

Abstract
4-Fluoro-threonine, the only fluoro amino acid of natural origin discovered so far, is an interesting target for both synthetic and theoretical investigations. In this work, we lay the foundation for spectroscopic characterization of 4-fluoro-threonine. First, we report a diastereoselective synthetic route, which is suitable to produce synthetic material for experimental characterization. The addition of the commercially available ethyl isocyanoacetate to benzyloxyacetaldehyde led to the corresponding benzyloxy-oxazoline, which was hydrolyzed and transformed into ethyl (4S*,5S*)-5-hydroxymethyl-2-oxo-4-oxazolidinecarboxylate in a few steps. Fluorination with diethylamino sulfur trifluoride (DAST) afforded ethyl (4S*,5S*)-5-fluoromethyl-2-oxo-4-oxazolidinecarboxylate, which was deprotected to give the desired diastereomerically pure 4-fluoro-threonine, in 8–10% overall yield. With the synthetic material in our hands, acid–base titrations have been carried out to determine acid dissociation constants and the isoelectric point, which is the testing ground for the theoretical analysis. We have used machine learning coupled with quantum chemistry at the state-of-the-art to analyze the conformational space of 4-fluoro-threonine, with the aim of gaining insights from the comparison of computational and experimental results. Indeed, we have demonstrated that our approach, which couples a last-generation double-hybrid density functional including empirical dispersion contributions with a model combining explicit first-shell molecules and a polarizable continuum for describing solvent effects, provides results and trends in remarkable agreement with experiments. Finally, the conformational analysis applied to fluoro amino acids represents an interesting study for the effect of fluorine on the stability and population of conformers.
Introduction
The key role of fluorine in medicinal chemistry1,2 is witnessed by its presence in a large number of drugs and its increasing importance in biological studies by the extensive use of 18F-labeled molecules in positron emission tomography.2,3 Indeed, the incorporation of fluorine in key positions of a molecular system can affect its conformational stability (see, e.g., the “gauche effect”),1a,1c,2,4 membrane permeability, metabolic stability, and binding affinity,1a,1c,2 thus representing a very useful and versatile tool for drug design.2b,5 These distinctive features are related to the intrinsic properties of the C–F bond (a highly polarized covalent bond with a large dipole moment and bond strength greater than that of C–H), which is able to establish interactions of charge/dipole nature with the environment, also showing hyperconjugative effects related to the presence of a low-energy C–F sigma antibonding orbital.2,4b,4c
The importance of fluorine and, especially, organofluorine compounds, has stimulated the development of a large number of interesting methodologies for fluorine incorporation,6−8 with fluorinated amino acids being particularly significant targets since their introduction in specific domains of proteins can improve their stability and folding9 as well as their biological activity.10 Indeed, the incorporation of fluoro amino acids, such as fluorinated proline,11 phenylalanine,12 and tyrosine13 in specific proteins, has been analyzed in detail. Fluorinated amino acids can also be introduced in peptides by chemical synthesis,14 ribosomal translation,15 or chemical ligation.16 It is from this perspective that the increasing importance of fluorination for pharmaceutical applications has been put forward.17 In this framework, a particular role is played by 4-fluoro-threonine (hereafter 4F-Thr), the only naturally occurring fluorinated amino acid. Quite recently, 4F-Thr has been proposed as an effective bioisostere for diagnostic and therapeutic use toward adenocarcinomas, one of the most malignant and aggressive cancers.18
While 4F-Thr was found in Streptomyces cattleya,19 its relevance to metabolic pathways is still unknown, although the antimicrobial activity of 4F-Thr has been demonstrated.20 Instead, the metabolic pathways leading to 4F-Thr have been fully elucidated,21 with the key enzymatic role in fluorine incorporation being played by fluorinase,22 which activates the fluoride ion for the conversion of S-adenosyl-l-methionine (SAM) into 5′-deoxy-5′-fluoroadenosine.23 The F-bearing intermediate fluoroacetaldehyde is converted into 4F-Thr through an enzymatic transaldolization,24 which predominantly proceeds via two oxidative mechanisms that either use H2O2 or O2 as an oxidant.25 Unlike the enzymatic activation of chloride, bromide, and iodide through the formation of electrophilic halogen intermediates, such as X+ equivalents or halogen radicals,26 the use of co-oxidants is not possible for generating the F+ ion, and an alternative strategy based on nucleophilic fluorination is thus required.27 Since fluorinase is involved in the challenging nucleophilic fluorination, it has to overcome the high solvation energy to strip solvating water, thus generating a “naked” fluoride species and making it accessible for nucleophilic substitution, which might proceed via hydrogen bonding and desolvation of the fluoride anion.28 This important feature was clarified by crystallization studies of fluorinase from S. cattleya with SAM and fluoride.29
4F-Thr has been examined as a fluorinated analogue of threonine (Thr), and it probably exerts its action when it replaces Thr in primary metabolic pathways, thus resulting in the production of other toxic fluorinated species.30 The structures of fluoro amino acids and their behavior require special computational methodologies to reach a deeper understanding of their structural and physicochemical properties. As a matter of fact, the behavior of flexible molecules in aqueous media needs to be described in terms of an ensemble of low-lying conformers experiencing local fluctuations. Therefore, an incomplete ensemble of conformers can easily lead to unsatisfactory description and modeling of physical–chemical properties. To overcome these limitations, a step further is required: quantum chemistry needs to be coupled with machine learning to obtain an effective exploration of the “flat” conformational space, where the adjective flat points out that conformational changes are accompanied by small energy variations. In this context, 4F-Thr, together with its intrinsic interest, represents an ideal playground for testing and validating the proposed strategy. However, to make an effective comparison between theoretical investigations and observed physical–chemical properties it is necessary to synthesize a sufficient amount of 4F-Thr. To the best of our knowledge, only four synthetic routes have been reported until now for 4F-Thr.31−34 Although the enantioenriched synthesis of 4F-Thr was reported in these papers, we decided to embark on the study of new approaches, designing different synthetic strategies. While an enantioenriched sample of 4F-Thr opens toward the possibility to compare calculated and experimental chiroptical properties,35 a direct diastereoselective approach to 4F-Thr can give us the possibility to address physical properties that do not need enantiopure samples (i.e., enantiomers feature the same behavior), but depend on the relative configuration of stereocenters (i.e., different diastereoisomers can be singled out by means of such properties). Indeed, this is the case for the acid dissociation constants of 4F-Thr. From a computational point of view, the pKa values can only be estimated by quantum-chemical computations that also take into account a large number of low-lying conformers, possibly present in the solution.
To summarize, in the present paper, we describe a new complete diastereoselective approach to racemic 4F-Thr and the exploitation of a computational strategy at the state-of-the-art for the comparison with physical–chemical data experimentally obtained on the racemic sample.
Results and Discussion
Diastereoselective Synthesis of Racemic 4-Fluoro-threonine
Detailed studies and advancements in possible applications of 4F-Thr have been suffering from the lack of available materials due to difficult or tedious syntheses, while drug design applications might be hampered by the lack of structural and physicochemical characterization. Based on these grounds, we have developed a new diastereoselective synthesis of 4F-Thr starting from commercially available precursors (Scheme 1), and this approach requires neither specific expertize nor extreme conditions. The strategy considered the transformation of hydroxymethyl-oxazolidinone with a fluorinating agent. The racemic but diastereomerically pure precursor of oxazolidinone was obtained by a copper-mediated cycloaddition to afford the corresponding oxazoline, starting from commercially available precursors. The salient points for the synthesis are detailed here, while some unsuccessful routes, strategies that gave byproducts, and detour on difficult transformations are fully reported and commented on in the Supporting Information.
Scheme 1. Retrosynthetic Diastereoselective Approach for Racemic 4-Fluoro-threonine.

In recent years, the formal [3 + 2] cycloaddition of α-isocyanoesters with carbonyl compounds has been investigated as a powerful methodology for the construction of chiral-substituted 2-oxazolines bearing two adjacent stereocenters.36 The synthetic approach to dl-threonine employing the addition of α-isocyanoacetamides to acetaldehyde as a key step was reported.37 According to the literature, upon careful selection of the base (KOH) and the solvent (MeOH), the reaction afforded the trans-5-methyl-4-N′-substituted-aminocarbonyl-2-oxazoline, which was easily hydrolyzed to dl-threonine in high yield. In addition to the use of organic or inorganic bases, Saegusa reported (in 1971) the catalytic use of copper salt in the addition of isocyanoesters to aldehydes.38 The cycloaddition in the presence of copper was further evaluated for diastereoselective reactions.39 Particularly interesting is that aliphatic aldehydes afforded trans-oxazolines using simple Cu(I) catalysts. Exploiting the conditions described by Kirchner,39b we employed the commercially available or simply prepared aldehyde 1 with ethyl isocyanoacetate (see Scheme 2).
Scheme 2. New Synthetic Approach to Racemic 4-Fluoro-threonine.

With respect to the reaction of isocyanocarboxylates with aldehydes promoted by Cu2O (described in the ′70s),38 the reaction is more diastereoselective if performed with CuCl in the presence of Ph3P as a ligand, thus obtaining only the trans diastereoisomer (d.r. > 99%, according to 1H NMR analysis of the crude mixture). Unfortunately, the oxazoline cannot be used as an effective protective group, and we observed the presence of the opened formamide amino alcohol product 3, in variable quantities from batch to batch. Upon flash chromatography, the mixture was hydrolyzed with methanolic HCl and then treated with triphosgene to afford the protected oxazolidinone 4 in good yield. Quantitative removal of the benzyl group by hydrogenation gave 5, which was then subjected to several conditions/reagents for fluorination (see the “Optimization of the Reaction Conditions” Section of the Supporting Information (SI) for the list of conditions/reagents employed). Unfortunately, all of the modern variants of fluorinating agents40 employed gave either extensive decompositions or complex mixtures of products, and in some cases, no reaction was observed at all. The best conditions developed so far for the transformation were based on the report by Scolastico on the use of diethylaminosulfur trifluoride (DAST).31 However, in our hands, the reaction was quite sensitive to scale up and gave the reported or slightly better yields (57%) only in a few cases. By a careful scale up (up to 350 mg of starting oxazolidinone), it was possible to reach 32% yield. To improve the yields and generality of the transformation, we have tested several fluorinating agents and conditions, but unfortunately, without success. The yields obtained are comparable to the results reported in the literature.31 The fluorinated product was then subjected to complete hydrolysis with aq. 6 M HCl to give the desired 4F-Thr as a single diastereoisomer.
To study the behavior of 4F-Thr, we needed the zwitterionic form of the amino acid and, to avoid the use of ion exchange resins, we treated the hydrochloride of 4F-Thr with an excess of propylene oxide.41 The desired compound was obtained in pure zwitterionic form after simple removal of solvent and excess of reagent.
Conformational Analysis
The conformational space of 4F-Thr was explored as detailed in the Computational Methods section, with all structures lying within 25 kJ mol–1 (with respect to the lowest energy minimum) being optimized at a high level of theory with bulk solvent effects considered. The relative electronic energy (ΔE) and free energy (ΔG) values of the low-lying conformers, as obtained by following the procedure described in the Computational Methods section, are reported in Figure 1 (only ΔE) and in Table S4 (both ΔE and ΔG) of the Supporting Information for the three possible pH-dependent forms in the solution: anionic (4F-Thr-A), zwitterionic (4F-Thr-Z), and cationic (4F-Thr-C) species.
Figure 1.
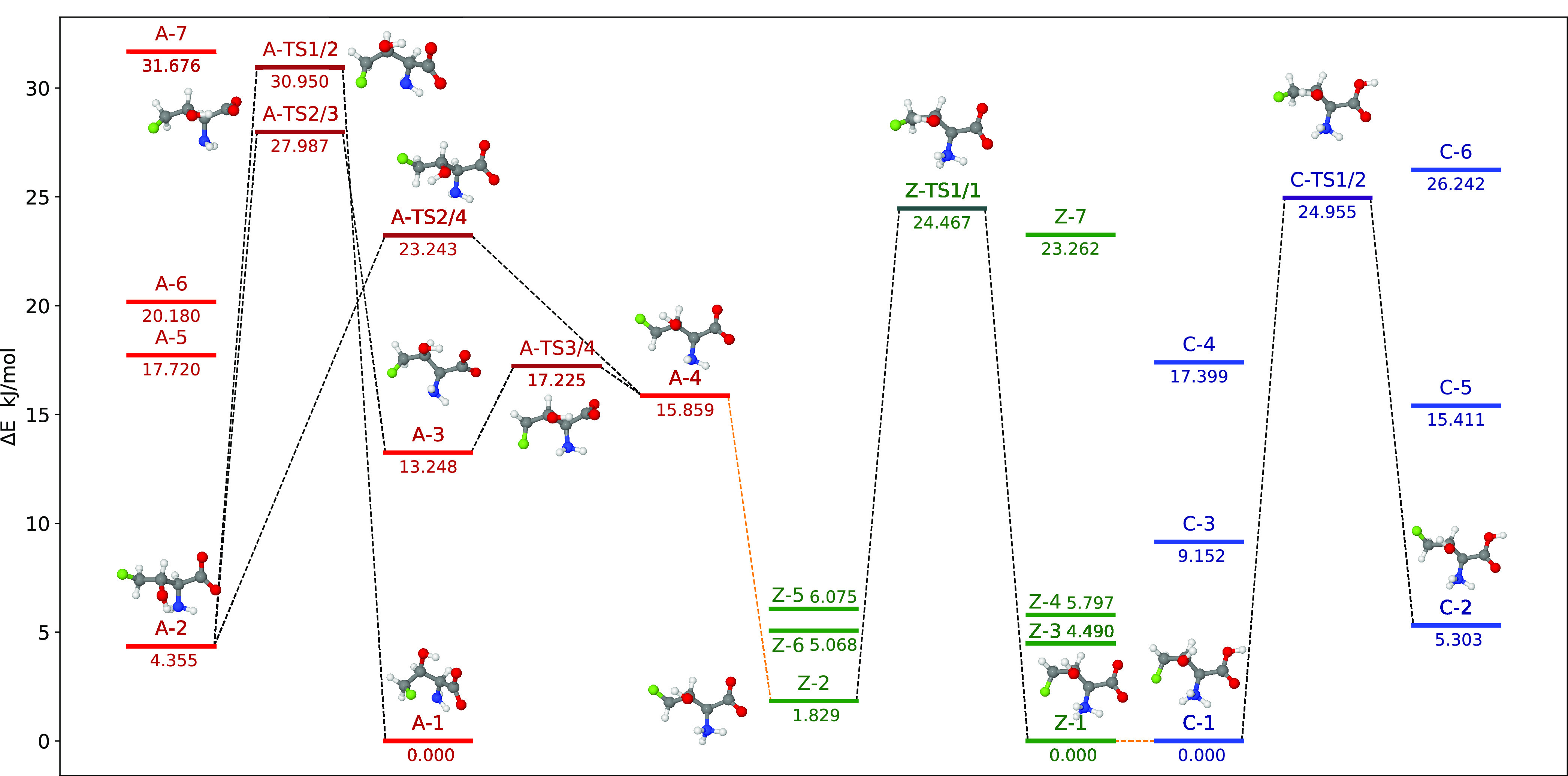
Conformers (within 32 kJ mol–1) and transition states ruling their interconversion for the anionic (red), zwitterionic (green), and cationic (blue) forms of 4F-Thr: the orange line connects structures upon acid dissociation. The relative energies are expressed with respect to the lowest energy minimum of each form. The structures of the most relevant conformers are also shown.
In analogy with other amino acids, the zwitterionic form of 4F-Thr is more stable than the corresponding neutral form in an aqueous solution at neutral pH values.42 The former can then undergo either protonation of the carboxylate moiety or deprotonation of the ammonium one, depending on the considered pH value. Figure 1 shows the relationships between the low-lying conformers of the three possible protonation states (orange lines) and sketches the transition states ruling the interconversion between the most relevant conformers. A more quantitative analysis of the similarity between different conformers (in terms of their root mean square displacements, rms) is provided in Tables S5–S9 of the Supporting Information.
Starting the analysis from the cationic species (blue energy levels in Figure 1), it can be observed that the lowest energy minimum (LEM) is stabilized by a strong interaction between the charged NH3+ moiety and both the electronegative atoms present in the molecule, namely, the carboxylic oxygen and the fluorine atom (N–H···O and N–H···F, 2.1152 and 2.2706 Å, respectively). In a second conformer, this latter interaction is replaced by the less stabilizing one between the fluorine atom and the hydroxyl hydrogen (N–H···O and O–H···F, 2.0602 and 2.4003 Å, respectively), with the two conformers interconverting through the rotation of the CH2F group, which is ruled by a transition state lying about 25 kJ mol–1 above the LEM. The zwitterionic form presents an analogous behavior (green levels), with the LEMs of the zwitterionic and cationic forms showing a good match. Therefore, we can conclude that deprotonation of the carboxylic group occurs without significant conformational rearrangements. The situation is more complicated for the deprotonation of the ammonium moiety leading to the anionic form (red levels), whose LEM does not show any close resemblance with its zwitterionic counterpart. Indeed, when the ammonium moiety becomes a noncharged amine group by deprotonation, the higher electronegativity of oxygen with respect to nitrogen leads to a stronger hydrogen bond of carboxylate oxygen with the alcoholic proton (C=O···H–O, 1.7007 Å) rather than with the amine one (C=O···H–N, 2.1174 Å). The best match was then found between the second conformer of the zwitterion (4F-Thr-Z-2) and the fourth conformer of the anion (4F-Thr-A-4). Looking at the possible interconversion paths, 4F-Thr-A-4 and 4F-Thr-A-3 are separated by an almost negligible energy barrier (about 1.5 kJ mol–1), whereas higher barriers connect 4F-Thr-A-4 and 4F-Thr-A-2 (about 7.5 kJ mol–1) as well as 4F-Thr-A-3 and 4F-Thr-A-2 (nearly 15 kJ mol–1). Though we were able to locate a direct path connecting 4F-Thr-A-2 to the LEM, the barrier was remarkably high (∼31 kJ mol–1).
Next, we compared the conformers of 4F-Thr with the low-lying conformers of the closely related Thr amino acid. As shown in Figure 2, there is a one-to-one correspondence between the low-lying conformers of 4F-Thr and Thr, except for the LEMs of the anionic forms, probably because of a particularly significant role of the electronegative fluorine atom in this situation.
Figure 2.

Correlations (black dashed line) between the low-energy conformers of 4F-Thr (4F-Thr-X-X labels) and Thr (Thr-X-X labels). The structures of the conformers used in the estimation of pKa1 are shown above the energy scheme, whereas those employed for pKa2 are shown below the energy scheme. The relative energies are expressed with respect to each lowest energy minimum.
Acid–Base Properties
From a computational point of view, once the most stable conformers and the possible paths of the proton dissociation in 4F-Thr are known, together with the related Thr structures (see Figure 2), it is possible to estimate their acid dissociation constants and address the differences. Under the same experimental conditions, the protonation free energies of 4F-Thr are given by
where ΔGTexp is the experimental protonation free energy of Thr and the first term on the right-hand side is evaluated as
and it is thus the difference of the computed free energies in the solution of the conformers of 4F-Thr and Thr, with D and P denoting the deprotonated and protonated forms, respectively. To evaluate ΔΔG4FT,Tcalc for the deprotonation of the carboxyl group (pKa1), the LEMs of the cationic and zwitterionic forms were used for Thr and 4F-Thr, while pKa2 was evaluated using the 4F-Thr-Z-2 conformer, which, as mentioned above, shows the best match with one of the low-lying conformers of the anionic form, the 4F-Thr-A-4 conformer. Since the latter almost freely interconverts to its more stable 4F-Thr-A-3 counterpart, this form was therefore employed to estimate the ΔG values. The same behavior is also observed for Thr, the Thr-A-4 conformer is separated by a negligible energy barrier from Thr-A-3.
In an attempt to take specific solvent effects into account, we have also performed some computations, including explicit water molecules. The first-solvation shells of 4F-Thr (Figure 3) and Thr (Figure S3) in different protonation states were built starting from clusters containing the solute and the water molecules close to aminic and carboxylic moieties. For the sake of consistency, the maximum number of water molecules found for any species was retained for all of the other ones, thus leading to a total of seven water molecules (roughly three close to the amine/ammonium and four close to the carboxyl/carboxylate moieties). Next, the geometries were fully optimized also taking into account bulk solvent effects by means of the polarizable continuum model. Additional tests showed that clusters containing seven explicit water molecules provide converged results and that the structures of the most stable conformers discussed above for the bare solutes embedded in a polarizable continuum are not significantly modified (additional details are given in the Computational Methods section).
Figure 3.
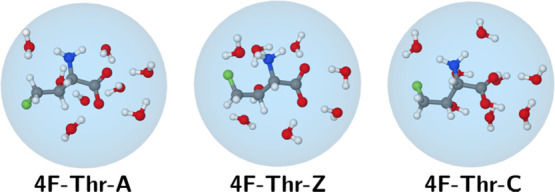
Clusters containing the water molecules of the first-solvation shell for the anionic (left), zwitterionic (middle), and cationic (right) forms of 4F-Thr.
The results reported in Table 1 show that, although explicit inclusion of first-shell water molecules slightly tunes the values of pKa1 and pKa2, the isoelectronic point (pI) and the general trends remain the same: as expected, both deprotonation processes of 4F-Thr are slightly favored with respect to Thr, in agreement with the presence of the electron-withdrawing fluorine atom.
Table 1. Experimental Acid Dissociation Constants and Isoelectric Point of Thr and 4F-Thr together with the Corresponding (Computed and Experimental) differences.
| Thr (lit.)a | Thr (exp.) | 4F-Thr (exp.) | 4F-Thr-Thr exp.b | 4F-Thr-Thr calc. (PCM) | 4F-Thr-Thr calc. (7W + PCM) | |
|---|---|---|---|---|---|---|
| pKa1 | 2.20 | 2.38 ± 0.02 | 2.26 ± 0.09 | –0.12 ± 0.11 | –0.16 | –0.36 |
| pI | 5.60 | 5.75 ± 0.09 | 5.43 ± 0.04 | –0.32 ± 0.13 | –0.40 | –0.41 |
| pKa2 | 8.96 | 9.1 ± 0.17 | 8.61 ± 0.02 | –0.49 ± 0.22 | –0.64 | –0.46 |
From ref (43).
Dissociation constants of both Thr and 4F-Thr were obtained in this work.
From an experimental point of view, the titration curves of 4F-Thr and Thr (for reference purposes) were recorded three times under the same conditions to allow the calculation of mean values and confidence intervals for the acid dissociation constants (pKa1, pKa2) and the isoelectric point (pI). The semiempirical method described in the following was used for the quantitative analysis of titration curves, with all experimental runs being normalized according to the initial pH of the solutions (dependent on the amount of concentrated HCl added, see the Experimental Section).
First, pKa2 was determined as the pH value corresponding to the second half-equivalence point, which coincides with a local maximum of the first derivative of the inverted titration curve. Next, we moved to the graphical determination of pKa1, starting from pKa2 and taking advantage of both the narrow volume ranges encompassing equivalence zones and the narrow pH ranges encompassing buffer zones. The value of pI was then evaluated from the pKa1 and pKa2 values using the Henderson–Hasselbalch equation, with the detailed experimental procedure being reported in the Experimental Section. To better address the role played by fluorination in the behavior of amino acids, we prefer to compare the experimental and computed differences between the values for 4F-Thr and Thr instead of their absolute values. The good agreement between the theory and experiment is not only apparent in Table 1 but also in Figure 4, which depicts the comparison between the experimental and simulated titration curves, with the latter being obtained from the charge balance equation (see ref (44)).
Figure 4.
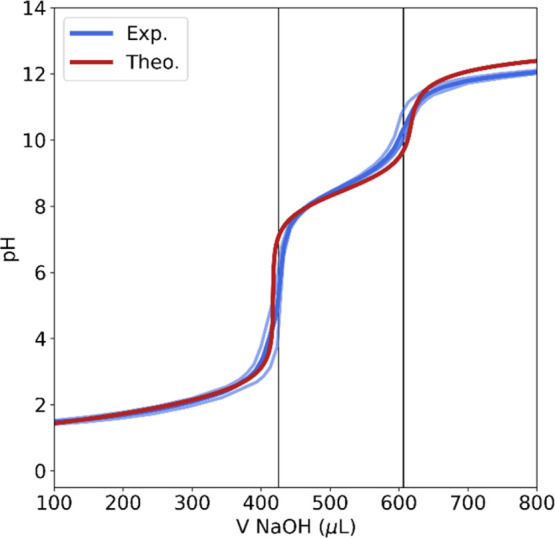
Experimental (light blue lines: the three different titration analyses. Blue line: their average plot) vs simulation (red line) of the titration curve of 4F-Thr. The two experimentally determined equivalent-point volumes are also highlighted by (black) vertical lines.
NMR Spectra
The effects of fluorine can also be addressed by analyzing the NMR chemical shifts of 4F-Thr and Thr (see also ref (45)), whose zwitterionic form spectra were obtained under the same experimental conditions (see the Experimental Section for details and the Supporting Information for the spectra). As expected, the fluorine-withdrawing properties cause a remarkable deshielded shift of the 1H and 13C signals of the −CH2F group with respect to those of the −CH3 moiety in Thr (1H(4F-Thr): 4.60 and 4.72 ppm vs 1H(Thr): 1.32 ppm; 13C(4F-Thr): 85.6 ppm vs 13C(Thr): 20.1 ppm), while the 1H (HCα, HCβ) and 13C (Cα, Cβ) chemical shifts are much closer when comparing 4F-Thr with Thr (1HCα(4F-Thr) and 1HCβ(4F-Thr): 3.87 and 4.35 ppm vs 1HCα(Thr) and 1HCβ (Thr): 3.57 and 4.24 ppm; 13Cα(4F-Thr) and 13Cβ(4F-Thr): 56.8 and 68.4 ppm vs 13Cα(Thr) and 13Cβ (Thr): 61.0 and 66.5 ppm). For 4F-Thr, the Cα–F (3JC–F: 4.4 Hz) and Cβ–F (2JC–F: 19.1 Hz) indirect spin–spin coupling constants lie within the typical range for the carbon–fluorine interaction. From a computational point of view, the chemical shifts of the low-lying conformations of both molecules (see the Supporting Information) are in good agreement with the experimental counterparts, thus confirming both the reliability of the conformational search and the inductive nature of the fluorine effect on the active nuclei.
Conclusions
A novel synthesis of 4F-Thr, the only natural amino acid containing fluorine, and an integrated experiment-theory approach for its characterization have been reported. The first step of the presented strategy is the development of novel, convenient, and straightforward diastereoselective synthetic routes for the racemic form of 4F-Thr, which have been complemented by acid–base titrations to determine the acid dissociation constants. In parallel, a comprehensive quantum-chemical investigation at the state-of-the-art allowed us to unveil the conformational behavior of the cationic, zwitterionic, and anionic forms of the title molecule as well as the relationships between them and with the most stable conformers of the parent Thr amino acid. Furthermore, the quantum-chemical study led to acid dissociation constants in good agreement with those experimentally derived.
To explore the potential energy surface of this flexible system, the methodology described in the Computational Methods section has been exploited; this relies on the integration of machine learning (genetic algorithm) and a two-stage approach to evaluate geometry and energy, as described in refs (46, 47).
At the same time, the effectiveness of the new synthetic approach allowed us to obtain remarkable quantities of 4F-Thr, thus paving the route toward further investigations both in the gas phase and in solution. In a more general perspective, the strategy pursued in this work offers, in our opinion, interesting clues for the study of other systems of biological and/or medicinal relevance.
Experimental Section
General Methods
Unless otherwise stated, common chemicals and solvents (HPLC grade or reagent grade) were purchased from commercial sources and used without further purification. Solvents are reported with the following abbreviations: cyclohexane (CyH), dichloromethane (DCM), diethyl ether (Et2O), ethanol (EtOH), ethyl acetate (EtOAc), methanol (MeOH), and tetrahydrofuran (THF). Plastic plates coated with a 0.2 mm thick layer of silica gel 60 F254 were used for thin-layer chromatography analyses (TLC), whereas flash column chromatography purification was carried out using silica gel 60 (230–400 mesh). NMR spectra were recorded at 25 °C in a 400 MHz spectrometer using the deuterated solvent as an internal deuterium lock. The residual protic signal of the solvent (7.26 ppm, for 1H-NMR), and the 13CDCl3 signal (77.16 ppm, for 13C-NMR), were used for spectra recorded in CDCl3. For NMR spectra recorded in D2O, a drop of methanol was added, and its signals were used as references for both 1H and 13C spectra (3.34 and 49.50 ppm, respectively). Trifluoroacetic acid signal (−76.55 ppm) was used as a reference for 19F NMR spectra. Chemical shifts are reported in parts per million (ppm) of the δ scale relative to TMS for 1H and 13C spectra and CFCl3 for 19F NMR spectra; coupling constants are in Hertz, and the multiplicity is as follows: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (doublet of doublets), dt (doublet of triplets), ddd (doublet of doublet of doublets), br (broad signal). Gas chromatography-mass spectrometry (GC-MS) spectra were obtained by EI ionization at 70 eV on a Hewlett-Packard 5971 with GC injection; they are reported as m/z (rel. intensity). High performance liquid chromatography-mass spectrometry (HPLC-MS) analyses were performed on an Agilent Technologies HP1100 instrument coupled with an Agilent Technologies MSD1100 single-quadrupole mass spectrometer using a Phenomenex Gemini C18 3 μm (100 × 3 mm2) column; mass spectrometric detection was performed in the full-scan mode from m/z 50 to 2500, a scan time of 0.1 s in the positive ion mode, ESI spray voltage of 4500 V, nitrogen gas of 35 psi, a drying gas flow rate of 11.5 mL min–1, a fragmentor voltage of 30 V.
2-(Benzyloxy)acetaldehyde (1)
A tube with a bubbler was charged with a solution of allyl benzyl ether (2 mL, 1.92 g, 12.9 mmol) in DCM (20 mL) and cooled at −78 °C. The mixture was fluxed with O2 for 1 min, then O3 was fluxed until the mixture became blue, maintaining the reaction temperature at −78 °C. Excess O3 was removed by fluxing O2 for 5 min. Dimethyl sulfide (9.5 mL, 8.04 g, 129 mmol) was added at −78 °C, and the reaction mixture was slowly warmed at room temperature and stirred overnight. Volatiles were removed under reduced pressure, and the crude aldehyde was purified by flash column chromatography (silica gel, 0–10% EtOAc in CyH), affording 1.15 g (7.7 mmol, 60% yield) of aldehyde as a pale yellow oil. TLC: Rf (CyH/EtOAc = 1:1) = 0.25. 1H NMR (400 MHz, CDCl3) δ: 9.73 (1H, s), 7.40–7.30 (5H, m), 4.64 (2H, s), 4.11 (2H, s). 13C NMR (101 MHz, CDCl3) δ: 200.6, 137.0, 128.8 (2C), 128.4, 128.2 (2C), 75.4, 73.8. EI-MS m/z: 107 (M – CH2CHO, 33), 91 (100), 77 (7), 65 (17), 51 (5). Spectroscopical data are in agreement with those reported in the literature.48
Ethyl 5-((Benzyloxy)methyl)-4,5-dihydrooxazole-4-carboxylate (2)
A dried three-neck round-bottom flask, equipped with a pressure-equalizing dropping funnel, was charged with CuCl (38 mg, 0.38 mmol), triphenylphosphine (100 mg, 0.38 mmol), and dry DCM (10 mL) under a N2 atmosphere. The mixture was stirred for 5 min, and then dry diisopropylethylamine (0.13 mL, 96 mg, 0.76 mmol) was added and the mixture was stirred for further 10 min. A solution of 1 (1.15 g, 7.7 mmol) and ethyl 2-isocyanoacetate (0.84 mL, 0.87 g, 7.7 mmol) in dry DCM (10 mL) was added dropwise at 0 °C. The dropping funnel was washed with two 5 mL portions of dry DCM to transfer all of the reagents inside the reaction flask, and the reaction mixture was stirred overnight at room temperature (or until completion by TLC). The solvent was removed under reduced pressure and the crude mixture was chromatographed (silica gel, 0–50% EtOAc in CyH), affording 1.55 g (5.9 mmol, 77% yield) of 2 as a pale yellow oil. TLC: Rf (CyH/Et2O = 1:4) = 0.41. 1H NMR (400 MHz, CDCl3) δ: 7.37–7.28 (5H, m), 6.93 (1H, d, J = 2.0 Hz), 4.85 (1H, ddd, J = 7.8, 5.0, 3.4 Hz), 4.60 (2H, s), 4.57 (1H, dd, J = 7.8, 2.0 Hz), 4.29–4.18 (m, 2H), 3.72 (1H, dd, J = 10.9, 3.4 Hz), 3.60 (1H, dd, J = 10.9, 5.0 Hz), 1.30 (3H, t, J = 7.2 Hz). 13C NMR (101 MHz, CDCl3) δ: 170.7, 156.5, 137.6, 128.6 (2C), 128.0, 127.8 (2C), 80.1, 73.6, 70.2, 69.3, 62.0, 14.3. EI-MS m/z: 190 (M – COOEt, 2), 172 (M – Bn, 2), 157 (8), 91 (100), 84 (35), 77 (5), 70 (6), 65 (13), 56 (8).
Compound 2 was obtained in a mixture with the corresponding formamide 3 (variable from 0 to 15%, batch dependent), derived from the hydrolysis of the oxazoline ring due to trace of water during the column chromatography process. The mixture of the two compounds could be used directly in the next step without any further purification. In the following, characteristic 1H NMR signals and ESI-MS data for compound 3 are reported. 1H NMR (400 MHz, CDCl3) δ: 8.27 (1H, s), 7.39–7.28 (5H, m), 6.39 (1H, d, J = 8.8 Hz, NH), 4.77 (1H, dd, J = 8.8, 2.6 Hz), 4.54 (2H, s), 4.38 (1H, dddd, J = 7.9, 4.2, 3.9, 2.6 Hz), 4.23 (2H, q, J = 7.1 Hz), 3.59 (1H, dd, J = 9.5, 4.2 Hz), 3.44 (1H, dd, J = 9.5, 7.9 Hz), 2.71 (1H, d, J = 3.9 Hz), 1.28 (3H, t, J = 7.1 Hz). ESI-MS m/z: 282.2 [M + H]+ (100); 304.2 [M + Na]+ (4).
Ethyl 5-((Benzyloxy)methyl)-2-oxooxazolidine-4-carboxylate (4)
A dried two-neck round-bottom flask was charged with 2 (1.55 g, 5.9 mmol) and dry MeOH (12 mL) under a N2 atmosphere. The mixture was cooled to 0 °C and HCl (3 M in methanol, 12 mL, 36 mmol) was slowly added. The reaction mixture was stirred at 0 °C for 30 min and then allowed to warm to room temperature and stirred overnight or until completion (TLC). The solvent was removed under reduced pressure, affording crude ethyl 2-amino-4-(benzyloxy)-3-hydroxybutanoate hydrochloride salt as a viscous amber oil, which was used in the next step without purification. The reaction crude was dissolved in dry DCM (15 mL) and triethylamine (2.67 mL, 1.94 g, 19.3 mmol) was added dropwise at 0 °C under a N2 atmosphere. The reaction mixture was stirred at 0 °C for 10 min, and then a solution of triphosgene (1.90 g, 6.4 mmol) in dry DCM (15 mL) was added dropwise. The reaction mixture was stirred at 0 °C for 30 min and then allowed to warm to room temperature and stirred overnight or until completion (TLC). The reaction mixture was cooled to 0 °C, and a saturated Na2CO3 aqueous solution was slowly added under vigorous stirring until basic pH was reached (pH = 9–10). Upon separation, the organic phase was washed with aqueous 1 M HCl and saturated NaHCO3 successively, and then dried on anhydrous Na2SO4. The solvent was removed under reduced pressure. Flash column chromatography of the crude (silica gel, CyH/Et2O = 1:1) afforded 1.02 g (3.7 mmol, 62%) of trans-4 as a pale yellow oil. TLC: Rf (Et2O) = 0.64. 1H NMR (400 MHz, CDCl3) δ: 7.33–7.22 (5H, m), 6.93 (1H, s), 4.71 (1H, ddd, J = 5.4, 4.4, 3.7 Hz), 4.58 (1H, d, J = 12.1H), 4.56 (1H, d, J = 12.1 Hz), 4.29 (1H, d, J = 5.4 Hz), 4.18 (2H, q, J = 7.1 Hz), 3.71 (1H, dd, J = 11.0, 3.7 Hz), 3.64 (1H, dd, J = 11.0, 4.4 Hz), 1.22 (3H, t, J = 7.1 Hz). 13C NMR (101 MHz, CDCl3) δ: 169.9, 158.4, 137.3, 128.3 (2C), 127.7, 127.5 (2C), 77.4, 73.3, 69.5, 62.0, 55.3, 13.8. EI-MS m/z: 206 (M – COOEt, 2), 173 (7), 130 (3), 107 (7), 100 (20), 91 (100), 86 (14), 77 (5), 65 (14), 56 (17).
The analytical sample of cis-4 was obtained through a similar Cu(I)-based experimental procedure (CuCl 5 mol %, triethylamine 5 mol %, THF),49 only partially diastereoselective (trans/cis = 85:15), to perform the complete NMR characterization of cis-4. TLC: Rf (Et2O) = 0.36. 1H NMR (400 MHz, CDCl3) δ: 7.36–7.26 (5H, m), 5.48 (1H, s), 4.94 (1H, dt, J = 9.3, 3.5 Hz), 4.52 (1H, d, J = 12.1 Hz), 4.50 (1H, d, J = 12.1 Hz), 4.48 (1H, d, J = 9.3 Hz), 4.09 (2H, q, J = 7.2 Hz), 3.80–3.78 (2H, m), 1.17 (3H, t, J = 7.2 Hz). 13C NMR (101 MHz, CDCl3) δ: 168.9, 158.4, 137.4, 128.5 (2C), 127.9, 127.7 (2C), 76.9, 73.7, 67.7, 62.2, 55.5, 14.0. EI-MS m/z: 206 (M – COOEt, 3), 173 (5), 158 (3), 130 (3), 107 (7), 100 (20), 91 (100), 86 (10), 77 (5), 65 (13), 56 (17).
Ethyl 5-(Hydroxymethyl)-2-oxooxazolidine-4-carboxylate (5)
A dried two-neck round-bottom flask was charged with a solution of trans-4 (0.83 g, 2.97 mmol) in EtOH (96% purity, 30 mL) under a N2 atmosphere. Palladium on carbon (10% Pd/C, 83 mg, 10% w/w in relation to the substrate) was added to the mixture. The flask was partially evacuated and refilled with H2 (balloon). The mixture was vigorously stirred at room temperature overnight or until completion (TLC). The reaction mixture was filtered over a Celite, washed with DCM, and the filtrate was evaporated under reduced pressure, affording 562 mg (2.97 mmol, quantitative yield) of spectroscopically pure 5 as a colorless oil. The latter solidified upon storage at −20 °C overnight, affording a white sticky solid. TLC: Rf (CyH/EtOAc = 1:4) = 0.22. 1H NMR (400 MHz, CDCl3) δ: 6.19 (1H, bs), 4.70 (1H, ddd, J = 5.6, 3.3, 2.4 Hz), 4.42 (1H, d, J = 5.6 Hz), 4.27 (2H, q, J = 7.1 Hz), 4.01 (1H, dd, J = 12.7, 2.4 Hz), 3.75 (1H, dd, J = 12.9, 3.3 Hz), 2.84 (1H, bs), 1.31 (3H, t, J = 7.1 Hz). 13C NMR (101 MHz, CDCl3) δ: 170.5, 159.1, 79.4, 77.4, 62.3, 55.1, 14.1. EI-MS m/z: 189 (M+, 2), 173 (7), 130 (3), 107 (7), 100 (20), 91 (100), 86 (14), 77 (5), 65 (14), 56 (17).
Ethyl 5-(Fluoromethyl)-2-oxooxazolidine-4-carboxylate (6)
A dried two-neck round-bottom flask was charged with 5 (712 mg, 3.76 mmol) and dry DCM (20 mL) under a N2 atmosphere. The reaction mixture was cooled at −78 °C and diethylaminosulfur trifluoride (0.99 mL, 1.2 g, 7.53 mmol) was added dropwise. The reaction mixture was stirred at −78 °C for 30 min, and then allowed to warm up to 0 °C and stirred 3 h until completion (TLC). The reaction vessel was cooled to −78 °C and a saturated NaHCO3 solution (10 mL) was added under vigorous stirring. The mixture was allowed to warm to room temperature under vigorous stirring. Upon extraction (DCM, 3 × 10 mL) and separation, the collected organic phases were dried on anhydrous Na2SO4 and the solvent was removed under reduced pressure. Flash column chromatography (silica gel, CyH/Et2O = 1:1) afforded 233 mg (1.2 mmol, 32% yield) of 6 as a pale yellow oil. TLC: Rf (CyH/EtOAc = 1:4) = 0.77. 1H NMR (400 MHz, CDCl3) δ: 5.76 (1H, bs), 4.82 (1H, dddd, J = 23.8, 5.6, 3.2, 2.8 Hz), 4.73 (1H, ddd, J = 47.6, 10.8, 2.8 Hz), 4.58 (1H, ddd, J = 46.1, 10.8, 3.2 Hz), 4.36 (1H, d, J = 5.6 Hz), 4.30 (2H, q, J = 7.2 Hz), 1.33 (3H, t, J = 7.2 Hz). 19F NMR (376.5 Hz, CDCl3) δ: −235 (dt, J = 46.9, 23.8 Hz). 13C NMR (101 MHz, CDCl3) δ: 169.4, 157.6, 81.9 (d, J = 177.7 Hz), 76.7 (d, J = 19.9 Hz), 62.8, 54.5 (d, J = 6.2 Hz), 14.2. EI-MS m/z: 191 (M+, 2), 118 (M – COOEt, 100), 101 (2), 85 (5), 74 (6), 54 (6).
2-Amino-4-fluoro-3-hydroxybutanoic Acid, Hydrochloride Salt (7)
A Schlenk tube was charged with 6 (432 mg, 2.26 mmol) and aqueous HCl (6 M, 4 mL, 24 mmol) under a N2 atmosphere. The reaction mixture was refluxed under vigorous stirring for 2 days, until complete conversion (1H NMR). The solvent was evaporated under reduced pressure, affording 352 mg of crude 7 (2 mmol, 90% yield) as a viscous amber oil, which was used as such in the next step. 1H NMR (400 MHz, D2O) δ: 4.76 (1H, ddd, J = 46.2, 10.5, 3.7 Hz), 4.64 (1H, ddd, J = 47.0, 10.5, 3.5 Hz), 4.47 (1H, dddd, J = 24.7, 3.9, 3.7, 3.5 Hz), 4.22 (1H, d, J = 3.9 Hz). 19F NMR (376.5 Hz, D2O) δ: −232 (dt, J = 46.7, 24.8 Hz). 13C NMR (101 MHz, D2O) δ: 170.9, 85.2 (d, J = 168.0 Hz), 67.8 (d, J = 19.2 Hz), 55.7 (d, J = 4.6 Hz). ESI-MS m/z: 138.2 [M – Cl]+ (100); 92.2 [M – (Cl + COOH)]+ (10).
4-Fluoro-threonine; 2-Amino-4-fluoro-3-hydroxybutanoic Acid (8)
A Schlenk tube was charged with 7 (352 mg, 2.03 mmol) and dry MeOH (10 mL) under a N2 atmosphere. Propylene oxide (1.42 mL, 1.22 g, 20.3 mmol) was added to the mixture, which was stirred at room temperature overnight. During this stage, a pale yellow precipitate formed. The solvent and excess propylene oxide were removed under reduced pressure. Recrystallization from MeOH afforded 171 mg (1.25 mmol, 61% yield) of spectroscopically pure 8 as a white powder. 1H NMR (400 MHz, D2O) δ: 4.72 (1H, ddd, J = 46.6, 10.8, 3.7 Hz), 4.60 (1H, ddd, J = 47.0, 10.8, 3.1 Hz), 4.35 (1H, dddd, J = 24.9, 4.7, 3.7, 3.1 Hz), 3.87 (1H, d, J = 4.7 Hz). 19F NMR (376.5 Hz, D2O) δ: −232 (dt, J = 46.8, 24.9 Hz). 13C NMR (101 MHz, D2O) δ: 172.5, 85.6 (d, J = 167.3 Hz), 68.4 (d, J = 19.1 Hz), 56.8 (d, J = 4.4 Hz). ESI-MS m/z: 138.2 [M + H]+ (100); 92.2 [M – COOH]+ (15). Spectroscopical data are in agreement with those reported in the literature.33
Acid–Base Titrations
To determine the main chemical properties (i.e., pKa1, pKa2, and pI) of 4F-Thr, a small amount of the racemic amino acid was used to prepare a stock solution to perform acid–base titrations. The approach described below was preliminarily performed with a stock solution of Thr, with the aim of assessing the quality of the experimental procedure. Since this method was found to be fully reliable in terms of repeatability, we focus our attention on the differences between Thr and 4F-Thr, also in view of the significant spread of the values available in the literature for Thr.
The titration curve of 4F-Thr was recorded three times in the same conditions to allow the calculation of mean values and confidence intervals for pKa1, pKa2, and pI. The titration was performed using an aqueous 0.05 M stock solution of 4F-Thr. A test tube equipped with a magnetic stirring bar was charged with 1 mL of such stock solution and a few microliters (thus, a negligible amount with respect to the total volume) of HCl 37% were added under vigorous stirring to lower the pH of the amino acid solution down to 1–2. This ensured that even the first buffer zone (corresponding to pKa1) could be properly mapped. The pH-meter electrode was inserted in the test tube until its bulb was completely soaked in the stirred solution. A 0.25 M solution of NaOH was used as a titrant. In both the early and late plateau regions of the titration curve (featuring low-slope values), the titrant was added 50 μL at a time, while 10 μL additions were performed in the central part, due to the presence of the two equivalence points, i.e., maximum-slope regions. All of the titrant additions were performed with a 100 μL Hamilton syringe, under proper stirring conditions.
The following semiempirical method was used for the quantitative analyses of titration curves. First, the pKa2 was determined as the pH value corresponding to the second half-equivalence point. Such specific titration point coincides with a local maximum of the first derivative of the inverted titration curve (i.e., the pH value on the x-axis and the titrant volume on the y-axis), referred to as the inverted derivative in the following. This approach is applicable only for the determination of pKa values corresponding to buffer zones encompassed between two equivalence points since this situation leads to a single easily identifiable maximum in the inverted derivative. Furthermore, such identification is even easier because buffer zones are usually characterized by a high sampling rate of both pH values and titrant volumes, leading to quite resolved inverted derivatives. Last but not least, a buffer zone, by definition, is encompassed within a very narrow pH range, thus leading to smaller errors in the determination of the corresponding pKa value. A similar approach could be used for pI, identifiable as the pH value corresponding to the first equivalence point. However, the sampling rate is not sufficiently high to enable easy identification of the equivalence point, even with the analysis of the first and second derivatives of the titration curve, due to their low resolution. Indeed, unlike buffer zones, the region encompassing an equivalence point suffers from tremendous pH changes even when dealing with very small titrant volume variations. Nevertheless, equivalence zones are encompassed within narrow ranges of the titrant volume, thus leading to small errors in volumetric analyses. Therefore, our approach moved to the graphical determination of the pKa1, starting from pKa2 and taking advantage of both the narrow volume ranges encompassing equivalence zones and the narrow pH ranges encompassing buffer zones. In particular, the titrant volume difference between the two equivalence points (which is supposed to experience almost negligible fluctuations as a consequence of the unavoidable small discrepancies between the observed and the real equivalence points) is the same observed between the two half-equivalence points. Such difference was determined graphically, upon identification of five linear-regime zones: three low-slope regions (the early and late titration plateau, and the mid buffer zone corresponding to pKa2) and two high-slope ones (the two equivalence zones). For each zone, the points were interpolated with linear functions and the intersection points between the adjacent linear regressions were determined (four points, overall). Two relevant line segments were then identified. On the one hand, the first one (bounded by the first two points) lies on the first pH jump and its midpoint’s abscissa corresponds to the titrant volume at the first equivalence point. On the other hand, the second line segment (bounded by the last two points) lies on the second pH jump and its midpoint’s abscissa corresponds to the titrant volume at the second equivalence point. The volume difference between the two equivalence points was hence determined and subtracted to the titrant volume corresponding to pKa2. This new titrant volume is the abscissa of the first half-equivalence point; therefore, the corresponding pH value is pKa1. The value of pI was determined starting from pKa1 and pKa2 using the Henderson–Hasselbalch equation.
Computational Methods
Conformational Analysis
The conformational space of 4F-Thr was explored using a recently proposed island model evolutionary algorithm (EA)46 coupled with the two-stage method to evaluate structures, which has been described in ref (47). For each form of 4F-Thr, a search using a pool of 30 (randomly generated) structures was carried out for 70 iterations. At each step, every new structure was evaluated performing a geometry optimization with the semiempirical PM7 method (constraining the dihedral angles, whose values were changed by the EA),50 followed by a single point energy evaluation at the B3LYP-D3(BJ)/6-31+G(d)51,52 level, also exploiting the conductor version of the polarizable, continuum model (CPCM)53 to take bulk effects into account (water ε = 78.3553). All structures lying within 25 kJ mol–1 above the lowest energy minimum were then reoptimized at a higher level of theory to achieve a better description of the stability of the 4F-Thr species. The double-hybrid B2PLYP functional,54,55 also including empirical dispersion (D3BJ),52 in conjunction with the jun-cc-pVTZ basis set56 (overall shortly denoted as B2) was employed, which is proven to provide reliable results in the characterization of conformers in solution57,58 at a limited computational cost. Relative free energies (ΔG) were determined by adding CPCM solvent effects together with the zero-point energy and thermal contributions, evaluated within the rigid-rotor harmonic-oscillator approximation at the B3LYP-D3(BJ)/jul-cc-pVDZ level, to relative electronic energies (ΔE). All quantum-chemical calculations have been performed with the G16.C01 version of the Gaussian suite of programs.59
The solvate structures were obtained from the lowest conformer geometries employing the solvation functionality, as implemented in the Proxima library.60 Then, only the first seven water molecules directly H-bonded to the solute were kept, and the structures were reoptimized at the B2 level of theory.
NMR Spectra
Chemical shifts and indirect J spin–spin coupling constants (values collected in the Supporting Information) have been computed at the B3LYP-D3(BJ)/jul-cc-pVDZ level of theory using B2PLYP-D3(BJ)/jun-cc-pVTZ reference geometries. Bulk solvent effects were again accounted for using CPCM. Only conformers within 10 kJ mol–1 above the global energy minimum were considered. To compute the 1H and 13C chemical shifts, methanol spectra were simulated at the same level of theory and then used as the reference (1H 3.34 ppm and 13C ppm 49.50 ppm). For the 19F nucleus, trifluoroacetic acid (TFA) was used as the reference (−76.55 ppm). Spin–spin coupling constants were simulated with both the one-step (spin–spin) and the two-step61 (mixed) procedures. All quantum-chemical computations have been performed using the G16.C01 version of the Gaussian suite of programs.59
Acknowledgments
This work has been supported by MIUR “PRIN 2017” (Grant Numbers: 20174SYJAF, 2017A4XRCA), the Italian Space Agency (ASI; “Life in Space” project, No. 2019-3-U.0), and the University of Bologna (RFO funds). The SMART@SNS Laboratory (http://smart.sns.it) is also acknowledged for providing high-performance computing facilities. Dr. Enrico Emer is fully acknowledged for precious and fruitful discussion concerning fluorination reactions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c01007.
Optimization of the reaction conditions; titration data of threonine and 4-fluorothreonine; conformational analysis results; simulated NMR chemical shifts; J spin–spin coupling constants and their comparison with the experimental data; calculated molecular structures and their cartesian coordinates in XYZ format; and copies of NMR spectra (PDF)
Author Contributions
⊥ S.P. and L.S. contributed equally to the work.
The authors declare no competing financial interest.
Supplementary Material
References
- a Böhm H. J.; Banner D.; Bendels S.; Kansy M.; Kuhn B.; Müller K.; Obst-Sander U.; Stahl M. Fluorine in Medicinal Chemistry. ChemBioChem 2004, 5, 637–643. 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]; b Bégué J. P.; Bonnet-Delpon D. Recent Advances (1995–2005) in Fluorinated Pharmaceuticals Based on Natural Products. J. Fluorine Chem. 2006, 127, 992–1012. 10.1016/j.jfluchem.2006.05.006. [DOI] [Google Scholar]; c Purser S.; Moore P. R.; Swallow S.; Gouverneur V. Fluorine in Medicinal Chemistry. Chem. Soc. Rev. 2008, 37, 320–330. 10.1039/B610213C. [DOI] [PubMed] [Google Scholar]; d O’Hagan D. Fluorine in Health Care: Organofluorine Containing Blockbuster Drugs. J. Fluorine Chem. 2010, 131, 1071–1081. 10.1016/j.jfluchem.2010.03.003. [DOI] [Google Scholar]
- a Shah P.; Westwell A. D. The Role of Fluorine in Medicinal Chemistry. J. Enzyme Inhib. Med. Chem. 2007, 22, 527–540. 10.1080/14756360701425014. [DOI] [PubMed] [Google Scholar]; b Gillis E. P.; Eastman K. J.; Hill M. D.; Donnelly D. J.; Meanwell N. A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- a Phelps M. E. Positron Emission Tomography Provides Molecular Imaging of Biological Processes. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 9226–9233. 10.1073/pnas.97.16.9226. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ametamey S. M.; Honer M.; Schubiger P. A. Molecular Imaging with PET. Chem. Rev. 2008, 108, 1501–1516. 10.1021/cr0782426. [DOI] [PubMed] [Google Scholar]
- a Wolfe S. The Gauche Effect. Some Stereochemical Consequences of Adjacent Electron Pairs and Polar Bonds. Acc. Chem. Res. 1972, 5, 102–111. 10.1021/ar50051a003. [DOI] [Google Scholar]; b O’Hagan D. Understanding Organofluorine Chemistry. An Introduction to the C–F Bond. Chem. Soc. Rev. 2008, 37, 308–319. 10.1039/B711844A. [DOI] [PubMed] [Google Scholar]; c Zimmer L. E.; Sparr C.; Gilmour R. Fluorine Conformational Effects in Organocatalysis: An Emerging Strategy for Molecular Design. Angew. Chem., Int. Ed. 2011, 50, 11860–11871. 10.1002/anie.201102027. [DOI] [PubMed] [Google Scholar]
- a Meanwell N. A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]; b Kirk K. Selective Fluorination in Drug Design and Development: An Overview of Biochemical Rationales. Curr. Top. Med. Chem. 2006, 6, 1447–1456. 10.2174/156802606777951073. [DOI] [PubMed] [Google Scholar]
- Richardson P. Fluorination Methods for Drug Discovery and Development. Expert Opin. Drug Discovery 2016, 11, 983–999. 10.1080/17460441.2016.1223037. [DOI] [PubMed] [Google Scholar]
- a O’Hagan D.; Deng H. Enzymatic Fluorination and Biotechnological Developments of the Fluorinase. Chem. Rev. 2015, 115, 634–649. 10.1021/cr500209t. [DOI] [PubMed] [Google Scholar]; b Odar C.; Winkler M.; Wiltschi B. Fluoro Amino Acids: A Rarity in Nature, yet a Prospect for Protein Engineering. Biotechnol. J. 2015, 10, 427–446. 10.1002/biot.201400587. [DOI] [PubMed] [Google Scholar]
- Moschner J.; Stulberg V.; Fernandes R.; Huhmann S.; Leppkes J.; Koksch B. Approaches to Obtaining Fluorinated α-Amino Acids. Chem. Rev. 2019, 119, 10718–10801. 10.1021/acs.chemrev.9b00024. [DOI] [PubMed] [Google Scholar]
- a Ayyadurai N.; Prabhu N. S.; Deepankumar K.; Kim A.; Lee S. G.; Yun H. Biosynthetic Substitution of Tyrosine in Green Fluorescent Protein with Its Surrogate Fluorotyrosine in Escherichia Coli. Biotechnol. Lett. 2011, 33, 2201–2207. 10.1007/s10529-011-0679-4. [DOI] [PubMed] [Google Scholar]; b Baker P. J.; Montclare J. K. Enhanced Refoldability and Thermoactivity of Fluorinated Phosphotriesterase. ChemBioChem 2011, 12, 1845–1848. 10.1002/cbic.201100221. [DOI] [PubMed] [Google Scholar]; c Steiner T.; Hess P.; Bae J. H.; Wiltschi B.; Moroder L.; Budisa N. Synthetic Biology of Proteins: Tuning GFPs Folding and Stability with Fluoroproline. PLoS One 2008, 3, e1680 10.1371/journal.pone.0001680. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Acevedo-Rocha C. G.; Hoesl M. G.; Nehring S.; Royter M.; Wolschner C.; Wiltschi B.; Antranikian G.; Budisa N. Non-Canonical Amino Acids as a Useful Synthetic Biological Tool for Lipase-Catalysed Reactions in Hostile Environments. Catal. Sci. Technol. 2013, 3, 1198–1201. 10.1039/C3CY20712A. [DOI] [Google Scholar]; e Deepankumar K.; Nadarajan S. P.; Ayyadurai N.; Yun H. Enhancing the Biophysical Properties of mRFP1 through Incorporation of Fluoroproline. Biochem. Biophys. Res. Commun. 2013, 440, 509–514. 10.1016/j.bbrc.2013.09.062. [DOI] [PubMed] [Google Scholar]
- a Dominguez M. A. Jr.; Thornton K. C.; Melendez M. G.; Dupureur C. M. Differential Effects of Isomeric Incorporation of Fluorophenylalanines into PvuII Endonuclease. Proteins 2001, 45, 55–61. 10.1002/prot.1123. [DOI] [PubMed] [Google Scholar]; b Parsons J. F.; Xiao G.; Gilliland G. L.; Armstrong R. N. Enzymes Harboring Unnatural Amino Acids: Mechanistic and Structural Analysis of the Enhanced Catalytic Activity of a Glutathione Transferase Containing 5-Flourotryptophan. Biochemistry 1998, 37, 6286–6294. 10.1021/bi980219e. [DOI] [PubMed] [Google Scholar]
- Renner C.; Alefelder S.; Bae J. H.; Budisa N.; Huber R.; Moroder L. Fluoroprolines as Tools for Protein Design and Engineering. Angew. Chem., Int. Ed. 2001, 40, 923–925. . [DOI] [PubMed] [Google Scholar]
- Woll M. G.; Hadley E. B.; Mecozzi S.; Gellman S. H. Stabilizing and Destabilizing Effects of Phenylalanine → F5-Phenylalanine Mutations on the Folding of a Small Protein. J. Am. Chem. Soc. 2006, 128, 15932–15933. 10.1021/ja0634573. [DOI] [PubMed] [Google Scholar]
- a Minnihan E. C.; Young D. D.; Schultz P. G.; Stubbe J. Incorporation of Fluorotyrosines into Ribonucleotide Reductase Using an Evolved, Polyspecific Aminoacyl-tRNA Synthetase. J. Am. Chem. Soc. 2011, 133, 15942–15945. 10.1021/ja207719f. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wilkins B. J.; Marionni S.; Young D. D.; Liu J.; Wang Y.; Di Salvo M. L.; Deiters A.; Cropp T. A. Site-Specific Incorporation of Fluorotyrosines into Proteins in Escherichia Coli by Photochemical Disguise. Biochemistry 2010, 49, 1557–1559. 10.1021/bi100013s. [DOI] [PubMed] [Google Scholar]
- Pandey A. K.; Naduthambi D.; Thomas K. M.; Zondlo N. J. Proline Editing: A General and Practical Approach to the Synthesis of Functionally and Structurally Diverse Peptides. Analysis of Steric versus Stereoelectronic Effects of 4-Substituted Prolines on Conformation within Peptides. J. Am. Chem. Soc. 2013, 135, 4333–4363. 10.1021/ja3109664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoesl M. G.; Budisa N. Recent Advances in Genetic Code Engineering in Escherichia coli. Curr. Opin. Biotechnol. 2012, 23, 751–757. 10.1016/j.copbio.2011.12.027. [DOI] [PubMed] [Google Scholar]
- Jäckel C.; Koksch B. Fluorine in Peptide Design and Protein Engineering. Eur. J. Org. Chem. 2005, 2005, 4483–4503. 10.1002/ejoc.200500205. [DOI] [Google Scholar]
- Johnson B. M.; Shu Y. Z.; Zhuo X.; Meanwell N. A. Metabolic and Pharmaceutical Aspects of Fluorinated Compounds. J. Med. Chem. 2020, 63, 6315–6386. 10.1021/acs.jmedchem.9b01877. [DOI] [PubMed] [Google Scholar]
- Olgun A. Selective Targeting of Signet Ring Cell Adenocarcinomas. Med. Hypotheses 2019, 133, 109380 10.1016/j.mehy.2019.109380. [DOI] [PubMed] [Google Scholar]
- Murphy C. D.; Schaffrath C.; O’Hagan D. Fluorinated Natural Products: The Biosynthesis of Fluoroacetate and 4-Fluorothreonine in Streptomyces cattleya. Chemosphere 2003, 52, 455–461. 10.1016/S0045-6535(03)00191-7. [DOI] [PubMed] [Google Scholar]
- Sanada M.; Miyano T.; Iwadare S.; Williamson J. M.; Arison B. H.; Smith J. L.; Douglas A. W.; Liesch J. M.; Inamine E. Biosynthesis of Fluorothreonine and Fluoroacetic Acid by the Thienamycin Producer, Streptomyces cattleya. J. Antibiot. 1986, 39, 259–265. 10.7164/antibiotics.39.259. [DOI] [PubMed] [Google Scholar]
- Deng H.; O’Hagan D.; Schaffrath C. Fluorometabolite Biosynthesis and the Fluorinase from Streptomyces cattleya. Nat. Prod. Rep. 2004, 21, 773–784. 10.1039/B415087M. [DOI] [PubMed] [Google Scholar]
- O’Hagan D. Recent Developments on the Fluorinase from Streptomyces cattleya. J. Fluorine Chem. 2006, 127, 1479–1483. 10.1016/j.jfluchem.2006.09.006. [DOI] [Google Scholar]
- O’Hagan D.; Schaffrath C.; Cobb S. L.; Hamilton J. T. G.; Murphy C. D. Biosynthesis of an Organofluorine Molecule. Nature 2002, 416, 279 10.1038/416279a. [DOI] [PubMed] [Google Scholar]
- Murphy C. D.; O’Hagan D.; Schaffrath C. Identification of a PLP-Dependent Threonine Transaldolase: A Novel Enzyme Involved in 4-Fluorothreonine Biosynthesis in Streptomyces cattleya. Angew. Chem., Int. Ed. 2001, 40, 4479–4481. . [DOI] [PubMed] [Google Scholar]
- Blasiak L. C.; Drennan C. L. Structural Perspective on Enzymatic Halogenation. Acc. Chem. Res. 2009, 42, 147–155. 10.1021/ar800088r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillancourt F. H.; Yeh E.; Vosburg D. A.; Garneau-Tsodikova S.; Walsh C. T. Nature’s Inventory of Halogenation Catalysts: Oxidative Strategies Predominate. Chem. Rev. 2006, 106, 3364–3378. 10.1021/cr050313i. [DOI] [PubMed] [Google Scholar]
- Wu J. Review of Recent Advances in Nucleophilic C–F Bond-Forming Reactions at sp3 Centers. Tetrahedron Lett. 2014, 55, 4289–4294. 10.1016/j.tetlet.2014.06.006. [DOI] [Google Scholar]
- Zechel D. L.; Reid S. P.; Nashiru O.; Mayer C.; Stoll D.; Jakeman D. L.; Warren R. A. J.; Withers S. G. Enzymatic Synthesis of Carbon–Fluorine Bonds. J. Am. Chem. Soc. 2001, 123, 4350–4351. 10.1021/ja005855q. [DOI] [PubMed] [Google Scholar]
- Cadicamo C. D.; Courtieu J.; Deng H.; Meddour A.; O’Hagan D. Enzymatic Fluorination in Streptomyces cattleya Takes Place with an Inversion of Configuration Consistent with an SN2 Reaction Mechanism. ChemBioChem 2004, 5, 685–690. 10.1002/cbic.200300839. [DOI] [PubMed] [Google Scholar]
- Wu L.; Deng H. Defluorination of 4-Fluorothreonine by Threonine Deaminase. Org. Biomol. Chem. 2020, 18, 6236–6240. 10.1039/d0ob01358g. [DOI] [PubMed] [Google Scholar]
- Scolastico C.; Conca E.; Prati L.; Guanti G.; Banfi L.; Berti A.; Farina P.; Valcavi U. Diastereo- And Enantioselective Synthesis of Fluorinated Threonines. Synthesis 1985, 1985, 850–855. 10.1055/s-1985-31363. [DOI] [Google Scholar]
- Shimizu M.; Yokota T.; Fujimori K.; Fujisawa T. Stereodivergent Synthesis of Fluorinated Threonine Derivatives in High Optical Purity. Tetrahedron: Asymmetry 1993, 4, 835–838. 10.1016/S0957-4166(00)80121-4. [DOI] [Google Scholar]
- Amin M. R.; Harper D. B.; Moloney J. M.; Murphy C. D.; Howard J. A. K.; O’Hagan D. A Short Highly Stereoselective Synthesis of the Fluorinated Natural Product (2S,3S)-4-Fluorothreonine. Chem. Commun. 1997, 1471–1472. 10.1039/A703121A. [DOI] [Google Scholar]
- Graham T. J. A.; Lambert R. F.; Ploessl K.; Kung H. F.; Doyle A. G. Enantioselective Radiosynthesis of Positron Emission Tomography (PET) Tracers Containing [18F]Fluorohydrins. J. Am. Chem. Soc. 2014, 136, 5291–5294. 10.1021/ja5025645. [DOI] [PubMed] [Google Scholar]
- The stereoselective synthesis of 4F-Thr is in progress with a new approach starting from commercially available methionine; work in progress.
- a Hayashi T.; Uozumi Y.; Yamazaki A.; Sawamura M.; Hamashima H.; Ito Y. Silver(I)-Catalyzed Asymmetric Aldol Reaction of Isocyanoacetate. Tetrahedron Lett. 1991, 32, 2799–2802. 10.1016/0040-4039(91)85090-R. [DOI] [Google Scholar]; b Gorla F.; Togni A.; Venanzi L. M.; Albinati A.; Lianza F. Synthesis of an Optically Active Platinum(II) Complex Containing a New Terdentate P–C–P Ligand and Its Catalytic Activity in the Asymmetric Aldol Reaction of Methyl Isocyanoacetate. X-Ray Crystal Structure of [2,6-Bis [(1′S,2′S)-1′-(Diphenylphosphino)-2′,3′-O-Isopropylidene-2′,3′-Dihydroxypropyl]phenyl] (η1-nitrato)platinum(II). Organometallics 1994, 13, 1607–1616. 10.1021/om00017a018. [DOI] [Google Scholar]; c Longmire J. M.; Zhang X.; Shang M. Synthesis and X-Ray Crystal Structures of Palladium(II) and Platinum(II) Complexes of the PCP-Type Chiral Tridentate Ligand (1R,1′R)-1,3-Bis[1-(Diphenylphosphino)Ethyl]Benzene. Use in the Asymmetric Aldol Reaction of Methyl Isocyanoacetate and Aldehydes. Organometallics 1998, 17, 4374–4379. 10.1021/om980461h. [DOI] [Google Scholar]; d Gosiewska S.; Veld M. H.; de Pater J. J. M.; Bruijnincx P. C. A.; Lutz M.; Spek A. L.; van Koten G.; Klein Gebbink R. J. M. Novel Enantiopure Non-C2-Symmetric NCN-Pincer Palladium Complexes with l-Proline Chiral Auxiliaries: mer η3-N,C,N versus Square Planar η4-N,C,N,O Coordination. Tetrahedron: Asymmetry 2006, 17, 674–686. 10.1016/j.tetasy.2005.12.040. [DOI] [Google Scholar]; e Yoon M. S.; Ryu D.; Kim J.; Ramesh R.; Ahn K. H. Cyclometalated Platinum(II) Complexes Derived from a Chiral Pyridine Ligand: Synthesis, Structure, and Catalytic Activity. Bull. Korean Chem. Soc. 2007, 28, 2045–2050. 10.5012/bkcs.2007.28.11.2045. [DOI] [Google Scholar]; f Xue M. X.; Guo C.; Gong L. Z. Asymmetric Synthesis of Chiral Oxazolines by Organocatalytic Cyclization of α-Aryl Isocyanoesters with Aldehydes. Synlett 2009, 2009, 2191–2197. 10.1055/s-0029-1217549. [DOI] [Google Scholar]; g Sladojevich F.; Trabocchi A.; Guarna A.; Dixon D. J. A New Family of Cinchona-Derived Amino Phosphine Precatalysts: Application to the Highly Enantio- and Diastereoselective Silver-Catalyzed Isocyanoacetate Aldol Reaction. J. Am. Chem. Soc. 2011, 133, 1710–1713. 10.1021/ja110534g. [DOI] [PubMed] [Google Scholar]; h Kim H. Y.; Oh K. Highly Diastereo- and Enantioselective Aldol Reaction of Methyl α-Isocyanoacetate: A Cooperative Catalysis Approach. Org. Lett. 2011, 13, 1306–1309. 10.1021/ol103104y. [DOI] [PubMed] [Google Scholar]; i Zhao M. X.; Zhou H.; Tang W. H.; Qu W. S.; Shi M. Cinchona Alkaloid-Derived Thiourea-Catalyzed Diastereo- and Enantioselective [3+2] Cycloaddition Reaction of Isocyanoacetates to Isatins: A Facile Access to Optically Active Spirooxindole Oxazolines. Adv. Synth. Catal. 2013, 355, 1277–1283. 10.1002/adsc.201300077. [DOI] [Google Scholar]; j Wang F.; Chen J.; Huang Y. Synthesis of Optically Active Oxazolines by an Organocatalytic Isocyanoacetate Aldol Reaction with α-Keto Esters. Synlett 2017, 28, 1300–1304. 10.1055/s-0036-1588718. [DOI] [Google Scholar]; k De La Campa R.; Ortín I.; Dixon D. J. Direct Catalytic Enantio- and Diastereoselective Ketone Aldol Reactions of Isocyanoacetates. Angew. Chem., Int. Ed. 2015, 54, 4895–4898. 10.1002/anie.201411852. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Martínez-Pardo P.; Blay G.; Muñoz M. C.; Pedro J. R.; Sanz-Marco A.; Vila C. Enantioselective Synthesis of Chiral Oxazolines from Unactivated Ketones and Isocyanoacetate Esters by Synergistic Silver/Organocatalysis. Chem. Commun. 2018, 54, 2862–2865. 10.1039/C8CC00856F. [DOI] [PubMed] [Google Scholar]; For an example using a chiral auxiliary, see:; m Hundscheid F. J. A.; Tandon V. K.; Rouwette P. H. F. M.; van Leusen A. M. Synthesis of Chiral Sulfonylmethyl Isocyanides, and Comparison of Their Propensities in Asymmetric Induction Reactions with Acetophenones. Tetrahedron 1987, 43, 5073–5088. 10.1016/S0040-4020(01)87684-5. [DOI] [Google Scholar]; n Martínez-Pardo P.; Blay G.; Vila C.; Sanz-Marco A.; MunÌoz M. C.; Pedro J. R. Enantioselective Synthesis of 5-Trifluoromethyl-2-Oxazolines under Dual Silver/Organocatalysis. J. Org. Chem. 2019, 84, 314–325. 10.1021/acs.joc.8b02808. [DOI] [PubMed] [Google Scholar]
- Ozaki Y.; Matsumoto K.; Miyoshi M. A Useful Synthetic Method of DL-Threonine Using α-Isocyanoacetamides. Agric. Biol. Chem. 1978, 42, 1565–1569. 10.1080/00021369.1978.10863205. [DOI] [Google Scholar]
- Saegusa T.; Ito Y.; Kixoshita H.; Tomita S. Synthetic Reactions by Complex Catalysts. XIX. Copper-Catalyzed Cycloaddition Reactions of Isocyanides. Novel Synthesis of Δ1-Pyrroline and Δ2-Oxazoline. J. Org. Chem. 1971, 36, 3316–3323. 10.1021/jo00821a011. [DOI] [Google Scholar]
- a Aouadi K.; Lajoix A. D.; Gross R.; Praly J. P. Multi-Step Synthesis and Biological Evaluation of Analogues of Insulin Secretagogue (2S,3R,4S)-4-Hydroxyisoleucine. Eur. J. Org. Chem. 2009, 2009, 61–71. 10.1002/ejoc.200800744. [DOI] [Google Scholar]; b Benito-Garagorri D.; Bocokić V.; Kirchner K. Copper(I)-Catalyzed Diastereoselective Formation of Oxazolines and N-Sulfonyl-2-Imidazolines. Tetrahedron Lett. 2006, 47, 8641–8644. 10.1016/j.tetlet.2006.10.040. [DOI] [Google Scholar]
- a Furuya T.; Kamlet A. S.; Ritter T. Catalysis for Fluorination and Trifluoromethylation. Nature 2011, 473, 470–477. 10.1038/nature10108. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yang X.; Wu T.; Phipps R. J.; Toste F. D. Advances in Catalytic Enantioselective Fluorination, Mono-, Di-, and Trifluoromethylation, and Trifluoromethylthiolation Reactions. Chem. Rev. 2015, 115, 826–870. 10.1021/cr500277b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Schöllkopf U.; Hartwig W.; Pospischil K. H.; Kehne H. Asymmetric Synthesis via Heterocyclic Intermediates; VII. Enantioselective Synthesis of (R)-α-Amino Acids Using (S)-O,O-Dimethyl-α-Methyldopa as Chiral Auxiliary Reagent. Synthesis 1981, 1981, 966–969. 10.1055/s-1981-29661. [DOI] [Google Scholar]; b Ito Y.; Sawamura M.; Kobayashi M.; Hayashi T. Asymmetric Aldol Reaction of α-Isocyanoacetamides with Aldehydes Catalyzed by a Chiral Ferrocenylphosphine-Gold(I) Complex. Tetrahedron Lett. 1988, 29, 6321–6324. 10.1016/S0040-4039(00)82336-9. [DOI] [Google Scholar]; c Schöllkopf U.; Groth U.; Gull M. -R.; Nozulak J. Asymmetrische Synthesen über heterocyclische Zwischenstufen, XVIII. Zur enantioselektiven Synthese von (2R)-Serin-methylestern oder (2R)-Serinen ausgehend vom Bislactimether von cyclo-(-L-Val-Gly-). Liebigs Ann. Chem. 1983, 1983, 1133–1151. 10.1002/jlac.198319830706. [DOI] [Google Scholar]
- a Bhagavan N. V.Amino Acids. In Medical Biochemistry, 4th ed.; Academic Press: Cambridge, MA, 2002; pp 17–33. [Google Scholar]; b Xu S.; Nilles J. M.; Bowen K. H. Zwitterion Formation in Hydrated Amino Acid, Dipole Bound Anions: How Many Water Molecules Are Required?. J. Chem. Phys. 2003, 119, 10696–10701. 10.1063/1.1620501. [DOI] [Google Scholar]
- Rumble J. R.; Bruno T. J.; Doa M.. CRC Handbook of Chemistry and Physics, 101st ed.; Rumble J. R., Ed.; CRC Press: Boca Raton, 2020. [Google Scholar]
- De Levie R. A General Simulator for Acid-Base Titrations. J. Chem. Educ. 1999, 76, 987–991. 10.1021/ed076p987. [DOI] [Google Scholar]
- Roberts G. C. K.; Jardetzky O. Nuclear Magnetic Resonance Spectroscopy of Amino Acids, Peptides, and Proteins. Adv. Protein Chem. 1970, 24, 447–545. 10.1016/S0065-3233(08)60246-6. [DOI] [PubMed] [Google Scholar]
- Mancini G.; Fusè M.; Lazzari F.; Chandramouli B.; Barone V. Unsupervised Search of Low-Lying Conformers with Spectroscopic Accuracy: A Two-Step Algorithm Rooted into the Island Model Evolutionary Algorithm. J. Chem. Phys. 2020, 153, 124110 10.1063/5.0018314. [DOI] [PubMed] [Google Scholar]
- Chandramouli B.; Del Galdo S.; Fusè M.; Barone V.; Mancini G. Two-Level Stochastic Search of Low-Energy Conformers for Molecular Spectroscopy: Implementation and Validation of MM and QM Models. Phys. Chem. Chem. Phys. 2019, 21, 19921–19934. 10.1039/C9CP03557E. [DOI] [PubMed] [Google Scholar]
- Nishizono N.; Akama Y.; Agata M.; Sugo M.; Yamaguchi Y.; Oda K. Synthesis of Thietane Nucleoside with an Anomeric Hydroxymethyl Group. Tetrahedron 2011, 67, 358–363. 10.1016/j.tet.2010.11.038. [DOI] [Google Scholar]
- Ito Y.; Matsuura T.; Saegusa T. ZnCl2 and CuCl Promoted Aldol Reactions of Isocyanoacetate with α,β-Unsaturated Carbonyl Compounds. Tetrahedron Lett. 1985, 26, 5781–5784. 10.1016/S0040-4039(00)98925-1. [DOI] [Google Scholar]
- Stewart J. J. P. Optimization of Parameters for Semiempirical Methods VI: More Modifications to the NDDO Approximations and Re-Optimization of Parameters. J. Mol. Model. 2013, 19, 1–32. 10.1007/s00894-012-1667-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Becke A. D. A New Mixing of Hartree-Fock and Local Density-Functional Theories. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]; b Lee C.; Yang W.; Parr R. G. Phys. Rev. B 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Cossi M.; Rega N.; Scalmani G.; Barone V. Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model. J. Comput. Chem. 2003, 24, 669–681. 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]
- Grimme S. Semiempirical Hybrid Density Functional with Perturbative Second-Order Correlation. J. Chem. Phys. 2006, 124, 034108 10.1063/1.2148954. [DOI] [PubMed] [Google Scholar]
- Biczysko M.; Panek P.; Scalmani G.; Bloino J.; Barone V. Harmonic and Anharmonic Vibrational Frequency Calculations with the Double-Hybrid B2PLYP Method: Analytic Second Derivatives and Benchmark Studies. J. Chem. Theory Comput. 2010, 6, 2115–2125. 10.1021/ct100212p. [DOI] [PubMed] [Google Scholar]
- Papajak E.; Zheng J.; Xu X.; Leverentz H. R.; Truhlar D. G. Perspectives on Basis Sets Beautiful: Seasonal Plantings of Diffuse Basis Functions. J. Chem. Theory Comput. 2011, 7, 3027–3034. 10.1021/ct200106a. [DOI] [PubMed] [Google Scholar]
- Fusè M.; Mazzeo G.; Longhi G.; Abbate S.; Masi M.; Evidente A.; Puzzarini C.; Barone V. Unbiased Determination of Absolute Configurations by Vis-à-Vis Comparison of Experimental and Simulated Spectra: The Challenging Case of Diplopyrone. J. Phys. Chem. B 2019, 123, 9230–9237. 10.1021/acs.jpcb.9b08375. [DOI] [PubMed] [Google Scholar]
- Paoloni L.; Mazzeo G.; Longhi G.; Abbate S.; Fusè M.; Bloino J.; Barone V. Toward Fully Unsupervised Anharmonic Computations Complementing Experiment for Robust and Reliable Assignment and Interpretation of IR and VCD Spectra from Mid-IR to NIR: The Case of 2,3-Butanediol and Trans-1,2-Cyclohexanediol. J. Phys. Chem. A 2020, 124, 1011–1024. 10.1021/acs.jpca.9b11025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, revision C.01; Gaussian, Inc.: Wallingford CT, 2016.
- Lazzari F.; Salvadori A.; Mancini G.; Barone V. Molecular Perception for Visualization and Computation: The Proxima Library. J. Chem. Inf. Model. 2020, 60, 2668–2672. 10.1021/acs.jcim.0c00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W.; Cheeseman J. R.; Frisch M. J. Calculation of Nuclear Spin–Spin Coupling Constants of Molecules with First and Second Row Atoms in Study of Basis Set Dependence. J. Chem. Theory Comput. 2006, 2, 1028–1037. 10.1021/ct600110u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.