

Abstract
Our insight into the diverse and complex nature of dilated cardiomyopathy (DCM) genetic architecture continues to evolve rapidly. The foundations of DCM genetics rest on marked locus and allelic heterogeneity. While DCM exhibits a Mendelian, monogenic architecture in some families, preliminary data from our studies and others suggests that at least 20–30% of DCM may have an oligogenic basis, meaning that multiple rare variants from different, unlinked loci, determine the DCM phenotype. It is also likely that low-frequency and common genetic variation contribute to DCM complexity, but neither has been examined within a rare variant context. Other types of genetic variation are also likely relevant for DCM, along with gene-by-environment interaction, now established for alcohol- and chemotherapy-related DCM. Collectively, this suggests that the genetic architecture of DCM is broader in scope and more complex than previously understood. All of this elevates the impact of DCM genetics research, as greater insight into the causes of DCM can lead to interventions to mitigate or even prevent it, and thus avoid the morbid and mortal scourge of human heart failure.
Keywords: dilated cardiomyopathy, genetics, human, genomics
Subject Terms: Cardiomyopathy, Heart Failure
I. Introduction.
Insight into the genetic basis of dilated cardiomyopathy (DCM) has increased remarkably in recent years. Initial research was fueled by the clinical recognition in the 1980s to 1990s that some DCM could be familial,1 with the inference that familial DCM could then have a genetic basis. Starting in the 1990s, the phenotype and pedigree relationships of these families provided the framework for the first discovery of genes with rare disease-causing variants.1 Since then, the molecular genetic basis of DCM has continued to emerge.2–10
Our understanding of the genetic architecture of DCM also continues to evolve. By genetic architecture, we mean a description of how genetic factors contribute to the development and manifestation of phenotype. Some cardiovascular genetic conditions, such as hypertrophic cardiomyopathy (HCM) or the Long QT syndrome, in most cases fit well into a simple Mendelian paradigm, in which one highly penetrant rare variant usually explains most genetic cause in an individual or family. In contrast, emerging evidence suggests that a substantial proportion of DCM (up to 20%11 in our preliminary data; up to 38% in an earlier European cohort12) may have an oligogenic basis, meaning that multiple rare (minor allele frequency [MAF] <0.1%) variants from different (unlinked) loci may determine the DCM phenotype.13 Moreover, the focus on these rare coding sequence variants in genes that disrupt protein structure or function, while by far the most fruitful investigation thus far, does not address low-frequency (MAF ≥0.1% but ≤5%) or common (MAF >5%) variation, non-coding sequence relevant for gene promoters or other regulatory regions, non-coding RNAs, or epigenetic phenomena, all of which may be relevant for DCM genetics. Indeed genome-wide association studies indicate that common variants are also relevant for DCM.14–19 Low-frequency variants, likely also relevant for DCM, have been evaluated in a single study.20 We also recognize, as described below in detail, that “variant” within the context of DCM usually means one or a few nucleotides, although larger rare structural variants may also be relevant. Increasing evidence of the involvement genetic factors outside of the simple Mendelian paradigm suggests that the genetic architecture of DCM is broader in scope and more complex than previously understood.
All of this elevates study of DCM genetics to a highly impactful role in broadening our understanding of the fundamental causes of human heart failure. Heart failure (HF), the multi-organ response to diminished cardiac output and decreased blood pressure, is a downstream late-phase event that signals a major decrement in left ventricular systolic function.21 While enormous effort continues to be applied to understand the pathogenesis and treatment of HF, we suggest that understanding the root cause of HF, specifically the myocardial dysfunction resulting from underlying DCM genetics, will provide foundational insights into generation of preventive or treatment options.
This progress has been made possible by dramatic advances in next-generation sequencing (NGS) technologies. Even though exomes or genomes can be sequenced and analyzed rapidly, identifying genes yet unknown to cause DCM still takes enormous time, effort and care to acquire the clinical, pedigree and experimental molecular genetic data to conclusively establish causal relationships. Even once a DCM is established, proving that any specific variant in such a gene causes DCM within a conventional Mendelian paradigm remains an arduous task.
What is also new is an appreciation that clinical DCM at times emerges following an environmental insult with an impact rendered more severe by underlying genetic risk. Situations in which the effect of an environmental exposure on phenotype is modified by the genetic makeup of an individual are termed gene-environment (GxE) interactions.22 GxE interactions in DCM are not surprising and have been previously examined.23 Viral infections24 or other inflammatory conditions have been postulated for years as triggers for DCM, and recent genetic evidence links exposure to alcohol25 or chemotherapeutic agents26 to the development of DCM. Thus, the extent of DCM and its emergence due to GxE interaction remains a key new research frontier.
Despite these gains, the probability of identifying genetic cause by conventional clinical genetic testing is still at best 35%,10 even with familial DCM, where multiple closely related individuals in a multigenerational pedigree have DCM without other known or usually detectable clinical cause, thus meeting a formal definition of “idiopathic” DCM. What are we missing? Is most of DCM a genetic condition? The heritable and familial nature of DCM2 would suggest that indeed it is, but clinical studies have suggested that familial DCM can be identified in less than one-third of probands.1 In non-familial DCM, the genetic basis remains uncertain. To clarify this key question the DCM Precision Medicine study has proposed to test the hypotheses that most of DCM, whether familial or non-familial, does indeed have a genetic basis.27
Even with our growing understanding of DCM genetic architecture, caveats remain. First, we only find what we look for, and DCM genetics thus far has focused primarily on rare variants in coding sequence. Moreover, even as we expand our knowledge of DCM genetics, we also recognize the wisdom of theoretical physicist Albert Einstein: “The larger the circle of light, the greater the perimeter of darkness around it.” This reflects the reality of DCM genetics. While we are grateful for the enormous progress of the past 30 years, we trust that the next 30 years will bring similarly monumental discoveries. Our goal must be to continue to pursue a greater understanding of DCM genetic risk and to develop new strategies to prevent, treat, and cure, human DCM and the HF that results from it. We must also abandon conventional thinking that may hinder novel insight, remembering the words of historian Daniel Boorstin who reminds us that “the greatest obstacle to discovery is the illusion of knowledge.”
II. Clinical DCM.
Definition of DCM.
The classic definition of DCM is that of left ventricular systolic dysfunction (LVSD) with left ventricular enlargement (LVE), after all usual clinically detectable causes (except genetic) have been excluded.28 This also serves as the clinical definition of idiopathic dilated cardiomyopathy (IDC). For decades, IDC has been the starting point for investigation of familial dilated cardiomyopathy (FDC) and DCM genetic discovery.29 We clarify that the use of DCM herein is synonymous with the more technical term IDC. Other causes of a non-idiopathic phenotype fitting generic LVSD and LVE (and at times, also included by authors in a generic use of the “DCM” moniker) include myocardial infarction (MI) from coronary artery disease (CAD), primary valvular heart disease (VHD), congenital or other structural heart disease (CHD), and endocrine disorders such as hypothyroidism, among others.29 From a clinical genetics standpoint, all of these causes of myocyte dysfunction stem from conditions beyond the myocyte itself, even if secondary to cardiac pathology (e.g., valvular or vascular from cardiac endocarditis, or CAD resulting in MI, respectively). Some causes, extrinsic to the myocardium, result from other organ dysfunction in a patient (e.g., hypothyroidism). Other causes stem from exposure to environmental toxins, such chemotherapy, chronic alcohol abuse, acute heavy metal exposure, or other extrinsic conditions.29
Most DCM is adult onset, most commonly occurring in midlife, but can also present in neonates, young children and adolescence as well as individuals in the last decades of life. DCM occasionally presents with neuromuscular or mitochondrial syndromic disease.30–32 Why rare variants in the same gene cause DCM onset over such a wide range of ages remains largely unexplained, although modifier variants, likely rare, low-frequency and common, as well as environmental conditions presumably interact. Such conditions we presume include not only usual physiology such as exercise, or pathophysiology such as hypertension, but also drugs, alcohol, radiation, and ambient conditions such as air, water and other environmental exposure. The totality of non-genetic factors has been termed the exposome, the sum of all non-genetic exposures, including dietary, physical, psychological stressors, and the increasingly extensive environmental chemical exposures, all of which interact with genetics.33
Differentiating DCM from the other classic cardiomyopathies, HCM, restrictive cardiomyopathy (RCM), or arrhythmogenic right ventricular cardiomyopathy (ARVC) continues to be essential for the clinical cardiologist. ARVC, classically defined at a disease of the right ventricle, has been established to also cause biventricular or left-sided disease that can present as DCM with early arrhythmia, and for this reason has also been termed Arrhythmogenic Cardiomyopathy.34 This is because, for clinical cardiovascular genetic practice, we must still rely on the correlation of phenotype with any genetic test results to make sense of a purported genetic etiology. This is a “phenotype-first” approach, where the indication for and interpretation of clinical genetic testing is driven by the cardiovascular phenotype of an affected individual.35 We predict that this approach will remain the standard for at least this decade because the genomes of ostensibly “normal” individuals (i.e., without known or detectable cardiovascular disease) carry rare variants, even in known DCM genes, that are unconnected to any phenotype. Questions of variant pathogenicity will become even more of an issue as clinical cardiologists are referred increasing numbers of patients who have undergone clinical exome sequencing for a non-cardiovascular condition and have had “secondary findings” found in a relevant gene for cardiovascular disease.36 The clinician is obligated to conduct screening for a presumptive phenotype.37 Of the 73 genes on the updated American College of Medical Genetics and Genomics (ACMG) secondary findings gene list, 33 are related to cardiovascular conditions, and of those 33, slightly over half are related to cardiomyopathies.38
Finally, a key point for cardiovascular clinicians is that DCM is not HF and HF is not DCM. This is an oft forgotten fact, that DCM can be asymptomatic for years and thus can occur well before the onset of HF, and that HF is a late-phase manifestation of DCM39 (Figure 1). This concept provides the foundational rationale for DCM clinical genetic practice: specifically, that DCM identified prior to the onset of HF in at-risk family members may be more responsive to treatment than later phase disease. That is, arresting the causal pathway to HF driven by DCM with family-based screening may delay or even prevent late phase disease, as has been shown preliminarily.40 This hypothesis – that medical intervention into very early DCM, well before the onset of HF, can prevent progression to fully defined DCM (Figure 1: Phase 1, Stage 1A,) – has not yet been formally tested or informed with gene-specific data. Nevertheless, inferential evidence as well as decades of clinical observation strongly support this concept. Ideally, formal testing of this hypothesis will occur during this decade.
Figure 1. The asymptomatic and symptomatic phases of DCM.
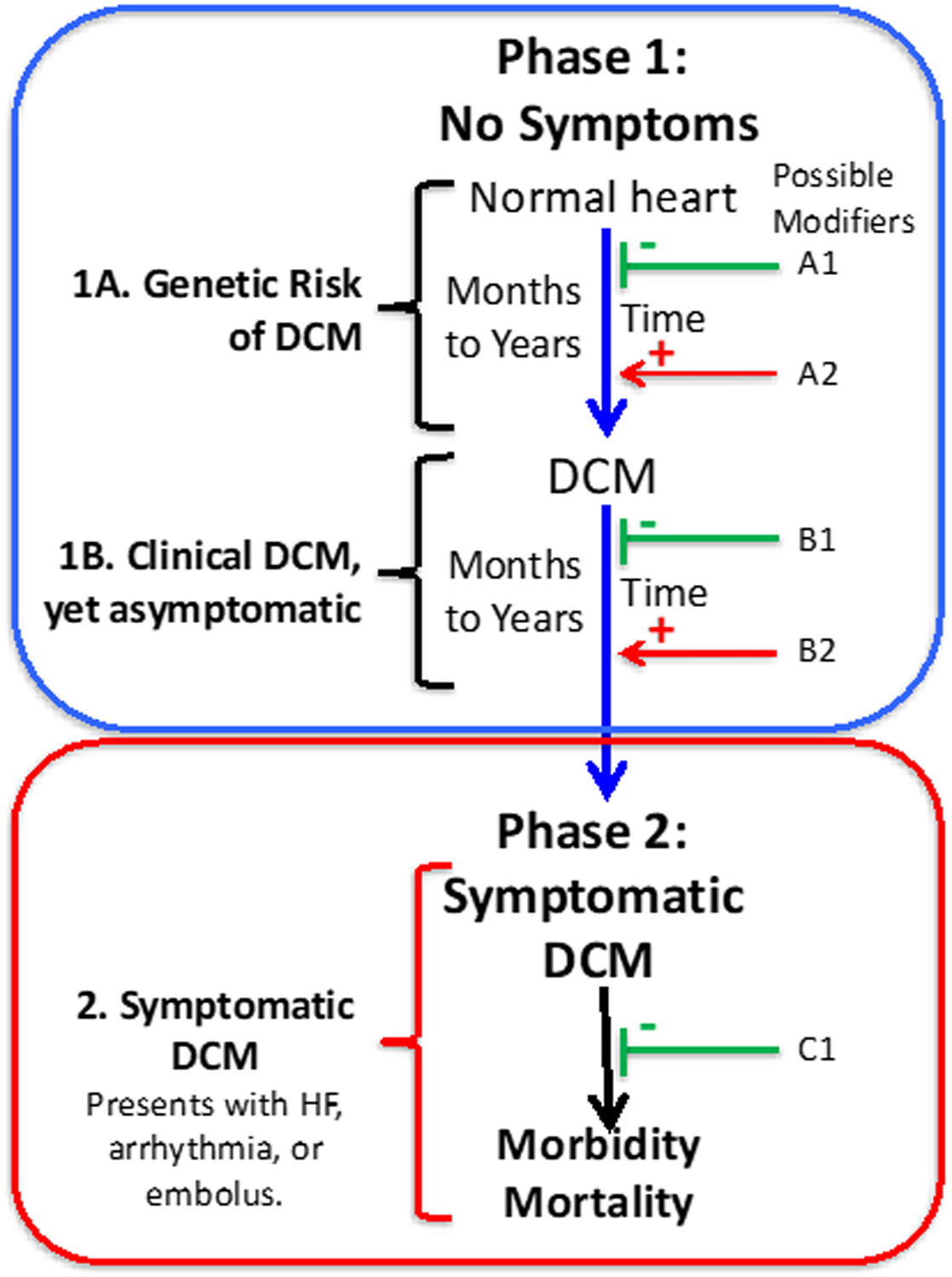
The causal pathway of DCM is illustrated, as adapted,16, 30 in adult-onset DCM. Phase 1 includes two periods, both asymptomatic. In the first period (1A), individuals who harbor rare DCM variants have risk of developing DCM over time. Genetic information identifies the individuals who would benefit from periodic clinical screening to detect early clinical disease. In Phase 1B, DCM can be clinically detected by appropriate imaging studies but remains asymptomatic, evading detection for years unless surveillance clinical screening is undertaken to detect it. In Phase 2, the individual with DCM develops symptoms, most commonly with HF, which triggers a medical evaluation with a diagnosis of HF and the eventual discovery of the underlying DCM. Factors that may diminish or block these causal pathways to DCM or HF are shown in green (A1, B1, C1)); environmental factors could include good nutrition, a low salt diet, low blood pressure, or drug therapy (e.g., angiotensin-converting enzyme inhibitors or β-blockers). Factors that may accelerate the causal pathway to DCM or HF are shown in red (A2, B2); examples include hypertension, alcohol use, or exposure to chemotherapy or other myocardial toxins. Genomic modifiers, both injurious or protective, include rare, low frequency or common variants that could be operative at all points. Detection of a “pre-DCM phenotype” that portends the transition from Phase 1A to Phase 1B could permit earlier introduction of conventional medical therapy in an effort to block the development of DCM. Following the transition to Phase 1B, all efforts need to be made to prolong this phase to avert late phase symptomatic disease in Phase 2.
III. DCM Molecular Genetics.
Variant classification.
Current clinical variant classification standards are defined by The American College of Medical Genetics and Genomics (ACMG): (P, pathogenic; LP, likely pathogenic; VUS, variant of uncertain significance; LB, likely benign; and B, benign).41 These categorizations are assigned by applying adjudication criteria using currently available clinical and experimental evidence that support a pathogenic and/or benign role, with central criteria including those specific to sequence consequence, as in the case of predicted null variation for genes that truncation is an established disease-causing mechanism, allele frequency, case-level and family segregation data, and functional evidence.41
Despite efforts to make gene- and disease-specific modifications to the foundational ACMG variant adjudication approach,11, 42 many variants in DCM genes are classified as VUS. This is because some adjudication criteria are often challenging to apply, including case evidence and experimental evidence, although tools such as CardioClassifier43 and the modified Polyphen prediction for sarcomere genes,44 may help with functional predictions in the absence of experimental evidence. However, the lack of publicly available case- and family-level data is a consistent barrier to moving a VUS to LP or P. When opportunities present to clinically study a family with a rare VUS(s), efforts to track how a variant is segregating with disease should be pursued to resolve clinical uncertainty whether clinically- or research-based. The practice of variant classification is both dynamic and probabilistic in nature and may change overtime as new data presents,45 which may result in clinically significant impacts.46
DCM gene variants meeting criteria for P or LP classifications predominantly manifest as nonsynonymous missense mutations in coding regions or splice junctions, or as nonsense or frameshift variants resulting in prematurely truncated protein products. Owing to its extensive coding regions, rare, pathogenic, truncating variants are routinely identified in Titin (TTN).47, 48 As the largest known protein, the canonical TTN sequence spans 363 exons alternatively spliced into several variably sized isoforms (Figure 2). Resulting protein products range in size between 5,604 (the small Novex-3 cardiac isoform) and 34,350 amino acids (the large N2BA cardiac isoform) and vary predominantly in elastic I-band regions. Truncating A-band TTN variants (TTNtvs), identified in 15–25% of families with DCM and 10–18% of sporadic cases,49 are now recognized as the most frequent monogenic cause of DCM.
Figure 2:

A. Schematic representation of the cardiac sarcomere. Depicted are actin and troponin-complex containing thin filaments and myosin thick filaments interacting with Z-disk and M-band anchored Titin. A-band, I-band, and H-band segments are indicated by horizontal bars. B. TTN exon and protein structure. The TTN canonical transcript (NM_001267550.1) is encoded by 363 exons alternatively spliced into several variably sized isoforms. N2BA (NM_001256850.1), N2B (NM_003319.4), Novex-1 (NM_133432.3), Novex-2 (NM_133437.3) and Novex-3 (NM_133379.3) isoforms are cardiac expressed, while N2BA (NM_133378.4) is expressed in skeletal muscle. Exon structure of each isoform is depicted. Exons are not drawn to scale. Grey color indicates exons spliced from the processed transcript. Exons encoding Ig-like (red, triangle), Z-repeat (pink, square), PEVK (blue, hexagon), fibronectin type III (yellow, star), and protein kinase (cyan, circle) domains are indicated by the indicated colors and symbols. For visual simplicity, symbols are used to mark only the first exon encoding each domain, the exon immediately following encoding of a different domain, or exons encoding multiple domains. Encoded proteins are depicted below each exon structure. Proteins are scaled to show relative proportions of each transcript occupied by Z-disk (grey), near Z-disk (yellow), I-band (blue), A-band (red), and M-band (grey-green) domains. Depicted colors match those in A. Boundaries and amino acid ranges are indicated for each isoform.
DCM Locus and allelic heterogeneity.
DCM has significant locus heterogeneity (Table 1). While modern DCM testing panels routinely offer sequencing of >50 genes, these represent only a subset of genes with suspected DCM involvement. DCM gene curation efforts led by a sub-working group of the Clinical Genome Resource (ClinGen)50 Cardiovascular Clinical Domain Working Group identified 267 loci with suggested involvement in DCM (Supplemental Table).10 The breadth of locus heterogeneity, alongside low individual gene prevalence, distinguishes DCM from HCM and ARVC, for which genetic variation in a handful of key genes, MYH7 and MYBPC3 in HCM, and PKP2, DSP, and DSG2 in ARVC, explain 80% or 40–60% of all cases, respectively.2
Table 1. Disease Validity Classifications for DCM Genes Considered Within a Monogenic Framework.
The 51 genes curated for DCM by the ClinGen DCM Gene Curation Panel are grouped by levels of evidence supporting a Mendelian relationship with the DCM phenotype.5 Genes meeting a classification of moderate, strong, or definitive, as defined by the ClinGen framework, have high levels of evidence that can be used for molecular diagnosis of DCM in the clinical setting. Genes with limited classifications have variable degrees of evidence, some of which may emerge as high evidence genes in the future. Genes in the minimal evidence category, assigned a “disputed” or “no known disease relationship” classification, are unlikely to accumulate evidence to reach a high classification standard. However, while highly unlikely to be a monogenic cause of DCM, it is possible that genes in this category may have a modest clinical effect as a genetic modifier of disease. Abbreviations: AD, autosomal dominant; AR; autosomal recessive; DCM, dilated cardiomyopathy; MOI, mode of inheritance; SD, semi-dominant (both AD and AR mechanisms reported).
| ClinGen DCM Gene-Disease Validity Classification | Gene | Protein | MOI | Function | |
|---|---|---|---|---|---|
| High Evidence Genes | Moderate | ACTC1 | Actin, alpha cardiac 1 | AD | Sarcomere |
| Moderate | ACTN2 | Alpha actinin 2 | AD | Sarcomere | |
| Definitive | BAG3 | BCL2-associated anthanogene 3 | AD | Co-chaperone/ Heat shock protein |
|
| Definitive | DES | Desmin | AD | Cytoskeleton | |
| Strong | DSP | Desmoplakin | AD | Desmosome | |
| Definitive | FLNC | Filamin C | AD | Sarcomere | |
| Moderate | JPH2 | Junctophilin 2 | SD | Junctional membrane | |
| Definitive | LMNA | Lamin A/C | AD | Nuclear envelope | |
| Definitive | MYH7 | Myosin heavy chain 7, beta | AD | Sarcomere | |
| Moderate | NEXN | Nexilin | AD | Cytoskeleton | |
| Definitive | PLN | Phospholamban | AD | Sarcoplasmic reticulum | |
| Definitive | RBM20 | RNA binding motif protein 20 | AD | Nucleus/RNA-binding | |
| Definitive | SCN5A | Sodium channel, voltage-gated type V, alpha | AD | Ion channel | |
| Definitive | TNNC1 | Troponin C, slow | AD | Sarcomere | |
| Moderate | TNNI3 | Troponin I | AD | Sarcomere | |
| Definitive | TNNT2 | Troponin T | AD | Sarcomere | |
| Moderate | TPM1 | Tropomyosin 1 | AD | Sarcomere | |
| Definitive | TTN | Titin | AD | Sarcomere | |
| Moderate | VCL | Metavinculin | AD | Cytoskeleton | |
| Low/ Variable Evidence Genes | Limited | ABCC9 | ATP-binding cassette, subfamily C, member 9 | AD | Ion channel, sarcomere |
| Limited | ANKRD1 | Ankyrin repeat domain 1 | AD | Sarcomere | |
| Limited | CSRP3 | Cysteine-rich protein 3 | AD | Cytoskeleton | |
| Limited | CTF1 | Cardiotrophin 1 | AD | Cytokine | |
| Limited | DSG2 | Desmoglein 2 | AD | Desmosome | |
| Limited | DTNA | Dystrobrevin, Alpha | AD | Sarcomere | |
| Limited | EYA4 | Eyes absent 4 | AD | Nucleus | |
| Limited | GATAD1 | GATA zinc finger domain-containing protein 1 | AR | Nucleus | |
| Limited | ILK | Integrin-linked kinase | AD | Cytoskeleton | |
| Limited | LAMA4 | Laminin, alpha 4 | AD | Extracellular matrix | |
| Limited | LDB3 | LIM domain binding 3 | AD | Cytoskeleton | |
| Limited | MYBPC3 | Myosin-binding protein C | AD | Sarcomere | |
| Limited | MYH6 | Myosin heavy chain 6, alpha | AD | Sarcomere | |
| Limited | MYL2 | Myosin light chain 2 | AD | Sarcomere | |
| Limited | MYPN | Myopalladin | AD | Sarcomere | |
| Limited | NEBL | Nebulette | AD | Sarcomere | |
| Limited | NKX2–5 | NL2 homeobox 5 | AD | Nucleus | |
| Limited | OBSCN | Obscurin | AD | Sarcomere | |
| Limited | PLEKHM2 | Pleckstrin homology domain-containing protein, family M, member 2 | AR | Cytoskeleton | |
| Limited | PRDM16 | PR domain-containing protein 16 | AD | Nucleus | |
| Limited | PSEN2 | Presenilin 2 | AD | Plasma membrane | |
| Limited | SGCD | Sarcoglycan, delta | AD | Cytoskeleton | |
| Limited | TBX20 | T-box 20 | AD | Nucleus | |
| Limited | TCAP | Telethonin | AD | Sarcomere | |
| Limited | TNNI3K | TNNI3-interacting kinase | AD | Sarcomere | |
| Minimal Evidence Genes | No known disease relationship | LRRC10 | Leucine-rich repeat-containing protein 10 | AR | Cytoskeleton |
| No known disease relationship | MIB1 | Mindbomb E3 ubiquitin protein ligase 1 | AD | Cytoskeleton | |
| Disputed | MYL3 | Myosin light chain 3 | AD | Cytoskeleton | |
| No known disease relationship | NPPA | Natriuretic peptide precursor A | AR | Hormone | |
| Disputed | PDLIM3 | PDZ and LIM domain protein 3 | AD | Cytoskeleton | |
| Disputed | PKP2 | Plakophilin 2 | AD | Desmosome | |
| Disputed | PSEN1 | Presenilin 1 | AD | Plasma membrane |
The vast majority of clinically-identified DCM variants are private to single families, reflecting an added level of allelic heterogeneity. Pathogenic variants are usually distributed throughout all coding exons, although mutational hotspots have been noted, such as a 5 amino acid region in exon 9 of RBM20.51 A recent signal-to-noise analysis of exome-detected TTNtvs identified several A- and I-band hotspots that were more likely to be spliced into a greater proportion of coding transcripts in patients with DCM.52 TTNtvs that were identified incidentally and TTNtvs identified in general population databases were less likely to be localized to these regions. Such examples serve as a reminder that, while most DCM variants are unique to individual families, their distribution within a given gene is likely to be dependent on ultimate impact on protein structure and function. While DCM-relevant TTN variants appear to cluster predominantly in the A-band, the large number of coding exons introduces additional potential for compound heterozygosity involving multiple pathogenic variants. Compound heterozygosity has previously been reported in instances of TTN-related muscular disease, including tibial53 and limb girdle muscular dystrophies.54 In the latter study, a TTNtv was identified alongside a splice junction variant previously reported in a patient with DCM. While the pathogenic significance of TTNtvs is well-established, the impact, if any, of TTN missense variants for DCM has not been established.
The preponderance of pathogenic A-band TTNtvs in DCM is likely a consequence of constitutive inclusion of these exons in cardiac-expressed transcripts, N2BA, N2B, and Novex isoforms 1 and 255 (Figure 2). Not all TTN transcripts are expressed in the heart, and whether cardiac-expressed exons are impacted by a given TTNtv is a key factor in determining the likelihood of its pathogenicity.56 However, while A-band TTNtvs are a major genetic contributor to DCM, they are also identified in an estimated 0.2–2% of the general population.47, 57
Diversity in genetic studies has been limited to European ancestry cohorts.
Although non-ischemic DCM has been cited as having higher prevalence and worse outcomes in individuals of African ancestry,58 most genetic investigations have included only European cohorts even though genome-wide association findings are enriched in individuals of African ancestry or Hispanic ethnicity.59 The need for greater inclusion in DCM genetic studies is highlighted by reports citing higher likelihood of VUS detection in African populations,60 increased LV diameter amongst African and Hispanic VUS carriers,61 and enrichment of TTNtvs in individuals of European ancestry.62 Studies designed to target enrollment of under-represented populations, such as the DCM Precision Medicine study,27 will help provide the genetic data needed to validate these or identify novel population differences.
Research versus clinical standards.
The standards by which a variant should be considered of interest differ between clinical settings focused on patient care and research settings focused on scientific discovery. Importantly, LP and P classifications are not recommended to be applied to variants identified in non-established DCM genes,41 even though such variants, of necessity classified as VUS, may ultimately be found to have clinical relevance. Likewise, restricting determinations of pathogenicity to particular variant types or gene regions, such as A-band TTNtvs, is likely to exclude genetically significant variants located elsewhere in the gene.63, 64 Such challenges are not unique to the study of genetic DCM, however, it is important to recognize both the utility and limitations of existing adjudication criteria.
IV. DCM Clinical Genetics
DCM Transmission Patterns.
The pattern of transmission of DCM genetic risk has largely been described in a classic autosomal dominant model, incurring a 50% risk to all first-degree relatives (FDRs) (parents, siblings, children) of an affected individual to share the same genetic predisposition.1 While less common, autosomal recessive, X-linked, and mitochondrial inheritance patterns have also been reported.29 Data suggests that familial and non-familial DCM have a similar genetic background,65, 66 in part due to the fact that determining a familial categorization depends on the clinical evaluation of family members and can be restricted by penetrance or small family size.
DCM Genetic Architecture.
Over past 3 decades dozens of genes have been suggested to have variants causal for DCM, and while most of this gene-specific discovery literature has been summarized previously,1–8, 10, 29 a broadly-based systematic effort to curate the vast amount of literature surrounding genetic causes of DCM had never been done. To remedy this, ClinGen50 assembled an international panel to evaluate gene-disease relationships for isolated, non-syndromic DCM.67 To develop an initial gene list, publicly available gene databases were interrogated, resulting in a set of 267 genes (Supplemental Table). A final list of 51 genes (Table 1) underwent rigorous curation of available literature to classify the degree to which the data supports an association with human DCM. This work classified 19 genes from 10 gene ontologies as having “moderate,” “strong,” or “definitive” evidence to support a single-gene, Mendelian cause of DCM (Figure 3). The resulting clinically relevant gene list is consistent with findings of recent case-control analyses, with high evidence genes also enriched in DCM cases.68
Figure 3. DCM genetic architecture spans ten gene ontologies.
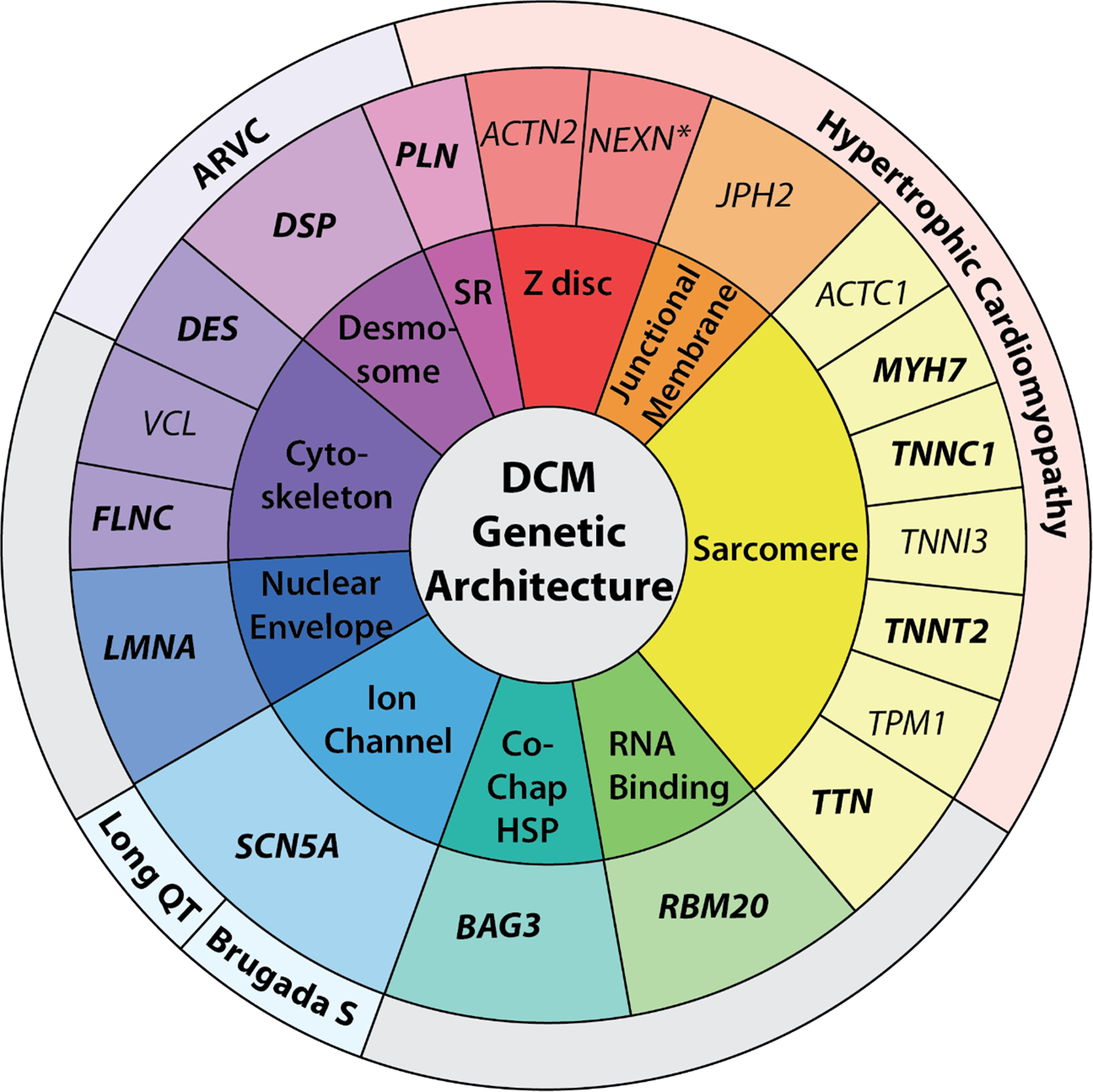
19 genes deemed highly clinically relevant (definitive and strong noted in bold; moderate in regular text) for DCM are in the middle ring. The outermost ring lists other high evidence phenotypic associations and the innermost risk provides an ontology classification for each gene, respectively. Of 19 DCM genes, 14 have also been classified as high levels of evidence in HCM, ARVC, LQTS, and/or Brugada Syndrome, except for NEXN, noted with an asterisk, which has limited evidence in HCM. Ontology abbreviations include: SR, sarcoplasmic reticulum; Co-Chap HSP, co-chaperone, heat shock protein. Figure from5.
For the 19 genes deemed highly clinically relevant, the ACMG variant interpretation criteria41 can be applied, where a P/LP variant in a clinically relevant gene can be considered a stand-alone cause of disease and, importantly, can be used for risk prediction in relatives. Notably, the ClinGen gene curation framework is designed specifically to evaluate gene-disease relationships under Mendelian-acting models, so that genes with high classifications (Definitive, Strong, Moderate) are usually considered to be clinically relevant for molecular diagnosis.
Genetic Evaluation of DCM
A genetic evaluation is recognized as a central component of the clinical management of DCM. A genetic evaluation is recommended to be family-based, including collection of medical and family history, clinical evaluation of at-risk relatives, and cascade genetic testing of variants in clinically relevant gene(s), when applicable. Specifically, when a P/LP variant is found in a high evidence, clinically relevant DCM gene, at-risk FDRs are recommended to be tested for this same variant for their own risk stratification to enable opportunity for early clinical screening and, if needed, intervention. For at-risk relatives found to share the same genetic predisposition as their family member with DCM, clinical surveillance including electrocardiography and imaging by echocardiogram or cardiac magnetic resonance (CMR) imaging is recommended at age-based intervals. For those that have normal clinical screening and do not harbor the DCM-causing variant, no further follow up is warranted.37
Even when a genetic cause is known, additional considerations in the risk assessment of the patient and family may be necessary.10 For example, some DCM-associated genes may have variable presentations, such as genes known to have conduction system abnormalities, as in DCM arising from P/LP variants in LMNA, DSP, SCN5A, FLNC, and/or DES genes. Others may require additional neuromuscular evaluation, as in the case of LMNA, some cases of TTN, and others. Further, reduced penetrance and variable expressivity may complicate the interpretation of data in the family.
With a large number of proposed genes, genetic testing panels for DCM include from 30 to >100 genes.67 However, despite the nearly universal use of large multi-gene NGS panels in DCM clinical genetic testing, current estimates show that genetic cause is only detected in about 20–35% of cases.11, 12 Even after tailoring ACMG variant adjudication criteria by the ClinGen MYH7 variant curation panel42 and further by the DCM Precision Medicine study,11 the majority of genetic results in DCM cases remain as variants of uncertain significance (VUS).
Notably, many clinically available genetic testing panels include genes with low degrees of evidence, representing “genes of uncertain significance” when evaluating through a Mendelian lens. Any variants identified in genes where the relationship and mechanism of action is not clearly defined should not exceed a classification greater than that of a VUS.41 Variants of uncertain significance in DCM genes are not recommended for use in clinical practice.37, 67, 69
More broadly, this gap in detecting genetic cause is likely driven by many factors, such as locus heterogeneity, illustrated by the extensive number of genes proposed relevant to the phenotype, with each DCM-associated gene accounting for only a small proportion of cause, the exception being TTNtvs. Still, TTN-related DCM is commonly shown to have reduced penetrance, suggesting a role of modifiers, which may include additional genetic burden of low-frequency or common variants and/or additional environmental insults.70 Further, significant allelic heterogeneity of variants in DCM genes also complicates clinical translation, with rare and private variants frequently reported.
Lastly, current variant interpretation standards are not designed for evaluating genetic variation under non-Mendelian mechanisms. This is highly relevant to DCM, as multiple variant architecture continues to be demonstrated in DCM in the literature.11–13 While a single variant acting in an autosomal dominant pattern is currently estimated to explain 20–40% of cases with detectable genetic cause, additional non-Mendelian models are also likely to be at play.
An Oligogenic Model of DCM
A multiple hit hypothesis, where multiple variants collectively contribute to DCM disease (Table 2), has been previously proposed.2, 10 As noted above, data supporting a multiple variant architecture has been replicated in exemplary pedigrees13 and cohort studies.11, 12 Citing key examples, in a large, multi-center DCM cohort, 38% of patients had multiple variants.12 In a LMNA-related cardiomyopathy study, 5 of 19 (26%) probands had multiple variants at play,13 and multiple rare variants were identified in 21% of cases in a pilot set of DCM probands from the DCM Precision Medicine Study.11
Table 2. Clinical impact of considering non-Mendelian models of DCM genetic architecture.
Broadening the scope of the genetic evaluation of DCM to consider multi-variant and gene-environment interaction models of disease has downstream implications for the approach to risk assessment as well as the ongoing management and care of DCM patients and families. However, any recommendations beyond those guideline-based are recommended to be conducted within an investigational environment, as mechanisms to define moderate-impact gene-disease relationships, to adjudicate disease-contributing variants that exceed a monogenic model, and to quantify environmental contributions to disease, in a clinical setting are needed to provide a tailored approach to care.
| Models of DCM Genetic Architecture | |||
|---|---|---|---|
| Components of a Genetic Evaluation | Mendelian Model | Multivariant Model | Gene-Environment Interaction (GxE) Model |
| Proband Medical History | • Exclude all other known clinical causes of DCM. | • Exclude all other known clinical causes of DCM. | • Comprehensive evaluation of environmental factors that may be contributing to DCM phenotype. |
| Family History (FHx) | • Identify a single lineage harboring the genetic risk. • ≥3 generations of FHx.57 |
• Seek evidence of bilineal inheritance. • Spectrum of penetrance and expression likely. • ≥3 generations of FHx. |
• Seek evidence of bilineal inheritance. • Comprehensive clinical and environmental data. • Spectrum of penetrance and expression expected. • >3 generations of FHx. |
| Pedigree Considerations | • Dominant features (e.g. males and females affected, male-to-male transmission, multiple generations with disease). • Variable expression and reduced penetrance may complicate interpretation. |
• May appear de novo or recessive, as proband may be the only individual with a complete DCM phenotype arising from a unique variant burden. • Variable age of onset may be observed depending on variant burden of each individual. • Relatives may have subtle/mild disease, be asymptomatic, or unaffected. |
• May appear de novo or recessive, as the proband may be the only individual with a complete DCM phenotype with sufficient burden of genetic and environmental factors • Variable age of onset as environmental factors more likely to occur in adulthood and multiple factors required to meet disease threshold • Relatives may have subtle/mild disease, be asymptomatic, or unaffected |
Pedigree Samples
|
 |
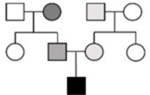 |
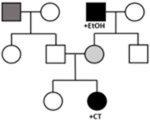 |
| Risk Counseling | • 50% chance to FDRs to share genetic predisposition. • Individuals with genetic predisposition may not develop disease at the same age or severity. |
• Discussion of disease threshold model, where multiple variants additively cause the phenotype. • 50% chance to FDRs to have each individual variant. • Reduced penetrance driven by variant burden. • Relatives may have mild or subclinical disease or be unaffected. |
• Discussion of disease threshold model, where multiple variants interact with environmental factor(s) to cause the phenotype. • 50% chance to FDRs to have each individual variant • Relatives may have mild or subclinical disease or be unaffected. • Environmental factors can change over time (e.g. age) • Motivational counseling strategies important in order to inspire preventive health behaviors, emphasizing on the role of environment and managing non-genetic risk factors as possible to mitigate risk. |
| Clinical Evaluation of Family | • ECHO or CMR, and ECG, of FDRs at baseline and repeated at age-defined intervals.28 | • ECHO or CMR, and ECG, of FDRs at baseline. • Above repeated at frequency recommended by clinician based on genetics and family presentation until data-driven guidelines available. |
• ECHO or CMR, and ECG, of FDRs at baseline. • Above repeated at frequency recommended by clinician based on genetics, environmental factors, and family presentation until data-driven guidelines available. |
| Genetic Testing (GT) of Proband | • 19 high evidence genes.5 | • At least 51 genes with varying degrees of human genetic evidence.5 • Engage in research to investigate DCM genetic architecture. |
• At least 51 genes with varying degrees of human genetic evidence.5 • Engage in research to investigate DCM genetic architecture, which may include calculation of polygenic risk. |
| Variant Interpretation | • Very rare variants (MAF <0.01% in all non-founder subpopulations111). • Case-level criteria to only include counts for strictly applied DCM phenotype.6 |
• Very rare, rare and low frequency variants (MAFs <0.01%, <0.05%, and <1%) • Case-level criteria to include idiopathic DCM and incomplete DCM phenotypes in counts (e.g. rEF only, LVE only, CSD only, etc) |
• Rare to common variation. • Case-level criteria for rare variants to include broad inclusion of phenotypes in case counts, including incomplete DCM phenotypes and GxE DCM phenotypes. |
| GT Considerations for FDRs | • GT of FDRs if P/LP variant(s) found in proband. • Discharge those with negative cascade GT from follow up.28 |
• Consider FDR GT for disease-contributing/causing variants, regardless of Mendelian classification. | • Consider FDR GT for disease-causing/contributing variants, regardless of Mendelian classification • Quantification of environmental risk burden (when data-driven approach available). |
| Clinical Recommendations | • Communicate risk with family • Baseline clinical screening of FDRs per guidelines. • Cascade GT of FDRs when applicable. • Continued surveillance of FDRs with genetic risk per guidelines.28 |
• Communicate risk with family. • Clinical screening of FDRs per guidelines. |
• Communicate risk with family. • Clinical screening of FDRs per guidelines. |
| Research Recommendations | • Research participation to study penetrance, expression, and additional genetic architecture of Mendelian DCM • This may also include investigational approaches to care. |
• Research participation to investigate DCM genetic architecture, which may also include investigational approaches to care: ○ Consider cascade GT of FDRs, and possibly second-degree relatives as indicated by pedigree, when relevant disease-causing/contributing variants are identified in the proband. ○ Relatives discharged from continued surveillance if all relevant variants are absent and clinical screening is negative, informed by clinical judgement. |
• Research participation to investigate DCM genetic architecture, which may also include investigational approaches to care: ○ Consider cascade GT that may well exceed FDRs when relevant disease-causing/ contributing variants are identified in the proband. ○ Quantification of environmental risk. ○ Consider continuing clinical surveillance in relatives if GxE burden is estimated to approach the disease threshold. ○ If not initially estimated to have a GxE burden warranting ongoing surveillance, consider repeating environmental risk quantification to re-estimate environmental risk burden; if elevated, surveillance recommendations may change as informed by clinical judgement. |
Under an oligogenic model, each variant in isolation may not be sufficient to cause disease, but multiple variants with low to moderate effect together incur sufficient burden for DCM development.2, 10 A detailed family history71 followed by comprehensive phenotypic assessment of at-risk relatives is essential to define the oligogenic architecture of DCM, and to clinically classify variants identified by genetic testing (Table 2). This is because disease-contributing variants, as opposed to disease-causing variants (classified as P/LP by current standards) may be inherited bilineally, that is, from both maternal and paternal sides of the proband’s pedigree, and in different combinations throughout the pedigree.2, 10 The differing variant burden creates clinical complexity across the pedigree, which may express in relatives as an incomplete DCM phenotype.
DCM Phenotypes Resulting from GxE Interaction
GxE interaction may also contribute to DCM. Initially thought to be of non-genetic cause, peripartum or pregnancy-associated cardiomyopathy (PPCM/PACM),72–74 chemotherapy-related (CTrCM),26 or alcohol-related cardiomyopathy (EtOHrCM),25 have since been shown to have a rare variant genetic background. Sequencing probands with DCM presenting after cardiotoxic exposure, including alcohol and chemotherapy, both having clear evidence of environmental impact, found an enrichment of TTNtvs.25, 26 Therefore, previously “phenotypes of uncertain genetic significance,” CTrCM and EtOHrCM, now have evidence of a rare variant genetic background. While rare variants in DCM genes have been detected in cases of PPCM/PACM,72–74 finding non-genetic environmental mechanisms in humans that are relevant for the PPCM/PACM phenotype has remained elusive.75, 76
Clinical translation of a complex genetic architecture
There are three prerequisites for genetic information to be able to be used for molecular diagnosis: (1) the phenotype is known to have genetic cause; (2) the relationship of genes with the phenotype is understood; and (3) the biological significance of a variant is clinically interpretable. As such, the DCM phenotype, including familial and non-familial DCM in addition to the GxE DCM phenotypes of CTrCM and EtOHrCM, has an established genetic background. In addition, gene-disease relationships have been authoritatively defined recently, with 19 genes currently rated to have high degrees of evidence in monogenic DCM (Table 2).
Despite establishing exemplary GxE interaction for DCM phenotypes, we currently lack a framework to clinically quantify the contribution of genetics or environmental impact in such cases. This presents a major challenge in the translation of genetic information into the care of the family. For GxE phenotypes arising in the setting of rare variants meeting Mendelian P/LP classification standards, proceeding with usual cascade clinical evaluation and genetic testing of family members may be pursued with some modifications (Table 2). For example, for relatives who test positive for a P/LP variant identified in the proband of their family with a GxE phenotype, a later onset penetrance or decreased disease severity may be anticipated if they do not share the environmental factors of the proband. However, when variant(s) are identified that do not meet the rigorous P/LP classification standards, we currently lack a framework to translate these disease-contributing variants to family-based clinical care. Until such guidance is established, disease-contributing variation will continue to be classified as VUS and lack clinical utility in DCM and in other established GxE DCM phenotypes, restricting the reach of genetic information to the care of this population.
To bridge these existing gaps to include multivariant and GxE models of DCM into clinical care, targeted efforts for gene discovery and a broadened approach to variant interpretation to define the role of disease-contributing variants of modest to moderate clinical effect is critically needed. Establishing non-Mendelian disease mechanisms in DCM will change the practice of genetics providers (Table 2). Further, it will enable an improved ability for molecular diagnosis, reducing the degree of uncertainty and lack of resolution for the many DCM patients and families that remain unsolved under current standards and approaches to genetic care.
V. Statistical Approaches to Discovery and Characterization of DCM Genes and Variants
Statistical Modeling of a Complex Genetic Architecture
Discoveries in human genetics are made and validated by correlation of phenotype with genotype. The conceptual underpinning of this process is a penetrance model, which provides a formal mathematical description of how genetic and environmental factors contribute to a particular observed trait in the population.22 The goal of discovery is to find which genes and variants contribute to this model; characterization then determines how genes and variants previously discovered contribute, possibly in the context of other factors. As reviewed above, the penetrance model for DCM is frequently characterized as simple Mendelian with age-dependent penetrance.2, 5 However, the qualifiers “reduced penetrance” and “variable expressivity” are typically added to accommodate substantial variation in presentation for carriers of the family’s variant in terms of age at onset and disease severity.2, 5 The necessity of such qualification, as well as the alternative oligogenic and GxE interaction models discussed above, suggests that DCM genetics may be better understood in the context of a more comprehensive multifactorial penetrance model.
Because DCM is defined by LVSD and LVE meeting certain thresholds, a threshold model involving underlying quantitative endophenotypes provides a useful framework (Figure 4).22, 77 This model accommodates the usual Mendelian paradigm in which the negative impact of a single rare LP/P rare variant on these quantitative endophenotypes eventually leads to disease, with age at onset and disease severity subject to random variation between individuals. However, it can also accommodate the additional complexity necessary to explain the full spectrum of phenotypic variation. We have previously reported that the presence or absence of additional rare variants modifies the severity of disease within families.2, 13, 78–80 Low-frequency and common variants may also play a role in DCM susceptibility; a recent study found a SNP-heritability estimate of 0.33 for DCM in African Americans,17 and heritability estimates above 0.30 have been found for left ventricular ejection fraction and size in large studies of various populations.18, 81, 82 When incorporated into a threshold model, these additional genetic factors can explain reduced penetrance and variable expressivity by modifying the impact of a single LP/P rare variant on the expected trajectory of the quantitative endophenotype (Figure 4). A threshold model also allows environmental factors to affect penetrance and expressivity through their modification of the expected trajectory of the quantitative endophenotypes determined by the genetic background.
Figure 4. A threshold model for DCM.
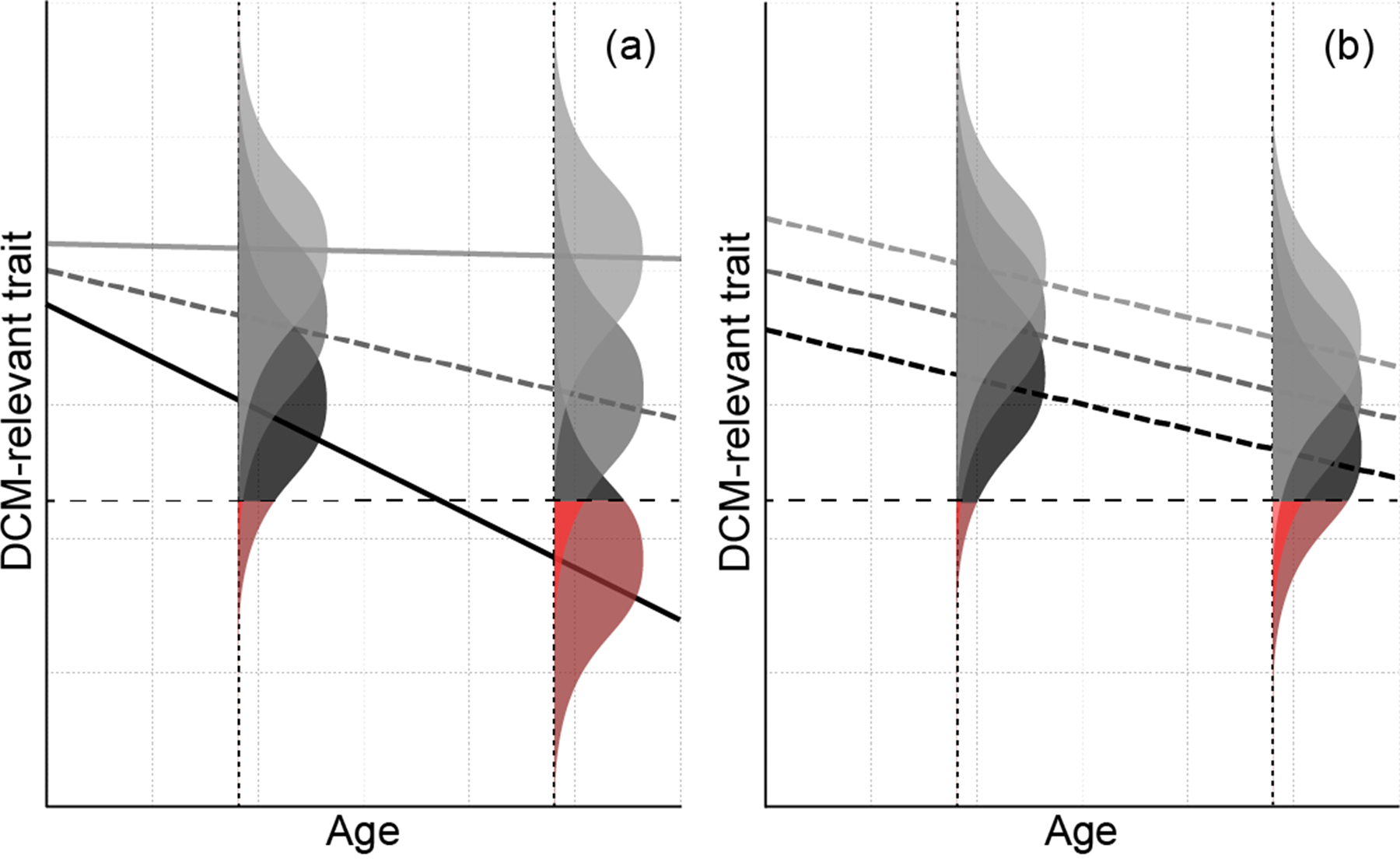
Lines denote the mean quantitative trait measurement (vertical axis) as a function of age (horizontal axis) in individuals with a particular genetic background. The distribution of the actual measurements in these individuals at a particular age arising from non-genetic variation is represented by the shaded bell curve centered on the mean for that age. The dashed horizontal line indicates the threshold below which an individual is considered to have DCM; the amount of total area in each bell curve shaded red indicates risk of DCM. Panel (a) illustrates how the number, or burden, of deleterious rare variants might impact the age trajectory with other factors held fixed. In the absence of such variants (light gray), neither the mean nor risk changes appreciably with age. While all groups have similar means at birth, individuals with higher burdens of deleterious rare variants (denoted by darker lines) have more rapid declines in the mean quantitative trait with age, which result in greater risk and more severe phenotypes, on average, at a given age as well as earlier onset. Panel (b) illustrates how polygenic effects arising from common variants might modify the average trajectory for the rare variant burden in the dashed middle curve from panel (a). In this example, the polygenic effect shifts the entire age trajectory up or down, either nearly eliminating the increased risk due to the rare variant burden (light gray) or exacerbating it (black). More complex descriptions of the age trajectory as a function of rare variants, common variants, and non-genetic factors are possible under this model; this example serves primarily to demonstrate that even a simple multifactorial model involving multiple genetic components can explain empirically observed age-dependent penetrance and variable expressivity.
Genotyping Assays
The genetic components of the penetrance model that can be interrogated in a particular study are determined in part by genotyping assay choice. High-coverage genome sequencing (GS) captures variation across the allele frequency spectrum in both coding and non-coding regions but entails significant upstream and downstream cost. As a result, more cost-effective assays tuned to specific subtypes of variants are frequently employed. Array-based genotyping directly targets common variants typically located in non-coding regions. However, the advent of large publicly available haplotype reference panels based on GS now allows accurate imputation of variants with MAFs as low as 0.1%, including those in coding regions, in diverse populations.83, 84 Private or extremely rare variants not represented in existing haplotype reference panels cannot be recovered by imputation, which leaves a niche for high-coverage exome sequencing (ES). Like GS, ES captures variation across the allele frequency spectrum but economizes on cost by focusing on variants that are likely to directly impact protein function at the expense of non-coding and structural variants.
Single Proband Designs
The availability of ES and GS has made variant discovery possible in clinical settings or “N = 1” studies involving a single proband. Doing so effectively with limited data requires focusing on a simple Mendelian penetrance model, which limits inquiry to rare or private variants with a high prior probability of being relevant to disease, typically in coding regions or genes with prior evidence of relevance to DCM. Candidate variants are prioritized and classified based on externally sourced biological annotations and epidemiological evidence, such as case counts, linkage or association studies, observed segregations, and population frequencies, and predictions from bioinformatics tools, to determine which might be relevant under the assumed penetrance model.11, 41–43
An important component of this approach is eliminating candidate rare variants that are too frequent in public reference samples to cause disease under the assumed Mendelian penetrance model.11, 41, 42 To operationalize this approach, maximum credible allele frequency thresholds for elimination can be set on the basis of disease prevalence as well as assumptions regarding penetrance and the fraction of cases attributable to a particular allele under Mendelian inheritance models.42, 85 In comparing the estimated allele frequency to the maximum credible value, sampling variability must be taken into account,85 which can be done using an appropriate binomial confidence interval to determine whether frequencies at or below the maximum credible allele frequency can be excluded with a high degree of confidence.11 In addition, the maximum of frequencies across non-founder populations, rather than the pooled frequency across all populations, is a better indicator of purifying selection.11, 85
Because of its reliance on population allele frequency filtering, this approach to discovery cannot be used to investigate the impact of low-frequency or more common risk alleles. Moreover, application to discovery of novel genes is limited as it may be difficult to separate relevant rare variants from the many rare and private variants that impact a protein product in each individual.86 As will be discussed subsequently, the availability of family data becomes crucial in adapting this approach to discovery of novel loci rather than novel variants in existing loci. Finally, statistical methods to characterize the contribution of the variant or gene to the penetrance model cannot be applied to a single proband.
Proband Cohort Designs
Through continuous ascertainment and genotyping of single cases, molecular labs and investigators have amassed large cohorts of unrelated individuals with DCM.27, 87, 88 Variant discovery in these cohorts can proceed using single-case approaches described above on each individual, and such studies are particularly useful for describing the contributions of various genes with established roles to DCM cases.65, 67 Such cohorts also present additional opportunities for variant and gene characterization, specifically determining whether a GxE or gene-gene interaction modifies the relative risk or odds ratio of a particular environmental or genetic factor.89, 90 However, such designs are valid only under the assumption that the interacting factors are independent in appropriate strata and do not allow for estimation of the main effects of genetic or environmental factors on disease risk.89, 90 Variant-level resolution for genes with high allelic heterogeneity may also not be possible for reasons discussed subsequently.
Associations of disease severity and quantitative endophenotypes with genetic factors may also be estimated in this design, although these should be interpreted with caution. In particular, because probands are explicitly selected on the basis of quantitative endophenotypes or factors determined by them, estimated associations may not reflect the actual effects of genetic factors in the general population.91 Thus, while such analyses can generalize to associations in the population of DCM cases and suggest gene- or variant-specific phenotypic signatures in this population, they do not necessarily provide unbiased estimates of penetrance model parameters necessary for gene characterization.
Case-control Designs
With a sample of unrelated probands and a similarly genotyped cohort of unrelated controls or public reference set, statistical association with case status can be used to identify genetic and environmental factors relevant to the penetrance model. Discovery and characterization of common variants is feasible with variant-at-a-time analyses in the context of the large sample sizes of genome-wide association studies (GWAS), and recent advances in imputation panels have extended this to low-frequency variants.83, 84 Four DCM-specific GWAS have been completed,14–17 and three loci meeting genome-wide significance have been identified in European ancestry subjects: an intronic SNP near to HSPB714 and two intronic SNPs in HGC22.16 In the one study conducted with African ancestry participants,17 a single intronic locus in CACNB4 met genome-wide significance.17 However, variants in genes such as TTN and LMNA that are individually rare or private collectively underlie a substantial proportion of DCM cases, suggesting that relevant genes may be missed by GWAS. Conducting an association study using ES and GS to assay these rare variants is thus a potentially attractive alternative. The single exome-wide association study (EWAS) in DCM, which was performed with an exome array rather than NGS, identified three established DCM genes (BAG3, FLNC, TTN) along with five others (FHOD3, MLIP, NMB-ALPK3, SLC39A8, ZBTB17-HSPB7).20
The principal challenge of association studies involving rare variants is that considering them individually leads to low power,92, 93 particularly in situations where heterogeneous rare alleles in a particular gene have similar effects in the penetrance model.92, 94–96 In such situations, correct simplifying assumptions about the effects of rare variants on the penetrance model scale can improve power, and several techniques for rare variant association testing are derived by exploiting such assumptions, either implicitly or explicitly. Pooling tests assume that rare variants in a specified grouping enter the penetrance model only through the presence or number (burden) of variant alleles, with weighting by external factors such as allele frequency sometimes used to allow heterogeneity in the effects of individual variants.92, 96 These assumptions are most tenable when the group of variants being pooled is carefully curated to eliminate those with low prior probability of biological relevance, such as by considering only loss-of-function variants in a particular gene. When neutral variants are included or there is substantial effect heterogeneity, the power of techniques based on pooling suffers, which has led to alternative approaches that explicitly model effect heterogeneity or are motivated by efficiently combining single-variant test statistics for locus-wide inference.92, 95, 97 Another class of techniques aims to find an optimal combination of other approaches.92, 94 Even with the most efficient of these techniques, large sample sizes are required to have reasonable power to discover new genes in most cases.93
When an investigator does not have access to an internal control set, association testing may be performed with external controls. Individual-level genotype data may be obtainable from sources such as dbGaP, but harmonization of genotype data obtained on differing platforms is a crucial step to avoid spurious associations, particularly with ES.98 Aggregate genotype data made available by projects such as ExAC99 and gnomAD100 can also be used to formally evaluate the association of specific variants or genes with disease risk.101, 102 As individual-level data are not available, applying this approach at the gene level requires translating the number of rare variant minor alleles into a mean per-individual burden or number of carriers102 under various simplifying assumptions. When applying this approach for discovery, rather than characterization, iterative calibration may also be required to reduce false positives and negatives.102
Case-control studies with individual-level data also provide an opportunity to characterize the effects of multiple genetic and environmental factors in the penetrance model concurrently. In fact, a logistic regression model for disease risk in the population arises under a threshold model when latent DCM liability has a standardized logistic distribution with mean given by a linear function of the predictors.103 A convenient feature of a logistic population model is that fitting it to a case-control sample still estimates the parameters in the population sample other than the intercept,104, 105 which leaves odds ratios interpretable as population parameters despite biased ascertainment of cases.
Confounding by ancestry, or population stratification, is an important consideration in case-control studies for discovery or characterization.22, 106 State-of-the-art approaches to adjusting for ancestry, such as including ancestry principal components as covariates in a logistic regression model,106 require access to individual-level genomic data. With aggregate control data, stratification within broad ancestry categories, which are determined on the basis of genomic clustering in gnomAD,100 may provide some degree of protection.
Population-Based Cross-sectional and Cohort Designs
Cross-sectional and cohort studies sample unrelated individuals or families from well-defined populations or strata in a way that does not depend on the phenotype of interest and then collect genomic, exposure, and phenotypic data, possibly with long-term follow-up. The UK Biobank107 is a canonical example of such a design, and a recent study used CMR measurements and array-based genotype data from this resource to identify 45 novel loci and characterize their contribution to both prevalent and incident DCM.18
The measured genotype model108 provides a statistical framework for estimating the parameters of complex population penetrance models like the threshold model considered here (Figure 4). In this sampling design, sampling units are independent realizations from the population penetrance model, meaning that inference can proceed within a generalized linear (mixed) model framework.109 Common variants can be considered individually in the linear predictor for discovery and characterization, or their joint contribution can be characterized parsimoniously using a polygenic risk score.110 The effects of rare variants, possibly in multiple classes, can also be included in the linear predictor, although parsimonious representations of groups of rare variants based on simplifying assumptions, as discussed above, remains essential for power and precision. The linear predictor can also include non-genetic factors, and the interactions between all types of factors can be modeled with standard approaches. Finally, the residual polygenic contribution to trait or liability variance, typically measured by heritability, is estimable in a mixed model setting using a genetic relationship matrix estimated from high-density genotype data111 or family pedigrees.108 In these models, control for confounding by ancestry remains essential and can be accomplished by including ancestry principal components in the linear predictor or fitting a mixed model using an estimated genetic relationship matrix,112 the latter of which also accounts for potential cryptic relatedness.
Despite its considerable flexibility, this design samples rare variants contributing to DCM at their population frequencies, and the paucity of variation in the resulting predictors limits the ability characterize the contribution of various genes to the penetrance model. For example, even truncating variants in TTN, a large gene with the most putative causal variants in idiopathic DCM, occurred in only 0.7% of ExAC controls.113 As a result, characterization of the effects of rare variation, both alone and in concert with common variants, in these designs has typically focused exclusively on TTN, even in the largest cohorts.18, 62, 113 Our ability to characterize the role of rare variation in other genes with this design may increase as cohort sizes grow but is likely to remain limited for the foreseeable future.
Additional Opportunities from Family Data
In the single-proband and proband cohort designs, the addition of family data can substantially strengthen the level of evidence and even provide attractive alternative approaches that address limitations of other designs. In the single-proband design, data on family members can demonstrate segregation of a candidate variant found in the proband with disease. The incremental value of such evidence is recognized by variant classification guidelines, which map higher numbers of segregations with disease to stronger evidence of pathogenicity.11, 41, 42 In a single family of adequate size, this approach can be formalized with parametric linkage analysis114 or exact techniques measuring the deviation from random segregation, although power may be limited. Even with family data, statistical methods to characterize the contribution of the variant or gene to the penetrance model may be difficult to apply due to the lack of independent sampling units.
Collection of family data on each proband in a cohort27 opens a variety of avenues for discovery and characterization. By virtue of their ascertainment through a proband, these families are enriched for DCM-relevant rare variants and DCM relative to families sampled from the general population. Linkage techniques, which examine the relationship between allele sharing in a given region and phenotypic similarity in families, can facilitate discovery by identifying regions harboring relevant rare variants even when the causal variants are family-specific and could not be detected by association.115, 116 In fact, such techniques have been a mainstay of research in DCM genetics.51, 117 The principal drawback of such techniques in the era before NGS was that linkage peaks tend to encompass large genomic regions that were not economical to assay with standard capillary sequencing.118 With the availability of ES and GS, linkage techniques applied to family data can provide a useful adjunct for filtering-based approaches using NGS data from probands.70, 119, 120 Moreover, a linkage marker panel can be constructed using less costly array-based genotypes on family members. Despite their robustness to allelic heterogeneity, the power of linkage techniques is reduced by locus heterogeneity, and approaches allowing for this heterogeneity should be considered where necessary.22, 70, 114
Because most DCM is a late-onset disease, some currently unaffected individuals within a multigenerational pedigree may not be able to contribute to analysis. Under a threshold model (Figure 4), changes in quantitative endophenotypes may manifest well before overt disease, which suggests using quantitative trait linkage techniques115, 116 to capture subclinical genetic effects. While the derivation of these techniques assumes population sampling of families, ascertaining families through a single proband can increase power provided that an appropriate correction is made to the model likelihood.115, 116 Ascertainment correction is required because the distribution of phenotypes in families with probands selected for having disease or extreme phenotypes will differ from that of families randomly sampled from the population.121
A similar ascertainment correction can allow use of a cohort of probands with family data as a viable alternative to population-based designs. Regardless of how the proband is selected, the conditional distribution of phenotypes in family members given the proband’s data is the same.121 As a result, the same analyses of DCM risk or quantitative endophenotypes performed in a population-based cross-sectional or cohort study can proceed in a cohort of families enriched for DCM-relevant rare variants and DCM using the appropriate conditional likelihood. Considerations for these analyses discussed above, including parsimonious representation of rare variants, and control of confounding by ancestry, apply in this setting as well. This approach has been applied to characterize the joint contributions of rare variants in LMNA and other genes to variation in quantitative endophenotypes.13 A drawback of this approach, however, is that families comprising a single proband or families with missing data on the proband cannot be used in the conditional likelihood.
In this context, family-based association tests122 can also be used as an alternative to a case-control design for gene discovery. These tests reject the appropriate null hypothesis only when a particular genetic marker is both linked to and associated with the disease or quantitative trait locus. As a result, an association cannot be found in the absence of linkage, and these methods are therefore robust to population stratification. However, a linked locus cannot be discovered with these techniques in the absence of association, which necessitates modifications to prevent power loss in the face of allelic heterogeneity arising from rare variants.123
Moving Beyond Classification for Characterizing Rare Variants
As is clear from the preceding discussion, the ACMG and related approaches to variant classification incorporate methods for variant discovery and characterization in a variant-at-a-time manner. Such a variant-at-a-time approach is appropriately designed for a penetrance model in which a single, highly penetrant variant with a clear Mendelian inheritance pattern causes disease in an individual or family. However, even in these circumstances, the variant-at-a-time approach makes it difficult for a variant to achieve LP/P status in situations with high allelic heterogeneity. In a gene in which private rare variants clustering in a certain functional domain cause disease, there may never be any case or segregation evidence for a particular variant outside of the family under study. Moreover, classification guidelines address age-dependent penetrance and variable expressivity in an ad hoc manner. Adapting classification approaches to a multifactorial model in order to address this shortcoming would require evaluating the potential impact of each variant in the context of other possibly relevant rare variants, the background of possibly relevant low-frequency variants, polygenic risk from common variants, and environmental factors.
In contrast, a threshold penetrance model based on underlying quantitative endophenotypes naturally translates a complex genetic architecture to empirical manifestations such as age-dependent penetrance and variable expressivity in a principled manner. In addition to suggesting subclinical changes in quantitative endophenotypes as a potentially fruitful target for future study, this framework allows the impacts of rare variants, low-frequency variants, common variants, and environmental factors to be modeled jointly in many designs. Joint modeling can yield improved mechanistic understanding, and fitted models can also be calibrated for risk prediction. The main drawback is that variant-level resolution for rare and low-frequency variants will likely not be achievable with these techniques, even with large sample sizes. Nonetheless, estimating the parameters of these models using relevant groupings of variants, such as TTN truncating variants, may provide the information necessary to classify or generate risk predictions when a novel variant in a particular class is observed.
VI. Major Challenges Yet to be Resolved in DCM genetics.
This list is not exhaustive but rather illustrative of challenges that remain for DCM genetics:
The utility of DCM genetics – does genotype drive prognosis or the efficacy of a particular therapy? Studies need to be designed to test these questions.
Is DCM amenable to gene therapy for intervention and cure?
What is the optimal approach to detect earliest clinical evidence of “pre-DCM”? Is this an imaging approach, such as CMR imaging, or the measurement of some biologic marker?
Will conventional drug treatment, effective for symptomatic DCM, prevent the development of “pre-DCM” in those who are genetically at-risk?
Can approaches be developed to assess the biological relevance of rare variants that with current clinical standards can only be classified as VUSs?
Can an integrated approach to risk prediction incorporating variants across the allele frequency spectrum as well as environmental factors be developed?
Supplementary Material
Sources of Funding.
This work is supported by the National Heart, Lung, and Blood Institute and National Human Genome Research Institute of the National Institutes of Health under awards R01HL128857 R01HL149423, and R01HL148581. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Non-standard Abbreviations and Acronyms
- ACMG
American College of Medical Genetics and Genomics
- ARVC
arrhythmogenic right ventricular cardiomyopathy
- B
benign
- CHD
congenital heart disease
- ClinGen
The Clinical Genome Resource
- CMR
cardiac magnetic resonance
- CTrCM
chemotherapy-related cardiomyopathy
- DCM
dilated cardiomyopathy
- EtOHrCM
alcohol-related cardiomyopathy
- Echo
echocardiogram
- ES
exome sequencing
- FDC
familial dilated cardiomyopathy
- FDR
first-degree relative
- GS
genome sequencing
- GT
genetic testing
- GxE
gene-environment
- HCM
hypertrophic cardiomyopathy
- HF
heart failure
- IDC
idiopathic dilated cardiomyopathy
- LB
likely benign
- LP
likely pathogenic
- LVE
left ventricular enlargement
- LVSD
left ventricular systolic dysfunction
- MAF
mean allele frequency
- MI
myocardial infarction
- NGS
next-generation sequencing
- P
pathogenic
- PACM
pregnancy-associated cardiomyopathy
- PPCM
peripartum cardiomyopathy
- RCM
restrictive cardiomyopathy
- TTNtvs
TTN truncating variants
- VHD
valvular heart disease
- VUS
variants of uncertain significance
Footnotes
Disclosures.
None.
References.
- 1.Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2005;45:969–81. [DOI] [PubMed] [Google Scholar]
- 2.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–47. [DOI] [PubMed] [Google Scholar]
- 3.Sweet M, Taylor MR, Mestroni L. Diagnosis, prevalence, and screening of familial dilated cardiomyopathy. Expert Opin Orphan Drugs. 2015;3:869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Japp AG, Gulati A, Cook SA, Cowie MR, Prasad SK. The Diagnosis and Evaluation of Dilated Cardiomyopathy. J Am Coll Cardiol. 2016;67:2996–3010. [DOI] [PubMed] [Google Scholar]
- 5.McNally EM, Mestroni L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ Res. 2017;121:731–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tayal U, Prasad S, Cook SA. Genetics and genomics of dilated cardiomyopathy and systolic heart failure. Genome Med. 2017;9:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fatkin D, Huttner IG, Kovacic JC, Seidman JG, Seidman CE. Precision Medicine in the Management of Dilated Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;74:2921–2938. [DOI] [PubMed] [Google Scholar]
- 8.Reichart D, Magnussen C, Zeller T, Blankenberg S. Dilated cardiomyopathy: from epidemiologic to genetic phenotypes: A translational review of current literature. J Intern Med. 2019;286:362–372. [DOI] [PubMed] [Google Scholar]
- 9.Yu J, Zeng C, Wang Y. Epigenetics in dilated cardiomyopathy. Curr Opin Cardiol. 2019;34:260–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jordan E, Hershberger RE. Considering complexity in the genetic evaluation of dilated cardiomyopathy. Heart. 2021;107:106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morales A, Kinnamon DD, Jordan E, et al. Variant Interpretation for Dilated Cardiomyopathy: Refinement of the American College of Medical Genetics and Genomics/ClinGen Guidelines for the DCM Precision Medicine Study. Circ Genom Precis Med. 2020;13:e002480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haas J, Frese KS, Peil B, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015;36:1123–35a. [DOI] [PubMed] [Google Scholar]
- 13.Cowan JR, Kinnamon DD, Morales A, Salyer L, Nickerson DA, Hershberger RE. Multigenic Disease and Bilineal Inheritance in Dilated Cardiomyopathy Is Illustrated in Nonsegregating LMNA Pedigrees. Circ Genom Precis Med. 2018;11:e002038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stark K, Esslinger UB, Reinhard W, et al. Genetic association study identifies HSPB7 as a risk gene for idiopathic dilated cardiomyopathy. PLoS Genet. 2010;6:e1001167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villard E, Perret C, Gary F, et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur Heart J. 2011;32:1065–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meder B, Ruhle F, Weis T, et al. A genome-wide association study identifies 6p21 as novel risk locus for dilated cardiomyopathy. Eur Heart J. 2014;35:1069–77. [DOI] [PubMed] [Google Scholar]
- 17.Xu H, Dorn GW 2nd, Shetty A, et al. A Genome-Wide Association Study of Idiopathic Dilated Cardiomyopathy in African Americans. J Pers Med. 2018;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pirruccello JP, Bick A, Wang M, et al. Analysis of cardiac magnetic resonance imaging in 36,000 individuals yields genetic insights into dilated cardiomyopathy. Nat Commun. 2020;11:2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tadros R, Francis C, Xu X, et al. Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nat Genet. 2021;53:128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Esslinger U, Garnier S, Korniat A, et al. Exome-wide association study reveals novel susceptibility genes to sporadic dilated cardiomyopathy. PLoS One. 2017;12:e0172995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piran S, Liu P, Morales A, Hershberger RE. Where genome meets phenome: rationale for integrating genetic and protein biomarkers in the diagnosis and management of dilated cardiomyopathy and heart failure. J Am Coll Cardiol. 2012;60:283–9. [DOI] [PubMed] [Google Scholar]
- 22.Thomas DC. Statistical methods in genetic epidemiology. Oxford; New York: Oxford University Press; 2004. [Google Scholar]
- 23.Hazebroek MR, Moors S, Dennert R, et al. Prognostic Relevance of Gene-Environment Interactions in Patients With Dilated Cardiomyopathy: Applying the MOGE(S) Classification. J Am Coll Cardiol. 2015;66:1313–23. [DOI] [PubMed] [Google Scholar]
- 24.Knowlton KU. Dilated Cardiomyopathy. Circulation. 2019;139:2339–2341. [DOI] [PubMed] [Google Scholar]
- 25.Ware JS, Amor-Salamanca A, Tayal U, et al. Genetic Etiology for Alcohol-Induced Cardiac Toxicity. J Am Coll Cardiol. 2018;71:2293–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia-Pavia P, Kim Y, Restrepo-Cordoba MA, et al. Genetic Variants Associated With Cancer Therapy-Induced Cardiomyopathy. Circulation. 2019;140:31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kinnamon DD, Morales A, Bowen DJ, Burke W, Hershberger RE, for the DCM Consortium. Toward Genetics-Driven Early Intervention in Dilated Cardiomyopathy: Design and Implementation of the DCM Precision Medicine Study. Circ Cardiovasc Genet. 2017;10:p. e001826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hershberger RE. The Dilated, Restrictive, and Infiltrative Cardiomyopathies. In: Libby P, Bonow RO, Mann DL, Tomaselli G, Bhatt D and Solomon SD, eds. Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine, 12th Edition: Elsevier; 2021. [Google Scholar]
- 29.Hershberger RE, Morales A, Siegfried JD. Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet Med. 2010;12:655–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kindel SJ, Miller EM, Gupta R, Cripe LH, Hinton RB, Spicer RL, Towbin JA, Ware SM. Pediatric cardiomyopathy: importance of genetic and metabolic evaluation. J Card Fail. 2012;18:396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rampersaud E, Siegfried JD, Norton N, Li D, Martin E, Hershberger RE. Rare variant mutations identified in pediatric patients with dilated cardiomyopathy. Prog Pediatr Cardiol. 2011;31:39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scaglia F, Towbin JA, Craigen WJ, et al. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics. 2004;114:925–31. [DOI] [PubMed] [Google Scholar]
- 33.Vermeulen R, Schymanski EL, Barabasi AL, Miller GW. The exposome and health: Where chemistry meets biology. Science. 2020;367:392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corrado D, Basso C, Judge DP. Arrhythmogenic Cardiomyopathy. Circ Res. 2017;121:784–802. [DOI] [PubMed] [Google Scholar]
- 35.Parikh VN. Promise and Peril of Population Genomics for the Development of Genome-First Approaches in Mendelian Cardiovascular Disease. Circ Genom Precis Med. 2021;14:e002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller DT, Lee K, Gordon AS, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2020 update: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;(in press). [DOI] [PMC free article] [PubMed]
- 37.Hershberger RE, Givertz MM, Ho CY, et al. Genetic Evaluation of Cardiomyopathy-A Heart Failure Society of America Practice Guideline. J Card Fail. 2018;24:281–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller DT, Lee K, Chung WK, et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;(in press). [DOI] [PubMed]
- 39.Morales A, Hershberger RE. The Rationale and Timing of Molecular Genetic Testing for Dilated Cardiomyopathy. Can J Cardiol. 2015;31:1309–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moretti M, Merlo M, Barbati G, Di Lenarda A, Brun F, Pinamonti B, Gregori D, Mestroni L, Sinagra G. Prognostic impact of familial screening in dilated cardiomyopathy. Eur J Heart Fail. 2010;12:922–7. [DOI] [PubMed] [Google Scholar]
- 41.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kelly MA, Caleshu C, Morales A, et al. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet Med. 2018;20:351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whiffin N, Walsh R, Govind R, et al. CardioClassifier: disease- and gene-specific computational decision support for clinical genome interpretation. Genet Med. 2018;20:1246–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jordan DM, Kiezun A, Baxter SM, et al. Development and validation of a computational method for assessment of missense variants in hypertrophic cardiomyopathy. Am J Hum Genet. 2011;88:183–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Westphal DS, Burkard T, Moscu-Gregor A, Gebauer R, Hessling G, Wolf CM. Reclassification of genetic variants in children with long QT syndrome. Mol Genet Genomic Med. 2020;8:e1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.VanDyke RE, Hashimoto S, Morales A, Pyatt RE, Sturm AC. Impact of variant reclassification in the clinical setting of cardiovascular genetics. J Genet Couns. 2021;30:503–512. [DOI] [PubMed] [Google Scholar]
- 47.Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Akinrinade O, Alastalo TP, Koskenvuo JW. Relevance of truncating titin mutations in dilated cardiomyopathy. Clin Genet. 2016;90:49–54. [DOI] [PubMed] [Google Scholar]
- 49.Tabish AM, Azzimato V, Alexiadis A, Buyandelger B, Knoll R. Genetic epidemiology of titin-truncating variants in the etiology of dilated cardiomyopathy. Biophys Rev. 2017;9:207–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rehm HL, Berg JS, Brooks LD, et al. ClinGen--the Clinical Genome Resource. N Engl J Med. 2015;372:2235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brauch KM, Karst ML, Herron KJ, de Andrade M, Pellikka PA, Rodeheffer RJ, Michels VV, Olson TM. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J Am Coll Cardiol. 2009;54:930–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Connell PS, Berkman AM, Souder BM, et al. Amino Acid-Level Signal-to-Noise Analysis Aids in Pathogenicity Prediction of Incidentally Identified TTN-Encoded Titin Truncating Variants. Circ Genom Precis Med. 2021;14:e003131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Savarese M, Vihola A, Oates EC, et al. Genotype-phenotype correlations in recessive titinopathies. Genet Med. 2020;22:2029–2040. [DOI] [PubMed] [Google Scholar]
- 54.Harris E, Topf A, Vihola A, et al. A ‘second truncation’ in TTN causes early onset recessive muscular dystrophy. Neuromuscul Disord. 2017;27:1009–1017. [DOI] [PubMed] [Google Scholar]
- 55.Gigli M, Begay RL, Morea G, Graw SL, Sinagra G, Taylor MR, Granzier H, Mestroni L. A Review of the Giant Protein Titin in Clinical Molecular Diagnostics of Cardiomyopathies. Front Cardiovasc Med. 2016;3:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roberts AM, Ware JS, Herman DS, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015;7:270ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Akinrinade O, Koskenvuo JW, Alastalo TP. Prevalence of Titin Truncating Variants in General Population. PLoS One. 2015;10:e0145284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bibbins-Domingo K, Pletcher MJ, Lin F, Vittinghoff E, Gardin JM, Arynchyn A, Lewis CE, Williams OD, Hulley SB. Racial differences in incident heart failure among young adults. N Engl J Med. 2009;360:1179–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morales J, Welter D, Bowler EH, et al. A standardized framework for representation of ancestry data in genomics studies, with application to the NHGRI-EBI GWAS Catalog. Genome Biol. 2018;19:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Landry LG, Rehm HL. Association of Racial/Ethnic Categories With the Ability of Genetic Tests to Detect a Cause of Cardiomyopathy. JAMA Cardiol. 2018;3:341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pottinger TD, Puckelwartz MJ, Pesce LL, et al. Pathogenic and Uncertain Genetic Variants Have Clinical Cardiac Correlates in Diverse Biobank Participants. J Am Heart Assoc. 2020;9:e013808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haggerty CM, Damrauer SM, Levin MG, et al. Genomics-First Evaluation of Heart Disease Associated With Titin-Truncating Variants. Circulation. 2019;140:42–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Akinrinade O, Helio T, Lekanne Deprez RH, et al. Relevance of Titin Missense and Non-Frameshifting Insertions/Deletions Variants in Dilated Cardiomyopathy. Sci Rep. 2019;9:4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Deo RC. Alternative Splicing, Internal Promoter, Nonsense-Mediated Decay, or All Three: Explaining the Distribution of Truncation Variants in Titin. Circ Cardiovasc Genet. 2016;9:419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hershberger RE, Norton N, Morales A, Li D, Siegfried JD, Gonzalez-Quintana J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ Cardiovasc Genet. 2010;3:155–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Spaendonck-Zwarts KY, van Rijsingen IA, van den Berg MP, et al. Genetic analysis in 418 index patients with idiopathic dilated cardiomyopathy: overview of 10 years’ experience. Eur J Heart Fail. 2013;15:628–36. [DOI] [PubMed] [Google Scholar]
- 67.Jordan E, Peterson L, Ai T, et al. An Evidence-based Assessment of Genes in Dilated Cardiomyopathy. Circulation. 2021;(in press). [DOI] [PMC free article] [PubMed]
- 68.Mazzarotto F, Tayal U, Buchan RJ, et al. Reevaluating the Genetic Contribution of Monogenic Dilated Cardiomyopathy. Circulation. 2020;141:387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hershberger RE, Givertz MM, Ho CY, et al. Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2018;20:899–909. [DOI] [PubMed] [Google Scholar]
- 70.Norton N, Li D, Rampersaud E, et al. Exome sequencing and genome-wide linkage analysis in 17 families illustrate the complex contribution of TTN truncating variants to dilated cardiomyopathy. Circ Cardiovasc Genet. 2013;6:144–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morales A, Cowan J, Dagua J, Hershberger RE. Family history: an essential tool for cardiovascular genetic medicine. Congest Heart Fail. 2008;14:37–45. [DOI] [PubMed] [Google Scholar]
- 72.Morales A, Painter T, Li R, Siegfried JD, Li D, Norton N, Hershberger RE. Rare variant mutations in pregnancy-associated or peripartum cardiomyopathy. Circulation. 2010;121:2176–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van Spaendonck-Zwarts KY, van Tintelen JP, van Veldhuisen DJ, van der Werf R, Jongbloed JD, Paulus WJ, Dooijes D, van den Berg MP. Peripartum cardiomyopathy as a part of familial dilated cardiomyopathy. Circulation. 2010;121:2169–75. [DOI] [PubMed] [Google Scholar]
- 74.Ware JS, Li J, Mazaika E, et al. Shared Genetic Predisposition in Peripartum and Dilated Cardiomyopathies. N Engl J Med. 2016;374:233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Falk RH, Hershberger RE. The Dilated, Restrictive, and Infiltrative Cardiomyopathies. In: Zipes DP, Libby P, Bonow RO, Mann DL and Tomaselli G, eds. Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine, 11th Edition: Elsevier; 2018. [Google Scholar]
- 76.Davis MB, Arany Z, McNamara DM, Goland S, Elkayam U. Peripartum Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2020;75:207–221. [DOI] [PubMed] [Google Scholar]
- 77.Falconer DS. Introduction to quantitative genetics. 3rd ed. New York: Wiley; 1989. [Google Scholar]
- 78.Hershberger RE. A glimpse into multigene rare variant genetics: triple mutations in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010;55:1454–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hershberger RE, Parks SB, Kushner JD, et al. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Translational Science. 2008;1:21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Parks SB, Kushner JD, Nauman D, et al. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J. 2008;156:161–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jin Y, Kuznetsova T, Bochud M, Richart T, Thijs L, Cusi D, Fagard R, Staessen JA. Heritability of left ventricular structure and function in Caucasian families. Eur J Echocardiogr. 2011;12:326–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bella JN, MacCluer JW, Roman MJ, et al. Heritability of left ventricular dimensions and mass in American Indians: The Strong Heart Study. J Hypertens. 2004;22:281–6. [DOI] [PubMed] [Google Scholar]
- 83.Kowalski MH, Qian H, Hou Z, et al. Use of >100,000 NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium whole genome sequences improves imputation quality and detection of rare variant associations in admixed African and Hispanic/Latino populations. PLoS Genet. 2019;15:e1008500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McCarthy S, Das S, Kretzschmar W, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Whiffin N, Minikel E, Walsh R, et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med. 2017;19:1151–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tennessen JA, Bigham AW, O’Connor TD, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pugh TJ, Kelly MA, Gowrisankar S, et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. 2014;16:601–8. [DOI] [PubMed] [Google Scholar]
- 88.Kushner JD, Nauman D, Burgess D, Ludwigsen S, Parks S, Pantely G, Burkett EL, Hershberger R. Clinical characteristics of 304 kindreds evaluated for familial dilated cardiomyopathy. J Cardiac Failure. 2006;12:422–29. [DOI] [PubMed] [Google Scholar]
- 89.Piegorsch WW, Weinberg CR, Taylor JA. Non-hierarchical logistic models and case-only designs for assessing susceptibility in population-based case-control studies. Stat Med. 1994;13:153–62. [DOI] [PubMed] [Google Scholar]
- 90.Yang Q, Khoury MJ, Sun F, Flanders WD. Case-only design to measure gene-gene interaction. Epidemiology. 1999;10:167–70. [PubMed] [Google Scholar]
- 91.Glymour MM, Greenland S. Causal Diagrams Modern epidemiology. 3 ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2008: 183–209. [Google Scholar]
- 92.Bomba L, Walter K, Soranzo N. The impact of rare and low-frequency genetic variants in common disease. Genome Biol. 2017;18:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Moutsianas L, Agarwala V, Fuchsberger C, et al. The power of gene-based rare variant methods to detect disease-associated variation and test hypotheses about complex disease. PLoS Genet. 2015;11:e1005165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee S, Emond MJ, Bamshad MJ, et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet. 2012;91:224–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Morris AP, Zeggini E. An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet Epidemiol. 2010;34:188–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kinnamon DD, Hershberger RE, Martin ER. Reconsidering association testing methods using single-variant test statistics as alternatives to pooling tests for sequence data with rare variants. PLoS One. 2012;7:e30238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Auer PL, Reiner AP, Wang G, et al. Guidelines for Large-Scale Sequence-Based Complex Trait Association Studies: Lessons Learned from the NHLBI Exome Sequencing Project. Am J Hum Genet. 2016;99:791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Walsh R, Thomson KL, Ware JS, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2016;19:192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Guo MH, Plummer L, Chan YM, Hirschhorn JN, Lippincott MF. Burden Testing of Rare Variants Identified through Exome Sequencing via Publicly Available Control Data. Am J Hum Genet. 2018;103:522–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Greene WH. Econometric analysis. 5th ed. Upper Saddle River, N.J.: Prentice Hall; 2003. [Google Scholar]
- 104.Agresti A Categorical data analysis. 2nd ed. New York: Wiley-Interscience; 2002. [Google Scholar]
- 105.Prentice RL, Pyke R. Logistic Disease Incidence Models and Case-Control Studies. Biometrika. 1979;66:403–411. [Google Scholar]
- 106.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. [DOI] [PubMed] [Google Scholar]
- 107.Sudlow C, Gallacher J, Allen N, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12:e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Boerwinkle E, Chakraborty R, Sing CF. The use of measured genotype information in the analysis of quantitative phenotypes in man. I. Models and analytical methods. Ann Hum Genet. 1986;50:181–94. [DOI] [PubMed] [Google Scholar]
- 109.Stroup W Generalized linear mixed models: modern concepts, methods and applications.: CRC Press; 2012. [Google Scholar]
- 110.Chatterjee N, Shi J, Garcia-Closas M. Developing and evaluating polygenic risk prediction models for stratified disease prevention. Nat Rev Genet. 2016;17:392–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yang J, Benyamin B, McEvoy BP, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yang J, Zaitlen NA, Goddard ME, Visscher PM, Price AL. Advantages and pitfalls in the application of mixed-model association methods. Nat Genet. 2014;46:100–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schafer S, de Marvao A, Adami E, et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet. 2017;49:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ott J Analysis of human genetic linkage. Baltimore: The Johns Hopkins University Press; 1991. [Google Scholar]
- 115.Amos CI, de Andrade M. Genetic linkage methods for quantitative traits. Stat Methods Med Res. 2001;10:3–25. [DOI] [PubMed] [Google Scholar]
- 116.Almasy L, Blangero J. Variance component methods for analysis of complex phenotypes. Cold Spring Harb Protoc. 2010;2010:pdb top77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, Horton SC, Rodeheffer RJ, Anderson JL. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005;293:447–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bailey-Wilson JE, Wilson AF. Linkage analysis in the next-generation sequencing era. Hum Hered. 2011;72:228–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Silva CT, Zorkoltseva IV, Amin N, et al. A Combined Linkage and Exome Sequencing Analysis for Electrocardiogram Parameters in the Erasmus Rucphen Family Study. Front Genet. 2016;7:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Smith KR, Bromhead CJ, Hildebrand MS, et al. Reducing the exome search space for mendelian diseases using genetic linkage analysis of exome genotypes. Genome Biol. 2011;12:R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Beaty TH, Liang KY, Rao DC. Robust inference for variance components models in families ascertained through probands: I. Conditioning on proband’s phenotype. Genet Epidemiol. 1987;4:203–210. [DOI] [PubMed] [Google Scholar]
- 122.Laird NM, Lange C. Family-Based Methods for Linkage and Association Analysis Advances in Genetics: Academic Press; 2008(60): 219–252. [DOI] [PubMed] [Google Scholar]
- 123.De G, Yip WK, Ionita-Laza I, Laird N. Rare variant analysis for family-based design. PLoS One. 2013;8:e48495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.