance of pathological biomarkers, including the accumulation of amyloid β (Aβ) plaques and the deposition of neurofibrillary tangles (NFTs). 1 Given that these hallmarks often indicate that irreversible progress of the disease has already taken place, a refined and economical screening method is urgently needed to capture the preclinical changes before the occurrence of biomarker abnormality or a full‐blown clinical symptom. 2 On top of traditional biomarkers, the Alzheimer's continuum offers more sensitivity by involving longitudinal changes on cognitive performance. 3 The National Institute on Aging and the Alzheimer's Association (NIA‐AA) has proposed a staging framework for cognitively normal (CN) individuals involving longitudinal changes on cognitive performance to characterize “preclinical” Alzheimer's disease (AD), in which it has incorporated subtle cognitive impairment (SCI) and biomarkers abnormality for the stage 3. 4 SCI, specifically, is defined by a cognitive transitional status involving pathological changes but not sufficiently severe to manifest conspicuous functional loss. 3 The assessment of SCI or cognitive decline can be reflected by reliable neuropsychological tests or composite scores, and it could be more advanced than or at least as sensitive as neuroimaging and cerebrospinal fluid (CSF) biomarkers in predicting clinical progression of AD. 5 , 6 , 7 , 8 Thus SCI is expected to be the optimal means of screening and monitoring the pathological and clinical progression of groups at high risk of AD.
Previous studies have mainly converged on the predictive relationships of cognitive impairment with clinical conversion, 5 , 9 or the influence of AD pathologies on subsequent cognition changes. 10 , 11 Gustavson et al. have reported that episodic memory assessed by extensive neuropsychological tests could sensitively predict the risk of progressing to mild cognitive impairment (MCI), compared favorably to the biomarker‐based prediction 12 . Yet, limitations existed because the population was based on the late middle age group and lacked sufficient data on the progression of pathological biomarkers. In addition, fewer studies have investigated the associations of cognitive impairment with later AD pathological changes. Only a single or small proportion of relevant biomarkers were covered, and the outcomes were inconsistent. 8 , 12 , 13 , 14 For example, Elman et al. have found that cognition at baseline could predict conversion to Aβ abnormality in a non‐demented cohort, 8 but the results were contra‐indicated with previous literature when the sample size was expanded. 13 The involvement of MCI might have also led to bias because other factors could influence the process of disease. 8 In addition, the exclusion of other pathologies, such as tau deposition and neurodegeneration, could also narrow the scope of evidence. Therefore, it remains unclear whether SCI could predict Alzheimer's pathologies and advancement.
The present study aimed to elucidate the neuropathological and clinical correlates of baseline SCI in elderly subjects with normal cognition. Subjects with baseline SCI were hypothesized to have a greater burden of AD pathologies and worse clinical progression. Considering that global and domain‐specific composite scores typically have less variability than raw scores from single neuropsychological tests, 15 , 16 we defined the baseline SCI category using modified preclinical Alzheimer's cognitive composite (mPACC) and theoretically derived domain‐specific (memory, executive, language, and visuospatial functioning) scores. Cross‐sectional and longitudinal relationships of baseline SCI with amyloid, tau, and neurodegeneration biomarkers were evaluated separately in CSF and positron emission tomography (PET).
RESEARCH IN CONTEXT
Systematic review: Authors reviewed the literature on correlations of cognitive impairment with Alzheimer's disease (AD) pathologies and clinical status. Most studies focused on the advancement of subjective cognitive decline (SCI), or the influence of pathologies on subsequent cognition changes, but rare studies evaluated the impact of SCI on pathological progression in the normal elderly. Relevant publications are appropriately cited.
Interpretation: Our findings provided insights into the predictive relationships of baseline SCI with AD pathologies and clinical conversion, and enabled cognitive measures at a very early stage to be informative for screening high‐risk populations.
Future directions: The article proposes that SCI could be a valid marker for the neuropathological and clinical progression of AD. Studies in larger cohorts with different races are required to ascertain thresholds of SCI in cognitively normal individuals. More comprehensive longitudinal data could help to investigate the effect of SCI on monitoring the therapeutic efficacy in the early phase.
2. METHODS
2.1. Participants
In this study, we selected cognitively normal (CN) participants from the Alzheimer's Disease Neuroimaging Initiative (ADNI) cohort (http://adni.loni.usc.edu). Beginning in 2004, the multicenter ADNI project was designed to help predict the early onset of MCI and AD. For each individual, the cognitive trajectories were collected, and the biomarkers were repeatedly assessed during the follow‐up period to track the pathology as the disease progressed. Written informed consent was obtained on human experimentation at each institution. (For detailed information, see http://adni.loni.usc.edu/study‐design.)
A total of 816 individuals with baseline normal cognition (or CN) were included (298 of them reported subjective memory complaints). They underwent a series of neuropsychological assessments: Mini‐Mental State Examination (MMSE), Clinical Dementia Rating ‐ Sum of Boxes (CDR‐SB), the Alzheimer's Disease Assessment Scale (ADAS), Category Fluency, WAIS‐R Digit Symbol, Boston Naming Test, Trail Making Test, Rey Auditory Verbal Learning Test, and so on. Participants with either eligible CSF or PET data were separately included in the exploration of relationships between SCI and AD biomarkers.
2.2. Cognitive assessment and definition of cognitive cutoffs
Cognition states were presented by mPACC scores and ADNI composite measures for memory (ADNI MEM), executive functioning (ADNI EF), language (ADNI LAN), and visuospatial (ADNI VS) domains. The mPACC is generated from the delayed recall from the ADAS ‐ Cognition and Logical Memory, MMSE, and Trail Making Test Part B time. It was designed for the detection of amyloid‐related cognition decline. The ADNI composite scores were evaluated from the ADNI neuropsychological battery using item response theory (IRT) methods. The metrics for these composite scores are defined to have a mean of 0 and standard deviation of 1, and the resulting data are provided and used in the analyses. Lower composite scores indicated worse cognition status. To define cognitive cutoffs for individuals with SCI, we further identified a cohort of participants with a follow‐up period of at least 7 years (n = 216). Among them, those who remained stable CN during the follow‐up period (n = 166) were used to calculate cognitive cutoffs indicating SCI. Here, we used the 10th percentile of composite scores, which were broadly acknowledged and utilized in research. 5 , 17 , 18 Cutoffs at the bottom 10th percentile were −2.547 for the mPACC score, 0.528 for the ADNI MEM score, 0.126 for the ADNI EF score, 0.199 for the ADNI LAN score, and −0.564 for the ADNI VS score. Individuals with baseline composite scores below the corresponding cutoffs were determined as SCI subjects (PACC SCI, MEM SCI, EF SCI, LAN SCI, and VS SCI, respectively). A group of participants was finally defined as multi‐domain SCI by the presence of at least two domain impairments in the four ADNI composite scores (MEM, EF, LAN, and VS scores).
2.3. CSF and PET imaging measures and biomarker cutoffs
Data sets of CSF and PET assessments were downloaded from ADNI in March 2020. CSF Aβ1‐42, phosphorylated tau (p‐tau)181 and total tau (t‐tau) were measured using the fully automated Roche Elecsys and cobas e 601 immuno‐assay analyzer system. The process of sample testing and quality control followed the acceptance criteria according to the Roche Protocol in the UPenn/ADNI Biomarker Laboratory, which was described in previous studies. 19 , 20 Baseline Aβ status (Aβ+/−) of participants was determined by the cutoff of 1098 pg/mL for CSF Aβ1‐42, 21 whereas p‐tau181 and t‐tau used 27 pg/mL and 300 pg/mL, 22 respectively.
All PET data used were from the UC Berkeley and Lawrence Berkeley National Laboratory. Brain amyloid burden used florbetapir (AV45) standardised uptake value ratios (SUVRs) were calculated by averaging across four cortical regions (frontal, anterior/posterior cingulate, lateral parietal, lateral temporal) and then divided by the whole cerebellum as reference region. Brain tau deposit was measured via the flortaucipir (AV‐1451) processing method. A composite set of pre‐defined regions of interest (metaROI) of bilateral entorhinal, amygdala, fusiform, inferior, and middle temporal regions were considered for tau PET assessment. 21 , 23 Brain neurodegeneration used hypometabolism assessed by fluorodeoxyglucose (FDG) PET, which was from the average of five metaROIs (left angular gyrus, right angular gyrus, bilateral posterior cingular, left inferior temporal gyrus, right inferior temporal gyrus). 24 The cutoffs for categories of brain amyloid, tau, and FDG PET were listed as below: 1.11 for florbetapir SUVR, 1.37 for flortaucipir metaROI SUVR, 21 and 1.21 for FDG PET. 25 (For concrete processing methods see http://adni.loni.usc.edu/methods.)
2.4. Statistical analyses
Data were presented mean (standard deviation [SD]) or number (%) when appropriate. Individual extreme values that exceeded the range of mean ± 3*SD were eliminated in analyses. We tested the differences in demographic and clinical characteristics between SCI and normal groups using the Wilcoxon test or Kruskal‐Wallis test (for continuous variables) and the chi‐square test or Cochran‐Mantel‐Haenszel test (for categorical variables). Spearman correlation and regression analyses were performed to explore the associations between CSF/PET biomarker levels and cognitive composite scores. Age, gender, years of education, and apolipoprotein E (APOE) genotype were included as covariates in all regression analyses.
To evaluate the risk of clinical progression in CN individuals with SCI at baseline, we plotted Kaplan‐Meier survival curves and computed hazard ratios in the Cox proportional‐hazards models. Clinical progression was defined as progressing to MCI/AD dementia and CDR‐SB > or = 0.5, 26 separately. Then, we assessed whether baseline SCI was associated with AD pathological progression in CN individuals. Considering that most of the tau PET and part of CSF data were obtained during the follow‐up, we redefined the time point when each participant first obtained the biomarker test as the new baseline in analyses of biomarkers. At the new baseline, those who have already converted to MCI or AD dementia were excluded from further analyses. Among CN individuals with normal categories of CSF or PET biomarkers at the redefined baseline, we used survival curves and Cox proportional‐hazards model analyses to test the risk of progression to abnormality in SCI subjects, based on the normal groups. Age, gender, years of education, and APOE genotype were empirically considered as confounding factors and adjusted in the multivariate models. The longitudinal relationships between SCI and annual changes of CSF/PET pathologic biomarkers were computed by linear mixed‐effects model analyses, and then compared between SCI and normal groups.
Potential predictive relationships between CSF and PET pathology over time were further explored via the cross‐lagged panel model (CLPM) analyses, 27 , 28 using three time points of longitudinal data (baseline, 24 months, and 48 months). The CLPM also investigated at the same time points whether baseline SCI predicted CSF biomarkers differently from it predicted PET biomarkers. Subjects included in CLPM analyses were demanded to possess baseline as well as follow‐up data from at least one of 24 and 48 months. The maximum‐likelihood estimator was adopted to implement CLPM analyses, bearing randomly missing data. Standardized β estimate and its P value were calculated for each association measured. All models were tested for goodness of fit via the root mean square error of approximation (RMSEA), normed fit index (NFI), incremental fit index (IFI), comparative fit index (CFI), Tucker–Lewis index (TLI) and chi‐square divided by its degrees of freedom (CMIN/DF). Good fit was indicated with model values of RMSEA < 0.06, NFI > 0.95, IFI > 0.95, CFI > 0.95, TLI > 0.90, and CMIN/DF < 5 29 , 30 .
All statistical analyses were conducted using R (version 3.6.3) software, except for the CLPM analyses that were performed with Amos (version 23.0) software. Statistical significance was considered as a two‐sided P value < .05.
3. RESULTS
3.1. Descriptive characteristics of the cohort
Of 816 CN individuals included in this study, 360 (44.1%) were male, and 230 (30.3% in those who provided genotype information) were APOE ɛ4 carriers (ɛ4+/− or ɛ4+/+) (see flowchart in the Supplementary material). The average (SD) age and education of this cohort were 72.9 (6.2) and 16.6 (2.6) years, respectively. Characteristics of participants grouped by cognition status at baseline were summarized in Table 1. SCI individuals were obviously older than those in the normal group (P < .05). Gender proportion did not differentiate between normal and SCI groups by language, executive, and visuospatial functioning. However, male patients were over‐represented in the PACC SCI (SCI defined by ADNI mPACC score) (P = .002) and ADNI memory SCI (SCI defined by ADNI MEM score) (P < .001) groups. In addition, SCI individuals were less educated than individuals in normal groups, which have reached or were close to a significant level. As for categorization for multiple cognitive domains, multi‐domain SCI participants were older, less educated, and had a larger proportion of male participants than the normal group (P < .001). There were no differences in the proportion of APOE genotype among all groups.
TABLE 1.
Descriptive features of cognitively normal individuals grouped by cognition composite scores
| Characteristics | mPACC | ADNImemory | ADNIexecutive | ADNIlanguage | ADNI visuospatial | Multi‐domain | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Normal | SCI | Normal | SCI | Normal | SCI | Normal | SCI | Normal | SCI | Normal | Single SCI | MD SCI | |
| No. | 679 | 137 | 656 | 160 | 673 | 143 | 667 | 149 | 699 | 117 | 466 | 197 | 153 |
| Age (year) | 72.3 (6.1) | 75.6* (6.3) | 72.4 (6.1) | 74.8* (6.4) | 72.0 (5.9) | 76.9* (6.0) | 72.5 (6.1) | 74.6* (6.6) | 72.6 (6.2) | 74.4* (6.5) | 71.7 (5.9) | 73.1 (6.0) | 76.1* (6.4) |
| Gender, male | 283 (41.7) | 77* (56.2) | 247 (37.7) | 113* (70.6) | 290 (43.1) | 70 (49.0) | 295 (44.2) | 65 (43.6) | 309 (44.2) | 51 (43.6) | 178 (38.2) | 101 (51.3) | 81* (52.9) |
| Education (year) | 16.8 (2.4) | 15.5* (3.0) | 16.6 (2.5) | 16.3 (2.6) | 16.7 (2.4) | 15.8* (2.9) | 16.8 (2.4) | 15.4* (2.9) | 16.6 (2.5) | 16.0 (2.9) | 16.9 (2.3) | 16.5 (2.5) | 15.5* (3.0) |
| Follow‐up (year) | 4.2 (3.8) | 3.9 (3.4) | 4.3 (3.8) | 3.5* (3.4) | 4.2 (3.8) | 3.9 (3.2) | 4.2 (3.8) | 3.6 (3.4) | 4.2 (3.7) | 3.6 (3.6) | 4.4 (3.8) | 4.0 (3.6) | 3.5 (3.2) |
| APOE ɛ4 carriers | 195 (30.8) | 35 (28.0) | 188 (30.8) | 42 (28.6) | 191 (30.7) | 39 (28.7) | 192 (30.9) | 38 (27.9) | 197 (30.4) | 33 (30.0) | 132 (30.4) | 57 (31.5) | 41 (28.7) |
| CSF biomarkers | |||||||||||||
| Aβ42 (pg/mL) | 1326.3 (601.9) | 1226.1 (628.6) | 1335.5 (595.1) | 1205.9* (645.7) | 1330.6 (590.9) | 1217.0* (672.1) | 1329.5 (600.2) | 1222.6 (631.7) | 1308.0 (608.1) | 1328.2 (600.7) | 1344.2 (579.1) | 1326.4 (643.2) | 1185.9* (633.9) |
| P‐tau (pg/mL) | 21.5 (9.3) | 21.9 (8.8) | 21.4 (9.2) | 22.3 (9.5) | 21.4 (9.1) | 22.4 (9.7) | 21.3 (9.0) | 22.8 (10.2) | 21.7 (9.6) | 20.7 (7.2) | 21.2 (9.0) | 21.5 (9.5) | 22.8 (9.5) |
| T‐tau (pg/mL) | 235.9 (88.1) | 239.1 (88.6) | 235.3 (89.0) | 241.0 (84.7) | 235.2 (88.3) | 242.1 (87.7) | 234.8 (87.0) | 243.9 (93.2) | 237.5 (90.4) | 230.3 (74.2) | 234.2 (88.9) | 235.6 (89.0) | 244.4 (85.0) |
| PET biomarkers | |||||||||||||
| florbetapir | 1.10 (0.17) | 1.14 (0.19) | 1.11 (0.17) | 1.12 (0.17) | 1.10 (0.16) | 1.14 (0.20) | 1.11 (0.17) | 1.12 (0.19) | 1.10 (0.16) | 1.15 (0.21) | 1.09 (0.15) | 1.13 (0.19) | 1.14 (0.20) |
| flortaucipir | 1.15 (0.10) | 1.23* (0.21) | 1.15 (0.10) | 1.20 (0.19) | 1.16 (0.12) | 1.20* (0.17) | 1.16 (0.12) | 1.17 (0.15) | 1.16 (0.13) | 1.16 (0.11) | 1.15 (0.11) | 1.17 (0.10) | 1.20* (0.20) |
| FDG | 1.31 (0.11) | 1.28* (0.13) | 1.31 (0.11) | 1.28* (0.13) | 1.32 (0.11) | 1.27* (0.12) | 1.31 (0.11) | 1.29 (0.13) | 1.31 (0.11) | 1.31 (0.11) | 1.32 (0.11) | 1.28 (0.10) | 1.29* (0.13) |
Note: Data are presented mean (standard deviation [SD]) or number (%) as appropriate. Characteristics of CSF and PET biomarkers are summarizations of participants with eligible CSF and PET data, respectively.
Abbreviation: PACC = preclinical Alzheimer's cognitive composite, SCI = subtle cognitive impairment, MD SCI = multi‐domain SCI.
* P value <.05 in comparisons between SCI and normal groups.
3.2. Associations of cognition with baseline amyloid, tau, and neurodegeneration biomarkers
Univariate relationships between cognitive composite scores and AD biomarkers were summarized and plotted in the Supplementary material. Lower scores of mPACC, ADNI memory, and executive function were correlated with increased brain burden of amyloidosis and tau pathology, and higher CSF p‐tau181 and t‐tau levels. Worse scores were linked to lower levels of CSF Aβ1‐42 and FDG PET. In addition, the composite score of language was inversely associated with tau PET and positively correlated with FDG PET, whereas no statistical relationship was observed of ADNI visuospatial function score with CSF or PET biomarkers.
Adjusted for age, gender, years of education and APOE genotype, we found that increased amyloid, tau PET uptake, and decreased FDG PET were correlated with worse mPACC score (standardized β estimate = ‐0.106, P = .024, β = ‐0.195, P < .001, and β = 0.150, P < .001, respectively). Lower ADNI memory score was merely related with higher tau PET uptake (β = ‐0.207, P < .001), whereas decreased executive score was correlated with increased amyloid (β = ‐0.111, P = .018), tau PET (β = ‐0.132, P = .022), and lower levels of CSF Aβ1‐42 (β = 0.126, P = .004) and FDG PET (β = 0.164, P = .001). The remaining results were settled in the Supplementary material.
At the redefined baseline, SCI groups except for visuospatial SCI had lower levels of CSF Aβ1‐42 than the normal groups, up to or close to statistical significance (Table 1). But no statistical difference was observed in group comparisons of CSF p‐tau181 and t‐tau. Amyloid PET showed a trend of increased cortical uptake only in the PACC SCI group (P = .062). Several types of SCI illustrated worse brain tau deposition and hypometabolism (Table 1).
3.3. Associations of SCI with longitudinal amyloid, tau and neurodegeneration changes
As shown in Figure 1, significant decreases of Aβ1‐42 level and increases of p‐tau181 and t‐tau were observed in the CSF of both normal and PACC SCI groups (SCI defined by ADNI mPACC score) (P < .05, Figure 1A‐C; other results see in the Supplementary material). CSF Aβ1‐42 and p‐tau181 showed trends of greater annual change rates in PACC SCI individuals compared to the normal (group‐wise difference: −1.49%, P = .134 and .95%, P = .074, respectively), which were close to statistical significance. Although analyses of CSF biomarker normal/abnormal (−/+) subgroups showed that PACC SCI individuals had faster rates in elevation of p‐tau181 level among T− subjects (group‐wise difference: 1.16%, P = .042) and a very slight trend toward the significance of a faster reduction of Aβ1‐42 level among A− subjects (group‐wise difference: −2.07%, P = .169), compared to normal individuals.
FIGURE 1.
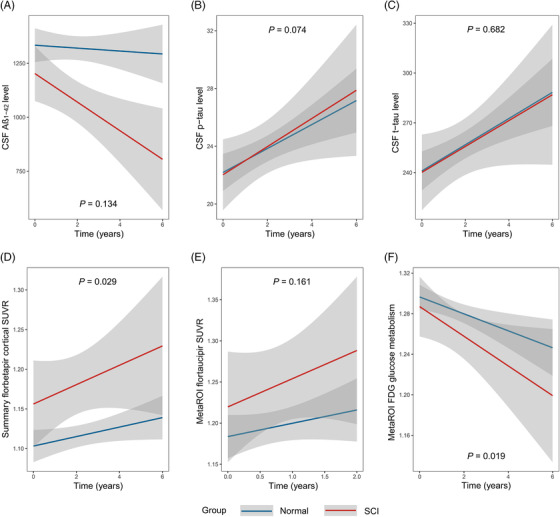
Longitudinal changes of amyloid, tau, and neurodegeneration between subtle cognitive impairment (SCI) and normal individuals. (A–E) Longitudinal changes of amyloid, tau, and neurodegeneration between PACC SCI (SCI defined by ADNI mPACC score) and normal individuals. (F) Longitudinal changes of amyloid, tau, and neurodegeneration between ADNI memory SCI (SCI defined by ADNI MEM score) and normal individuals.
In assessing longitudinal changes of PET‐imaging biomarkers, incremental uptakes of amyloid AV45 PET tracer and tau AV1451 PET tracer were observed over time in the whole cohort, as well as decreased glucose metabolism assessed by FDG PET (P < .05, Figure 1D‐F; detailed results see in the Supplementary material). Furthermore, the PACC SCI group showed a significantly higher annual rate of brain amyloid burden than the normal group (group‐wise difference: 0.54%, P = .029). In addition, ADNI memory SCI individuals presented a faster decrease in glucose metabolism (group‐wise difference: −0.75%, P = .019). In subgroup analyses of PET biomarker normal/abnormal (−/+) groups, SCI of A− individuals had a faster growth rate over normal subjects (group‐wise difference: 0.49%, P = .077), which approached but failed to achieve a customary level of statistical significance. We did not perform subgroup analyses of tau PET and FDG PET biomarkers due to the small sample size of biomarker abnormal groups (n = 59 and n = 15, respectively) and possible underpowered detection.
3.4. Associations of SCI with pathological and clinical progression
PACC SCI individuals (SCI defined by ADNI mPACC score) showed higher conversion risk from CSF/PET A− to A+, based on normal subjects (Figure 2A,D and Table 2). These analyses were conducted in the subgroups with normal levels of baseline CSF/PET biomarkers. Biomarkers did not show any statistical difference between normal and SCI individuals in these subgroups (Supplementary material). Multivariate models met the criteria for proportional hazard assumption (Schoenfeld global test P = .101 and.195, respectively). In addition, SCI subjects seemed to have higher conversion rates from PET T− to T+, but did not reach statistical significance (Figure 2E). In addition, SCI individuals grouped by ADNI memory composite score (SCI defined by ADNI MEM score) also illustrated a potentially increased conversion risk from PET N− to N+ (Figure 2F). Survival results of other comparisons did not illustrate positive outcomes (P > .20).
FIGURE 2.
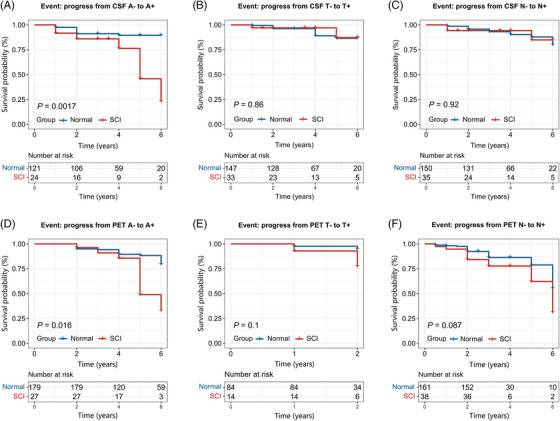
Kaplan‐Meier curves showing survival probability of pathological progression. (A–C) Progression from CSF biomarker normal to abnormal. (D–F) Progression from PET imaging biomarker normal to abnormal. CSF Aβ status (Aβ +/−) of participants was determined by the cutoff of 1098 pg/mL for CSF Aβ1‐42, 21 whereas p‐tau181 and t‐tau used 27 pg/mL and 300 pg/mL 22 , respectively. The cutoffs for categories of brain amyloid, tau, and FDG PET were listed as below: 1.11 for florbetapir SUVR, 1.37 for flortaucipir metaROI SUVR, 21 and 1.21 for FDG PET. 25 In Figure 2A‐E, SCI individuals were defined by abnormal ADNI mPACC score, whereas in Figure 2F, SCI individuals were defined by abnormal ADNI MEM score.
TABLE 2.
Progression risk of SCI and normal individuals
| Unadjusted | Adjusted | |||||
|---|---|---|---|---|---|---|
| Short‐term progression rate | Long‐term progression rate | HR (95% CI) | P | HR (95% CI) | P | |
| Progress to MCI/AD dementia a | ||||||
| Normal | 8.9% | 15.1% | ref. | – | ref. | – |
| SCI | 25.0% | 32.4% | 3.06 (2.04–4.59) | <.001 | 2.59 (1.68–4.00) | <.001 |
| Progress to CDR‐SB > or = 0.5 a | ||||||
| Normal | 17.8% | 28.4% | ref. | – | ref. | – |
| SCI | 45.0% | 54.1% | 2.86 (2.11–3.89) | <.001 | 2.43 (1.75–3.37) | <.001 |
| Progress from CSF A− to A+ b | ||||||
| Normal | 8.3% | 9.1% | ref. | – | ref. | – |
| SCI | 12.5% | 29.2% | 4.06 (1.57–10.51) | .004 | 3.84 (1.36–10.80) | .011 |
| Progress from PET A− to A+ b | ||||||
| Normal | 5.6% | 12.8% | ref. | – | ref. | – |
| SCI | 7.4% | 25.9% | 2.79 (1.18–6.59) | .020 | 2.62 (1.08–6.38) | .034 |
Abbreviation: SCI = subtle cognitive impairment, MCI = mild cognitive impairment, AD = Alzheimer's disease, A− = amyloid biomarker normal, A+ = amyloid biomarker abnormal, HR = hazard ratio.
Note: Hazard ratios (95% CI) calculated using Cox regression analyses, in unadjusted and adjusted models corrected for baseline age, gender, APOE ε4 status, and years of education.
Short‐term = 5 years, long‐term = 10 years;
Short‐term = 3 years, long‐term = 6 years.
Overall, CN individuals presented a progression rate to MCI/AD dementia of 18.2% and that to CDR‐SB > or = 0.5 of 33.1% followed by 10 years. Compared with normal, PACC SCI individuals showed higher rates of clinical progression by 10 years and 5 years (Table 2). Survival curves with results of log‐rank tests were exhibited in Figure 3 and the Supplementary material. Age at baseline, gender, years of education, and APOE genotype were adjusted as covariates in the multivariate model analyses, which met the proportional hazard assumption via Schoenfeld residuals technique (Schoenfeld global test P = .098 and.202, respectively). SCI groups showed a higher risk of clinical progression based on the normal group (results of PACC SCI are presented in Table 2). Considering the influence of baseline mild neurodegenerative pathologies possibly existing in some of the participants, we further conducted the multivariable Cox regression analyses adding the adjustment of baseline FDG PET. SCI individuals remained higher risk conversion to MCI/dementia (PACC: hazard ratio [HR] 1.88, 95% confidence interval [CI] 1.12‐3.17, P = .017; ADNI memory: HR 2.92, 95% CI 1.79‐4.75, P < .001) and to CDR‐SB > or = 0.5 (PACC: HR 2.14, 95% CI 1.46‐3.12, P < .001; ADNI memory: HR 2.16, 95% CI 1.48‐3.16, P < .001) compared with the normal individuals, in the adjusted model of baseline age, gender, APOE ε4 status, years of education, and level of FDG PET.
FIGURE 3.
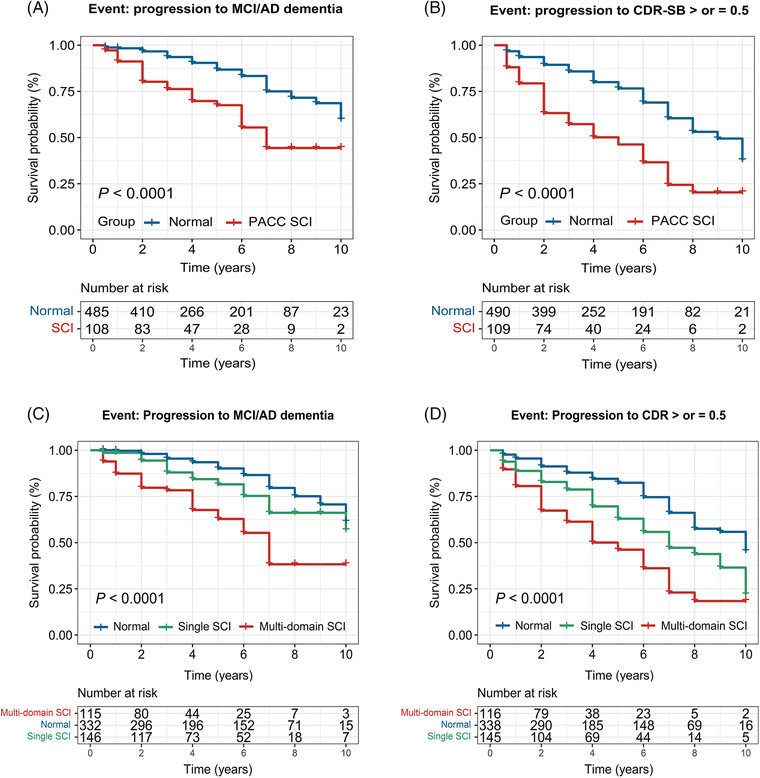
Kaplan‐Meier curves showing survival probability of clinical progression. (A) Progression from cognitively normal participants to mild cognitive impairment or AD dementia. (B) Progression from cognitively normal participants to incident prodromal stage of AD indicated by a CDR‐global score of 0.5
3.5. Predictive relationships between CSF and PET biomarkers
Time‐specific correlations among composite cognitive scores, CSF Aβ1‐42, and amyloid AV45 PET were tested by CLPM analyses in a subset of 151 CN individuals, who provided baseline and at least one follow‐up (24‐month and/or 48‐month) biomarker measurements. Data were well fitted with the model for the relationships between CSF Aβ1‐42 and amyloid PET (CMIN/DF = 1.490, NFI = 0.994, IFI = 0.998, CFI = 0.998, TLI = 0.990, and RMSEA = 0.057). Lower CSF Aβ1‐42 level at baseline predicted higher amyloid AV45 PET uptake at 24 months, and lower CSF Aβ1‐42 at 24 months predicted higher AV45 PET uptake at 48 months (standardized β estimate: −0.07, P = .024 and −.08, P = .032, respectively; Figure 4).
FIGURE 4.
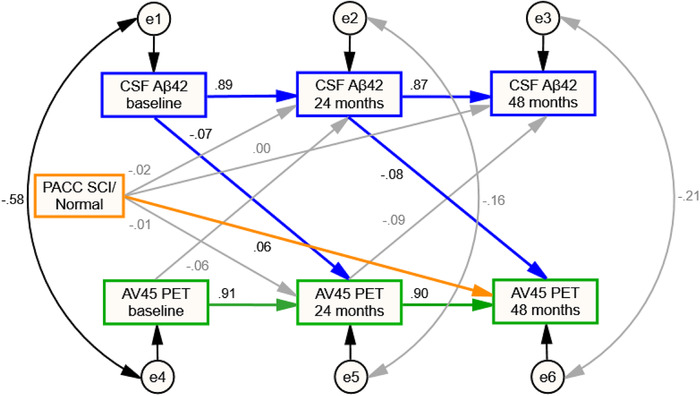
Cross‐lagged panel model testing predictive relationships between CSF and PET amyloid biomarkers. Brain amyloid burden measured by PET and CSF Aβ1‐42 level were modeled across three waves (baseline, 24 months, and 48 months) with baseline SCI/normal category as the control variable. Rectangles, observed variables; small circles, residual variances; thin gray arrows, nonsignificant longitudinal predictions or correlations between residuals; black arrows, significant covariate associations or correlations between residuals; green arrows, significant longitudinal predictions from brain amyloid PET uptake to a variable at a subsequent time point; blue arrows, significant longitudinal predictions from CSF Aβ1‐42 levels to a variable at a subsequent time point; orange arrows, significant or close to statistically significant longitudinal predictions from baseline SCI/normal category to a variable at a subsequent time point. Standardized coefficients (β) are displayed for all predictions
We also tested the influence of baseline SCI on separate CSF and PET biomarkers progression at the same follow‐up points. Based on the normal group, PACC SCI individuals tended to have greater AV45 PET uptake at 48 months, which was very close to statistical significance (standardized β estimate: 0.06, P = .067; Figure 4). Similar prediction relationships were not found in CSF data (P > .20; Figure 4).
Other models were fitted modestly (RMSEA < 0.10, NFI > 0.95, IFI > 0.95, CFI > 0.95, TLI > 0.90, and CMIN/DF < 5.5). Decreased CSF Aβ1‐42 level at baseline was associated with higher CSF p‐tau181 and t‐tau level at 24 months (−0.06, P < .001 and −.05, P = .003, respectively), whereas increased AV45 PET uptake at baseline showed correlations with higher CSF p‐tau181 and t‐tau level at 24 months (0.03, P = .065 and 0.03, P = .098, respectively).
4. DISCUSSION
In the present study, we provided amplified evidence for the associations of SCI with Alzheimer's pathologies and clinical progression among CN individuals. CN with SCI possessed a faster accumulation of brain amyloidosis and a higher risk of conversion to Aβ positivity, suggesting a predictive link of SCI with amyloidosis progression. In addition, SCI showed a potential for predicting the longitudinal increase of p‐tau signature and the decrease of brain glucose metabolism. Furthermore, baseline SCI individuals were at a greater risk of clinical conversion. All these findings support that SCI can provide imperative information for early detection and intervention.
Our study is the first to report the predictive relationship of baseline SCI in cognitively normal individuals with the progress of amyloid pathology. It crucially replenishes the gap in current research and further elaborates the impact of cognition on subsequent pathological and clinical progression. 5 , 8 We found greater conversion proportions to Aβ positivity in SCI individuals compared with normal, whether in CSF or PET imaging. This result is in concordance with the recent findings from a non‐demented cohort. 8 However, it should be noted that we defined SCI using more acceptable and strict cutoffs based on the 10th percentile of baseline CN participants in our sample of pure CN subjects, 5 , 31 because it enables cognition status, evaluated via non‐invasive cognitive measures, to be informative in a very early phase, and also provides more objective criteria for population selection of disease‐modifying therapy and clinical trials compared with subjective memory decline. 9 , 10
Faster increments of brain amyloid burden were correlated with PACC SCI as well as SCI on the language domain, suggesting that the relationships between cognitive decline and AD pathologies were not restricted to memory alone. 32 Compared to normal groups, we found that SCI individuals had a lower CSF Aβ1‐42 level at the baseline, whereas CSF Aβ1‐42 change rates did not show distinct differences. The findings indicated that the amyloid pathology would or may already have been developed in SCI individuals. It also suggested a dominant role of subthreshold amyloid changes that surpasses the impact of cognition in the pathological process. 8 In any case, our data demonstrated a predictive relationship of CSF Aβ1‐42 on amyloid PET and supported the temporal evolution of the first appearance of CSF abnormality followed by PET abnormality. 33 Although CSF and PET Aβ biomarkers track the same protein, they deliver unique information about the underlying AD pathogenesis process. 34 , 35 The different performance of baseline SCI in predicting subsequent CSF and PET pathologies was consistent with the temporal evolution, in which abnormality initially appeared in CSF, followed by PET. 33 It is reasonable to speculate that SCI performed a more accurate prediction of amyloid PET than CSF. Studies with longer follow‐up and larger sample sizes are needed to verify this conjecture.
Baseline SCI was also proved to have the potential for predicting the advancement of tau pathology. In the whole cohort, we found that the CSF p‐tau of the PACC SCI subjects had a higher growth trend compared to that of the normal group. This longitudinal trend was significant in subjects with CSF p‐tau level under the threshold despite that compared with the normal group, SCI individuals did not present higher conversion rates to suprathreshold. This result supports a previous finding that some states of soluble p‐tau (p‐tau181 and p‐tau217) change synchronizing with the initial accumulation of Aβ aggregates in the early phase. 36 We, therefore, can argue that SCI could be utilized as an early predictor of longitudinal changes of tau pathology. On the other hand, even though PACC SCI individuals showed a slightly higher risk of progressing to tau abnormality presented by PET, this result must be interpreted with more caution due to the greater uptake of SCI groups compared to normal groups at baseline. In addition, CSF tau in our data showed modest but not tight associations with tau PET tracer, which is in contrast to previous studies reporting good accordance. 37 , 38 However, it provides evidence to the hypothesis, in which tau status in CSF and PET may represent a different facet of progressive tau pathology. 39 , 40
Individuals with baseline memory SCI compared with the normal had lower brain glucose metabolism over time, indicating a possible link between SCI and neurodegeneration. Only a slight trend of difference was found between memory SCI/normal groups in conversion rates from normal to abnormal. One possible explanation for this finding is that the short follow‐up period (6 years) cannot lead to adequate cases of abnormality conversion, for it restricted us to track the neurodegenerative process sufficiently.
Overall, baseline SCI was the harbinger of greater rates of clinical progression, whether in diagnostic implication or the global assessment of cognitive and functional impairment. 41 All types of SCI groups, except for the visuospatial functioning domain, showed a higher risk of clinical progression, which was in conformity with previous findings. 5 , 42 Moreover, multi‐domain SCI was more prone to advance than single domain SCI. In addition, SCI individuals remained at higher risk of clinical conversion adjusted for age and FDG PET, indicating that SCI still has the potential for predicting clinical progression, after excluding the possible influence of mild neurodegenerative pathologies related to age. Although these composite scores on cognition all showed some predictive power in clinical progression, the PACC and ADNI memory scores seemed to show different sensitivities to Aβ‐related pathology and neurodegeneration, respectively. Future research should compare these cognition composite scores in detecting early SCI of various domains.
There were several limitations to be acknowledged. The employment of tau‐PET tracer 18F‐AV1451 restricted the enrollment of CN subjects in recent years. Most ADNI participants have progressed to MCI or AD dementia at first scan. The remaining shared a short average follow‐up time, leading to less powerful observations. In addition, the thresholds selected for tau PET and CSF biomarkers in the present study were not the only ones recognized. There are other calculated cutoffs and equally valid methods for defining metaROIs.
5. CONCLUSIONS
Together, our findings provide valuable insights into the correlations of subtle cognitive impairment with AD‐related pathologies. In the preclinical stage, baseline SCI will likely be a powerful marker of clinical and pathological progression. It may also provide benefits for screening the population at high risk for AD.
CONFLICT OF INTEREST
None of the authors has financial disclosures or conflicts of interest.
ACKNOWLEDGMENTS
This study was supported by grants from the National Natural Science Foundation of China (91849126 and 81771148), Shanghai Technology and Science Key Project in Healthcare (No. 17441902100), Shanghai Municipal Science and Technology Major Project (No. 2018SHZDZX03), and ZHANGJIANG LAB, Tianqiao and Chrissy Chen Institute, and the State Key Laboratory of Neurobiology and Frontiers Center for Brain Science of Ministry of Education, Fudan University. Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (US Department of Defense award number W81XWH‐12‐2‐0012). Data used in the preparation for this article were derived from the ADNI database (http://adni.loni.usc.edu/). The authors express appreciation to contributors of the ADNI database. The ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Shen X‐N, Kuo K, Yang Yu‐X,et al. Alzheimer's Disease Neuroimaging Initiative. Subtle cognitive impairment as a marker of Alzheimer's pathologies and clinical progression in cognitively normal individuals. Alzheimer's Dement. 2021;13:e12198. 10.1002/dad2.12198
REFERENCES
- 1. Jack CR Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9:119‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sperling R, Mormino E, Johnson K. The evolution of preclinical Alzheimer's disease: implications for prevention trials. Neuron. 2014;84:608‐622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jack CR Jr, Bennett DA, Blennow K, et al. NIA‐AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:280‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Toledo JB, Bjerke M, Chen K, et al. Memory, executive, and multidomain subtle cognitive impairment: clinical and biomarker findings. Neurology. 2015;85:144‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jedynak BM, Lang A, Liu B, et al. A computational neurodegenerative disease progression score: method and results with the Alzheimer's disease neuroimaging initiative cohort. Neuroimage. 2012;63:1478‐1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gomar JJ, Bobes‐Bascaran MT, Conejero‐Goldberg C, Davies P, Goldberg TE. Utility of combinations of biomarkers, cognitive markers, and risk factors to predict conversion from mild cognitive impairment to Alzheimer disease in patients in the Alzheimer's disease neuroimaging initiative. Arch Gen Psychiatry. 2011;68:961‐969. [DOI] [PubMed] [Google Scholar]
- 8. Elman JA, Panizzon MS, Gustavson DE, et al. Amyloid‐beta positivity predicts cognitive decline but cognition predicts progression to amyloid‐beta positivity. Biol Psychiatry. 2020;87:819‐828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Buckley RF, Maruff P, Ames D, et al. Subjective memory decline predicts greater rates of clinical progression in preclinical Alzheimer's disease. Alzheimers Dement. 2016;12:796‐804. [DOI] [PubMed] [Google Scholar]
- 10. Vogel JW, Varga Dolezalova M, La Joie R, et al. Subjective cognitive decline and beta‐amyloid burden predict cognitive change in healthy elderly. Neurology. 2017;89:2002‐2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pontecorvo MJ, Devous MD, Kennedy I, et al. A multicenter longitudinal study of flortaucipir (18F) in normal ageing, mild cognitive impairment and Alzheimer's disease dementia. Brain. 2019;142:1723‐1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gustavson DE, Elman JA, Sanderson‐Cimino M, et al. Extensive memory testing improves prediction of progression to MCI in late middle age. Alzheimers Dement (Amst). 2020;12:e12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mattsson N, Insel PS, Donohue M, et al. Predicting reduction of cerebrospinal fluid beta‐amyloid 42 in cognitively healthy controls. JAMA Neurol. 2015;72:554‐560. [DOI] [PubMed] [Google Scholar]
- 14. Jonaitis EM, Koscik RL, Clark LR, et al. Measuring longitudinal cognition: individual tests versus composites. Alzheimers Dement (Amst). 2019;11:74‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dowling NM, Hermann B, La Rue A, Sager MA. Latent structure and factorial invariance of a neuropsychological test battery for the study of preclinical Alzheimer's disease. Neuropsychology. 2010;24:742‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koscik RL, La Rue A, Jonaitis EM, et al. Emergence of mild cognitive impairment in late middle‐aged adults in the wisconsin registry for Alzheimer's prevention. Dement Geriatr Cogn Disord. 2014;38:16‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Knopman DS, Jack CR Jr, Wiste HJ, et al. Short‐term clinical outcomes for stages of NIA‐AA preclinical Alzheimer disease. Neurology. 2012;78:1576‐1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vos SJ, Xiong C, Visser PJ, et al. Preclinical Alzheimer's disease and its outcome: a longitudinal cohort study. Lancet Neurol. 2013;12:957‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bittner T, Zetterberg H, Teunissen CE, et al. Technical performance of a novel, fully automated electrochemiluminescence immuno‐assay for the quantitation of beta‐amyloid (1‐42) in human cerebrospinal fluid. Alzheimers Dement. 2016;12:517‐526. [DOI] [PubMed] [Google Scholar]
- 20. Hansson O, Seibyl J, Stomrud E, et al. CSF biomarkers of Alzheimer's disease concord with amyloid‐beta PET and predict clinical progression: a study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement. 2018;14:1470‐1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Meyer PF, Binette AP, Gonneaud J, Breitner JCS, Villeneuve S. Characterization of alzheimer disease biomarker discrepancies using cerebrospinal fluid phosphorylated tau and AV1451 positron emission tomography. JAMA Neurol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Blennow K, Shaw LM, Stomrud E, et al. Predicting clinical decline and conversion to Alzheimer's disease or dementia using novel Elecsys Abeta(1‐42), pTau and tTau CSF immuno‐assays. Sci Rep. 2019;9:19024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ossenkoppele R, Rabinovici GD, Smith R, et al. Discriminative accuracy of [18F]flortaucipir positron emission tomography for Alzheimer disease vs other neurodegenerative disorders. JAMA. 2018;320:1151‐1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Landau SM, Harvey D, Madison CM, et al. Associations between cognitive, functional, and FDG‐PET measures of decline in AD and MCI. Neurobiol Aging. 2011;32:1207‐1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Landau SM, Harvey D, Madison CM, et al. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology. 2010;75:230‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Coley N, Andrieu S, Jaros M, Weiner M, Cedarbaum J, Vellas B. Suitability of the clinical dementia rating‐sum of boxes as a single primary endpoint for Alzheimer's disease trials. Alzheimers Dement. 2011;7:602‐610.e2. [DOI] [PubMed] [Google Scholar]
- 27. Hamaker EL, Kuiper RM, Grasman RP. A critique of the cross‐lagged panel model. Psychol Methods. 2015;20:102‐116. [DOI] [PubMed] [Google Scholar]
- 28. Kenny DA, Harackiewicz JM. Cross‐lagged panel correlation—practice and promise. J Appl Psychol. 1979;64:372‐379. [Google Scholar]
- 29. Hu L‐T, Bentler PM. Cutoff criteria for fit indexes in covariance structure analysis: conventional criteria versus new alternatives. Struct Equ Modeling. 1999;6:1‐55. [Google Scholar]
- 30. Bentler PM. Comparative fit indexes in structural models. Psychol Bull. 1990;107:238‐246. [DOI] [PubMed] [Google Scholar]
- 31. Jack CR Jr, Knopman DS, Weigand SD, et al. An operational approach to National Institute on Aging‐Alzheimer's Association criteria for preclinical Alzheimer disease. Ann Neurol. 2012;71:765‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. La Joie R, Perrotin A, Egret S, et al. Qualitative and quantitative assessment of self‐reported cognitive difficulties in non‐demented elders: association with medical help seeking, cognitive deficits, and beta‐amyloid imaging. Alzheimers Dement (Amst). 2016;5:23‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Blennow K, Mattsson N, Scholl M, Hansson O, Zetterberg H. Amyloid biomarkers in Alzheimer's disease. Trends Pharmacol Sci. 2015;36:297‐309. [DOI] [PubMed] [Google Scholar]
- 34. Mattsson N, Insel PS, Donohue M, et al. Independent information from cerebrospinal fluid amyloid‐beta and florbetapir imaging in Alzheimer's disease. Brain. 2015;138:772‐783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krance SH, Cogo‐Moreira H, Rabin JS, Black SE, Swardfager W. Reciprocal predictive relationships between amyloid and tau biomarkers in Alzheimer's disease progression: an empirical model. J Neurosci. 2019;39:7428‐7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barthelemy NR, Li Y, Joseph‐Mathurin N, et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer's disease. Nat Med. 2020;26:398‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brier MR, Gordon B, Friedrichsen K, et al. Tau and Abeta imaging, CSF measures, and cognition in Alzheimer's disease. Sci Transl Med. 2016;8:338ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chhatwal JP, Schultz AP, Marshall GA, et al. Temporal T807 binding correlates with CSF tau and phospho‐tau in normal elderly. Neurology. 2016;87:920‐926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. La Joie R, Bejanin A, Fagan AM, et al. Associations between [(18)F]AV1451 tau PET and CSF measures of tau pathology in a clinical sample. Neurology. 2018;90:e282‐e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mattsson N, Smith R, Strandberg O, et al. Comparing (18)F‐AV‐1451 with CSF t‐tau and p‐tau for diagnosis of Alzheimer disease. Neurology. 2018;90:e388‐e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Williams MM, Storandt M, Roe CM, Morris JC. Progression of Alzheimer's disease as measured by clinical dementia rating sum of boxes scores. Alzheimers Dement. 2013;9:S39‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Papp KV, Buckley R, Mormino E, et al. Clinical meaningfulness of subtle cognitive decline on longitudinal testing in preclinical AD. Alzheimers Dement. 2020;16:552‐560. [DOI] [PMC free article] [PubMed] [Google Scholar]