

Abstract
A palladium-catalysed intramolecular allylic (hetero)arylation strategy for the synthesis of fused cyclopentenes incorporated with all-carbon quaternary and spiro centres is described. The method is straightforward, shows broad scope, proceeds in synthetically useful yields, and provides a rare means to construct complex cyclopentanoids. The reaction is believed to involve a kinetically unfavourable 5-endo-trig carbocyclisation of the tethered (π-allyl)palladium system. Further, this method was successfully applied as the key step in the total synthesis of diterpene natural products taiwaniaquinone H and dichroanone.
A palladium-catalysed intramolecular allylic (hetero)arylation strategy for the synthesis of fused cyclopentenes incorporated with all-carbon quaternary and spiro centres is described.
Introduction
Cyclopentannulated arenes and heteroarenes are the primary molecular architectures of many bioactive natural products and drug candidates; they also find potential applications in medicinal chemistry and in materials science.1 Among them are a sizeable number of bioactive molecules that encompass quaternary carbon atoms on one or more stereogenic centers.2 Some of the representative bioactive molecules possessing fused cyclopentane scaffolds and featuring at least one all-carbon quaternary/spiro centre are shown in Fig. 1.3
Fig. 1. Some of the representative biologically active molecules under the purview of this work.

Among several methods for the synthesis of cyclopenta-fused arenes and heteroarenes, to our knowledge, a disconnection based on 5-endo-trig cyclisation mode is yet to be realised. Baldwin's rules describe 5-endo-trig cyclisation as a stereoelectronically disfavoured pathway owing to the geometric constraints of the reactive functional groups not being able to achieve the Burgi–Dunitz angle.4
Palladium-catalysed allylic alkylations have been the subject of intensive investigations subsequent to the seminal contributions of Tsuji and Trost, Scheme 1a.5 The construction of nearly all types of rings can be assumed by employing Tsuji–Trost chemistry.6 However, Pd-catalysed 5-endo-trig carbocyclisation leading to the formation of cyclopentanes is quite uncommon, Scheme 1b.7 Against this background, Malacria in 1998 reported an example depicting Pd-catalysed and silicon-directed 5-endo-trig cyclisation for the formation of a silylated cyclopentene.8 Recently, the Tius group demonstrated Pd-catalysed transformation of diketoesters to 2-hydroxycyclopentenones.9 While the reaction is considered to be an intramolecular allylic alkylation reaction,10 the possibility of Pd-catalysed anionic 5-endo-trig cyclisation may not be ruled out.7,11 A mechanistically distinct event on a similar substrate design was reported by Oestreich and co-workers, where palladium catalyses 5-endo-trig cyclisation of 1-(1H-indolyl)prop-2-enones for the formation of 3H-pyrrolo[1,2-a]indole-3-ones.12
Scheme 1. Pd-catalysed 5-endo-trig allylic arylation for fused cyclopentenes incorporated with a quaternary/spiro centre: this work.

Having developed several catalytic strategies for the synthesis of various types of cyclopropanoids and cyclopentanoids,13 we envisioned that appropriately substituted (hetero)aryl allyl acetates A under palladium catalysis will generate (π-allyl)palladium species B, which, by undergoing 3-exo-trig cyclisation will deliver vinyl cyclopropanes C (path-a), Scheme 1c.14 On the other hand, a possibility for the formation of fused cyclopentenes D may also arise if B undergoes rather unfavorable 5-endo-trig cyclisation (path-b). It is interesting to note that whenever competition arises, palladium catalysis usually favours the 3-exo-trig process over 5-endo-trig cyclisation; however, this fact may derive from kinetic preference.15
Here, we describe a general and straightforward Pd(ii)-catalysed 5-endo-trig allylic (hetero)arylation of (hetero)aryl allyl acetates A that provides ready access to a wide range of indenes and cyclopentene-fused heteroarenes D incorporated with an all-carbon quaternary or a spiro centre, which, to our knowledge, have not been achieved thus far, as shown in Scheme 1c. At the beginning of the study, it was expected that the formation of C may outweigh the formation of D based on the considerations that the 5-endo-trig cyclisation mode is disfavoured (especially over 3-exo-trig process)14–16 and also that the steric encumbrance at C-3 (of B) may further discourage the formation of D.
Results and discussion
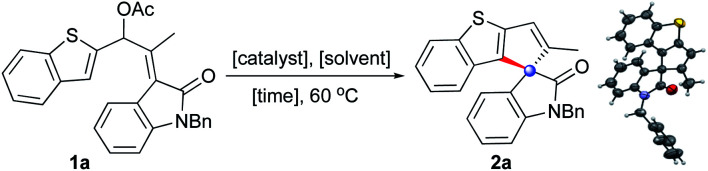
In order to verify the hypothesis presented in Scheme 1c, the oxindole-based allyl acetate 1a was prepared and the reaction was evaluated with respect to the solvent, temperature, and catalyst, Table 1. Initial screening with Pd(0) catalysts provided mixed results. For example, Pd(PPh3)4 showed no reaction while Pd2(dba)3 provided encouraging results by generating the desired product 2a, although in moderate yield (entries 1 and 2). The structure of 2a was readily deduced from the NMR data, and was eventually confirmed by the single-crystal X-ray diffraction analysis of 2a.17
Optimisation of the reaction parametersa,b,c.
| ||||
|---|---|---|---|---|
| Entry | Catalyst (10 mol%) | Solvent | Time (h) | Yieldd (%) |
| 1 | Pd(PPh3)4 | 1,2-DCE | 48 | — |
| 2 | Pd2(dba)3 | 1,2-DCE | 48 | 48 |
| 3 | Pd(PPh3)2Cl2 | 1,2-DCE | 48 | — |
| 4 | Pd(OAc)2 | 1,2-DCE | 48 | 30 |
| 5 | PdCl2 | 1,2-DCE | 8 | 82 |
| 6 | PdCl2 | Toluene | 30 | 43 |
| 7e | PdCl2 | DMF | 10 | — |
| 8 | PdCl2 | MeCN | 48 | 35 |
| 9 | PdCl2 | MeNO2 | 6 | 73 |
| 10 | PdCl2 | 1,4-Dioxane | 36 | 63 |
| 11f | PdCl2 | 1,2-DCE | 18 | 75 |
| 12 | — | 1,2-DCE | 120 | — |
| 13g | PdCl2 | 1,2-DCE | 22 | 76 |
| 14h | PdCl2 | 1,2-DCE | 16 | 74 |
Reaction conditions: see the ESI for details.
Yield of the reaction at 30 °C (for 36 h) is 53%; yield at 45 °C (for 12 h) is 62%.
No reaction was observed with either Ni(cod)2 or [Ir(cod)Cl]2.
Isolated yield after column chromatography.
1a decomposed.
5 mol% PdCl2 was employed.
In the presence of 1 equiv. of 2,6-di-tert-butyl-4-methylpyridine (DTBMP).
In the presence of 1 equiv. of N,N-dimethylaminopyridine (DMAP).
Among Pd(ii) catalysts, interestingly, Pd(OAc)2 provided 2a in poor yield, but the reaction catalysed by PdCl2 furnished 2a in good yield in a short reaction time (entries 3–5). As part of our efforts to improve the efficiency of the reaction, a brief solvent screening was undertaken, which did not provide any better result (entries 6–10). Lowering the catalyst loading to 5 mol% prolonged the reaction time and resulted in less yield of the product (entry 11). Further, no reaction was observed in the absence of any catalyst (entry 12) and the reactions which were performed in the presence of 2,6-di-tert-butyl-4-methylpyridine (DTBMP) or N,N-dimethylaminopyridine (DMAP), to rule out the possibility of any traces of HCl catalyzing the reaction, delivered 2a in good yields (entries 13 and 14).
Thus, an unusual Pd-catalysed 5-endo-trig spirannulation of oxindoles was established based on rational designing of the precursor. Unlike several palladium-catalysed reactions, intriguingly, this reaction does not require any external additives, oxidants, bases, or ligands.18
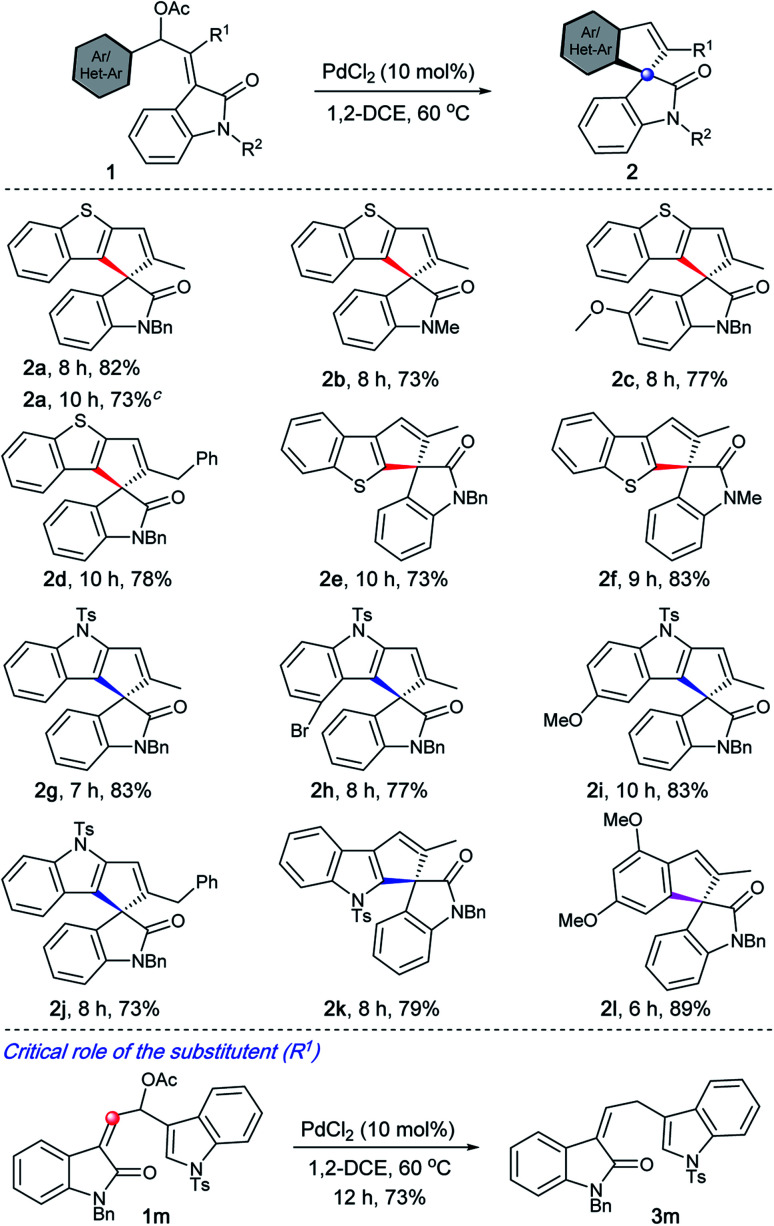
Under the optimised conditions, the generality of the method was evaluated. It is evident from Table 2 that a wide range of heteroarene-fused spirocyclopentene oxindoles possessing electronically diverse substituents can be obtained in good to excellent yields (2a–2k). The method can also be extended to the synthesis of indeno spiroxindoles such as 2l. In particular, the reaction works efficiently from both C-2 and C-3 of benzothiophenes (2a–2dvs.2e–2f), indoles (2g–2jvs.2k), and arenes (2l) indicating that the reaction may not be involving a C–H activation pathway.19
Generality and scope: spiroxindolesa,b.
|
Reaction conditions: see the ESI for details.
Isolated yield after column chromatography.
With the OBoc substrate instead of OAc.
Spirocyclopentane oxindoles are known to be privileged structures with regard to their structural complexity and their biological activity.20 In this context, it is worth highlighting that 2k represents part of the structure of natural products such as similisines and spiroindimicins (see Fig. 1) whereas 2e and 2f serve as their sulphur analogues.3 On the other hand, 2l represents a XEN 907 analogue.3
A critical role of the substituent (R1) was realised during the evaluation of the substrate scope, Table 2. For example, when 1m was treated under the optimised conditions, only the deacylated product 3m was realised.21
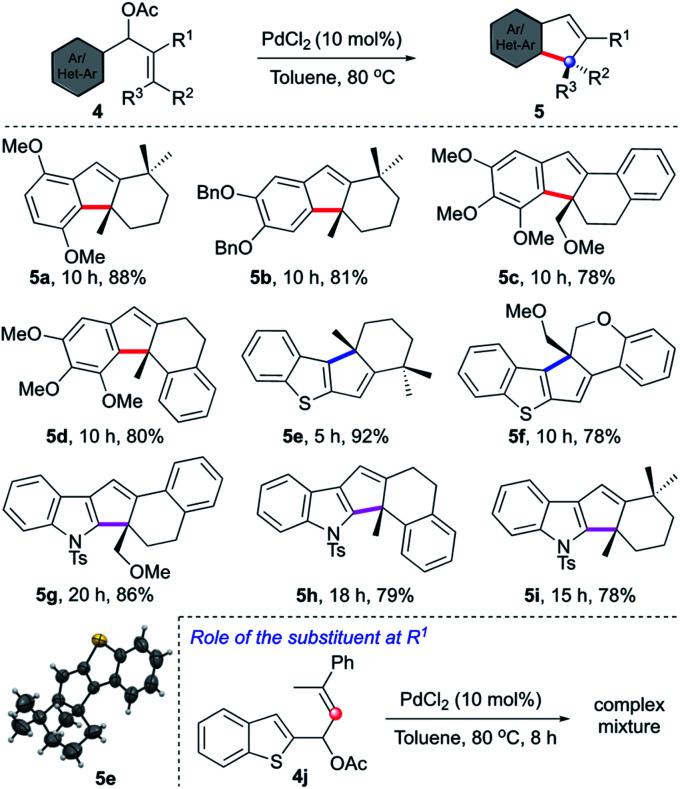
Having established an unprecedented approach for the synthesis of oxindoles bearing a spiro center, we intended to extend the concept for the creation of an all-carbon quaternary center, Table 3. It was expected that the substrate design 4 will provide cyclopentene-fused arenes and heteroarenes 5 having a quaternary carbon. Indeed, after brief optimisation of the reaction conditions, we could achieve the synthesis of natural product-like tri-, tetra- and pentacyclic indenes (5a–5d) and cyclopentannulated heteroarenes (5e–5i) in good to excellent yields. The structure of the representative compound (5e) was unambiguously confirmed by X-ray diffraction analysis.17 The reaction of 4j (where R1 = H) under the prototypical conditions generated a complex mixture, thereby signifying the role of the substituent (R1) in the outcome of the reaction. Nevertheless, some of the merits of this strategy are: (i) neutral conditions, (ii) easily accessible starting compounds, (iii) occurrence of numerous natural products and pharmaceutically relevant compounds with the kind of molecular architecture accessed herein.1,3
Substrate scope: cyclopentene-fused arenes and heteroarenesa,b.
|
Reaction conditions: see the ESI for details.
Chromatographic yields.
We then turned our attention to establish the mechanism. Based on the experimental evidence22 and related literature reports,18 a plausible mechanism is proposed in Scheme 2. The reaction begins with the formation of an (η3-allyl)palladium complex E,23 which reorganises itself into F owing to steric considerations. The intermediate F is now poised to undergo an ene-type 5-endo-trig cyclisation to generate another π-allylpalladium species G, which upon rearomatisation via deprotonation24 delivers 2 (or 5) and regenerates the active catalyst. The measurement of the kinetic isotope effect (KIE)25 strongly suggested that the new C–C bond-forming cyclisation event most likely involved an ene-type allylic arylation step rather than a C–H activation pathway. Otherwise, typical metal-catalysed C–H functionalisation processes involve C–H bond cleavage in the rate determining step.26
Scheme 2. Plausible reaction mechanism.

Next, we decided to apply the current strategy in the total synthesis of taiwaniaquinone H 9 and dichroanone 10, Scheme 3.3 Taiwaniaquinoids are a family of diterpenoid natural products possessing a [6,5,6]-abeo-abietane skeleton with an all-carbon quaternary stereocenter at the pseudobenzylic position. Some of the members of taiwaniaquinoids exhibit potent cytotoxic activity against KB epidermoid carcinoma cancer cells, antitumor activity, aromatase inhibitory activity, to mention a few.27 Owing to their diverse bioactivity profiles and complex structural features, taiwaniaquinoids have attracted the attention of synthetic chemists.3b,28
Scheme 3. Acid-free synthesis of bioactive natural products taiwaniaquinone H and dichroanone.

Our strategy for the synthesis of 9 and 10 involved palladium-catalysed 5-endo-trig cyclisation of 7 to 8 as the key step, Scheme 3. Accordingly, the desired acetate 7 was synthesised by the addition of the lithiated 6 to β-cyclocitral followed by acylation of the intermediate alcohol.3b The reaction of 7 under the prototypical conditions described in Table 3 generated 8 in excellent yield. We then accomplished the total synthesis of taiwaniaquinone H 9 in 65% yield by performing ceric ammonium nitrate (CAN)-mediated oxidative transformation of 8. Subsequently, the total synthesis of dichroanone 10 was also achieved in 66% yield by subjecting 9 to methanolic KOH conditions. As such, the synthesis of 8 also represents the formal synthesis of taiwaniaquinol B.28 The synthetic methodology delineated herein is general and can pave the way for the acid-free synthesis of several other related taiwaniaquinoids as well.
Conclusions
An efficient Pd(ii)-promoted 5-endo-trig cyclisation of (hetero)aryl allyl acetates to spirocyclopentene oxindoles, indenes and cyclopentene-fused heteroarenes is presented.29 A hallmark of this strategy is its ability to construct a quaternary or a spiro carbon center from easily accessible starting compounds under neutral conditions. Since the method represents one of the very few examples for highly efficient synthesis of complex cyclopentanoids under oxidant, base, additive or ligand-free conditions, it should find practical applications in the synthesis of natural products, pharmaceutically relevant compounds and materials. We are in the process of extending this work for the creation of new spiro structures, and applying the current strategy in the total synthesis of the spiroindimicin and polyveoline family of natural products. The results will be communicated in due course.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
IISER Mohali is thanked for funding, and for the NMR, mass and departmental X-ray facilities. S. S. V. R. thanks the DST for the Swarnajayanti fellowship (DST/SJF/CSA-01/2017-18), and the SERB for the Core Research Grant (CRG/2018/000016). B. S. thanks the UGC, and S. K. B., and K. K. thank IISER Mohali for the research fellowships.
Electronic supplementary information (ESI) available. CCDC 1965370 and 1965374. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01932a
Notes and references
- (a) Somei M. Yamada F. Nat. Prod. Rep. 2005;22:73. doi: 10.1039/B316241A. [DOI] [PubMed] [Google Scholar]; (b) Cacchi S. Fabrizi G. Chem. Rev. 2005;105:2873. doi: 10.1021/cr040639b. [DOI] [PubMed] [Google Scholar]; (c) Kochanowska-Karamyan A. J. Hamann M. T. Chem. Rev. 2010;110:4489. doi: 10.1021/cr900211p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ie Y. Nishida K. Karakawa M. Tada H. Aso Y. J. Org. Chem. 2011;76:6604. doi: 10.1021/jo200890b. [DOI] [PubMed] [Google Scholar]; (e) Gabriele B. Mancuso R. Veltri L. Chem.–Eur. J. 2016;22:5056. doi: 10.1002/chem.201503933. [DOI] [PubMed] [Google Scholar]; (f) Chanda T. Singh M. S. Org. Biomol. Chem. 2016;14:8895. doi: 10.1039/C6OB01648K. [DOI] [PubMed] [Google Scholar]; (g) Satpathi B. Mondal A. Ramasastry S. S. V. Chem.–Asian J. 2018;13:1642. doi: 10.1002/asia.201800389. [DOI] [PubMed] [Google Scholar]; (h) Vivekanand T. Satpathi B. Bankar S. K. Ramasastry S. S. V. RSC Adv. 2018;8:18576. doi: 10.1039/C8RA03480J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The construction of quaternary/spiro carbon centres is challenging even in the context of modern organic synthesis, see: ; (a) Hong A. Y. Stoltz B. M. Eur. J. Org. Chem. 2013;14:2745. doi: 10.1002/ejoc.201201761. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Quasdorf K. W. Overman L. E. Nature. 2014;516:181. doi: 10.1038/nature14007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Prusov E. V. Angew. Chem., Int. Ed. 2017;56:14356. doi: 10.1002/anie.201706629. [DOI] [PubMed] [Google Scholar]; (d) Xu P.-W. Yu J.-S. Chen C. Cao Z.-Y. Zhou F. Zhou J. ACS Catal. 2019;9:1820. doi: 10.1021/acscatal.8b03694. [DOI] [Google Scholar]
- For dichroanone and taiwaniaquinones, see: ; (a) Majetich G. Shimkus J. M. J. Nat. Prod. 2010;73:284. doi: 10.1021/np9004695. [DOI] [PubMed] [Google Scholar]; (b1) Kakde B. N. Parida A. Kumari P. Bisai A. Tetrahedron Lett. 2016;57:3179. doi: 10.1016/j.tetlet.2016.06.030. [DOI] [Google Scholar]; . For dasyscyphins, see:; (c1) Zhang L. Xie X. Liu J. Qi J. Ma D. She X. Org. Lett. 2011;13:2956. doi: 10.1021/ol201047m. [DOI] [PubMed] [Google Scholar]; . For polyveoline and similisines, see:; (d1) Dhiman S. Ramasastry S. S. V. Org. Lett. 2015;17:5116. doi: 10.1021/acs.orglett.5b02632. [DOI] [PubMed] [Google Scholar]; . For spiroindimicins, see:; (e1) Zhang W. Liu Z. Li S. Yang T. Zhang Q. Ma L. Tian X. Zhang H. Huang C. Zhang S. Ju J. Shen Y. Zhang C. Org. Lett. 2012;14:3364. doi: 10.1021/ol301343n. [DOI] [PubMed] [Google Scholar]; . For XEN 907 and analogues, see:; (f) Chowdhury S. Chafeev M. Liu S. Sun J. Raina V. Chui R. Young W. Kwan R. Fu J. Cadieux J. A. Bioorg. Med. Chem. Lett. 2011;21:3676. doi: 10.1016/j.bmcl.2011.04.088. [DOI] [PubMed] [Google Scholar]
- (a) Baldwin J. E. J. Chem. Soc., Chem. Commun. 1976:734. doi: 10.1039/C39760000734. [DOI] [Google Scholar]; (b) Baldwin J. E. Cutting J. Dupont W. Kruse L. Silberman L. Thomas R. C. J. Chem. Soc., Chem. Commun. 1976:736. doi: 10.1039/C39760000736. [DOI] [Google Scholar]; (c) Gilmore K. Manoharan M. Wu J. I. Schleyer P. V. R. Alabugin I. V. J. Am. Chem. Soc. 2012;134:10584. doi: 10.1021/ja303341b. [DOI] [PubMed] [Google Scholar]; (d) Alabugin I. V. Gilmore K. Chem. Commun. 2013;49:11246. doi: 10.1039/C3CC43872D. [DOI] [PubMed] [Google Scholar]
- (a) Tsuji J. Takahashi H. Morikawa M. Tetrahedron Lett. 1965;6:4387. doi: 10.1016/S0040-4039(00)71674-1. [DOI] [Google Scholar]; (b) Trost B. M. Fullerton T. J. J. Am. Chem. Soc. 1973;95:292. doi: 10.1021/ja00782a080. [DOI] [Google Scholar]
- (a) Trost B. M. Acc. Chem. Res. 1996;29:355. doi: 10.1021/ar9501129. [DOI] [Google Scholar]; (b) Jellerichs G. Kong J.-R. Krische M. J. J. Am. Chem. Soc. 2003;125:7758. doi: 10.1021/ja0301469. [DOI] [PubMed] [Google Scholar]; (c) Kazmaier U. Curr. Org. Chem. 2003;7:317. doi: 10.2174/1385272033372888. [DOI] [Google Scholar]; (d) Trost B. M. Crawley M. L. Chem. Rev. 2003;103:2921. doi: 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]; (e) Miyabe H. Takemoto Y. Synlett. 2005;11:1641. [Google Scholar]; (f) You S.-L. Dai L.-X. Angew. Chem., Int. Ed. 2006;45:5246. doi: 10.1002/anie.200601889. [DOI] [PubMed] [Google Scholar]; (g) Braun M. Meier T. Angew. Chem., Int. Ed. 2006;45:6952. doi: 10.1002/anie.200602169. [DOI] [PubMed] [Google Scholar]; (h) Ibrahem I. Còrdova A. Angew. Chem., Int. Ed. 2006;45:1952. doi: 10.1002/anie.200504021. [DOI] [PubMed] [Google Scholar]; (i) Lu Z. Ma S. Angew. Chem., Int. Ed. 2008;47:258. doi: 10.1002/anie.200605113. [DOI] [PubMed] [Google Scholar]; (j) Jensen T. Fristrup P. Chem.–Eur. J. 2009;15:9632. doi: 10.1002/chem.200901935. [DOI] [PubMed] [Google Scholar]; (k) Weaver J. D. Recio III A. Grenning A. J. Tunge J. A. Chem. Rev. 2011;111:1846. doi: 10.1021/cr1002744. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Wanner M. J. Claveau E. van Maarseveen J. H. Hiemstra H. Chem.–Eur. J. 2011;17:13680. doi: 10.1002/chem.201103150. [DOI] [PubMed] [Google Scholar]; (m) Watsona I. D. G. Toste F. D. Chem. Sci. 2012;3:2899. doi: 10.1039/C2SC20542D. [DOI] [Google Scholar]; (n) Majumdar K. C. Sinha B. Synthesis. 2013;45:1271. doi: 10.1055/s-0032-1316918. [DOI] [Google Scholar]; (o) Kammerer C. Prestat G. Madec D. Poli G. Acc. Chem. Res. 2014;47:3439. doi: 10.1021/ar500178n. [DOI] [PubMed] [Google Scholar]; (p) Liu Y. Han S.-J. Liu W.-B. Stoltz B. M. Acc. Chem. Res. 2015;48:740. doi: 10.1021/ar5004658. [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Trost B. M. Tetrahedron. 2015;71:5708. doi: 10.1016/j.tet.2015.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]; (r) Fernandes R. A. Nallasivam J. L. Org. Biomol. Chem. 2019;17:8647. doi: 10.1039/C9OB01725A. [DOI] [PubMed] [Google Scholar]
- Pd-catalyzed anionic rearrangement of vinyl cyclopropanes to cyclopentenes, see: ; (a) Morizawa Y. Oshima K. Nozaki H. Tetrahedron Lett. 1982:2871. doi: 10.1016/S0040-4039(00)88436-1. [DOI] [Google Scholar]; (b) Ahmar M. Gazes B. Gore J. Tetrahedron. 1987;43:3453. doi: 10.1016/S0040-4020(01)81636-7. [DOI] [Google Scholar]; (c) Hiroi K. Arinaga Y. Tetrahedron Lett. 1994;35:153. doi: 10.1016/0040-4039(94)88188-X. [DOI] [Google Scholar]
- For a single example of silicon-directed Pd-catalyzed 5-endo-trig cyclization, see: ; Thorimbert S. Malacria M. Tetrahedron Lett. 1998;39:9659. doi: 10.1016/S0040-4039(98)02208-4. [DOI] [Google Scholar]
- For the Tius-Nazarov reaction, see: ; (a) Bee C. Leclerc E. Tius M. A. Org. Lett. 2003;5:4927. doi: 10.1021/ol036017e. [DOI] [PubMed] [Google Scholar]; (b) Cai Z. Harmata M. Org. Lett. 2010;12:5668. doi: 10.1021/ol102478h. [DOI] [PubMed] [Google Scholar]; (c1) Shimada N. Stewart C. Bow W. F. Jolit A. Wong K. Zhou Z. Marcus M. A. Angew. Chem., Int. Ed. 2012;51:5727. doi: 10.1002/anie.201201724. [DOI] [PMC free article] [PubMed] [Google Scholar]; . For related Pd-catalyzed Nazarov-type reactions, see:; (d) Subramanium S. S. Handa S. Miranda A. J. Slaughter L. M. ACS Catal. 2011;1:1371. doi: 10.1021/cs200449g. [DOI] [Google Scholar]; (e) Shao J. Hu P. Hong G. Fang M. Li X. Xu X. Synlett. 2014;25:1009. doi: 10.1055/s-0033-1339115. [DOI] [Google Scholar]
- Atesin T. A. Martinez G. M. Flores D. Organometallics. 2017;36:3589. doi: 10.1021/acs.organomet.7b00494. [DOI] [Google Scholar]
- For formal anionic 5-endo-trig cyclizations, see: ; (a) Beak P. Burg D. A. J. Org. Chem. 1989;54:1647. doi: 10.1021/jo00268a028. [DOI] [Google Scholar]; (b) Batson W. A. Sethumadhavan D. Tius M. A. Org. Lett. 2005;7:2771. doi: 10.1021/ol050970x. [DOI] [PubMed] [Google Scholar]; (c) Johnston C. P. Kothari A. Sergeieva T. Okovytyy S. I. Jackson K. E. Paton R. S. Smith M. D. Nat. Chem. 2015;7:171. doi: 10.1038/nchem.2150. [DOI] [PubMed] [Google Scholar]
- Kandukuri S. R. Schiffner J. A. Oestreich M. Angew. Chem., Int. Ed. 2012;51:1265. doi: 10.1002/anie.201106927. [DOI] [PubMed] [Google Scholar]
- Some selected references: ; (a) Dhiman S. Ramasastry S. S. V. Chem. Commun. 2015;51:557. doi: 10.1039/C4CC08174A. [DOI] [PubMed] [Google Scholar]; (b) Dhiman S. Ramasastry S. S. V. Org. Lett. 2015;17:5116. doi: 10.1021/acs.orglett.5b02632. [DOI] [PubMed] [Google Scholar]; (c) Satpathi B. Ramasastry S. S. V. Angew. Chem., Int. Ed. 2016;55:1777. doi: 10.1002/anie.201510457. [DOI] [PubMed] [Google Scholar]; (d) Raghu M. Grover J. Ramasastry S. S. V. Chem.–Eur. J. 2016;22:18316. doi: 10.1002/chem.201604562. [DOI] [PubMed] [Google Scholar]; (e) Bankar S. K. Singh B. Tung P. Ramasastry S. S. V. Angew. Chem., Int. Ed. 2018;57:1678. doi: 10.1002/anie.201711797. [DOI] [PubMed] [Google Scholar]; (f) Mondal A. Hazra R. Grover J. Raghu M. Ramasastry S. S. V. ACS Catal. 2018;8:2748. doi: 10.1021/acscatal.8b00397. [DOI] [Google Scholar]; (g) Mishra U. K. Patel K. Ramasastry S. S. V. Org. Lett. 2019;21:175. doi: 10.1021/acs.orglett.8b03537. [DOI] [PubMed] [Google Scholar]; (h) Satpathi B. Dutta L. Ramasastry S. S. V. Org. Lett. 2019;21:170. doi: 10.1021/acs.orglett.8b03658. [DOI] [PubMed] [Google Scholar]; (i) Patel K. Mishra U. K. Mukhopadhyay D. Ramasastry S. S. V. Chem.–Asian J. 2019;14:4568. doi: 10.1002/asia.201901108. [DOI] [PubMed] [Google Scholar]
- 3-Exo-trig closures have extremely low activation barriers, see: ; Fiser B. Cuerva J. M. Gomez-Bengoa E. Organometallics. 2018;37:390. doi: 10.1021/acs.organomet.7b00812. [DOI] [Google Scholar]
- According to Trost, cyclopropanes are normally preferred with carbon nucleophiles, but this may be a kinetic preference and does not test the feasibility of five-membered ring formation via a 5-endo-trig cyclization, for details see: ; Trost B. M. Scanlan T. S. J. Am. Chem. Soc. 1989;111:4988. doi: 10.1021/ja00195a068. [DOI] [Google Scholar]
- For a recent example where the 3-exo-trig mode is preferred over 5-endo-trig, see: ; Braun J. Ariëns M. I. Matsuo B. T. de Vries S. van Wordragen E. D. H. Ellenbroek B. D. Velde C. M. L. V. Orru R. V. A. Ruijter E. Org. Lett. 2018;20:6611. doi: 10.1021/acs.orglett.8b02232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- X-ray crystallographic data of compounds 2a and 5e are provided in the ESI.† CCDC 1965370 (2a) and 1965374 (5e).
- (a) Yuan F.-Q. Gao L.-X. Han F.-S. Chem. Commun. 2011;47:5289. doi: 10.1039/C1CC10953G. [DOI] [PubMed] [Google Scholar]; (b) Yuan F.-Q. Sun F.-Y. Han F.-S. Tetrahedron. 2012;68:6837. doi: 10.1016/j.tet.2012.06.038. [DOI] [Google Scholar]
- Considering the comparable efficiency with arenes and heteroarenes, the reaction can be better described as an ene-type process. Further, it is well-established that heteroarenes respond differently (from C-2 and C-3) under C–H activation conditions. For example, see: ; (a) Lane B. S. Sames D. Org. Lett. 2004;6:2897. doi: 10.1021/ol0490072. [DOI] [PubMed] [Google Scholar]; (b) Grimster N. P. Gauntlett C. Godfrey C. R. A. Gaunt M. J. Angew. Chem., Int. Ed. 2005;44:3125. doi: 10.1002/anie.200500468. [DOI] [PubMed] [Google Scholar]; (c) Stuart D. R. Villemure E. Fagnou K. J. Am. Chem. Soc. 2007;129:12072. doi: 10.1021/ja0745862. [DOI] [PubMed] [Google Scholar]; (d) Cambeiro X. C. Ahlsten N. Larrosa I. J. Am. Chem. Soc. 2015;137:15636. doi: 10.1021/jacs.5b10593. [DOI] [PubMed] [Google Scholar]; (e) Zhang S. Niu Y.-H. Ye X.-S. Org. Lett. 2017;19:3608. doi: 10.1021/acs.orglett.7b01583. [DOI] [PubMed] [Google Scholar]
- Some selected recent studies: ; (a) Desrosiers J.-N. Hie L. Biswas S. Zatolochnaya O. V. Rodriguez S. Lee H. Grinberg N. Haddad N. Yee N. K. Garg N. K. Senanayake C. H. Angew. Chem., Int. Ed. 2016;55:11921. doi: 10.1002/anie.201606955. [DOI] [PubMed] [Google Scholar]; (b) Samineni R. Madapa J. Srihari P. Mehta G. Org. Lett. 2017;19:3119. doi: 10.1021/acs.orglett.7b01233. [DOI] [PubMed] [Google Scholar]; (c) Wang L. Li S. Blümel M. Puttreddy R. Peuronen A. Rissanen K. Enders D. Angew. Chem., Int. Ed. 2017;56:8516. doi: 10.1002/anie.201704210. [DOI] [PubMed] [Google Scholar]; (d) Wang C.-C. Huang J. Li X.-H. Kramer S. Lin G.-Q. Sun X.-W. Org. Lett. 2018;20:2888. doi: 10.1021/acs.orglett.8b00927. [DOI] [PubMed] [Google Scholar]; (e) Chaudhari P. D. Hong B.-C. Wen C.-L. Lee G.-H. ACS Omega. 2019;4:655. doi: 10.1021/acsomega.8b03049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mass analysis of the crude reaction mixture also did not indicate the formation of the required product. See the ESI† for details
- Experimental observations indicated the pronounced effect of the substitution at R1 (see Tables 2 and 3). We believe that R1 plays a dual role – electronic and steric. R1 stabilises cationic π-allylpalladium intermediates such as E and G. Especially, the carbon connected to R1 bears a formal positive charge in G; therefore the tertiary carbocation is stabilized. The steric role of R1 is also shown in Scheme 2. We believe that R1 is responsible in converting E to F. In order for this reaction to happen, the attainment of form F is necessary
- The substrate itself may reduce Pd(ii) to Pd(0) during the reaction (analogous to Wacker-type process), and consequently, the in situ formed Pd(0) serves as the active metal center to promote the subsequent allylation reaction. The formation of Pd black in our reactions is an indicative of this proposition. For a discussion on substrate-induced reduction of Pd(ii), see: ; Beletskaya I. P. Cheprakov A. V. Chem. Rev. 2000;100:3009. doi: 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]; and references cited therein
- For deprotonation pathways in Pd-catalyzed reactions, see: ; (a) Garcia-Cuadrado D. de Mendoza P. Braga A. A. C. Maseras F. Echavarren A. M. J. Am. Chem. Soc. 2007;129:6880. doi: 10.1021/ja071034a. [DOI] [PubMed] [Google Scholar]; (b) Gorelsky S. I. Lapointe D. Fagnou K. J. Am. Chem. Soc. 2008;130:10848. doi: 10.1021/ja802533u. [DOI] [PubMed] [Google Scholar]; (c) Liegault B. Petrov I. Gorelsky S. I. Fagnou K. J. Org. Chem. 2010;75:1047. doi: 10.1021/jo902515z. [DOI] [PubMed] [Google Scholar]
- The kH/kD was found to be 0.51. See the ESI† for details
- For the study of kinetic isotope effects in organometallic reactions, see: ; (a) Jones W. D. Acc. Chem. Res. 2003;36:140. doi: 10.1021/ar020148i. [DOI] [PubMed] [Google Scholar]; (b) Li J.-J. Giri R. Yu J.-Q. Tetrahedron. 2008;64:6979. doi: 10.1016/j.tet.2008.03.026. [DOI] [Google Scholar]; (c) García-Rubia A. Urones B. Arrayas R. G. Carretero J. C. Chem.–Eur. J. 2010;16:9676. doi: 10.1002/chem.201001126. [DOI] [PubMed] [Google Scholar]; (d) Gomez-Gallego M. Sierra M. A. Chem. Rev. 2011;111:4857. doi: 10.1021/cr100436k. [DOI] [PubMed] [Google Scholar]; (e) Simmons E. M. Hartwig J. F. Angew. Chem., Int. Ed. 2012;51:3066. doi: 10.1002/anie.201107334. [DOI] [PubMed] [Google Scholar]
- (a) Minami T. Iwamoto M. Ohtsu H. Ohishi H. Tanaka R. Yoshitake A. Planta Med. 2002;68:742. doi: 10.1055/s-2002-33787. [DOI] [PubMed] [Google Scholar]; (b) Chang C. I. Chang J. Y. Kuo C. C. Pan W. Y. Kuo Y. H. Planta Med. 2005;71:72. doi: 10.1055/s-2005-837754. [DOI] [PubMed] [Google Scholar]; (c) Katoh T. Akagi T. Noguchi C. Kajimoto T. Node M. Tanaka R. Nishizawa M. Ohtsu H. Suzuki N. Saito K. Bioorg. Med. Chem. 2007;15:2736. doi: 10.1016/j.bmc.2007.01.031. [DOI] [PubMed] [Google Scholar]
- Majetich G. Shimkus J. M. Tetrahedron Lett. 2009;50:3311. doi: 10.1016/j.tetlet.2009.02.074. [DOI] [Google Scholar]; and references cited therein
- To rule out PdCl2 acting as a Lewis acid, the reaction of 1e was performed with Lewis acids such as MgBr2, InCl3, and ZnCl2. These reactions hardly gave any desired product. See the ESI† for details
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.