

Abstract
Preventing and delaying emergence of drug-resistance is an essential goal of antimalarial drug development. Monotherapy and highly mutable drug targets have each facilitated resistance, and both are undesirable in effective long-term strategies against multi-drug resistant (MDR) malaria. Heme remains an immutable and vulnerable target, since it is not parasite-encoded and its detoxification during haemoglobin degradation, critical to parasite survival, can be subverted by drug-heme interaction as in the case of quinolines and many other drugs1–5. We describe here a novel antimalarial chemotype which combines the heme-targeting character of acridones, together with a chemosensitizing component that counteracts resistance to quinoline antimalarial drugs. Beyond the essential intrinsic characteristics common to deserving candidate antimalarials (high potency in vitro against both pan-sensitive and MDR P. falciparum, efficacy and safety in vivo after oral administration, inexpensive synthesis and favourable physicochemical properties), our initial lead, T3.5 (3-chloro-6-(2-diethylamino-ethoxy)-10-(2-diethylamino-ethyl)-acridone), demonstrates unique synergistic properties. In addition to “verapamil-like” chemosensitization to chloroquine and amodiaquine against quinoline-resistant parasites, T3.5 also results in an apparently mechanistically distinct synergism with quinine and with piperaquine. This latter synergy, evident in both quinoline-sensitive and quinoline-resistant parasites, has been demonstrated both in vitro and in vivo. In summary, this innovative acridone design merges intrinsic potency and resistance-counteracting functions in one molecule, and represents a novel strategy to expand, enhance, and sustain effective antimalarial drug combinations.
While feeding within the host red blood cell, malaria parasites ingest and degrade vast amounts of haemoglobin as a source of amino acids, consequently releasing toxic free heme as a by-product6. The parasites protect themselves from the toxic insult by converting heme into an insoluble crystalline material termed “hemozoin” within the acidic digestive vacuole (DV)7,8. Because both haemoglobin degradation and heme detoxification are essential for parasite survival, these processes are important targets for antimalarial drug development2. The process of heme detoxification is widely believed to be the primary target of quinoline antimalarials (e.g., chloroquine and quinine), and it remains one of the most attractive and durable drug development targets1,3–5, particularly because the complexity of the DV environment and the immutable nature of the heme molecule likely have delayed the development of quinoline resistant malaria for decades (chloroquine) or centuries (quinine) in the past9.
The now-evident resistance to chloroquine is directly associated with mutations in the gene encoding the DV membrane protein Plasmodium falciparum Chloroquine Resistance Transporter (PfCRT), which results in reduced drug concentration at the target without altering the heme target itself10–13. In this case, in contrast to drug resistance on the basis of protein target mutations, the target remains vulnerable and the organism susceptible if access to the target can be restored. For this reason chemosensitizers (or so called “resistance reversal agents”) that interact with PfCRT to “reverse” quinoline resistance have been studied, but have largely failed to gain traction as candidate components of antimalarial combinations. Beyond the challenge of achieving adequate potency and safety, quinoline chemosensitizers have also lacked intrinsic antimalarial efficacy, and thus when combined with a quinoline antimalarial, would effectively result in monotherapy14–16. Because of the critical value of heme detoxification as a drug target and the appeal of preserving or restoring quinoline efficacy, we have sought to develop an intrinsically active antimalarial that also restores quinoline sensitivity to MDR parasites.
Exploiting the acridone chemical structure, we engineered a novel scaffold with both features incorporated into one molecule: 1) a heme-targeting tricyclic mainframe with an ionizable side chain to promote accumulation in the DV; and 2) a chemosensitization moiety at the 10(N) position to counteract quinoline resistance (Figure 1). The rigid tricyclic aromatic acridone core promotes π-π stacking for heme binding. The side chain at the 6-position engages one of the propionates of heme in an ionic bond and promotes acid trapping in the DV. The side chain attachment at the central nitrogen atom provides a hydrogen bond acceptor needed for the chemosensitization function. This feature is a well-established component of the pharmacophore for effective chloroquine chemosensitizers17,18, including previously described acridone chemosensitizers16, and further confirmed by the ineffectiveness as a quinoline chemosensitizer of 3-chloro-6-(2-diethylamino-ethoxy)-10H-acridone (T2), an intrinsically potent acridone derivative lacking the essential side chain attached to the central nitrogen atom (Supplementary Table 1).
Figure 1.
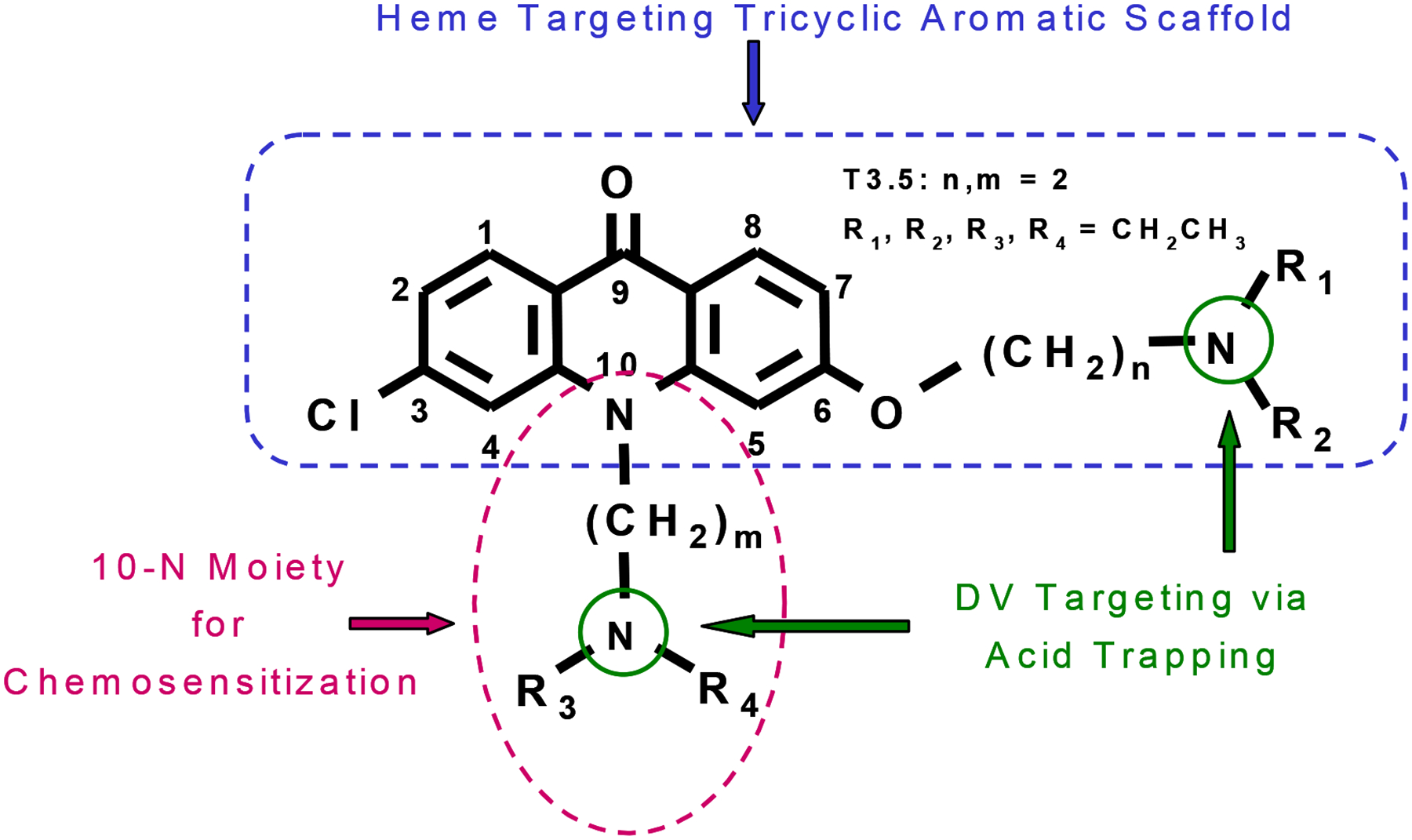
Generalized chemical structure of dual-function acridone derivatives. The rigid tricyclic aromatic acridone core promotes π-π stacking for heme binding. The side chain attachment at the central nitrogen atom provides a hydrogen bond acceptor needed for the chemosensitization function, and together with the side chain at the 6-position facilitate accumulation in the digestive vacuole (DV) via acid trapping.
Twelve compounds synthesized following this rational design approach were initially screened for intrinsic activity (Supplementary Table 2). In vitro antimalarial potency was demonstrated against a panel of chloroquine sensitive and MDR strains of P. falciparum with different geographic and genetic backgrounds. Results from testing of our initial lead compound T3.5, 3-chloro-6-(2-diethylamino-ethoxy)-10-(2-diethylamino-ethyl)-acridone, are shown in Table 1. In vivo intrinsic antimalarial efficacy of T3.5 against patent infection in mice was demonstrated using once-daily oral dosing for three days in two murine models. A dose of 100 mg/kg/day T3.5 diminished P. berghei parasitemia by 95%; against P. yoelii dose-response testing showed ED50 and ED90 values of 56 and 88 mg/kg/day, respectively (Table 2). Initial high dose testing (256 mg/kg/day orally or 200 mg/kg/day intraperitoneally) was curative. No overt toxicity or behaviour change was observed in the assessment of general measures of animal well-being (weight, grooming, or locomotor activity), and there has been no apparent in vitro mammalian cell cytotoxicity against the proliferation of murine splenic lymphocytes or human foreskin fibroblast cells (Supplementary Table 3). Apparent structural similarities between T3.5 and tricyclic antidepressants led us to assess the activity of T3.5 in a model of cloned biogenic amine transporters. Unlike cyclic antidepressants, T3.5 had no significant affinity with serotonin, dopamine, or norepinephrine transporters (Supplementary Table 4).
Table 1.
Intrinsic in vitro antimalarial activity against P. falciparum.
| Compound | IC50* (nM) vs. P. falciparum | |||
|---|---|---|---|---|
| D6** | Dd2** | 7G8** | Tm90-C2B** | |
| T3.5 | 44.8 ± 5.2 | 77.3 ± 6.0 | 85.9 ± 6.8 | 71.3 ± 7.4 |
| Chloroquine | 8.4 ± 1.9 | 124.7 ± 9.9 | 235.7 ± 25.6 | 122.7 ± 10.5 |
| Quinine | 19.4 ± 1.1 | 87.2 ± 9.7 | 77.3 ± 6.9 | 55.8 ± 5.5 |
Values are the means ± standard errors of the mean (S.E.M.) from eight independent experiments, each in quadruplicate, utilizing MSF assay with 0.2% parasitemia and 2% hematocrit.
D6 (Africa): chloroquine sensitive; Dd2 (Indochina): multi-drug resistant; 7G8 (Brazil): multi-drug resistant; Tm90-C2B (Thailand): multi-drug (including atovaquone and anti-folate) resistant.
Table 2.
Synergism of T3.5/quinine combination in vivo against patent infection of P. yoelii in mice.
| Effect | Dose (mg/kg/day)* | ||
|---|---|---|---|
| T3.5 alone | Quinine alone | T3.5:Quinine Combination | |
| ED50 | 56 ± 7 | 39 ± 4 | 14:14 ± 4:4 |
| ED75 | 70 ± 11 | 57 ± 9 | 19:19 ± 4:4 |
| ED90 | 88 ± 20 | 85 ± 25 | 24:24 ± 7:7 |
Values are the means ± standard deviation (S.D.).
The in vitro interactions of T3.5 with other antimalarials were assessed using a rigorous fixed-ratio combination strategy16,19. In combination with five prototypical quinoline derivatives, T3.5 proved synergistic with chloroquine, amodiaquine, quinine, or piperaquine, but not with mefloquine against the MDR P. falciparum strain Dd2 (Figure 2A and 2B). As illustrated by the isobologram in Figure 2A, there was no synergy in the additive interaction between T3.5 and chloroquine against the chloroquine sensitive parasite D6. In contrast, the synergy with quinine is distinct. Classic verapamil-like quinoline chemosensitizers (including earlier acridones16) modulate drug sensitivity only in drug-resistant parasites, the effect is more pronounced in the “Old World” (Asian/African) phenotype than in the “New World” (American/Oceanic) phenotype, and often micro-molar concentrations are required14–16. As shown in Figure 2C (solid line), the T3.5/quinine combination is entirely different, demonstrating equal synergy in both Dd2 (Indochina) and 7G8 (Brazil) strains of P. falciparum, and more remarkably, synergy against quinine-sensitive D6 (Africa) (mean FIC indices 0.50, 0.49, and 0.64, respectively). It is noteworthy that similar synergy characteristics were observed for T3.5/piperaquine combination as well (Figure 2C, dashed line).
Figure 2.

Isobolograms of the in vitro interaction of A) T3.5/chloroquine against MDR P. falciparum strain Dd2 and chloroquine sensitive P. falciparum strain D6 (mean FIC indices are 0.72 and 0.97, respectively); B) T3.5/amodiaquine, T3.5/mefloquine, T3.5/quinine and T3.5/piperaquine combinations against MDR strain Dd2 (mean FIC indices are 0.73, 1.25, 0.50, and 0.60, respectively); C) T3.5/quinine (solid line) combination against chloroquine sensitive strain D6 and MDR strains Dd2 and 7G8 (mean FIC indices are 0.64, 0.50, and 0.49, respectively); T3.5/piperaquine (dashed line) combination against chloroquine sensitive strain D6 and MDR strains Dd2 and 7G8 (mean FIC indices are 0.66, 0.60, and 0.51, respectively). The mean FIC indices ± standard errors of the mean (S.E.M.) were derived from three independent experiments. The x-axis represents the FICs of quinolines, and y-axis represents the FICs of T3.5. The diagonal line (FIC index = 1) indicates the hypothetical additive drug effect. A concave curve (FIC index < 1) below the diagonal line typically indicates synergy of the combination, while a convex curve (FIC index > 1) above the diagonal line indicates antagonism.
The synergy between T3.5 and quinine was also observed in vivo against patent infection with quinine-sensitive P. yoelii (K). In combination, substantial reductions in the effective dosage of T3.5 and quinine were noted. For example, the values of ED90 for T3.5 and quinine alone were 88 and 85 mg/kg/day, respectively, but the same effect was achieved by combining less than 1/3 of those doses (Table 2).
The profile of the interaction between T3.5 and PfCRT was assessed using novel PfCRT mutant lines with various point mutations at codon 76, previously utilized to distinguish characteristics of verapamil-like chemosensitizers12,13,20–22. With chloroquine, the interaction with T3.5 mirrored that of verapamil, however, the chemosensitization pattern of T3.5 with quinine clearly differs (Table 3). In contrast to verapamil-like chemosensitizers that are more intrinsically potent against P. falciparum with a K76N mutation, the intrinsic efficacy of T3.5 is comparable against all four PfCRT mutant lines. Unlike verapamil, T3.5 is synergistic with quinine against the quinine-hypersensitive mutant line 106/176I. Moreover, in contrast to verapamil, T3.5 potentiated quinine activity against the chloroquine-sensitive parent line Sudan 106/1K76. In fact, T3.5 demonstrated chemosensitization to quinine against more than two-dozen strains of P. falciparum with wide variations in quinoline resistance profiles (data not shown). Such broad synergistic interaction with quinine has not been previously described.
Table 3.
Effect of PfCRT position 76 mutations on chemosensitization of quinine by T3.5 and verapamil in P. falciparum.
| Compound | IC50* (nM) P. falciparum | |||
|---|---|---|---|---|
| 106/1K76** | 106/176T*** | 106/176N*** | 106/176I*** | |
| Verapamil alone | 25800 ± 128 | 1779 ± 105 | 632 ± 78 | 8630 ± 312 |
| T3.5 alone | 93.6 ± 12 | 86.9 ± 8.5 | 134 ± 15 | 100.8 ± 18 |
| Chloroquine alone | 15.9 ± 1.4 | 188 ± 2.3 | 147 ± 6.8 | 158 ± 4.4 |
| Chloroquine + 500 nM verapamil | 17.2 ± 1.6 | 51.2 ± 3.2 | 39.9 ± 5.4 | 47.7 ± 5.6 |
| Chloroquine + 25 nM T3.5 | 14.5 ± 2.1 | 67.8 ± 9.8 | 74.3 ± 6.8 | 58.5 ± 6.1 |
| Quinine alone | 112 ± 18 | 204 ± 19 | 186 ± 21 | 20.8 ± 2.1 |
| Quinine + 500 nM verapamil | 118 ± 10 | 71.4 ± 8.9 | 53.5 ± 4.6 | 32.1 ± 2.4 |
| Quinine + 25 nM T3.5 | 55.6 ± 4.0 | 56.7 ± 5.3 | 71.2 ± 6.8 | 5.5 ± 0.8 |
Values are the means ± standard errors of the mean (S.E.M.) from three independent experiments, each in quadruplicate, utilizing the MSF assay with 0.2% parasitemia and 2% hematocrit.
Sudan 106/1K76 is known to have 7 of the 8 mutations in the pfcrt gene necessary for chloroquine resistance.
Mutant line selected with chloroquine pressure from 106/1K76.
Investigations of the proposed mechanisms of intrinsic activity confirmed the interaction of T3.5 with heme, interference with hemozoin formation, and drug accumulation within the DV. Examination of Giemsa-stained blood smears after T3.5 exposure revealed a dose-related failure of parasite progression to schizogeny and markedly diminished hemozoin content in both P. yoelii infected mice and in T3.5 exposed P. falciparum (D6 and Dd2) in vitro (Supplementary Figure 1). Quantitative measurement of hemozoin production correlates well with microscopy (Supplementary Figure 2). Intracellular hemozoin incorporated during 24 hours of incubation with 500 nM T3.5 decreased from 0.78 to 0.06 fmole per parasitized erythrocyte in the D6 strain, and from 1.09 to 0.25 fmole in the Dd2 strain. In addition, T3.5 inhibits β-hematin formation in vitro (Supplementary Table 5) and interacts with heme to form a soluble complex with strong affinity at pH 4.8 (Supplementary Figure 3). Visual localization of T3.5 inside the P. falciparum infected erythrocyte by confocal fluorescence microscopy indicates uptake and accumulation in the DV (Figure 3 and Supplementary Methods).
Figure 3.

Confocal microscopy of localized T3.5 fluorescence in two intraerythrocytic P. falciparum trophozoites. Fluorescence was determined in live P. falciparum-infected erythrocytes using a laser (emission line 351 nm) scanning confocal microscope. The figure shows the intrinsic fluorescence of T3.5 (blue) superimposed on the brightfield transmission image of the infected cells.
There is current consensus that combination therapy is essential to delay the emergence of drug resistance and preserve the long-term usefulness of new antimalarials23. The search for alternatives to artemisinin-based combinations has elevated interest in developing “dual-function” antimalarials with both intrinsic and chemosensitizing efficacy24,25. Some existing DV-active compounds exhibit intrinsic efficacy against Plasmodium parasites regardless of the chloroquine resistance profile. Because these compounds are likely to interact or interfere with PfCRT, there has been speculation that they might also exhibit chemosensitization in combination with quinolines26,27, but no such activity has been demonstrated. The present study provides the first evidence of such dual functionality in a single molecule, and offers a powerful approach to combination therapy.
If a global effort to eradicate malaria is to be successful, the drug therapy component of that effort must address the gaps and weaknesses in the armamentarium of currently available therapies. Affordability, safety in the most vulnerable, and low susceptibility to drug resistance adaptations each represent unmet needs. In contrast to other drug classes (e.g., respiratory inhibitors and anti-folates), development of drug resistance to DV-active drugs that target heme-processing has been slow (e.g., chloroquine) or of low order (e.g., quinine), and this remains the only identified immutable parasite target. For older drugs in this class, cost is very low, there is extensive experience with their use in children and during pregnancy, and short-course therapy is facilitated by very long elimination times. Paradoxically then, although the failure of chloroquine is at the core of the global drug-resistance crisis, these drugs actually characterize the ideals now sought in new antimalarial drugs for both treatment and intermittent prophylaxis. The concept described in this paper specifically aims to exploit the strengths of such compounds by making possible a new combination therapy strategy. The ability to maintain the efficacy of newer drugs (e.g., piperaquine) and to restore the efficacy of older drugs (e.g., chloroquine) represents a uniquely powerful tool, and one ideally suited to achieve the broadest possible benefit as a renewed malaria eradication effort proceeds.
Methods Summary
In vitro antimalarial activity was determined by the malaria SYBR Green I-based fluorescence (MSF) assay described previously28with slight modification16. In vivo efficacy was determined using once-daily oral dosing for three days against patent infection in two murine models. In vitro interaction of T3.5 and other antimalarial agents was assessed by isobolar analysis using fixed-ratio combination, where drugs were diluted in fixed ratios of starting concentrations predetermined to generate well-defined concentration response curves16,19. Effect of PfCRT mutations on drug interaction was determined by a modified MSF method described previously13,16.
Detailed methods and synthesis procedures are provided in Supplementary Information
Supplementary Material
Acknowledgements
We thank Dennis Kyle from Walter Reed Army Institute of Research for the generous gift of P. falciparum parasite Tm90-C2B, and the Malaria Research and Reference Resource Center for supplying P. falciparum parasites D6, Dd2, and 7G8. We thank Andrew P Waters and Chris J Janse from Leiden University for the donation of GFP labeled P. berghei strain ANKA. We thank Lorraine Jones-Brando and Robert Yolken for the cytotoxicity (HFF) data. We are grateful to Anda Cornea from Oregon Regional Primate Center for the confocal imaging. We thank Katherine Liebman, Cheryl Hudson, Steve Burgess, and David Peyton for compound characterization. We acknowledge financial support from the Merit Review Program of the Department of Veterans Affairs. United States patent applications have been filed by the U.S. Department of Veterans Affairs to protect the intellectual property described in this report.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Competing Interests statement The authors declare that they have no competing financial interests.
References
- 1.Yayon A, Cabantchik ZI & Ginsburg H Identification of the acidic compartment of Plasmodium falciparum-infected human erythrocytes as the target of the antimalarial drug chloroquine. Embo J 3, 2695–700 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Olliaro PL & Goldberg DE The Plasmodium digestive Vacuole: Metabolic Headquarters and Choice Drug Target. Parasitol Today 11, 294–7 (1995). [DOI] [PubMed] [Google Scholar]
- 3.Sullivan DJ Jr., Gluzman IY, Russell DG & Goldberg DE On the molecular mechanism of chloroquine’s antimalarial action. Proc Natl Acad Sci U S A 93, 11865–70 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wellems TE Plasmodium chloroquine resistance and the search for a replacement antimalarial drug. Science 298, 124–6 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Kumar S, Guha M, Choubey V, Maity P & Bandyopadhyay U Antimalarial drugs inhibiting hemozoin (beta-hematin) formation: a mechanistic update. Life Sci 80, 813–28 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Krugliak M, Zhang J & Ginsburg H Intraerythrocytic Plasmodium falciparum utilizes only a fraction of the amino acids derived from the digestion of host cell cytosol for the biosynthesis of its proteins. Mol Biochem Parasitol 119, 249–56 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Pagola S, Stephens PW, Bohle DS, Kosar AD & Madsen SK The structure of malaria pigment beta-haematin. Nature 404, 307–10 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Egan TJ Haemozoin formation. Mol Biochem Parasitol 157, 127–36 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Foley M & Tilley L Quinoline antimalarials: mechanisms of action and resistance. Int J Parasitol 27, 231–40 (1997). [DOI] [PubMed] [Google Scholar]
- 10.Fitch CD Chloroquine resistance in malaria: a deficiency of chloroquine binding. Proc Natl Acad Sci U S A 64, 1181–7 (1969). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krogstad DJ et al. Efflux of chloroquine from Plasmodium falciparum: mechanism of chloroquine resistance. Science 238, 1283–5 (1987). [DOI] [PubMed] [Google Scholar]
- 12.Fidock DA et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell 6, 861–71 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cooper RA et al. Alternative mutations at position 76 of the vacuolar transmembrane protein PfCRT are associated with chloroquine resistance and unique stereospecific quinine and quinidine responses in Plasmodium falciparum. Mol Pharmacol 61, 35–42 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Martin SK, Oduola AM & Milhous WK Reversal of chloroquine resistance in Plasmodium falciparum by verapamil. Science 235, 899–901 (1987). [DOI] [PubMed] [Google Scholar]
- 15.van Schalkwyk DA & Egan TJ Quinoline-resistance reversing agents for the malaria parasite Plasmodium falciparum. Drug Resist Updat 9, 211–26 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Kelly JX et al. Design, synthesis, and evaluation of 10-N-substituted acridones as novel chemosensitizers in Plasmodium falciparum. Antimicrob Agents Chemother 51, 4133–40 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhattacharjee AK, Kyle DE & Vennerstrom JL Structural analysis of chloroquine resistance reversal by imipramine analogs. Antimicrob Agents Chemother 45, 2655–7 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhattacharjee AK, Kyle DE, Vennerstrom JL & Milhous WKA 3D QSAR pharmacophore model and quantum chemical structure--activity analysis of chloroquine(CQ)-resistance reversal. J Chem Inf Comput Sci 42, 1212–20 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Fivelman QL, Adagu IS & Warhurst DC Modified fixed-ratio isobologram method for studying in vitro interactions between atovaquone and proguanil or dihydroartemisinin against drug-resistant strains of Plasmodium falciparum. Antimicrob Agents Chemother 48, 4097–102 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waller KL et al. Chloroquine resistance modulated in vitro by expression levels of the Plasmodium falciparum chloroquine resistance transporter. J Biol Chem 278, 33593–601 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Cooper RA, Hartwig CL & Ferdig MT pfcrt is more than the Plasmodium falciparum chloroquine resistance gene: a functional and evolutionary perspective. Acta Trop 94, 170–80 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Cooper RA et al. Mutations in transmembrane domains 1, 4 and 9 of the Plasmodium falciparum chloroquine resistance transporter alter susceptibility to chloroquine, quinine and quinidine. Mol Microbiol 63, 270–82 (2007). [DOI] [PubMed] [Google Scholar]
- 23.(WHO), W. H. O. Guidlines for the treatment of malaria 2006. 1108 (2006). [Google Scholar]
- 24.Biot C & Chibale K Novel approaches to antimalarial drug discovery. Infect Disord Drug Targets 6, 173–204 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Egan TJ & Kaschula CH Strategies to reverse drug resistance in malaria. Curr Opin Infect Dis 20, 598–604 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Biot C et al. Insights into the mechanism of action of ferroquine. Relationship between physicochemical properties and antiplasmodial activity. Mol Pharm 2, 185–93 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Burgess SJ et al. A chloroquine-like molecule designed to reverse resistance in Plasmodium falciparum. J Med Chem 49, 5623–5 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P & Riscoe M Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob Agents Chemother 48, 1803–6 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.