

Abstract
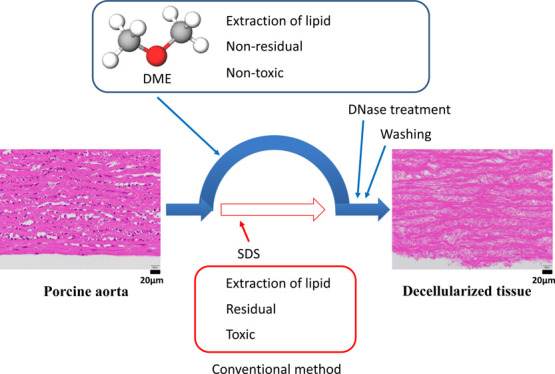
Porcine aortic tissue was decellularized by subcritical dimethyl ether (DME) used as an alternative to the surfactant sodium dodecyl sulfate. The process included three steps. For the first step, lipids were extracted from the porcine aorta using subcritical DME at 23 °C with a DME pressure of 0.56 MPa. Next, DME was evaporated from the aorta under atmospheric pressure and temperature. The second step involved DNA fragmentation by DNase, which was primarily identical to the common method. For the third step, similar to the common method, DNA fragments were removed by washing with water and ethanol. After 3 days of DNase treatment, the amount of DNA remaining in the porcine aorta was 40 ng/dry-mg, which was lower than the standard value of 50 ng/mg-dry. Hematoxylin and eosin staining showed that most cell nuclei were removed from the aorta. These results demonstrate that subcritical DME eliminates the need to utilize surfactants.
Introduction
Currently, organ transplantation is a treatment method used for severe organ failure. Organ transplantation is complicated by many ethical issues, a lack of transplantable organs, and rejection due to immunity. Therefore, to overcome these obstacles, the patient’s own cells can be cultured three-dimensionally in a scaffold to create an organ that does not cause rejection. Decellularization of porcine tissue to create a scaffold is expected to solve the problem of donor deficiency as the porcine aorta is similar in tissue structure to the human aorta and is suitable for transplantation in humans.
Decellularization methods are classified into chemical methods, such as treatment with an acid or alkali, detergent, or enzyme digestion.1−5 The most widely used detergent treatments have the advantage of being easy to use. Typical detergent treatment includes three steps: extraction of lipids by sodium dodecyl sulfate (SDS), DNA fragmentation by DNase, and removal of DNA fragments by washing with water and ethanol. For tissues, SDS has a very strong degreasing action. However, SDS has been found to cause inflammation at the point of contact. In addition, it has an extremely high affinity for proteins and can cause protein denaturation. However, complete removal of toxic surfactants from the tissue is difficult even after repeated washing since they show high affinity to the extracellular matrix.6 If SDS remains in the scaffold, there is a concern that cell cultures in the scaffold will be inhibited or that function may be impaired in vivo following transplantation; therefore, SDS should be removed completely following decellularization treatment. Moreover, SDS significantly damages the extracellular matrix, which is composed of proteins. In summary, the drawbacks of SDS include a long treatment time, alteration of mechanical properties, and residual toxicity.7−15 Decellularization without SDS is difficult, whereas high-hydrostatic pressure (HHP) technology can achieve almost complete decellularization of porcine blood vessels and corneal tissue.8,16 However, the HHP technology is limited by the need for special equipment that can withstand extremely high pressures over 600 MPa. There is also a decellularization method using supercritical carbon dioxide.6,17 In this method, supercritical carbon dioxide is saturated with ethanol, an entrainer, to remove phospholipids, thereby avoiding the use of SDS. However, collagen and other proteins can be cross-linked by alcohol, so it is necessary to consider the concentration and type of alcohol to be used as well as surfactants. Furthermore, the use of alcohol in substances that will ultimately be implanted in the human body may not be desirable for religious reasons.
Apart from the above methods, we and other researchers have succeeded in extracting lipids and functional materials from various wet microalgae, sludge, and plants using subcritical dimethyl ether (DME).18−30 Subcritical DME extracted both polar and nonpolar lipids. In the case of microalgae, the quantity of lipids extracted by DME was almost identical to that extracted by a methanol and chloroform mixture used for measuring the total amount of lipids in living tissues. In the case of Euglena sp., which, like animals, has no cell wall, the molecular weight distribution of the lipids extracted by subcritical DME was the same as those obtained using hexane.18 Therefore, in the absence of a cell wall, all lipids were extracted completely. Furthermore, DME has the advantage of not remaining in the extraction residue.
These results are due to the unique chemical and physical properties of DME. Although DME is the simplest form of ether, its characteristics are very different from those of other ethers such as di“e”thyl ether. The standard boiling point of DME is very low at −24.8 °C.31 Due to this low boiling point, very little DME remains in the extraction residue. DME is a weak polar solvent and partially mixes with water.32,33 Therefore, the samples in the previous studies did not require drying as pretreatment for extraction. European Food Safety Authority recognizes DME as a safe food processing solvent and considers DME to be of no safety concern when used as an extraction solvent under the intended conditions of use and with the proposed residual limits of 3 mg/kg in defatted protein products and 9 μg/kg specifically in gelatin.34 In the United States, DME is used as a propellant to coat the surface of sweets with chocolate.35 In addition, as shown in the Supporting Information of this study, bioassays have confirmed that DME dissolved in water is not toxic to microorganisms. Moreover, it exhibits resistance to autoxidation, unlike other alkyl ethers.36
Based on these findings, it was conceived that DME could be used instead of SDS to extract lipids from wet porcine tissues and that DME would not remain in the tissues. In this study, lipid extraction using DME was attempted to prepare decellularized porcine aortic tissues. After the aorta was decellularized by DME extraction and DNase treatment, the success of decellularization was assessed. This paper focuses specifically on the preparation of decellularized porcine aortic tissues without SDS using the DME extraction technique.
The criteria required for decellularized tissue are as follows: no visible cell nuclei are seen on hematoxylin and eosin staining. The amount of residual DNA should be less than 50 ng/mg-dry. Fragment of residual DNA should be less than 200 bp. In this study, we will determine whether decellularized tissues generated using DME meet these criteria.5,37
Results and Discussion
Lipid Extraction by Subcritical DME
Figure 1 shows the efficiency of lipid extraction from porcine aortas using DME as 1.65 wt % lipids were extracted with subcritical DME 150 times the dry weight of the aorta. The porcine aorta was removed from the extraction column after the amount of extracted lipids reached a constant level. At that time, 90.1% of the water contained in the initial aorta was also extracted and removed. Figure 2 reveals the appearance of the porcine aorta before and after treatment with subcritical DME; after treatment, the aorta becomes white and dry.
Figure 1.

Amount of lipids extracted by subcritical DME extraction from porcine aortas.
Figure 2.
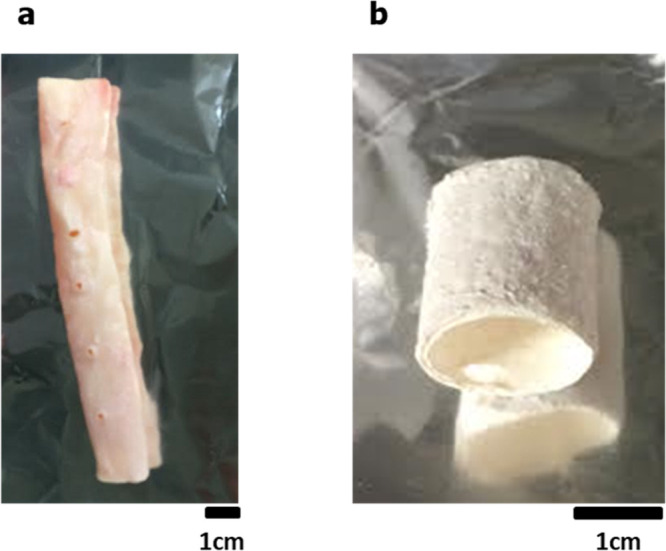
Porcine aorta before (a) and after (b) subcritical DME treatment.
Residual DME in Lipid Extracted Tissue
Figure 3 shows the results of a gas chromatography/mass spectrometry (GC/MS) measurement of the residue in the DME-treated sample. Some peaks were detected, but the areas were very small and those detected peaks were of hexamethyl cyclotrisiloxane, which is completely unrelated to this experiment. It is thought that substances used in liquid chromatography in the same room, or impurities in the column, were detected. Thus, no residual DME was detected in the aorta using a headspace GC/MS because DME was depressurized to atmospheric pressure and completely evaporated when the aorta was removed from the extraction column. In previous studies, when subcritical DME was applied to extract lipids from microalgae and macroalgae,23,38 no DME was detected in the extraction residues, and the present results are consistent with this. Thus, instead of SDS, subcritical DME was able to remove lipids from the porcine aorta.
Figure 3.

GC spectra of air in a closed vial with DME-treated porcine aorta.
FTIR Spectra
The Fourier transform infrared spectroscopy (FTIR) spectra obtained from the original and the DME-extracted porcine aortas are shown in Figure 4. Both of original (red curve) and DME-extracted (green curve) porcine aortas show the typical specific amide bands of proteins, which was almost the same as porcine gelatine shown in a previous study.39 The amide I band peaking at 1633 cm–1 is due to C=O stretching vibration, while the amide II band at 1548 cm–1 is assigned to C–N stretching and N–H bending vibration.39 In the case of the original porcine aorta, the presence of a large peak at 3280 cm–1 due to the OH group of the adsorbed water is characteristic. In the porcine aorta after DME extraction, this large peak disappears due to the removal of water, and therefore, the peaks at 2918 and 2850 cm–1, which were buried under the large peak in the red curve, appear as inflection points in the black curve. Absorptions at around 2918 and 2850 cm–1 come from the C–H group vibration of protein, which is commonly found in any original porcine and bovine gelatines.39 In the DME-extracted porcine aortas, the same characteristics as gelatine with a cross-linked structure were observed at 1736 cm–1.40 The absorbance is due to C=N stretching vibration caused by Schiff-base reactions, which is formed between amino groups and carbonyl groups of the gelatine as a result of the dehydration reaction.40
Figure 4.

FTIR spectra of porcine aorta before (a), after (b), and rewetting after (c) subcritical DME treatment.
Here, FTIR spectra were also measured on porcine aorta that had been wetted with water as they then undergo a DNase solution treatment and washing process, so they eventually become wet after DME extraction. The FTIR spectrum of DME-extracted porcine aorta after wetting shows that the absorbance at 1736 cm–1 due to C=N has completely disappeared, as shown in the blue curve in Figure 4, and is almost completely consistent with that of the original porcine aorta. This result indicates that although the extraction of fats and oils with DME results in temporary cross-linking due to dehydration of gelatine, the reaction is reversible and the cross-linking is eliminated by rewetting, so that no chemical change occurs from the original porcine aorta.
Hematoxylin–Eosin Staining
Following treatment with DNase for various periods and washing, the decellularized porcine aortic tissues were evaluated by hematoxylin–eosin staining (Figure 5). Compared with the untreated porcine aortic tissue, as shown in Figure 5a, more cell nuclei were visible in samples treated with only subcritical DME extraction (Figure 5b). After 1 day of DNase treatment (Figure 5c), almost all cell nuclei were removed as few were observed. As shown in Figure 5d–f, no cell nuclei were observed following DNase treatment for 3–7 days after DME extraction, clearly indicating that the cell nuclei were completely removed. These results clarified that although subcritical DME does not remove cell nuclei, it can remove the cell nuclei without using SDS when combined with DNase treatment and washing. The DNase treatment time after DME extraction was around 1–3 days, which is the borderline between the success or failure of cell nucleus removal. In previous studies of decellularization of porcine annulus fibrosus using SDS, collagen fiber fractures occurred when SDS was used, but no such collagen fiber fractures were observed in the samples prepared in this study.41
Figure 5.

Hematoxylin–eosin staining: (a) untreated. (b) DME extraction only. (c–f) DNase treatment for 1 (c), 3 (d), 5 (e), and 7 (f) days following DME extraction.
Quantification of Residual DNA
An example of UV spectra is shown in Figure 6, where the 260 nm/280 nm absorbance ratio is approximately 2/1, a condition that removes most of the protein and allows correct quantification of the DNA.42 The amount of residual DNA in the DME-extracted samples with or without further DNase treatment and washing is shown in Figure 6. In the case of DME extraction alone, the amount of residual DNA was 1704 ng/mg-dry, which was slightly decreased from the control of 2295 ng/mg-dry. In other words, subcritical DME may have some ability to extract and remove DNA with low efficacy. For decellularization, DME extraction must be combined with DNase treatment and washing. The amount of residual DNA after 1 day of DNase treatment was 32% compared to the control, indicating that DNA was easily removed and degraded rapidly. After 3 days of DNase treatment, the amount of residual DNA was 40 ng/mg-dry; this was below the target value of 50 ng/mg-dry,43−46 which is favorable for decellularization. However, as shown in Figure 7, error is present when measuring the amount of the DNA residue; therefore, 5 day DNase treatment is desirable to ensure that the result is below the regulation. After 7 days of DNase treatment, the residual DNA amount was 8 ng/mg-dry, and almost complete removal of DNA was achieved.
Figure 6.

UV spectra for quantification of residual DNA.
Figure 7.

Residual DNA amounts in the porcine aortas.
DNA Fragment Distribution
The distribution of DNA fragments remaining in the samples detected by agarose gel electrophoresis is shown in Figure 8. The gel lanes are as follows: the leftmost lane indicates the DNA fragment length standards; (a) original porcine aorta sample; (b) sample subjected to lipid removal by DME; (c) DME followed by DNase treatment for 1 day; and (d–f), DME followed by DNase treatment for 3, 5, and 7 days, respectively.
Figure 8.

Fragments of residual DNA in the samples detected by agarose gel electrophoresis: (a) untreated. (b) DME extraction only. (c–f) DNase treatment for 1 (c), 3 (d), 5 (e), and 7 (f) days following DME extraction.
As shown in Figure 8a,b, in the original sample and DME-treated samples, DNA was detected in the wide range from >1000 to <200 bp. In other words, subcritical DME was incapable of fragmenting DNA. The luminescence intensity of the DME-treated samples was higher than that of the original sample. Since DME has the ability to cleave hydrogen bonds,47 it is possible that DME cleaves the hydrogen bonds between the phosphate groups of the DNA and the surrounding polar material, leaving the phosphate groups of the DNA free to more easily bind to the fluorescent dye. When treated with DNase for 1 day after DME extraction, no DNA was detected, revealing a complete degradation to <100 bp, as shown in (c). Also, when DNase was administered for longer periods, no DNA was detected, as shown in (d–f). That is, although 1 day of DNase treatment was sufficient to fragment DNA to <200 bp, the results of residual fragmented DNA, as shown in Figure 7, suggest that further fragmentation by extended DNase treatment is required to remove the fragmented DNA by washing. The result of DNA fragmentation is consistent with the result of hematoxylin–eosin staining. In other words, the combination of lipid extraction by subcritical DME and DNA fragmentation by DNase ensures that the porcine aorta can be decellularized without the use of SDS.
In the future, it will be necessary to clarify whether the decellularization method using subcritical DME can be applied to tissues other than the aorta. Whether cells can safely grow in scaffolds created with subcritical DME should also be examined. Furthermore, the mechanical properties of the decellularized tissue may have been altered as the collagen was temporarily cross-linked by the subcritical DME, so its mechanical properties should also be investigated.
Conclusions
Subcritical DME extracted lipids from porcine aorta, after which DME had evaporated at room temperature due to its low boiling point and therefore did not remain in the aorta. DNA was not fragmented by DME extraction and was completely fragmented to <200 bp by DNase treatment for 1 day. After 3 days of DNase treatment, the amount of DNA remaining in the porcine aorta was 40 ng/dry-mg, which was lower than the target value of 50 ng/mg-dry. DNase treatment for 7 days after DME extraction resulted in a more complete removal of DNA. Furthermore, hematoxylin and eosin staining showed that most cell nuclei were removed from the porcine aorta. This means that after a few tens of minutes of lipid extraction with subcritical DME, porcine aortas can be decellularized by DNase treatment for at least 5 days and washing. These results show that introducing subcritical DME into the conventional method of decellularization of porcine aortas eliminates the need to utilize surfactants.
Materials and Methods
Materials
Fresh porcine aortas were obtained from a local slaughterhouse (Tokyo Shibaura Organ Co. Ltd., Tokyo, Japan). The pig aorta used in the paper is not from pigs slaughtered specifically for this experiment but from pigs slaughtered for meat processing. The aortas were excised and cut into 3 cm sections. The water content of porcine aortas was 60.2 wt %, which was determined by the weight difference before and after heating at 107 °C until the weight remained constant. Lipids were trimmed with a knife from wet aortic tissue and stored immediately at 4 °C in phosphate-buffered saline without Ca2+ or Mg2+ for transport to the laboratory for further processing.
Decellularization by DME
The decellularization protocol comprised three steps: (1) lipid extraction by subcritical DME, (2) DNase treatment, and (3) washing. The main difference between this method and the conventional method is the first step, which uses subcritical DME instead of SDS. Steps two and three are identical to those of the conventional decellularization method using SDS. The series of steps from lipid extraction to analysis were repeated three times to check their reproducibility.
Lipid Extraction by Subcritical DME
DME extraction was performed according to the following protocol, as described in a previous study of lipid extraction from microalgae.24 From a storage vessel filled with subcritical DME (volume: 500 mL; TVS-1-500, Taiatsu Techno Corporation, Saitama, Japan), subcritical DME was supplied to the extraction column. When the storage vessel was heated to 35 °C, the vapor pressure of saturated DME in the storage vessel increased. The liquified DME (Spray Work Air Can 420D) used for extraction was manufactured by Tamiya Incorporated (Shizuoka, Japan). Pressurized subcritical DME was pumped out from the storage vessel by its higher vapor pressure and cooled rapidly in a 1/16 in. SUS tube (1 m length) connected to the extraction column. The DME conditions were 23 °C and 0.56 MPa at the inlet of the extraction column. The DME flow rate was adjusted to 10 (±1) mL/min using a manual flow control valve (1315G4Y, Swagelok, Hyogo, Japan) attached to the outlet of the extraction column. 5.48 (±0.1) g of the wet aortic tissue was loaded into a 96 cm3 glass pressure vessel as the extraction column (cylindrical shape with a narrow lower end; inner diameter, 27.0 mm; length, 238 mm; customized HPG-96-3, Taiatsu Techno Corp.). Since DME flows from bottom to top, the voids around the aorta in the column were filled with cotton and glass beads to secure the aorta so that it would not be shaken in the extraction column by the flow force of the DME. In the extraction column, lipids and water were extracted by the subcritical DME. The outlet of the extraction column was connected to an empty 96 cm3 pressure vessel (HPG-96-3, Taiatsu Techno Corp.) via a connection SUS tube. The used subcritical DME flows into the empty pressure vessel, which is composed of transparent glass and has a volume memory printed similar to that of a measuring cylinder. The DME flow rate was controlled with the volume memory. When an appropriate amount (around 30 mL) of DME was stored in the vessel, the manual flow control valve was stopped, and then, the vessel was quickly replaced with a new vessel. The pressure-reducing valve of the old vessel was opened to reduce internal pressure. DME was evaporated by decompression, after which the extracted lipids and water remained in the vessel. Extracted water in the vessel was evaporated and separated from lipids by heating at 107 °C, and water and lipid weights were determined. Finally, after DME had flowed for 60 min, the extraction column was opened to evaporate any DME remaining in the extraction column, and then, the aorta was obtained.
DNase Treatment
The DNase treatment has been slightly modified based on a previous study.41 DNA was fragmented using 30 mL of DNase saline solution on 1.0 g of the DME-extracted porcine aorta. The saline solution was prepared with deionized water containing 0.9% NaCl and 1% penicillin and streptomycin (Thermo Fisher Scientific, Kanagawa, Japan). An enzyme solution was prepared by adding 0.2% DNase (Roche Diagnostics, Tokyo, Japan) and 0.05 mol/L MgCl2·6H2O (Wako, Osaka, Japan) to the saline. The prepared DNase solution was handled in a clean bench while avoiding contact with ambient air. After DME extraction, the porcine aorta was shaken in the DNase–saline solution at 4 °C for 1–7 days.
Washing
After DNase treatment, samples were washed with 80/20 (v/v) ethanol/saline for 1 h. Then, the samples were immersed in fresh 80/20 (v/v) ethanol/saline containing antibiotics and stored at 4 °C. Washing and storage were repeated daily for 3 days. Then, the samples were immersed in saline containing antibiotics at 4 °C for 1 day.
Analysis of Treated Tissue
Analysis of the treated tissues follows intact the typical methods of previous decellularization studies,48 except for the quantification of residual DME.
Quantification of Residual DME
The amount of DME remaining in the porcine aortas was detected by a GC/MS head space system according to the following protocol, as described in previous studies.23,38 One day after DME extraction, 0.10 g of the porcine aortas was placed in a 1 mL vial. GC/MS analysis was carried out using an Agilent 7890A GC system connected to an Agilent 5975C mass spectrometer with a silica capillary column [HP-5MS; 30 m × 0.25 mm (internal diameter) × 0.25 μm, Agilent Technologies Tokyo Ltd., Hachioji, Japan]. For GC, the oven temperature was initially set at 40 °C for 5 min, and then, it was allowed to increase to 260 °C at a rate of 5 °C min–1.
FTIR Spectra
To investigate the cross-linked structure of the DME-extracted aorta, FTIR spectra of the original aorta and the DME-extracted aorta were obtained using attenuated total reflection-FTIR (100 scans, PerkinElmer Spectrum Two, PerkinElmer Japan K.K., Yokohama, Japan). The DME-extracted aorta has had water removed, so the aorta with water added again was also measured.
Hematoxylin–Eosin Staining
The decellularized porcine aortas were stained with 1% hematoxylin–eosin. The slides were sliced and observed by an optical microscope.
Quantification of Residual DNA
DNA was removed and purified from the DME-extracted porcine aortas. First, 5 mg of DME-extracted porcine aortic tissue was mixed with 200 μL of proteinase K solution [the ratio of 1 M Tris-HCl aqueous solution (pH 7.8), 0.5 M ethylenediaminetetraacetic acid aqueous solution (EDTA, pH 8.0), proteinase K, water, and SDS = 1:2:2:95:0.5 v/v/v/v/w] in a microtube at 55 °C for 1 day and was prepared in a liquid-like state. Tris-HCl and EDTA were purchased from Nippon Gene Co., Ltd. (Tokyo, Japan). Proteinase K was purchased from Takara Bio Inc. (Kusatsu, Japan). DNA was removed from tissues by phenol/chloroform extraction and purified by ethanol precipitation.48
To quantify DNA, 1 μL of the TE buffer aqueous solution containing dissolved DNA was measured by UV spectrophotometry at 260 nm (NanoDrop microvolume spectrophotometer and fluorometer, Thermo Fisher Scientific, Kanagawa, Japan). No peak was observed at 265 nm corresponding to phenols. Furthermore, since the ratio of the intensity at 260 and 280 nm was approximately 2:1, it was confirmed that the protein could be removed almost completely. When the 260 nm/280 nm absorbance ratio is less than 2, the amount of residual DNA is less than the measured amount of DNA because contained protein was detected as DNA, which means that for the purpose of this study, the DNA removal is strictly judged.42
DNA Fragment Distribution
The fragment distributions of the DNA solutions obtained in the previous section were determined by an agarose gel electrophoresis system (Mini-Slab size electrophoresis system with integrated power supply, WSE-1150 PageRunAce, Atto Corporation, Tokyo, Japan). Precast polyacrylamide gels (EHR-R12.5L e-PAGEL HR) were employed with a gel/buffer composition of polyacrylamide/Tris-HCl buffer with 12.5% gel. DNA was separated at molecular weights ranging from 100 to >1000 bp. The DNA solution was mixed with loading dye buffer (WSE-7040 EzApply DNA, Atto Corporation) at a ratio of 5:1 (v/v). Then, the DNA fragments were dyed by a molecular weight marker (WSE-7030 EzDNA Ladder, Atto Corporation) and fluorescent stain reagent (WSE-7130 EzFluoroStain DNA, Atto Corporation).
Acknowledgments
This research was supported by Japan Society for the Promotion of Science KAKENHI (grant numbers 17K06922 and JP20K05192 to H.K.), Science and Technology Research Partnership for Sustainable Development Program of Japan Science and Technology Agency (JST), and Japan International Cooperation Agency (JICA) (grant number JPMJSA1505 to H.K.), Tatematsu Foundation, and Ito Foundation. The authors are grateful to Prof. Akio Kishida (Tokyo Medical and Dental University) for their kind help and constructive suggestions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c01549.
The authors declare no competing financial interest.
Supplementary Material
References
- Badylak S. F. Xenogeneic extracellular matrix as a scaffold for tissue reconstruction. Transplant Immunol. 2004, 12, 367–377. 10.1016/j.trim.2003.12.016. [DOI] [PubMed] [Google Scholar]
- McFetridge P. S.; Daniel J. W.; Bodamyali T.; Horrocks M.; Chaudhuri J. B. Preparation of porcine carotid arteries for vascular tissue engineering applications. J. Biomed. Mater. Res., Part A 2004, 70, 224–234. 10.1002/jbm.a.30060. [DOI] [PubMed] [Google Scholar]
- Ketchedjian A.; Jones A. L.; Krueger P.; Robinson E.; Crouch K.; Wolfinbarger L.; Hopkins R. Recellularization of decellularized allograft scaffolds in ovine great vessel reconstructions. Ann. Thorac. Surg. 2005, 79, 888–896. 10.1016/j.athoracsur.2004.09.033. [DOI] [PubMed] [Google Scholar]
- Rieder E.; Kasimir M.-T.; Silberhumer G.; Seebacher G.; Wolner E.; Simon P.; Weigel G. Decellularization protocols of porcine heart valves differ importantly in efficiency of cell removal and susceptibility of the matrix to recellularization with human vascular cells. J. Thorac. Cardiovasc. Surg. 2004, 127, 399–405. 10.1016/j.jtcvs.2003.06.017. [DOI] [PubMed] [Google Scholar]
- Rana D.; Zreiqat H.; Benkirane-Jessel N.; Ramakrishna S.; Ramalingam M. Development of decellularized scaffolds for stem cell-driven tissue engineering. J. Tissue Eng. Regener. Med. 2017, 11, 942–965. 10.1002/term.2061. [DOI] [PubMed] [Google Scholar]
- Sawada K.; Terada D.; Yamaoka T.; Kitamura S.; Fujisato T. Cell removal with supercritical carbon dioxide for acellular artificial tissue. J. Chem. Technol. 2008, 83, 943–949. 10.1002/jctb.1899. [DOI] [Google Scholar]
- Wu P.; Nakamura N.; Kimura T.; Nam K.; Fujisato T.; Funamoto S.; Higami T.; Kishida A. Decellularized porcine aortic intima-media as a potential cardiovascular biomaterial. Interact. Cardiovasc. Thorac. Surg. 2015, 21, 189–194. 10.1093/icvts/ivv113. [DOI] [PubMed] [Google Scholar]
- Funamoto S.; Nam K.; Kimura T.; Murakoshi A.; Hashimoto Y.; Niwaya K.; Kitamura S.; Fujisato T.; Kishida A. The use of high-hydrostatic pressure treatment to decellularize blood vessels. Biomaterials 2010, 31, 3590–3595. 10.1016/j.biomaterials.2010.01.073. [DOI] [PubMed] [Google Scholar]
- Sullivan D. C.; Mirmalek-Sani S.-H.; Deegan D. B.; Baptista P. M.; Aboushwareb T.; Atala A.; Yoo J. J. Decellularization methods of porcine kidneys for whole organ engineering using a high-throughput system. Biomaterials 2012, 33, 7756–7764. 10.1016/j.biomaterials.2012.07.023. [DOI] [PubMed] [Google Scholar]
- Keane T. J.; Swinehart I. T.; Badylak S. F. Methods of tissue decellularization used for preparation of biologic scaffolds and in vivo relevance. Methods 2015, 84, 25–34. 10.1016/j.ymeth.2015.03.005. [DOI] [PubMed] [Google Scholar]
- Crapo P. M.; Gilbert T. W.; Badylak S. F. An overview of tissue and whole organ decellularization processes. Biomaterials 2011, 32, 3233–3243. 10.1016/j.biomaterials.2011.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J. K.; Agnihotri A. K.; Titus J. S.; Torchiana D. F. A stentless trileaflet valve from a sheet of decellularized porcine small intestinal submucosa. Ann. Thorac. Surg. 2005, 80, 704–707. 10.1016/j.athoracsur.2004.08.063. [DOI] [PubMed] [Google Scholar]
- Korossis S. A.; Wilcox H. E.; Watterson K. G.; Kearney J. N.; Ingham E.; Fisher J. In-vitro assessment of the functional performance of the decellularized intact porcine aortic root. J. Heart Valve Dis. 2005, 14, 408–422. [PubMed] [Google Scholar]
- Gilbert T. W.; Sellaro T. L.; Badylak S. F. Decellularization of tissues and organs. Biomaterials 2006, 27, 3675–3683. 10.1016/j.biomaterials.2006.02.014. [DOI] [PubMed] [Google Scholar]
- Prasertsung I.; Kanokpanont S.; Bunaprasert T.; Thanakit V.; Damrongsakkul S. Development of acellular dermis from porcine skin using periodic pressurized technique. J. Biomed. Mater. Res., Part B 2008, 85, 210–219. 10.1002/jbm.b.30938. [DOI] [PubMed] [Google Scholar]
- Sasaki S.; Funamoto S.; Hashimoto Y.; Kimura T.; Honda T.; Hattori S.; Kobayashi H.; Kishida A.; Mochizuki M. In vivo evaluation of a novel scaffold for artificial corneas prepared by using ultra high hydrostatic pressure to decellularize porcine corneas. Mol. Vision 2009, 15, 2022–2028. [PMC free article] [PubMed] [Google Scholar]
- Gil-Ramírez A.; Rosmark O.; Spégel P.; Swärd K.; Westergren-Thorsson G.; Karin A.; Callerfelt L.; Rodríguez-Meizoso I. Pressurized carbon dioxide as a potential tool for decellularization of pulmonary arteries for transplant purposes. Sci. Rep. 2020, 10, 4031. 10.1038/s41598-020-60827-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda H.; Li P.; Goto M.; Makino H. Energy-saving lipid extraction from wet Euglena gracilis by low boiling point solvent dimethyl ether. Energies 2015, 8, 610–620. 10.3390/en8010610. [DOI] [Google Scholar]
- Kanda H.; Li P.; Ikehara T.; Yasumoto-Hirose M. Lipids extracted from several species of natural blue-green microalgae by dimethyl ether: extraction yield and properties. Fuel 2012, 95, 88–92. 10.1016/j.fuel.2011.11.064. [DOI] [Google Scholar]
- Kanda H.; Li P.; Yoshimura T.; Okada S. Wet extraction of hydrocarbons from Botryococcus braunii by dimethyl ether as compared with dry extraction by hexane. Fuel 2013, 105, 535–539. 10.1016/j.fuel.2012.08.032. [DOI] [Google Scholar]
- Kanda H.; Morita M.; Makino H.; Takegami K.; Yoshikoshi A.; Oshita K.; Takaoka M.; Morisawa S.; Takeda N. Deodorization and Dewatering of Biosolid by Using Dimethyl Ether. Water Environ. Res. 2011, 83, 23–25. 10.2175/106143010x12609736966847. [DOI] [PubMed] [Google Scholar]
- Li P.; Kanda H.; Makino H. Simultaneous production of bio-solid fuel and bio-crude from vegetal biomass using liquefied dimethyl ether. Fuel 2014, 116, 370–376. 10.1016/j.fuel.2013.08.020. [DOI] [Google Scholar]
- Kanda H.; Wahyudiono; Machmudah S.; Goto M. Direct extraction of lutein from wet macroalgae by liquefied dimethyl ether without any pretreatment. ACS Omega 2020, 5, 24005–24010. 10.1021/acsomega.0c03358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda H.; Hoshino R.; Murakami K.; Wahyudiono; Zheng Q.; Goto M. Lipid Extraction from Microalgae Covered with Biomineralized Cell Walls using Liquefied Dimethyl Ether. Fuel 2020, 262, 116590. 10.1016/j.fuel.2019.116590. [DOI] [Google Scholar]
- Subratti A.; Lalgee L. J.; Jalsa N. K. Liquified dimethyl ether (DME): A green solvent for the extraction of hemp (Cannabis sativa L.) seed oil. Sustainable Chem. Pharm. 2019, 12, 100144. 10.1016/j.scp.2019.100144. [DOI] [Google Scholar]
- Subratti A.; Lalgee L. J.; Jalsa N. K. Efficient extraction of black cumin (Nigella sativa L.) seed oil containing thymol, using liquefied dimethyl ether (DME). J. Food Process. Preserv. 2019, 43, e13913 10.1111/jfpp.13913. [DOI] [Google Scholar]
- Catchpole O. J.; Grey J. B.; Perry N. B.; Burgess E. J.; Redmond W. A.; Porter N. G. Extraction of Chili, Black Pepper, and Ginger with Near-Critical CO2, Propane, and Dimethyl Ether: Analysis of the Extracts by Quantitative Nuclear Magnetic Resonance. J. Agric. Food Chem. 2003, 51, 4853–4860. 10.1021/jf0301246. [DOI] [PubMed] [Google Scholar]
- Boonnoun P.; Shotipruk A.; Kanda H.; Goto M. Optimization of rubber seed oil extraction using liquefied dimethyl ether. Chem. Eng. Commun. 2019, 206, 746–753. 10.1080/00986445.2018.1522502. [DOI] [Google Scholar]
- Wang Q.; Oshita K.; Takaoka M.; Shiota K. Influence of water content and cell disruption on lipid extraction using subcritical dimethyl ether in wet microalgae. Bioresour. Technol. 2021, 329, 124892. 10.1016/j.biortech.2021.124892. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Oshita K.; Takaoka M. Effective lipid extraction from undewatered microalgae liquid using subcritical dimethyl ether. Biotechnol. Biofuels 2021, 14, 17. 10.1186/s13068-020-01871-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.; Zhou Y.; Lemmon E. W. An equation of state for the thermodynamic properties of dimethyl ether. J. Phys. Chem. Ref. Data 2011, 40, 023104. 10.1063/1.3582533. [DOI] [Google Scholar]
- Holldorff H.; Knapp H. Binary vapor-liquid-liquid equilibrium of dimethyl ether-water and mutual solubilities of methyl chloride and water: experimental results and data reduction. Fluid Phase Equilib. 1988, 44, 195–209. 10.1016/0378-3812(88)80111-0. [DOI] [Google Scholar]
- Tallon S.; Fenton K. The solubility of water in mixtures of dimethyl ether and carbon dioxide. Fluid Phase Equilib. 2010, 298, 60–66. 10.1016/j.fluid.2010.07.009. [DOI] [Google Scholar]
- Scientific Opinion on the safety of use of dimethyl ether as an extraction solvent under the intended conditions of use and the proposed maximum residual limits. EFSA J. 2015, 13, 4174. 10.2903/j.efsa.2015.4174. [DOI] [Google Scholar]
- Varlet V.; Smith F.; Augsburger M. New trends in the kitchen: propellants assessment of edible food aerosol sprays used on food. Food Chem. 2014, 142, 311–317. 10.1016/j.foodchem.2013.07.036. [DOI] [PubMed] [Google Scholar]
- Naito M.; Radcliffe C.; Wada Y.; Hoshino T.; Liu X.; Arai M.; Tamura M. A comparative study on the autoxidation of dimethyl ether (DME) comparison with diethyl ether (DEE) and diisopropyl ether (DIPE). J. Loss Prev. Process Ind. 2005, 18, 469–473. 10.1016/j.jlp.2005.07.001. [DOI] [Google Scholar]
- Crapo P. M.; Gilbert T. W.; Badylak S. F. An overview of tissue and whole organ decellularization processes. Biomaterials 2011, 32, 3233–3243. 10.1016/j.biomaterials.2011.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino R.; Ogawa M.; Murakami K.; Wahyudiono; Kanda H.; Goto M. Extraction of lipids from wet Arthrospira platensis by liquefied dimethyl ether. Solvent Extr. Res. Dev., Jpn. 2017, 24, 47–60. 10.15261/serdj.24.47. [DOI] [Google Scholar]
- Pradini D.; Juwono H.; Madurani K. A.; Kurniawan F. A preliminary study of identification halal gelatin using quartz crystal microbalance (QCM) sensor. Mal. J. Fund. Appl. Sci. 2018, 14, 325–330. 10.11113/mjfas.v14n3.942. [DOI] [Google Scholar]
- Kim S.; Kang Y.; Krueger C. A.; Sen M.; Holcomb J. B.; Chen D.; Wenke J. C.; Yang Y. Sequential delivery of BMP-2 and IGF-1 using a chitosan gel with gelatin microspheres enhances early osteoblastic differentiation. Acta Biomater. 2012, 8, 1768–1777. 10.1016/j.actbio.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.; Xu B.; Yang Q.; Li X.; Ma X.; Xia Q.; Zhang Y.; Zhang C.; Wu Y.; Zhang Y. Comparison of decellularization protocols for preparing a decellularized porcine annulus fibrosus scaffold. PLoS One 2014, 9, e86723 10.1371/journal.pone.0086723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasel J. A. Validity of nucleic acid purities monitored by 260nm/280nm absorbance ratios. Biotechniques 1995, 18, 62–63. [PubMed] [Google Scholar]
- Nagata S.; Hanayama R.; Kawane K. Autoimmunity and the clearance of dead cells. Cell 2010, 140, 619–630. 10.1016/j.cell.2010.02.014. [DOI] [PubMed] [Google Scholar]
- Manfredi A. A.; Capobianco A.; Bianchi M. E.; Rovere-Querini P. Regulation of dendritic- and T-cell fate by injury-associated endogenous signals. Crit. Rev. Immunol. 2009, 29, 69–86. 10.1615/critrevimmunol.v29.i1.30. [DOI] [PubMed] [Google Scholar]
- Brown B. N.; Valentin J. E.; Stewart-Akers A. M.; McCabe G. P.; Badylak S. F. Macrophage phenotype and remodeling outcomes in response to biologic scaffolds with and without a cellular component. Biomaterials 2009, 30, 1482–1491. 10.1016/j.biomaterials.2008.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Raoof M.; Chen Y.; Sumi Y.; Sursal T.; Junger W.; Brohi K.; Itagaki K.; Hauser C. J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda H.; Katsube T.; Hoshino R.; Kishino M.; Wahyudiono; Goto M. Ethanol-free antisolvent crystallization of glycine by liquefied dimethyl ether. Heliyon 2020, 6, e05258 10.1016/j.heliyon.2020.e05258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negishi J.; Funamoto S.; Kimura T.; Nam K.; Higami T.; Kishida A. Porcine radial artery decellularization by high hydrostatic pressure. J. Tissue Eng. Regener. Med. 2015, 9, E144–E151. 10.1002/term.1662. [DOI] [PubMed] [Google Scholar]
- Al-Awadhi H.; Al-Hasan R. H.; Radwan S. S. Comparison of the potential of coastal materials loaded with bacteria for bioremediating oily sea water in batch culture. Microbiol. Res. 2002, 157, 331–336. 10.1078/0944-5013-00168. [DOI] [PubMed] [Google Scholar]
- Banat I. M.; Hassan E. S.; El-Shahawi M. S.; Abu-Hilal A. H. Post-Gulf-War assessment of nutrients, heavy metal ions, hydrocarbons, and bacterial pollution levels in the United Arab Emirates coastal waters. Environ. Int. 1998, 24, 109–116. 10.1016/s0160-4120(97)00127-x. [DOI] [Google Scholar]
- Silva-Castro G. A.; Uad I.; Gónzalez-López J.; Fandiño C. G.; Toledo F. L.; Calvo C. Application of selected microbial consortia combined with inorganic and oleophilic fertilizers to recuperate oil-polluted soil using land farming technology. Clean Technol. Environ. Policy 2012, 14, 719–726. 10.1007/s10098-011-0439-0. [DOI] [Google Scholar]
- Yamamoto T.; Arakawa K.; Takahashi Y.; Sumiyoshi M. Antimicrobial activities of low molecular weight polymers synthesized through soap-free emulsion polymerization. Eur. Polym. J. 2018, 109, 532–536. 10.1016/j.eurpolymj.2018.10.047. [DOI] [Google Scholar]
- Balouiri M.; Sadiki M.; Ibnsouda S. K. Methods for in vitro evaluating antimicrobial activity: a review. J. Pharm. Anal. 2016, 6, 71–79. 10.1016/j.jpha.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa K.; Sugino F.; Kodama K.; Ishii T.; Kinashi H. Cyclization mechanism for the synthesis of macrocyclic antibiotic lankacidin in Streptomyces rochei. Chem. Biol. 2005, 12, 249–256. 10.1016/j.chembiol.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Yamamoto T.; Arakawa K.; Furuta R.; Teshima A. Antimicrobial activities of polymers synthesized through soap-free emulsion polymerization using a cationic initiator and styrene derivative monomers. Chem. Lett. 2018, 47, 1402–1404. 10.1246/cl.180762. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.