

Abstract
1,4,7,10-Tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid (DOTA) is a prominent chelating ligand used in imaging contrast agents and radiopharmaceuticals. The present study explores the stabilities, structures, and bonding properties of its complexes with trivalent actinides (Ac, U, Np, Pu, Am, Cm, Cf) using density functional theory and relativistic multireference calculations. For reference purposes, the La- and Lu-DOTA complexes are also included. Similar to La3+, the large An3+ ions prefer the TSAP conformer of the ligand. The An–ligand bonding is mainly electrostatic, with minor charge transfer contributions to the An 6d orbitals. For the assessment of the thermodynamic stabilities in aqueous solution, PCM radii to use in conjunction with the SMD solvation model were developed. Basically, the thermodynamic stability of the DOTA complexes increases along the An row but with notable counteracting of spin–orbit coupling.
Introduction
1,4,7,10-Tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid (DOTA)1,2 is a distinguished carrier of metal-based imaging contrast agents and radiopharmaceuticals3 on the basis of the high thermodynamic stabilities of these chelate complexes and their kinetic inertness in physiological conditions. Its compound with gadolinium is known as the MRI contrast agent Dotarem,4 while europium(III), ytterbium(III), and thulium(III) complexes with DOTA derivatives were considered as paramagnetic chemical exchange transfer (PARACEST) agents.5−7 DOTA was also tested with γ-emitting radiometals such as 99mTc and 111In for diagnostic tumor targeting.8 However, the most eminent recent application of this chelating agent emerged in targeted α-therapy (TAT).9,10
TAT is based on the short range and high energy of α-particles emitted from radioactive isotopes. By appropriate biological targeting vectors (antibody or peptide), the chelate complexes can be carried to and thus selectively destroy solid tumors and metastates.11−15 Prominent examples include the treatment of metastatic castration-resistant prostate cancer,16−18 acute myeloid leukemia,19,20 neuroendocrine tumors,21 and tumors expressing mesothelin.22 Presently, DOTA (or its derivative) is a primary choice for chelating 213Bi, 225Ac, and the β-emitting 177Lu isotopes used in radiotherapy.23,24 These agents passed successfully already the first clinical tests.24,25 Aiming for further improvement, the field is the subject of intense current research.9,10,23,24,26,27
New possibilities can emerge from the application of other actinide (An) radioisotopes. As the stability of DOTA complexes increases generally with decreasing metal size,28,29 the smaller An ions in the middle of the An row can be expected to form more stable complexes with DOTA compared to Ac3+. From the actinides, the medical application of 227Th is already at an advanced clinical test level with the HOPO chelator.10,18,30−33 The α-emitting 230U isotope, due to its short half-life of 20.83 days, can be a promising agent for TAT.34 However, a proper complexation of this An is challenging due to its reversible redox properties (a recent study of U-DOTA with axially bonded H2O and hydroxide found the UIV/UIII redox couple to be quasi-reversible35) and the under in vivo conditions very stable UO22+ form.36,37 Last but not the least, the slow neutron-emitting 252Cf isotope should be mentioned to be suitable for brachytherapy or internal radiation therapy.38,39
The aim of the present study is to comparatively analyze DOTA complexes with trivalent An (U, Np, Pu, Am, Cm, Cf) and to elucidate the trend in their stability and bonding properties. For this reason, the other frequent oxidation states of U, Np, and Pu are not included here. On the other hand, data of La-DOTA and Lu-DOTA are included for reference purposes. For these latter complexes, reliable experimental data on their structures40,41 and stability28,42,43 are available, which are used to validate the theoretical methods chosen for the present study.
The DOTA complexes of lanthanides (Ln) are well explored in the literature.2,28,42,44 In contrast, little is known about the An-DOTA complexes. Because of its TAT application, Ac-DOTA is the most known.45−51 In addition, there are a few recent studies on the stability constants of Am- and Cm-DOTA,47,52 water coordination in Cm-DOTA,47,52 and on the aqueous chemistry of An(IV)-DOTA complexes (An = Th, U, Np, Pu).35,53,54
Results and Discussion
Structure
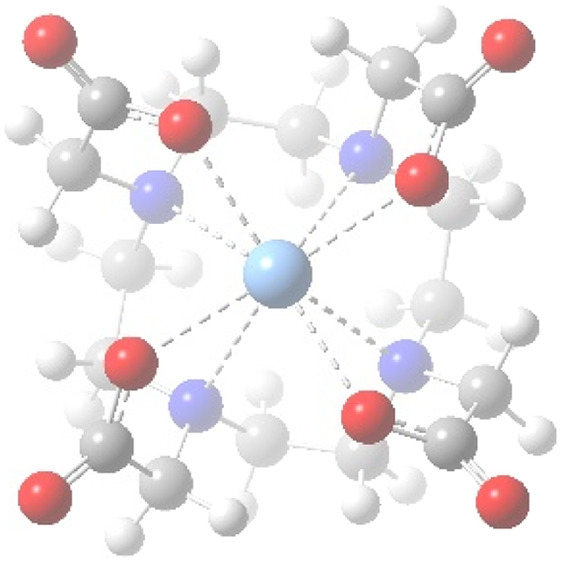
The DOTA ligand is a 12-membered tetraazamacrocycle containing four pendant carboxylate arms (Figure 1a). The structure of the chelate complex depends on the size of the metal centers.2 Large metal ions (lanthanides and heavy metals) are coordinated to the four N atoms of the cyclen ring and to the four O atoms from the COO– arms, leaving sufficient free space for a 9th coordination,40,41,55−60 e.g., to a H2O molecule (Figure 1b). Other metals (e.g., Ca, Bi, and Sc) were shown to have a coordination number of CN = 8,56,61,62 while the coordination of small transition metals is limited to the four N atoms and two or three carboxylates (CN = 6, 7).57,63−65
Figure 1.
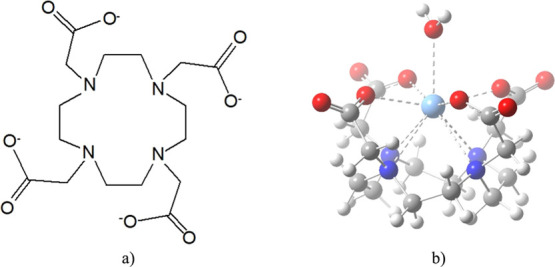
(a) DOTA4– ligand and (b) structure of the Ac(DOTA)(H2O)− complex.
The DOTA ligand can appear as two diastereoisomeric pairs of enantiomers in its octacoordinated complexes.44 One stereochemical element is the macrocycle, where the binding of the metal to the four N atoms fixes the four ethylene groups either in a δ- or a λ-gauche orientation (δδδδ or λλλλ, respectively). The four COO– groups represent the other stereochemical element and can form two opposite helical arrangements, Δ and Λ. Thus, the diastereoisomeric pairs are Λ(δδδδ)/Δ(λλλλ) where the ring and the arms have opposite helicities and Δ(δδδδ)/Λ(λλλλ) with the same ring and acetate helicity. The former conformers possess a square antiprismatic coordination geometry (designated as SAP or M in the literature), while the latter ones give a twisted square antiprismatic structure (designated as TSAP or m).
The conformational properties of Ln-DOTA complexes have been studied extensively. The crystal structures have been determined for the La,40 Ce, Pr, Nd, Dy, Ho,56 Eu,55,56 Gd,57,66 Tm,56 and Lu41 complexes. These studies revealed that the complexes from Pr to Lu (except for Tm) crystallize in the SAP geometry, while the complexes with the larger La and Ce crystallize in the TSAP geometry. The regular conformations and the switch between Ce and Pr were ascribed to the Ln3+-contraction, namely, how the Ln3+ ions match the different sizes and shapes of the cavities (determined primarily by the carboxylate arms) of the two DOTA conformers. The exceptional eight-coordinate TSAP crystal structure of Tm-DOTA was ascribed to the missing H2O ligand,56 which at the 9th coordination site supports the SAP geometry in the crystal. According to NMR studies, in solution, the Ln-DOTA complexes present mixtures of two conformers.67,68 The ratio of the SAP and TSAP conformers depends somewhat on the concentration of inorganic salts in the solution, temperature, and pressure, but essentially, the trend in the crystal state was found in solution, too. The H2O ligand at the 9th coordination site is involved in a rapid exchange process with the solvent H2O molecules, hence the observed C4 symmetry.43,69
Early quantum chemical calculations performing geometry optimization at the Hartree–Fock (HF) level70 and B3LYP single-point energy calculations on HF-optimized geometries71 reproduced the trend of the increasing SAP preference along the Ln row. The small free energy differences between the two conformers (within 10 kJ/mol in aqueous solution43) could quite well be reproduced at the B3LYP/6-311G**//HF/3-21 level using the C-PCM solvation model.72 From the actinides, theoretical results are available only for Ac-DOTA: in these recent DFT calculations modeling the aqueous solution, the TSAP conformer proved to be the most stable one.48,49 Accordingly, for the other large An3+ ions, the preference of the TSAP conformer can be expected.
The present TPSSh/TZ calculations using the SMD solvation model with PCM radii73,74 reproduced well the experimental ΔG°298 differences of the TSAP and SAP conformers for the La and Lu complexes43 (Figure 2). They are also in accord with previous reports on solvent effects enhancing the stability of the TSAP conformer in water:71,75 in the crystal, the switch from TSAP to SAP was observed between Ce and Pr,56 while in solution, it was observed between Nd and Sm.43 However, while being pleased about the shown performance of the present computational level, the possibility of fortuitous error cancellation of the theoretical models for this complex chemical system should be kept in mind. Nevertheless, the computed preference of the TSAP conformers for the large early actinides agrees with the expectations.
Figure 2.
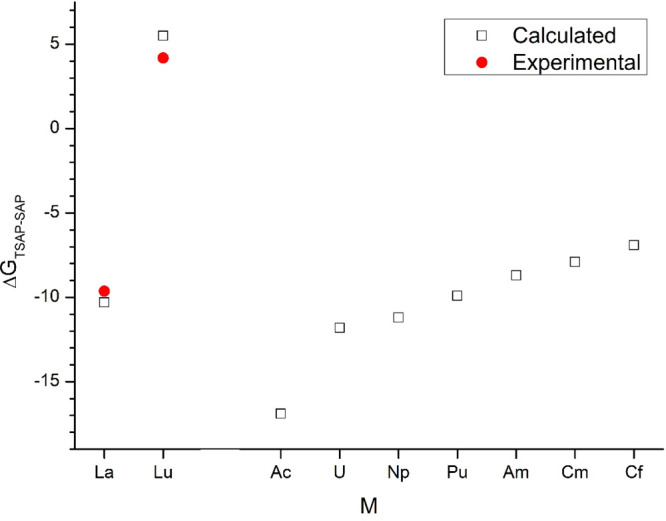
Calculated ΔG°298 differences (kJ/mol) of the TSAP and SAP conformers of M(DOTA)(H2O)− complexes. Calculated at the TPSSh/TZ geometries using the SMD solvation model in conjunction with PCM ionic radii.
On the other hand, the marked preference of the TSAP geometry for the Cf complex is somewhat unexpected in view of its size. The ionic radius of six-coordinate Cf3+ is 1.09 Å, comparable to the one of Sm3+ (1.098 Å).76 The Sm-DOTA complex is known to appear in the SAP form in aqueous solution.43,71 Hence, strictly on the basis of the ionic size from the literature, Cf3+ would be expected to prefer the SAP conformer, too. However, the abovementioned radii refer to six-coordinate metal ions, while in the M-DOTA complexes, they are nine-coordinate (which M3+ radii are not available). Furthermore, the bonding interactions of lanthanides can somewhat deviate from those of the actinides; hence, a comparison on the basis of ionic radii alone does not apply.
Based on the found preference of the TSAP conformer for actinide complexes, the following analyses are restricted on these isomers using the TPSSh/TZ optimized structures.
Figure 3 demonstrates an excellent correlation between the M3+ ionic radii76 and the computed M–O and M–N bond distances in the DOTA complexes. The H2O ligand at the 9th coordination site has a small influence on the M–DOTA interaction, as seen in the slight increase (0.03–0.04 Å) of the M–O and M–N bond distances upon coordination of H2O. The H2O ligand is bonded relatively weakly to the metal: the M–OH2 bonds are longer by 0.09–0.17 Å than the M–O bonds with DOTA. The main reason is the neutral nature of the H2O oxygen, in contrast to the anionic carboxylate oxygens of DOTA. Another effect is the probable electrostatic repulsion with the adjacent carboxylate arms (cf. Figure 1b).
Figure 3.
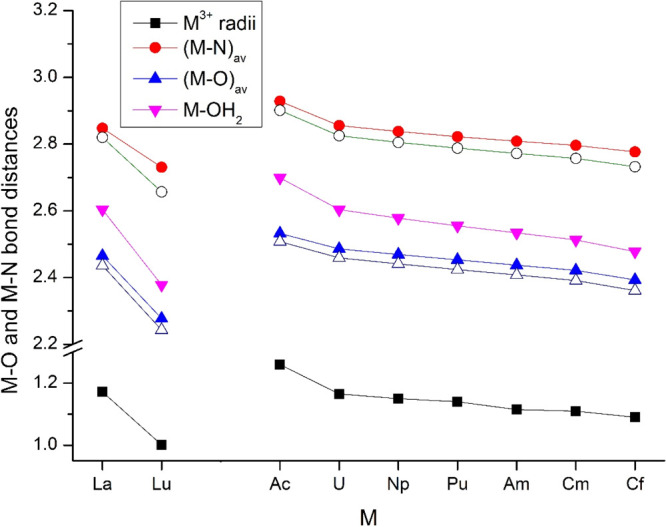
Comparison of M–O and M–N bond distances (Å) of M(DOTA)(H2O)− (filled symbols) and M(DOTA)− (empty symbols) complexes optimized at the TPSSh/TZ level as well as the M3+ ionic radii.76 The slightly differing values originating from the C2 symmetry of M(DOTA)(H2O)− structures are averaged.
Obviously, the M–O interaction with the anionic carboxylate oxygens is the strongest component of the M–DOTA bonding. The Wiberg bond indices77 are ca. 0.2 for this interaction (cf. Table S3 in the Supporting Information). The Wiberg indices for the M–N interactions are ca. 0.1, while those for the M–OH2 interaction are around 0.14, in agreement with the relations of the respective bond distances in Figure 3.
Relativistic Multireference Calculations
In order to verify the multiconfigurational nature and the role of the 5f subshell in the metal–ligand interactions, CASSCF calculations were performed on the An(DOTA)(H2O)− and An(DOTA)− TSAP structures. Selected characteristics of the spin–orbit (SO) and spin–orbit–free (SF) ground states are compiled in Table 1. In addition, the SO energies were utilized as correction terms in the evaluation of the thermodynamic stabilities (vide infra).
Table 1. Selected Characteristics of the Spin–Orbit (SO) and Spin–Orbit–Free (SF) Ground States of the An(DOTA)(H2O)− Complexes.
| An | statea | SO composition (%)b | SF composition (%)c |
|---|---|---|---|
| Ac | 1S0 | 1A (100) | 1A (100) |
| U | 4I9/2 | 4A (26) + 4B (25) + 4A (17) + 4B (14) | 4A (83) |
| Np | 5I4 | 5A (33) + 5A (29) | 5A (82) |
| Pu | 6H5/2 | 6B (29) + 6B (28) + 6A (15) | 6B (71) |
| Am | 7F0 | 6B (32) + 6B (31) | 6B (100) |
| Cm | 8S7/2 | 8A (100) | 8A (100) |
| Cf | 6H | -d | 7A (55) |
Term symbol of the An state.
Composition of the SO ground state from the SF states (lowest-energy ones from the C2 irreps).
Symmetry and contribution of the main electron configuration of the SF ground state.
The hyphen means that SO calculation could not be performed with the Cf pseudopotential in MOLCAS.
The electronic characters (term symbols) of An in the SO ground states of the complexes correspond to those of the free An3+ ions.78 The ligand splitting effects on the ground-state multiplet in An(DOTA)(H2O)− molecules in terms of SO-CASPT2 energies are given in Table S2 of the Supporting Information.
The SO ground states of Ac(DOTA)(H2O)− and Cm(DOTA)(H2O)− are composed entirely (100%) of the SF ground state. The other actinides form their SO ground state as mixtures of the lowest-energy SF states. In contrast, the SF ground states contain mostly one dominant (70–100%) electron configuration, the only exception being Cf(DOTA)(H2O)−. The major electron configurations of the SF ground states are generally strongly mixed and cannot be characterized by specific magnetic quantum numbers of the 5f electrons.
Bonding
Table 2 presents selected results from the QTAIM analysis of CASSCF wave functions of the M(DOTA)(H2O)− complexes. Additional data are given in Table S3 of the Supporting Information. As Cf was modeled with a different (pseudopotential) basis, the data of Cf(DOTA)(H2O)− should be considered cautiously in the trends.
Table 2. Selected Resultsa from the QTAIM Analysis of CASSCF Wave Functions of the M(DOTA)(H2O)− Complexes.
| M3+ | qM | CT1 | CT2 | DI1 | DI2 | DI3 | ρ(M–O) | ∇2ρ(M–O) |
|---|---|---|---|---|---|---|---|---|
| Ac | 2.54 | 0.44 | 0.02 | 0.25 | 0.15 | 0.13 | 0.048 | 0.19 |
| U | 2.46 | 0.51 | 0.03 | 0.26 | 0.17 | 0.14 | 0.051 | 0.21 |
| Np | 2.45 | 0.52 | 0.03 | 0.26 | 0.17 | 0.14 | 0.051 | 0.22 |
| Pu | 2.43 | 0.54 | 0.03 | 0.26 | 0.17 | 0.14 | 0.052 | 0.23 |
| Am | 2.42 | 0.55 | 0.03 | 0.26 | 0.17 | 0.14 | 0.052 | 0.24 |
| Cm | 2.42 | 0.55 | 0.03 | 0.26 | 0.17 | 0.14 | 0.053 | 0.25 |
| Cf | 2.39 | 0.58 | 0.03 | 0.26 | 0.17 | 0.14 | 0.053 | 0.24 |
| La | 2.48 | 0.50 | 0.02 | 0.25 | 0.15 | 0.13 | 0.050 | 0.19 |
| Lu | 2.45 | 0.52 | 0.03 | 0.23 | 0.14 | 0.10 | 0.055 | 0.29 |
Bader charge of M (qM, e); charge transfer to M from DOTA (CT1, e) and from H2O (CT2, e); delocalization indices between M–ODOTA (DI1, e), M – OH2O (DI2, e), and M–NDOTA (CT3, e), the ones with DOTA were averaged because of the C2 symmetry. Averaged electron density (ρ, au) and Laplacian of this electron density (∇2ρ, au) at the bond critical points of the M–ODOTA interactions. Additional topological data are given in Table S3.
The main bonding interaction between M and the ligands is the electrostatic attraction between the strongly positive M (Bader charges around +2.4 e) and the partially negative oxygens. Further evidences for this dominant ionic interaction are the very low electron densities (ρ) and the positive Laplacian values of ρ at the M–O and M–N bond critical points (BCPs) (cf. Table 2 and Table S3). The minor covalent part of the M–ligand bonding includes charge transfer interactions between M and Tp (CT) and energy-degeneracy-driven orbital overlap.79,80 The former interaction can quantitatively be estimated from the Bader charges: the net CT from DOTA to M amounts to ca. 0.5 e, while that from H2O to M amounts to 1 order of magnitude smaller (0.03 e). The considerable difference is determined by the anionic character and multiple number (four) of the DOTA carboxylate oxygen donors. The concept of energy-degeneracy-driven covalency is based on the mixing of metal and ligand orbitals on the basis of their close energies.79,80 Recently, an increasing trend of 5f and ligand orbital mixing has been shown in various An compounds79,81−85 due to decreasing 5f orbital energies across the An row. Unfortunately, the computational tools applied in the present study do not facilitate a straightforward analysis of energy-driven covalency in the title complexes.
The Bader charges of M correspond to a slightly decreasing ionic character along the An row (and between La and Lu). The same gentle trend appears also in the increasing electron densities at the BCPs.
Another important integral property is the delocalization index (DI), which corresponds to the number of electrons forming the covalent (donor–acceptor) bonding between M and the donor atoms of the ligands. The DI values of ca. 0.25, 0.16, and 0.14 e for M–ODOTA, M–OH2O, and M–NDOTA, respectively (cf. Table 2), correlate well with the Wiberg bond indices77 of ca. 0.21, 0.14 and 0.09 (given in detail in Table S3), respectively. As can be seen, despite the marginal charge transfer from the neutral H2O ligand to M, a significant number of electrons are participating in its orbital interactions with M.
Inspection of the CASSCF orbitals indicated that the 5f electrons occupy nonbonding orbitals mostly with pure 5f (in few cases with minor 6d contributions) character. 5f populations could not be recognized as notable contributors in any bonding orbitals. The lack of 5f orbital participation in the bonding in the An-DOTA complexes justifies the applicability of 5f-in-core pseudopotentials. The An–ligand bonding is manifested by the 6d orbitals of An that, due to their symmetries, are better suited to interact with the deformed C4-type DOTA ligand than 5f. It should be noted that there are no pure metal–ligand bonding molecular orbitals, but ones mixed with other intra-DOTA orbital interactions. The four characteristic molecular orbitals of the U(DOTA)− complex containing significant U–DOTA bonding are shown in Figure 4.
Figure 4.

Characteristic molecular orbitals of U(DOTA)− from CASSCF calculations: (a) U–O; (b) U–N; (c,d) U–O,N bonding.
Thermodynamics in Aqueous Solution
The relative thermodynamic stabilities are assessed with respect to the Lu-DOTA complexes according to the following reactions (utilizing the more stable SAP isomer for the Lu while the TSAP one for the other complexes):
| 1 |
| 2 |
Experimental data are available for DOTA complexes of La, Lu, Ac, Am, and Cm. For La- and Lu-DOTA, the values of the stability constants from potentiometric titration (lgK = 22.9 and 25.4, respectively)28 and from a spectrophotometric study (20.7 and 23.5, respectively)42 differ considerably due to the differences in the experimental conditions. However, the relative stabilities calculated from these two sets of data are in reasonable agreement (ΔG°298 = 14.3 and 16.0 kJ/mol, respectively). The stability constants of Am- and Cm-DOTA from solvent extraction experiments proved to be nearly the same; in fact, the difference is well within the experimental uncertainties (lgK = 23.95 ± 0.09 and 24.02 ± 0.11, respectively).52 The slightly larger value of Cm-DOTA is in agreement with the expected larger stability of the latter complex on the basis of ionic radii. A distinct decreasing trend in the stability along the An row was demonstrated by the thermodynamic data of Ac-DOTA and Cm-DOTA in ref (47) obtained from samples with various Cm3+:DOTA ratios using time-resolved laser fluorescence spectroscopy detection in the case of Cm-DOTA while separation of free Ac3+ and the complex by instant thin layer chromatography followed by detection using gamma spectroscopy in the case of Ac-DOTA. The lgK values (25 °C, I = 0.1 M, approximately comparable to the experimental conditions in ref (28)) were 16.4 and 19.0 for Ac- and Cm-DOTA, respectively. The reported corresponding ΔG°298 values are −93.9±2.1 and −108.3±13.7 kJ/mol, respectively.
The ΔG°298 values from the present TPSSh/DZ + SMD(aq) calculations are presented in Figure 5. The thermal contributions were calculated using the rigid rotor harmonic oscillator approximation and neglecting the electronic contributions. Because the electronic contributions are expected to be similar for the M3+ ions and the M-DOTA complexes, they should more or less cancel each other in eqs 1 and 2.
Figure 5.
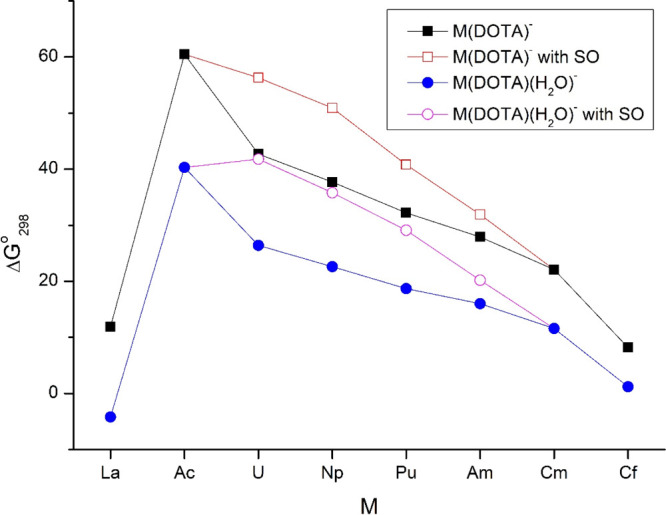
Relative thermodynamic stabilities of M(DOTA)− and M(DOTA)(H2O)− complexes on the basis of the exchange reaction M-DOTA(TSAP) + Lu3+ → Lu-DOTA(SAP) + M3+. The data given by unfilled symbols include spin–orbit coupling.
The value of 11.9 kJ/mol for La(DOTA)− from eq 1 is in good agreement with the experimentally obtained stability difference between the La- and Lu-DOTA complexes (vide supra). In contrast, the −4.2 kJ/mol value for the La(DOTA)(H2O)− model (eq 2) would correspond to a higher stability of the La complex with respect to the Lu one, which is in contradiction with experimental observations.28,42 Hence, the M(DOTA)− model structures (eq 1) seem to be more appropriate for assessment of the relative stabilities in aqueous solution. The better performance of this model can also be related to the observed reversible bonding of the H2O molecule at the 9th coordination site, i.e., its rapid exchange with the solvent H2O molecules.43,69
The trend of increasing thermodynamic stabilities of the studied An-DOTA complexes along the An row (lower ΔG°298) is in agreement with the abovementioned limited experimental information.47,52 It should be noted that the SO corrections taken from the SO-CASPT2 calculations decrease the calculated relative stabilities: the reason is the larger stabilization of the An3+ ions than that of the respective complexes by the SO interaction. The SO effect could not be evaluated in the present study for the Cf-DOTA complexes, but it can be expected to increase its ΔG°298 value, too.
The calculated ΔG°298 difference of Ac(DOTA)− and Cm(DOTA)− is considerably overestimated compared to the experimental value from ref (47) (38.4 vs 14.4 ± 15.8 kJ/mol, vide supra), though the large experimental uncertainty smoothens the discrepancy. Part of the deviation may still stem from the neglect of electronic contribution in the calculated ΔG°298 data. While this contribution should be negligible for La3+, Lu3+, and Ac3+ due to the lack of low-lying electronic states, it may have some relevance for Cm3+ where the excitation of the 5f7 configuration results in medium-energy excited states.
Computational Details
The density functional theory (DFT) computations were performed with the Gaussian09 suite of programs.86 On the basis of the reported good experience with the TPSSh meta-hybrid functional87 for the molecular geometries and relative stabilities of various lanthanide complexes,73,74,88,89 this functional was considered at the first place in conjunction with the 4/5f-in-core (large-core) quasi-relativistic pseudopotentials of the Stuttgart–Cologne group (La-ECP46MWB, Lu-ECP60MWB,90,91 Ac-ECP78MWB, U-ECP81MWB, Np-ECP82MWB, Pu-ECP83MWB, Am-ECP84MWB, Cm-ECP85MWB, Cf-ECP87MWB92) and double-zeta valence basis sets ([7s6p5d]/[5s4p3d] for the lanthanides,90 [7s6p5d]/[4s3p3d] for the actinides,92 and 6-31G** for the light atoms). This DFT level is denoted in the paper as TPSSh/DZ. Comparative calculations were performed with a larger basis set of triple-zeta quality using contracted [8s7p6d3f3g]/[6s5p5d3f2g] valence basis sets for the lanthanides,90,93,94 [7s6p5d2f]/[5s4p4d2f] for the actinides,92 and 6-311+G** for the light atoms (denoted as TPSSh/TZ). In contrast to DZ, this basis set has f polarization functions, which can have relevance for the bonding and electronic properties, particularly of the An complexes. Calculations with 5f-in-valence (small-core) pseudopotentials (ECP60MWB)95,96 failed because of SCF convergence problems already in the case of the Ac complex (with the simplest An electronic structure).
The performance of the above DFT levels was tested on the experimental X-ray diffraction structural data of the Lu- and La-DOTA complexes40,41 taking advantage of their significantly different Ln3+ ion radii and the covered two DOTA conformers.44 In addition, some other DFT functionals (B3PW91,97,98 BH&HLYP,99 B3LYP,97,100 BP86,101,102 PBE,103 M062X,104 and wB97XD105) and basis sets for the light atoms (6-31+G**, cc-pVDZ, cc-pVTZ) were also probed.
The model structure selected for the present study required some considerations. The Lu-DOTA complex, similar to the other late small-sized Ln3+ ions, has a H2O ligand in the first coordination sphere.41,55−58,66 In contrast, the nine-coordinate structure of solid La-DOTA (similar to Ce56) is formed by bridging of a carboxylate O atom of a neighboring complex molecule.40 Accordingly, La-DOTA has a polymeric structure in the crystal, yet the molecular structure of the La(DOTA)− unit is expected to be close to the one in the La(DOTA)(H2O)− species. Therefore, for the present theoretical study, the M(DOTA)(H2O)− model was used for both the La and An complexes. The H2O molecule was constrained in a vertical arrangement in order to facilitate C2 symmetry (being, e.g., advantageous for the multireference calculations). Though this structure is a second-order saddle-point on the potential energy surface, due to the very weak interaction, it is only slightly higher in energy than the global minimum. Moreover, this arrangement resembles more that in the crystal and polar solvents, where this H2O is involved rather in interactions with the environment than with the DOTA ligand in the same complex.
The computed La/Lu–O and La/Lu–N distances from the benchmark study are compiled in the Supporting Information (Table S1). The general feature of all tested levels is the underestimation of the M–O and overestimation of the M–N distances compared to the solid-phase data. Such deviations are understandable in view of the missing intermolecular interactions of both the carboxylate groups and the H2O ligand in the computed isolated molecules. The stronger M–O interactions in the isolated molecule pull the metal away from the N donors toward the free site of DOTA, i.e., closer to the carboxylate O donors. The larger underestimation of the Lu–Ow distance is in agreement with the larger effect of intermolecular interactions on the weakly bonded H2O ligand. The modeling of such effects is difficult: it required for solvated Gd(DOTA)(H2O)− a mixed cluster/continuum approach including explicitly two second-sphere water molecules.106
Nevertheless, the data confirm the superior performance of the TPSSh functional over the other probed DFT ones as well as that of the 6-311+G** basis set. Comparison of the TPSSh/DZ and TPSSh/TZ levels revealed a slightly better performance of TPSSh/TZ for the geometry. However, calculations of basis set superposition error (BSSE) using the counterpoise method107 failed for some complexes at the TPSSh/TZ level because of SCF convergence problems. There were no such problems with the smaller DZ basis; thus, the TPSSh/DZ level was used to assess the solution stability ratios (similar to previous studies on related complexes73,74,89) where the BSSE corrections were shown to play an important role.
The solvent effects were taken into account using the polarizable continuum model (PCM)108,109 with radii and nonelectrostatic terms for Truhlar and co-workers’ SMD solvation model.110 For La3+, Lu3+, Ac3+, and Cf3+, the PCM radii from the literature were used.73,74 For the other An3+ ions, no such radii are available; therefore, they were developed in the present study (Table 3) on the basis of literature hydration free energies.111 No scaling factor (α = 1.0) was applied for the PCM radii in the present study.
Table 3. PCM Radii (Å) of the M3+ Ions Used in Conjunction with the SMD Solvation Model.
The multiconfigurational nature of the complexes was studied by single-point relativistic complete active space self-consistent field (CASSCF)112 calculations in order to verify the ground electronic states and their character. For these single-point calculations, the TPSSh/TZ reference geometries were used taking advantage of their C2 symmetry.
The scalar relativistic effects were taken into account with the second-order Douglas–Kroll–Hess Hamiltonian.113,114 The used atomic natural orbital type all-electron basis sets developed for relativistic calculations (ANO-RCC) had the contraction schemes of [26s23p17d13f5g3h]/[8s7p5d3f1g] for Ac, U, Np, Pu, Am, and Cm,115 [8s4p3d1f]/[2s1p] for H,116 and [14s9p4d3f2g]/[3s2p1d] for C, N, and O117 corresponding to valence double-zeta plus polarization quality. As the ANO-RCC basis is not available for Cf, these calculations were performed using the small-core quasi-relativistic pseudopotential of the Stuttgart–Cologne group (ECP60MWB) in conjunction with a contacted [12s11p10d8f]/[8s7p6d4f] valence basis95,96 and Dunning’s cc-pVDZ basis118 for the light atoms.
In conformity with the large size of the complex, the active space consisted of the seven 5f orbitals occupied by the 5f electrons of the An3+ ions. In the state-averaged calculations, all the states of the ground-state spin multiplicities were considered, i.e., 34, 35, 21, 7, 1, and 21 roots for U, Np, Pu, Am, Cm, and Cf, respectively.
Dynamic electron correlation was considered by second-order perturbation theory calculations on the basis of the CASSCF wave functions (CASPT2).119,120 Spin–orbit (SO) effects were taken into account by using the complete active space state interaction (CASSI) method,121 which allows CASSCF wave functions for different electronic states to interact under the influence of a spin–orbit Hamiltonian. Dynamic electron correlation was represented by the CASPT2 energies used in the spin–orbit Hamiltonian (SO-CASPT2). The CASSI treatment was not applicable in the case of the quasi-relativistic pseudopotential of Cf. The above multireference calculations were performed by means of the MOLCAS 8.2 code.122,123
Bonding analysis was performed on the basis of the CASSCF wave functions. The molecular orbitals were visualized using the Luscus software.124 Bader atomic charges, delocalization indices, and topological properties in terms of Quantum Theory of Atoms in Molecules (QTAIM)125 were obtained with the Multiwfn code using medium quality grid.126 The wfn input files for the latter analyses were obtained with the Molden2AIM utility program127 from the Molden input files generated by MOLCAS.
Conclusions
The DOTA ligand forms stable complexes with the studied trivalent actinides (Ac, U, Np, Pu, Am, Cm, Cf). The thermodynamic stability in aqueous solution increases along the An row, with the ΔG°298 values without SO correction showing good correlation with the An3+ ionic radii. The SO effects stabilize the An3+ ions somewhat more than the An(DOTA)− complexes, worsening the correlation but still keeping the gradual decreasing trend. The increasing stability across the An row is in line with results on An complexes with other chelating ligands.79,81,83 The found trend supports the applicability of the DOTA ligand for heavy actinides.
A further characteristic of the solvated complexes is the preference of the TSAP conformers for all the An involved in the study. The above solution-phase results were achieved with PCM radii used in conjunction with the SMD solvation model, evaluated in the present study for U3+, Np3+, Pu3+, Am3+, and Cm3+.
The relativistic multireference calculations at the SO-CASPT2/DZ level confirmed the electronic ground states of the complexes to be the same as in the An3+ ions. Worthwhile, 5f populations were not found in the bonding molecular orbitals, justifying the applicability of 5f-in-core pseudopotentials. The An–ligand bonding is mainly electrostatic, with minor charge transfer contributions to the An 6d orbitals according to the (deformed) C4 symmetric nature of the complexes.
Acknowledgments
The author thanks Dr. A. Morgenstern for advice.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c01292.
Benchmark results using the X-ray diffraction reference data of La- and Lu-DOTA; SO-CASPT2 energy levels of the ground-state multiplets in An(DOTA)(H2O)− molecules (An = U, Np, Pu); selected Wiberg bond indices and electron density and Laplacian values of the electron density data at M–O and M–N bond critical points; input Gaussian09 file of the SMD solvation calculation on Pu(DOTA)− using the PCM radii of Pu; Cartesian coordinates of the M(DOTA)− and M(DOTA)(H2O)− complexes optimized at the TPSSh/TZ level (PDF)
The author declares no competing financial interest.
Supplementary Material
References
- De Lén-Rodŕguez L. M.; Kovacs Z. The Synthesis and Chelation Chemistry of DOTA-Peptide Conjugates. Bioconjugate Chem. 2008, 19, 391–402. 10.1021/bc700328s. [DOI] [PubMed] [Google Scholar]
- Viola-Villegas N.; Doyle R. P. The coordination chemistry of 1,4,7,10-tetraazacyclododecane-N,N’,N″,N’″-tetraacetic acid (H4DOTA): Structural overview and analyses on structure-stability relationships. Coord. Chem. Rev. 2009, 253, 1906–1925. 10.1016/j.ccr.2009.03.013. [DOI] [Google Scholar]
- Magerstädt M.; Gansow O. A.; Brechbiel M. W.; Colcher D.; Baltzer L.; Knop R. H.; Girton M. E.; Naegele M. Gd(DOTA): An alternative to Gd(DTPA) as a T1,2 relaxation agent for NMR imaging or spectroscopy. Magn. Reson. Med. 1986, 3, 808–812. 10.1002/mrm.1910030517. [DOI] [PubMed] [Google Scholar]
- Chapon C.; Lemaire L.; Franconi F.; Marescaux L.; Legras P.; Denizot B.; Le Jeune J.-J. Assessment of myocardial viability in rats: Evaluation of a new method using superparamagnetic iron oxide nanoparticles and Gd-DOTA at high magnetic field. Magn. Reson. Med. 2004, 52, 932–936. 10.1002/mrm.20210. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Trokowski R.; Sherry A. D. A Paramagnetic CEST Agent for Imaging Glucose by MRI. J. Am. Chem. Soc. 2003, 125, 15288–15289. 10.1021/ja038345f. [DOI] [PubMed] [Google Scholar]
- Chauvin T.; Durand P.; Bernier M.; Meudal H.; Doan B.-T.; Noury F.; Badet B.; Beloeil J.-C.; Tóth É. Detection of Enzymatic Activity by PARACEST MRI: A General Approach to Target a Large Variety of Enzymes. Angew. Chem., Int. Ed. 2008, 47, 4370–4372. 10.1002/anie.200800809. [DOI] [PubMed] [Google Scholar]
- Yoo B.; Pagel M. D. A PARACEST MRI Contrast Agent To Detect Enzyme Activity. J. Am. Chem. Soc. 2006, 128, 14032–14033. 10.1021/ja063874f. [DOI] [PubMed] [Google Scholar]
- Chen J.; Cheng Z.; Miao Y.; Jurisson S. S.; Quinn T. P. α-Melanocyte-stimulating hormone peptide analogs labeled with technetium-99m and indium-111 for malignant melanoma targeting. Cancer 2002, 94, 1196–1201. 10.1002/cncr.10284. [DOI] [PubMed] [Google Scholar]
- Tafreshi N. K.; Doligalski M. L.; Tichacek C. J.; Pandya D. N.; Budzevich M. M.; El-Haddad G.; Khushalani N. I.; Moros E. G.; McLaughlin M. L.; Wadas T. J.; Morse D. L. Development of Targeted Alpha Particle Therapy for Solid Tumors. Molecules 2019, 24, 4314. 10.3390/molecules24234314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscher M.; Bakos G.; Benešová M. Atomic Nanogenerators in Targeted Alpha Therapies: Curie’s Legacy in Modern Cancer Management. Pharmaceuticals 2020, 13, 76. 10.3390/ph13040076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couturier O.; Supiot S.; Degraef-Mougin M.; Faivre-Chauvet A.; Carlier T.; Chatal J.-F.; Davodeau F.; Cherel M. Cancer radioimmunotherapy with alpha-emitting nuclides. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 601–614. 10.1007/s00259-005-1803-2. [DOI] [PubMed] [Google Scholar]
- Brechbiel M. W. Targeted α-therapy: past, present, future?. Dalton Trans. 2007, 4918–4928. 10.1039/b704726f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.-S.; Brechbiel M. W. An overview of targeted alpha therapy. Tumor Biol. 2012, 33, 573–590. 10.1007/s13277-011-0286-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson S. Radium-223 Therapy of Bone Metastases in Prostate Cancer. Semin. Nucl. Med. 2016, 46, 544–556. 10.1053/j.semnuclmed.2016.07.007. [DOI] [PubMed] [Google Scholar]
- Morgenstern A.; Apostolidis C.; Kratochwil C.; Sathekge M.; Krolicki L.; Bruchertseifer F. An overview of targeted alpha therapy with 225Actinium and 213Bismuth. Curr. Radiopharm. 2018, 11, 200–208. 10.2174/1874471011666180502104524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratochwil C.; Bruchertseifer F.; Giesel F. L.; Weis M.; Verburg F. A.; Mottaghy F.; Kopka K.; Apostolidis C.; Haberkorn U.; Morgenstern A. 225Ac-PSMA-617 for PSMA-targeted α-radiation therapy of metastatic castration-resistant prostate cancer. J. Nucl. Med. 2016, 57, 1941–1944. 10.2967/jnumed.116.178673. [DOI] [PubMed] [Google Scholar]
- Kratochwil C.; Bruchertseifer F.; Rathke H.; Bronzel M.; Apostolidis C.; Weichert W.; Haberkorn U.; Giesel F. L.; Morgenstern A. Targeted α-therapy of metastatic castration-resistant prostate cancer with 225Ac-PSMA-617: Dosimetry estimate and empiric dose finding. J. Nucl. Med. 2017, 58, 1624–1631. 10.2967/jnumed.117.191395. [DOI] [PubMed] [Google Scholar]
- Hammer S.; Hagemann U. B.; Zitzmann-Kolbe S.; Larsen A.; Ellingsen C.; Geraudie S.; Grant D.; Indrevoll B.; Smeets R.; von Ahsen O.; Kristian A.; Lejeune P.; Hennekes H.; Karlsson J.; Bjerke R. M.; Ryan O. B.; Cuthbertson A. S.; Mumberg D. Preclinical Efficacy of a PSMA-Targeted Thorium-227 Conjugate (PSMA-TTC), a Targeted Alpha Therapy for Prostate Cancer. Clin. Cancer Res. 2020, 26, 1985–1996. 10.1158/1078-0432.CCR-19-2268. [DOI] [PubMed] [Google Scholar]
- Jurcic J. G.; Rosenblat T. L.; McDevitt M. R.; Pandit-Taskar N.; Carrasquillo J. A.; Chanel S. M.; Zikaras K.; Frattini M. G.; Maslak P. M.; Cicic D.; Larson S. M.; Scheinberg D. A. Targeted Alpha-Particle Nano-Generator Actinium-225 (225Ac)-Lintuzumab (Anti-CD33) in Acute Myeloid Leukemia (AML). Clin. Lymphoma, Myeloma Leuk. 2013, 13, S379–S380. 10.1016/j.clml.2013.07.088. [DOI] [Google Scholar]
- Jurcic J. G.; Ravandi F.; Pagel J. M.; Park J. H.; Smith B. D.; Douer D.; Levy M. Y.; Estey E.; Kantarjian H. M.; Earle D.; Cicic D.; Scheinberg D. A. Phase I trial of α-particle therapy with actinium-225 (225Ac)-lintuzumab (anti-CD33) and low-dose cytarabine (LDAC) in older patients with untreated acute myeloid leukemia (AML.). J. Clin. Oncol. 2015, 33, 7050–7050. 10.1200/jco.2015.33.15_suppl.7050. [DOI] [Google Scholar]
- Kratochwil C.; Bruchertseifer F.; Giesel F.; Apostolidis C.; Haberkorn U.; Morgenstern A. Ac-225-DOTATOC - an Empiric Dose Finding for Alpha Particle Emitter Based Radionuclide Therapy of Neuroendocrine Tumors. J. Nucl. Med. 2015, 56, 1232. [Google Scholar]
- Hagemann U. B.; Ellingsen C.; Schuhmacher J.; Kristian A.; Mobergslien A.; Cruciani V.; Wickstroem K.; Schatz C. A.; Kneip C.; Golfier S.; Smeets R.; Uran S.; Hennekes H.; Karlsson J.; Bjerke R. M.; Ryan O. B.; Mumberg D.; Ziegelbauer K.; Cuthbertson A. S. Mesothelin-Targeted Thorium-227 Conjugate (MSLN-TTC): Preclinical Evaluation of a New Targeted Alpha Therapy for Mesothelin-Positive Cancers. Clin. Cancer Res. 2019, 25, 4723–4734. 10.1158/1078-0432.CCR-18-3476. [DOI] [PubMed] [Google Scholar]
- Chakravarty R.; Siamof C. M.; Dash A.; Cai W. Targeted α-therapy of prostate cancer using radiolabeled PSMA inhibitors: a game changer in nuclear medicine. Am. J. Nucl. Med. Mol. Imaging 2018, 8, 247–267. [PMC free article] [PubMed] [Google Scholar]
- Jadvar H. Targeted α-Therapy in Cancer Management: Synopsis of Preclinical and Clinical Studies. Cancer Biother. Radiopharm. 2020, 35, 475–484. 10.1089/cbr.2019.3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern A.; Apostolidis C.; Bruchertseifer F. Supply and Clinical Application of Actinium-225 and Bismuth-213. Seminars in Nuclear Medicine 2020, 50, 119–123. 10.1053/j.semnuclmed.2020.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruigrok E. A. M.; van Weerden W. M.; Nonnekens J.; de Jong M. The Future of PSMA-Targeted Radionuclide Therapy: An Overview of Recent Preclinical Research. Pharmaceutics 2019, 11, 560. 10.3390/pharmaceutics11110560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czerwińska M.; Bilewicz A.; Kruszewski M.; Wegierek-Ciuk A.; Lankoff A. Targeted Radionuclide Therapy of Prostate Cancer-From Basic Research to Clinical Perspectives. Molecules 2020, 25, 1743. 10.3390/molecules25071743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacheris W. P.; Nickle S. K.; Sherry A. D. Thermodynamic study of lanthanide complexes of 1,4,7-triazacyclononane-N,N’,N″-triacetic acid and 1,4,7, 10-tetraazacyclododecane-N,N’,N″,N’’’-tetraacetic acid. Inorg. Chem. 1987, 26, 958–960. 10.1021/ic00253a038. [DOI] [Google Scholar]
- Clarke E. T.; Martell A. E. Stabilities of trivalent metal ion complexes of the tetraacetate derivatives of 12-, 13- and 14-membered tetraazamacrocycles. Inorg. Chim. Acta 1991, 190, 37–46. 10.1016/S0020-1693(00)80229-7. [DOI] [Google Scholar]
- Henriksen G.; Bruland Ø. S.; Larsen R. H. Thorium and Actinium Polyphosphonate Compounds As Bone-seeking Alpha Particle-emitting Agents. Anticancer Res. 2004, 24, 101–105. [PubMed] [Google Scholar]
- Dahle J.; Borrebæk J.; Jonasdottir T. J.; Hjelmerud A. K.; Melhus K. B.; Bruland Ø. S.; Press O. W.; Larsen R. H. Targeted cancer therapy with a novel low-dose rate α-emitting radioimmunoconjugate. Blood 2007, 110, 2049–2056. 10.1182/blood-2007-01-066803. [DOI] [PubMed] [Google Scholar]
- Dahle J.; Larsen R. H. Targeted alpha-particle therapy with 227Th-labeled antibodies. Curr. Radiopharm. 2008, 1, 209–214. 10.2174/1874471010801030209. [DOI] [Google Scholar]
- Abbas N.; Heyerdahl H.; Bruland Ø. S.; Borrebæk J.; Nesland J.; Dahle J. Experimental α-particle radioimmunotherapy of breast cancer using 227Th-labeled p-benzyl-DOTA-trastuzumab. EJNMMI Res. 2011, 1, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern A.; Apostolidis C.; Bruchertseifer F.; Capote R.; Gouder T.; Simonelli F.; Sin M.; Abbas K. Cross-sections of the reaction 232Th(p,3n)230Pa for production of 230U for targeted alpha therapy. Appl. Radiat. Isot. 2008, 66, 1275–1280. 10.1016/j.apradiso.2008.02.066. [DOI] [PubMed] [Google Scholar]
- Dovrat G.; Illy M.-C.; Berthon C.; Lerner A.; Mintz M. H.; Maimon E.; Vainer R.; Ben-Eliyahu Y.; Moiseev Y.; Moisy P.; Bettelheim A.; Zilbermann I. On the Aqueous Chemistry of the UIV-DOTA Complex. Chem. – Eur. J. 2020, 26, 3390–3403. 10.1002/chem.201905357. [DOI] [PubMed] [Google Scholar]
- Montavon G.; Apostolidis C.; Bruchertseifer F.; Repinc U.; Morgenstern A. Spectroscopic study of the interaction of U(VI) with transferrin and albumin for speciation of U(VI) under blood serum conditions. J. Inorg. Biochem. 2009, 103, 1609–1616. 10.1016/j.jinorgbio.2009.08.010. [DOI] [PubMed] [Google Scholar]
- Montavon G.; Repinc U.; Apostolidis C.; Bruchertseifer F.; Abbas K.; Morgenstern A. Investigation of para-sulfonatocalix[n]arenes [n = 6, 8] as potential chelates for 230U. Dalton Trans. 2010, 39, 1366–1374. 10.1039/B918426K. [DOI] [PubMed] [Google Scholar]
- Schlea C. S.; Stoddard D. H. Californium Isotopes Proposed for Intracavity and Interstitial Radiation Therapy with Neutrons. Nature 1965, 206, 1058–1059. 10.1038/2061058a0. [DOI] [PubMed] [Google Scholar]
- Wang C.-K. C.Progress in Californium-252 Neutron Brachytherapy. In Brachytherapy; Kishi K., Ed.; InTech: Rijeka, Croatia, 2012; pp. 33–58. [Google Scholar]
- Aime S.; Barge A.; Benetollo F.; Bombieri G.; Botta M.; Uggeri F. A Novel Compound in the Lanthanide(III) DOTA Series. X-ray Crystal and Molecular Structure of the Complex Na[La(DOTA)La(HDOTA)]·10H2O. Inorg. Chem. 1997, 36, 4287–4289. 10.1021/ic9704501. [DOI] [Google Scholar]
- Aime S.; Barge A.; Botta M.; Fasano M.; Ayala J. D.; Bombieri G. Crystal structure and solution dynamics of the lutetium(III) chelate of DOTA. Inorg. Chim. Acta 1996, 246, 423–429. 10.1016/0020-1693(94)03964-X. [DOI] [Google Scholar]
- T́th Ė.; Br̈cher E. Stability constants of the lanthanide(III)-1,4,7,10-tetraazacyclododecane-N,N’,N″,N’″-tetraacetate complexes. Inorg. Chim. Acta 1994, 221, 165–167. 10.1016/0020-1693(94)03964-X. [DOI] [Google Scholar]
- Aime S.; Botta M.; Fasano M.; Marques M. P. M.; Geraldes C. F. G. C.; Pubanz D.; Merbach A. E. Conformational and Coordination Equilibria on DOTA Complexes of Lanthanide Metal Ions in Aqueous Solution Studied by 1H-NMR Spectroscopy. Inorg. Chem. 1997, 36, 2059–2068. 10.1021/ic961364o. [DOI] [PubMed] [Google Scholar]
- Peters J. A.; Djanashvili K.; Geraldes C. F. G. C.; Platas-Iglesias C. The chemical consequences of the gradual decrease of the ionic radius along the Ln-series. Coord. Chem. Rev. 2020, 406, 213146. 10.1016/j.ccr.2019.213146. [DOI] [Google Scholar]
- Deal K. A.; Davis I. A.; Mirzadeh S.; Kennel S. J.; Brechbiel M. W. Improved in vivo stability of actinium-225 macrocyclic complexes. J. Med. Chem. 1999, 42, 2988–2992. 10.1021/jm990141f. [DOI] [PubMed] [Google Scholar]
- McDevitt M. R.; Ma D.; Simon J.; Frank R. K.; Scheinberg D. A. Design and synthesis of 225Ac radioimmunopharmaceuticals. Appl. Radiat. Isot. 2002, 57, 841–847. 10.1016/S0969-8043(02)00167-7. [DOI] [PubMed] [Google Scholar]
- Kannengießer S.Optimization of the Synthesis of Ac-225-labelled DOTA-Radioimmunoconjugates for Targeted Alpha Therapy based on Investigations on the Complexation of Trivalent Actinides by DOTA. Ph. D. Thesis, Ruprecht-Karls-Universität Heidelberg, 2013.
- Wilson J. J.; Ferrier M.; Radchenko V.; Maassen J. R.; Engle J. W.; Batista E. R.; Martin R. L.; Nortier F. M.; Fassbender M. E.; John K. D.; Birnbaum E. R. Evaluation of nitrogen-rich macrocyclic ligands for the chelation of therapeutic bismuth radioisotopes. Nucl. Med. Biol. 2015, 42, 428–438. 10.1016/j.nucmedbio.2014.12.007. [DOI] [PubMed] [Google Scholar]
- Khabibullin A. R.; Karolak A.; Budzevich M. M.; McLaughlin M. L.; Morse D. L.; Woods L. M. Structure and properties of DOTA-chelated radiopharmaceuticals within the 225Ac decay pathway. MedChemComm 2018, 9, 1155–1163. 10.1039/C8MD00170G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele N. A.; Wilson J. J. Actinium-225 for targeted α therapy: Coordination chemistry and current chelation approaches. Cancer Biother. Radiopharm. 2018, 33, 336–348. 10.1089/cbr.2018.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern A.; Lilley L. M.; Stein B. W.; Kozimor S. A.; Batista E. R.; Yang P. Computer-Assisted Design of Macrocyclic Chelators for Actinium-225 Radiotherapeutics. Inorg. Chem. 2021, 60, 623–632. 10.1021/acs.inorgchem.0c02432. [DOI] [PubMed] [Google Scholar]
- Thakur P.; Conca J. L.; Choppin G. R. Complexation studies of Cm(III), Am(III), and Eu(III) with linear and cyclic carboxylates and polyaminocarboxylates. J. Coord. Chem. 2011, 64, 3214–3236. 10.1080/00958972.2011.616927. [DOI] [Google Scholar]
- Tamain C.; Dumas T.; Hennig C.; Guilbaud P. Coordination of Tetravalent Actinides (An=ThIV, UIV, NpIV, PuIV) with DOTA: From Dimers to Hexamers. Chem. – Eur. J. 2017, 23, 6864–6875. 10.1002/chem.201700493. [DOI] [PubMed] [Google Scholar]
- Kent G. T.; Wu G.; Hayton T. W. Synthesis and Crystallographic Characterization of the Tetravalent Actinide-DOTA Complexes [AnIV(κ8-DOTA)(DMSO)] (An = Th, U). Inorg. Chem. 2019, 58, 8253–8256. 10.1021/acs.inorgchem.9b00736. [DOI] [PubMed] [Google Scholar]
- Spirlet M. R.; Rebizant J.; Desreux J. F.; Loncin M. F. Crystal and molecular structure of sodium aqua(1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetato)europate(III) tetrahydrate Na+(EuDOTA.H2O)−.4H2O, and its relevance to NMR studies of the conformational behavior of the lanthanide complexes formed by the macrocyclic ligand DOTA. Inorg. Chem. 1984, 23, 359–363. 10.1021/ic00171a018. [DOI] [Google Scholar]
- Benetollo F.; Bombieri G.; Calabi L.; Aime S.; Botta M. Structural Variations Across the Lanthanide Series of Macrocyclic DOTA Complexes: Insights into the Design of Contrast Agents for Magnetic Resonance Imaging. Inorg. Chem. 2003, 42, 148–157. 10.1021/ic025790n. [DOI] [PubMed] [Google Scholar]
- Chang C. A.; Francesconi L. C.; Malley M. F.; Kumar K.; Gougoutas J. Z.; Tweedle M. F.; Lee D. W.; Wilson L. J. Synthesis, characterization, and crystal structures of M(DO3A) (M = iron, gadolinium) and Na[M(DOTA)] (M = Fe, yttrium, Gd). Inorg. Chem. 1993, 32, 3501–3508. 10.1021/ic00068a020. [DOI] [Google Scholar]
- Parker D.; Pulukkody K.; Smith F. C.; Batsanov A.; Howard J. A. K. Structures of the yttrium complexes of 1,4,7,10-tetraazacyclododecane-N,N’,N″,N’″-tetraacetic acid (H4dota) and N,N″-bis(benzylcarbamoylmethyl)diethylenetriamine-N,N’,N″-triacetic acid and the solution structure of a zirconium complex of H4dota. J. Chem. Soc., Dalton Trans. 1994, 689–693. 10.1039/DT9940000689. [DOI] [Google Scholar]
- Benetollo F.; Bombieri G.; Aime S.; Botta M. A holmium complex of a macrocyclic ligand (DOTA) and its isostructural europium analogue. Acta Cryst. C 1999, 55, 353–356. 10.1107/S0108270198014450. [DOI] [Google Scholar]
- Burai L.; T́th É.; Moreau G.; Sour A.; Scopelliti R.; Merbach A. E. Novel macrocyclic EuII complexes: Fast water exchange related to an extreme M-Owater distance. Chem. – Eur. J. 2003, 9, 1394–1404. 10.1002/chem.200390159. [DOI] [PubMed] [Google Scholar]
- Anderson O. P.; Reibenspies J. H. {Ca(OH2)3[Ca(DOTA)].7.7H2O}n. Acta Cryst. C 1996, 52, 792–795. 10.1107/S0108270195013916. [DOI] [Google Scholar]
- Csajbók É.; Baranyai Z.; Bányai I.; Brücher E.; Király R.; Müller-Fahrnow A.; Platzek J.; Radüchel B.; Schäfer M. Equilibrium, 1H and 13C NMR spectroscopy, and X-ray diffraction studies on the complexes Bi(DOTA)− and Bi(DO3A-Bu). Inorg. Chem. 2003, 42, 2342–2349. 10.1021/ic0261272. [DOI] [PubMed] [Google Scholar]
- Riesen A.; Zehnder M.; Kaden T. A. Metal complexes of macrocyclic ligands. Part XXIII. Synthesis, properties, and structures of mononuclear complexes with 12- and 14-membered tetraazamacrocycle-N,N′,N″,N‴-tetraacetic Acids. Helv. Chim. Acta 1986, 69, 2067–2073. 10.1002/hlca.19860690830. [DOI] [Google Scholar]
- Riesen A.; Zehnder M.; Kaden T. A. Structures of two Zn2+ complexes with two tetraaza macrocyclic tetraacetates. Acta Cryst. C 1991, 47, 531–533. 10.1107/S0108270190009581. [DOI] [Google Scholar]
- Heppeler A.; André J. P.; Buschmann I.; Wang X.; Reubi J.-C.; Hennig M.; Kaden T. A.; Maecke H. R. Metal-ion-dependent biological properties of a chelator-derived somatostatin analogue for tumour targeting. Chem. – Eur. J. 2008, 14, 3026–3034. 10.1002/chem.200701264. [DOI] [PubMed] [Google Scholar]
- Dubost J. P.; Leger J. M.; Langlois M. H.; Meyer D.; Schaefer M. Crystallography Structure d’un agent de contraste utilisé en imagerie de résonance magnétique le complexe DOTA Gd C16H24N4O8NaGd, 5H2O. C. R. Seances Acad. Sci., Ser. 2 1991, 312, 349–354. [Google Scholar]
- Desreux J. F. Nuclear magnetic resonance spectroscopy of lanthanide complexes with a tetraacetic tetraaza macrocycle. Unusual conformation properties. Inorg. Chem. 1980, 19, 1319–1324. 10.1021/ic50207a042. [DOI] [Google Scholar]
- Aime S.; Botta M.; Ermondi G. NMR study of solution structures and dynamics of lanthanide(III) complexes of DOTA. Inorg. Chem. 1992, 31, 4291–4299. 10.1021/ic00047a016. [DOI] [Google Scholar]
- Hoeft S.; Roth K. Struktur und Dynamik von Lanthanoid-Tetraazacyclododecantetraacetat-(DOTA–)Komplexen in L̈sung. Chem. Ber. 1993, 126, 869–873. 10.1002/cber.19931260404. [DOI] [Google Scholar]
- Vaira M. D.; Stoppioni P. Theoretical investigation on the geometries of DOTA and DOTA-like complexes and on the transition states of their conformational equilibria. New J. Chem. 2002, 26, 136–144. 10.1039/b106168m. [DOI] [Google Scholar]
- Cosentino U.; Villa A.; Pitea D.; Moro G.; Barone V.; Maiocchi A. Conformational Characterization of Lanthanide(III)-DOTA Complexes by ab Initio Investigation in Vacuo and in Aqueous Solution. J. Am. Chem. Soc. 2002, 124, 4901–4909. 10.1021/ja017666t. [DOI] [PubMed] [Google Scholar]
- Barone V.; Cossi M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. 10.1021/jp9716997. [DOI] [Google Scholar]
- Regueiro-Figueroa M.; Esteban-Gómez D.; de Blas A.; Rodríguez-Blas T.; Platas-Iglesias C. Understanding Stability Trends along the Lanthanide Series. Chem. – Eur. J. 2014, 20, 3974–3981. 10.1002/chem.201304469. [DOI] [PubMed] [Google Scholar]
- Kovács A. Theoretical Study of Actinide Complexes with Macropa. ACS Omega 2020, 5, 26431–26440. 10.1021/acsomega.0c02873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purgel M.; Baranyai Z.; de Blas A.; Rodríguez-Blas T.; Bányai I.; Platas-Iglesias C.; Tóth I. An NMR and DFT Investigation on the Conformational Properties of Lanthanide(III) 1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetraacetate Analogues Containing Methylenephosphonate Pendant Arms. Inorg. Chem. 2010, 49, 4370–4382. 10.1021/ic100177n. [DOI] [PubMed] [Google Scholar]
- Shannon R. D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Cryst. 1976, 32, 751–767. 10.1107/S0567739476001551. [DOI] [Google Scholar]
- Wiberg K. B. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. 10.1016/0040-4020(68)88057-3. [DOI] [Google Scholar]
- Carnall W. T.A systematic analysis of the spectra of trivalent actinide chlorides in D3h site symmetry; Argonne National Laboratory: 1989. [Google Scholar]
- Kelley M. P.; Su J.; Urban M.; Luckey M.; Batista E. R.; Yang P.; Shafer J. C. On the Origin of Covalent Bonding in Heavy Actinides. J. Am. Chem. Soc. 2017, 139, 9901–9908. 10.1021/jacs.7b03251. [DOI] [PubMed] [Google Scholar]
- Pace K. A.; Klepov V. V.; Berseneva A. A.; zur Loye H.-C. Covalency in Actinide Compounds. Chem. – Eur. J. 2021, 27, 5835–5841. 10.1002/chem.202004632. [DOI] [PubMed] [Google Scholar]
- Kelley M. P.; Deblonde G. J. P.; Su J.; Booth C. H.; Abergel R. J.; Batista E. R.; Yang P. Bond Covalency and Oxidation State of Actinide Ions Complexed with Therapeutic Chelating Agent 3,4,3-LI(1,2-HOPO). Inorg. Chem. 2018, 57, 5352–5363. 10.1021/acs.inorgchem.8b00345. [DOI] [PubMed] [Google Scholar]
- Su J.; Batista E. R.; Boland K. S.; Bone S. E.; Bradley J. A.; Cary S. K.; Clark D. L.; Conradson S. D.; Ditter A. S.; Kaltsoyannis N.; Keith J. M.; Kerridge A.; Kozimor S. A.; Löble M. W.; Martin R. L.; Minasian S. G.; Mocko V.; La Pierre H. S.; Seidler G. T.; Shuh D. K.; Wilkerson M. P.; Wolfsberg L. E.; Yang P. Energy-Degeneracy-Driven Covalency in Actinide Bonding. J. Am. Chem. Soc. 2018, 140, 17977–17984. 10.1021/jacs.8b09436. [DOI] [PubMed] [Google Scholar]
- Chandrasekar A.; Ghanty T. K. Uncovering Heavy Actinide Covalency: Implications for Minor Actinide Partitioning. Inorg. Chem. 2019, 58, 3744–3753. 10.1021/acs.inorgchem.8b03358. [DOI] [PubMed] [Google Scholar]
- Berryman V. E. J.; Shephard J. J.; Ochiai T.; Price A. N.; Arnold P. L.; Parsons S.; Kaltsoyannis N. Quantum chemical topology and natural bond orbital analysis of M-O covalency in M(OC6H5)4 (M = Ti, Zr, Hf, Ce, Th, Pa, U, Np). Phys. Chem. Chem. Phys. 2020, 22, 16804–16812. 10.1039/D0CP02947E. [DOI] [PubMed] [Google Scholar]
- Cooper S.; Kaltsoyannis N. Covalency in AnCl3 (An = Th - No). Dalton Trans. 2021, 50, 1478–1485. 10.1039/D0DT03699D. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Keith T.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas O.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford CT, 2010. [Google Scholar]
- Tao J.; Perdew J. P.; Staroverov V. N.; Scuseria G. E. Climbing the density functional ladder: Nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. 10.1103/PhysRevLett.91.146401. [DOI] [PubMed] [Google Scholar]
- Roca-Sabio A.; Regueiro-Figueroa M.; Esteban-Gómez D.; de Blas A.; Rodríguez-Blas T.; Platas-Iglesias C. Density functional dependence of molecular geometries in lanthanide(III) complexes relevant to bioanalytical and biomedical applications. Comput. Theor. Chem. 2012, 999, 93–104. 10.1016/j.comptc.2012.08.020. [DOI] [Google Scholar]
- Thiele N. A.; Woods J. J.; Wilson J. J. Implementing f-Block Metal Ions in Medicine: Tuning the Size Selectivity of Expanded Macrocycles. Inorg. Chem. 2019, 58, 10483–10500. 10.1021/acs.inorgchem.9b01277. [DOI] [PubMed] [Google Scholar]
- Dolg M.; Stoll H.; Savin A.; Preuss H. Energy-adjusted pseudopotentials for the rare earth elements. Theor. Chim. Acta 1989, 75, 173–194. 10.1007/BF00528565. [DOI] [Google Scholar]
- Dolg M.; Stoll H.; Preuss H. A combination of quasirelativistic pseudopotential and ligand field calculations for lanthanoid compounds. Theor. Chim. Acta 1993, 85, 441–450. 10.1007/BF01112983. [DOI] [Google Scholar]
- Moritz A.; Cao X.; Dolg M. Quasirelativistic energy-consistent 5f-in-core pseudopotentials for trivalent actinide elements. Theor. Chem. Acc. 2007, 117, 473–481. 10.1007/s00214-006-0180-7. [DOI] [Google Scholar]
- Yang J.; Dolg M. Valence basis sets for lanthanide 4f-in-core pseudopotentials adapted for crystal orbital ab initio calculations. Theor. Chem. Acc. 2005, 113, 212–224. 10.1007/s00214-005-0629-0. [DOI] [Google Scholar]
- Weigand A.; Cao X.; Yang J.; Dolg M. Quasirelativistic f-in-core pseudopotentials and core-polarization potentials for trivalent actinides and lanthanides: molecular test for trifluorides. Theor. Chem. Acc. 2010, 126, 117–127. 10.1007/s00214-009-0584-2. [DOI] [Google Scholar]
- K̈chle W.; Dolg M.; Stoll H.; Preuss H. Energy-Adjusted Pseudopotentials for the Actinides. Parameter Sets and Test Calculations for Thorium and Thorium Monoxide. J. Chem. Phys. 1994, 100, 7535–7542. 10.1063/1.466847. [DOI] [Google Scholar]
- Cao X.; Dolg M.; Stoll H. Valence basis sets for relativistic energy-consistent small-core actinide pseudopotentials. J. Chem. Phys. 2003, 118, 487–496. 10.1063/1.1521431. [DOI] [Google Scholar]
- Becke A. D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Perdew J. P. In Electronic Structure of Solids; Ziesche, P; Eschrig H., Eds.; Akademie Verlag: Berlin, 1991; pp. 11. [Google Scholar]
- Becke A. D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. 10.1063/1.464304. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Phys. Rev. A 1988, 38, 3098. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- Perdew J. P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822. 10.1103/PhysRevB.33.8822. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Chai J.-D.; Head-Gordon M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- Esteban-Gómez D.; de Blas A.; Rodríguez-Blas T.; Helm L.; Platas-Iglesias C. Hyperfine coupling constants on inner-sphere water molecules of GdIII-based MRI contrast agents. ChemPhysChem 2012, 13, 3640–3650. 10.1002/cphc.201200417. [DOI] [PubMed] [Google Scholar]
- Boys S. F.; Bernardi F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. 10.1080/00268977000101561. [DOI] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- Scalmani G.; Frisch M. J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110. 10.1063/1.3359469. [DOI] [PubMed] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Choppin G. R.; Jensen M. P.. Actinides in Solution: Complexation and Kinetics. In The Chemistry of the Actinide and Transactinide Elements; Morss L. R., Edelstein N. M., Fuger J., Eds.; Springer: Dordrecht, 2006; Vol. 4; pp. 2539–2540. [Google Scholar]
- Roos B. O. In Advances in Chemical Physics, Ab Initio Methods in Quantum Chemistry - II; Lawley K. P., Ed.; John Wiley & Sons Ltd.: Chichester, 1987; pp. 399–446. [Google Scholar]
- Douglas N.; Kroll N. M. Quantum Electrodynamical Corrections to the Fine Structure of Helium. Ann. Phys. 1974, 82, 89–155. 10.1016/0003-4916(74)90333-9. [DOI] [Google Scholar]
- Hess B. A. Relativistic Electronic-Structure Calculations Employing a Two-Component No-Pair Formalism with External-Field Projection Operators. Phys. Rev. A 1986, 33, 3742–3748. 10.1103/PhysRevA.33.3742. [DOI] [PubMed] [Google Scholar]
- Roos B. O.; Lindh R.; Malmqvist P.-Å.; Veryazov V.; Widmark P.-O. New Relativistic ANO Basis Sets for Actinide Atoms. Chem. Phys. Lett. 2005, 409, 295–299. 10.1016/j.cplett.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Widmark P.-O.; Malmqvist P.-A.; Roos B. O. Density matrix averaged atomic natural orbital (ANO) basis sets for correlated molecular wave functions. Theor. Chim. Acta 1990, 77, 291–306. 10.1007/BF01120130. [DOI] [Google Scholar]
- Roos B. O.; Lindh R.; Malmqvist P.-Å.; Veryazov V.; Widmark P.-O. Main Group Atoms and Dimers Studied with a New Relativistic ANO Basis Set. J. Phys. Chem. A 2004, 108, 2851–2858. 10.1021/jp031064+. [DOI] [Google Scholar]
- Dunning T. H. Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. 10.1063/1.456153. [DOI] [Google Scholar]
- Andersson K.; Malmqvist P.-A.; Roos B. O.; Sadlej A. J.; Wolinski K. Second-Order Perturbation Theory with a CASSCF Reference Function. J. Phys. Chem. 1990, 94, 5483–5488. 10.1021/j100377a012. [DOI] [Google Scholar]
- Andersson K.; Malmqvist P.-Å.; Roos B. O. Second-Order Perturbation Theory with a Complete Active Space Self-Consistent Field Reference Function. J. Chem. Phys. 1992, 96, 1218–1226. 10.1063/1.462209. [DOI] [Google Scholar]
- Roos B. O.; Malmqvist P.-Å. Relativistic Quantum Chemistry: The Multiconfigurational Approach. Phys. Chem. Chem. Phys. 2004, 6, 2919–2927. 10.1039/b401472n. [DOI] [Google Scholar]
- Karlström G.; Lindh R.; Malmqvist P.-Å.; Roos B. O.; Ryde U.; Veryazov V.; Widmark P.-O.; Cossi M.; Schimmelpfennig B.; Neogrady P.; Seijo L. MOLCAS: A Program Package for Computational Chemistry. Comput. Mater. Sci. 2003, 28, 222–239. 10.1016/S0927-0256(03)00109-5. [DOI] [Google Scholar]
- Aquilante F.; Autschbach J.; Carlson R. K.; Chibotaru L. F.; Delcey M. G.; de Vico L.; Galván I. F.; Ferré N.; Frutos L. M.; Gagliardi L.; Garavelli M.; Giussani A.; Hoyer C. E.; Manni G. L.; Lischka H.; Ma D.; Malmqvist P. Å.; Müller T.; Nenov A.; Olivucci M.; Pedersen T. B.; Peng D.; Plasser F.; Pritchard B.; Reiher M.; Rivalta I.; Schapiro I.; Segarra-Martí J.; Stenrup M.; Truhlar D. G.; Ungur L.; Valentini A.; Vancoillie S.; Veryazov V.; Vysotskiy V. P.; Weingart O.; Zapata F.; Lindh R. MOLCAS 8: New Capabilities for Multiconfigurational Quantum Chemical Calculations Across the Periodic Table. J. Comput. Chem. 2016, 37, 506–541. 10.1002/jcc.24221. [DOI] [PubMed] [Google Scholar]
- Kovačević G.; Veryazov V. Luscus: molecular viewer and editor for MOLCAS. Aust. J. Chem. 2015, 7, 16. 10.1186/s13321-015-0060-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader R. F. W.Atoms in Molecules. A Quantum Theory; Oxford University Press: Oxford, 1990. [Google Scholar]
- Lu T.; Chen F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- Zou W.; Nori-Shargh D.; Boggs J. E. On the Covalent Character of Rare Gas Bonding Interactions: A New Kind of Weak Interaction. J. Phys. Chem. A 2013, 117, 207–212. 10.1021/jp3104535. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.