

Abstract
Segmental injuries to peripheral nerves (PNs) too often result in lifelong disability or pain syndromes due to a lack of restorative treatment options. For injuries beyond a critical size, a bridging device must be inserted to direct regeneration. PN allografts from immunologically incompatible donors are highly effective bridging devices but are not a regular clinical option because of the expense and health risks of systemic immunosuppression (ISN). We have developed a method to deliver a single administration of ISN localized around a PN allograft that circumvents the risks of systemic ISN. Localized ISN was provided by regulatory T cells (Tregs), a potently immunosuppressive cell type, that was delivered around a PN allograft with a poly(ethylene glycol) norbornene (PEGNB) degradable hydrogel. Tregs are released from the hydrogel over 14 d, infiltrate the graft, suppress the host immune response and facilitate regeneration of the recipient rats equal to the autograft control. Furthermore, this method was effective in a segmental PN defect that included a branch point, for which there currently exist no treatment options. These results show that localized delivery of immunosuppressive cells for PN allografts is an effective new strategy for treating segmental PN defects that can also be used to regenerate complex nerve structures.
Keywords: Nerve regeneration, Regulatory T cells, Branched peripheral nerve, Allografts, PEGNB
1. Introduction
Injuries to peripheral nerves (PNs) affect hundreds of thousands of new individuals in the United States each year [1]. These injuries often leave patients with motor disability, sensory aberrations and pain syndromes [2,3]. When a PN is bisected and a gap of more than a few millimeters is created, a bridging device must be inserted for regeneration to occur. The degree of regeneration depends on how well surviving axons cross the bridging device, re-enter the distal nerve structure and navigate to their target tissues, which may involve long distances and many branch points. The slow rate of axonal regeneration and misrouting of axons at branch points are major impediments to functional recovery.
The best current option for a bridging device is an autologous nerve, such as the sural branch of sciatic nerve or the dorsal cutaneous branch of the ulnar nerve. These are sensory nerves that are an acceptable tradeoff for partial restoration of motor function but are not ideal for regeneration of motor or mixed PNs, which contain both motor and sensory axons [4–6]. To offset concerns of donor site morbidity, non-exact physical matching and sensorimotor mixmatching of sensory autografts, scaffolds of degradable conduits and decellularized allografts have been developed as bridging devices. Although approved for small PN segmental defects, they lack the bioactivity of sensory autografts and are fractionally effective in comparison with autografts [7–14]. Many innovative methods are being developed to bioactivate such scaffolds, but none are yet equivalent to sensory autografts.
Furthermore, treatment of a PN defect that includes a branch point is particularly complex. With an organization similar to vasculature, PNs extend in large bundles out from the spinal cord and progressively branch into smaller fibers that innervate motor and sensory targets [15]. In the human sciatic nerve alone, there are 10 major branch points and numerous minor branch points until the nerves reach their enervation targets [15]. When an injury affects a branched structure, it is conceivable that surgeons could anastomose two autologous nerve sections to the larger proximal stump that are then connected to the distal branches, similar to what occurs in linear defects when the autologous nerve is significantly smaller in diameter than the affected nerve [16]. However, there are no reports of this practice or its efficacy for branched defects. 3D printed conduits for branched defects have been developed [17], but nothing has been as of yet tested in vivo.
PN allografts are highly effective bridging devices for PN defects but currently are limited by the need for systemic immunosuppression (ISN). Allografts can be from mixed nerves, can be anatomically matched to the implant site and better recapitulate fascicular organization, all of which lead to superior regeneration [4–7,18,19]. Three other factors make PN allografts amenable for clinical use; Graft procurement is facilitated because immunological matching is not necessary, surgeons often wait weeks to months from the time of initial injury to specifically address the damaged PN and PN allografts can be stored and retain their regenerative potential [20,21]. The primary limitation preventing widespread use of PN allografts is ISN. Systemic ISN carries substantial risks to the patient, such as susceptibility to pathogens, nephrotoxicity and lymphoproliferative disorders [22]. Systemic ISN is also expensive and is generally reserved for quantity of life, rather than quality of life, scenarios [23].
PNs have unique immunological properties that may be leveraged to circumvent the risks and expense of systemic ISN. Observations across multiple species illustrate that temporary systemic ISN is sufficient to stimulate full regeneration with a PN allograft [24–31]. Thus, achieving long-term tolerance isn’t necessary for PN allografts. This informed our hypothesis that a sufficient delay might be achieved by localized ISN instead of systemic ISN to allow for PN allograft regeneration. Localized ISN would circumvent or alleviate concerns about systemic ISN and has long been a goal of transplantation therapy.
Here we provided localized ISN with regulatory T cells (Tregs), which are an immunosuppressive sub-population of CD4+ T cells [32]. Tregs suppress antigen-presenting cells (APCs) and effector T cells and have shown temporary efficacy in clinical models of immune suppression [33,34]. In our study, Tregs were placed around 2-cm branched PN allografts via a degradable hydrogel carrier at the time of graft placement. The poly(ethylene glycol) norbornene (PEGNB) hydrogel delivered the Tregs over a period of 14 d. No additional immunosuppressive interventions were administered. PN regeneration in animals that received PEGNB with Tregs was equal to the regeneration in rats that received mixed autografts. This new, highly effective strategy for PN regeneration suggests that localized ISN is possible for PN allografts and also is capable of promoting regeneration through branched PN defects.
2. Materials and methods
2.1. Animal acquisition and care
The animals were acquired, cared for and used in accordance with the NIH Guide for the Care and Use of Laboratory Animals and followed the protocol approved by the University of Wyoming IACUC. Rats were housed at ambient temperature with stable humidity and a natural day-night cycle, with free access to rodent laboratory food and water. Lewis and SD rats were obtained from Charles River Laboratories. Wilmington, MA, USA. SD rats constitutively expressing GFP, strain SD-Tg (UBC-EGFP) 2BalRrrc (common name SD-EGFP), were obtained from the Rat Resource and Research Center (RRRC), Columbia, MO, USA. Animals were housed jointly in cages until 1 week before surgery, at which point they were kept in individual cages for the duration of the study.
2.2. Isolation, expansion and immunophenotyping of rat tregs
Spleens were harvested from SD rats or from heterozygotes obtained by breeding SD-EGFP animals with SD animals. Tregs were purified from the spleens by magnetic-activated cell sorting with phycoerythrin (PE)-conjugated anti-CD4 (eBioscience, cat. no. 12–0040-82), anti-PE multisorting microbeads (Miltenyl; cat. no. 130–040-100), biotin-labeled anti-CD25 (Biotin; cat. no. 102004) and Streptavidin MicroBeads (Miltenyl, cat. no. 130–048-102). Isolated Tregs were expanded in RPMI 1640 (Sigma, cat. no. R8758) supplemented with 400 U/mL interleukin-2 (IL-2) (R&D systems, cat. no. P60568), 1 × non-essential amino acids (Gibco, cat. no.11140050), 1 × sodium pyruvate (Gibco, cat. no. 11360070) and 10% fetal bovine serum (FBS) (Life Technologies, cat. no. 10437028) in a plate coated with 5 μg/mL of goat anti−rat IgG1 (Thermo Fisher Scientific, cat. no. A-11006) and 5 μg/mL of purified NA/LE mouse anti−rat CD28-superagonist (BD Biosciences, cat. no. 554992) [35].
For immunophenotyping, cells were washed with PBS (Sigma-Aldrich, cat. no. P4417) and labeled with PE-conjugated mouse anti−rat CD4 (1:50) (eBioscience, cat. no. 12–0040-82) and with mouse anti−rat CD25 (1:50) (Biolegend, cat. no. 202111) and Alexa Fluor 488−conjugated goat anti−mouse IgG1 (1–10 μg/mL) (Thermo Fisher Scientific, cat. no. A-21121). Cells were permeabilized with 0.1% Triton X-100 and labeled with FoxP3-APC (1:100) (eBioscience, cat. no. 17–5773-80) and DAPI (Invitrogen; cat. no. D1306, 0.01 mg/mL). Labeled cells were mounted on coverslips with Fluoroshield (Sigma-Aldrich, cat. no. F6182) and imaged by fluorescence microscopy (Zeiss 710 confocal microscope). Counts were performed manually by individuals blinded to the experimental groups.
2.3. In vitro suppression assay
Expanded Tregs were co-cultured with isolated splenocytes from Lewis rats (allograft recipient) stimulated with 5 μg/mL concanavalin A (conA) (Sigma, cat. no. L7647). Co-culture was carried out in medium consisting of RPMI 1640, 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin (Thermo Fisher Scientific, cat. no. 15140122) and 50 μM β-mercaptoethanol in 96-well plates. Tregs were mixed with 20,000 conA-stimulated splenocytes at the indicated ratios, and proliferation of stimulated splenocytes was measured by Alamar blue assay (Thermo Fisher Scientific, cat. no. DAL1100). Measurements of Tregs alone and conA-stimulated splenocytes alone were taken to calculate the individual influence of these cell types from the co-culture assay.
2.4. Synthesis of PEGNB hydrogels and treg biocompatibility and escape
PEGNB hydrogels were prepared following established protocols [36]. To prepare PEGNB hydrogel−forming solution, a 1:1 M ratio between thiol and -ene functional groups was maintained for effective crosslinking. PEGNB was mixed with di-thiol linker and photo initiator LAP (lithium phenyl-2,4,6-trimethylbenzoylphosphinate) (Sigma Aldrich, cat. no. 900889) to a final concentration of 10% (w/w) 20k 4-arm PEGNB (Sigma Aldrich, cat. no. 808466), 10 mM di-thiol linker (1.5k, Sigma-Aldrich cat. no. 704369) and 0.1% (w/w) LAP. 5 × 106 Tregs were mixed with 450 μl hydrogel-forming solution before polymerization. Hydrogels were formed via photopolymerization by exposure to UV light at 100 mW/cm2 for 20 s [36]. Viability of encapsulated cells was assessed using the LIVE/DEAD Viability/Cytotoxicity Kit (Thermo Fisher Scientific, cat. no. L3224).
To quantify Treg escape, 5 × 106 Tregs were encapsulated in 10% (w/w) bulk hydrogel and placed on a 40-μm cell strainer (Sigma, cat. no. CLS431751). The cell strainer with hydrogel was placed in a 6-well plate containing 8 mL of medium so that the hydrogel was submerged under the medium. The cell strainer containing the cell-laden hydrogel was then transferred to a new well each day. Cells that escaped from the hydrogel were stained with 0.2% trypan blue (Thermo Fisher Scientific; cat. no. 15250061), and live cells were counted with a hematocytometer.
To quantify the Tregs remaining in the PEGNB hydrogel, 5 × 105 Tregs were encapsulated in PEGNB as previously discussed. On days 1, 7 and 14, cell-laden hydrogels were lysed in 1% SDS supplemented with proteinase K in a tissue grinder on ice, then were centrifuged at 2600 g for 5 min. Supernatant was collected for measuring dsDNA content using Quant-iT Picogreen dsDNA Reagent (Thermofisher Scientific; cat. no. P11495) following the manufacture’s instruction.
2.5. Harvest and storage of nerve allografts
Animals were euthanized via cervical dislocation while under isoflurane anesthesia. PN grafts of the sciatic nerve were harvested from each flank and placed into PBS and stored on ice until implantation. Implantation occurred between 3 and 8 h after harvest.
2.6. Surgical procedure and post-operative care
Surgery was performed using previously published methods [10]. Briefly, Lewis rats were anesthetized with isoflurane and their hind limb was shaved. They were kept on a 38 °C heating pad and given intraperitoneal injections at the future incision site that consisted of bupivicaine (2 mg/kg) and baytril (5 mg/kg), and the surgical site was washed three times with iodine and isopropyl alcohol. Vaseline was placed on their eyes to prevent drying, sterile drapes were placed over the surgical site and the foot pinch test was performed to verify the level of anesthesia. Under sterile conditions, the skin was incised with a scalpel and the gluteus superficialis and biceps femoralis muscles were gently dissected to reveal the sciatic nerve. A 2-cm section of the sciatic nerve was removed with scissors, beginning 1 mm distal to the external obturator tendon and extending past the tibial-peroneal branch point. For autograft groups, this section of nerve was resutured with 9–0 sutures back into the created defect exactly as it was prior to removal. Allografts from SD donors were likewise sutured into place, connecting the peroneal to peroneal and tibial to tibial segments. Nerves were sutured at the epineurium layer under a surgical microscope using 9–0 sutures. 4.5 × 106 Tregs were prepared within 450 μl of PEGNB as described above was pipetted into the implant site and polymerized in place by UV illumination (100 mW/cm2) for 20 s. This quantity of hydrogel completely covered the entire length of the PN allografts and portions of the proximal and distal boundary with host PN. The muscle and skin were then sequentially sutured using 6–0 sutures [10].
For post-operative analgesia, rats received bupranorphine (0.03 mg/kg) twice per day for the first 3 d after surgery and acetaminophen (64 mg/kg/day) once per day for 7 d after surgery. Rats were fitted with Elizabethan collars (Kent Scientific, cat. no. EC404VS) for the first 3–7 d after surgery. No incidences of autophagy occurred.
2.7. Electromyography (EMG) to measure CMAPs
CMAP recording was performed on all recipient rats prior to surgery and at the designated time points afterward. CMAPs were recorded using a Viking NCS EMG EP IOM System as described [10,37]. The three highest amplitudes or shortest latencies were averaged for each animal at each time point.
2.8. Tissue collection at endpoint
Under isoflurane anesthesia, nerves were surgically exposed and fixed in situ with Trump’s fixative (4% paraformaldehyde, 1% glutaraldehyde, 1.16 g/100 mL NaH2PO4 in PBS) for 30 min. Nerves were then removed and placed in Trumps fixative, and the animals were sacrificed by cervical dislocation.
2.9. Sectioning and histology
Sciatic nerves were harvested, kept in Trump’s fixative solution overnight and then were incubated in 20% sucrose solution overnight at 4 °C for cryoprotection. Samples were embedded in Tissue-Tek® O.C.T. Compound, Sakura® Finetek (VWR, cat. no. 25608–930) and frozen at −20 °C. Nerves were cryosectioned longitudinally (8–10 μm thick), and the resulting sections were placed on Superfrost Plus glass slides (Thermo Fisher Scientific, cat. no. 9951LPLUS).
Immunofluorescence of frozen sections was performed for PE-conjugated anti−rat CD4 (1:50). Frozen sections were washed three times with PBS, followed by a 1 h incubation in blocking solution (5% BSA with 0.1% of Triton X-100) at room temperature and were then incubated with the antibody diluted in blocking solution for 1 h at room temperature. The sections were washed three times with PBS and, lastly, were incubated with DAPI (Thermo Fisher Scientific, cat. no. 62248) for 5 min. Sections were analyzed by photomicroscopy using an EVOS FL Color Imaging System (Life Technologies) microscope.
2.10. Imaging and densitometry
First, from each of the 12 animals used for the in vivo Allograft + GFP + Treg analysis, 50 images from the donor/boundary/host region were taken using the EVOS FL microscope at 4 × and 10 × magnification, to cover a region of 9 mm in length. These images were used for intensity density measurement and tracking of the GFP + Tregs. Second, from each of these same images at least 10 frames were taken with the same microscopy setup at 4 ×, 10 × and 20 × magnification of the proximal stump/junction region to analyze CD4 intensity density measurements. Photomicrographs were analyzed using confocal and EVOS FL microscopes and ImageJ software (NIH). Finally, the mean gray value of each antibody/marker (integrated density, %) was obtained with the measurement function of ImageJ software as described [38]. From each photomicrograph, the average of the mean gray values from multiple photomicrographs was used for analyses. This method was used to quantify the abundance of positive cells for each antibody/marker in the nerves.
2.11. Nerve morphometry
Nerve segments were embedded and stained with toluidine blue according to our previous protocol [39] or stained with Masson’s Trichrome. Toluidine blue sections quantified are 4 mm proximal from the peroneal-tibial branch point and sections for Masson’s Trichrome are 2 mm proximal from the branch point. Quantification of regeneration from images was performed by individuals blinded to the experimental groups.
3. Results
3.1. Isolation, expansion and characterization of tregs
Tregs were isolated from Sprague Dawley (SD) rat spleens by magnetic-activated cell sorting for CD4 and CD25 and were cultured with IL-2 and surface-adsorbed anti-CD28 superagonist antibody. Tregs were expanded in vitro for up to 12 d prior to use in experiments (Fig. S1A). Immunophenotyping the expanded cells showed the population to be > 98% CD4+, > 98% CD25 + and > 89% FoxP3+ (Fig. S1B, C). To determine the functional characteristics of the cells, Tregs were placed in co-culture with splenocytes isolated from Lewis rats and stimulated with concanavalin A (conA). As shown in Fig. S1D, Tregs inhibited expansion of conA-stimulated splenocytes, consistent with previous characterization of Treg functionality [32].
3.2. Encapsulation and release of tregs from PEGNB hydrogel
While Tregs delivered via intravenous infusion do circulate and localize to tissue grafts, they may not do so in sufficient numbers at the target site, have the potential to be active at non-target sites and billions of cells are required [34,40,41]. To increase the consistency of their target site accumulation, we developed a hydrogel for localized and controlled delivery of Tregs at the site and time of graft placement. PEG-based hydrogels possess excellent versatility and biocompatibility for cell immobilization and delivery applications [42]. We adapted a PEGNB hydrogel that forms thiol-ene crosslinks in the presence of a dithiol PEG. Biocompatibility was evaluated by assessing the viability of PEGNB encapsulated Tregs compared to standard 2-dimensional (2D) culture conditions. This assessment did not include IL-2 or anti-CD28 in the medium, as these factors are available only in vivo following effector immune cell infiltration of the area. Results showed improved Treg survival in PEGNB hydrogels compared to 2D cultures at 7 d and 14 d (Fig. 1A).
Fig. 1.

Biocompatibility and release of Tregs from the PEGNB hydrogel. (A) Viability of Tregs encapsulated in PEGNB hydrogel over time, using viability in 2D culture as a control. (B) Cumulative release profile of 5 × 106 Tregs from 500 μl of PEGNB hydrogel. (C) Percentage of Tregs in PEGNB hydrogel over time measured by DNA content within PEGNB. N ≥ 3, **p < 0.005 by ANOVA and Tukey’s post-hoc test. Error bars depict s.e.m.
To quantify release, 5 × 106 Tregs were encapsulated in 500 μl of PEGNB and placed in a cell strainer with 40-μm pores in a 6-well tissue culture dish. The cell strainer was shifted to a fresh well each day, and the number of viable Tregs left in the well bottom was quantified. As shown in Figs. 1B and 2% (∼100,000) Tregs escaped the first day, 6–10% (∼300,000–500,000) escaped per day on days 2–9 and 2–6% escaped per day on days 10–14. By day 14, the hydrolytically degrading gel had lost most of its structural integrity. Quantification of the DNA in the hydrogel at 7 and 14 d showed that 57 and 0.17% of Tregs remained in the hydrogel respectively at these days (Fig. 1C). Overall, 84% of the encapsulated Tregs escaped and were viable over the 14 d period. This release profile is consistent with the goal of ensuring that sufficient Tregs are on-site throughout the two-week time frame over which immune cells begin to infiltrate allogeneic PN tissue grafts [43].
3.3. In vivo experiments with a 2-cm branched nerve defect
An enabling feature of PN allografts is the potential to serve as bridging devices for complex nerve structures. Our in vivo experiments to test the effectiveness of locally delivered Tregs used a 2-cm defect that encompassed the peroneal-tibial branch point of the sciatic nerve (Fig. 2A). Allografts were harvested from donor SD rats and sutured into recipient Lewis rats, where a corresponding 2-cm segment of the sciatic nerve had been removed. The PEGNB hydrogel with Tregs was polymerized in the graft site after the graft had been sutured into place (Fig. 2B and C). No additional immunosuppressive interventions were administered. Control groups included the allograft with and without the PEGNB vehicle and an autograft control.
Fig. 2.

Surgical placement of branched PN allograft and Tregs within PEGNB hydrogel. (A) A 2-cm PN allograft that includes the peroneal-tibial branch point of the sciatic nerve harvested from a donor SD rat. (B) Image of the implant site within a recipient Lewis rat after the PN allograft was sutured into place. (C) Graft site after the PEGNB hydrogel containing Tregs was polymerized into place.
Compound muscle action potentials (CMAPs) are a standard clinical method for measuring PN function and were used to quantify functional regeneration [44]. CMAPs were measured for the distal enervation muscle targets in the foot of the peroneal and tibial branches of the sciatic nerve. CMAP amplitudes, which measure the quantity of electrically conductive fibers, first began to be detectible for the peroneal branch 8 wk after surgery (Fig. 3A). By 12 wk, the Treg-treated group showed peroneal CMAP amplitudes statistically equivalent to the autograft positive control and greater than other groups. This trend was maintained for all additional time points.
Fig. 3.

CMAP amplitudes of regenerated sciatic nerve. (A) Amplitudes of the peroneal branch of the sciatic nerve. (B) Amplitudes of the tibial branch of the sciatic nerve. N = 6 rats per group, *p < 0.05, **p < 0.005 by two-way ANOVA with Tukey’s post hoc test. Error bars depict s.e.m.
CMAP amplitudes of the tibial branch of the sciatic nerve in Treg-treated animals were also statistically equivalent to the autograft positive control and greater than the allograft negative control at the study endpoint (Fig. 3B). Interestingly, tibial amplitudes for allograft negative control and vehicle-treated animals were relatively higher in relation to the autograft as compared with peroneal amplitudes. CMAP latencies, which measure the time for the fastest nerve fibers to conduct between the stimulation and recording sites, showed longer conduction times only for the negative control group of allograft without vehicle compared to other treatment groups (Fig. S2). Together, the CMAP data indicates that of all the treatment groups, only the Treg-treated animals showed equivalency to autograft in both the number of conductive fibers and speed of conduction.
3.4. Morphometry of regenerated nerve within the grafts
To assess the morphometry of the regenerated nerves, toluidine blue staining of 1-μm-thick cross-sections was performed on the center section of the graft prior to the branch point (Fig. 4A–D). The morphometry showed that nerves from animals with allograft ± vehicle had axons spread over a large area within the nerve cable similar to autograft, but there was a higher abundance of non-nerve tissue within allograft ± vehicle compared to autograft. All Treg-treated allografts displayed a distinct morphometry where all axons were highly condensed within an area of the graft (Fig. 4C). This corresponded to a much greater axonal density for Treg-treated groups (Fig. 4E). While there was a trend for greater percent myelination for the Treg-treated group, this was not statically significant (p = 0.063) (Fig. 4F). Masson’s trichrome stain is used to indicate fibrous tissue and the degree of stain on Treg-treated PNs suggests that the non-nerve tissue within the epineurium is inconsistent with fibrous tissue (Fig. 4G, Fig S3). Quantification showed that the percentage of non-nerve tissue was higher in Treg-treated animals (Fig. 4H).
Fig. 4.
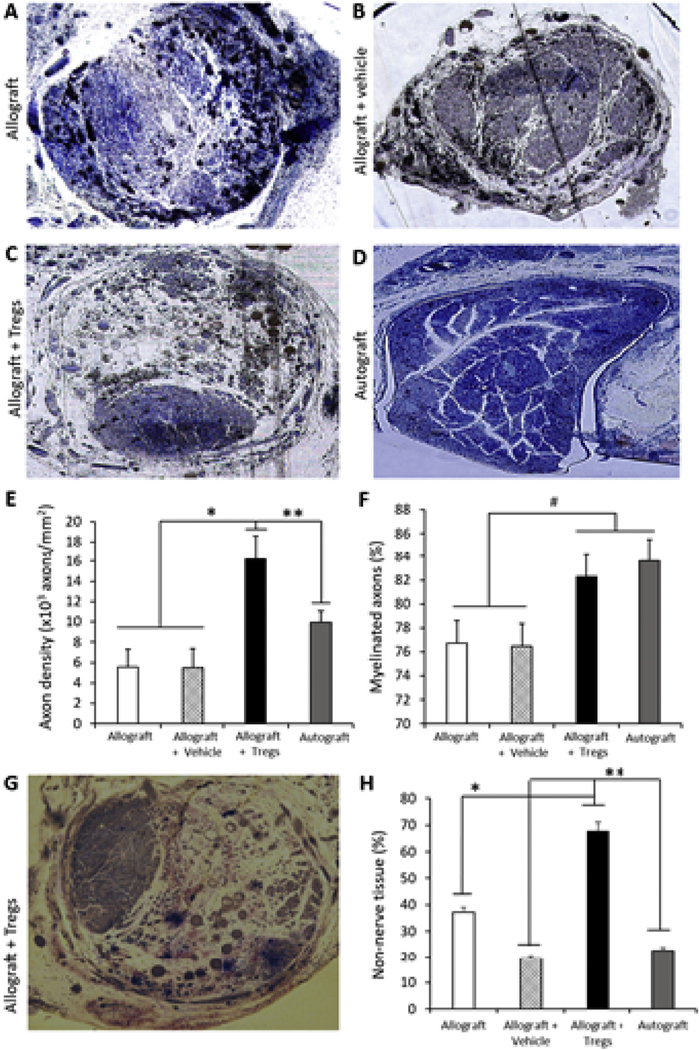
Morphometry of nerves within grafts at 20 weeks after surgery. (A−D) Representative images (10 × ) of nerve cross-sections from each of the four groups stained with toluidine blue. (E) Percentage of myelinated axons within grafts from the different treatment groups. (F) Axonal density of nerve cables within grafts. (G) Representative image (10 × ) of nerve cross-sections from Allograft + Tregs group stained with Masson’s trichrome. (H) Percentage of non-nerve tissue within the epineurium. N = 6 rats per group, #p = 0.063, *p < 0.05, **p < 0.005 by two-way ANOVA with Tukey’s post hoc test. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.5. Treg infiltration into the PN allografts
To evaluate the extent that Tregs migrated into the allografts, Lewis recipients with branched PN allografts from SD donors were administered SD-EGFP Tregs within the PEGNB hydrogel. Tissue was harvested at 3, 7, 14 and 21 d after surgery and assessed for GFP fluorescence at the proximal donor-host boundary. At 3 d, a large degree of GFP fluorescence was observed at the donor-host boundary, within both the graft and host nerve (Fig. 5A). By 7 d, the GFP signal had decreased relative to 3 d at the donor-host boundary but appeared to be more diffusely distributed along the length of the graft (Fig. 5B). At 14 d and 21 d, GFP fluorescence had increased within both the donor and host nerve structures (Fig. 5C and D).
Fig. 5.

Localization and abundance of Tregs within PN allografts. GFP + Tregs from SD-EGFP animals were implanted within PEGNB gels around SD PN allografts within recipient Lewis rats. (A−D) Images of GFP fluorescence in PN allografts harvested at day 3, 7, 14 and 21 after implantation. Boxes indicate the donor-host boundary identified through sutures. Dashed lines are the outline of the PN. Scale bar = 1 mm. (E) Quantification of the localization of GFP + Tregs at the donor-host boundary and out to 4 mm in either direction. (F) Quantification of the abundance of GFP fluorescence in a 9-mm segment of the PN, including 4 mm of the graft, 1 mm of the donor-host boundary and 4 mm of the host nerve. N = 3, #p = 0.039, **p = 0.0022 by Tukey’s multivariable test. Error bars depict s.e.m.
Quantification of densitometry shows the changing distribution of GFP intensity relative to the donor-host boundary over time (Fig. 5E), as the GFP-expressing Tregs spread from the donor-host boundary into both the host and donor PN. This showed a significant increase in overall intensity of GFP in the 9 mm segment of the nerves analyzed at 21 d relative to 7 d (Fig. 5F). As the PEGNB hydrogel had completely degraded by 14 d in vivo, this suggests that the increase in GFP intensity at later time points was due to proliferation of the surviving GFP + Tregs within the nerve rather than increased infiltration of Tregs released from the PEGNB hydrogel.
To evaluate Treg migration to other tissues, we collected the spleens from the Lewis rats that received GFP + Tregs with PN allografts. Isolated pockets of GFP-expressing cells were observed at 3 d and 7 d in the red pulp of the spleen (Fig. S4A, B). GFP-expressing cells were not observed in the white pulp. The abundance of GFP-expressing cells at 14 d and 21 d was progressively less than at previous days (Fig. S4B–D). This indicates that some of the Tregs locally implanted around the PN graft entered the circulation and entered the spleen but did not endure and proliferate in the spleen as was the case for Tregs within the PN allograft (Fig. 5).
3.6. Effects of tregs on host-derived CD4+ T cells
Tregs suppress the proliferation and stimulate apoptosis of effector T cells [33]. To assess the functional impact of locally delivered Tregs, the abundance of host-derived CD4+ cells within the proximal 5 mm of the graft was analyzed. As Tregs are also CD4+, the co-label of GFP was used to distinguish host-derived CD4+ cells from Tregs in the Treg-treated animals. Densitometry showed a sustained reduction in the quantity of CD4+ GFP− cells in Treg-treated grafts at all timepoints (Fig. 6), indicating a significant functional impact of the Tregs in the PN allografts.
Fig. 6.

Abundance of host-derived CD4+ cells in PN allografts after implantation. (A, B) Images of PN allografts harvested 14 d after implantation and immunolabeled for CD4. (C, D). Images of PN allografts treated with GFP + Tregs harvested 14 d after implantation and immunolabeled for CD4. Yellow indicates implanted GFP + CD4+ Tregs. Scale bars in A and C = 400 μm, B and D = 100 μm. (E) Quantification of the CD4 densitometry between allografts with and without Tregs, in which GFP was used to distinguish CD4+ Tregs from host-derived CD4+ T cells. N = 3, **p < 0.005, ***p < 0.0005 by Tukey’s multivariable test. Error bars depict s.e.m. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
4. Discussion and conclusion
A bridging device is required when PN segmental defects are more than several millimeters in length. Scaffolds as bridging devices for segmental PN defects were developed to replace autologous sensory nerves. Although conduits and decellularized allografts can be effective for stimulating regeneration of shorter PN segmental defects, these scaffolds are not as effective as autografts for short defects and cannot be used for long defects [45], for which an autograft is still the only clinical option. The inclusion of cells, neurotrophic factors and small molecules, electrical stimulation, material advances and new fabrication methods are all being investigated to improve scaffold performance [46]. Whereas many of these innovative advances show promise, none have yet shown the potential to match sensory nerve autografts. Mixed PN allografts are already as effective as autografts, can be used for long defects and have many advantages as bridging devices but are not routinely used because of the risks and expense of systemic ISN [26].
The primary advancement of our study is that a single localized application of Tregs delivered by PEGNB around the graft was sufficient to replace systemic ISN for PN allografts. Without any additional ISN, PN allografts treated with Tregs showed regeneration equivalent to mixed autograft at 20 wks, where CMAPs were the primary measure of functional regeneration. The survival and engraftment of Tregs via localized delivery has also been observed in a case of pancreatic cell transplantation, where the locally delivered Tregs improved the survival time of the transplanted islet cells [47]. In other studies, Tregs are delivered via intravenous bolus dose [33].
In our study, the positive control group was an autologous graft that was cut out and immediately sutured back in place in the exact pre-injury configuration. This was necessary due to the branched nature of the defect and is distinct from studies on linear defects where the autograft is reversed within the defect site. Clinical studies with sensory autografts are not exactly matched in terms of mixed vs sensory, diameter and fascicular organization, which leads to inferior regeneration compared to mixed grafts [4–6]. Thus, the positive control of the mixed nerve directly sutured into the defect in exactly the same configuration in this case represents the best possible regeneration.
PNs appear to have immunological features that may make localized ISN more feasible for PN allografts than for transplantation of other tissues. A substantial degree of regeneration occurs following PN allografts even in the absence of any immunosuppressive therapy [4,29,43,48–50] (Fig. 3). Immune rejection of allogeneic skin grafts, by comparison, occurs within 2 weeks and the entire graft loses integrity and fails [51]. Within PN allografts, donor-derived cells survive for longer but are eventually eliminated and replaced by host cells [43,50]. Potential reasons for this disparity are differences in APCs within skin versus PNs [38,52] and because PN allografts serve only as a temporary scaffold for regeneration of host axons, which are the functional component for regeneration.
Within PNs, Schwann cells are the most important support cell for stimulating axonal regeneration, yet they also appear to be the most immunogenic [53,54]. Despite this, allogeneic Schwann cells persist in PN grafts for long periods of time even without ISN but are ultimately replaced by host-derived Schwann cells [20,49,50]. The mechanisms by which Schwann cells can temporarily evade the immune response and still promote regeneration are yet to be defined.
The second advancement of our study is the test on a critical size defect that included a branch point. Allografts are uniquely positioned to be used for such defects, as scaffolds are not available for branched defects and harvesting branched sensory autografts would increase donor-site morbidity. As regenerating axons reach branch points, incorrect routing can result in complete pruning of the axon or aberrant sensorimotor connections [55]. PN grafts from motor or mixed PNs contain biological cues that better stimulate regeneration of motor axons that sensory grafts lack [56]. These cues also promote correct routing of motor axons at branch points [57,58]. Consequently, it is likely that if a segmental defect encompasses a branch point where sensory and motor axons diverge, a PN allograft harvested from the corresponding PN region from a donor would be the ideal bridging device.
Differences in control group peroneal and tibial CMAPs potentially indicate the circumstances when ISN is most important. Tibial CMAP amplitudes in negative controls were much more similar to those of autograft and Treg-treated allografts as compared with peroneal CMAP amplitudes (Fig. 3A and B). While the CMAPs were taken from the most distal muscles in the foot enervated by the nerves, the tibial anterior muscle is the most proximal motor enervation target of the tibial fibers and is closer to the defect than those of the peroneal fibers. Motor axons that initially reach their targets rapidly recruit other motor axons in a process called preferential motor reinnervation [59]. The difference in the degree of regeneration in the allograft negative control between the tibial and peroneal may suggest that preferential motor reinnervation occurred for tibial axons at the expense of peroneal axons in the absence of Tregs, suggesting Tregs or other methods of ISN are most critical for axons that have longer distances to regenerate after the graft. Additional tests would need to be performed to confirm this.
Tregs that were applied locally to the PN allograft infiltrated and remained in the PN allograft. While some Tregs did enter the spleen, their presence in the spleen was temporary and localized to the red but not white pulp. This is indicative of localized ISN but is not a direct test of the ability of animals to combat and generate immunological memory to a pathogen away from the area of Treg administration. It is also presently unclear how long the Tregs remain in the recipient. Tregs proliferate in response to signaling from effector T cells and APCs, which may persist in the graft for long periods. Tregs are also capable of transdifferentiation under some circumstances into effector T cells [60], which may become a concern. Finally, although we used a Treg population consistent with current clinical applications [33], there is growing diversity in the number of subpopulations, degree of allospecificity and source of Tregs that may be more or less advantageous for PN allografts.
Application of this new strategy in the clinic is facilitated by advancements in donor tissue procurement and screening [61], the excellent safety record of Tregs in clinical studies [62], and the prior regulatory approval of PEG-based hydrogels [63]. However, additional pre-clinical development and assessment are needed in large-animal models, in which direct comparison to sensory autografts can occur, autologous Tregs can be tested and the strategy can be assessed for larger nerve gaps.
Supplementary Material
Acknowledgments
The authors would like to thank Zhaojie Zhang at the University of Wyoming Jenkins Microscopy facility for assistance with imaging and densitometry, Richard Czaikowski for toluidine blue histology, Daniel Burns for quantification of nerve morphometry and Huong Pham for assistance in assessing Treg viability in hydrogels. Funding: Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number P20GM103432, the National Science Foundation Faculty Early Career Development (CAREER) Program (BBBE 1254608), the Universty of Wyoming Engineering Initiative and by the Universty of Wyoming Startup funds. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the National Science Foundation or the University of Wyoming.
Footnotes
Conflicts of interest
Authors have been granted and have filed several patents related to this study: patent 10,064,938 and patent applications 16/035,362, 16/035,355 and 16/049,343.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.biomaterials.2019.04.015.
Data availability
All the processed data required to reproduce these findings were shared at this time in the Material and Methods section and Supplementary data.
References
- [1].Wiberg M., Terenghi G., Will it be possible to produce peripheral nerves? Surg. Technol. Int 11 (2003) 303–310. [PubMed] [Google Scholar]
- [2].Frostick SP, The physiological and metabolic consequences of muscle denervation, Int. Angiol 14 (1995) 278–287. [PubMed] [Google Scholar]
- [3].Evans GR, Challenges to nerve regeneration, Semin. Surg. Oncol 19 (2000) 312–318. [DOI] [PubMed] [Google Scholar]
- [4].Nichols CM, et al. , Effects of motor versus sensory nerve grafts on peripheral nerve regeneration, Exp. Neurol 190 (2004) 347–355 10.1016/j.expneurol.2004.08.003. [DOI] [PubMed] [Google Scholar]
- [5].Brenner MJ, et al. , Repair of motor nerve gaps with sensory nerve inhibits regeneration in rats, Laryngoscope 116 (2006) 1685–1692 10.1097/01.mlg.0000229469.31749.91. [DOI] [PubMed] [Google Scholar]
- [6].Moradzadeh A., et al. , The impact of motor and sensory nerve architecture on nerve regeneration, Exp. Neurol 212 (2008) 370–376 10.1016/j.expneurol.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gaudin R., et al. , Approaches to peripheral nerve repair: generations of biomaterial conduits yielding to replacing autologous nerve grafts in craniomaxillofacial surgery, BioMed, Res. Intell. 2016 (2016) 3856262 10.1155/2016/3856262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Arslantunali D., Dursun T., Yucel D., Hasirci N., Hasirci V., Peripheral nerve conduits: technology update, Med. Devices 7 (2014) 405–424 10.2147/MDER.S59124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].de Boer R., et al. , Rat sciatic nerve repair with a poly-lactic-co-glycolic acid scaffold and nerve growth factor releasing microspheres, Microsurgery 31 (2011) 293–302 10.1002/micr.2086. [DOI] [PubMed] [Google Scholar]
- [10].Clements BA, et al. , Design of barrier coatings on kink-resistant peripheral nerve conduits, J. Tissue Eng 7 (2016) 2041731416629471 10.1177/2041731416629471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dalamagkas K., Tsintou M., Seifalian A., Advances in peripheral nervous system regenerative therapeutic strategies: a biomaterials approach, Mater. Sci. Eng. C 65 (2016) 425–432 10.1016/j.msec.2016.04.048. [DOI] [PubMed] [Google Scholar]
- [12].Isaacs J., Browne T., Overcoming short gaps in peripheral nerve repair: conduits and human acellular nerve allograft, Hand 9 (2014) 131–137 10.1007/s11552-014-9601-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lin MY, Manzano G., Gupta R., Nerve allografts and conduits in peripheral nerve repair, Hand, Clinic 29 (2013) 331–348 10.1007/s11552-014-9601-6. [DOI] [PubMed] [Google Scholar]
- [14].de Ruiter GC, Malessy MJ, Yaszemski MJ, Windebank AJ, Spinner RJ, Designing ideal conduits for peripheral nerve repair, Neurosurg, Focus 26 (2009) E5 10.3171/FOC.2009.26.2.E5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Paulsen F., Waschke J., Sobotta Atlas of Human Anatomy, fourteenth ed., Urban & Fischer; 2, 2013. pg 345, 349, 350. [Google Scholar]
- [16].Lee SK, Wolfe SW SW, Peripheral nerve injury and repair, J. Am. Acad. Orthop. Surg 8 (2000) 243–252. [DOI] [PubMed] [Google Scholar]
- [17].Johnson BN, et al. , 3D printed anatomical nerve regeneration pathways, Adv. Funct. Mater 39 (2015) 6205–6217 10.1002/adfm.201501760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jaeger SH, Singer DI, Whitenack SH, Mandel S., Nerve injury complications. Management of neurogenic pain syndromes, Hand Clin. 2 (1986) 217–234. [PubMed] [Google Scholar]
- [19].Ray WZ, Mackinnon SE, Management of nerve gaps: autografts, allografts, nerve transfers, and end-to-side neurorrhaphy, Exp. Neurol 223 (2010) 77–85 10.1016/j.expneurol.2009.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Evans PJ, et al. , Cold preserved nerve allografts: changes in basement membrane, viability, immunogenicity, and regeneration, Muscle Nerve 21 (1998) 1507–1522. [DOI] [PubMed] [Google Scholar]
- [21].Evans PJ, et al. , Regeneration across preserved peripheral nerve grafts, Muscle Nerve 18 (1995) 1128–1138. [DOI] [PubMed] [Google Scholar]
- [22].Sen A., Callisen H., Libricz S., Patel B., Complications of solid organ transplantation: cardiovascular, neurologic, renal, and gastrointestinal, Crit. Care Clin 35 (2019) 169–186 10.1016/j.ccc.2018.08.011. [DOI] [PubMed] [Google Scholar]
- [23].Adams DH, Sanchez-Fueyo A., Samuel D., From immunosuppression to tolerance, J. Hepatol. (Amst.) 62 (2015) S170–S185 10.1016/j.jhep.2015.02.042. [DOI] [PubMed] [Google Scholar]
- [24].Atchabahian A., et al. , Indefinite survival of peripheral nerve allografts after temporary Cyclosporine A immunosuppression, Restor. Neurol. Neurosci 13 (1998) 129–139. [PubMed] [Google Scholar]
- [25].Mackinnon SE, Doolabh VB, Novak CB, Trulock EP, Clinical outcome following nerve allograft transplantation, Plast. Reconstr. Surg 107 (2001) 1419–1429. [DOI] [PubMed] [Google Scholar]
- [26].Siemionow M., Sonmez E., Nerve allograft transplantation: a review, J. Reconstr. Microsurg 23 (2007) 511–520 10.1055/s-2007-1022694. [DOI] [PubMed] [Google Scholar]
- [27].Midha R., et al. , Comparison of regeneration across nerve allografts with temporary or continuous cyclosporin A immunosuppression, J. Neurosurg 78 (1993) 90–100. [DOI] [PubMed] [Google Scholar]
- [28].Auba C., Hontanilla B., Arcocha J., Gorria O., Peripheral nerve regeneration through allografts compared with autografts in FK506-treated monkeys, J. Neurosurg 105 (2006) 602–609 10.3171/jns.2006.105.4.602. [DOI] [PubMed] [Google Scholar]
- [29].Udina E., Gold BG, Navarro X., Comparison of continuous and discontinuous FK506 administration on autograft or allograft repair of sciatic nerve resection, Muscle Nerve 29 (2004) 812–822 10.1002/mus.20029. [DOI] [PubMed] [Google Scholar]
- [30].Mackinnon SE, Hudson AR, Clinical application of peripheral nerve transplantation, Plast. Reconstr. Surg 90 (1992) 695–699. [DOI] [PubMed] [Google Scholar]
- [31].Mackinnon SE, Nerve allotransplantation following severe tibial nerve injury. Case report, J. Neurosurg 84 (1996) 671–676. [DOI] [PubMed] [Google Scholar]
- [32].Sakaguchi S., Sakaguchi N., Asano M., Itoh M., Toda M., Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases, J. Immunol 155 (1995) 1151–1164. [PubMed] [Google Scholar]
- [33].Safinia N., Scotta C., Vaikunthanathan T., Lechler RI, Lombardi G., Regulatory T cells: serious contenders in the promise for immunological tolerance in transplantation, Front. Immunol 6 (2015) 438 10.3389/fimmu.2015.00438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Singer BD, King LS, D’Alessio FR, Regulatory T cells as immunotherapy, Front. Immunol 5 (2014) 46 10.3389/fimmu.2014.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Beyersdorf N., Balbach K., Hünig T., Kerkau T., Large-scale expansion of rat CD4+ CD25+ T reg cells in the absence of T-cell receptor stimulation, Immunology 119 (2006) 441–450 10.1111/j.1365-2567.2006.02455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jiang Z., Xia B., McBride R., Oakey J., A microfluidic-based cell encapsulation platform to achieve high long-term cell viability in photopolymerized PEGNB hydrogel microspheres, J. Mater. Chem. B 5 (2017) 173–180 10.1039/C6TB02551J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Varejao AS, Melo-Pinto P., Meek MF, Filipe VM, Bulas-Cruz J., Methods for the experimental functional assessment of rat sciatic nerve regeneration, Neurol. Res 26 (2004) 186–194 10.1179/016164104225013833. [DOI] [PubMed] [Google Scholar]
- [38].Schneider CA, Rasband WS, Eliceiri KW, NIH Image to ImageJ: 25 years of image analysis, Nat. Methods 9 (2012) 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ghnenis AB, Czaikowski RE, Zhang ZJ, Bushman JS, Toluidine blue staining of resin-embedded sections for evaluation of peripheral nerve morphology, JoVE 137 (2018), 10.3791/58031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bluestone JA, et al. , Type 1 diabetes immunotherapy using polyclonal regulatory T cells, Sci. Transl. Med 7 (2015) 315ra189 10.1126/scitranslmed.aad4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Arellano B., Graber DJ, Sentman CL, Regulatory T cell-based therapies for autoimmunity, Discov. Med 22 (2016) 73–80. [PMC free article] [PubMed] [Google Scholar]
- [42].Fischer P., Tibbitt M., Kloxin A., Anseth K., Oakey J., Photodegradable hydrogels for selective capture and release of Mammalian cells, Biomed. Sci. Instrum 50 (2014) 62–67. [PubMed] [Google Scholar]
- [43].Roballo KCS, Bushman J., Evaluation of the host immune response and functional recovery in peripheral nerve autografts and allografts, Transpl. Immunol 53 (2019) 61–71 10.1016/j.trim.2019.01.003. [DOI] [PubMed] [Google Scholar]
- [44].Mallik A., Weir AI, Nerve conduction studies: essentials and pitfalls in practice, J. Neurol. Neurosurg. Psychiatry 76 (2005) ii23–ii31 10.1136/jnnp.2005.069138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Safa B., Buncke G., Autograft substitutes: conduits and processed nerve allografts, Hand Clin. 32 (2016) 127–140, 10.1016/j.hcl.2015.12.012. [DOI] [PubMed] [Google Scholar]
- [46].Jones S., Eisenberg HM, Jia X., Advances and future applications of augmented peripheral nerve regeneration, Int. J. Mol. Sci 17 (2016) E1494 10.3390/ijms17091494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Takemoto N., Konagaya S., Kuwabara R., Iwata H., Coaggregates of regulatory T cells and islet cells allow long-term graft survival in liver without immunosuppression, Transplantation 99 (2015) 942–947 10.1097/TP.0000000000000579. [DOI] [PubMed] [Google Scholar]
- [48].Carlson TL, Wallace RD, Konofaos P., Cadaveric nerve allograft: single center’s experience in a variety of peripheral nerve injuries, Ann. Plast. Surg 80 (2018) S328–S332 10.1097/SAP.0000000000001470. [DOI] [PubMed] [Google Scholar]
- [49].Katsube K., et al. , Successful nerve regeneration and persistence of donor cells after a limited course of immunosuppression in rat peripheral nerve allografts, Transplantation 66 (1998) 772–777. [DOI] [PubMed] [Google Scholar]
- [50].Midha R., Mackinnon SE, Becker LE, The fate of Schwann cells in peripheral nerve allografts, J. Neuropathol. Exp. Neurol 53 (1994) 316–322. [DOI] [PubMed] [Google Scholar]
- [51].Benichou G., et al. , Immune recognition and rejection of allogeneic skin grafts, Immunotherapy 3 (2011) 757–770 10.2217/imt.11.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Smyth LA, et al. , Tolerogenic donor-derived dendritic cells risk sensitization in vivo owing to processing and presentation by recipient APCs, J. Immunol 190 (2013) 4848–4860 10.4049/jimmunol.1200870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Yang Y., et al. , Downregulating immunogenicity of Schwann cells via inhibiting a potential target of class II transactivator (CIITA) gene, Biosci.trends 7 (2013) 50–55. [PubMed] [Google Scholar]
- [54].Samuel NM, Mirsky R., Grange JM, Jessen KR, Expression of major histo-compatibility complex class I and class II antigens in human Schwann cell cultures and effects of infection with Mycobacterium leprae, Clin. Exp. Immunol 68 (1987) 500–509. [PMC free article] [PubMed] [Google Scholar]
- [55].Madison RD, Robinson GA, Chadaram SR, The specificity of motor neurone regeneration (preferential reinnervation), Acta Physiol. 189 (2007) 201–206 10.1111/j.1748-1716.2006.01657.x. [DOI] [PubMed] [Google Scholar]
- [56].Martini R., Schachner M., Brushart TM, The L2/HNK-1 carbohydrate is preferentially expressed by previously motor axon-associated Schwann cells in reinnervated peripheral nerves, J. Neurosci 14 (1994) 7180–7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Irintchev A., et al. , Glycomimetic improves recovery after femoral injury in a nonhuman primate, J. Neurotrauma 28 (2011) 1295–1306 10.1089/neu.2011.1775. [DOI] [PubMed] [Google Scholar]
- [58].Martini R., Xin Y., Schmitz B., Schachner M., The L2/HNK-1 carbohydrate epitope is involved in the preferential outgrowth of motor neurons on ventral roots and motor nerves, Eur. J. Neurosci 4 (1992) 628–639. [DOI] [PubMed] [Google Scholar]
- [59].Brushart TM, Motor axons preferentially reinnervate motor pathways, J. Neurosci 13 (1993) 2730–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Josefowicz SZ, Lu LF, Rudensky AY, Regulatory T cells: mechanisms of differentiation and function, Annu. Rev. Immunol 30 (2012) 531–564 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Datta N., Yersiz H., Kaldas F., Azari K., Procurement strategies for combined multiorgan and composite tissues for transplantation, Curr. Opin. Organ Transplant 20 (2015) 121–126 10.1097/MOT.0000000000000172. [DOI] [PubMed] [Google Scholar]
- [62].Gliwinski M., Iwaszkiewicz-Grzes D., Trzonkowski P., Cell-based therapies with T regulatory cells, BioDrugs : clinical immunotherapeutics, BioDrugs. 31 (2017) 335–347 10.1007/s40259-017-0228-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Liechty WB, Kryscio DR, Slaughter BV, Peppas NA, Polymers for drug delivery systems, Annu. Rev. Chem. Biomol. Eng 1 (2010) 149–173 10.1146/annurev-chembioeng-073009-100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.