

Abstract
The ability of tumor cells to adapt to changes in oxygen tension is essential for tumor development. Low oxygen concentration influences cellular metabolism and, thus, affects proliferation, migration, and invasion. A focal point of the cell’s adaptation to hypoxia is the transcription factor HIF1α (hypoxia-inducible factor 1 alpha), which affects the expression of specific gene networks involved in cellular energetics and metabolism. This review illustrates the mechanisms by which HIF1α-induced metabolic adaptation promotes angiogenesis, participates in the escape from immune recognition, and increases cancer cell antioxidant capacity. In addition to hypoxia, metabolic inhibition of 2-oxoglutarate-dependent dioxygenases regulates HIF1α stability and transcriptional activity. This phenomenon, known as pseudohypoxia, is frequently used by cancer cells to promote glycolytic metabolism to support biomass synthesis for cell growth and proliferation. In this review, we highlight the role of the most important metabolic intermediaries that are at the center of cancer’s biology, and in particular, the participation of these metabolites in HIF1α retrograde signaling during the establishment of pseudohypoxia. Finally, we will discuss how these changes affect both the development of cancers and their resistance to treatment.
Keywords: Hypoxia, pseudo-hypoxia, metabolites, TCA
1. Introduction
A prominent feature of cancer development is the low oxygen tension in the center of solid tumors. Hypoxia is a crucial microenvironmental factor that defines a tumor’s growth and aggressiveness. Oxygen concentration influences a cell’s preference for specific metabolic pathways, determining its proliferation and invasiveness rate. Under hypoxia, tumor cells prefer glycolysis as an ATP source. Though less energy-efficient than oxidative phosphorylation, glycolysis generates metabolic intermediates for cell growth, proliferation, and adhesion molecule expression [1]. Thus, the induction of glycolytic metabolism under reduced oxygen concentration represents a significant advantage to cancer cells.
Since metabolic reprogramming under hypoxia benefits tumor growth and metastasis, many cancer cells have developed the ability to activate and utilize hypoxia-dependent pathways even in the presence of oxygen, a phenomenon that is known as pseudohypoxia [2, 3]. Otto Warburg reported one of the earliest examples of this behavior more than 80 years ago. Specifically, he observed that tumor cells consume more glucose and produce more lactate than normal cells, displaying high glycolytic activity supported by increased expression of enzymes that allow rapid conversion of glucose into lactate, bypassing mitochondrial oxygen reduction even when oxygen was available.
The transcription factor hypoxia-inducible factor 1α (HIF1α) plays a fundamental role in the metabolic adaptation associated with hypoxia and pseudohypoxia (Figure 1). First identified by its ability to induce the erythropoietin gene in response to hypoxia [4], HIF1α has a half-life of minutes. Specific prolyl-4-hydroxylases constantly hydroxylate HIF1α, which is then recognized by an E3 ubiquitin ligase complex, polyubiquitinated, and degraded by the proteasome [5]. Prolyl hydroxylases (PHDs), a class of 2-oxoglutarate (α-ketoglutarate)-dependent dioxygenases (2OGDDs), use molecular oxygen as a substrate for catalysis with a Km of about 250 μM [5]. When oxygen concentrations fall below the Km, PHD activity is inhibited, and thus HIF1α is stabilized. HIF1α stabilization can also occur under normoxic conditions (pseudo-hypoxia), independently of oxygen concentrations. In addition to oxygen, PHDs require α-ketoglutarate (α-KG) as well as the cofactors Fe2+ and ascorbate. Modification of any of these components affects their activity and, consequently, HIF1α stability [5, 6]. Importantly, a series of metabolites are responsible for competitive inhibition of PHDs, thus stabilizing HIF1α [2, 7] (Figure 1). Finally, increased deubiquitinase activity can also contribute to HIF1α stabilization under normoxia [8].
Figure 1. Schematic representation of metabolic pathways in normoxia and hypoxia/pseudohypoxia.
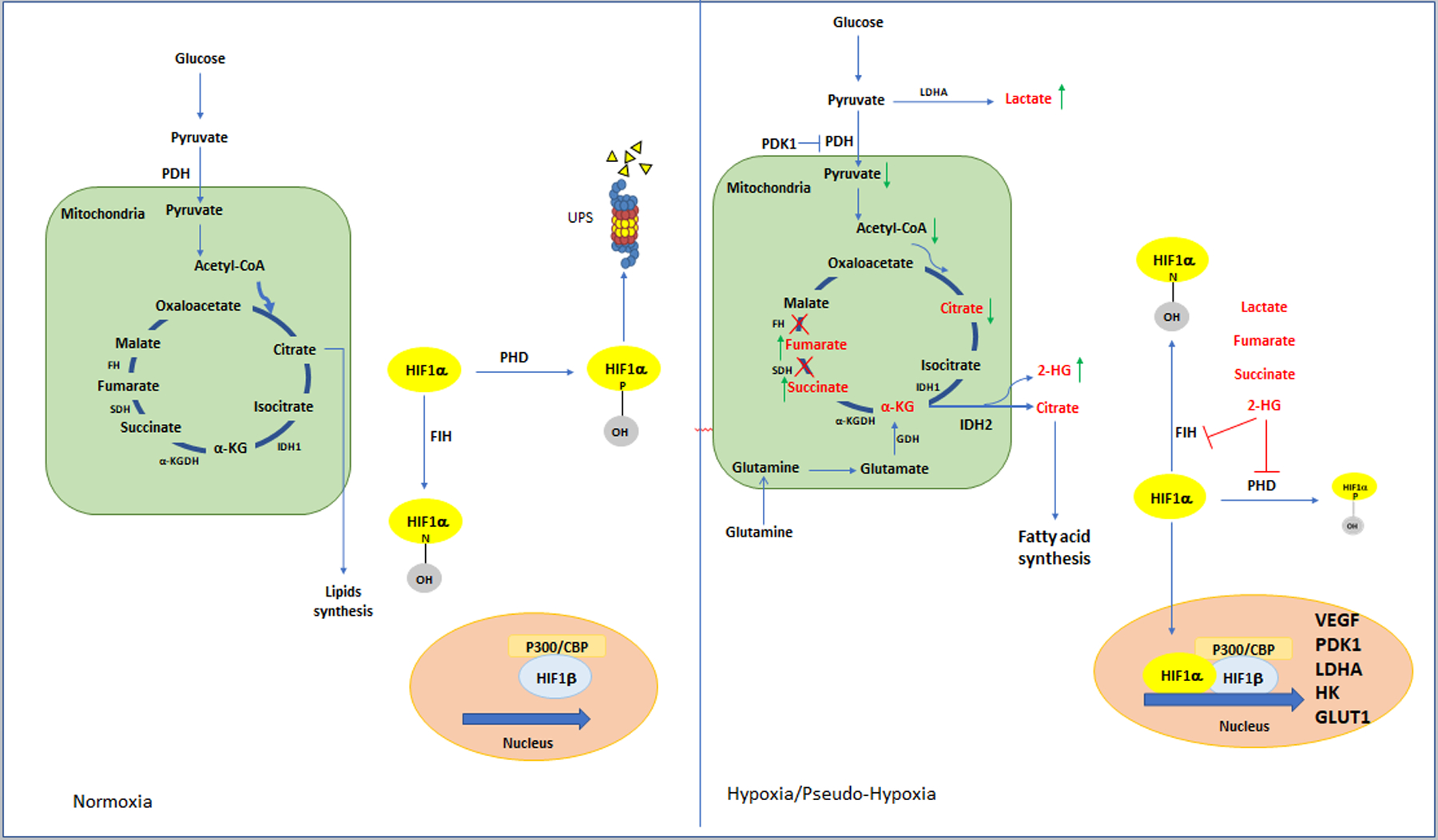
Through changes in metabolic pathways and levels of different metabolic intermediates, tumor cells can adapt to the oxygen conditions of the environment. The metabolites that change under hypoxia/pseudohypoxia conditions in cells are shown in red. Green arrows indicate metabolites that change in cancer cells. The red crosses show those enzymes that decrease in cancer cells.
Stable HIF1α dimerizes with HIF1β and interacts with transcriptional coactivators via the C-terminal transactivation domain (CAD). Finally, the HIF1α-containing complex binds to hypoxia response elements (HREs) to activate the transcription of a variety of genes [9].
The Factor-inhibiting hypoxia-inducible factor (FIH) is another enzyme that inhibits HIF1α activity. By hydroxylation of asparagine 803 within the CAD, the association of HIF1α with its coactivators is impeded, thereby reducing HIF1α activity [10]. FIH, also a member of the 2OGDD family of oxidases, displays a higher affinity for oxygen than PHDs. With an approximate Km of ∼90–200 μM, FIH is more active than PHDs under hypoxic conditions [11] and Figure 1. Like PHDs, in addition to oxygen, FIH uses α-KG as a substrate and requires Fe2+ and ascorbate as cofactors [11] (Figure 1).
HIF1α-targeted genes participate in many cellular functions, ranging from metabolic adaptation to oxygen and nutrient deprivation to angiogenesis, cell proliferation, apoptosis, adhesion, migration, and survival. Among the genes under HIF1α regulation are those that encode enzymes necessary to process the incoming glucose, such as the glucose transporters GLUT1 and GLUT3, the enzymes hexokinase 1 and 2 (HK1 and HK2), and phosphoglycerate kinase 1 (PGK1) [9]. Importantly, HIF1α-mediated transcription of the inhibitor of pyruvate dehydrogenase (PDH), pyruvate dehydrogenase kinase 1 (PDK1), inhibits the TCA [12]. In this manner, HIF1α orchestrates the metabolic changes that allow cells to adapt to oxygen deprivation (Figure1). During tumor development, HIF1α plays a central role in the tumor’s metabolic adaption to its microenvironment. For example, tumor growth depends on the cell’s ability to shift glucose metabolism from the more efficient oxidative phosphorylation to the less efficient glycolytic pathway to maintain the necessary ATP levels and the production of metabolic intermediates to maintain cellular functions. As previously mentioned, HIF1α regulates GLUT1 and HK2, which are necessary to increase the glucose consumption that characterizes tumor metabolism. Furthermore, HIF1α regulates the expression of vascular endothelial growth factor (VEGF) [13], which stimulates neovascularization to promote tumor growth.
Metabolites resulting from glycolysis and mitochondrial metabolism fulfill functions as substrates for chemical reactions and act as signaling molecules [7, 14]. Furthermore, metabolites can inhibit enzymes through competitive inhibition or post-translational modification [7, 15, 16]. These ubiquitous molecules’ functions are primarily dependent on their intracellular and extracellular concentrations and their sub compartmental localization [14, 15]. Significantly, the signaling functions of these metabolic intermediates extend beyond self-regulatory roles and include cell communication and sensing of micro-environmental conditions to elicit stress responses and cellular adaptation.
In this review, we focus on the fundamentals of how hypoxia affects tumor growth and will discuss how the intermediate metabolites citrate, α-KG, 2HG, succinate, fumarate, pyruvate, and lactate are generated in response to metabolic changes (Table 1). Additionally, we will discuss how these changes affect both the development of cancers and their resistance to different treatments.
Table 1.
Role of metabolite accumulation in cancer.
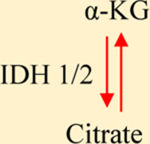
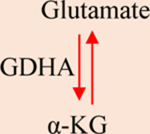
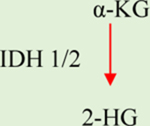
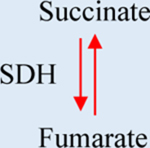
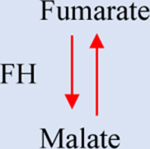
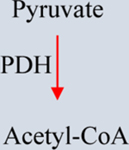
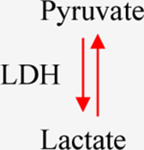
a-KG: Alpha-ketoglutarate. IDH1/2: Isocitrate dehydrogenase 1/2. GDHA: Glutamate dehydrogenase. HIF1a: hypoxia-inducible factor 1. 2-HG: 2-hydroxyglutarate. SDH: Succinate dehydrogenase. TET: Ten-eleven translocation methylcytosine dioxygenase. FH: Fumarate hydratase. PDH: Pyruvate dehydrogenase. LDH: Lactate dehydrogenase.
2. Citrate
Citrate participates in mitochondrial bioenergetics and is an intermediate for acetyl-CoA transfer to the cytosol, where it participates in fatty-acid synthesis, integrating mitochondrial function and lipid metabolism. Additionally, citrate can directly activate AKT, ERK, and MMP2/9 [17].
In normoxia, citrate synthesis mostly occurs in the TCA cycle by condensation of acetyl-CoA and oxaloacetate, a reaction catalyzed by the enzyme citrate synthetase. Under hypoxia, diminished activity of pyruvate dehydrogenase (PDH) decreases acetyl-CoA production, forcing the cell to use alternative pathways to maintain citrate levels [18]. One of these pathways is the carboxylation of glutamine-derived α-ketoglutarate by isocitrate dehydrogenase 2 (IDH2) to form isocitrate, which yields citrate after isomerization [18] (Table 1). In addition to biosynthetic pathways, cells can uptake citrate through the plasma membrane citrate carrier SLC13A5, a variant of the mitochondrial citrate transporter SLC25A1 [19, 20]. Once synthesized, citrate can be further oxidized in the TCA cycle or transported out of the mitochondria by the SLC25A1and the citrate-malate transport system [19, 20]. In the cytosol, citrate is converted to oxaloacetate and acetyl-CoA by ATP citrate lyase (ACLY) [21, 22]. Oxaloacetate is used to synthesize amino acids, while acetyl-CoA is converted by acetyl-CoA carboxylase into malonyl-CoA and used to extend fatty acid chains [22, 23]. Lipogenesis is enhanced in cancer, which requires maintaining adequate citrate levels within the cell [23, 24]. Since many tumors generate limited citrate via TCA, various cancers use the IDH2-dependent pathway [18, 24]. Once citrate moves out of the mitochondria, glutamine replenishes the α-KG necessary to synthesize more citrate in the mitochondria [23, 24]. This cycle ensures the synthesis of lipids and amino acids and produces cofactors such as NADH necessary for biosynthesis and essential to prevent oxidative damage [25, 26]. Acetyl-CoA produced from citrate metabolism is crucial for histone acetylation, which in cancer activates the transcription of genes involved in glucose metabolism [27, 28].
Given the importance of citrate, malignant tumors often have mutations that regulate citrate levels. Mutations of citrate synthase (CS) have been described in pancreatic [29], ovarian [30], cervical [31], and breast cancer [31]. Increased CS activity induces a glycolytic switch, promoting cancer cell proliferation, migration, and invasion [32]. In the case of SLC25A1, amplification of the chromosome region that carries the gene results in the up-regulation of SLC25A1 and the intracellular citrate in breast, lung, and [33] hepatic [34] cancers. In addition, the activity of ACLY is frequently up-regulated in liver [35], lung [36], breast [37], gastric [38], prostate [39], and colon [39] cancers.
Although citrate is an essential metabolite for the survival and development of tumor cells, it also plays a role as a negative regulator of glycolysis [28], and is also described as an antitumor metabolite [32]. Several studies have demonstrated that citrate inhibits proliferation and promotes apoptosis in multiple cancer cell lines [40]. Furthermore, oral administration of citrate reduces tumor growth in various mouse models of breast and lung cancer [41], which coincides with increased leukocyte infiltration [41]. When administered to patients, citrate supplementation decreases chemoresistance to cisplatin [41]. Collectively, these data imply a more active role for this metabolite in cancer.
3. Alpha-ketoglutarate
Alpha-ketoglutarate (α-KG, aka 2-oxoglutarate) is a metabolic intermediary that participates in amino acid synthesis, ATP production, and reducing equivalent generation [42]. As mentioned earlier, α-KG is a substrate for 2OGDDs [43]. As such, α-KG concentration regulates the activity of the members of the 2OGDD family of enzymes which are responsible for a variety of cellular functions such as collagen synthesis, proteasome-mediated HIF1α degradation, and histone and DNA demethylation. By regulating this class of dioxygenases, α-KG directly influences gene expression associated with cancer. Furthermore, the ten-eleven translocation (TET) methylcytosine dioxygenases that oxidize 5-methylcytosine to participate in DNA demethylation, the AlkB enzymes that repair DNA alkylation damage, and the JmjCs that demethylate histones are also members of the 2OGDDs family of enzymes [43], and their chemical modifications of chromatin and histones play a fundamental role in the regulation of genome function and, therefore, in cellular physiology. Thus, changes in the concentration of α-KG can impact the epigenetic and transcriptional regulation of genes in tumor cells.
α-KG is produced by oxidative decarboxylation of isocitrate (catalyzed by IDH) or the oxidative deamination of glutamate (catalyzed by glutamate dehydrogenase) derived from glutamine (Table 1). In the TCA cycle, α-KG is decarboxylated to succinyl-CoA and CO2 by α-KG dehydrogenase (α-KGDH). Under hypoxia and pseudohypoxia, α-KG is generated in anaplerotic reactions, and α-KGDH activity is reduced [1, 44]. Additionally, NAD+/NADH and NADP+/NADPH ratios and acidification can affect α-KG levels [25, 45]. These parameters relate to the cell’s redox state and are altered in various malignancies [46–48]. Furthermore, IDH activity is NADPH-dependent [18]. NADH inhibits α-KGDH by allosteric inhibition of its subunits [45].
Reduced α-KG slows down hydroxylation reactions involving PHDs, resulting in HIF1α accumulation [3]. The Km of the PHDs for α-KG is ~55–60μM, close to its physiological concentration [5]. Thus, small modifications in α-KG levels interfere with PHD activity and HIF1α levels. Reduced α-KG also slows down the hydroxylation reaction involving FIH, increasing HIF1α transcription activity [49]. FIH inhibits HIF1-α signaling, hydroxylating an asparagine residue (Asn803) in the C-terminal transactivation domain (CAD) of HIF1α. This modification blocks the binding with the coactivator p300 / CREB-binding protein (p300 / CBP) necessary for the function of HIF1α [49]. Consequently, the low levels of α-KG in cancer cells lead to increased HIF1α-regulated genes, such as LDH, HK2, VEGF, and GLUT1[9].
The role of α-KG in cancer has been extensively studied, and evidence supports the theory that increases in intracellular α-KG could have antitumor effects [42]. For example, treatment of the liver cancer cell line Hep3B with exogenous α-KG induces HIF1α degradation under hypoxia [50]. Similarly, recent studies demonstrate that α-KG supplementation prevents tumor growth and metastasis of triple-negative breast cancer (TNBC) by switching from glycolytic to oxidative metabolism [51]. Additionally, in a murine xenograft model of lung cancer, intraperitoneal injection of α-KG inhibited tumor growth and angiogenesis [52]. Finally, various drugs that inhibit α-KGDH activate TETs with anti-metastasis and antitumor effects [53, 54]. These studies demonstrate the potential role of α-KG in the treatment of cancer.
4. 2-Hydroxyglutarate
Mutations of IDH1/2 in cancer result in the production of D(R)-2-hydroxyglutarate (D-2-HG) (Table 1), an oncometabolite that acts as a competitive inhibitor of 2OGDDs [55]. Mutations of the IDH1-2 genes are found in multiple human tumors [56–62]. The D-2-HG enantiomer, L-2-HG, was recently identified as an abnormal α-KG metabolism product under hypoxia [63]. The increase of both enantiomers of 2-HG is associated with increased malignancy in various cancers, particularly in aggressive glioma [57, 59].
2-HGs inhibit 2OGDDs, including TETs, AlkBs, PHDs, and FIH [7, 64, 65]. For example, in RCC tumors, high L-2HG correlates with reduced levels of 5-hydroxymethylcytosine (5hmC), consistent with TET enzyme inhibition, and reconstitution of L2HGDH lowered 2-HG and increased 5hmC levels while also suppressing in vitro tumor phenotypes [64]. Additionally, 2-HG can support tumorigenesis by inhibiting the repair of DNA alkylation damage through competitive inhibition of the AlkB (Alkylation repair Homolog) family of Fe(II)- and α-ketoglutarate-dependent dioxygenases [66]. While 2-HG is a weak inhibitor of AlkB proteins, a 2-HG increase of up to 373-fold has been observed in glioma patients, resulting in competitive inhibition of AlkBs promoting microevolution glioma, possibly by elevating the intra-cancerous mutation rate [66]. Furthermore, because 2-HG is a known inhibitor of PHDs and FIH, it may be required for HIF1α stabilization and affect the expression of genes required to maintain glycolytic metabolism, angiogenesis, and metastasis [67].
Recently, two mutant IDH inhibitors, Enasidenib and Ivosidenib, have been FDA-approved to treat relapsed or refractory acute myeloid leukemia, and their efficacy in other cancers are in various stages of investigation. Patient-derived bone marrow blasts treated with Enasidenib demonstrate inhibited cellular proliferation and reversal of the histone hypermethylation associated with the IDH2 mutation [61]. Other drugs that target the inhibition of mutated IDH1/2 have been generated and are in preclinical and early clinical studies. In all, mutIDH1/2 and 2-HGs are attractive therapeutic targets for cancer.
5. Succinate
At the crossroads of various metabolic routes, succinate is associated with branched-chain amino acid metabolism, the synthesis of heme, the use of ketone bodies, and the GABA shunt [15]. Additionally, succinate participates in signal transduction by means of protein succinylation, a recently discovered post-translational modification [68].
During the TCA cycle, succinate is generated by the α-KGDH complex and succinyl-CoA synthetase, which progressively metabolize α-KG to succinate in two successive reactions. In normoxia, succinate is converted to fumarate by the enzyme succinate dehydrogenase (SDH) (Table 1). SDH participates in both the TCA and the electron transport chain connecting the two metabolic pathways. SDH loss of function is associated with the nuclear stabilization of HIF1α and antineoplastic resistance [69].
Frequently, succinate accumulates in cancer cells [70], inhibiting PHDs, and stabilizing HIF1α [3]. Likewise, the exogenous addition of succinate stabilizes HIF1α and increases the growth and proliferation of glioblastoma cells [71]. Elevated levels of succinate caused by SDH loss-of-function are associated with impaired JmjC and TET activity, leading to dysregulation of proliferation and migration genes [72], loss of the Electron transport chain complex II [15, 73], and increased ROS production [69]. Cancer cells-secreted succinate can also act in a paracrine manner. A recent study showed that secreted tumor-derived succinate activates the succinate receptor (SUCNR1) and induces polarization of tumor-associated macrophages contributing to the immunosuppressive tumor microenvironment [74].
Protein succinylation has emerged as a novel PTM in which succinyl is added to lysine and, to a lesser extent, arginine or histidine residues [16] to alter protein activity and localization. Succinylation activates Pyruvate kinase isoform M2 (PKM2) and mediates its translocation to the mitochondria [75]. Furthermore, the succinylation of the calcium-binding protein S100A10 increases the invasion and migration of human gastric carcinoma [76]. Recent data indicate that histone succinylation might modulate gene expression [77] and that aberrant chromatin hypersuccinylation contributes to DNA double-strand break repair [78]. Consequently, it is not surprising that increases in chromatin succinylation promote tumor growth in renal [79], colon [80], gastrointestinal [81], and thyroid cancers [82].
The emerging roles of succinate in the hypoxic response and cancer development extend beyond metabolism involving gene transcription changes and epigenetics, making it an attractive therapeutic target. More studies on succinate and the enzymes involved in its metabolism are necessary to establish its potential role as a therapeutic target in specific cancers.
6. Fumarate
Fumarate is a metabolic intermediate of both the TCA and the urea cycles [83]. The urea cycle is closely related to the TCA and derives nitrogen through the transamination of oxaloacetate to form aspartate, and returns fumarate to the TCA [83]. Furthermore, fumarate is also related to the metabolism of amino acids phenylalanine and tyrosine through a series of degradation reactions that metabolize these amino acids into fumarate and acetoacetate [84]. Additionally, fumarate participates in the purine nucleotide cycle, which generates ammonia and fumarate from aspartate, using inosine monophosphate, adenylosuccinate, and adenosine monophosphate in catalytic amounts [85].
In normoxia, fumarate is formed within the TCA by SDH-mediated oxidation of succinate. However, this reaction can be reversed in hypoxia, leading to succinate accumulation and continued NADH oxidation by complex I [86]. Replenishment of NAD+ allows ATP synthesis and glycolysis to be maintained with reduced lactate production for an extended period before the onset of acidosis. In an aerobic environment, most fumarate is converted to malate by fumarate hydratase (FH), or fumarase (Table 1), which is found in the cytoplasm and mitochondria [87]. In mitochondria, FH catalyzes the hydration of fumarate to generate malate as part of the TCA. This pathway is essential for cellular energy production, and it also acts as a key metabolic center for generating macromolecular precursors [87]. In the cytosol, FH participates in multiple pathways, such as the urea and the purine nucleotide cycles [88].
Defects in fumarate metabolism are associated with various cancers, such as hereditary leiomyomatosis and renal cell cancer (HLRCC), a tumor predisposition syndrome caused by heterozygous germline FH mutations [89, 90]. Fumarate competitively inhibits 2OGDDs, leading to HIF1α stabilization and pseudohypoxia, which is associated with tumor growth and metastasis [90]. Additionally, FH knockout mice develop renal cysts characterized by elevated nuclear HIF1α expression and increased transcription of HIF1α target genes GLUT1 and VEGF [91]. Understanding these processes will be vital in determining which of the fumarate pathways are viable targets for cancer development and progression prevention.
7. Pyruvate
As the final product of glycolysis, pyruvate is the linchpin that connects glycolytic and oxidative metabolism. It also plays a pivotal role in hypoxia as it is converted to lactate, generating NAD+ to support glycolysis and ATP synthesis.
As a starting point for gluconeogenesis [92] and , as an essential acetyl-CoA source, lipogenesis, pyruvate can be incorporated into glucose or fatty acids. Additionally, pyruvate can be used to generate non-essential amino acids [93]. Through acetyl-CoA, pyruvate can participate in gene regulation via histone acetylation [94].
In cancer, pyruvate is often metabolized in anaerobic processes independently of oxygen concentration. Instead of fully oxidizing pyruvate in the TCA cycle, pyruvate is used to generate metabolites, such as lactate and alanine [95]. In several human tumor-derived cell lines, exogenous pyruvate increases proliferation and stabilizes HIF1α [96, 97]. Additionally, in head and neck squamous carcinoma, high pyruvate production is associated with increased invasion [95], and blood pyruvate levels are frequently elevated in leukemia, lymphoma, and other malignant tumors such as renal cell carcinoma [95, 98, 99]. Increasing pyruvate levels may allow tumor cells to maintain high glycolytic activity during increased anabolic demand [95]. Particularly, proliferating cells usually have an increased need to synthesize various metabolites such as amino acids and lipids [100]. Pyruvate, through anaplerotic reactions, may contribute carbon to these synthetic activities [101].
Enzymes regulating pyruvate are altered in many cancers. For example, PKM can dynamically switch between tetramers that efficiently generate ATP and pyruvate and less active dimers leading to the accumulation of glycolytic metabolites that promote biosynthetic pathways. In many cancers, the dimeric form of PKM2 predominates [102]. Additionally, PKM2 is frequently upregulated in cancers, and PKM2 inhibitors, alone or in combination with antineoplastic drugs, restrain glycolysis, gene transcription, and cellular proliferation in melanoma, non-small cell lung carcinoma (NSCLC), and lung adenocarcinoma cells [103, 104]. Furthermore, PKM2 inhibitors, in combination with antineoplastic drugs, showed greater efficacy inhibiting the growth of bladder cancer in both in vitro and in vivo models [105]. Relevant to hypoxia, PKM2 interacts directly with HIF1α in the nucleus, leading to transcriptional activation of previously discussed target genes, promoting angiogenesis and a metabolic shift towards glycolysis [106]. In all, PKM2 activity is vital for cancer progression and adaption to the environment, making it a prime drug target for treating various cancers, especially those with drug-resistant phenotypes.
In several cancers, PDH inhibition diverts pyruvate from entering the TCA (Table 1). For example, in NSCLCs, PDH protein expression is reduced, and this coincides with increased HIF1α stabilization [107]. Decreased PDH activity in tumors has also been linked to over-expression of PDKs (four isoforms exist) that phosphorylate PDH and inhibit its activity [108]. Additionally, PDH inhibition in cancer cells is associated with the normoxic stabilization of HIF1α by glycolytic metabolites, which could be reversed by knockdown of PDK1 [109]. PDK1 is a HIF1α-regulated gene, and the accumulation of glycolytic metabolites may promote HIF1α activation and a feed-forward loop promoting tumor progression [109]. Since PDH activity is altered in many cancers, agents that ameliorate this effect are warranted. Dichloroacetate (DCA) is a PDK-2 inhibitor used to treat congenital lactic acidosis and mitochondrial disorders [110]. DCA increases pyruvate flux into the mitochondria, promoting glucose oxidation over glycolysis and increasing susceptibility to mitochondrial-induced cell death. [110]. DCA effectively inhibits the growth and survival of various cancer cell lines; however, DCA is a more potent inhibitor of PDK2, possibly making it better suited for cancers that express the PDK2 isoform, such as glioblastomas [110, 111]. Preclinical studies involving a small patient cohort and patient-derived cells demonstrated that DCA could be beneficial in treating glioblastoma multiforme by increasing PDH activity and apoptosis while reducing HIF1α-mediated p53 activation and angiogenesis, both in vivo and in vitro [111]. Studies suggest that DCA, combined with other therapeutic agents, might sensitize tumors by inhibiting autophagy [112], making them more susceptible to mitochondrial-dependent apoptosis [110]. DCA increased cancer cell sensitivity to platinum compounds in various ovarian, lung, bone, and colon cancer cell lines [113]. In radioresistant medulloblastoma cells, DCA treatment alters mitochondrial respiration and glycolysis, suppresses the cancer stem cell phenotype, and reverses radioresistance [114]. Additionally, DCA, in conjunction with Metformin, synergistically inhibits the growth and enhances the apoptosis of ovarian cancer [115], glioma [116], and lung cancer cells [117]. Multiple preclinical and clinical trials are currently ongoing to determine the safety and efficacy of DCA and other PDK inhibitors alone and in conjunction with other therapeutic agents in various cancers.
Beyond pyruvate’s conversion to lactate during hypoxia, it can also be carboxylated by the pyruvate carboxylase (PC), an enzyme commonly associated with biosynthetic pathways, particularly gluconeogenesis [118]. In this anaplerotic reaction, pyruvate is converted to oxaloacetate, which is crucial for replenishing tricarboxylic acid cycle intermediates when they are used for biosynthetic processes [119]. This pathway is upregulated in NSCLC and breast cancer and correlates with poor prognostic outcomes, as it regulates proliferation and tumorigenesis, even under normoxic conditions [120, 121]. Unfortunately, the availability of PC inhibitors is limited and serves as an obstacle for clinical investigation. However, the small molecule ZY-444 that binds to PC, inhibiting its activity, suppresses the wnt/β-catenin/Snail signaling, decreasing proliferation and metastasis of breast tumors [122].
Finally, pyruvate also can act as an antioxidant by a non-enzymatic reaction with hydrogen peroxide [123]. It is important to note that many antineoplastic agents, such as cisplatin, induce ROS-dependent apoptosis activation, and pyruvate might interfere with their effectiveness.
In summary, pyruvate is a key metabolite that mediates the adaptive response of both cancerous and non-transformed cells to hypoxia, as the conversion of pyruvate to lactate is required for the maintenance of cellular homeostasis in hypoxia. The altered fate of pyruvate promotes the stabilization of HIF1α, reinforces glycolysis, stimulates angiogenesis, and inhibits apoptosis. Thus, more therapeutic options that target mutations in pyruvate metabolism are needed. Additionally, pyruvate’s antioxidant abilities may interfere with current therapeutic options, and thus methods for addressing this problem should be further explored.
8. Lactate
Lactate is the end product of anaerobic glycolysis, formed by directly reducing pyruvate by LDH (Table 1). Indeed, elevated lactate production is the main feature of the Warburg phenotype [124], which is characterized by a high rate of glucose consumption and lactate production in tumors, even in the presence of oxygen. This phenomenon is a direct consequence of defects in cellular respiration, overexpression of glycolytic enzymes and transporters, and oncogene activation and tumor suppressor gene inactivation. The influence of lactate on the tumor microenvironment and subsequent signaling contributes to malignancy [125]. Lactate regulates many signaling pathways essential for gene transcription, migration, angiogenesis, cellular repair, and immunosuppression, processes commonly required for tumorigenesis [93, 125].
Under hypoxia or pseudohypoxia, a concomitant increase in HIF1α activity upregulates PDK1 and lactate dehydrogenase A, further diverting pyruvate metabolism towards lactate formation [126, 127]. LDH subunits are upregulated in various cancers, and selective inhibitors decrease the growth of various colon, breast, liver, melanoma, and large cell lung cancer cell lines [128]. As lactate is generated, cytosolic acidification increases, which can inhibit glycolysis; thus, lactate is transported across the plasma membrane and excreted through monocarboxylate transporters (MCTs) [93, 127, 129]. Lactate is then released, where it can act on surrounding cells, or transported by the bloodstream to the liver for gluconeogenesis or excreted by the kidney.
MCTs play a pivotal role in regulating lactate efflux. While MCT1 is expressed in most tissues at low levels, MCT2, 3, and 4 usually have a more tissue-specific distribution, with MCT4 being primarily expressed in highly glycolytic tissue [125, 130]. MCT1 and MCT4 are upregulated in cancer, and loss of their lactate transporter activity is associated with decreased glycolysis and tumor growth [125, 130]. Notably, the MCT4 gene is a direct transcriptional target of HIF1α, and along with MCT1, participates in the shuttling of lactate between cells of the tumor microenvironment [125, 130].
Clinically, high lactate levels have been associated with more aggressive tumors with a higher probability of metastasis and increased mortality [129, 131]. In these cancers, where aerobic glycolysis persists, the excess lactate excreted by tumors creates an acidic environment that contributes to metastasis and angiogenesis [130].
Intracellularly, lactate regulates the cell’s hypoxic response by stabilizing HIF1α, HIF2α, and N-myc downstream-regulated gene 3 (NDRG3) via PHD inhibition [125]. Under hypoxia, NDRG3 activates the Raf/ERK pathway, further stimulating tumor growth and angiogenesis [132]. Extracellularly, cancer cell-derived lactate stimulates endothelial migration [133, 134] and triggers ECM production in fibroblasts, promoting tumor growth [127].
Additionally, lactate can exert its effects on the tumor microenvironment by promoting immunosuppression. Various studies indicate that lactate interferes with cytotoxic T cell-derived interferon γ signaling [135], activates the IL-23/IL-17 pathway [136], and inhibits natural killer cell function [137], and high lactate levels induce natural killer cell apoptosis [130]. The lactate-rich tumor microenvironment also promotes macrophage polarization toward an M2-like phenotype by post-translational modification of histones via lysine lactylation [138]. These alterations impair the immune response, allowing cancer cells to escape the immune system’s antitumor response and, consequently, cell death.
Elevated lactate levels also correlate to tumor resistance to therapy. In multiple breast cancer cell lines resistant to Paclitaxel, inhibition of LDH led to the resensitization of the cells to the antineoplastic agent [139]. Lactate concentrations also positively correlate with radioresistance [140]. Furthermore, the lactate-mediated metabolic crosstalk between cancer cells and cancer-associated fibroblasts can aid in developing non-cell-autonomous mechanisms of chemoresistance. In a co-culture model, lactate stimulated cancer-associated fibroblasts to produce hepatocyte growth factor, which activated cMet-receptor tyrosine kinase-dependent signaling in cancer cells, promoting sustained resistance to tyrosine receptor kinase inhibitors [141].
Overall, lactate is an oncometabolite that plays an essential role in cancer development and progression by regulating cancer cell metabolism and tumor microenvironment. In cancer, many enzymes that regulate lactate metabolism, signaling, and shuttling are mutated, making them prime targets for the creation of oncogenic therapies.
9. Conclusion
Metabolic intermediates are small molecules essential for the maintenance of cellular homeostasis (Figure 1). These molecules can act as substrates, cofactors, second messengers, and enzymatic inhibitors. During hypoxia or pseudohypoxia, cellular energy metabolism changes include an increase in glucose consumption, inhibition of the electron transport chain, and the TCA cycle’s incomplete activity. This metabolic reprogramming alters the levels of numerous metabolites (Figure 1). These molecules directly aid the development of the tumor or contribute to the stabilization of HIF1α, exacerbating hypoxia-mediated signaling pathways. Thus, understanding these mechanisms of action helps understand how cancer develops and how we can create new, more efficient therapies.
Mutations in the enzymes responsible for the synthesis or utilization of these molecules are common in cancer and are usually associated with increased tumor malignancy. Not surprisingly, various pharmacological strategies have focused on regulating such enzymes, aiming to regulate the intracellular levels of these metabolites. Thus, a varied battery of inhibitors of these enzymes has been proposed as drugs for cancer treatment. Recent studies show that many of these drugs would have positive effects on specific types of cancer. Furthermore, the use of metabolites such as citrate and pyruvate, which have shown some antineoplastic effects, has been proposed as a therapeutic strategy. Finally, several of these metabolites and mutations in the enzymes that metabolize them have been proposed as malignancy markers. High lactate levels, for example, are linked to malignancy. In the case of mutations in FH and SDH, it is known that specific mutations are related to a worse prognosis. Studying these mutations in patient tumors opens a path to personalized diagnosis.
Acknowledgement
This study was supported by The National Heart, Lung, and Blood Institute of the National Institutes of Health under award HL095070 and by the American Heart Association grant AHA 19POST3438089.
Abbreviations
- 2-HG
2-hydroxyglutarate
- 2OGDD
2-oxoglutarate-dependent dioxygenase
- α-KG
Alpha-ketoglutarate
- α-KGDH
Alpha-ketoglutarate dehydrogenase
- ACLY
ATP citrate lyase
- AlkB
Alkylation repair homolog
- CIT
Mitochondrial citrate transporter
- CoA
Coenzyme A
- DCA
Dichloroacetate
- FIH
The Factor-inhibiting hypoxia-inducible factor
- FH
Fumarate Hydratase
- GABA
Gamma-Aminobutyric acid
- GDHA
Glutamate Dehydrogenase
- GLUT
Glucose transporter
- HLRCC
Hereditary leiomyomatosis and renal cell cancer
- HRE
Hypoxia response element
- HK
Hexokinase
- HIF1α
Hypoxia-inducible factor 1 alpha
- IDH
Isocitrate dehydrogenase
- IL
Interleukin
- LDH
Lactate dehydrogenase
- MCT
Monocarboxylate transporter
- MPP
Matrix Metallopeptidase
- NDRG3
N-myc downstream-regulated gene 3
- NSCLC
non-small cell lung carcinoma
- PC
Pyruvate carboxylase
- PDH
Pyruvate dehydrogenase (PDH)
- PDK1
Pyruvate dehydrogenase kinase 1 (PDK1)
- PHD
Prolyl hydroxylase
- PKM1/2
Pyruvate kinase M1/2
- ROS
Reactive oxygen species
- SDH
Succinate dehydrogenase
- SLC25A1 (CTP)
Solute Carrier Family 25 Member 1 (Inner membrane (mitochondrial) citrate transport protein)
- SLC13A5 (PMCT)
Solute Carrier Family 13 Member 5 (Plasma Membrane Na⁺-Coupled Citrate Transporter)
- TCA
tricarboxylic acid cycle
- TET
Ten-eleven translocation methylcytosine dioxygenase
- TNBC
Triple-negative breast cancer
- VEGF
Vascular endothelial growth factor
Footnotes
Declaration of competing interest
The authors declare no competing financial interests.
References
- [1].DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB, Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis, Proc Natl Acad Sci U S A, 104 (2007) 19345–19350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Iommarini L, Porcelli AM, Gasparre G, Kurelac I, Non-Canonical Mechanisms Regulating Hypoxia-Inducible Factor 1 Alpha in Cancer, Front Oncol, 7 (2017) 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].MacKenzie ED, Selak MA, Tennant DA, Payne LJ, Crosby S, Frederiksen CM, Watson DG, Gottlieb E, Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells, Mol Cell Biol, 27 (2007) 3282–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wang GL, Semenza GL, General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia, Proc Natl Acad Sci U S A, 90 (1993) 4304–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hirsila M, Koivunen P, Gunzler V, Kivirikko KI, Myllyharju J, Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor, J Biol Chem, 278 (2003) 30772–30780. [DOI] [PubMed] [Google Scholar]
- [6].Markolovic S, Wilkins SE, Schofield CJ, Protein Hydroxylation Catalyzed by 2-Oxoglutarate-dependent Oxygenases, J Biol Chem, 290 (2015) 20712–20722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yang M, Soga T, Pollard PJ, Oncometabolites: linking altered metabolism with cancer, J Clin Invest, 123 (2013) 3652–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Schober AS, Berra E, DUBs, New Members in the Hypoxia Signaling clUb, Front Oncol, 6 (2016) 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dengler VL, Galbraith M, Espinosa JM, Transcriptional regulation by hypoxia inducible factors, Crit Rev Biochem Mol Biol, 49 (2014) 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK, FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor, Genes Dev, 16 (2002) 1466–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tarhonskaya H, Hardy AP, Howe EA, Loik ND, Kramer HB, McCullagh JS, Schofield CJ, Flashman E, Kinetic Investigations of the Role of Factor Inhibiting Hypoxia-inducible Factor (FIH) as an Oxygen Sensor, J Biol Chem, 290 (2015) 19726–19742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC, HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption, Cell Metab, 3 (2006) 187–197. [DOI] [PubMed] [Google Scholar]
- [13].Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL, Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1, Mol Cell Biol, 16 (1996) 4604–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Haas R, Cucchi D, Smith J, Pucino V, Macdougall CE, Mauro C, Intermediates of Metabolism: From Bystanders to Signalling Molecules, Trends Biochem Sci, 41 (2016) 460–471. [DOI] [PubMed] [Google Scholar]
- [15].Tretter L, Patocs A, Chinopoulos C, Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis, Biochim Biophys Acta, 1857 (2016) 1086–1101. [DOI] [PubMed] [Google Scholar]
- [16].Weinert BT, Scholz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA, Choudhary C, Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation, Cell Rep, 4 (2013) 842–851. [DOI] [PubMed] [Google Scholar]
- [17].Peng M, Yang D, Hou Y, Liu S, Zhao M, Qin Y, Chen R, Teng Y, Liu M, Intracellular citrate accumulation by oxidized ATM-mediated metabolism reprogramming via PFKP and CS enhances hypoxic breast cancer cell invasion and metastasis, Cell Death Dis, 10 (2019) 228. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [18].Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB, Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability, Proc Natl Acad Sci U S A, 108 (2011) 19611–19616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sun J, Aluvila S, Kotaria R, Mayor JA, Walters DE, Kaplan RS, Mitochondrial and Plasma Membrane Citrate Transporters: Discovery of Selective Inhibitors and Application to Structure/Function Analysis, Mol Cell Pharmacol, 2 (2010) 101–110. [PMC free article] [PubMed] [Google Scholar]
- [20].Gnoni GV, Priore P, Geelen MJ, Siculella L, The mitochondrial citrate carrier: metabolic role and regulation of its activity and expression, IUBMB Life, 61 (2009) 987–994. [DOI] [PubMed] [Google Scholar]
- [21].Granchi C, ATP citrate lyase (ACLY) inhibitors: An anti-cancer strategy at the crossroads of glucose and lipid metabolism, Eur J Med Chem, 157 (2018) 1276–1291. [DOI] [PubMed] [Google Scholar]
- [22].Costello LC, Franklin RB, ‘Why do tumour cells glycolyse?’: from glycolysis through citrate to lipogenesis, Mol Cell Biochem, 280 (2005) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Williams NC, O’Neill LAJ, A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation, Front Immunol, 9 (2018) 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chen M, Huang J, The expanded role of fatty acid metabolism in cancer: new aspects and targets, Precis Clin Med, 2 (2019) 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Xiao W, Wang RS, Handy DE, Loscalzo J, NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism, Antioxid Redox Signal, 28 (2018) 251–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kulikova VA, Gromyko DV, Nikiforov AA, The Regulatory Role of NAD in Human and Animal Cells, Biochemistry (Mosc), 83 (2018) 800–812. [DOI] [PubMed] [Google Scholar]
- [27].Verdone L, Caserta M, Di Mauro E, Role of histone acetylation in the control of gene expression, Biochem Cell Biol, 83 (2005) 344–353. [DOI] [PubMed] [Google Scholar]
- [28].Iacobazzi V, Infantino V, Citrate--new functions for an old metabolite, Biol Chem, 395 (2014) 387–399. [DOI] [PubMed] [Google Scholar]
- [29].Schlichtholz B, Turyn J, Goyke E, Biernacki M, Jaskiewicz K, Sledzinski Z, Swierczynski J, Enhanced citrate synthase activity in human pancreatic cancer, Pancreas, 30 (2005) 99–104. [DOI] [PubMed] [Google Scholar]
- [30].Chen L, Liu T, Zhou J, Wang Y, Wang X, Di W, Zhang S, Citrate synthase expression affects tumor phenotype and drug resistance in human ovarian carcinoma, PLoS One, 9 (2014) e115708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lin CC, Cheng TL, Tsai WH, Tsai HJ, Hu KH, Chang HC, Yeh CW, Chen YC, Liao CC, Chang WT, Loss of the respiratory enzyme citrate synthase directly links the Warburg effect to tumor malignancy, Sci Rep, 2 (2012) 785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Huang L, Wang C, Xu H, Peng G, Targeting citrate as a novel therapeutic strategy in cancer treatment, Biochim Biophys Acta Rev Cancer, 1873 (2020) 188332. [DOI] [PubMed] [Google Scholar]
- [33].Hlouschek J, Hansel C, Jendrossek V, Matschke J, The Mitochondrial Citrate Carrier (SLC25A1) Sustains Redox Homeostasis and Mitochondrial Metabolism Supporting Radioresistance of Cancer Cells With Tolerance to Cycling Severe Hypoxia, Front Oncol, 8 (2018) 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kaplan RS, Morris HP, Coleman PS, Kinetic characteristics of citrate influx and efflux with mitochondria from Morris hepatomas 3924A and 16, Cancer Res, 42 (1982) 4399–4407. [PubMed] [Google Scholar]
- [35].Yahagi N, Shimano H, Hasegawa K, Ohashi K, Matsuzaka T, Najima Y, Sekiya M, Tomita S, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Nagai R, Ishibashi S, Kadowaki T, Makuuchi M, Ohnishi S, Osuga J, Yamada N, Co-ordinate activation of lipogenic enzymes in hepatocellular carcinoma, Eur J Cancer, 41 (2005) 1316–1322. [DOI] [PubMed] [Google Scholar]
- [36].Migita T, Narita T, Nomura K, Miyagi E, Inazuka F, Matsuura M, Ushijima M, Mashima T, Seimiya H, Satoh Y, Okumura S, Nakagawa K, Ishikawa Y, ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer, Cancer Res, 68 (2008) 8547–8554. [DOI] [PubMed] [Google Scholar]
- [37].Yancy HF, Mason JA, Peters S, Thompson CE 3rd, Littleton GK, Jett M, Day AA, Metastatic progression and gene expression between breast cancer cell lines from African American and Caucasian women, J Carcinog, 6 (2007) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Varis A, Wolf M, Monni O, Vakkari ML, Kokkola A, Moskaluk C, Frierson H Jr., Powell SM, Knuutila S, Kallioniemi A, El-Rifai W, Targets of gene amplification and overexpression at 17q in gastric cancer, Cancer Res, 62 (2002) 2625–2629. [PubMed] [Google Scholar]
- [39].Halliday KR, Fenoglio-Preiser C, Sillerud LO, Differentiation of human tumors from nonmalignant tissue by natural-abundance 13C NMR spectroscopy, Magn Reson Med, 7 (1988) 384–411. [DOI] [PubMed] [Google Scholar]
- [40].Icard P, Lincet H, The reduced concentration of citrate in cancer cells: An indicator of cancer aggressiveness and a possible therapeutic target, Drug Resist Updat, 29 (2016) 47–53. [DOI] [PubMed] [Google Scholar]
- [41].Ren JG, Seth P, Ye H, Guo K, Hanai JI, Husain Z, Sukhatme VP, Citrate Suppresses Tumor Growth in Multiple Models through Inhibition of Glycolysis, the Tricarboxylic Acid Cycle and the IGF-1R Pathway, Sci Rep, 7 (2017) 4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zdzisinska B, Zurek A, Kandefer-Szerszen M, Alpha-Ketoglutarate as a Molecule with Pleiotropic Activity: Well-Known and Novel Possibilities of Therapeutic Use, Arch Immunol Ther Exp (Warsz), 65 (2017) 21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Islam MS, Leissing TM, Chowdhury R, Hopkinson RJ, Schofield CJ, 2-Oxoglutarate-Dependent Oxygenases, Annu Rev Biochem, 87 (2018) 585–620. [DOI] [PubMed] [Google Scholar]
- [44].Fendt SM, Bell EL, Keibler MA, Olenchock BA, Mayers JR, Wasylenko TM, Vokes NI, Guarente L, Vander Heiden MG, Stephanopoulos G, Reductive glutamine metabolism is a function of the alpha-ketoglutarate to citrate ratio in cells, Nat Commun, 4 (2013) 2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Vatrinet R, Leone G, De Luise M, Girolimetti G, Vidone M, Gasparre G, Porcelli AM, The α-ketoglutarate dehydrogenase complex in cancer metabolic plasticity, Cancer Metab, 5 (2017) 3–3.28184304 [Google Scholar]
- [46].Moreira JD, Hamraz M, Abolhassani M, Bigan E, Peres S, Pauleve L, Nogueira ML, Steyaert JM, Schwartz L, The Redox Status of Cancer Cells Supports Mechanisms behind the Warburg Effect, Metabolites, 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Santidrian AF, Matsuno-Yagi A, Ritland M, Seo BB, LeBoeuf SE, Gay LJ, Yagi T, Felding-Habermann B, Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression, J Clin Invest, 123 (2013) 1068–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Desquiret-Dumas V, Gueguen N, Leman G, Baron S, Nivet-Antoine V, Chupin S, Chevrollier A, Vessieres E, Ayer A, Ferre M, Bonneau D, Henrion D, Reynier P, Procaccio V, Resveratrol induces a mitochondrial complex I-dependent increase in NADH oxidation responsible for sirtuin activation in liver cells, J Biol Chem, 288 (2013) 36662–36675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wong BW, Kuchnio A, Bruning U, Carmeliet P, Emerging novel functions of the oxygen-sensing prolyl hydroxylase domain enzymes, Trends Biochem Sci, 38 (2013) 3–11. [DOI] [PubMed] [Google Scholar]
- [50].Matsumoto K, Imagawa S, Obara N, Suzuki N, Takahashi S, Nagasawa T, Yamamoto M, 2-Oxoglutarate downregulates expression of vascular endothelial growth factor and erythropoietin through decreasing hypoxia-inducible factor-1alpha and inhibits angiogenesis, J Cell Physiol, 209 (2006) 333–340. [DOI] [PubMed] [Google Scholar]
- [51].Tseng CW, Kuo WH, Chan SH, Chan HL, Chang KJ, Wang LH, Transketolase Regulates the Metabolic Switch to Control Breast Cancer Cell Metastasis via the alpha-Ketoglutarate Signaling Pathway, Cancer Res, 78 (2018) 2799–2812. [DOI] [PubMed] [Google Scholar]
- [52].Matsumoto K, Obara N, Ema M, Horie M, Naka A, Takahashi S, Imagawa S, Antitumor effects of 2-oxoglutarate through inhibition of angiogenesis in a murine tumor model, Cancer Sci, 100 (2009) 1639–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Atlante S, Visintin A, Marini E, Savoia M, Dianzani C, Giorgis M, Surun D, Maione F, Schnutgen F, Farsetti A, Zeiher AM, Bertinaria M, Giraudo E, Spallotta F, Cencioni C, Gaetano C, alpha-ketoglutarate dehydrogenase inhibition counteracts breast cancer-associated lung metastasis, Cell Death Dis, 9 (2018) 756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Stuart SD, Schauble A, Gupta S, Kennedy AD, Keppler BR, Bingham PM, Zachar Z, A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process, Cancer Metab, 2 (2014) 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM, Cancer-associated IDH1 mutations produce 2-hydroxyglutarate, Nature, 462 (2009) 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, Pollock R, O’Donnell P, Grigoriadis A, Diss T, Eskandarpour M, Presneau N, Hogendoorn PC, Futreal A, Tirabosco R, Flanagan AM, IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours, J Pathol, 224 (2011) 334–343. [DOI] [PubMed] [Google Scholar]
- [57].Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A, Analysis of the IDH1 codon 132 mutation in brain tumors, Acta Neuropathol, 116 (2008) 597–602. [DOI] [PubMed] [Google Scholar]
- [58].Borger DR, Tanabe KK, Fan KC, Lopez HU, Fantin VR, Straley KS, Schenkein DP, Hezel AF, Ancukiewicz M, Liebman HM, Kwak EL, Clark JW, Ryan DP, Deshpande V, Dias-Santagata D, Ellisen LW, Zhu AX, Iafrate AJ, Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping, Oncologist, 17 (2012) 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, Felsberg J, Wolter M, Mawrin C, Wick W, Weller M, Herold-Mende C, Unterberg A, Jeuken JW, Wesseling P, Reifenberger G, von Deimling A, Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas, Acta Neuropathol, 118 (2009) 469–474. [DOI] [PubMed] [Google Scholar]
- [60].Murugan AK, Bojdani E, Xing M, Identification and functional characterization of isocitrate dehydrogenase 1 (IDH1) mutations in thyroid cancer, Biochem Biophys Res Commun, 393 (2010) 555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, Stone RM, DeAngelo DJ, Levine RL, Flinn IW, Kantarjian HM, Collins R, Patel MR, Frankel AE, Stein A, Sekeres MA, Swords RT, Medeiros BC, Willekens C, Vyas P, Tosolini A, Xu Q, Knight RD, Yen KE, Agresta S, de Botton S, Tallman MS, Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia, Blood, 130 (2017) 722–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, Rabinowitz JD, Carroll M, Su SM, Sharp KA, Levine RL, Thompson CB, The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate, Cancer Cell, 17 (2010) 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Oldham WM, Clish CB, Yang Y, Loscalzo J, Hypoxia-Mediated Increases in L-2-hydroxyglutarate Coordinate the Metabolic Response to Reductive Stress, Cell Metab, 22 (2015) 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Shim EH, Livi CB, Rakheja D, Tan J, Benson D, Parekh V, Kho EY, Ghosh AP, Kirkman R, Velu S, Dutta S, Chenna B, Rea SL, Mishur RJ, Li Q, Johnson-Pais TL, Guo L, Bae S, Wei S, Block K, Sudarshan S, L-2-Hydroxyglutarate: an epigenetic modifier and putative oncometabolite in renal cancer, Cancer Discov, 4 (2014) 1290–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IK, Li XS, Woon EC, Yang M, McDonough MA, King ON, Clifton IJ, Klose RJ, Claridge TD, Ratcliffe PJ, Schofield CJ, Kawamura A, The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases, EMBO Rep, 12 (2011) 463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Reiter-Brennan C, Semmler L, Klein A, The effects of 2-hydroxyglutarate on the tumorigenesis of gliomas, Contemp Oncol (Pozn), 22 (2018) 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Nadtochiy SM, Schafer X, Fu D, Nehrke K, Munger J, Brookes PS, Acidic pH Is a Metabolic Switch for 2-Hydroxyglutarate Generation and Signaling, J Biol Chem, 291 (2016) 20188–20197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Zhang Z, Tan M, Xie Z, Dai L, Chen Y, Zhao Y, Identification of lysine succinylation as a new post-translational modification, Nat Chem Biol, 7 (2011) 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Saito Y, Ishii KA, Aita Y, Ikeda T, Kawakami Y, Shimano H, Hara H, Takekoshi K, Loss of SDHB Elevates Catecholamine Synthesis and Secretion Depending on ROS Production and HIF Stabilization, Neurochem Res, 41 (2016) 696–706. [DOI] [PubMed] [Google Scholar]
- [70].Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, Hargreaves IP, Heales SJ, Chung YL, Griffiths JR, Dalgleish A, McGrath JA, Gleeson MJ, Hodgson SV, Poulsom R, Rustin P, Tomlinson IP, Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations, Hum Mol Genet, 14 (2005) 2231–2239. [DOI] [PubMed] [Google Scholar]
- [71].Pistollato F, Abbadi S, Rampazzo E, Viola G, Della Puppa A, Cavallini L, Frasson C, Persano L, Panchision DM, Basso G, Hypoxia and succinate antagonize 2-deoxyglucose effects on glioblastoma, Biochem Pharmacol, 80 (2010) 1517–1527. [DOI] [PubMed] [Google Scholar]
- [72].Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, Zhao S, Ye D, Xiong Y, Guan KL, Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors, Genes Dev, 26 (2012) 1326–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Rutter J, Winge DR, Schiffman JD, Succinate dehydrogenase - Assembly, regulation and role in human disease, Mitochondrion, 10 (2010) 393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Wu JY, Huang TW, Hsieh YT, Wang YF, Yen CC, Lee GL, Yeh CC, Peng YJ, Kuo YY, Wen HT, Lin HC, Hsiao CW, Wu KK, Kung HJ, Hsu YJ, Kuo CC, Cancer-Derived Succinate Promotes Macrophage Polarization and Cancer Metastasis via Succinate Receptor, Mol Cell, 77 (2020) 213–227 e215. [DOI] [PubMed] [Google Scholar]
- [75].Qi H, Ning X, Yu C, Ji X, Jin Y, McNutt MA, Yin Y, Succinylation-dependent mitochondrial translocation of PKM2 promotes cell survival in response to nutritional stress, Cell Death Dis, 10 (2019) 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wang C, Zhang C, Li X, Shen J, Xu Y, Shi H, Mu X, Pan J, Zhao T, Li M, Geng B, Xu C, Wen H, You Q, CPT1A-mediated succinylation of S100A10 increases human gastric cancer invasion, J Cell Mol Med, 23 (2019) 293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Smestad J, Erber L, Chen Y, Maher LJ 3rd, Chromatin Succinylation Correlates with Active Gene Expression and Is Perturbed by Defective TCA Cycle Metabolism, iScience, 2 (2018) 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Li L, Shi L, Yang S, Yan R, Zhang D, Yang J, He L, Li W, Yi X, Sun L, Liang J, Cheng Z, Shi L, Shang Y, Yu W, SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability, Nat Commun, 7 (2016) 12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Agaimy A, [Succinate dehydrogenase (SDH)-deficient renal cell carcinoma], Pathologe, 37 (2016) 144–152. [DOI] [PubMed] [Google Scholar]
- [80].Wang H, Chen Y, Wu G, SDHB deficiency promotes TGFbeta-mediated invasion and metastasis of colorectal cancer through transcriptional repression complex SNAIL1-SMAD3/4, Transl Oncol, 9 (2016) 512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Nannini M, Astolfi A, Urbini M, Indio V, Santini D, Heinrich MC, Corless CL, Ceccarelli C, Saponara M, Mandrioli A, Lolli C, Ercolani G, Brandi G, Biasco G, Pantaleo MA, Integrated genomic study of quadruple-WT GIST (KIT/PDGFRA/SDH/RAS pathway wild-type GIST), BMC Cancer, 14 (2014) 685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Ni Y, Seballos S, Ganapathi S, Gurin D, Fletcher B, Ngeow J, Nagy R, Kloos RT, Ringel MD, LaFramboise T, Eng C, Germline and somatic SDHx alterations in apparently sporadic differentiated thyroid cancer, Endocr Relat Cancer, 22 (2015) 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Shambaugh GE 3rd, Urea biosynthesis I The urea cycle and relationships to the citric acid cycle, Am J Clin Nutr, 30 (1977) 2083–2087. [DOI] [PubMed] [Google Scholar]
- [84].Matthews DE, An overview of phenylalanine and tyrosine kinetics in humans, J Nutr, 137 (2007) 1549S–1555S; discussion 1573S-1575S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Stepinski J, Bizon D, Piec G, Angielski S, The purine nucleotide cycle activity in renal cortex and medulla, Am J Kidney Dis, 14 (1989) 307–309. [DOI] [PubMed] [Google Scholar]
- [86].Bisbach CM, Hass DT, Robbings BM, Rountree AM, Sadilek M, Sweet IR, Hurley JB, Succinate Can Shuttle Reducing Power from the Hypoxic Retina to the O2-Rich Pigment Epithelium, Cell Rep, 31 (2020) 107606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Yang M, Soga T, Pollard PJ, Adam J, The emerging role of fumarate as an oncometabolite, Front Oncol, 2 (2012) 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Yogev O, Yogev O, Singer E, Shaulian E, Goldberg M, Fox TD, Pines O, Fumarase: a mitochondrial metabolic enzyme and a cytosolic/nuclear component of the DNA damage response, PLoS Biol, 8 (2010) e1000328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Lehtonen HJ, Kiuru M, Ylisaukko-Oja SK, Salovaara R, Herva R, Koivisto PA, Vierimaa O, Aittomaki K, Pukkala E, Launonen V, Aaltonen LA, Increased risk of cancer in patients with fumarate hydratase germline mutation, J Med Genet, 43 (2006) 523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, Merino M, Trepel J, Zbar B, Toro J, Ratcliffe PJ, Linehan WM, Neckers L, HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability, Cancer Cell, 8 (2005) 143–153. [DOI] [PubMed] [Google Scholar]
- [91].Pollard PJ, Spencer-Dene B, Shukla D, Howarth K, Nye E, El-Bahrawy M, Deheragoda M, Joannou M, McDonald S, Martin A, Igarashi P, Varsani-Brown S, Rosewell I, Poulsom R, Maxwell P, Stamp GW, Tomlinson IP, Targeted inactivation of fh1 causes proliferative renal cyst development and activation of the hypoxia pathway, Cancer Cell, 11 (2007) 311–319. [DOI] [PubMed] [Google Scholar]
- [92].Landau BR, Gluconeogenesis and pyruvate metabolism in rat kidney, in vitro, Endocrinology, 67 (1960) 744–751. [DOI] [PubMed] [Google Scholar]
- [93].Adeva-Andany M, Lopez-Ojen M, Funcasta-Calderon R, Ameneiros-Rodriguez E, Donapetry-Garcia C, Vila-Altesor M, Rodriguez-Seijas J, Comprehensive review on lactate metabolism in human health, Mitochondrion, 17 (2014) 76–100. [DOI] [PubMed] [Google Scholar]
- [94].Sutendra G, Kinnaird A, Dromparis P, Paulin R, Stenson TH, Haromy A, Hashimoto K, Zhang N, Flaim E, Michelakis ED, A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation, Cell, 158 (2014) 84–97. [DOI] [PubMed] [Google Scholar]
- [95].Roudier E, Perrin A, Considering the role of pyruvate in tumor cells during hypoxia, Biochim Biophys Acta, 1796 (2009) 55–62. [DOI] [PubMed] [Google Scholar]
- [96].Diers AR, Broniowska KA, Chang CF, Hogg N, Pyruvate fuels mitochondrial respiration and proliferation of breast cancer cells: effect of monocarboxylate transporter inhibition, Biochem J, 444 (2012) 561–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Lu H, Forbes RA, Verma A, Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis, J Biol Chem, 277 (2002) 23111–23115. [DOI] [PubMed] [Google Scholar]
- [98].Gao H, Dong B, Liu X, Xuan H, Huang Y, Lin D, Metabonomic profiling of renal cell carcinoma: high-resolution proton nuclear magnetic resonance spectroscopy of human serum with multivariate data analysis, Anal Chim Acta, 624 (2008) 269–277. [DOI] [PubMed] [Google Scholar]
- [99].Schaur RJ, Georgiopulos E, Fink E, Kronberger L, Schauenstein E, [Citrate values in the whole blood of patients with malignant tumors of various locations], Med Klin, 71 (1976) 900–903. [PubMed] [Google Scholar]
- [100].Hosios AM, Vander Heiden MG, The redox requirements of proliferating mammalian cells, J Biol Chem, 293 (2018) 7490–7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Owen OE, Kalhan SC, Hanson RW, The key role of anaplerosis and cataplerosis for citric acid cycle function, J Biol Chem, 277 (2002) 30409–30412. [DOI] [PubMed] [Google Scholar]
- [102].Desai S, Ding M, Wang B, Lu Z, Zhao Q, Shaw K, Yung WK, Weinstein JN, Tan M, Yao J, Tissue-specific isoform switch and DNA hypomethylation of the pyruvate kinase PKM gene in human cancers, Oncotarget, 5 (2014) 8202–8210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Shankar Babu M, Mahanta S, Lakhter AJ, Hato T, Paul S, Naidu SR, Lapachol inhibits glycolysis in cancer cells by targeting pyruvate kinase M2, PLoS One, 13 (2018) e0191419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Vander Heiden MG, Christofk HR, Schuman E, Subtelny AO, Sharfi H, Harlow EE, Xian J, Cantley LC, Identification of small molecule inhibitors of pyruvate kinase M2, Biochem Pharmacol, 79 (2010) 1118–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Su Q, Tao T, Tang L, Deng J, Darko KO, Zhou S, Peng M, He S, Zeng Q, Chen AF, Yang X, Down-regulation of PKM2 enhances anticancer efficiency of THP on bladder cancer, J Cell Mol Med, 22 (2018) 2774–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, Cole RN, Pandey A, Semenza GL, Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1, Cell, 145 (2011) 732–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Koukourakis MI, Giatromanolaki A, Sivridis E, Gatter KC, Harris AL, Tumor G Angiogenesis Research, Pyruvate dehydrogenase and pyruvate dehydrogenase kinase expression in non small cell lung cancer and tumor-associated stroma, Neoplasia, 7 (2005) 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Popov KM, Kedishvili NY, Zhao Y, Gudi R, Harris RA, Molecular cloning of the p45 subunit of pyruvate dehydrogenase kinase, J Biol Chem, 269 (1994) 29720–29724. [PubMed] [Google Scholar]
- [109].McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, Wu H, Schell MJ, Tsang TM, Teahan O, Zhou S, Califano JA, Jeoung NH, Harris RA, Verma A, Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells, J Biol Chem, 283 (2008) 22700–22708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Michelakis ED, Webster L, Mackey JR, Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer, Br J Cancer, 99 (2008) 989–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC, Metabolic modulation of glioblastoma with dichloroacetate, Sci Transl Med, 2 (2010) 31ra34. [DOI] [PubMed] [Google Scholar]
- [112].Lu X, Zhou D, Hou B, Liu QX, Chen Q, Deng XF, Yu ZB, Dai JG, Zheng H, Dichloroacetate enhances the antitumor efficacy of chemotherapeutic agents via inhibiting autophagy in non-small-cell lung cancer, Cancer Manag Res, 10 (2018) 1231–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Olszewski U, Poulsen TT, Ulsperger E, Poulsen HS, Geissler K, Hamilton G, In vitro cytotoxicity of combinations of dichloroacetate with anticancer platinum compounds, Clin Pharmacol, 2 (2010) 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Sun L, Moritake T, Ito K, Matsumoto Y, Yasui H, Nakagawa H, Hirayama A, Inanami O, Tsuboi K, Metabolic analysis of radioresistant medulloblastoma stem-like clones and potential therapeutic targets, PLoS One, 12 (2017) e0176162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Li B, Li X, Ni Z, Zhang Y, Zeng Y, Yan X, Huang Y, He J, Lyu X, Wu Y, Wang Y, Zheng Y, He F, Dichloroacetate and metformin synergistically suppress the growth of ovarian cancer cells, Oncotarget, 7 (2016) 59458–59470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Kolesnik DL, Pyaskovskaya ON, Yurchenko OV, Solyanik GI, Metformin enhances antitumor action of sodium dichloroacetate against glioma C6, Exp Oncol, 41 (2019) 123–129. [DOI] [PubMed] [Google Scholar]
- [117].Kolesnik DL, Pyaskovskaya ON, Gorbach O, Solyanik GI, Metformin enhances cytotoxic action of dichloroacetate against Lewis lung carcinoma cells in vitro, Exp Oncol, 42 (2020) 35–39. [DOI] [PubMed] [Google Scholar]
- [118].Jitrapakdee S, Vidal-Puig A, Wallace JC, Anaplerotic roles of pyruvate carboxylase in mammalian tissues, Cell Mol Life Sci, 63 (2006) 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Brunengraber H, Roe CR, Anaplerotic molecules: current and future, J Inherit Metab Dis, 29 (2006) 327–331. [DOI] [PubMed] [Google Scholar]
- [120].Phannasil P, Thuwajit C, Warnnissorn M, Wallace JC, MacDonald MJ, Jitrapakdee S, Pyruvate Carboxylase Is Up-Regulated in Breast Cancer and Essential to Support Growth and Invasion of MDA-MB-231 Cells, PLoS One, 10 (2015) e0129848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Sellers K, Fox MP, Bousamra M 2nd, Slone SP, Higashi RM, Miller DM, Wang Y, Yan J, Yuneva MO, Deshpande R, Lane AN, Fan TW, Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation, J Clin Invest, 125 (2015) 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Lin Q, He Y, Wang X, Zhang Y, Hu M, Guo W, He Y, Zhang T, Lai L, Sun Z, Yi Z, Liu M, Chen Y, Targeting Pyruvate Carboxylase by a Small Molecule Suppresses Breast Cancer Progression, Adv Sci (Weinh), 7 (2020) 1903483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].O’Donnell-Tormey J, Nathan CF, Lanks K, DeBoer CJ, de la Harpe J, Secretion of pyruvate. An antioxidant defense of mammalian cells, J Exp Med, 165 (1987) 500–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Warburg O, On the origin of cancer cells, Science, 123 (1956) 309–314. [DOI] [PubMed] [Google Scholar]
- [125].Baltazar F, Afonso J, Costa M, Granja S, Lactate Beyond a Waste Metabolite: Metabolic Affairs and Signaling in Malignancy, Front Oncol, 10 (2020) 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Eales KL, Hollinshead KE, Tennant DA, Hypoxia and metabolic adaptation of cancer cells, Oncogenesis, 5 (2016) e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Hirschhaeuser F, Sattler UG, Mueller-Klieser W, Lactate: a metabolic key player in cancer, Cancer Res, 71 (2011) 6921–6925. [DOI] [PubMed] [Google Scholar]
- [128].Kim EY, Chung TW, Han CW, Park SY, Park KH, Jang SB, Ha KT, A Novel Lactate Dehydrogenase Inhibitor, 1-(Phenylseleno)-4-(Trifluoromethyl) Benzene, Suppresses Tumor Growth through Apoptotic Cell Death, Sci Rep, 9 (2019) 3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Sola-Penna M, Metabolic regulation by lactate, IUBMB Life, 60 (2008) 605–608. [DOI] [PubMed] [Google Scholar]
- [130].de la Cruz-Lopez KG, Castro-Munoz LJ, Reyes-Hernandez DO, Garcia-Carranca A, Manzo-Merino J, Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches, Front Oncol, 9 (2019) 1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Walenta S, Mueller-Klieser WF, Lactate: mirror and motor of tumor malignancy, Semin Radiat Oncol, 14 (2004) 267–274. [DOI] [PubMed] [Google Scholar]
- [132].Lee GY, Chun YS, Shin HW, Park JW, Potential role of the N-MYC downstream-regulated gene family in reprogramming cancer metabolism under hypoxia, Oncotarget, 7 (2016) 57442–57451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Beckert S, Farrahi F, Aslam RS, Scheuenstuhl H, Konigsrainer A, Hussain MZ, Hunt TK, Lactate stimulates endothelial cell migration, Wound Repair Regen, 14 (2006) 321–324. [DOI] [PubMed] [Google Scholar]
- [134].Vegran F, Boidot R, Michiels C, Sonveaux P, Feron O, Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis, Cancer Res, 71 (2011) 2550–2560. [DOI] [PubMed] [Google Scholar]
- [135].Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, Renner K, Timischl B, Mackensen A, Kunz-Schughart L, Andreesen R, Krause SW, Kreutz M, Inhibitory effect of tumor cell-derived lactic acid on human T cells, Blood, 109 (2007) 3812–3819. [DOI] [PubMed] [Google Scholar]
- [136].Shime H, Yabu M, Akazawa T, Kodama K, Matsumoto M, Seya T, Inoue N, Tumor-secreted lactic acid promotes IL-23/IL-17 proinflammatory pathway, J Immunol, 180 (2008) 7175–7183. [DOI] [PubMed] [Google Scholar]
- [137].Husain Z, Huang Y, Seth P, Sukhatme VP, Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells, J Immunol, 191 (2013) 1486–1495. [DOI] [PubMed] [Google Scholar]
- [138].Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, Ding J, Czyz D, Hu R, Ye Z, He M, Zheng YG, Shuman HA, Dai L, Ren B, Roeder RG, Becker L, Zhao Y, Metabolic regulation of gene expression by histone lactylation, Nature, 574 (2019) 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Zhou M, Zhao Y, Ding Y, Liu H, Liu Z, Fodstad O, Riker AI, Kamarajugadda S, Lu J, Owen LB, Ledoux SP, Tan M, Warburg effect in chemosensitivity: targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol, Mol Cancer, 9 (2010) 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Sattler UG, Meyer SS, Quennet V, Hoerner C, Knoerzer H, Fabian C, Yaromina A, Zips D, Walenta S, Baumann M, Mueller-Klieser W, Glycolytic metabolism and tumour response to fractionated irradiation, Radiother Oncol, 94 (2010) 102–109. [DOI] [PubMed] [Google Scholar]
- [141].Apicella M, Giannoni E, Fiore S, Ferrari KJ, Fernandez-Perez D, Isella C, Granchi C, Minutolo F, Sottile A, Comoglio PM, Medico E, Pietrantonio F, Volante M, Pasini D, Chiarugi P, Giordano S, Corso S, Increased Lactate Secretion by Cancer Cells Sustains Non-cell-autonomous Adaptive Resistance to MET and EGFR Targeted Therapies, Cell Metab, 28 (2018) 848–865 e846. [DOI] [PubMed] [Google Scholar]