

Abstract
Immune-based therapies have revolutionized cancer treatments. Cardiovascular sequelae from these treatments, however, have emerged as critical complications, representing new challenges in cardio-oncology. Immune therapies include a broad range of novel drugs, from antibodies and other biologics, including immune checkpoint inhibitors and bispecific T-cell engagers, to cell-based therapies, such as chimeric-antigen receptor (CAR) T-cell therapies. The recognition of immunotherapy-associated cardiovascular side effects has also catapulted new research questions revolving around the interactions between the immune and cardiovascular systems, and the signaling cascades affected by T cell activation, cytokine release, and immune system dysregulation. Here, we review the specific mechanisms of immune activation from immunotherapies and the resulting cardiovascular toxicities associated with immune activation and excess cytokine production.
Keywords: Cardio-oncology, immuno-oncology, immune checkpoint inhibitors, CAR T-cell therapy, bispecific T-cell engager, cytokine release syndrome, cytokine storm, COVID-19, cardiotoxicity, immunology, oncology
Mechanisms of Cardiovascular Toxicities Associated with Immunotherapies.
While initially tested in refractory cancers, immune-based therapies have become standard of care for many cancer types and are approved in the front-line setting in certain cancers, often combined with traditional chemotherapy regimens. Nearly 50% of all patients with advanced cancer are eligible for immune-based therapies [1]. Immunotherapies include immune checkpoint inhibitors, chimeric antigen receptor (CAR) T-cell therapy, bispecific T-cell engaging antibodies, cytokines (high dose interleukin (IL)-2 and IFN-α), and monoclonal antibody therapy (Table 1) [2]. Although these therapies generally function by activating the immune system to eradicate cancer cells, the specific mechanisms of immune activation are unique with each drug, a concept that is relevant to both anti-tumor efficacy and toxicity.
Table 1:
Types of Immunotherapy
| Immunotherapy | Examples | Mechanism of Action | Cancer targets |
|---|---|---|---|
| CAR T-cell therapy | - Axicabtagene ciloleucel (Yescarta) - Tisagenlecleucel (Kymriah) - Brexucabtagene autoleucel (Tecartus) |
- Adoptive cell transfer - T cell-mediated killing of cancer cells |
- Relapsed or refractory (r/r) non-Hodgkin lymphoma (diffuse large B cell lymphoma (DLBCL), primary mediastinal B cell lymphoma, high grade B cell lymphoma, transformed follicular lymphoma,) - r/r DLBCL - Mantle Cell Lymphoma |
| Bispecific antibodies | - Blinatumomab (Blincyto) - AMG 420 - IMCgp100 (Tebentafusp) - AFM13 - MGD006 |
- CD19- and CD3 targeting bispecific antibody - BCMA and CD3 - GP100 and CD3 - CD30 and CD16A - CD123 and CD3 |
- Relapsed or refractory B-cell precursor ALL - Multiple myeloma - Metastatic uveal melanoma - CD30-positive Hodgkin’s lymphoma - Acute Myeloid Leukemia |
| Immune checkpoint inhibitors | See Table 2 | - Block checkpoint proteins from interacting with partner proteins, allowing T cell- mediated killing of cancer cells | See Table 2 |
| Monoclonal antibody therapya | Alemtuzumab (Lemtrada) Rituximab (Rituxan) Elotuzumab (Empliciti) Daratumumab (Darzalex) |
- Monoclonal antibody, binds to CD52 (protein on surface of mature lymphocytes); CD52-bearing lymphocytes targeted for destruction - Chimeric monoclonal antibody, targets CD20 (primarily found on surface of immune B cells); promotes B cell elimination - SLAMF7 (CD319)-directed immunostimulatory antibody - Anti-CD38, promotes apoptosis |
- Chronic lymphocytic leukemia - Non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, GPA and MPA - Relapsed multiple myeloma - Multiple myeloma, diffuse large B cell lymphoma, follicular lymphoma, mantle cell lymphoma |
| Cytokinesb | High-dose IL-2 | - Promotes expansion of natural killer (NK) and T lymphocytes | Advanced renal cell carcinoma (RCC), metastatic melanoma |
| Recombinant interferon-α | - Potent anti-angiogenic activity [152] | Hairy cell leukemia Follicular non-Hodgkin lymphoma Melanoma AIDS-related Kaposi’s sarcoma Chronic myelogenous leukemia (CML) |
Selected examples of monoclonal antibodies
Selected examples of high-dose cytokines
Overview of Immune Checkpoints
Immune checkpoints are regulatory molecules that attenuate T cell activation, prevent unchecked immune system activation, and guard against autoimmunity [3]. Immune tolerance is carefully regulated by co-stimulatory and co-inhibitory signals that maintain immune homeostasis. Activation of T lymphocytes requires at least two stimulatory signals: recognition of specific antigenic peptides presented on major histocompatibility complexes (MHC) by the T-cell receptor (TCR), and co-stimulatory pathways mediated by T cell surface markers. MHC recognition alone is insufficient to activate naïve T cells. In the absence of the second stimulatory signal, the T cell undergoes apoptosis or becomes anergic [4]. Of the positive co-stimulatory molecules, CD28 is the most effective co-stimulatory T cell surface glycoprotein. CD28 binds to CD80 (B7–1) and CD86 (B7–2) membrane proteins, which are located on various immune cells, including antigen-presenting cells (APCs), monocytes, and B cells [5]. CD28 activates numerous downstream signaling cascades, including PI3K, NF-kB, AP-1, resulting in enhanced cytokine production and T cell activation, proliferation, and migration [6].
T cell activation is also attenuated by other surface markers. Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) (CD152) is a protein receptor that is expressed exclusively on regulatory CD4+ and CD8+ T cells, is induced following T cell activation via CD28 and TCR signaling and functions as a critical negative regulator of T cell activation [3] [7]. CTLA-4 shares 31% amino acid homology with CD28, and also binds to CD80/CD86 membrane proteins. However, CTLA-4 has a higher binding affinity to CD80/CD86, resulting in preferential binding and counteracting the stimulatory activity of CD28. CTLA-4 binding to CD80/CD86 leads to cell cycle arrest at the G1 phase, reduces cytokine production, and attenuates the immune response [8]. Thus, CTLA-4 acts as an “off” signal when bound to CD80/CD86 on the surface of APCs, blocking naïve T cell activation in secondary lymphoid organs. These inhibitory functions maintain homeostasis of immune responses by dampening TCR signaling and protecting the host from autoimmunity [6]. Indeed, CTLA-4 knockout mice experience fatal autoimmunity with multi-organ involvement, including fulminant myocarditis, within 3–4 weeks of age due to unrestrained immune activation [9] [10].
Programmed cell death-1 (PD-1) (CD279) is also an inhibitory receptor that functions to maintain T cell homeostasis and suppress T cell inflammatory activity, especially in non-lymphoid tissues. PD-1 is expressed on activated T cells, B cells, macrophages, dendritic cells, and monocytes. PD-1 is structurally homologous to CD28/CTLA-4 and binds two ligands: programed cell death ligand 1 (PD-L1) and programed cell death ligand 2 (PD-L2), which are expressed on APCs and target non-lymphoid cells in the periphery, including the heart [3]. The PD-1/PD-L1 axis serves to attenuate T cell inflammatory activity in response to pro-inflammatory cytokines (e.g., IFN-γ) and immune activation. Cancer cells can co-opt the suppressive function of immune checkpoints, thus reducing anti-tumor activity and establishing cancer immune evasion (Figure 1A).
Figure 1. Mechanisms of immunotherapy.

A. Immune checkpoint inhibitors. B. Bispecific T cell engager (TCE). C. Chimeric antigen receptor (CAR) T-cell therapy. Abbreviation: antigen-presenting cell (APC); chimeric antigen receptor (CAR); cluster of differentiation (CD); cytotoxic T-lymphocyte associated protein 4 (CTLA-4); major histocompatibility complex (MHC); programmed cell death protein-1 (PD-1); programmed death-ligand 1 (PD-L1); single-chain fragment variable region (scFv); T cell receptor (TCR).
Immune Checkpoint Inhibitors
Immune checkpoint inhibitors (ICI) are monoclonal antibodies that target and functionally block immune checkpoints, thus preventing transmission of an “off signal” to immune cells and releasing T cell-mediated responses directed towards cancer cells [4] [11]. ICIs include antibodies against CTLA-4 (ipilimumab), PD-1 (pembrolizumab, nivolumab, cemiplimab), and PD-L1 (atezolizumab, avelumab, durvalumab). Anti-CTLA-4 antibodies block the binding between CTLA-4 and its ligands, CD80/CD86, allowing the co-stimulatory surface protein CD28 to bind to these ligands, resulting in T cell activation. The downstream stimulatory signaling pathways lead to increased T cell proliferation, differentiation, and cytokine production, which mediate anti-tumor activity (Figure 1A). Anti-PD-1 and anti-PD-L1 antibodies block the interaction between PD-1 and PD-L1, thus releasing the immunosuppressive and cancer immune evasion effects of PD-1/PD-L1 binding. CTLA-4 and PD-1 inhibition have distinct and non-redundant roles in T cell activation. Therefore, there is increased anti-tumor activity when these agents are used in combination.
ICIs were initially tested in advanced malignancies (e.g., metastatic melanoma, lung cancer), resulting in profound responses and long-term remissions in a subset of patients [12]. They were then tested in multiple cancer types and since 2011, have been FDA-approved for over 17 different malignancies. It is expected that their indications for use will continue to expand in the coming years (Table 2) [13]. For example, anti-PD-1 agents are now used as first-line therapy in many cancers, including lung and renal cancers, and melanoma. They have also been used in combination with anti-CTLA-4 agents, cytotoxic chemotherapy, and small molecule inhibitors to augment anti-tumor activity and induce durable responses.
Table 2:
Immune checkpoint inhibitors
| Checkpoint | Checkpoint inhibitor | Malignancies with FDA approved use |
|---|---|---|
| CTLA-4 | Ipilimumab (Yervoy) | Melanoma (unresectable or metastatic); monotherapy, in combination with ipilimumab; adjuvant |
| Renal cell carcinoma (advanced or metastatic); combination with nivolumab | ||
| Microsatellite instability-high or mismatch repair deficient metastatic colorectal cancer; monotherapy or in combination with nivolumab | ||
| Hepatocellular carcinoma (in combination with ipilimumab) | ||
| PD-1 | Nivolumab (Opdivo) | Melanoma (unresectable or metastatic), monotherapy pr in combination with ipilimumab; adjuvant |
| Squamous NSCLC (metastatic) | ||
| Renal cell carcinoma (advanced) | ||
| Classical Hodgkin’s lymphoma (relapsed) | ||
| Head and neck squamous cell carcinoma (recurrent or metastatic) | ||
| Urothelial carcinoma (advanced or metastatic) | ||
| Microsatellite instability-high or mismatch repair deficient metastatic CRC; monotherapy or in combination with ipilimumab | ||
| Hepatocellular carcinoma (monotherapy or in combination with ipilimumab) | ||
| Renal cell carcinoma (advanced or metastatic); monotherapy or in combination with ipilimumab | ||
| Pembrolizumab (Keytruda) | Melanoma (unresectable or metastatic) | |
| NSCLC (metastatic) | ||
| Head or neck squamous cell carcinoma (recurrent or metastatic) | ||
| Classical Hodgkin’s lymphoma (refractory) | ||
| Urothelial carcinoma (locally advanced or metastatic) | ||
| Microsatellite instability-high or mismatch repair deficient solid tumors and colorectal cancer | ||
| Gastric or gastroesophageal junction adenocarcinoma (locally advanced or metastatic) | ||
| Cervical cancer (recurrent or metastatic) | ||
| Primary mediastinal large B cell lymphoma (refractory) | ||
| Hepatocellular carcinoma | ||
| Merkel cell carcinoma (recurrent or metastatic) | ||
| Cutaneous squamous cell carcinoma (advanced) | ||
| Cemiplimab (Libtayo) | Cutaneous squamous cell carcinoma (advanced) | |
| PD-L1 | Atezolizumab (Tecentriq) | Urothelial carcinoma (locally advanced or metastatic) |
| NSCLC (metastatic) | ||
| Small cell lung cancer (metastatic) | ||
| Avelumab (Bavencio) | Merkel cell carcinoma (metastatic) | |
| Urothelial carcinoma (locally advanced or metastatic) | ||
| Advanced renal cell carcinoma | ||
| Durvalumab (Imfinzi) | Urothelial carcinoma (locally advanced or metastatic) Unresectable stage III non-small cell lung cancer |
|
| NSCLC (unresectable) | ||
| Select examples of emerging ICI in clinical trials | ||
| LAG-3 (CD223) | BMS-986016 (Relatlimab) LAG525 (IMP701) REGN3767 TSR033 MK-4280 TSR-033 Sym022 |
Clinical studies pending for advanced hematologic and solid tumors, including NSCLC (metastatic), RCC (advanced), metastatic or unresectable melanoma, microsatellite high (MSI-H) and non-MSI-H colon cancer, gastro-esophageal cancer, glioblastoma, gliosarcoma, Hodgkin’s lymphoma, DLBCL |
| TIM-3 | BMS-986258 TSR-022 (Cobolimab) BGB-A425 MBG453 LY3415244 |
Clinical studies pending for advanced cancer |
| VISTA | CA-170 (oral PD-L1, PD-L2, VISTA checkpoint antagonist) JNJ-61610588 |
Clinical studies pending for advanced cancer |
There are additional immune checkpoints that have emerged as potential targets for cancer immunotherapy. These include lymphocyte activation gene-3 (LAG-3), V-domain immunoglobulin-containing suppressor of T cell activation (VISTA), T cell immunoglobulin and mucin domain 3 (TIM-3), and T cell immunoreceptor with Ig and ITIM domains (TIGIT), among others [14]. These checkpoints also suppress T cell activation; clinical trial investigation of these agents and others are ongoing (Table 2).
Due to increased T cell activation, ICIs can cause not only anti-tumor responses, but also inflammatory side effects known as immune-related adverse events (irAE). irAE can affect nearly every organ system, underlining the important role of immune checkpoints in the maintenance of immune self-tolerance [15] [16]. The factors predisposing certain patients to ICI-associated toxicities, as well as the factors mediating the severity of such manifestations, remain unclear. In most cases, irAE are reversible and responsive to corticosteroids. However, fatal irAE can occur, with an incidence of 0.3–1.3% [17]. Immune-related hepatitis, encephalitis, pneumonitis, and myocarditis are most commonly associated with fatal outcomes.
Though only occurring about 1% of the time (incidence estimates 0.27%−1.14%), myocarditis has the highest fatality rate (39.7%) compared to other irAE [18] [19]. In addition, ICI-associated myocarditis frequently fails to respond to steroids and other immunosuppressants [17] [20] [21] [22]. The majority of ICI-mediated cardiotoxicity occurs within the first 3 months of therapy and the median time from initiation of therapy is 17–34 days. However, late presentations have been described. ICI-associated myocarditis occurs more frequently in patients who receive combination therapy (e.g., CTLA-4 inhibitor plus PD-1 inhibitor), and is more likely to be severe than myocarditis from ICI monotherapy, possibly due to the non-redundant, additive roles of CTLA-4 and PD-1 in immune regulation [23]. In a recently described mouse model, genetic absence of Pdcd1 (encoding PD-1) and Ctla4 haploinsufficiency recapitulate various features of ICI-associated myocarditis, including myocardial infiltration by T cells and severe electrocardiographic abnormalities (i.e., sinus node dysfunction, sinus arrest, and atrioventricular conduction block). Therapeutic intervention with abatacept (recombinant CTLA-4 immunoglobulin) rescues the fatal myocarditis in this mouse model, providing mechanistic support for inhibition of T cell co-stimulation mediated by CTLA-4 as a treatment for ICI-associated myocarditis [24].
Besides myocarditis, cardiovascular irAE also include vasculitis, pericarditis, and arrhythmias (e.g., supraventricular and ventricular tachycardia, heart block) (Figure 2). Patients can present with a wide spectrum of symptoms and signs, ranging from angina and dyspnea to cardiac arrest and fulminant myocarditis. Clinically, ICI-associated myocarditis is arrhythmogenic and is associated with myositis and a myasthenia-gravis-like syndrome, possibly due to T cell targeting of a shared antigen between skeletal and cardiac muscle [22] [23] [25] [26] [27]. Based on retrospective data, cancer patients treated with ICI seem to have a 3-fold higher risk for cardiovascular events (myocardial infarction, coronary revascularization, and ischemic stroke) compared to controls, though the mechanisms are unknown [28]. Future randomized prospective clinical trials with close monitoring of cardiovascular toxicities are needed to better understand the full spectrum of cardiotoxicities associated with ICI therapy [29]. The importance of closely monitoring for cardiovascular irAE with ICI is underscored by testing of these drugs in multiple myeloma, where the FDA placed investigations on hold due to increased toxicity-related mortality, including significant cardiovascular irAE [30].
Figure 2. Proposed mechanisms of cardiovascular toxicities associated with immunotherapies.
Immunotherapy can affect numerous components of the cardiovascular system, including the myocardium, blood vessels, electrical system and pericardium. Activated T lymphocytes (CD4+ and CD8+ T cells) following immune checkpoint inhibitors (ICI) have been shown to infiltrate into cardiomyocytes and skeletal myocytes, leading to local inflammation and damage. Additional theoretical mechanisms of ICI-mediated adverse effects include recognition of self-antigens by auto-antibodies and T cell-mediated injury of PD-L1-expressing stressed cardiomyocytes and endothelial cells. CAR T-cells can have “off-target, off-tumor” effects, as well as theoretical “on-target, off-tumor” effects. Pro-inflammatory cytokines can be upregulated following T cell-engaging therapies, leading to cytokine release syndrome, which is considered an “on-tumor, on-target” effect. Pro-inflammatory cytokines can have negative inotropic and cytotoxic effects on cardiomyocytes, resulting in decreased myocardial contractility, cardiomyopathy, and hypotension. Abbreviation: nitric oxide (NO); reactive oxygen species (ROS); T cell receptor (TCR)
Mechanisms of ICI-associated cardiovascular irAE
The mechanisms of cardiovascular irAE are incompletely understood [18]. Pathologically, ICI-myocarditis is characterized by myocardial infiltration of macrophages and T lymphocytes associated with myocyte death, consistent with the pathological definition of lymphocytic myocarditis (Figure 2) [23]. ICI-associated myocarditis is associated with infiltration of CD4+ and CD8+ T cells and CD68+ macrophages into the myocardium and conduction system. Interestingly, there is a paucity of other immune cells, such as B cells. The question remains: how is cellular tolerance disrupted, and what drives T cell infiltration into the myocardium? Identical T lymphocyte clones are observed in the heart, skeletal muscle, and tumor within the same patient [23], with a possible explanation being cross-reactivity of tumor antigens with those in the skeletal muscle and cardiac muscle, leading to infiltration of activated T cells, macrophages, and monocytes into normal muscle. The exact mechanisms by which T cells damage myocardium are likely multifactorial. The heart could experience “bystander” injury by direct T cell cytotoxic killing of myocytes, fibroblasts, endothelial, and mesothelial cells, coupled with elevated levels of pro-inflammatory cytokines from activated immune cells [31]. Indeed, proliferation of activated T cells can increase production of pro-inflammatory cytokines, including IL-1a, IL-2, IFNα2 and IL-17, in the setting of ICI therapy [32] [33]. The emergence of myocarditis and other cardiovascular irAE have peaked interest in the role of immune checkpoints in the heart. In pre-clinical studies, mice with genetic deletion of CTLA-4 or PD-1 have evidence of T lymphocyte accumulation in the heart and immune-mediated myocardial disease, underscoring a critical role for immune checkpoints in the heart [8], [9], [34].
Theoretically, there may be additional mechanisms for cardiovascular irAE: 1.) Expression of PD-L1 on normal cells (e.g., myocytes, endothelial cells) can lead to direct binding of anti-PD1-mAb to these antigens and T-cell mediated lysis of target cells. PD-L1 is normally expressed at very low levels in myocytes and endothelial cells, though inflammation (e.g., IFN-γ) and ischemia-reperfusion injury can increase expression level of PD-L1 [34] [35], [36] [37]. Furthermore, pericardial samples from patients with ICI-associated pericardial toxicity also show expression of PD-L1, suggesting that increased expression of these immune checkpoints might heighten the risk of irAE [38]. Though cardiac PD-L1 expression likely normally serves a protective role against autoreactivity by downregulating self-reactive T cell responses, increased PD-L1 expression could lead to pathologic T cell autoreactivity in the setting of immune checkpoint inhibition. 2.) Up-regulation of pre-existing auto-antibodies with subsequent recognition of self-antigens and immune-mediated attack of normal tissues. 3.) Heightened systemic activation of T cell activity against antigens present on both normal cardiac tissue and malignant cells, leading to self-antigen specific immune responses. 4.) Sub-clinical or “smoldering” microbial-induced inflammation prior to ICI therapy might become unleased by checkpoint blockade, resulting in T cell infiltration. Though this mechanism has been described in ICI-associated encephalitis and colitis, its causal relationship to cardiac irAE is currently unknown [39], [40].
Bispecific T-cell engagers
Bispecific T-cell engagers (TCE) are engineered antibodies that contain two different antigen binding sites. One binding site is generally directed against the CD3 receptor, which is the signal transduction domain of the TCR that activates cytotoxic T lymphocytes. The second is directed against a specific antigen of malignant cells (e.g., CD19, CD20, CD123, HER2, EpCAM, PSMA, etc.) [41] [42]. Binding of tumor cells directs specific effectors of the immune system to tumor cells, leading to selective lysis of tumor cells and perforin-mediated death (Figure 1B). All cells expressing the antigen are targeted for destruction. Bispecific TCE can also have non-cancer cell targets, including VEGF, TNF, and clotting factors IXa and X [43].
Blinatumomab is a TCE that is composed from two single-chain variable fragments joined together by a short glycine-serine linker. The first antigen-binding site is directed against CD3 and the second antigen-binding site is directed against the CD19 protein on the surface of B-cells, allowing endogenous T cells to recognize and destroy CD19+ lymphoma cells (Figure 1B) [44]. CD19 is a cell surface protein that is broadly expressed on the surface of more than 90% of B-cell precursor ALL blasts. Blinatumomab is FDA approved for high-risk refractory or relapsed B-cell ALL and MRD-positive B cell ALL in children and adults (Table 1). It has been shown to significantly reduce risk of death, increase long-lasting complete remission rates, and increase rate of event-free survival compared to standard chemotherapy in patients with refractory B-lineage ALL [45], [46].
Blinatumomab is associated with various adverse events, affecting virtually all treated patients. Adverse events often develop in the first few days following the first cycle of treatment and vary in severity from mild to severe, with the former including pyrexia, febrile neutropenia, and cytopenias. Common cardiovascular adverse events include hypertension (5–8%), hypotension (12–14%), and sinus tachycardia (5–6%), which are usually mild [45] [47], [48]. Serious cardiovascular adverse events are rare (<0.5%), and include myocardial infarction (MI), atrial arrhythmias, congestive heart failure, cardiac arrest, and pericardial effusion [45]. Dose-limiting toxicities in a phase 1 dose escalation study of blinatumomab were all related to excess immune system activation, but also included gastrointestinal hemorrhage, cardiac arrest associated with hypotonia and muscle weakness, and fatal heart failure [46]. Fatal adverse events arising from therapy occur in 3% of patients [36] [46] [49].
Chimeric-antigen receptor (CAR) T-cell therapy
Chimeric antigen receptor (CAR) T-cell therapy is a form of cell-based immunotherapy that uses gene transfer technology to modify autologous or allogenic T lymphocytes to stably express a single-chain fragment variable region (scFv) domain linked to the signaling domain of the T cell receptor (TCR) [50]. These engineered T lymphocytes target specific tumor antigens resulting in antitumor activity (Figure 1C). CD28 and/or 4–1BB costimulatory domains are built into the CAR T construct with the purpose of making the CAR T-cell fully activated and functional without dependence on macrophages. The CAR T manufacturing process starts with the removal of mature T cells from the body by leukapheresis, genetic engineering of T cells to express a specific CAR, cultured to achieve a pre-specified dose, and re-infusion into the patient, usually after a lymphodepleting disease-specific chemotherapy regimen is given (most commonly with fludarabine and cyclophosphamide). After infusion, the CAR T-cells multiply in the body, engaging and killing cells that express the target antigen (e.g., CD19). This immune-mediated action of T cells often includes the mobilization of other myeloid cells, which can also produce cytokines adding to systemic inflammation. CAR T-cell driven immune mediated antitumor activity is often accompanied by complete eradication of cancer cells leading to durable remissions [50].
While theoretically CAR T-cells can be made to target any tumor antigen, the FDA has approved three CAR T-cell therapies: axicabtagene ciloleucel (axi-cel), tisagenlecleucel (t-cel), and brexucabtagene autoleucel, which are autologous T cells transduced with scFv that is directed against the CD19 epitope (Table 1) [51] [52]. These adoptive T cell therapies have been shown to produce durable remissions in children and adults with relapsed refractory ALL, large B-cell lymphoma, and mantle cell lymphoma, which typically have extremely poor prognosis [52] [53] [54] [55]. CAR T-cell therapy products targeting BCMA have also achieved durable responses in patients with refractory multiple myeloma, and FDA approval is expected [56]. The successful deployment of CAR T therapy in hematological malignancies has opened the way for expansion to multiple other cancer types, including solid tumors. However, there are multiple challenges associated with targeting solid tumor surface antigens including lack of lineage-restricted target expression and widespread expression of target antigens in normal tissues [57].
Cardiovascular complications can occur in CAR T-cell therapy and include LV systolic dysfunction, myocardial injury (as evidenced by elevated troponin, ST segment changes on ECG), supraventricular arrhythmias (e.g., sinus tachycardia, atrial fibrillation), pericardial effusion, as well as possibly more life-threatening sequelae such as cardiogenic or vasodilatory shock, refractory hypotension, cardiomyopathy, and cardiac arrest [58]. The most commonly observed arrhythmias and conduction abnormalities are sinus tachycardia, atrial fibrillation, and QT prolongation [59]. A retrospective analysis of 137 patients who received CAR T-cell therapy showed that cardiovascular manifestations are frequent in patients with supraphysiologic immune activation, known as cytokine release syndrome (CRS), as described in detail below. These cardiovascular toxicities affect 59% of patients with at least mild (≥ grade 2) CRS (Table 3). Troponin elevation occurred in 54% of tested cases and decreased LV ejection fraction (LVEF) in 28% of cases where cardiac function was assessed. In those who experienced an adverse cardiovascular event, defined as CV mortality, new or decompensated heart failure, and new arrhythmias, 95% had evidence of troponin increase [60]. Similarly, in a retrospective study of 145 adult patients, 72% of patients experienced CRS, which was associated with a significant rate (21% of patients) of major cardiovascular events (MACE), defined as cardiovascular death, symptomatic heart failure, nonfatal acute coronary syndrome, nonfatal ischemic stroke, and de novo cardiac arrhythmia. Elevated baseline creatinine and ≥ grade 3 CRS were independently associated with MACE [61].
Table 3:
Cytokine Release Syndrome (CRS) Grading Scales [153]
| CRS | Lee criteria [154] | CTCAE [59] | Penn [155] | ASTCT [156] | Treatment Considerations |
|---|---|---|---|---|---|
| Grade 1 (Mild) | Not life-threatening, fever, constitutional symptoms (e.g., nausea, fatigue, myalgias, headache) | Mild reaction, tachycardia | Prodromal syndrome Mild reaction treated with supportive care |
Temperature ≥ 38° C | Monitoring Supportive care (e.g., antipyretics, antiemetics) Infusion interruption not recommended Assess for other causes (e.g., infection) |
| Grade 2 (Moderate) | Oxygen requirement < 40% OR Hypotension responsive to fluids or low dose of one vasopressor Grade ≥ 2 organ toxicity |
Stable dysrhythmias Hypotension responsive to fluid resuscitation or low dose vasopressors for < 24 hours Prophylactic medications indications for < 24 hours |
Moderate reaction – hypoxia, fever, mild hypotension. Signs of organ dysfunction (elevated Cr, LFTs) | Temperature ≥ 38° C Hypotension not requiring vasopressor OR hypoxia requiring low-flow nasal cannula or blow-by (≤ 6 L/minute. |
Supportive care Hospitalization for management of CRS-related symptoms IV fluids or low dose vasopressors Therapy interruption recommended Consider tocilizumab +/− corticosteroids |
| Grade 3 (Severe) | Oxygen requirement >= 40% OR Hypotension requiring high dose or multiple vasopressors Grade ≥ 3 organ toxicity OR Grade ≥ 4 transaminitis |
Prolonged reaction Unstable dysrhythmias Shock: requiring high dose vasopressors or multiple vasopressors for > 24 hours Signs of hypoperfusion Recurrence of symptoms following initial improvement |
Hospitalization required for management of organ dysfunction Hypotension treated with IV fluids or vasopressors Coagulopathy requiring FFP or cryoprecipitate Hypoxia requiring supplemental oxygen |
Temperature ≥ 38° C with hypotension requiring a vasopressor or hypoxia Requiring high-flow nasal cannula, facemask, nonrebreather mask, or Venturi mask. |
Supportive care Hospitalization required High dose or multiple vasopressors. Mechanical ventilation Tocilizumab +/− corticosteroids (second line) |
| Grade 4 (Life-threatening) | Requirement for ventilator support OR Grade ≥ 4 organ toxicity (excluding transaminitis) |
Life-threatening dysrhythmias Refractory shock requiring multiple vasopressors Ventilator support indicated Cardiomyopathy: LVEF < 20% |
Resistant CRS, no clinical improvement within 12–18 hours or worsening at any time despite prior management. Hypotension requiring high dose vasopressors Hypoxia requiring mechanical ventilation |
Temperature ≥ 38° C with hyotension requiring multiple vasopressors (excluding vasopressin). OR hypoxia requiring positive pressure (e.g., CPAP, BiPAP, intubation and mechanical ventilation) |
Supportive care Multiple vasopressors Mechanical ventilation Tocilizumab Corticosteroids Consider third line therapy or alternative measures |
| Grade 5 | Death | Death | Death | Death |
Based on CRS experience (Guidelines from NCI Experimental Transplantation and Immunology Branch), consider IL-6 inhibitor (e.g., tocilizumab) use if: LVEF < 40% by echocardiogram, NE requirement >=2 ug/min for 48 hours since the first administration of NE, SBP of 90mmHg that cannot be maintained by NE, O2 requirement of FiO2 > 50% or more for more than 2 hours continuously [157]. ASTCT consensus guidelines: Low flow: ≤ 6 L/minute
However, it is important to note the potential bias in such retrospective analyses since only some patients undergo cardiovascular evaluation, presumably patients with suspicion of cardiovascular issues. In the above study, for example, only 29 patients (out of 137) had follow-up echocardiograms, with 8 patients having evidence of cardiomyopathy. Moreover, cardiomyopathy in these cases may represent a measure of overall “sickness.” Unbiased, prospective studies are needed to better determine relevant cardiovascular issues in CAR T-cell treated populations. Further assessment of cardiovascular toxicities in a real-world population of patients treated with axicabtagene ciloleucel and tisagenlecleucel-T showed arrhythmias, cardiorenal syndrome, stress cardiomyopathy, LV dysfunction, and cardiac arrest in a small number of patients, with most of these events occurring in the context of CRS [62]. This study has significant limitations in that the total number of patients without cardiovascular issues is unclear.
Mechanisms of cardiovascular toxicities from T cell-based therapies.
The molecular mechanisms of cardiovascular toxicity from T cell-based therapies are incompletely understood. While some of these mechanisms may be more theoretical, each may become relevant given the explosion of T cell-based therapies and the increasing number of possible targets. Toxicities can result from an “on-target, on-tumor” effects (triggered by excessive cytokine release), “on-target, off-tumor” (resulting from direct attack by T cells of normal tissue that share a common antigen with the tumor) or “off-target, off-tumor” effect (where the transduced T cell unexpectedly attacks an antigen other than the intended tumor antigen) (Figure 2).
In clinical practice, an “off-target, off-tumor” toxicity could result from recognition of an unknown epitope expressed by normal tissue. An early example was observed after treatment of two patients with engineered T cells expressing affinity-enhanced TCR against MAGE-A3, a cancer germline antigen expressed in multiple tumor types (Figure 2). Both patients developed fatal cardiogenic shock and heart failure, with significant elevation of troponin as well as elevation of cytokines, including IFN-γ, IL-6, and IL-8. Autopsy from both patients showed T cell infiltration into the myocardium and cardiac myonecrosis, consistent with myocarditis [63]. In these cases, MAGE-A3 expression was not detected in the heart. Instead, the TCR directed to the MAGE-A3 epitope also recognized titin, a unrelated sarcomeric protein expressed by the heart, with titin mutations being a common cause of dilated cardiomyopathy [64]. These cases highlight that TCR recognition of an unrelated peptide can lead to dramatic off target cardiovascular complications, underlining the importance of targeting epitopes expressed exclusively on malignant cells to avoid reactivity with normal tissues.
The example of the cardiotoxicity seen with MAGE-A3 directed T cells, also introduces a greater need for consideration of additional mechanisms of cardiovascular toxicity from T cell-engaging therapies. Theoretically, these may also include mispairing of introduced and endogenous TCR α/β chains resulting in novel specificities. T cell-engaging therapies might cause alloreactivity against the host cardiac myocytes, whereby there is a primary T cell response against allelic variants of MHC molecules that were not encountered during thymic development, manifesting as graft versus host disease. These therapies are also associated with T cell infiltration into the myocardium through unclear mechanisms [63]. More globally, the injury could be due to direct T cell-mediated cytotoxicity or from “bystander injury,” whereby the cardiovascular system is damaged by pro-inflammatory cytokines (e.g., IL-6, IL-8, TNF-α). Indeed, excess activation of the immune system with resulting cytokine release syndrome is thought to be a major contributor to side effects of CAR T-cell therapy and a major driver of toxicity, as described below.
Cytokine release syndrome.
Theoretically, any immune-based therapy can result in a systemic heightened immune response, leading to excessive or even uncontrolled release of proinflammatory cytokines, commonly referred to as cytokine release syndrome (CRS). Cytokines are secreted glycoproteins and membrane-bound proteins that act as chemical messengers of intercellular signaling. They regulate homeostasis of the immune system and have important roles in cell proliferation, differentiation, and inflammation. Cytokines are produced by activated T lymphocytes, macrophages, monocytes, and endothelial cells, and act via specific membrane-bound receptors [65]. Numerous types of cytokines, including interferons (IFN), interleukins (IL), chemokines, colony-stimulating factors, and tumor necrosis factors (TNF), can be elevated following T cell-mediated therapies.
The frequency of CRS varies depending on the type of immune therapy and degree of immune activation. CRS affects 2–19% of patients treated with blinatumomab, though is usually mild to moderate, does not require cessation of treatment, and lasts on average 6.5 days (95% CI, 5–16 days) [45] [66]. CRS is common in patients treated with CAR T-cell therapy, affecting 58–100% of patients, and usually starts between 1–12 days after first infusion (median: 5 days) [55], [67], [68]. In contrast, CRS is uncommon following ICI therapy. Based on safety reports of ICI-related adverse drug reactions gathered by VigiBase, CRS cases range from 0.05–0.14%, with the highest cases reported with anti-PD1/PD-L1 antibody use [69].
Though CRS is common with the administration of T cell-mediated therapies, the specifics of how dysregulation of downstream signaling pathways leads to cardiovascular toxicities are still poorly understood. CRS is thought to occur due to the release of supraphysiologic levels of cytokines by activated lymphocytes and myeloid cells (e.g., macrophages, dendritic cells, monocytes) related to immune activation following tumor antigen recognition, reflecting an “on-target, on-tumor” side effect from immune system stimulation. The factors that predispose only certain individuals to moderate to severe CRS include high disease burden, higher initial dose of T cell engaging therapies (e.g., peak CAR T-cell expansion by copies/μg), and degree of T cell activation [53] [70].
Clinical Manifestations of CRS.
Clinically, inflammatory symptoms of CRS range from mild and self-limited (e.g., fever, chills, myalgias, malaise) to life-threatening (e.g., severe hypotension, vascular leak, hypoxic respiratory failure, acute thrombosis, and encephalopathy syndrome). Though different CRS grading scales exist, the severity of CRS symptoms is based on a 5-point scale, where grade 1 symptoms are mild and grade 5 represents death (Table 3). CRS is often self-limited, though can lead to life-threatening symptoms that require aggressive supportive care and management [71].
CRS was initially described in the 1980s following administration of high dose interleukin-2 (IL-2) in patients with metastatic cancer. Though treatment led to cancer response in a subset of patients, systemic toxicities including hypotension, capillary permeability, interstitial pulmonary edema, and acute kidney injury were observed, which usually subsided after cessation of IL-2 therapy [72]. CRS is also observed in graft-versus-host disease, which can cause cytokine dysregulation (e.g., elevated levels of IL-1, IL-2, TNFα), following allogeneic bone marrow transplantation [73].
Immune effector cell-associated neurotoxicity syndrome (ICANS), also referred to as CAR T-cell-related encephalopathy syndrome (CRES), is a distinct neurotoxic adverse event associated with CAR T-cell therapy and CRS. ICANS can have heterogenous symptoms, ranging from mild to life-threatening. The pathophysiology of ICANS is not fully understood, though potentially related to disruption of the blood brain barrier and subsequent influx of cytokines and inflammatory cells. A recent study using single-cell RNA sequencing analysis demonstrates that CD19 is expressed in human brain mural cells that are critical for blood-brain-barrier integrity, suggesting that this cell population may contribute to the neurotoxicity of CD19-directed CAR T-cells [74]. Further details of ICANS are reviewed elsewhere [75].
As discussed above, cardiovascular issues can arise in the setting of CRS and include LV systolic dysfunction or arrhythmias; however, the overall significance of cardiovascular events is unknown. It is unclear, for example, what the clinically significant implications of mild, potentially reversible LV dysfunction would be, if any. In this regard, LV dysfunction or elevated myocardial biomarkers in CRS might be similar to a clinical scenario in which patients develop demand ischemia (i.e., oxygen supply and demand mismatch) or stress-induced cardiomyopathy rather than direct myocardial injury. The cardiovascular issues associated with CRS also seem distinct from ICI-associated myocarditis, which is an overwhelming toxicity with a high fatality rate. These issues need further exploration with carefully designed prospective studies, especially given the explosion of novel cancer immunotherapies that are anticipated to be introduced clinically in the coming years.
Pro-inflammatory cytokines in CRS
Early data from clinical trials of immunotherapies implicate pro-inflammatory cytokines in the pathogenesis of CRS. In adult patients with B-lineage ALL treated with blinatumomab, FACS-based analysis shows polyclonal T cell activation (based on surface expression of the immediate early activation marker CD69) and proliferation of CD 8+ and CD 4+ T lymphocytes [76]. There is significant increase of the T-cell effector memory subset, including up-regulation of antigen-specific CD8+ T cells [71]. Blinatumomab treatment also promotes the release of numerous cytokines, including IL-2, IL-6, IL-10, and TNF-α [76]. Patients with CRS have higher peak levels of IFN-γ (mean 886 vs 135 pg/mL), IL-10 (mean 2279 vs 427 pg/mL), IL-6 (mean 17167 vs 926 pg/mL), and TNF-α (mean 144 vs 21 pg/mL) [46].
Severe CRS (grade 4–5) following CAR T-cell therapy is associated with an increase in twenty-four circulating cytokines based on retrospective data from 39 children and 12 adults with refractory or recurrent ALL treated with CTL019 at a single institution [70]. 48 out of the 51 patients (94%) developed CRS, with the majority experiencing mild to moderate symptoms. Twenty patients (39%) required vasoactive medications – the majority of these patients had distributive shock, though one patient experienced cardiogenic shock. 14 patients (27%) experienced severe CRS (grade 4–5), nine patients required mechanical ventilation, and three adult patients died from CRS-related complications. Serial measurements of 43 cytokines and biomarkers in these patients found that the peak measurements of 24 cytokines were associated with severe CRS compared to mild to moderate disease. These cytokines included those released from activated T cells (e.g., IL-6, IFN-γ, sIL2Ra, GM-CSF) and activated monocytes/macrophages (IL1RA, IL-10, IL-6, IP10, MIG, IFN-α, MIP1-α, MIP1-β, sILR). Based on cytokine profiling in patients who developed CRS, IFN-γ, soluble gp130 (sgp130), and sIL1RA predicted development of severe CRS. Other studies have shown that patients who develop severe CRS have higher peak levels of IL-6 than those with milder disease [53] [77].
Pre-clinical models of CRS
The clinical emergence of CRS has accelerated efforts to model the syndrome pre-clinically. In one elegant model, human T cells that had been matured in humanized mice (and thus were xenotolerant) were injected into leukemia-bearing humanized mice, resulting in anti-tumor activity but also an early, severe CRS-like syndrome [78]. These mice also experienced neurotoxicity, characterized by generalized paralysis and seizures. Monocyte depletion in these tumor-bearing, humanized mice before CAR T-cell infusion suppressed CRS but also attenuated tumor elimination by the CAR T-cells. In another mouse model of symptoms of CRS, intraperitoneal tumor growth allowed for a sufficient tumor burden to accumulate and for severe CRS to develop in immunodeficient mice [79]. In both mouse models, CRS was characterized by reduced activity, malaise, and weight loss within 2–3 days of CAR T-cell administration. Several pro-inflammatory cytokines and chemokines, including IL-6, mIL-6, mCCL2, mG-CSF, and hIL-2, were produced by activated myeloid cells and correlated with CRS severity, consistent with the clinical syndrome in patients.
Both models provide a means to better interrogate the role of cytokines in CRS, as well as possible therapeutic interventions. Interestingly, IL-1 preceded IL-6 induction in one of the studies, and while inhibiting either IL-1 and IL-6 attenuated CRS, only mice treated with IL-1 receptor blockade were free of neurotoxicity [78]. In the second model, IL-6 and IL-1 were produced by endogenous murine macrophages, rather than by infused human CAR T-cells. In addition to IL-6, both IL-1 and nitric oxide are critical determinants of CRS in this model. Treatment with the IL-1 receptor antagonist (anakinra) and iNOS inhibitors abrogated CRS-related morbidity and mortality. Similarly, these benefits were seen in mice that received CAR T-cells that constitutively produced IL-1 receptor antagonist. These models provide tremendous insights into the mechanisms of CRS and may be particularly conducive in terms of understanding cardiovascular toxicities of these therapies.
Treatment of CRS with immune-modulating therapies
Mild to moderate cases of CRS are self-limited and improve with supportive measures. However, severe cases of CRS require aggressive intervention, including mechanical ventilation and vasopressor support, as well as immune-modulating therapies to blunt the inflammatory response (Table 3) [59].
Based on early clinical experience with CAR T-cell therapy, IL-6 is considered a central mediator of CRS toxicity. IL-6 is a pro-inflammatory cytokine that is released from activated lymphocytes, dendritic cells, macrophages, and endothelial cells during the acute phase of the immune response. Under normal conditions, IL-6 is expressed at very low levels (on the order of pg/mL) and has roles in neutrophil trafficking, angiogenesis, autoantibody production, and B cell differentiation [80]. It is a major inducer of the hepatic acute phase response (e.g., ferritin, CRP, hepcidin, fibrinogen). Circulating levels of IL-6 increase markedly following T cell-mediated therapies, as well as in other hyper-inflammatory states (e.g., sepsis, graft versus host disease) [53], [77] [81] [82].
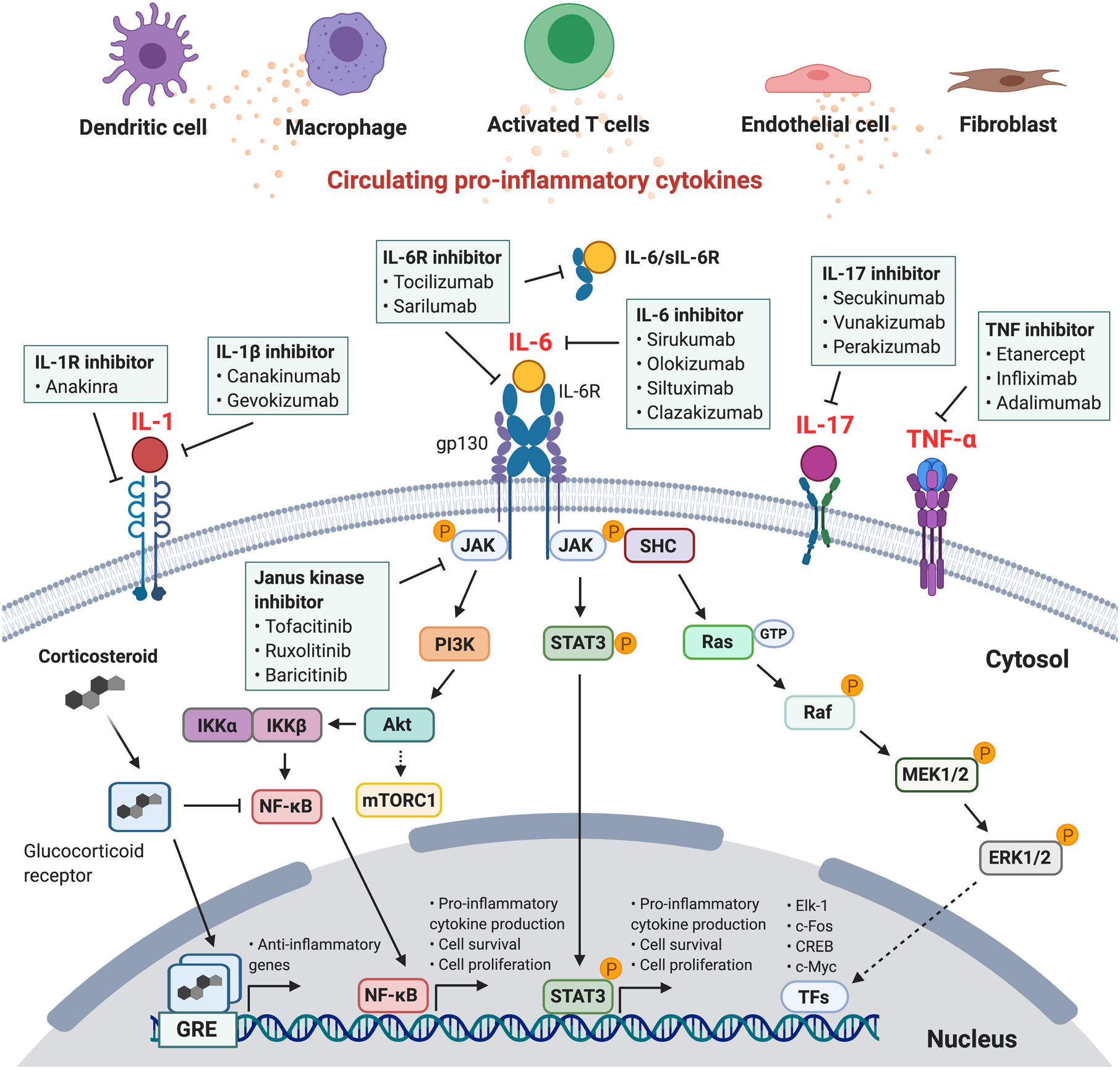
IL-6 signaling occurs via the membrane-bound or soluble IL-6 receptor (sIL-6R), also known as classical or trans-IL-6 signaling, respectively. In the classic signaling pathway, IL-6 binds to the IL-6 receptor (IL-6R) on the cell surface (Figure 3). This complex then interacts with the membrane-bound glycoprotein 130 receptor subunit (gp130). Though gp130 is ubiquitously expressed, membrane-bound IL-6R expression is limited to a few cell types (e.g., leukocytes, megakaryocytes, hepatocytes, barrier epithelial cells), thus limiting classic IL-6 signaling to these specific cell types. In contrast, in trans-IL-6 signaling, circulating sIL-6R and IL-6 bind to form a complex that then associates with membrane-bound gp130 [70], leading to activation of the trans-IL-6 signaling pathway on potentially all cells. Both classic and trans-IL-6 signaling leads to downstream signaling with gp130, activating intracellular signaling via the JAK/STAT3, PI3K/Akt, and Ras/mitogen-activated protein signaling pathways. These pathways augment cytokine release and up-regulate pro-inflammatory transcription targets (Figure 3) [83].
Figure 3. Pro-inflammatory cytokines, IL-6 cytokine intracellular signaling cascades, and immunomodulating therapies that inhibit intracellular cytokine signaling.

Cytokines are produced by numerous cell types, including activated T lymphocytes, dendritic cells, and macrophages. IL-6 signaling occurs via the membrane-bound or soluble IL-6 receptor (sIL-6R), also known as classical or trans-IL-6 signaling, respectively. IL-6R interacts with the membrane-bound glycoprotein 130 receptor subunit (gp130), leading to downstream pro-inflammatory signaling cascades via the JAK/STAT3, PI3K/Akt, and Ras/mitogen-activated protein signaling pathways. Various cytokines act via membrane-bound receptors that can be inhibited by targeted immunomodulating monoclonal antibodies. Glucocorticoids bind to the intracellular glucocorticoid receptor. The complex translocates to the nucleus and binds to the glucocorticoid-responsive element (GRE) complex as a homodimer, regulating the transcription of anti-inflammatory genes. Abbreviations: extracellular signal-regulated protein kinase (ERK); IκB kinase (IKK); Interleukin-6 (IL-6); Janus kinase (JAK); mammalian target of rapamycin (mTOR); mitogen-activated protein kinase (MAPK); nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB); phosphoinositide 3-kinase (PI3K); SHC adaptor protein (SHC); signal transducer and activator of transcription (STAT); transcription factors (TFs)
Tocilizumab is a recombinant human monoclonal antibody specifically binds soluble and membrane-bound IL-6 receptors (IL-6R), disrupting both classic and trans-signaling (Figure 3). Tocilizumab interferes with the cytokine feedback loop, blocks the inflammatory response, and decreases circulating levels of IL-6. Tocilizumab is primarily indicated for juvenile idiopathic arthritis and rheumatoid arthritis. However, the landmark studies that demonstrated the efficacy of CAR T-cell therapy also described the efficacy of IL-6 signaling blockade using tocilizumab in the management of severe CRS. In an initial study of two children with relapsed and refractory B-cell ALL, both developed CRS with grade 3–4 adverse events. The first patient (patient 1), a 7-year-old girl, experienced febrile neutropenia (peak temperature 40.7°C), hypotension requiring vasopressor support, acute vascular leak syndrome, and ARDS. Patient 2, a 10-year-old girl, developed encephalopathy and elevated LDH and ferritin levels. Both patients had markedly elevated inflammatory markers, including IL-6. Due to life-threatening symptoms that were refractory to standard treatment with steroids, patient 1 received tocilizumab, which reversed CRS, including resolution of fever and weaning of vasopressors within 24 hours. Patient 2 improved with supportive care and did not require glucocorticoids or anti-cytokine therapy [84].
In a subsequent study, 30 patients (25 children, 5 adults) with refractory ALL developed CRS following CAR T-cell therapy. Of these, 27% had severe symptoms, including encephalopathy, hypotension, and hypoxic respiratory failure. These patients were successfully treated with tocilizumab, which led to defervescence within hours and stabilization of hemodynamic parameters within 24–36 hours [53] [70]. Additional studies have also assessed the effectiveness of tocilizumab in treating severe CRS secondary to T cell-engaging therapies, including those with cardiovascular events [60] [85] [86] [87]. In patients with CRS-mediated CV events, each 12 hour delay of tocilizumab administration increases the risk of CV events by 1.7-fold [60].
Due to its clinical success in the management of severe CRS, tocilizumab was FDA approved for CRS in 2018 and is currently considered the front-line treatment for severe CRS secondary to CAR T-cell therapy. It is frequently administered in patients with CRS grade ≥ 2 that is refractory to IV fluids or low dose vasopressor support (Table 3). Importantly, tocilizumab does not appear to compromise efficacy of CAR T-cell therapy. The success of IL-6R antagonism in treating CRS highlights the central role of IL-6 signaling in other hyperinflammatory syndromes. Siltuximab is a chimeric mouse-human antibody that directly targets circulating IL-6, rather than its receptor and prevents binding of IL-6 with both membrane-bound and soluble IL-6 receptors (Figure 3). It is currently FDA approved for idiopathic multicentric Castleman’s disease. While siltuximab may be a reasonable alternative agent for treatment of CRS, clinical evidence in the setting of CAR T-cell therapy is lacking. Specifically, it is unclear whether siltuximab is equivalent to tocilizumab or if it can be used in a situation where the latter is ineffective [88].
In addition to IL-6 inhibition, other potential targetable cytokine pathways for managing CRS include IL-1 and TNF-α (Figure 3). Pre-clinical studies demonstrate that IL-1R antagonism with anakinra, might be a therapeutic option for CRS-associated neurotoxicity due to its ability to cross the blood brain barrier [78]. There are clinical studies assessing the effectiveness of IL-1R antagonism as a preventative strategy for severe neurotoxicity and CRS in patients following CAR T-cell therapy. Though TNF-α is often elevated in CRS, inhibition with etanercept has not reliably demonstrated benefit in severe CRS in the setting of CAR T-cell therapy [84]. Corticosteroids can also inhibit the immune response. However, due to concern about theoretically reducing CAR T-cell persistence and thus compromising CAR T-cell effectiveness, corticosteroids are considered a second line therapy for CRS that is refractory to anti-IL-6R therapy.
There are limited data in the treatment of steroid-refractory immune mediated adverse events from ICIs with cytokine signaling blockade. In a case cohort of 87 patients treated with nivolumab (anti-PD-1 mAb), 34 of these patients (39.1%) received tocilizumab due to steroid-refractory irAE, including for pneumonitis, cerebritis, hepatitis, and immune-mediated coagulopathy. Treatment was associated with significant decrease in CRP from a median of 109.3 mg/L to 19.2 mg/L, as well as clinical improvement in the majority of patients (79.4%) [89]. The IL-17 receptor inhibitor, secukinumab, has been used successfully to treat immune-mediated skin and gastrointestinal toxicities associated with pembrolizumab [90]. The role of these inhibitors of cytokine signaling in cardiovascular toxicities remains to be determined.
Cytokines and cardiovascular toxicity
Cardiovascular toxicities from excess cytokines in humans
High dose cytokines have anti-neoplastic and pro-apoptotic properties and were the first approved “immunotherapies” for cancer. They are also associated with cardiovascular toxicities. Single agent high-dose IL-2 can induce durable responses in select patients with melanoma or kidney cancer, though response rates are generally low and use is frequently limited by toxicities. Of the cardiovascular toxicities, hemodynamic alterations are most common; these are likely related to capillary leak syndrome, leading to systemic hypotension, volume overload, and hypoxic respiratory failure [91] [92]. Pulmonary edema is common, affecting up to 53% of patients [93]. IL-2 treatment can also cause transient, acute reductions in biventricular systolic function [94], and has been associated with increasing the risk of myocardial infarction and supraventricular arrhythmias [95] [96]. Though rare (1.5% of 652 consecutive patients), high-dose IL-2 therapy can have fatal complications, attributed to severe cardiorespiratory failure, acute MI with transmural myocardial necrosis, and myocarditis. In a case series of autopsy findings, 6 of 8 patients had diffuse, patchy myocarditis with a predominantly lymphocytic inflammatory infiltrate and cardiomyocyte necrosis [97]. Similarly, high dose IFN-α is approved for a number of malignancies and hepatitis C, though with low response rate and relative unfavorable toxicity profile [98]. Cardiovascular side effects, though uncommon, are usually dose dependent and include systemic hypotension, dilated cardiomyopathy, congestive heart failure, arrhythmias (atrial fibrillation, non-sustained VT, AV block), and MI [99]. In patients with hepatitis C treated with IFN-α, 3.2% of patients experienced cardiovascular adverse events, including arrhythmias (atrial fibrillation, AV block), coronary artery vasospasm, and transient LV dysfunction. In general, these toxicities are not life-threatening and resolve after discontinuation of therapy [100].
Additional cytokines in clinical cancer immunotherapy are still in their infancy or have been stymied by significant dose-limiting toxicities. In a phase I clinical study of patients with GI cancer, recombinant human interleukin-1β (IL-1β), alone or in combination with 5-FU, caused hypotension as a dose-limiting side effect in 60% of patients [101]. In the first-in-human clinical trial using recombinant human IL-15, patients experienced dose-limiting toxicities including high fever and hypotension. These clinical toxicities were associated with marked elevations in several cytokines, including IL-6, IL-8, IFN-γ, and TNF-α [102]. Early clinical studies with adoptively transferred human tumor-infiltrating lymphocytes genetically engineered to secrete single-chain IL-12 showed dose-related toxicities including life-threatening hypotension [103]. In addition, dose-dependent cardiovascular side effects have been observed in patients with metastatic melanoma and locally advanced soft tissue sarcomas treated with TNF-α – these reversible side effects include hypotension, coronary artery thrombosis, volume overload, and dilated cardiomyopathy [104]. Multiple clinical trials with cytokines in combination with immune checkpoint inhibitors are currently underway to determine if cytokines could affect NK and T lymphocyte function and overcome resistance mechanisms associated with ICI use.
Elevated levels of pro-inflammatory cytokines have also been associated with chronic heart failure, myocardial infarction, cardiomyopathies, cardiac transplant rejection, and atherosclerosis, though it is unclear if elevated cytokines directly contribute to cardiac pathology or if they are a biomarker of overall disease severity [105]. Elevated levels of TNF-α, IL-1, IL-6, IL-15, and IL-17 correlate to decreased functional status in patients with chronic heart failure [106] [107]. The role of cytokines in chronic cardiovascular disease are reviewed elsewhere [108] [109] [110] [111].
Pre-clinical models of cardiovascular toxicities from excess pro-inflammatory cytokines
While the mechanisms by which excess cytokines and CRS lead to cardiovascular perturbations are not entirely clear in humans, pre-clinical studies offer insights into pathogenesis of cytokine-mediated cardiac dysfunction. Pro-inflammatory cytokines, including TNF-α, IL-1B, IL-2, and IL-6 can have cardio-depressant, cytotoxic, and apoptotic effects [112]. The negative inotropic effects of cytokines have been studied in numerous contexts, including in vivo (whole animal), ex vivo (heart, isolated papillary muscles), and in vitro models. Sustained cytokine expression (>30 minutes) can decrease myocyte contractility and β-adrenergic receptor responsiveness by increasing nitric oxide generation, increasing synthesis of reactive oxygen species, and reducing β-adrenergic receptor responsiveness.
TNF-α
TNF-α is a pro-inflammatory cytokine that causes left ventricular dysfunction and hemodynamic changes over acute and chronic scales. In conscious dogs, a single intravenous infusion over 1 hour of recombinant human TNF-α decreased mean arterial pressure and LVEF in a dose-dependent manner. These changes occurred within 2 hours and up to 2 days post-infusion, with hemodynamic parameters normalizing after 7–10 days [113]. The negative inotropic effects of TNF-α in a dog model have been replicated in several studies, demonstrating that these effects can last from hours to days [114]. Pre-clinical models have shown that circulating TNF-α contributes to cardiac dysfunction over more chronic time scales. In a rodent model that exposed animals to levels of TNF-α seen in patients with chronic heart failure (~80–100 U/mL), TNF-α infusion over 15 days was associated with cardiac dysfunction, LV dilation, and depression of cardiac myocyte shortening. These effects were partially reversible with a soluble TNF-α antagonist [115]. Due to absence of heart tissue histology in these studies, it is unclear if TNF-α exposure alone leads to T lymphocyte infiltration and lymphocytic myocarditis.
In primary cardiomyocytes isolated from rats, TNF-α exposure increases ROS production, decreases Ca2+ channel reactivity to electrical stimulation, and decreases contractility. Induction of the anti-oxidant, glutathione, following treatment with NAC decreases ROS levels and improves contractility, suggesting that the negative inotropic effects of TNF-α are at least partially due to increased ROS [116]. In cultured rat cardiomyocytes, TNF-α causes concentration dependent depression of maximum extent and peak velocity of myocyte shortening in vitro [117].
Cardiomyocyte over-expression of TNF-α can result in cardiomyopathy and myocarditis. Transgenic mice over-expressing TNF-α specifically in cardiomyocytes develop dilated cardiomyopathy, with severity directly correlated to degree of TNF-α expression [118]. In another transgenic mouse model of murine TNF-α driven by the murine α-myosin heavy chain promoter, mice develop severe dilated cardiomyopathy, myocyte apoptosis, transmural myocarditis with a predominantly lymphocyte infiltrate, biventricular fibrosis, atrial thrombosis, and early mortality, demonstrating that myocyte production of TNF-α is sufficient to cause severe cardiac disease [119].
Furthermore, TNF-α has direct cytotoxic and pro-apoptotic effects. For example, in isolated adult rat cardiac myocytes, TNF-α expression is directly associated with cardiomyocyte apoptosis [120] [121]. The precise downstream signaling cascades that result in myocyte apoptosis are not well understood. These pre-clinical studies suggest that upregulation of TNF-α in the setting of ischemia-reperfusion injury, chronic heart failure, and other states of chronic inflammation could contribute to persistent cardiac dysfunction and myocyte death [122].
IL-1B
Reductions in LV function have been observed in intracoronary injection of IL-1B coated microspheres in a dog model. In adult guinea pig ventricular myocytes, exposure to human recombinant IL-1B for > 1 hour decreased β-adrenergic control of cardiac Ca2+ channels by increasing nitric oxide synthase. Increased nitric oxide synthesis blunts β-adrenergic signaling.
The impaired contractility was improved with IL-1 receptor antagonism and inhibition of nitric oxide synthase with NG-monomethyl-L-arginine [123]. Both IL-1B and TNF-α reduced contractility in an ex vivo model of isolated rat hearts by increasing NO content and production of peroxynitrite (ONOO-), a reactive nitrogen species created from the reaction of NO with superoxide. Myocardial function improved following treatment with a NO synthase inhibitor (N(G)-nitro-L-arginine) and the superoxide scavenger, tiron [124].
IL-2
In isolated hamster papillary muscles, IL-2 inhibited contractility in a concentration-dependent, reversible manner. These pro-inflammatory cytokines induced myocardial nitric oxide synthase, and treatment with the nitric oxide synthase inhibitor, NG-monomethyl-L-arginine (L-NMMA) improved myocyte contractility [125].
IL-6
In a rat model, a single bolus of IL-6 altered myocardial hemodynamics, leading to a transient reduction of MAP and LVESP. These effects were abolished with administration of an eNOS (neuronal synthase) inhibitor, suggesting that induction of NO synthesis is regulated by IL-6 and causes negative cardiac inotropism [126]. In single ventricular myocytes isolated from adult rats, IL-6 increased phosphorylation of STAT3 and ERK1/2 within minutes of exposure, and subsequently increased de novo synthesis of iNOS and NO production. iNO contributed to decreased cardiac contractility after 2 hours of incubation with IL-6. These effects were abrogated with JAK2 inhibition [127].
Taken together, the above results clearly suggest that pro-inflammatory cytokines can have negative inotropic and cytotoxic effects on cardiomyocytes. In vitro experiments, however, fail to recapitulate the complexities of in vivo exposure, including non-cell autonomous effects. It is also unclear whether classic myocarditis (myocardial immune infiltration with subsequent myocyte death) occurs in CRS due to the absence of histology in these pre-clinical studies. However, as the oncologic use of immunotherapies continues to grow, it is increasingly critical to better understand the effects of pro-inflammatory cytokines on the cardiovascular system.
Cytokine-Driven Hyperinflammatory Syndromes: Insights on Cytokine Release Syndrome in COVID-19
The CRS-associated cardiovascular toxicities arising from cancer immunotherapies are also observed in other cytokine-driven hyperinflammatory syndromes, also known as cytokine storm. Numerous infectious and non-infectious causes cause cytokine-driven inflammatory syndromes. Non-infectious causes include autoimmune disease (e.g., multiple sclerosis, rheumatoid arthritis), malignancies, graft versus host disease, macrophage activation syndrome (MAS), and hemophagocytic lymphohistiocytosis (HLH). MAS and HLH are severe systemic hyperinflammatory syndromes, often secondary to malignancy, rheumatologic disease, or severe viral infection [70] [85]. Severe complications from certain infections can also occur in the context of supraphysiologic immune activation. Examples include Middle East respiratory syndrome coronavirus (MERS-CoV), severe acute respiratory syndrome coronavirus (SARS-CoV), and avian flu, among others (Figure 4). Notably, cytokine storm is seen in a subset of patients with severe coronavirus disease-2019 (COVID-19) due to severe acute respiratory syndrome-coronavirus 2 (SARS-CoV-2) (Figure 4). The overlapping features of CRS and cytokine storm in COVID-19 are highlighted here to demonstrate how the clinical experience with immunotherapy-mediated CRS could motivate therapeutic considerations in COVID-19.
Figure 4. Cardiovascular toxicities associated with infectious and non-infectious cytokine-driven hyperinflammatory syndromes.

Cytokine-driven hyperinflammatory syndromes have various infectious and non-infectious causes. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a β-coronavirus that causes coronavirus disease-2019 (COVID-19). SARS-CoV-2 infection can result in T cell activation, immune dysregulation, and excess cytokine production, known as cytokine storm, which is associated with severe COVID-19 illness. CAR T-cell therapy also causes supraphysiologic immune activation and cytokine release syndrome, characterized by excess cytokine production from macrophages, monocytes, and dendritic cells. Overlapping cardiovascular toxicities associated with these cytokine-driven hyperinflammatory syndromes include cardiac injury, cardiomyopathy, arrhythmias, thrombosis, and vascular leak. Treatment considerations include immunomodulators. Abbreviations: chemokine receptor 6 (CCR6); chimeric antigen receptor (CAR); human leukocyte antigen-DR isotype (HLA-DR); interleukin (IL); macrophage inflammatory protein (MIP); Middle East Respiratory Syndrome (MERS); monocyte chemoattractant protein-1 (MCP-1); tumor necrosis factor (TNF); *: therapies under active clinical investigation.
While severe SARS-CoV-2 infection primarily leads to hypoxic respiratory failure, cardiovascular manifestations include capillary leak syndrome, hypercoagulability, cardiomyopathy, arrhythmias, acute heart failure, and cardiac arrest, with significant overlap with CRS [128] [129]. Severely ill patients can have newly reduced LV function and elevated cardiac biomarkers, which portend worse outcomes [130]. Emerging retrospective data suggest a positive correlation of troponin elevation (a marker of cardiac injury) with elevated IL-6 and D-dimer levels during hospitalization suggesting cytokine-mediated toxicity [131]. In some cases, cardiac MRI has shown biventricular myocardial interstitial edema and late gadolinium enhancement, which can be coupled with active lymphocytic inflammation on endomyocardial biopsy, consistent with acute myopericarditis [132] [133] [134]. Some patients with acute LV systolic dysfunction have subsequent normalization of LV function, demonstrating that the cardiomyopathy can be reversible [135], [136].
Our current understanding of COVID-19 pathogenesis continues to evolve, and the mechanisms of cardiovascular complications in COVID-19 are likely multifactorial [137]. Some patients with severe illness experience persistent anti-viral immune responses, resulting in massive production of inflammatory cytokines, which is a hallmark of cytokine storm. In this subset of patients with cytokine storm, there is evidence of circulating T cell-activation based on flow cytometry. This includes elevated levels of HLA-DR and CD38 double-positive fractions, increased concentration of the highly inflammatory CCR6+ Th17 in CD4+ T cells and inflammatory myocytes, and higher concentration of perforin and granulysin positive CD8+ cells [133] [138]. Patients requiring ICU-level of care have higher levels of inflammatory cytokines, including IL-1β, IL2, IL-6, IL7, IL10, MCP1, MIP1a, and TNF-α [139]. Elevated levels of pro-inflammatory cytokines are frequently associated with moderate to severe illness, including cardiomyopathy, cardiac injury, respiratory failure, and increased mortality [140] [141] [135] [142]. Future studies are needed to better understand the mechanisms of immune- and cytokine-mediated cardiovascular injury following SARS-CoV-2 infection.
Similar to CRS, the pro-inflammatory cytokine, IL-6, seems to be an important mediator of severe COVID-19 illness in patients with cytokine storm and is associated with increased mortality [143], raising the possibility that inhibitors of IL-6-IL-6R signaling, as well as other disease-modifying anti-rheumatic drugs (e.g., JAK inhibitors, IL-1 receptor antagonists) can be effective [144], [145]. Their use is currently being explored in randomized controlled trials. Systemic corticosteroids might improve outcomes and reduce mortality in a subset of critically ill patients with COVID-19 after the first week of illness, further supporting that excess inflammation has an important role in severe cases of COVID-19 [146] [147]. High-quality randomized controlled trials are needed to conclusively determine the role of immunomodulating therapies in cytokine storm in COVID-19.
Finally, pediatric patients with COVID-19 can develop a rare hyperinflammatory condition called multisystem inflammatory syndrome in children (MIS-C) [148], characterized by elevated pro-inflammatory cytokines (e.g., IL-6). These patients experience persistent fevers, gastrointestinal symptoms, lymphadenopathy, arterial vasculitis, and cardiovascular complications, including cardiomyopathy, thrombosis, and refractory hypotension [149]. MIS-C has similarities with severe Kawasaki disease, MAS/HLH, CRS, and toxic shock syndrome, though the acute cardiac complications in MIS-C seem to be more severe than typically seen in Kawasaki disease and CRS [150]. Interestingly, MIS-C typically develops 3–4 weeks after SARS-CoV-2 infection, suggesting that MIS-C might be due to a delayed, antibody-mediated response and antibody-dependent enhancement of viral entry. In these patients, IVIG, steroids, aspirin, and immunomodulatory agents (e.g., infliximab, anakinra, tocilizumab) have been used with favorable outcomes, though randomized controlled clinical trials are needed to establish optimal treatment regimens [151].
Conclusions.
Immunotherapies, including immune checkpoint inhibitors and adoptive T cell therapies, have revolutionized cancer treatment over the past several years, especially for metastatic and refractory malignancies with historically limited treatment options. Cardiovascular toxicities can arise from these therapies, suggesting critical interactions between the cardiovascular and adaptive immune systems, and underlining the need to better understand the mechanisms of T cell- and cytokine-mediated injury to the myocardium and cardiovascular system. The development of pre-clinical models are necessary to better delineate the basic biology of these cardiovascular toxicities. Future research is needed to better describe these potential mechanisms and provide insight into best practices in the management of cardiovascular irAE. Irrespective of the precise mechanisms of ICI-associated toxicities, the widespread use of ICI for multiple cancer types has motivated the development of multi-disciplinary clinical teams that include cardiologists to provide care for the diverse irAE that can occur in these patients. As clinical experience with cancer immunotherapies and newer cytokine-driven hyperinflammatory syndromes, including COVID-19, continues to expand, collaborative multi-disciplinary approaches from various subspecialties, including cardiology, oncology, and immunology will be vital to dissect the cardiovascular effects of immune system dysregulation.
ACKNOWLEDGEMENTS
Figures created with BioRender.com
SOURCES OF FUNDING
Alan H. Baik: NIH T32 HL007731-28
Olalekan O. Oluwole: None
Douglas B. Johnson: Research funding: BMS and Incyte
Joe-Elie Salem: None
Nina Shah: Research Funding: Celgene/BMS, Janssen, Bluebird Bio, Sutro Biopharma, Teneobio, Poseida, Nektar
Katy Tsai: Institutional research funding from Array/Pfizer, Oncosec, Parker Institute for Cancer Immunotherapy, Regeneron, Replimune
Javid J. Moslehi: Research Support: Dr. Moslehi is supported by NIH (R01HL41446 and R01HL155990).
Nonstandard Abbreviations and Acronyms:
- APC
antigen-presenting cell
- CCR6
chemokine receptor 6
- CAR
chimeric antigen receptor
- CD
cluster of differentiation
- COVID-19
coronavirus disease-2019
- CRS
cytokine release syndrome
- CTLA-4
cytotoxic T-lymphocyte associated protein 4
- HLA-DR
human leukocyte antigen-DR isotype
- ICI
immune checkpoint inhibitor
- IL
interleukin
- irAE
immune related adverse event
- JAK
Janus kinase
- mAb
monoclonal antibody
- MAPK
mitogen-activated protein kinase
- MIP
macrophage inflammatory protein
- MCP-1
monocyte chemoattractant protein-1
- MHC
major histocompatibility complex
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- PD-1
programmed cell death protein-1
- PI3K
phosphoinositide 3-kinase
- scFc
single-chain fragment variable region
- STAT
signal transducer and activator of transcription
- TCE
T cell engager
- TCR
T cell receptor
- TNF
tumor necrosis factor
Footnotes
DISCLOSURES
Alan H. Baik: None
Olalekan O. Oluwole: Serves on advisory board with Pfizer, Kite, Spectrum, and Bayer. Received honoraria from Pfizer.
Douglas B. Johnson: Serves on advisory boards for Array Biopharma, Bristol-Myers Squibb, Catalyst Biopharma, Iovance, Jansen, Merck, and Novartis.
Joe-Elie Salem: Serves on board for Bristol-Myers Squibb
Nina Shah: Advisory role: GSK, Amgen, Indapta Therapeutics, Sanofi, BMS, CareDx, Kite, Karyopharm
Katy Tsai: None
Javid J. Moslehi: Served on an advisory boards for Pfizer, Novartis, Bristol-Myers Squibb, Deciphera, Audentes Pharmaceuticals, Nektar, Takeda, Ipsen, Myokardia, AstraZeneca, GlaxoSmithKline, Intrexon, and Regeneron
REFERENCES
- [1].Haslam A, Gill J, and Prasad V, “Estimation of the Percentage of US Patients With Cancer Who Are Eligible for Immune Checkpoint Inhibitor Drugs,” JAMA Netw Open, vol. 3, no. 3, p. e200423, March. 2020, doi: 10.1001/jamanetworkopen.2020.0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].June CH and Sadelain M, “Chimeric Antigen Receptor Therapy,” N. Engl. J. Med, vol. 379, no. 1, pp. 64–73, 05 2018, doi: 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Schildberg FA, Klein SR, Freeman GJ, and Sharpe AH, “Coinhibitory Pathways in the B7-CD28 Ligand-Receptor Family,” Immunity, vol. 44, no. 5, pp. 955–972, 17 2016, doi: 10.1016/j.immuni.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wei SC, Duffy CR, and Allison JP, “Fundamental Mechanisms of Immune Checkpoint Blockade Therapy,” Cancer Discov, vol. 8, no. 9, pp. 1069–1086, 2018, doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- [5].Esensten JH, Helou YA, Chopra G, Weiss A, and Bluestone JA, “CD28 Costimulation: From Mechanism to Therapy,” Immunity, vol. 44, no. 5, pp. 973–988, 17 2016, doi: 10.1016/j.immuni.2016.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bluestone JA and Anderson M, “Tolerance in the Age of Immunotherapy,” N. Engl. J. Med, vol. 383, no. 12, pp. 1156–1166, 17 2020, doi: 10.1056/NEJMra1911109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Krummel MF and Allison JP, “CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells,” J. Exp. Med, vol. 183, no. 6, pp. 2533–2540, June. 1996, doi: 10.1084/jem.183.6.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Walunas TL et al. , “CTLA-4 can function as a negative regulator of T cell activation,” Immunity, vol. 1, no. 5, pp. 405–413, August. 1994, doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- [9].Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, and Sharpe AH, “Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4,” Immunity, vol. 3, no. 5, pp. 541–547, November. 1995, doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- [10].Waterhouse P et al. , “Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4,” Science, vol. 270, no. 5238, pp. 985–988, November. 1995, doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- [11].Ribas A and Wolchok JD, “Cancer immunotherapy using checkpoint blockade,” Science, vol. 359, no. 6382, pp. 1350–1355, 23 2018, doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hodi FS et al. , “Improved survival with ipilimumab in patients with metastatic melanoma,” N. Engl. J. Med, vol. 363, no. 8, pp. 711–723, August. 2010, doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Weinmann SC and Pisetsky DS, “Mechanisms of immune-related adverse events during the treatment of cancer with immune checkpoint inhibitors,” Rheumatology (Oxford), vol. 58, no. Suppl 7, pp. vii59–vii67, 01 2019, doi: 10.1093/rheumatology/kez308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Andrews LP, Yano H, and Vignali DAA, “Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: breakthroughs or backups,” Nat. Immunol, vol. 20, no. 11, pp. 1425–1434, 2019, doi: 10.1038/s41590-019-0512-0. [DOI] [PubMed] [Google Scholar]
- [15].Postow MA, Sidlow R, and Hellmann MD, “Immune-Related Adverse Events Associated with Immune Checkpoint Blockade,” N Engl J Med, vol. 378, no. 2, pp. 158–168, January. 2018, doi: 10.1056/NEJMra1703481. [DOI] [PubMed] [Google Scholar]
- [16].Johnson DB, Chandra S, and Sosman JA, “Immune Checkpoint Inhibitor Toxicity in 2018,” JAMA, vol. 320, no. 16, pp. 1702–1703, 23 2018, doi: 10.1001/jama.2018.13995. [DOI] [PubMed] [Google Scholar]
- [17].Wang DY et al. , “Fatal Toxic Effects Associated With Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis,” JAMA Oncol, vol. 4, no. 12, pp. 1721–1728, 01 2018, doi: 10.1001/jamaoncol.2018.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hu J-R et al. , “Cardiovascular toxicities associated with immune checkpoint inhibitors,” Cardiovasc. Res, vol. 115, no. 5, pp. 854–868, 15 2019, doi: 10.1093/cvr/cvz026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Salem J-E et al. , “Cardiovascular toxicities associated with immune checkpoint inhibitors: an observational, retrospective, pharmacovigilance study,” Lancet Oncol, vol. 19, no. 12, pp. 1579–1589, 2018, doi: 10.1016/S1470-2045(18)30608-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mahmood SS et al. , “Myocarditis in Patients Treated With Immune Checkpoint Inhibitors,” J. Am. Coll. Cardiol, vol. 71, no. 16, pp. 1755–1764, 24 2018, doi: 10.1016/j.jacc.2018.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Geraud A et al. , “Clinical Pharmacology and Interplay of Immune Checkpoint Agents: A Yin-Yang Balance,” Annual Review of Pharmacology and Toxicology, September. 2020, doi: 10.1146/annurev-pharmtox-022820-093805. [DOI] [PubMed] [Google Scholar]
- [22].Salem J-E et al. , “Abatacept for Severe Immune Checkpoint Inhibitor–Associated Myocarditis,” N Engl J Med, vol. 380, no. 24, pp. 2377–2379, June. 2019, doi: 10.1056/NEJMc1901677. [DOI] [PubMed] [Google Scholar]
- [23].Johnson DB et al. , “Fulminant Myocarditis with Combination Immune Checkpoint Blockade,” N. Engl. J. Med, vol. 375, no. 18, pp. 1749–1755, November. 2016, doi: 10.1056/NEJMoa1609214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wei SC et al. , “A genetic mouse model recapitulates immune checkpoint inhibitor-associated myocarditis and supports a mechanism-based therapeutic intervention,” Cancer Discov, p. CD-20–0856, November. 2020, doi: 10.1158/2159-8290.CD-20-0856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Anquetil C et al. , “Immune Checkpoint Inhibitor-Associated Myositis: Expanding the Spectrum of Cardiac Complications of the Immunotherapy Revolution,” Circulation, vol. 138, no. 7, pp. 743–745, 14 2018, doi: 10.1161/CIRCULATIONAHA.118.035898. [DOI] [PubMed] [Google Scholar]
- [26].Johnson DB et al. , “Neurologic toxicity associated with immune checkpoint inhibitors: a pharmacovigilance study,” Journal for Immunotherapy of Cancer, vol. 7, no. 1, p. 134, 22 2019, doi: 10.1186/s40425-019-0617-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Allenbach Y et al. , “Immune checkpoint inhibitor-induced myositis, the earliest and most lethal complication among rheumatic and musculoskeletal toxicities,” Autoimmunity Reviews, vol. 19, no. 8, p. 102586, August. 2020, doi: 10.1016/j.autrev.2020.102586. [DOI] [PubMed] [Google Scholar]
- [28].Drobni ZD et al. , “Association Between Immune Checkpoint Inhibitors with Cardiovascular Events and Atherosclerotic Plaque,” Circulation, October. 2020, doi: 10.1161/CIRCULATIONAHA.120.049981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Waliany S et al. , “Immune Checkpoint Inhibitor Cardiotoxicity: Understanding Basic Mechanisms and Clinical Characteristics and Finding a Cure,” Annu. Rev. Pharmacol. Toxicol, August. 2020, doi: 10.1146/annurev-pharmtox-010919-023451. [DOI] [PubMed] [Google Scholar]
- [30].Gormley NJ and Pazdur R, “Immunotherapy Combinations in Multiple Myeloma — Known Unknowns,” N Engl J Med, vol. 379, no. 19, pp. 1791–1795, November. 2018, doi: 10.1056/NEJMp1803602. [DOI] [PubMed] [Google Scholar]
- [31].Grabie N, Lichtman AH, and Padera R, “T cell checkpoint regulators in the heart,” Cardiovasc. Res, vol. 115, no. 5, pp. 869–877, 15 2019, doi: 10.1093/cvr/cvz025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lim SY et al. , “Circulating Cytokines Predict Immune-Related Toxicity in Melanoma Patients Receiving Anti-PD-1–Based Immunotherapy,” Clin Cancer Res, vol. 25, no. 5, pp. 1557–1563, March. 2019, doi: 10.1158/1078-0432.CCR-18-2795. [DOI] [PubMed] [Google Scholar]
- [33].Tarhini AA et al. , “Baseline circulating IL-17 predicts toxicity while TGF-β1 and IL-10 are prognostic of relapse in ipilimumab neoadjuvant therapy of melanoma,” J Immunother Cancer, vol. 3, p. 39, 2015, doi: 10.1186/s40425-015-0081-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tarrio ML, Grabie N, Bu D, Sharpe AH, and Lichtman AH, “PD-1 protects against inflammation and myocyte damage in T cell-mediated myocarditis,” J. Immunol, vol. 188, no. 10, pp. 4876–4884, May 2012, doi: 10.4049/jimmunol.1200389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rodig N et al. , “Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis,” Eur. J. Immunol, vol. 33, no. 11, pp. 3117–3126, November. 2003, doi: 10.1002/eji.200324270. [DOI] [PubMed] [Google Scholar]
- [36].Baban B, Liu JY, Qin X, Weintraub NL, and Mozaffari MS, “Upregulation of Programmed Death-1 and Its Ligand in Cardiac Injury Models: Interaction with GADD153,” PLoS ONE, vol. 10, no. 4, p. e0124059, 2015, doi: 10.1371/journal.pone.0124059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Grabie N et al. , “Endothelial programmed death-1 ligand 1 (PD-L1) regulates CD8+ T-cell mediated injury in the heart,” Circulation, vol. 116, no. 18, pp. 2062–2071, October. 2007, doi: 10.1161/CIRCULATIONAHA.107.709360. [DOI] [PubMed] [Google Scholar]
- [38].Altan M et al. , “Immune Checkpoint Inhibitor–Associated Pericarditis,” Journal of Thoracic Oncology, vol. 14, no. 6, pp. 1102–1108, June. 2019, doi: 10.1016/j.jtho.2019.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Dubin K et al. , “Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis,” Nature Communications, vol. 7, p. 10391, February. 2016, doi: 10.1038/ncomms10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Johnson DB et al. , “A case report of clonal EBV-like memory CD4+ T cell activation in fatal checkpoint inhibitor-induced encephalitis,” Nature Medicine, vol. 25, no. 8, pp. 1243–1250, 2019, doi: 10.1038/s41591-019-0523-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Slaney CY, Wang P, Darcy PK, and Kershaw MH, “CARs versus BiTEs: A Comparison between T Cell-Redirection Strategies for Cancer Treatment,” Cancer Discov, vol. 8, no. 8, pp. 924–934, 2018, doi: 10.1158/2159-8290.CD-18-0297. [DOI] [PubMed] [Google Scholar]
- [42].Sedykh S, Prinz V, Buneva V, and Nevinsky G, “Bispecific antibodies: design, therapy, perspectives,” DDDT, vol. Volume 12, pp. 195–208, January. 2018, doi: 10.2147/DDDT.S151282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Pipe SW et al. , “Efficacy, safety, and pharmacokinetics of emicizumab prophylaxis given every 4 weeks in people with haemophilia A (HAVEN 4): a multicentre, open-label, non-randomised phase 3 study,” Lancet Haematol, vol. 6, no. 6, pp. e295–e305, June. 2019, doi: 10.1016/S2352-3026(19)30054-7. [DOI] [PubMed] [Google Scholar]
- [44].Bargou R et al. , “Tumor regression in cancer patients by very low doses of a T cell-engaging antibody,” Science, vol. 321, no. 5891, pp. 974–977, August. 2008, doi: 10.1126/science.1158545. [DOI] [PubMed] [Google Scholar]
- [45].Kantarjian H et al. , “Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia,” N Engl J Med, vol. 376, no. 9, pp. 836–847, March. 2017, doi: 10.1056/NEJMoa1609783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].von Stackelberg A et al. , “Phase I/Phase II Study of Blinatumomab in Pediatric Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia,” J. Clin. Oncol, vol. 34, no. 36, pp. 4381–4389, 20 2016, doi: 10.1200/JCO.2016.67.3301. [DOI] [PubMed] [Google Scholar]
- [47].Przepiorka D et al. , “FDA Approval: Blinatumomab,” Clin. Cancer Res, vol. 21, no. 18, pp. 4035–4039, September. 2015, doi: 10.1158/1078-0432.CCR-15-0612. [DOI] [PubMed] [Google Scholar]
- [48].Gökbuget N et al. , “Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia,” Blood, vol. 131, no. 14, pp. 1522–1531, 05 2018, doi: 10.1182/blood-2017-08-798322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Topp MS et al. , “Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study,” The Lancet Oncology, vol. 16, no. 1, pp. 57–66, January. 2015, doi: 10.1016/S1470-2045(14)71170-2. [DOI] [PubMed] [Google Scholar]
- [50].Kalos M et al. , “T Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced Leukemia,” Science Translational Medicine, vol. 3, no. 95, pp. 95ra73–95ra73, August. 2011, doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rafiq S, Hackett CS, and Brentjens RJ, “Engineering strategies to overcome the current roadblocks in CAR T cell therapy,” Nat Rev Clin Oncol, vol. 17, no. 3, pp. 147–167, March. 2020, doi: 10.1038/s41571-019-0297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang ML et al. , “Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma,” The New England Journal of Medicine, vol. 369, no. 6, pp. 507–516, August. 2013, doi: 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Maude SL et al. , “Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia,” N Engl J Med, vol. 371, no. 16, pp. 1507–1517, October. 2014, doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Locke FL et al. , “Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial,” Lancet Oncol, vol. 20, no. 1, pp. 31–42, 2019, doi: 10.1016/S1470-2045(18)30864-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Schuster SJ et al. , “Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma,” N. Engl. J. Med, vol. 380, no. 1, pp. 45–56, 03 2019, doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- [56].Raje N et al. , “Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma,” N. Engl. J. Med, vol. 380, no. 18, pp. 1726–1737, 02 2019, doi: 10.1056/NEJMoa1817226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yeku O, Li X, and Brentjens RJ, “Adoptive T-Cell Therapy for Solid Tumors,” Am Soc Clin Oncol Educ Book, vol. 37, pp. 193–204, 2017, doi: 10.1200/EDBK_180328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ganatra S et al. , “Chimeric Antigen Receptor T-Cell Therapy for Cancer and Heart,” Journal of the American College of Cardiology, vol. 74, no. 25, pp. 3153–3163, December. 2019, doi: 10.1016/j.jacc.2019.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Gutierrez C et al. , “Management of the Critically Ill Adult Chimeric Antigen Receptor-T Cell Therapy Patient: A Critical Care Perspective,” Crit. Care Med, vol. 46, no. 9, pp. 1402–1410, 2018, doi: 10.1097/CCM.0000000000003258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Alvi RM et al. , “Cardiovascular Events Among Adults Treated With Chimeric Antigen Receptor T-Cells (CAR-T),” Journal of the American College of Cardiology, vol. 74, no. 25, pp. 3099–3108, December. 2019, doi: 10.1016/j.jacc.2019.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lefebvre B, Kang Y, Smith AM, Frey NV, Carver JR, and Scherrer-Crosbie M, “Cardiovascular Effects of CAR T Cell Therapy: A Retrospective Study,” JACC CardioOncol, vol. 2, no. 2, pp. 193–203, June. 2020, doi: 10.1016/j.jaccao.2020.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Salem J-E, Ederhy S, Lebrun-Vignes B, and Moslehi JJ, “Cardiac Events Associated With Chimeric Antigen Receptor T-Cells (CAR-T): A VigiBase Perspective,” J. Am. Coll. Cardiol, vol. 75, no. 19, pp. 2521–2523, May 2020, doi: 10.1016/j.jacc.2020.02.070. [DOI] [PubMed] [Google Scholar]
- [63].Linette GP et al. , “Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma,” Blood, vol. 122, no. 6, pp. 863–871, August. 2013, doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Herman DS et al. , “Truncations of Titin Causing Dilated Cardiomyopathy,” N Engl J Med, vol. 366, no. 7, pp. 619–628, February. 2012, doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, and Katze MG, “Into the eye of the cytokine storm,” Microbiol. Mol. Biol. Rev, vol. 76, no. 1, pp. 16–32, March. 2012, doi: 10.1128/MMBR.05015-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Apel A et al. , “Safety and efficacy of blinatumomab: a real world data,” Ann Hematol, vol. 99, no. 4, pp. 835–838, April. 2020, doi: 10.1007/s00277-019-03854-0. [DOI] [PubMed] [Google Scholar]
- [67].Nastoupil LJ et al. , “Standard-of-Care Axicabtagene Ciloleucel for Relapsed or Refractory Large B-Cell Lymphoma: Results From the US Lymphoma CAR T Consortium,” J. Clin. Oncol, p. JCO1902104, May 2020, doi: 10.1200/JCO.19.02104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Oluwole OO and Davila ML, “At The Bedside: Clinical review of chimeric antigen receptor (CAR) T cell therapy for B cell malignancies,” J. Leukoc. Biol, vol. 100, no. 6, pp. 1265–1272, 2016, doi: 10.1189/jlb.5BT1115-524R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ceschi A, Noseda R, Palin K, and Verhamme K, “Immune Checkpoint Inhibitor-Related Cytokine Release Syndrome: Analysis of WHO Global Pharmacovigilance Database,” Front. Pharmacol, vol. 11, p. 557, May 2020, doi: 10.3389/fphar.2020.00557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Teachey DT et al. , “Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia,” Cancer Discov, vol. 6, no. 6, pp. 664–679, 2016, doi: 10.1158/2159-8290.CD-16-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Topp MS et al. , “Targeted Therapy With the T-Cell–Engaging Antibody Blinatumomab of Chemotherapy-Refractory Minimal Residual Disease in B-Lineage Acute Lymphoblastic Leukemia Patients Results in High Response Rate and Prolonged Leukemia-Free Survival,” JCO, vol. 29, no. 18, pp. 2493–2498, June. 2011, doi: 10.1200/JCO.2010.32.7270. [DOI] [PubMed] [Google Scholar]
- [72].Rosenberg SA et al. , “A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone,” N. Engl. J. Med, vol. 316, no. 15, pp. 889–897, April. 1987, doi: 10.1056/NEJM198704093161501. [DOI] [PubMed] [Google Scholar]
- [73].Ferrara JL, Abhyankar S, and Gilliland DG, “Cytokine storm of graft-versus-host disease: a critical effector role for interleukin-1,” Transplant. Proc, vol. 25, no. 1 Pt 2, pp. 1216–1217, February. 1993. [PubMed] [Google Scholar]
- [74].Parker KR et al. , “Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies,” Cell, September. 2020, doi: 10.1016/j.cell.2020.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Hunter BD and Jacobson CA, “CAR T-cell associated neurotoxicity: Mechanisms, clinicopathologic correlates, and future directions,” J. Natl. Cancer Inst, February. 2019, doi: 10.1093/jnci/djz017. [DOI] [PubMed] [Google Scholar]
- [76].Klinger M et al. , “Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab,” Blood, vol. 119, no. 26, pp. 6226–6233, June. 2012, doi: 10.1182/blood-2012-01-400515. [DOI] [PubMed] [Google Scholar]
- [77].Neelapu SS et al. , “Kte-C19 (anti-CD19 CAR T Cells) Induces Complete Remissions in Patients with Refractory Diffuse Large B-Cell Lymphoma (DLBCL): Results from the Pivotal Phase 2 Zuma-1,” Blood, vol. 128, no. 22, p. LBA-6-LBA-6, December. 2016, doi: 10.1182/blood.V128.22.LBA-6.LBA-6. [DOI] [Google Scholar]
- [78].Norelli M et al. , “Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells,” Nat. Med, vol. 24, no. 6, pp. 739–748, 2018, doi: 10.1038/s41591-018-0036-4. [DOI] [PubMed] [Google Scholar]
- [79].Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, and Sadelain M, “CAR T cell–induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade,” Nat Med, vol. 24, no. 6, pp. 731–738, June. 2018, doi: 10.1038/s41591-018-0041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Rose-John S, “Interleukin-6 Family Cytokines,” Cold Spring Harb Perspect Biol, vol. 10, no. 2, 01 2018, doi: 10.1101/cshperspect.a028415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Tanaka T, Narazaki M, and Kishimoto T, “Immunotherapeutic implications of IL-6 blockade for cytokine storm,” Immunotherapy, vol. 8, no. 8, pp. 959–970, 2016, doi: 10.2217/imt-2016-0020. [DOI] [PubMed] [Google Scholar]
- [82].Nägele V et al. , “Changes in clinical laboratory parameters and pharmacodynamic markers in response to blinatumomab treatment of patients with relapsed/refractory ALL,” Experimental Hematology & Oncology, vol. 6, p. 14, 2017, doi: 10.1186/s40164-017-0074-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Ogura H et al. , “Interleukin-17 Promotes Autoimmunity by Triggering a Positive-Feedback Loop via Interleukin-6 Induction,” Immunity, vol. 29, no. 4, pp. 628–636, October. 2008, doi: 10.1016/j.immuni.2008.07.018. [DOI] [PubMed] [Google Scholar]
- [84].Grupp SA et al. , “Chimeric antigen receptor-modified T cells for acute lymphoid leukemia,” N. Engl. J. Med, vol. 368, no. 16, pp. 1509–1518, April. 2013, doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Fitzgerald JC et al. , “Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia,” Crit. Care Med, vol. 45, no. 2, pp. e124–e131, February. 2017, doi: 10.1097/CCM.0000000000002053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Frey NV et al. , “Refractory Cytokine Release Syndrome in Recipients of Chimeric Antigen Receptor (CAR) T Cells,” Blood, vol. 124, no. 21, pp. 2296–2296, December. 2014, doi: 10.1182/blood.V124.21.2296.2296. [DOI] [Google Scholar]
- [87].Teachey DT et al. , “Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy,” Blood, vol. 121, no. 26, pp. 5154–5157, June. 2013, doi: 10.1182/blood-2013-02-485623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Turtle CJ et al. , “Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated With CD19-Specific Chimeric Antigen Receptor–Modified T Cells After Failure of Ibrutinib,” JCO, vol. 35, no. 26, pp. 3010–3020, September. 2017, doi: 10.1200/JCO.2017.72.8519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Stroud CR et al. , “Tocilizumab for the management of immune mediated adverse events secondary to PD-1 blockade,” J Oncol Pharm Pract, vol. 25, no. 3, pp. 551–557, April. 2019, doi: 10.1177/1078155217745144. [DOI] [PubMed] [Google Scholar]
- [90].Esfahani K and Miller WH, “Reversal of Autoimmune Toxicity and Loss of Tumor Response by Interleukin-17 Blockade,” N Engl J Med, vol. 376, no. 20, pp. 1989–1991, May 2017, doi: 10.1056/NEJMc1703047. [DOI] [PubMed] [Google Scholar]
- [91].Rosenberg SA et al. , “Observations on the Systemic Administration of Autologous Lymphokine-Activated Killer Cells and Recombinant Interleukin-2 to Patients with Metastatic Cancer,” N Engl J Med, vol. 313, no. 23, pp. 1485–1492, December. 1985, doi: 10.1056/NEJM198512053132327. [DOI] [PubMed] [Google Scholar]
- [92].Schechter D and Nagler A, “Recombinant interleukin-2 and recombinant interferon α immunotherapy cardiovascular toxicity,” American Heart Journal, vol. 123, no. 6, pp. 1736–1739, June. 1992, doi: 10.1016/0002-8703(92)90856-Q. [DOI] [PubMed] [Google Scholar]
- [93].Mann H, Ward JH, and Samlowski WE, “Vascular leak syndrome associated with interleukin-2: chest radiographic manifestations.,” Radiology, vol. 176, no. 1, pp. 191–194, July. 1990, doi: 10.1148/radiology.176.1.2353090. [DOI] [PubMed] [Google Scholar]
- [94].Ognibene FP et al. , “Interleukin-2 administration causes reversible hemodynamic changes and left ventricular dysfunction similar to those seen in septic shock,” Chest, vol. 94, no. 4, pp. 750–754, October. 1988, doi: 10.1378/chest.94.4.750. [DOI] [PubMed] [Google Scholar]
- [95].Rosenberg SA, “Treatment of 283 Consecutive Patients With Metastatic Melanoma or Renal Cell Cancer Using High-Dose Bolus Interleukin 2,” JAMA, vol. 271, no. 12, p. 907, March. 1994, doi: 10.1001/jama.1994.03510360033032. [DOI] [PubMed] [Google Scholar]
- [96].White RL et al. , “Cardiopulmonary toxicity of treatment with high dose interleukin-2 in 199 consecutive patients with metastatic melanoma or renal cell carcinoma,” Cancer, vol. 74, no. 12, pp. 3212–3222, December. 1994, doi: 10.1002/1097-0142(19941215)74:12<3212::aid-cncr2820741221>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- [97].Kragel AH, Travis WD, Steis RG, Rosenberg SA, and Roberts WC, “Myocarditis or acute myocardial infarction associated with interleukin-2 therapy for cancer,” Cancer, vol. 66, no. 7, pp. 1513–1516, October. 1990, doi: 10.1002/1097-0142(19901001)66:7<1513::aid-cncr2820660713>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- [98].Cameron MJ et al. , “Interferon-mediated immunopathological events are associated with atypical innate and adaptive immune responses in patients with severe acute respiratory syndrome,” J. Virol, vol. 81, no. 16, pp. 8692–8706, August. 2007, doi: 10.1128/JVI.00527-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Sonnenblick M, Rosenmann D, and Rosin A, “Reversible cardiomyopathy induced by interferon,” BMJ, vol. 300, no. 6733, pp. 1174–1175, May 1990, doi: 10.1136/bmj.300.6733.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Teragawa H, Hondo T, Amano H, Hino F, and Ohbayashi M, “Adverse effects of interferon on the cardiovascular system in patients with chronic hepatitis C,” Jpn Heart J, vol. 37, no. 6, pp. 905–915, November. 1996, doi: 10.1536/ihj.37.905. [DOI] [PubMed] [Google Scholar]
- [101].Crown J et al. , “A phase I trial of recombinant human interleukin-1 beta alone and in combination with myelosuppressive doses of 5-fluorouracil in patients with gastrointestinal cancer,” Blood, vol. 78, no. 6, pp. 1420–1427, September. 1991. [PubMed] [Google Scholar]
- [102].Conlon KC et al. , “Redistribution, Hyperproliferation, Activation of Natural Killer Cells and CD8 T Cells, and Cytokine Production During First-in-Human Clinical Trial of Recombinant Human Interleukin-15 in Patients With Cancer,” JCO, vol. 33, no. 1, pp. 74–82, January. 2015, doi: 10.1200/JCO.2014.57.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Zhang L et al. , “Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma,” Clin. Cancer Res, vol. 21, no. 10, pp. 2278–2288, May 2015, doi: 10.1158/1078-0432.CCR-14-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Hegewisch S, “TNF-induced cardiomyopathy,” The Lancet, vol. 335, no. 8684, pp. 294–295, February. 1990, doi: 10.1016/0140-6736(90)90115-L. [DOI] [PubMed] [Google Scholar]
- [105].Latini R et al. , “Cytokines in acute myocardial infarction: selective increase in circulating tumor necrosis factor, its soluble receptor, and interleukin-1 receptor antagonist,” J. Cardiovasc. Pharmacol, vol. 23, no. 1, pp. 1–6, January. 1994. [PubMed] [Google Scholar]
- [106].Torre-Amione G, Kapadia S, Benedict C, Oral H, Young JB, and Mann DL, “Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: a report from the Studies of Left Ventricular Dysfunction (SOLVD),” J. Am. Coll. Cardiol, vol. 27, no. 5, pp. 1201–1206, April. 1996, doi: 10.1016/0735-1097(95)00589-7. [DOI] [PubMed] [Google Scholar]
- [107].Koller-Strametz J, Pacher R, Frey B, Kos T, Woloszczuk W, and Stanek B, “Circulating tumor necrosis factor-alpha levels in chronic heart failure: relation to its soluble receptor II, interleukin-6, and neurohumoral variables,” J. Heart Lung Transplant, vol. 17, no. 4, pp. 356–362, April. 1998. [PubMed] [Google Scholar]
- [108].Guo L, Liu M-F, Huang J-N, Li J-M, Jiang J, and Wang J-A, “Role of interleukin-15 in cardiovascular diseases,” J. Cell. Mol. Med, May 2020, doi: 10.1111/jcmm.15296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Allam G, Abdel-Moneim A, and Gaber AM, “The pleiotropic role of interleukin-17 in atherosclerosis,” Biomed. Pharmacother, vol. 106, pp. 1412–1418, October. 2018, doi: 10.1016/j.biopha.2018.07.110. [DOI] [PubMed] [Google Scholar]
- [110].Paulus WJ, “Cytokines and heart failure,” Heart Fail Monit, vol. 1, no. 2, pp. 50–56, 2000. [PubMed] [Google Scholar]
- [111].Mann DL, “The emerging role of innate immunity in the heart and vascular system: for whom the cell tolls,” Circ. Res, vol. 108, no. 9, pp. 1133–1145, April. 2011, doi: 10.1161/CIRCRESAHA.110.226936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Prabhu SD, “Cytokine-Induced Modulation of Cardiac Function,” Circulation Research, vol. 95, no. 12, pp. 1140–1153, December. 2004, doi: 10.1161/01.RES.0000150734.79804.92. [DOI] [PubMed] [Google Scholar]
- [113].Natanson C et al. , “Endotoxin and tumor necrosis factor challenges in dogs simulate the cardiovascular profile of human septic shock.,” The Journal of Experimental Medicine, vol. 169, no. 3, pp. 823–832, March. 1989, doi: 10.1084/jem.169.3.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Pagani FD, Baker LS, Hsi C, Knox M, Fink MP, and Visner MS, “Left ventricular systolic and diastolic dysfunction after infusion of tumor necrosis factor-alpha in conscious dogs,” J. Clin. Invest, vol. 90, no. 2, pp. 389–398, August. 1992, doi: 10.1172/JCI115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Bozkurt B et al. , “Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats,” Circulation, vol. 97, no. 14, pp. 1382–1391, April. 1998, doi: 10.1161/01.cir.97.14.1382. [DOI] [PubMed] [Google Scholar]
- [116].Cailleret M et al. , “N-acetylcysteine prevents the deleterious effect of tumor necrosis factor-(alpha) on calcium transients and contraction in adult rat cardiomyocytes,” Circulation, vol. 109, no. 3, pp. 406–411, January. 2004, doi: 10.1161/01.CIR.0000109499.00587.FF. [DOI] [PubMed] [Google Scholar]
- [117].Kumar A, Thota V, Dee L, Olson J, Uretz E, and Parrillo JE, “Tumor necrosis factor alpha and interleukin 1beta are responsible for in vitro myocardial cell depression induced by human septic shock serum.,” The Journal of Experimental Medicine, vol. 183, no. 3, pp. 949–958, March. 1996, doi: 10.1084/jem.183.3.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Franco F et al. , “Magnetic resonance imaging and invasive evaluation of development of heart failure in transgenic mice with myocardial expression of tumor necrosis factor-alpha,” Circulation, vol. 99, no. 3, pp. 448–454, January. 1999, doi: 10.1161/01.cir.99.3.448. [DOI] [PubMed] [Google Scholar]
- [119].Bryant D et al. , “Cardiac failure in transgenic mice with myocardial expression of tumor necrosis factor-alpha,” Circulation, vol. 97, no. 14, pp. 1375–1381, April. 1998, doi: 10.1161/01.cir.97.14.1375. [DOI] [PubMed] [Google Scholar]
- [120].Jarrah AA et al. , “SDF-1 induces TNF-mediated apoptosis in cardiac myocytes,” Apoptosis, vol. 23, no. 1, pp. 79–91, 2018, doi: 10.1007/s10495-017-1438-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Krown KA et al. , “Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death,” J. Clin. Invest, vol. 98, no. 12, pp. 2854–2865, December. 1996, doi: 10.1172/JCI119114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Olivetti G et al. , “Apoptosis in the Failing Human Heart,” N Engl J Med, vol. 336, no. 16, pp. 1131–1141, April. 1997, doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- [123].Rozanski GJ and Witt RC, “IL-1 inhibits beta-adrenergic control of cardiac calcium current: role of L-arginine/nitric oxide pathway,” Am. J. Physiol, vol. 267, no. 5 Pt 2, pp. H1753–1758, November. 1994, doi: 10.1152/ajpheart.1994.267.5.H1753. [DOI] [PubMed] [Google Scholar]
- [124].Ferdinandy P, Danial H, Ambrus I, Rothery RA, and Schulz R, “Peroxynitrite is a major contributor to cytokine-induced myocardial contractile failure,” Circ. Res, vol. 87, no. 3, pp. 241–247, August. 2000, doi: 10.1161/01.res.87.3.241. [DOI] [PubMed] [Google Scholar]
- [125].Finkel MS, Oddis CV, Jacob TD, Watkins SC, Hattler BG, and Simmons RL, “Negative inotropic effects of cytokines on the heart mediated by nitric oxide,” Science, vol. 257, no. 5068, pp. 387–389, July. 1992, doi: 10.1126/science.1631560. [DOI] [PubMed] [Google Scholar]
- [126].Comini L, Pasini E, Bachetti T, Dreano M, Garotta G, and Ferrari R, “Acute haemodynamic effects of IL-6 treatment in vivo: involvement of vagus nerve in NO-mediated negative inotropism,” Cytokine, vol. 30, no. 5, pp. 236–242, June. 2005, doi: 10.1016/j.cyto.2005.01.009. [DOI] [PubMed] [Google Scholar]
- [127].Yu X, Kennedy RH, and Liu SJ, “JAK2/STAT3, not ERK1/2, mediates interleukin-6-induced activation of inducible nitric-oxide synthase and decrease in contractility of adult ventricular myocytes,” J. Biol. Chem, vol. 278, no. 18, pp. 16304–16309, May 2003, doi: 10.1074/jbc.M212321200. [DOI] [PubMed] [Google Scholar]
- [128].Madjid M, Safavi-Naeini P, Solomon SD, and Vardeny O, “Potential Effects of Coronaviruses on the Cardiovascular System: A Review,” JAMA Cardiol, March. 2020, doi: 10.1001/jamacardio.2020.1286. [DOI] [PubMed] [Google Scholar]
- [129].Baldi E et al. , “Out-of-Hospital Cardiac Arrest during the Covid-19 Outbreak in Italy,” N Engl J Med, vol. 383, no. 5, pp. 496–498, July. 2020, doi: 10.1056/NEJMc2010418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Guo T et al. , “Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19),” JAMA cardiology, vol. 5, no. 7, pp. 811–818, 01 2020, doi: 10.1001/jamacardio.2020.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Li C et al. , “Longitudinal correlation of biomarkers of cardiac injury, inflammation, and coagulation to outcome in hospitalized COVID-19 patients,” J. Mol. Cell. Cardiol, vol. 147, pp. 74–87, August. 2020, doi: 10.1016/j.yjmcc.2020.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Inciardi RM et al. , “Cardiac Involvement in a Patient With Coronavirus Disease 2019 (COVID-19),” JAMA Cardiol, March. 2020, doi: 10.1001/jamacardio.2020.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Xu Z et al. , “Pathological findings of COVID-19 associated with acute respiratory distress syndrome,” The Lancet Respiratory Medicine, p. S221326002030076X, February. 2020, doi: 10.1016/S2213-2600(20)30076-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Puntmann VO et al. , “Outcomes of Cardiovascular Magnetic Resonance Imaging in Patients Recently Recovered From Coronavirus Disease 2019 (COVID-19),” JAMA Cardiol, July. 2020, doi: 10.1001/jamacardio.2020.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Zeng J-H et al. , “First case of COVID-19 complicated with fulminant myocarditis: a case report and insights,” Infection, April. 2020, doi: 10.1007/s15010-020-01424-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Hu H, Ma F, Wei X, and Fang Y, “Coronavirus fulminant myocarditis treated with glucocorticoid and human immunoglobulin,” European Heart Journal, p. ehaa190, March. 2020, doi: 10.1093/eurheartj/ehaa190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Topol EJ, “COVID-19 can affect the heart,” Science, September. 2020, doi: 10.1126/science.abe2813. [DOI] [PubMed] [Google Scholar]
- [138].Zhou Y et al. , “Pathogenic T cells and inflammatory monocytes incite inflammatory storm in severe COVID-19 patients,” National Science Review, p. nwaa041, March. 2020, doi: 10.1093/nsr/nwaa041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Huang C et al. , “Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China,” The Lancet, vol. 395, no. 10223, pp. 497–506, February. 2020, doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Wu F et al. , “A new coronavirus associated with human respiratory disease in China,” Nature, vol. 579, no. 7798, pp. 265–269, March. 2020, doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Henry BM, de Oliveira MHS, Benoit S, Plebani M, and Lippi G, “Hematologic, biochemical and immune biomarker abnormalities associated with severe illness and mortality in coronavirus disease 2019 (COVID-19): a meta-analysis,” Clinical Chemistry and Laboratory Medicine (CCLM), vol. 0, no. 0, April. 2020, doi: 10.1515/cclm-2020-0369. [DOI] [PubMed] [Google Scholar]
- [142].Yuan J et al. , “The correlation between viral clearance and biochemical outcomes of 94 COVID-19 infected discharged patients,” Inflamm. Res, March. 2020, doi: 10.1007/s00011-020-01342-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Grifoni E et al. , “Interleukin-6 as prognosticator in patients with COVID-19,” Journal of Infection, vol. 81, no. 3, pp. 452–482, September. 2020, doi: 10.1016/j.jinf.2020.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Toniati P et al. , “Tocilizumab for the treatment of severe COVID-19 pneumonia with hyperinflammatory syndrome and acute respiratory failure: A single center study of 100 patients in Brescia, Italy,” Autoimmun Rev, p. 102568, May 2020, doi: 10.1016/j.autrev.2020.102568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Xu X et al. , “Effective treatment of severe COVID-19 patients with tocilizumab,” Proc. Natl. Acad. Sci. U.S.A, vol. 117, no. 20, pp. 10970–10975, 19 2020, doi: 10.1073/pnas.2005615117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].The RECOVERY Collaborative Group, “Dexamethasone in Hospitalized Patients with Covid-19 — Preliminary Report,” N Engl J Med, p. NEJMoa2021436, July. 2020, doi: 10.1056/NEJMoa2021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].The WHO Rapid Evidence Appraisal for COVID-19 Therapies (REACT) Working Group et al. , “Association Between Administration of Systemic Corticosteroids and Mortality Among Critically Ill Patients With COVID-19: A Meta-analysis,” JAMA, September. 2020, doi: 10.1001/jama.2020.17023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Deza Leon MP et al. , “COVID-19–Associated Pediatric Multisystem Inflammatory Syndrome,” Journal of the Pediatric Infectious Diseases Society, p. piaa061, May 2020, doi: 10.1093/jpids/piaa061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Ahmed M et al. , “Multisystem inflammatory syndrome in children: A systematic review,” EClinicalMedicine, p. 100527, September. 2020, doi: 10.1016/j.eclinm.2020.100527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Shulman ST, “Pediatric COVID-associated Multi-system Inflammatory Syndrome (PMIS),” J Pediatric Infect Dis Soc, May 2020, doi: 10.1093/jpids/piaa062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Belhadjer Z et al. , “Acute heart failure in multisystem inflammatory syndrome in children (MIS-C) in the context of global SARS-CoV-2 pandemic,” Circulation, May 2020, doi: 10.1161/CIRCULATIONAHA.120.048360. [DOI] [PubMed] [Google Scholar]
- [152].Spaapen RM et al. , “Therapeutic activity of high-dose intratumoral IFN-β requires direct effect on the tumor vasculature,” J. Immunol, vol. 193, no. 8, pp. 4254–4260, October. 2014, doi: 10.4049/jimmunol.1401109. [DOI] [PubMed] [Google Scholar]
- [153].Porter D, Frey N, Wood PA, Weng Y, and Grupp SA, “Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel,” J Hematol Oncol, vol. 11, no. 1, p. 35, 02 2018, doi: 10.1186/s13045-018-0571-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Lee DW et al. , “Current concepts in the diagnosis and management of cytokine release syndrome,” Blood, vol. 124, no. 2, pp. 188–195, July. 2014, doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Porter DL et al. , “Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia,” Sci Transl Med, vol. 7, no. 303, p. 303ra139, September. 2015, doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Lee DW et al. , “ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells,” Biol. Blood Marrow Transplant, vol. 25, no. 4, pp. 625–638, 2019, doi: 10.1016/j.bbmt.2018.12.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Brudno JN and Kochenderfer JN, “Toxicities of chimeric antigen receptor T cells: recognition and management,” Blood, vol. 127, no. 26, pp. 3321–3330, 30 2016, doi: 10.1182/blood-2016-04-703751. [DOI] [PMC free article] [PubMed] [Google Scholar]