

Abstract
Aims:
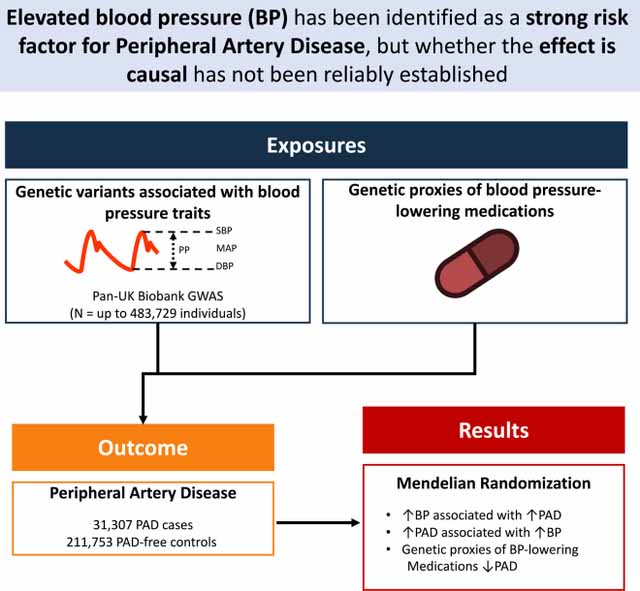
We aimed to estimate the effect of blood pressure (BP) traits and BP-lowering medications (via genetic proxies) on peripheral artery disease.
Methods and Results:
GWAS summary statistics were obtained for BP, peripheral artery disease (PAD), and coronary artery disease (CAD). Causal effects of BP on PAD were estimated by two-sample Mendelian Randomization using a range of pleiotropy-robust methods. Increased SBP, DBP, MAP, and PP each significantly increased risk of PAD (SBP OR 1.20 [1.16–1.25] per 10mmHg increase, p = 1 × 10−24; DBP OR 1.27 [1.18–1.35], p = 4 × 10−11; MAP OR 1.26 [1.19–1.33], p = 6 × 10−16; PP OR 1.31 [1.24–1.39], p = 9 × 10−23). The effects of SBP, DBP, and MAP were greater for CAD than PAD (SBP Ratio of ORs 1.06 [1.0 – 1.12], p = 0.04; MAP ROR 1.15 [1.06–1.26], p = 8.6 × 10−4; DBP ROR 1.21 [1.08–1.35], p = 6.9 × 10−4). Considered jointly, both PP and MAP directly increased risk of PAD (PP OR 1.26 [1.17–1.35], p = 3 × 10−10; MAP OR 1.14 [1.06–1.23], p = 2 × 10−4). The effects of antihypertensive medications were estimated using genetic instruments. SBP-lowering via beta-blocker- (OR 0.74 per 10mmHg decrease in SBP; 95% CI 0.65–0.84; p = 5 × 10−6), loop diuretic- (OR 0.66 [0.48–0.91], p = 0.01), and thiazide diuretic- (OR 0.57 [0.41, 0.79], p = 6 × 10−4) associated variants were protective of PAD.
Conclusions:
Higher BP is likely to cause PAD. BP-lowering through beta blockers, loop diuretics, and thiazide diuretics (as proxied by genetic variants) was associated with decreased risk of PAD. Future study is needed to clarify the specific mechanisms by which BP influences PAD.
Keywords: Blood pressure, Genetics, Atherosclerosis, Peripheral Artery Disease
Graphical Abstract
INTRODUCTION:
Peripheral artery disease (PAD) is a common manifestation of atherosclerotic cardiovascular disease (ASCVD), estimated to affect more than 12 million individuals in the United States, and more than 120 million individuals worldwide.1,2 PAD shares a number of risk factors with other forms ASCVD like coronary artery disease (CAD) and ischemic stroke.3 These risk factors include smoking, diabetes, hypertension, hyperlipidemia, and obesity.2–4 Observational studies have identified hypertension as one of the strongest risk factors for incident and prevalent PAD 5–11, although these studies may be limited by residual environmental confounding or reverse-causality. While randomized controlled trials of antihypertensive medications have demonstrated broad protection from coronary artery disease and death from cardiovascular causes, whether lower blood pressure reduces risk of PAD specifically has not been reliably established. Similarly, the relative effect of blood pressure on PAD has not been fully investigated.
Recent genome-wide association studies (GWAS) of PAD and blood pressure including more than 700,000 individuals have identified hundreds of genetic variants associated with these traits.12,13 The Mendelian randomization (MR) framework (under certain assumptions) can leverage this genetic variation (which is randomly assorted during meiosis, mimicking a randomized trial), to provide unconfounded causal estimates of the relationship between traits.14 MR assumes that genetic variants are likely to be independent of many confounders of the exposure-outcome relationship. This assumption is plausible because genetic variants are randomly inherited by offspring from parents during meiosis and conception, analogous to treatment allocation in a randomized trial. Because large, randomized trials evaluating the relationship between treatment of hypertension and PAD outcomes may be unfeasible, other study designs are needed to fill this evidence gap. Here, we leverage population-scale genetic variation within the Mendelian randomization framework to 1) establish the relationship between blood pressure and risk of PAD, 2) quantify differences in the effect of blood pressure on CAD and PAD risk, and 3) estimate the effect of blood pressure lowering (using genetic proxies of antihypertensive medications) on PAD risk.
METHODS:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Study Exposures
Trans-ancestry blood pressure GWAS of systolic blood pressure (SBP), diastolic blood pressure (DBP), mean arterial pressure (MAP), and pulse pressure (PP) were obtained from the Pan UK Biobank resource (https://pan.ukbb.broadinstitute.org/). These studies included up to 483,729 participants (262,223 male and 221,506 female; 420,136 European, 8,842 Central/South Asian, 6,614 African, 2,705 East Asian, 1,590 Middle Eastern, and 980 American [Hispanic/Latino]). BP measurements included both automated and manual measurements, and were adjusted for antihypertensive medication use. Details of genotyping and quality control, as well as links to download summary statistics can be found at https://pan.ukbb.broadinstitute.org/.
Ancestry-specific blood pressure effect estimates were obtained by first identifying genetic variants associated with each BP trait in the trans-ancestry Pan UK Biobank BP GWAS studies, and then extracting the effect estimates and standard errors for the corresponding variants in European and African ancestry-specific analyses. European ancestry-specific blood pressure effect estimates were obtained by first identifying from the 2018 Evangelou et al. International Consortium for Blood Pressure + UK Biobank GWAS, which included measurements of SBP, DBP, and PP in up to 757,601 individuals.15 Full GWAS summary statistics for the European ancestry blood pressure genome wide association study are publicly available and may be downloaded from the NHLBI GRASP catalog (https://grasp.nhlbi.nih.gov/FullResults.aspx). African ancestry-specific blood pressure effect estimates were obtained from BioVU (Vanderbilt University) and UK Biobank, and combined using fixed-effects inverse variance-weighted meta-analysis, including up to 16,784 individuals across both studies (Supplemental Methods).
Study Outcomes
The 2019 Million Veteran Program (MVP) genome wide association study of Peripheral Artery Disease by Klarin et al. identified 31,307 PAD cases (24,009 European; 7,373 African; 1,925 Hispanic) and 211,753 PAD-free controls .12 This study defined cases and controls based on electronic health record phenotyping within the Veterans Affairs (VA) Healthcare System and was validated against ankle brachial index measurement and manual chart review. The current analyses included trans-ancestry, European, and African-specific GWAS results. MVP PAD genome wide association study summary statistics are available on dbGAP (Accession phs001672.v2.p1).
We considered coronary artery disease (as another manifestation of atherosclerosis traditionally associated with elevations in blood pressure) for comparison. Genome-wide association study summary statistics for CAD were obtained from the Nikpay et al. 2015 CARDIoGRAMplusC4D 1000 Genomes-based GWAS.13 This study was a meta-analysis including 60,801 CAD cases and 123,504 controls, with genotypes imputed using the 1000 Genomes phase 1 version 3 reference. Summary statistics were downloaded from www.cardiogramplusc4d.org/data-downloads/.
Mendelian Randomization
Two-sample Mendelian randomization analyses were performed in R using the TwoSampleMR package (https://github.com/MRCIEU/TwoSampleMR).16 Genetic instruments for BP traits were constructed using variants that were in linkage equilibrium, physically separate (r2 < 0.001, distance = 10,000kb; 1000 Genomes reference panel), and associated with each trait at genome-wide significance (p < 5×10−8).. For bi-directional MR analysis, additional instruments were constructed for CAD and PAD using the same procedure. F-statistics were calculated for each variant using the formula F = beta2/SE2.The primary MR analyses used inverse-variance weighting with random effects. The MR-Egger intercept test was used to evaluate for evidence of horizontal pleiotropy. Leave-one-out, single-SNP, and funnel-plot diagnostic MR analyses were performed. Sensitivity analyses were performed using MR methods that make different assumptions about the presence of pleiotropy (weighted median, penalized weighted median, and weighted mode) 17. Multivariable MR (MVMR) was used in additional sensitivity analyses to jointly estimate the direct effects of blood pressure traits, again using genetic instruments based on variants that were in linkage equilibrium, physically separate (r2 < 0.001, distance = 10,000kb), and associated with any exposure at genome-wide significance (p < 5×10−8), weighted by the effect of each SNP on each exposure.18 MR-Steiger was performed to test the correct direction of effect.19 Effect estimates were scaled to correspond to a 10mmHg change in blood pressure.
Antihypertensive Drug MR
MR analyses were performed to estimate the effect of 10mmHg lowering of blood pressure by antihypertensive drugs. Genetic instruments consisted of variants that were associated with each BP trait at genome-wide significance and located near (+/− 200kb) or within genes encoding protein targets of 12 antihypertensive medication classes, with effect estimates for each genetic variant derived for each BP trait from the trans-ancestry BP GWAS.20,21 The primary analysis focused on the systolic blood pressure lowering effect, with sensitivity analyses considering the remaining BP traits (DBP, PP, MAP). Inverse-variance weighted, weighted-median, penalized weighted median, and weighted mode two-sample MR was performed, with MR-Egger intercept test used to assess for horizontal pleiotropy. For instruments with only 1 variant, Wald-ratio MR was performed.
Statistical Analysis
The primary analysis of the effect of blood pressure on PAD was performed using two-sample MR considering trans-ancestry BP exposures and trans-ancestry outcomes. We performed two additional ancestry-specific sensitivity analyses. First, we performed an analysis considering trans-ancestry BP exposures and ancestry-specific PAD outcomes. Second, due to the lack of genetic variants associated with BP traits as genome-wide significance in African-specific BP GWAS, we also performed a three-sample MR analysis. Here, genetic variants associated with BP were obtained from the trans-ancestry BP GWAS, with corresponding effect estimates and standard errors obtained from the European- and African-specific BP GWAS. Instruments were then filtered to include only those with F-statistic >10 to minimize weak instrument bias.14 Heterogeneity across ancestries was assessed using I2 and Cochran’s Q. The ratio of odds ratios (ROR) was used to compare effects of each BP trait on PAD and CAD.22 For all analyses we used Bonferroni adjustment for four BP traits, with p-values < 0.05/4 = 0.0125 considered significant. All statistical analyses were performed using R version 3.6.2 (R Foundation for Statistical Computing, Vienna, Austria).
RESULTS:
Effects of Genetic Variation in Blood Pressure on PAD: Mendelian Randomization
We performed two-sample Mendelian randomization using summary statistics from trans-ancestry genome-wide association studies to estimate the effect of genetic variation in blood pressure traits on PAD. Genetic instruments for blood pressure contained between 259 and 333 independent genetic variants, with F-statistics ranging from 29 to 938 (consistent with low risk of weak-instrument bias) (Supplemental Tables 1-2).
In inverse-variance weighted analyses, each genetically-proxied 10mmHg increase in SBP, DBP, MAP, and PP significantly increased the risk of PAD (SBP OR 1.20 [1.16–1.25] per 10mmHg increase, p = 1 × 10−24; DBP OR 1.27 [1.18–1.35], p = 4 × 10−11; MAP OR 1.26 [1.19–1.33], p = 6 × 10−16; PP OR 1.31 [1.24–1.39], p = 9 × 10−23) (Figure 1, Supplemental Table 3). The MR-Egger bias intercept term was p > 0.05 for all trait-outcome pairs (Supplemental Table 3). The results remained robust in sensitivity analyses using MR methods that make different assumptions about the presence of pleiotropy (Supplemental Table 3). MR-Steiger confirmed the directionality of all associations.
Figure 1: Effect of BP Traits on PAD.
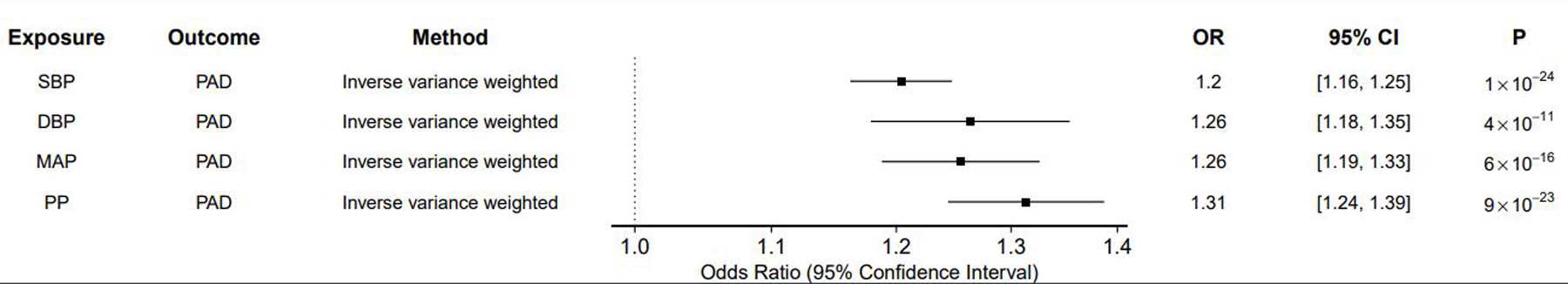
In inverse variance-weighted Mendelian randomization analyses, elevations in each blood pressure trait increased risk of peripheral artery disease. Results scaled to reflect odds of outcome per 10mmHg increase in blood pressure. n SNP = number of single nucleotide polymorphisms in the exposure instrument; OR = odds ratio; 95% CI = 95% confidence interval; P = p-value SBP = Systolic blood pressure; DBP = Diastolic blood pressure; MAP = mean arterial pressure; PP = Pulse pressure; PAD = Peripheral artery disease.
When considering trans-ancestry genetic instruments for BP and ancestry-specific PAD outcomes, SBP, DBP, MAP, and PP were significantly associated with PAD in a European-specific population, while only SBP and PP were associated with PAD in an African-specific population (Supplemental Figure 1A-B; Supplemental Tables 4-5). In these ancestry-specific analyses, we detected heterogenous effects of BP on PAD across all BP measures (I2 ranging from 87 to 90%, Cochran’s p < 0.05). Effects remained heterogeneous when considering both ancestry-specific BP effects and ancestry-specific PAD outcomes (Supplemental Figure 2; Supplemental Tables 6-7).
For comparison, we estimated the effects of SBP, DBP, MAP, and PP on CAD. As with PAD, each BP trait was significantly associated with CAD (Supplemental Tables 1-3). The effects of SBP, DBP, and MAP were greater for CAD than PAD (SBP Ratio of ORs 1.06 [1.0 – 1.12], p = 0.04; MAP ROR 1.15 [1.06–1.26], p = 8.6 × 10−4; DBP ROR 1.21 [1.08–1.35], p = 6.9 × 10−4) (Figure 2).
Figure 2: Effects of BP Traits on PAD vs. CAD.
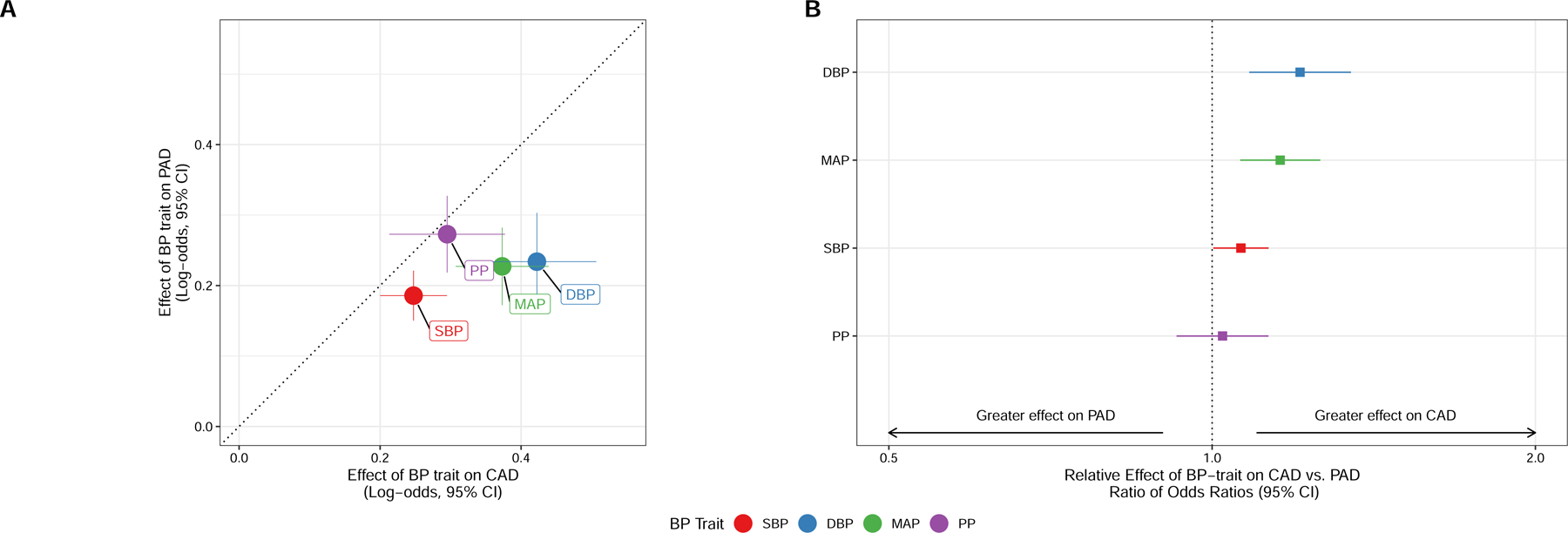
Comparison of the effects of BP traits on CAD and PAD. A) Log-odds effect estimates for CAD and PAD, with dotted line representing equal effects on both ASCVD outcomes, and crosshairs representing 95% confidence intervals for each effect estimate. B) Comparison of effect of each BP trait on PAD vs. CAD using the ratio of odds ratios test, with ROR > 1 representing greater effect on CAD and ROR < 1 representing greater effect on PAD.
Effects of Genetic Liability to PAD on Blood Pressure: Mendelian Randomization
Because stiffening of peripheral vessels may affect BP, the possibility of reverse-causation exists in assessment of the relationship between BP and PAD. To test for the presence of reverse-causation, we next perormed bi-directional MR analyses. Genetic instruments for PAD were selected and used to estimate the effect of genetic liability to PAD on blood pressure traits (Figure 3; Supplemental Tables 8-9). In inverse-variance weighted analysis, genetic liability to PAD increased SBP (beta = 0.28 mmHg per 1 log-odds increase in risk of PAD [0.16–0.41], p = 8 × 10−4), MAP (beta = 0.13 [0.027–0.24], p = 0.01), and PP (beta = 0.22 [0.13–0.32], p = 7 × 10−6). The MR-Egger bias intercept term was p > 0.05 for all analyses, indicating no positive evidence for bias. Results were consistent in sensitivity analysis applying MR methods making different assumptions about the presence of pleiotropy (Supplemental Table 9). For comparison, genetic liability CAD was not associated with BP traits after accounting for multiple testing (Supplemental Figure 2; Supplemental Tables 8-9). MR-Steiger confirmed the direction of effect for all associations.
Figure 3: Effects of PAD and CAD on BP traits.
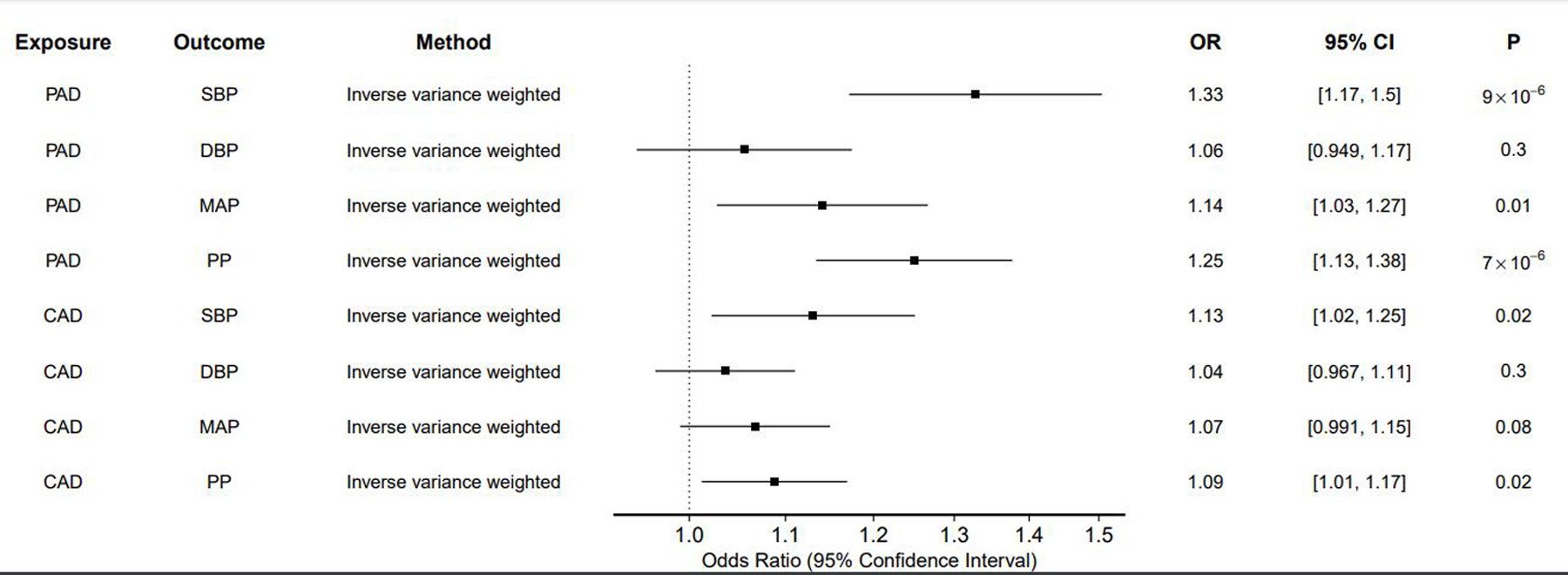
The effect of liability to PAD and CAD on each BP trait was estimated using inverse variance-weighted two-sample MR.
Multivariable Mendelian Randomization
Because blood pressure traits are highly correlated and unlikely to affect cardiovascular outcomes in isolation, we performed multivariable MR to jointly estimate the direct effects of blood pressure (as reflected by MAP), and arterial stiffness (as reflected by PP) trait on PAD. Considered jointly, each 10mmHg increase in both PP and MAP increased risk of PAD (PP OR 1.26 [1.17–1.35], p = 3 × 10−10; MAP OR 1.14 [1.06–1.23], p = 2 × 10−4) (Supplemental Figure 3).
Antihypertensive Drug Mendelian Randomization
In MR analyses designed to proxy the BP-lowering effects of antihypertensive medications, we identified several medications with protective effects on PAD. Genetically-proxied SBP-lowering via beta-blockers (OR 0.74 per 10mmHg decrease in SBP; 95% CI 0.65–0.84; p = 5 × 10−6), loop diuretics (OR 0.66 [0.48–0.91], p = 0.01), and thiazide diuretics (OR 0.57 [0.41, 0.79], p = 6 × 10−4) were associated with decreased risk of PAD (Figure 4; Supplemental Tables 10-11). The MR-Egger bias intercept term was p > 0.05 for all analyses, indicating no positive evidence for bias. Results were similar using MR methods making different assumptions about the presence of pleiotropy (Supplemental Tables 10-11). When considering genetically-proxied DBP and MAP-lowering effects we detected an additional protective association for renin-inhibitors (DBP OR 0.34 [0.17–0.67], p = 0.001; MAP OR 0.52 [0.29–0.91], p = 0.02) (Supplemental Tables 10-11). We did not detect any significant associations between genetically-proxied drug effects on PP-lowering and PAD.
Figure 4: Effect of SBP-lowering via genetic proxies of antihypertensive medications.
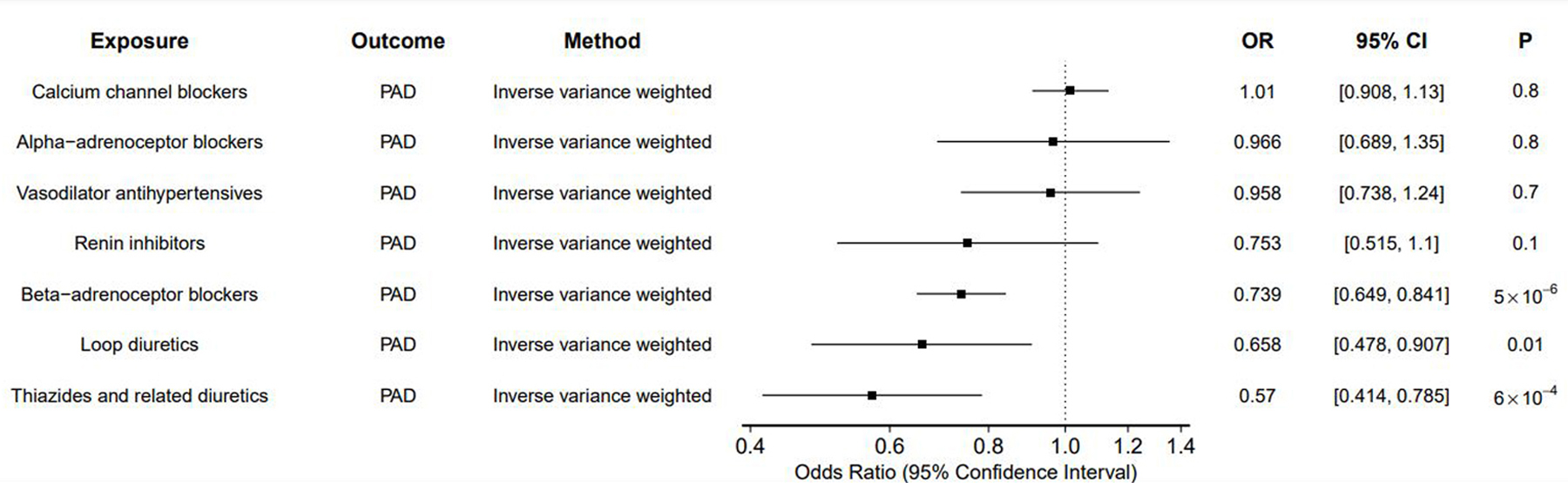
Inverse variance weighted MR was performed to estimate the SBP-lowering effect of genetic proxies of antihypertensive medications on PAD.
DISCUSSION
This Mendelian randomization study leveraged natural genetic variation to examine the relationship between blood pressure and both PAD. The principal findings were: 1) Lifetime exposure to elevated SBP, DBP, MAP, and PP all increased risk of PAD; 2) Elevated BP more strongly increased risk of CAD compared to PAD; 3) PAD led to small but significant increases in SBP, MAP, and PP; 4) Based on genetic proxies, beta-blockers, loop diuretics, thiazide diuretics, and renin-inhibitors were associated with decreased PAD risk. There are several implications from the results of this study.
First, this study supports observational findings that elevated blood pressure is associated with increased risk of PAD. Multiple observational studies have identified elevated SBP and clinical diagnosis of hypertension as strong risk factors for PAD, while the relationship between DBP and PAD has remained less clear.5–11,23–25 Unlike other observational studies, our MR study leveraged genetic variants as instrumental variables for SBP, DBP, MAP and PP. Because genetic variants are randomly inherited by offspring from their parents, mimicking a trial randomizing individuals to a lifetime of increased blood pressure, the Mendelian randomization framework is less susceptible to residual environmental confounding than traditional observational studies.14 The finding of our MR analysis that elevated SBP increases risk of both PAD and CAD is consistent with prior studies. We also find a strong effect of DBP on both PAD and CAD, clarifying discrepant findings in prior observational studies. Similarly, our multivariable MR findings demonstrate that both MAP and PP, reflecting pressure and pulsatility/stiffness respectively, both influence PAD risk. Overall, the MR findings of our study are consistent with a causal relationship between blood pressure traits and PAD.
Next, we found that elevated SBP, DBP, and MAP each increased risk of CAD more than PAD. These findings are in contrast to a prior observational analysis that found that SBP or DBP had similar effects on CAD and PAD.7 While broad recommendations for lifestyle modification and treatment of ASCVD risk factors are clearly important at both the population level and individual level, understanding the impact of interventions on specific ASCVD outcomes may further inform treatment and prevention guidelines and discussions with patients. Particularly in light of our recent finding that smoking more strongly increases risk of PAD in comparison to CAD or ischemic stroke 26, this study adds further nuance to the relationship between traditional ASCVD risk factors and specific ASCVD outcomes.
Our finding that increased pulse pressure increases PAD risk is consistent with findings from multiple prior observational studies.27–30 Because increased pulse pressure is a marker of increased arterial stiffness and may be caused by PAD, the observational studies investigating the relationship between these traits may have been limited by the possibility of reverse causality. Using bi-directional MR we were able to overcome this limitation, finding elevated PP to be a risk factor for PAD, and PAD to be a risk factor for increased PP. Similarly, when jointly considering MAP and PP we detected direct effects of both traits on PAD, suggesting that both arterial pressure (as measured by MAP) and arterial stiffness (as reflected by PP) directly influence development of PAD. Further study is warranted to determine the specific mechanisms by which these traits impact PAD, though our findings suggest Interventions targeting both traits may be useful in reducing the burden of PAD.
Finally, we used antihypertensive drug MR to estimate the effect of 10mmHg lowering of blood pressure by different classes of medication. In this analysis, we identified protective effects of several antihypertensive medications. A Cochrane Review found poor evidence for the use of antihypertensive medications specifically for PAD, though recognized the large benefit of these medications for prevention of cardiovascular events and mortality more broadly.31 Our results provide a genetic basis for considering future trials focused on beta-blockers, loop diuretics, and thiazide diuretics. While small beneficial genetic effects may compound over a lifetime leading to protection from ASCVD, the effects of antihypertensive medications occur on a much shorter timescale. Our findings do not exclude meaningful beneficial effects of other potent antihypertensive medications on risk of PAD, particularly given the strong overall causal effects of each BP trait on PAD. The optimal antihypertensive regimens for prevention/treatment of PAD remains unclear and may represent a focus for future effectiveness studies.
The overall findings of our study have implications for PAD prevention and treatment guidelines. The current 2016 American Heart Association/American College of Cardiology (AHA/ACC) and 2017 European Society of Cardiology (ESC) PAD guidelines make strong recommendations for the treatment of hypertension to prevent cardiovascular events.3,4 The trials cited to support these recommendations focused on cardiovascular events broadly, or differences in safety and efficacy between different antihypertensive classes, rather than PAD-specific outcomes.32–40 Our MR study provides strong evidence consistent with a casual effect of increased blood pressure on PAD. In the absence of large, randomized trials of antihypertensive medications focused on PAD-specific outcomes, these results add support for current guideline recommendations, and suggest possible medication classes that warrant further study specifically for PAD. Recent studies like SPRINT and multi-society BP guidelines have suggested that aggressive BP-lowering may be associated with improved outcomes in individuals at high ASCVD risk.41,42 While our current analyses do not provide a specific BP-lowering target that minimizes risk of PAD, future analyses leveraging participant-level data may help identify treatment thresholds. Our current results may help calibrate the overall expected benefit that programs to treat hypertension may have on the global burden of PAD.
This study has several limitations. Although we considered trans-ancestry studies of blood pressure and PAD, the underlying populations were primarily composed of individuals of European ancestry. We performed extensive ancestry-specific analyses, identifying heterogenous effects of BP on PAD. Whether these findings reflect biological differences in the pathogenesis of elevated blood pressure and PAD that vary by ancestry or reflect limitations of our current understanding of the genetic basis of these traits remains uncertain. Further study of BP and ASCVD genetics in diverse ancestral populations is necessary to improve the generalizability of our findings. Similarly, the stronger associations between BP traits and CAD in comparison to PAD may reflect pathophysiological differences in the risk factors for atherosclerosis across diverse vascular beds or may be due to differences in sampling or ascertainment of the underlying PAD and CAD GWAS studies. Mendelian randomization relies on a number of assumptions in order for causal estimates to be valid.14 While we have employed multiple MR methods and sensitivity analyses to assess for and address potential violations of these assumptions, we cannot completely exclude the possibility of confounding. Future study on the role of hypertension treatment in the prevention and treatment of PAD focused on PAD-specific outcomes is warranted.
Overall, we find strong evidence consistent with a causal effect of blood pressure traits on PAD, although find a stronger effect of SBP, DBP, and MAP on CAD in comparison to PAD. We identify genetic proxies of antihypertensive medications associated with decreased PAD risk, which may be prioritized for future study.
Supplementary Material
HIGHLIGHTS.
Although Peripheral Artery Disease (PAD) is a common manifestation of Atherosclerotic Cardiovascular Disease, the causal impact of blood pressure (BP) on risk of PAD has remained uncertain
In this Mendelian randomization study, increases in BP were robustly associated with increased risk of PAD
Genetic proxies of several antihypertensive medication classes were associated with decreased risk of PAD
Overall, this study provides evidence consistent with a causal association between BP and PAD, and prioritizes medications for future studies that consider PAD-specific outcomes
ACKNOWLEDGEMENTS:
We thank the participants of the VA Million Veterans Program and VA MVP collaborators (Supplemental Acknowledgements).
FUNDING:
This work was supported by US Department of Veterans Affairs grants IK2-CX001780 (S.M.D), MVP-DOE2 (S.M.D/P.S.T) and I01-BX003362 (P.S.T/K.M.C). This research is based on data from the MVP, Office of Research and Development, Veterans Health Administration. This publication does not represent the views of the Department of Veterans Affairs or the United States government. This work was also supported by the National Institute of Diabetes and Digestive and Kidney Diseases DK101478 (B.F.V), and a Linda Pechenik Montague Investigator Award (B.F.V). D.G. was supported by the British Heart Foundation Centre of Research Excellence (RE/18/4/34215) at Imperial College London and a National Institute for Health Research Clinical Lectureship at St. George’s, University of London (CL-2020–16-001). The Medical Research Council (MRC) and the University of Bristol support the MRC Integrative Epidemiology Unit [MC_UU_00011/1]. N.M.D is supported by a Norwegian Research Council Grant number 295989. J.N.H. is supported by K12 HD04348. The dataset(s) used for the African ancestry BP analyses were obtained from Vanderbilt University Medical Center’s BioVU which is supported by institutional funding, private agencies, and federal grants, including: NIH funded Shared Instrumentation Grant S10RR025141; CTSA grants UL1TR002243, UL1TR000445, and UL1RR024975; investigator-led projects that include U01HG004798, R01NS032830, RC2GM092618, P50GM115305, U01HG006378, U19HL065962, R01HD074711; and additional funding sources listed at https://victr.vumc.org/biovu-funding/.
Footnotes
DISCLOSURES:
D.G is employed part-time by Novo Nordisk. S.M.D has received grants from RenalytixAI and personal fees from Calico Labs outside the submitted work.
DATA AVAILABILITY:
Summary statistics for blood pressure genome wide association studies are available from https://pan.ukbb.broadinstitute.org/ and the NHLBI GRASP catalog (https://grasp.nhlbi.nih.gov/FullResults.aspx). MVP PAD genome wide association study summary statistics are available on dbGAP (Accession phs001672.v2.p1). Data on coronary artery disease have been contributed by CARDIoGRAMplusC4D investigators and may been downloaded from www.cardiogramplusc4d.org/data-downloads/.
REFERENCES:
- 1.James SL, Abate D, Abate KH, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 Diseases and Injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018;1789–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Criqui MH, Aboyans V. Epidemiology of Peripheral Artery Disease. Circ Res 2015;116:1509–1526. [DOI] [PubMed] [Google Scholar]
- 3.Gerhard-Herman MD, Gornik HL, Barrett C, et al. 2016 Lower Extremity Peripheral Arterial Disease Guidelines. J Am Coll Cardiol 2017;69:e71–e126. [DOI] [PubMed] [Google Scholar]
- 4.Aboyans V, Ricco J-B, Bartelink M-LEL, et al. 2017 ESC Guidelines on the Diagnosis and Treatment of Peripheral Arterial Diseases, in collaboration with the European Society for Vascular Surgery (ESVS): Document covering atherosclerotic disease of extracranial carotid and vertebral, mesenteric, renal,. Eur Heart J 2017;39:763–816. [DOI] [PubMed] [Google Scholar]
- 5.Bainton D, Sweetnam P, Baker I, Elwood P. Peripheral vascular disease: consequence for survival and association with risk factors in the Speedwell prospective heart disease study. Heart 1994;72:128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Criqui MH, Vargas V, Denenberg JO, Ho E, Allison M, Langer RD, Gamst A, Bundens WP, Fronek A. Ethnicity and peripheral arterial disease: The San Diego population study. Circulation 2005;112:2703–2707. [DOI] [PubMed] [Google Scholar]
- 7.Gerald F, Housley E, Riemersma RA, Macintyre CCA, Cawood EHH, Prescott RJ, Ruckley CV. Smoking, lipids, glucose intolerance, and blood pressure as risk factors for peripheral atherosclerosis compared with ischemic heart disease in the Edinburgh artery study. Am J Epidemiol 1992;135:331–340. [DOI] [PubMed] [Google Scholar]
- 8.Hooi JD, Kester ADM, Stoffers HEJH, Overdijk MM, Van Ree JW, Knottnerus JA. Incidence of and risk factors for asymptomatic peripheral arterial occlusive disease: A longitudinal study. Am J Epidemiol 2001;153:666–672. [DOI] [PubMed] [Google Scholar]
- 9.Dagenais GR, Maurice S, Robitaille NM, Gingras S, Lupien PJ. Intermittent claudication in Quebec men from 1974–1986: the Quebec Cardiovascular Study. Clin Invest Med 1991;14:93–100. [PubMed] [Google Scholar]
- 10.Garg PK, Biggs ML, Carnethon M, Ix JH, Criqui MH, Britton KA, Djoussé L, Sutton-Tyrrell K, Newman AB, Cushman M, Mukamal KJ. Metabolic syndrome and risk of incident peripheral artery disease: The cardiovascular health study. Hypertension 2014;63:413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Howard DPJ, Banerjee A, Fairhead JF, Hands L, Silver LE, Rothwell PM. Population-Based Study of Incidence, Risk Factors, Outcome, and Prognosis of Ischemic Peripheral Arterial Events: Implications for Prevention. Circulation 2015;132:1805–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klarin D, Lynch J, Aragam K, et al. Genome-wide association study of peripheral artery disease in the Million Veteran Program. Nat Med 2019;25:1274–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nikpay M, Goel A, Won HH, et al. A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet 2015;47:1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davies NM, Holmes M V., Davey Smith G. Reading Mendelian randomisation studies: A guide, glossary, and checklist for clinicians. BMJ 2018;362:k601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Evangelou E, Warren HR, Mosen-Ansorena D, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet 2018;50:1412–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hemani G, Zheng J, Elsworth B, et al. The MR-base platform supports systematic causal inference across the human phenome. Elife 2018;7:e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet Epidemiol 2016;40:304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanderson E, Davey Smith G, Windmeijer F, Bowden J. An examination of multivariable Mendelian randomization in the single-sample and two-sample summary data settings. Int J Epidemiol 2019;48:713–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hemani G, Tilling K, Davey Smith G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet 2017;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gill D, Georgakis MK, Koskeridis F, Jiang L, Feng Q, Wei W-Q, Theodoratou E, Elliott P, Denny JC, Malik R, Evangelou E, Dehghan A, Dichgans M, Tzoulaki I. Use of Genetic Variants Related to Antihypertensive Drugs to Inform on Efficacy and Side Effects. Circulation 2019;140:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walker VM, Kehoe PG, Martin RM, Davies NM. Repurposing antihypertensive drugs for the prevention of Alzheimer’s disease: a Mendelian randomization study. Int J Epidemiol 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Altman DG, Bland JM. Interaction revisited: The difference between two estimates. BMJ 2003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meijer WT, Grobbee DE, Hunink MGM, Hofman A, Hoes AW. Determinants of peripheral arterial disease in the elderly: The Rotterdam Study. Arch Intern Med 2000;160:2934–2938. [DOI] [PubMed] [Google Scholar]
- 24.Newman AB, Siscovick DS, Manolio TA, Polak J, Fried LP, Borhani NO, Wolfson SK. Ankle-arm index as a marker of atherosclerosis in the cardiovascular health study. Circulation 1993;88:837–845. [DOI] [PubMed] [Google Scholar]
- 25.McGee DL. Update on Some Epidemiologic Features of Intermittent Claudication: The Framingham Study. J Am Geriatr Soc 1985;33:13–18. [DOI] [PubMed] [Google Scholar]
- 26.Levin MG, Klarin D, Assimes TL, Freiberg MS, Ingelsson E, Lynch J, Natarajan P, O’Donnell C, Rader DJ, Tsao PS, Chang K-M, Voight BF, Damrauer SM, Program VMV. Genetics of Smoking and Risk of Atherosclerotic Cardiovascular Diseases: A Mendelian Randomization Study. medRxiv 2020;2020.04.07.20053447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mao Y, Huang Y, Yu H, Xu P, Yu G, Yu J, Zhan Y. Incidence of peripheral arterial disease and its association with pulse pressure: A prospective cohort study. Front Endocrinol (Lausanne) 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhan Y, Yu J, Chen R, Sun Y, Fu Y, Zhang L, Li S, Zhang F, Hu D. Prevalence of low ankle brachial index and its association with pulse pressure in an elderly Chinese population: a cross-sectional study. J Epidemiol 2012;22:454–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korhonen P, Kautiainen H, Aarnio P. Pulse pressure and subclinical peripheral artery disease. J Hum Hypertens 2014;28:242–5. [DOI] [PubMed] [Google Scholar]
- 30.Kiuchi S, Hisatake S, Watanabe I, Toda M, Kabuki T, Oka T, Dobashi S, Ikeda T. Pulse Pressure and Upstroke Time Are Useful Parameters for the Diagnosis of Peripheral Artery Disease in Patients With Normal Ankle Brachial Index. Cardiol Res 2016;7:161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lane DA, Lip GYH. Treatment of hypertension in peripheral arterial disease. Cochrane Database Syst. Rev 2013;2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ostergren J, Sleight P, Dagenais G, Danisa K, Bosch J, Qilong Y, Yusuf S, HOPE study investigators. Impact of ramipril in patients with evidence of clinical or subclinical peripheral arterial disease. Eur Heart J 2004;25:17–24. [DOI] [PubMed] [Google Scholar]
- 33.Yusuf S. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med 2000;342:145–153. [DOI] [PubMed] [Google Scholar]
- 34.Yusuf S, Teo KK, Pogue J, Dyal L, Copland I, Schumacher H, Dagenais G, Sleight P, Anderson C. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008;358:1547–1559. [DOI] [PubMed] [Google Scholar]
- 35.Bavry AA, Anderson RD, Gong Y, Denardo SJ, Cooper-Dehoff RM, Handberg EM, Pepine CJ. Outcomes Among hypertensive patients with concomitant peripheral and coronary artery disease: findings from the INternational VErapamil-SR/Trandolapril STudy. Hypertens (Dallas, Tex 1979) 2010;55:48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zanchetti A, Julius S, Kjeldsen S, et al. Outcomes in subgroups of hypertensive patients treated with regimens based on valsartan and amlodipine: An analysis of findings from the VALUE trial. J Hypertens 2006;24:2163–8. [DOI] [PubMed] [Google Scholar]
- 37.Diehm C, Pittrow D, Lawall H. Effect of nebivolol vs. hydrochlorothiazide on the walking capacity in hypertensive patients with intermittent claudication. J Hypertens 2011;29:1448–56. [DOI] [PubMed] [Google Scholar]
- 38.Espinola-Klein C, Weisser G, Jagodzinski A, Savvidis S, Warnholtz A, Ostad M-A, Gori T, Munzel T. β-Blockers in patients with intermittent claudication and arterial hypertension: results from the nebivolol or metoprolol in arterial occlusive disease trial. Hypertens (Dallas, Tex 1979) 2011;58:148–54. [DOI] [PubMed] [Google Scholar]
- 39.Paravastu SCV, Mendonca DA, Da Silva A. Beta blockers for peripheral arterial disease. Cochrane database Syst Rev 2013;CD005508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Group TAO and C for the ACR, Coordinators TAO and, Antihypertensive T, Treatment L. Major Outcomes in High-Risk Hypertensive Patients Randomized to Angiotensin-Converting Enzyme Inhibitor or Calcium Channel Blocker vs Diuretic. JAMA J Am Med Assoc 2002;288:2981–2997. [DOI] [PubMed] [Google Scholar]
- 41.A Randomized Trial of Intensive versus Standard Blood-Pressure Control. N Engl J Med 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults. J Am Coll Cardiol 2017; [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.