

Abstract
Objective:
High-density lipoprotein (HDL) can exert both anti-inflammatory and pro-inflammatory effects in macrophages due to its ability to induce cholesterol depletion. Because cholesterol depletion also increases sheddase activity of the membrane protease ADAM17 (ADAM metallopeptidase domain 17) in other cells, we examined if ADAM17 plays a role in HDL’s effects on inflammatory processes in macrophages in vitro and in vivo.
Approach and Results:
Sorted peritoneal macrophages from human APOA1 transgenic LDL receptor-deficient (APOA1Tg; Ldlr−/−) mice with and without myeloid cell-targeted ADAM17-deficiency were studied in parallel with wildtype and ADAM17-deficient bone marrow-derived macrophages stimulated with HDL in vitro. HDL increased ADAM17 expression and activity in macrophages. Furthermore, ADAM17-deficient macrophages exhibited reduced expression of ATP-binding cassette A1 (ABCA1) and reduced cholesterol efflux, and were cholesterol loaded. This was caused by the absence of shedding of TNFα, a major ADAM17 substrate. Sorted thioglycollate-elicited peritoneal macrophages freshly isolated from APOA1Tg; Ldlr−/− mice, which have higher HDL levels than Ldlr−/− controls, showed reduced expression of interferon-inducible genes in response to lipopolysaccharide or interferon-β, but exacerbated pro-inflammatory responses to lipopolysaccharide for Tnfa, Cxcl1, Ccl2 and Il1b, phenocopying cells stimulated with HDL in vitro. These effects were all prevented in ADAM17-deficient macrophages, and associated with lower concentrations of large HDL particles in APOA1Tg; Ldlr−/− mice with myeloid cell-targeted ADAM17-deficiency.
Conclusions:
The increased cholesterol loading of ADAM17-deficient macrophages prevents both anti- and pro-inflammatory responses of HDL. Our findings demonstrate a novel role for ADAM17 in maintaining cholesterol efflux in macrophages, thereby regulating the immune functions of these cells.
Keywords: ADAM17, Cholesterol efflux, High-density lipoprotein, Inflammation, Macrophages
Subject terms: Inflammation, Lipids and Cholesterol, Mechanisms
Introduction
High-density lipoprotein-cholesterol (HDL-C) is inversely associated with atherosclerotic cardiovascular disease (CVD).1, 2 However, a causal relationship between HDL-C and CVD has been challenged by Mendelian randomization studies3, 4 and by the failure or mediocre success of HDL-C-elevating treatments in cardiovascular outcome trials of statin-treated subjects.5, 6 More recently, a U-shaped association has been demonstrated between HDL-C and all-cause mortality, suggesting that, at very high levels, HDL-C may associate with increased cardiovascular events and all-cause death.7 Due to the lack of compelling evidence that HDL-C is in the causal pathway for CVD, attention has been turned toward the functionality of the HDL particle.
The most established functional property of HDL is its ability to remove cholesterol from myeloid cells through aqueous diffusion, scavenger receptor BI and the cholesterol transporters ABCA1 and ABCG1 (ATP-binding cassette transporters A1 and G1).8–10 The cholesterol efflux capacity of HDL in macrophages was indeed found to be superior to HDL-C levels in distinguishing between patients at increased risk for coronary artery disease and control individuals, highlighting the importance of understanding HDL’s functions.11, 12
Cholesterol efflux by HDL changes the macrophage plasma membrane lipid composition,10 which can modulate the effects of toll-like receptor (TLR) and type I interferon (IFN) stimulation, important pathways in the innate immune responses to pathogens.13–16 There have been recent interesting but contradictory findings on the role of HDL in modulating macrophage inflammatory responses, as some studies show an anti-inflammatory response to HDL17–20 and others describe pro-inflammatory effects of HDL or apolipoprotein A1 (APOA1), the main structural protein of HDL.21, 22 Ito and colleagues observed that cholesterol depletion by cyclodextrin suppresses ABCA1 expression and lipopolysaccharide (LPS)-induced inflammatory genes in bone marrow-derived macrophages (BMDMs).23 Similarly, Suzuki et al. observed anti-inflammatory responses to HDL on LPS-induced IFN signaling in macrophages.17 Contrary, work by van der Vorst and colleagues demonstrated that cholesterol depletion in macrophages by cyclodextrin or HDL can exacerbate pro-inflammatory responses to TLR ligands in macrophages.21 Moreover, Smoak et al. reported pro-inflammatory effects of APOA1 in macrophages.22
To better understand the effects of HDL on inflammatory responses in macrophages, we recently systematically examined global transcriptomic effects of HDL. Our studies revealed that HDL can exert both pro-inflammatory and anti-inflammatory responses in macrophages in vitro and that these responses are temporally different and mediated by distinct intracellular signaling pathways.24, 25 Moreover, we showed that both the pro-inflammatory and anti-inflammatory effects of HDL are mediated by cholesterol depletion. The anti-inflammatory response was due in part to suppression of type I IFN signaling while the late pro-inflammatory response to HDL was due to activation of an IRE1a (inositol-requiring enzyme 1a)/ASK1 (apoptosis signal-regulating kinase 1)/p38 MAPK (p38 mitogen-activated protein kinase) pathway.24, 25 Our studies also suggested that the anti-inflammatory effects of HDL predominate over pro-inflammatory effects in myeloid cells in lesions of atherosclerosis.25
ADAM metallopeptidase domain 17 (ADAM17), also known as TACE [tumor necrosis factor-α (TNFα)-converting enzyme] is a transmembrane protease critically involved in shedding of many cell surface proteins, including inflammatory cytokines/chemokines and their receptors. Over 80 substrates of ADAM17 have been identified and the list is still growing.26 Several of these substrates, such as TNFα and its receptors, ICAM-1, and VCAM-1 have been implicated in atherosclerosis.26 In mouse models, global reduction of ADAM17 expression results in increased lesion development through a mechanism involving enhanced TNF receptor 2 signaling.27 Contrary, endothelial cell-targeted ADAM17-deficiency reduces atherosclerosis, while myeloid cell-targeted ADAM17-deficiency has been shown to cause larger lesions with fewer macrophages and a more stable phenotype.28 Other studies are consistent with the hypothesis that ADAM17 in myeloid cells mediates primarily detrimental effects on atherosclerosis. For example, ADAM17 promotes tissue inflammation and limits synthesis of pro-resolving mediators and efferocytosis by cleaving MerTK29, 30 and promotes lesion macrophage proliferation.31
We reasoned that because ADAM17 is activated by plasma membrane lipid perturbations and cholesterol depletion in other cell types,32–35 ADAM17 might have a hitherto unknown role in mediating HDL’s effects in macrophages. Our findings show that HDL induces ADAM17 and that this mechanism serves to boost the functional effects of HDL through ABCA1 in macrophages.
Materials and Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Mice
All animal studies were approved by the Animal Care and Use Committee of the University of Washington (protocol 3154-01). Wild type C57BL/6J (stock number 000664) and APOA1Tg (stock number 001927) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Mice with myeloid cell-targeted deletion of ADAM17 (ADAM17M−/−) were generated by crossing Adam17fl/fl mice31 to mice expressing Cre recombinase under control of the Lyz2 promoter (Lyz2Cre/Cre). Lyz2Cre/Cre; Adam17Wt/Wt littermates were used as controls. We crossed APOA1Tg mice with Ldlr−/− mice to generate APOA1Tg; Ldlr−/− mice and Ldlr−/− littermates. All mice were on the C57BL/6J background and males and females 8–16 weeks of age were studied.
Bone marrow transplant study
To investigate the role of myeloid cell ADAM17 in HDL’s actions in vivo, chimeric mice were generated by whole body irradiation (10 Gy) of male APOA1Tg; Ldlr−/− and Ldlr−/− recipient mice. Femurs and tibias were removed from male Lyz2Cre/Cre; Adam17Wt/Wt or Lyz2Cre/Cre; Adam17fl/fl mice. Bone marrow was flushed out with sterile PBS (phosphate buffered saline, pH: 7.5) and purified of erythrocytes by ACK lysis buffer. Recipient mice were reconstituted with 5 × 106 bone marrow cells injected retro-orbitally within 24 hr post-irradiation. Bone marrow was allowed to engraft for 8 weeks before the mice were used for experiments. During recovery, the mice were treated with an antibiotic (2 mg/ml neomycin) added to the drinking water for the first two weeks after irradiation. After 8 weeks of recovery, the mice were sedated to collect whole blood for analysis of leukocyte numbers by a Hemavet automatic cell counter (Drew Scientific Inc., Miami Lakes, FL). Chimerism was confirmed by loss of Adam17 mRNA as well as cell surface expression of CD115 (an ADAM17 substrate) on blood monocytes (see Figure IA in the online-only Data Supplement for gating strategy). Plasma lipids were determined by colorimetric assays, according to the manufacturers’ instructions: triglycerides (Sigma-Aldrich, St. Louis, MO) and cholesterol (FUJIFILM Wako Diagnostics U.S.A., Mountain View, CA). Cholesterol lipoprotein profiles were analyzed as described previously.36
Thioglycollate-elicited peritoneal macrophages (1 ml of 3% thioglycollate per mouse, i.p.) were isolated by lavage 4 days after thioglycollate injection. Thioglycollate-elicited peritoneal macrophages identified as F4/80+ cells were stained with BODIPY 493/503 (Invitrogen, 1:1000 dilution) to measure neutral lipid content (see Figure IB in the online-only Data Supplement for gating strategy). The macrophage population was purified by depletion of unwanted non-macrophage cells using a negative selection macrophage isolation kit (Miltenyi Biotec Inc., Auburn, CA; 130-110-434). The sorted cells consisted of ~90% macrophages, identified as F4/80+ cells (Figure IC in the online-only Data Supplement). Sorted macrophages were further purified by 1 hr adhesion onto tissue culture plates before stimulation with LPS (ultrapure LPS; 10 ng/ml; List Biological Laboratories Inc.; NC9633766) or mouse recombinant IFNβ (1 ng/ml; R&D systems). At the end of the treatments, gene expression was measured by real-time PCR. To validate the benefits of macrophage sorting and subsequent adhesion purification versus adhesion purification only, we compared gene expression of different cellular markers. The magnetic sorting method with subsequent adhesion purification resulted in lower expression of the B-cell marker Cd19, as compared with purification by adhesion alone (Figure ID in the online-only Data Supplement), indicating that magnetic sorting plus cell adhesion purification results in a cleaner macrophage population than adhesion purification alone. We therefore used sorted and adhesion purified macrophages for subsequent experiments involving thioglycolate-elicited peritoneal macrophages.
Isolation, culture and stimulation of macrophages
Bone marrow-derived macrophages (BMDMs) were isolated and cultured as described previoulsy.37 After 7 days, BMDMs were treated with HDL isolated from APOA1Tg; Ldlr−/− mice in RPMI1640 medium containing 2% FBS for 18 hr, followed by washing of the cells and stimulation with ultrapure LPS, recombinant mouse IFNβ, TNFα (20 ng/ml; R&D systems; 410-MT), R848 (2 μg/ml, InvivoGen; tlrl-r848) or Poly:IC (10 μg/ml, Tocris Bioscience; 4287) for the indicated periods of time in the presence of 30% L929-conditioned medium. In some experiments, instead of HDL the cells were pre-incubated for 18 h with LDL isolated from plasma of APOA1Tg; Ldlr−/− mice, or with human wildtype APOA1 or an APOA1 C-terminal deletion mutant (1–184).38 APOA1 C-terminal deletion mutants have a severely reduced ability of inducing cholesterol efflux through ABCA1.39 In other experiments, chemical inhibitors were added before HDL treatment and LPS stimulation. These included: a p38 MAPK inhibitor (SB202190; 10 μM, Santa Cruz Biotechnology, Inc., Dallas, TX), ASK1 inhibitor (Selonsertib 10 μM, Selleck Chemicals Llc; Houston, TX) and an ADAM17 inhibitor (TAPI-1; 20 μM, Enzo Life Sciences, Inc., Farmingdale, NY). For some experiments, BMDMs were treated with the cholesterol-depleting agent methyl-β-cyclodextrin (MβCD; 10 μM, Sigma) or with acetylated LDL (AcLDL; 50 μg/ml human acetylated LDL; Kalen Biomedical, Germantown, MD; product number 770201) to prevent cholesterol depletion before stimulation of the cells with HDL and LPS. In TNFα neutralization experiments, BMDMs were treated with a neutralizing anti-TNFα antibody (10 μg/ml; R&D systems; MAB4101) or isotype control (10 μg/ml; R&D systems; MAB005) for 4 hr to block the response of shed TNFα.
For isolation of thioglycollate-elicited macrophages, peritoneal cells were collected from Ldlr−/− or APOA1Tg; Ldlr−/− male and female mice 4 days after thioglycollate injection, and purified by negative isolation, as described above. Isolated and sorted macrophages were further purified by adhesion (1 hr) before stimulation with LPS or IFNβ in RPMI1640 supplemented with 2% FBS for indicated periods of time. After stimulation with LPS or IFNβ, cells were lysed in RNA lysis buffer for RNA isolation or collected in Western lysis buffer (25 mM Tris-HCL pH 7.4, 150 mM NaCl, 2 mM EDTA, 10 mM Na2SO4, 1% Triton-X100) supplemented with phosphatase inhibitors and protease inhibitors, including 50 μM GM6001 to prevent ADAM17 autoproteolysis for protein analysis. The duration of the treatments is described in the figure legend of each experiment.
Quantitative PCR and ELISAs
Quantitative PCR was performed as described previously.40 Primers for qPCR assays are provided in Table SI. TNFα, CCL2 (Invitrogen) and CXCL1 (R&D systems) ELISAs were performed on conditioned media, according to the manufacturers’ instructions. Results were normalized to total cellular protein.
Immunoblot analysis
BMDMs were washed twice in PBS and lysed with lysis buffer supplemented with phosphatase inhibitors and protease inhibitors, including 50 μM GM6001 to prevent ADAM17 autoproteolysis.41 Cellular protein quantity was measured with the Pierce BCA protein assay kit (Thermo Fisher Scientific), and cell lysates were then subjected to SDS-PAGE on 10% gels with a protein ladder (Bio-Rad Laboratories, Hercules, CA), and transferred to nitrocellulose membranes (Bio-Rad). The membranes were blocked in 5% non-fat dry milk (Bio-Rad) in Tris-buffered saline with 0.1% Tween-20 (Thermo Fisher Scientific). To monitor the active form of ADAM17,31 cell lysates were added to neutravidin-agarose and incubated end-over-end for 2 hrs. Following 4 washes with lysis buffer supplemented with 300 mM NaCl, the biotinylated proteins were eluted using peptide-N-glycosidase F (PNGase F) denaturation buffer with boiling. Samples were treated with 500 units of PNGase F (New England BioLabs) and incubated for 1 hr at 37°C, followed by SDS gel separation and immunoblot analysis of ADAM17.
The primary and secondary antibodies and the dilution which they were used were: p-p38 MAPK (Cell Signaling; 9211, 1:1000 dilution), p38 MAPK (Cell Signaling; 9212, 1:2000 dilution), p-STAT1 (Cell Signaling; 9167, 1:1000 dilution), STAT1 (Cell Signaling; 9172, 1:2000 dilution), ADAM17 (Cell Signaling; 3976, 1:1000 dilution), β-actin (Sigma-Aldrich; A1978, 1:10000 dilution), α-tubulin (Sigma-Aldrich; T5168, 1:5000 dilution), anti-rabbit IgG HRP-linked antibody (Cell Signaling, 1:3000 dilution), anti-mouse IgG HRP-linked antibody (GE Healthcare, 1:10000 dilution). For detection of horseradish peroxidase (HRP) activity, the Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific) was used.
Flow cytometry
BMDMs were cultured on non-tissue culture plates (Thermo Fisher Scientific) and gently dislodged from the plates using ice-cold PBS containing 2 mM EDTA to avoid damage to cell surface antigens. BMDMs were centrifuged, washed, and resuspended in FACS buffer (0.1% BSA, 5 mM EDTA). Cell suspensions were preincubated with anti–CD16/CD32 mAb (eBiosciences; 14-0161, 1:4000 dilution) to block FcγRII/III receptors and stained on ice. The cells were stained for 30 min with a PE-labeled TREM2 antibody (Invitrogen; MA5-28225, 1:200 dilution), a PE-Cy7-labeled CD36 clone HM36 (Biolegend; 102615, 1:200 dilution), an APC-labeled MerTK clone DS5MMER (eBiosciences; 17-5751-80, 1:200 dilution), FITC-labeled VLDL receptor antibody (Abcam; ab75591, 1:200 dilution) or the neutral lipid dye BODIPY 493/503 (1:1000 dilution).
Thioglycollate-elicited peritoneal cells were collected from APOA1Tg; Ldlr−/− mice and Ldlr−/− littermates using PBS containing ultrapure 2 mM EDTA (Invitrogen). Cell suspensions were preincubated with anti–CD16/CD32 mAb to block FcγRII/III receptors, and were stained on ice. The cells were stained for 30 min with fluorochrome-conjugated monoclonal antibodies in the following color staining combinations: PE-Cy7-labeled F4/80 clone BM8 (eBiosciences; 25-4801-82, 1:2000 dilution), TLR4-PE/Cy7 clone SA15-21 (Biolegend; 145408, 1:200 dilution), V500-labeled CD11b clone M1/70 (BD Horizon; 1:1000 dilution), APC-labeled MerTK clone DS5MMER (eBiosciences; 17-5751-80, 1:200 dilution) and PE-labeled TREM2 (Invitrogen; MA5-28225, 1:200 dilution) or BODIPY 493/503 (1:1000 dilution). In separate sets of experiments, retro-orbital blood was harvested and purified of erythrocytes. Fluorescently labeled antibodies were added after addition of a viability dye (diluted 1:2; eBioscience, catalog 65-0863) and an Fc blocking step (eBioscience, catalog 14-0161). The cells were then stained for 30 min with PE-labeled CD115 clone AFS98 (eBiosciences; 12-1152-83, 1:1000 dilution). Cells were kept at 4°C during all steps in the staining protocol. All samples were analyzed on a FACSCanto II (BD Biosciences) or FACScan™ system (Becton Dickinson), running FlowJo software (Becton Dickinson).
Cholesterol and triglyceride assays
Cells were washed twice with ice-cold PBS and lysed with hypotonic buffer (PBS containing 10 mM sodium cholate, pH 7.4). The samples were centrifuged at 5,000 × g at 4 °C for 10 min to remove cell debris. The collected supernatants were used to measure total cholesterol mass using an Amplex Red Cholesterol Assay Kit (Life Technologies, Cat # A12216) according to the supplier’s instructions. In addition, triglyceride content in the samples was determined by using a colorimetric assay from Sigma-Aldrich (St Louis, MO) and normalized to milligrams of protein in the extracted supernatants.
Lipid raft, BODIPY and filipin staining
For lipid raft staining, BMDMs from C57BL/6J WT mice or peritoneal macrophages from Ldlr−/− and APOA1Tg; Ldlr−/− mice were plated on eight-chambered Lab-Tek borosilicate cover glass (Thermo Fisher Scientific, Nunc) at a density of 1 × 105 cells per well. Staining was performed using the cholera toxin subunit B (Thermo Fisher Scientific) according to the manufacturer’s protocol. Pictures were collected using a Nikon A1R Confocal microscope at 60X magnification and quantified using Image J analysis software. Corrected total cell fluorescence (CTCF) was calculated by using the formula, CTCF = Integrated Density – (area of selected cell X mean fluorescence of background readings).
Lipid droplets were identified with the neutral lipid dye BODIPY 493/503 (Invitrogen) using confocal microscopy. BMDMs from ADAM17-deficient mice and C57BL/6J controls were plated on eight-chambered Lab-Tek borosilicate cover glass. Pictures were collected using a Nikon A1R Confocal microscope at 60X magnification.
To determine free cholesterol accumulation, BMDMs were fixed in 4% paraformaldehyde for 10 min at room temperature, rinsed using glycine/PBS, and stained with 0.25 mg/ml filipin for 2 hr at room temperature. The cells were viewed in PBS by fluorescence microscopy (Keyence All-in-One Fluorescence Microscope BZ-X800) using a UV filter set (340–380 nm excitation) at 20X magnification.
HDL and LDL isolation from mouse plasma
HDL (density 1.125–1.21 g/ml) and LDL (density 1.019–1.063 g/ml) were isolated from EDTA plasma of APOA1Tg; Ldlr−/− mice by density gradient ultracentrifugation according to described methods.17, 40, 42
Quantification of HDL particle size and concentration
HDL particle concentration (HDL-P) and size distribution were measured using calibrated ion mobility analysis (cIMA).25, 43, 44 Three species of HDL particles were detected in APOA1Tg; Ldlr−/− mice by cIMA analysis, and were classified as small (S-HDL, average diameter 8.4 nm), medium (M-HDL, average diameter 9.5 nm), or large (L-HDL, average diameter 10.8 nm). The total HDL particle (T-HDL) concentration was calculated as the sum total of all 3 particle populations.
Cholesterol efflux assay
The cholesterol efflux capacity of HDL (100 μg/ml, 4 hr) was determined in C57BL/6J wild-type and ADAM17-deficient BMDMs radiolabeled with [3H]-cholesterol and incubated with cyclic AMP to induce expression of ABCA1 and ABCG1 and an acyl-coenzyme A:cholesterol acyltransferase inhibitor (2 μg/ml).11 In some experiments, cells were treated with either TNFα (20 ng/ml), a goat anti-mouse blocking TNFα antibody (R&D systems, AF-410-SP) or control IgG (R&D systems, AB-108-C) before measuring cholesterol efflux capacity of HDL. Cholesterol efflux was calculated as the percentage of radiolabeled cholesterol in the medium at the end of the incubation divided by the total radioactivity of the medium and cells.44
CRISPR/Cas9-mediated deletion of TREM2
A pSpCas9(BB)-2A-Puro (PX459) V2.0 plasmid was purchased from Addgene (Plasmid #62988). The single guide RNA (sgRNA) oligonucleotides for Trem2 (sgRNA1; CTGCAGCACCGTGGTGTTGA and sgRNA2; AAGGCCTGGTGTCGGCAGCT) were designed using the Broad CRISPR algorithm. sgRNAs were cloned into the sgRNA vector using a BsmBI restriction digest.
CRISPR/Cas9-mediated deletion of Trem2 was performed by transfection of Cas9 complexed with the Trem2-specific guide RNAs in BMDMs using lipofectamine 3000, following the manufacturer’s protocol (Thermo Fisher Scientific). Successfully transduced cells were selected using puromycin at 5 μg/ml. Trem2 knockdown was confirmed by flow cytometry using a PE-labeled TREM2 (Invitrogen; MA5-28225, 1:200 dilution) antibody.
Statistical analyses
All data are presented as mean ± SEM. In cell culture experiments, the sample size (n) represents the number of individually differentiated primary macrophage cultures in each experiment. In thioglycollate-elicited peritoneal macrophage experiments from the bone marrow transplant study, sample size (n) represents the number of individual mice in each experiment. The statistical parameters (n, mean, SEM, and statistical tests used) can be found within the figure legends. Tests for normality (Shapiro-Wilk) and equal variance (Brown-Forsythe) were performed for each of the data sets. To define differences between two datasets, unpaired two-tailed Mann-Whitney tests were used (datasets without equal variance or datasets too small to verify normal distribution). To assess differences between three or more groups, one-way ANOVA with Tukey’s multiple comparisons tests or two-way ANOVA followed by Sidak’s multiple comparison tests was used. When the criteria were not met for analyses by parametric test, Brown-Forsythe ANOVA followed by Dunnett’s multiple comparison tests or Kruskal-Wallis followed by Dunn’s multiple comparison tests was used. The criterion for significance was set at p < 0.05. Statistical analyses were performed using GraphPad Prism, version 8.4.3 (San Diego, CA).
See the online supplement for additional details.
Results
In vivo HDL priming exacerbates both pro-inflammatory and anti-inflammatory responses in macrophages
We recently demonstrated that reconstituted HDL (rHDL; CSL-111) induces both pro-inflammatory and anti-inflammatory responses to LPS in macrophages through cholesterol efflux and that these responses are both temporally different and mediated by distinct signaling pathways.24, 25 The pro-inflammatory effects of HDL in isolated macrophages are mediated by IRE1a/ASK1/p38 MAPK signaling whereas the anti-inflammatory effects in the same macrophages are due to reduced cell surface expression of TLR4 and to suppression of interferon receptor signaling.25
In the present study, we first sought to expand on our previously published observations using Ldlr−/− transgenic mice expressing human APOA1 (APOA1Tg), whose HDL levels are higher than those of Ldlr−/− mice,45 and whose HDL profile resembles that of humans, with small, medium and large HDL particle populations. Wildtype mice and Ldlr−/− mice have only one HDL population.45 Sorted thioglycollate-elicited peritoneal macrophages from APOA1Tg; Ldlr−/− mice showed increases in LPS-induced Tnfa, Cxcl1, Ccl2 and Il1b gene expression, as compared with macrophages from Ldlr−/− littermate controls (Figure 1A). To confirm that changes in pro-inflammatory genes translated to increased cytokine/chemokine secretion, we stimulated macrophages with LPS for different periods of time (4–20 hr). As shown in Figure 1B, macrophages from APOA1Tg; Ldlr−/− mice exhibited increases in secretion of TNFα and CXCL1 (8 hr and 20 hr), as compared with macrophages from Ldlr−/− mice.
Figure 1. Pro-inflammatory and anti-inflammatory responses in macrophages from APOA1Tg; Ldlr−/− mice mimic those of HDL.
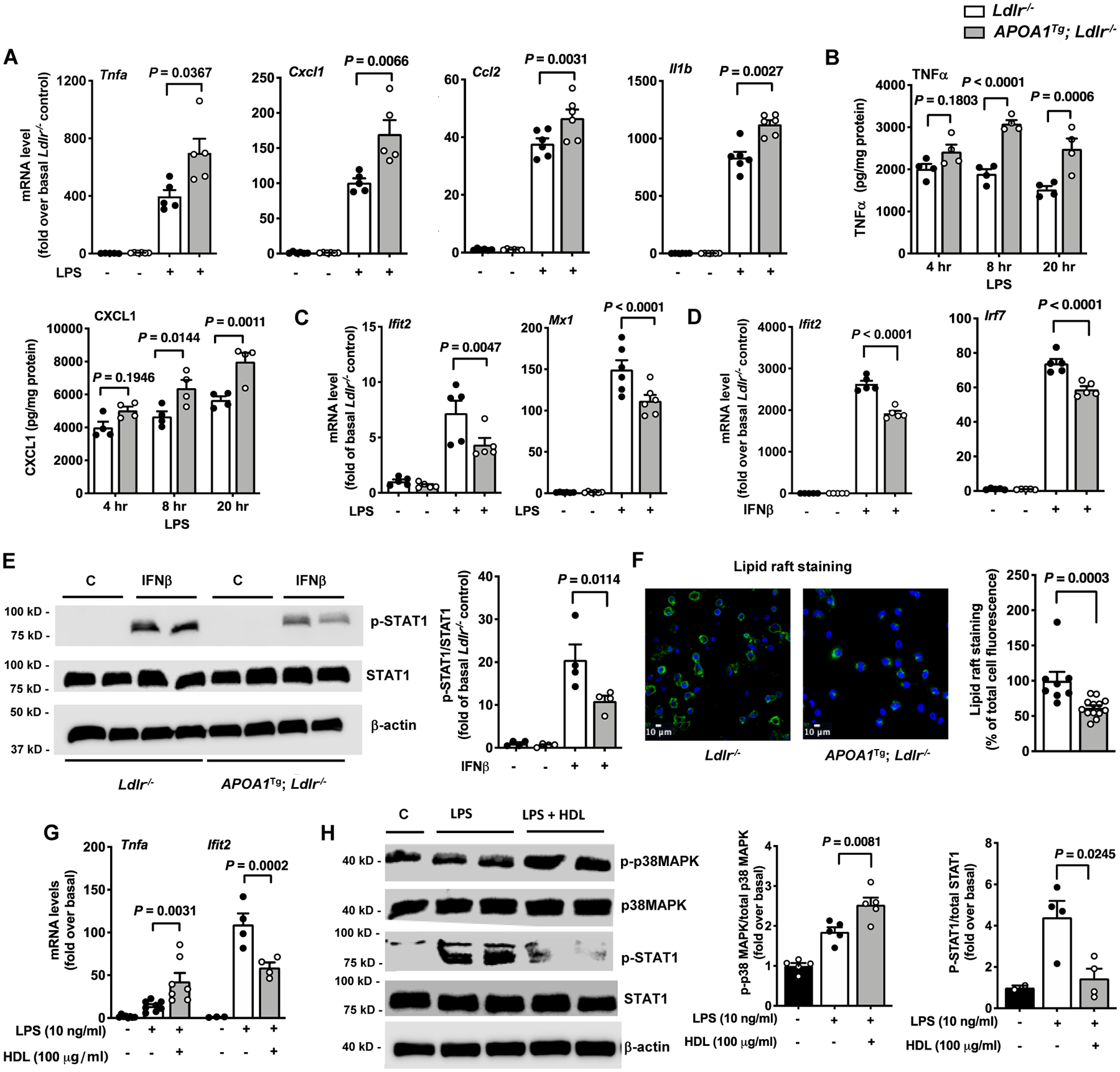
Peritoneal cells from male Ldlr−/− and APOA1Tg; Ldlr−/− control littermate mice were collected 4 days after thioglycolate injection. Macrophages were isolated from other cell types using a macrophage isolation kit and were further purified by a 1 hr adhesion protocol before stimulation with LPS or IFNβ. A. Effect of LPS (10 ng/ml, 4 hr) on inflammatory gene expression in macrophages isolated from Ldlr−/− and APOA1Tg; Ldlr−/− mice (n=5). B. Conditioned media were collected at indicated times from LPS-stimulated macrophages to analyze the release of TNFα and CXCL1 (n=4). C. Effect of LPS on the type 1 IFN-inducible genes Ifit2 and Mx1 analyzed in peritoneal macrophages from Ldlr−/− and APOA1Tg; Ldlr−/− mice (n=5–6). D. Effect of IFNβ on Ifit2 and Irf7 mRNA levels analyzed in peritoneal macrophages from Ldlr−/− and APOA1Tg; Ldlr−/− mice (n=5). E. Effect on STAT1 phosphorylation in IFNβ-stimulated peritoneal macrophages isolated from Ldlr−/− and APOA1Tg; Ldlr−/− mice (n=4). Phospho-STAT1 band intensity was normalized to that of total STAT1 and quantified (bar graph on the right). β-actin was used as an additional loading control. F. Representative photos and quantification (right) of lipid raft staining in thioglycolate-elicited peritoneal macrophages from Ldlr−/− and APOA1Tg; Ldlr−/− mice (n=7–13). G. BMDMs from female C57BL/6J mice were pre-treated with HDL (100 μg/ml) for 18 hr. The cells were then stimulated with LPS (10 ng/ml) in the absence of HDL for 4 hr, and inflammatory gene expression (Tnfa and Ifit2) was determined using qPCR (n=4–7). H. BMDMs from male C57BL/6J mice were pre-treated with HDL for 18 hr. The cells were then stimulated with LPS (10 ng/ml) in the absence of HDL for 4 hr to determine the phosphorylation status of p38 MAPK and STAT1 by immunoblot (n=4–5). Band intensities of p-p38 MAPK and p-STAT1 were normalized to those of total p38MAPK and STAT1, respectively, and quantified (bar graphs on the right). Data are shown as mean ± SEM. Tests for normality (Shapiro-Wilk) and equal variance (Brown-Forsythe) were performed for each of the data sets. P values were determined accordingly by Kruskal-Wallis followed by Dunn’s multiple comparison tests (A), one-way ANOVA followed by Tukey’s multiple comparison tests (C – Ifit2, D, E, H), Brown-Forsythe ANOVA followed by Dunnett’s multiple comparison tests (C - Mx1), two-way ANOVA followed by Sidak’s multiple comparison test (B, G), or unpaired two-tailed nonparametric Mann-Whitney test (F). Data are representative of at least three independent experiments performed in replicates.
In contrast, peritoneal macrophages from APOA1Tg; Ldlr−/− showed significant reductions in LPS-induced gene expression of Ifit2 and Mx1, consistent with the anti-inflammatory effect on the interferon signaling pathway (Figure 1C). Additionally, thioglycollate-elicited peritoneal macrophages from APOA1Tg; Ldlr−/− mice showed repressed expression of Ifit2, Mx1 and Irf7 in response to IFNβ (Figure 1D), confirming the anti-inflammatory effect on interferon signaling.
The pro-inflammatory and anti-inflammatory responses to LPS and IFNβ in thioglycollate-elicited peritoneal macrophages from APOA1Tg; Ldlr−/− mice were only observed in purer macrophage populations sorted by negative selection in addition to adhesion purification, as compared with 1 hr adhesion purification alone (Figure ID and IE in the online-only Data Supplement). These results indicate that the presence of other cell types in thioglycollate-elicited peritoneal macrophage preparations interfere with the results. The presence of B-cells is a likely confounder because the B-cell marker Cd19 was higher in cells purified by adhesion only (Figure ID in the online-only Data Supplement).
Consistent with anti-inflammatory effects downstream of IFNAR (interferon α and β receptor) activation, macrophages from APOA1Tg; Ldlr−/− mice exhibited reduced STAT1 (signal transducer and activator of transcription 1) phosphorylation in response to IFNβ stimulation (Figure 1E). The different responses to LPS of macrophages from APOA1Tg; Ldlr−/− mice were not due to altered cell surface expression of TLR4 (Figure IF in the online-only Data Supplement). Importantly, in the absence of LPS or IFNβ stimulation, macrophages from APOA1Tg; Ldlr−/− mice did not display altered pro-inflammatory or anti-inflammatory phenotypes (Figure 1A–1D). Previously we have shown that resident peritoneal macrophages from APOA1Tg; Ldlr−/− mice are less lipid loaded as compared with macrophages from Ldlr−/− mice.25 This was further confirmed by a reduced lipid raft staining in thioglycolate-elicited peritoneal macrophages from APOA1Tg; Ldlr−/− mice as compared with macrophages from Ldlr−/− mice (Figure 1F). These results indicate that in vivo cholesterol depletion of macrophages results in pro-inflammatory and anti-inflammatory responses to LPS and IFNβ.
We next isolated HDL from APOA1Tg; Ldlr−/− mice to characterize its pro-and anti-inflammatory potential in macrophages. HDL’s functionality was tested by first determining its anti-inflammatory potential in endothelial cells. The protective effect of HDL on endothelial cells was established in a monocyte adhesion assay, in which pre-treatment of HDL inhibited TNFα-induced monocyte adhesion (Figure IIA in the online-only Data Supplement). We then investigated the effect of HDL on inflammatory phenotypes of macrophages. BMDMs were pre-treated with HDL followed by stimulation with LPS for 4 hr in the absence of HDL. Similar to what we observed previously,25 pre-treatment with HDL had a pro-inflammatory effect as it increased LPS-induced Tnfa gene expression (Figure 1G) and secretion of TNFα and CXCL1 (Figure IIB in the online-only Data Supplement). Consistent with previous studies,21 the HDL-induced pro-inflammatory response was not restricted to TLR4 stimulation, as HDL also increased TLR3 and TLR7/8 mediated gene expression of Tnfa, but did not alter the effect of TNFα (Figure IIC and IID in the online-only Data Supplement). Additionally, HDL pretreatment inhibited LPS-induced gene expression of Ifit2 (Figure 1G), confirming the anti-inflammatory response of the interferon signaling pathway.
Importantly, the effects of native HDL were associated with increased p38MAPK phosphorylation and a reduction of STAT1 phosphorylation (Figure 1H), as we showed previously for reconstituted HDL.25 Furthermore, HDL treatment significantly reduced the lipid raft content in BMDMs, similar to what we observed in macrophages from APOA1Tg; Ldlr−/− mice. HDL’s effect on lipid rafts was comparable to that of the cholesterol depleting agent methyl-β-cyclodextrin (MβCD, Figure IIE in the online-only Data Supplement). Overall, these results are consistent with the notion that HDL’s ability to induce cholesterol depletion drives both pro- and anti-inflammatory responsiveness in macrophages in vitro and in vivo.
Next, we investigated whether LDL or APOA1 would have effects similar to those of HDL on pro- and anti-inflammatory processes. LDL slightly enhanced the effect of LPS-induced Tnfa and Cxcl1 gene expression, but did not affect the LPS-induced interferon pathway (Figure IIF in the online-only Data Supplement), indicating that the effects of LDL are distinct from those of HDL.
Strikingly, contrary to HDL’s pro-inflammatory effects, free APOA1 did not increase LPS-induced gene expression of Tnfa and Cxcl1 and exhibited a marked anti-inflammatory effect (Figure IIF in the online-only Data Supplement). Moreover, APOA1 suppressed HDL’s pro-inflammatory responses in macrophages (Figure IIG in the online-only Data Supplement), strongly arguing that the effects of HDL and APOA1 are mediated through distinct mechanisms. In order to further investigate the mechanism of APOA1’s anti-inflammatory effect, we used an APOA1 C-terminal deletion mutant (1–184). APOA1 C-terminal deletion mutants have a poor ability to mediate cholesterol efflux through ABCA1.39 The APOA1 mutant was much less potent in inducing anti-inflammatory effects in wildtype macrophages than was full-length APOA1 (Figure IIH in the online-only Data Supplement).
Together, these findings show that native HDL induces both pro- and anti-inflammatory effects in macrophages, consistent with our previous findings on reconstituted HDL, and that the pro-inflammatory capacity of HDL is mediated through mechanisms distinct from those of free APOA1.
ADAM17 is required for pro- and anti-inflammatory responses to HDL in macrophages
Because previous studies suggested that depletion of cholesterol promotes TNFα shedding from endothelial cells by redistribution of ADAM17 from lipid rafts,32, 33 we next investigated the role of ADAM17 in HDL’s pro- and anti-inflammatory effects in macrophages. HDL markedly increased ADAM17 gene and protein expression in these cells (Figure 2A–B). Similarly, cholesterol depletion by MβCD increased expression of ADAM17 (Figure IIIA in the online-only Data Supplement) while cholesterol loading using acetylated LDL blunted the HDL-mediated increase in ADAM17 expression (Figure 2C), indicating that cholesterol depletion drives ADAM17 induction. HDL-mediated induction of ADAM17 was not dependent on ASK1/p38 MAPK activation, because inhibition of p38 MAPK or ASK1 did not prevent HDL-induced ADAM17 expression (Figure IIIB–C in the online-only Data Supplement).
Figure 2. The pro-inflammatory and anti-inflammatory responses of HDL in macrophages are dependent on ADAM17.
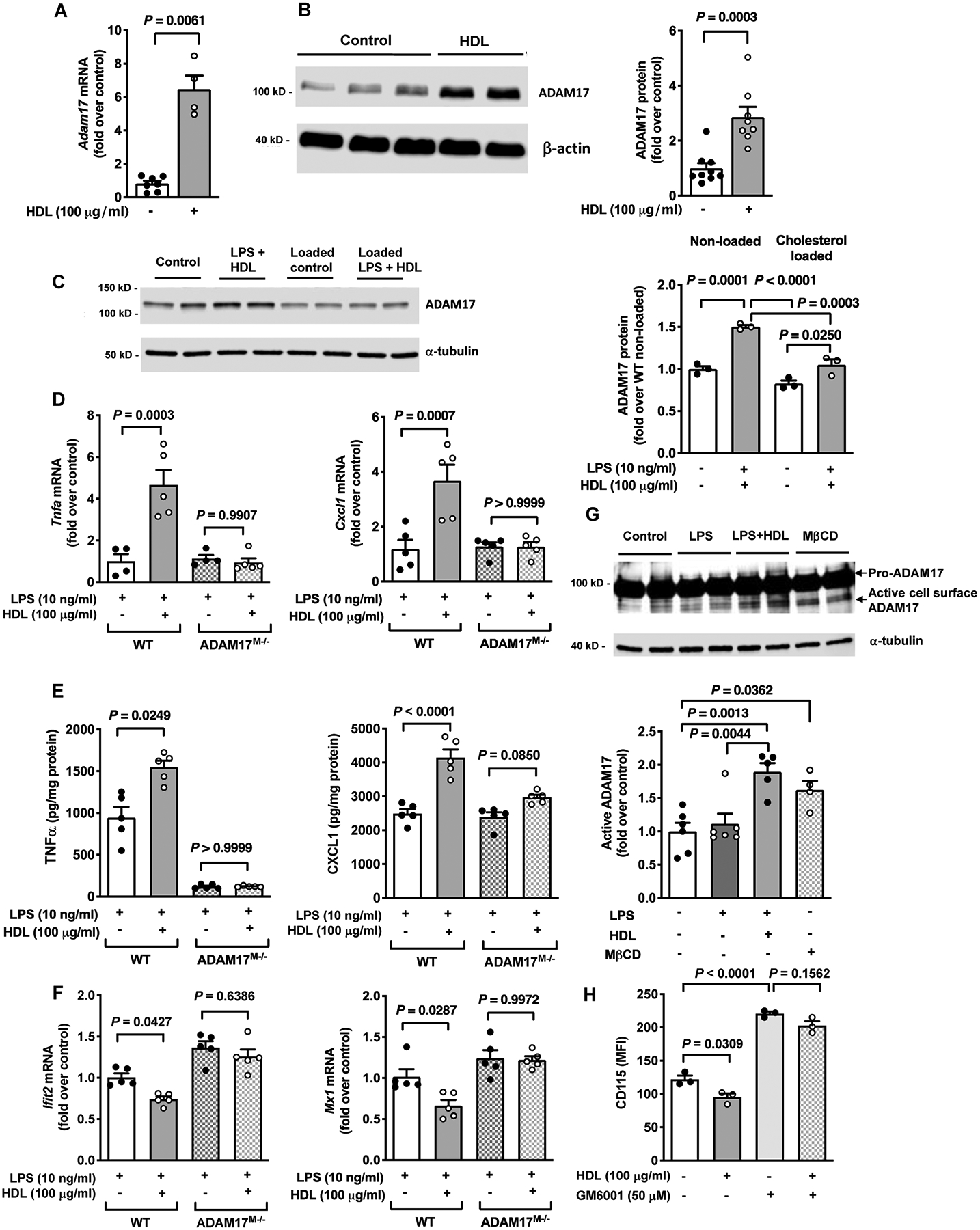
A. Gene expression of Adam17 was determined after an 18 hr treatment with HDL (100 μg/ml) in BMDMs from C57BL/6J mice (n=4–7). B. Protein expression of ADAM17 was determined after an 18 hr treatment with HDL (100 μg/ml) in BMDMs from C57BL/6J mice (n=8–9). C. BMDMs from male C57BL/6J mice were loaded with cholesterol using acetylated LDL (AcLDL; 50 μg/ml, 48 hr). The cells were then primed with HDL (100 μg/ml) for 18 hr, washed and stimulated with LPS (10 ng/ml) for 4 hr. Protein expression of ADAM17 was analyzed at the end of the treatment (n=3). D. WT and ADAM17-deficient BMDMs were primed with HDL (100 μg/ml) for 18 hr, washed and stimulated with LPS (10 ng/ml) for 4 hr. At the end of treatment, samples were used to analyze gene expression of Tnfa and Cxcl1 (n=4–5). E. Conditioned media were used to analyze release of TNFα and CXCL1 (n=5). F. WT or ADAM17-deficient BMDMs were primed with HDL (100 μg/ml) for 18 hr, washed and stimulated with IFNβ (1 ng/ml, 4 hr). At the end of treatment, samples were used to analyze gene expression of Ifit2 and Mx1 (n=5). G. Active cell surface ADAM17 was determined after an 18 hr stimulation with HDL in BMDMs. Cells were surface biotinylated, followed by purification of biotinylated proteins and analysis of cell lysates by immunoblot for active ADAM17 (quantification below the blot). H. BMDMs were pre-treated with the metalloproteinase inhibitor GM6001 (50 μM, 30 min) before the HDL (100 μg/ml for 18 hr) treatment. At the end of the stimulation, the cells were used to analyze cell surface expression of CD115 by flow cytometry (n=3; experiment performed twice with similar results). Data are shown as mean ± SEM. Tests for normality (Shapiro-Wilk) and equal variance (Brown-Forsythe) were performed for each of the data sets. P values were determined accordingly by nonparametric two-tailed Mann-Whitney test (A, B), one-way ANOVA followed by Tukey’s multiple comparison tests (C, D, E - CXCL1, F, G, H) or Brown-Forsythe ANOVA followed by Dunnett’s multiple comparison tests (E - TNFα). Unless otherwise stated, data are representative of at least three independent experiments performed in replicates in BMDMs from male mice.
To assess whether the pro-inflammatory and anti-inflammatory responses of HDL depend on ADAM17, we analyzed BMDMs from mice with myeloid cell-targeted ADAM17 deletion (ADAM17M−/−) and wildtype (WT) C57BL/6J controls. As shown in Figure 2D, pre-treatment with HDL significantly increased LPS-induced Tnfa and Cxcl1 mRNA expression in BMDMs from WT mice, as expected. Strikingly, cells deficient in ADAM17 exhibited no differences in these LPS responses when pre-incubated with HDL (Figure 2D). Furthermore, ADAM17-deficiency completely blocked the LPS/HDL-induced secretion of TNFα, consistent with TNFα being a major ADAM17 substrate and that LPS increases its release through ADAM17-mediated shedding (Figure 2E). However, ADAM17-deficiency also prevented HDL’s effect on CXCL1 secretion, without affecting the LPS response (Figure 2E). In addition, HDL’s anti-inflammatory response on IFNβ-induced activation of interferon signaling (Ifit2 and Mx1) was also absent in ADAM17-deficient macrophages, as compared with BMDMs from WT mice (Figure 2F).
We next determined whether HDL’s effect on inflammatory macrophage responses is due to an increase in ADAM17’s enzymatic activity. HDL increased cell surface levels of active forms of ADAM17 (Figure 2G) as well as enhanced CD115 shedding as a measure of ADAM17 activity31 in BMDMs (Figure 2H). Moreover, the ADAM17 activity inhibitor (TAPI-1) mimicked the effect of ADAM17-deficiency and prevented HDL’s pro-inflammatory and anti-inflammatory responses (Figure IIID and IIIE in the online-only Data Supplement). Together, these results indicate that HDL’s pro-inflammatory and anti-inflammatory activities in macrophages depend on the presence of active ADAM17.
Myeloid cell targeted ADAM17-deficiency prevents pro- and anti-inflammatory responses in APOA1Tg mice
To investigate the role of ADAM17 in vivo, bone marrow from ADAM17M−/− mice and WT littermates (controls) was transplanted into male APOA1Tg Ldlr−/− and Ldlr−/− recipient mice (Figure 3A). As shown in Figure 3B, thioglycollate-elicited peritoneal macrophages from APOA1Tg; Ldlr−/− mice transplanted with WT bone marrow showed a significant increase in Adam17 gene expression as compared with macrophages from Ldlr−/− mice transplanted with WT bone marrow (Figure 3B), consistent with the stimulatory effect of HDL on ADAM17 (Figure 2A–B). Similarly, resident peritoneal cells from APOA1Tg Ldlr−/− mice exhibited elevated gene expression of Adam17 as compared with cells from Ldlr−/− mice (Figure IVA in the online-only Data Supplement). Both Ldlr−/− and APOA1Tg Ldlr−/− mice receiving bone marrow from ADAM17M−/− mice exhibited a 95% reduction in Adam17 mRNA levels (Figure 3B), confirming near-complete chimerism. Moreover, blood leukocyte preparations from both APOA1Tg Ldlr−/− and Ldlr−/− mice with myeloid cell-targeted ADAM17-deficiency exhibited higher cell surface levels of the ADAM17 substrate CD11531 (Figure 3C) consistent with a reduced ADAM17 sheddase activity in blood myeloid cells.
Figure 3. Myeloid cell-targeted ADAM17-deficiency suppresses pro- and anti-inflammatory responses in macrophages from APOA1Tg; Ldlr−/− mice.
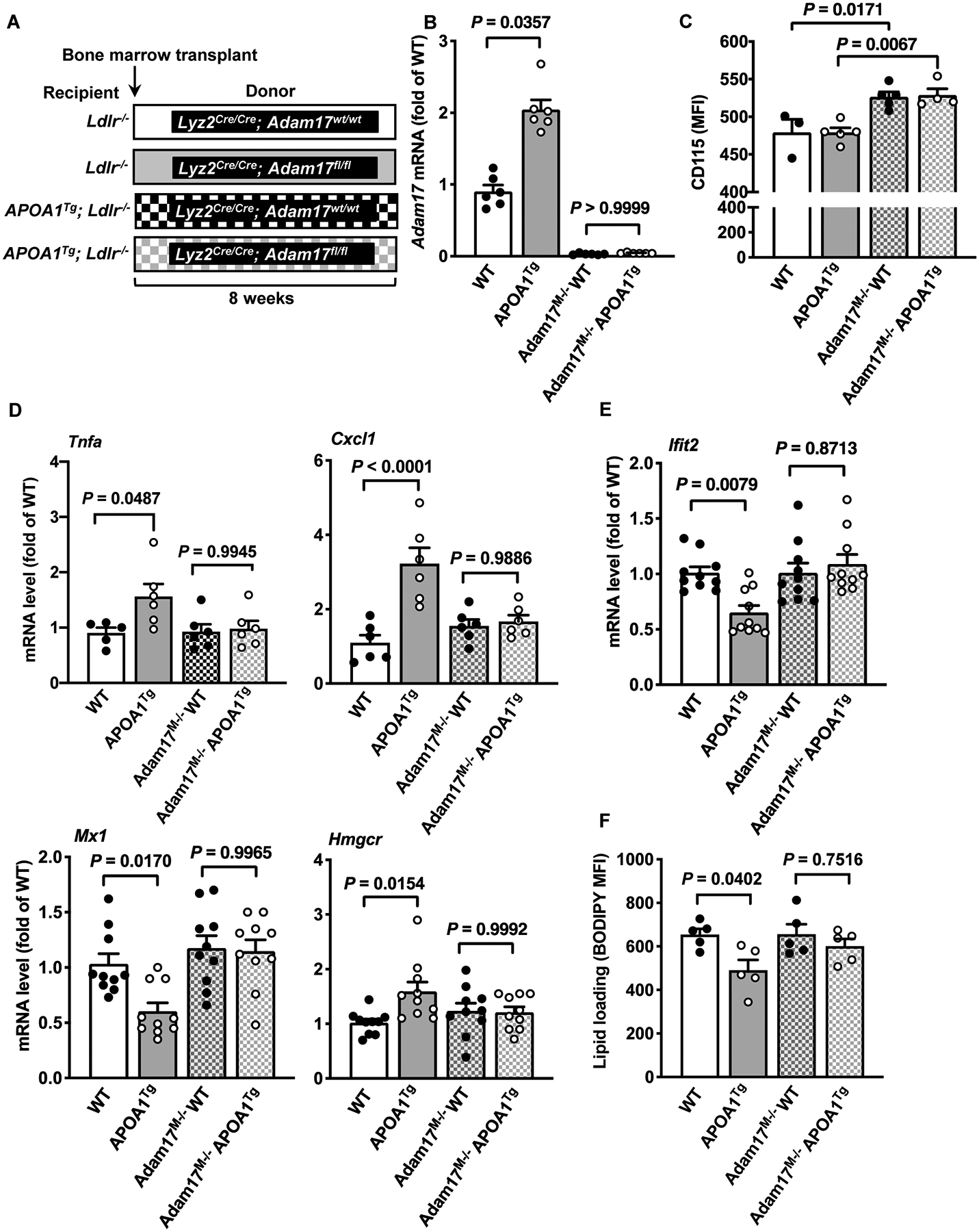
A. Bone marrow was harvested from myeloid cell-targeted ADAM17-deficient mice or WT C57BL/6J littermates and transplanted into lethally irradiated (10 Gy) male Ldlr−/− or APOA1Tg; Ldlr−/− recipient littermates. After 8 weeks of recovery, peritoneal cells from Ldlr−/− and APOA1Tg; Ldlr−/− recipient mice were collected 4 days after thioglycolate injection, macrophages were purified from other cell types using a macrophage isolation kit followed by a 1 hr adhesion purification, and RNA was harvested for further analysis. B. Adam17 mRNA levels analyzed in macrophages from Ldlr−/− and APOA1Tg; Ldlr−/− recipient mice (n=5–6). C. Blood leukocytes from Ldlr−/− and APOA1Tg; Ldlr−/− recipient mice stained with an anti-CD115 antibody and analyzed using flow cytometry (n=3–5). D. Macrophages from Ldlr−/− and APOA1Tg; Ldlr−/− mice stimulated with LPS (10 ng/ml, 4 hr). At the end of the treatment, the cells were used to analyze Tnfa mRNA and Cxcl1 (n=5). E. Macrophages from Ldlr−/− and APOA1Tg; Ldlr−/− mice were stimulated with IFNβ (1 ng/ml, 4 hr). At the end of the treatment, the cells were used to analyze gene expression of Ifit2, Mx1 and Hmgcr (n=10). F. Macrophages from Ldlr−/− and APOA1Tg; Ldlr−/− recipient mice stained with BODIPY® 493/503 and analyzed using flow cytometry for neutral lipids (n=5). Data are shown as mean ± SEM. Tests for normality (Shapiro-Wilk) and equal variance (Brown-Forsythe) were performed for each of the data sets. P values were determined accordingly by one-way ANOVA followed by Tukey’s multiple comparison tests (C, D, E, F) or Brown-Forsythe ANOVA followed by Dunnett’s multiple comparisons tests (B). One significant outlier (Grubb’s test) was excluded in the WT group in D (Tnfa).
We next stimulated thioglycollate-elicited purified macrophages from the bone marrow chimeric mice either with LPS or IFNβ. Macrophages from APOA1Tg Ldlr−/− mice transplanted with bone marrow from WT mice exhibited significant increases in LPS-induced Tnfa and Cxcl1 gene expression, as compared with macrophages from Ldlr−/− mice, but these responses were absent in ADAM17-deficient macrophages from APOA1Tg Ldlr−/− mice (Figure 3D). In addition, macrophages from APOA1Tg; Ldlr−/− mice transplanted with bone marrow from WT mice exhibited significant decreases in IFNβ-induced Ifit2 and Mx1 gene expression, as compared with macrophages from Ldlr−/− mice (Figure 3E). Again, myeloid cell-targeted ADAM17-deficiency prevented these effects (Figure 3E). Consistent with the pro- and anti-inflammatory responses of HDL being dependent on the lipid status of the macrophage, reduced neutral lipid content as indicated by reduced BODIPY staining and increased expression of Hmgcr (which is induced by cholesterol depletion) were observed in macrophages from APOA1Tg Ldlr−/− mice as compared with macrophages from Ldlr−/− mice transplanted with WT bone marrow (Figure 3E–F). Myeloid cell-targeted ADAM17-deficiency prevented the reduction in macrophage neutral lipid content and the increase in Hmgcr in APOA1Tg Ldlr−/− mice (Figure 3E–F).
Importantly, the effects of myeloid cell ADAM17-deficiency were not confounded by changes in plasma lipids, leukocytosis or numbers of peritoneal cells (Figure IVB–H in the online-only Data Supplement). Overall, these results indicate that ADAM17 is required to maintain the in vivo cholesterol depletion status in macrophages from APOA1Tg mice obligatory for the downstream effects on inflammatory responses.
ADAM17-deficiency increases cholesterol content in macrophages without enhancing cholesterol synthesis or uptake
Our previous studies demonstrated that cholesterol loading of macrophages prevented both the pro- and anti-inflammatory effects of HDL.25 To further investigate how ADAM17-deficiency prevents HDL’s immune effects on macrophages, we determined the lipid content in WT and ADAM17-deficient macrophages. Imaging of neutral lipids using BODIPY revealed that ADAM17-deficiency significantly increases lipid content in macrophages under baseline conditions (Figure 4A). This was further confirmed by flow cytometry, showing that macrophages from ADAM17M−/− mice have 3-fold higher levels of neutral lipids than those of WT mice (Figure 4B). Because increased BODIPY staining could be an indication of increased cholesterol and/or triglyceride levels, we next analyzed the triglyceride and cholesterol content. As shown in Figure 4C, triglyceride levels were similar between WT and ADAM17-deficient macrophages. Consistent with the higher BOPIPY-positive lipid content, ADAM17-deficient macrophages had significantly higher levels of free cholesterol, measured by filipin staining both in the presence and absence of HDL pre-treatment, as compared with macrophages from WT mice (Figure 4D). Additionally, prolonged (24–48 hr) inhibition of ADAM17 activity significantly increased the total cholesterol content in macrophages (Figure 4E), indicating that the sheddase function of ADAM17 acts to suppress macrophage cholesterol levels.
Figure 4. ADAM17-deficiency induces cholesterol loading and prevents HDL-mediated cholesterol depletion.
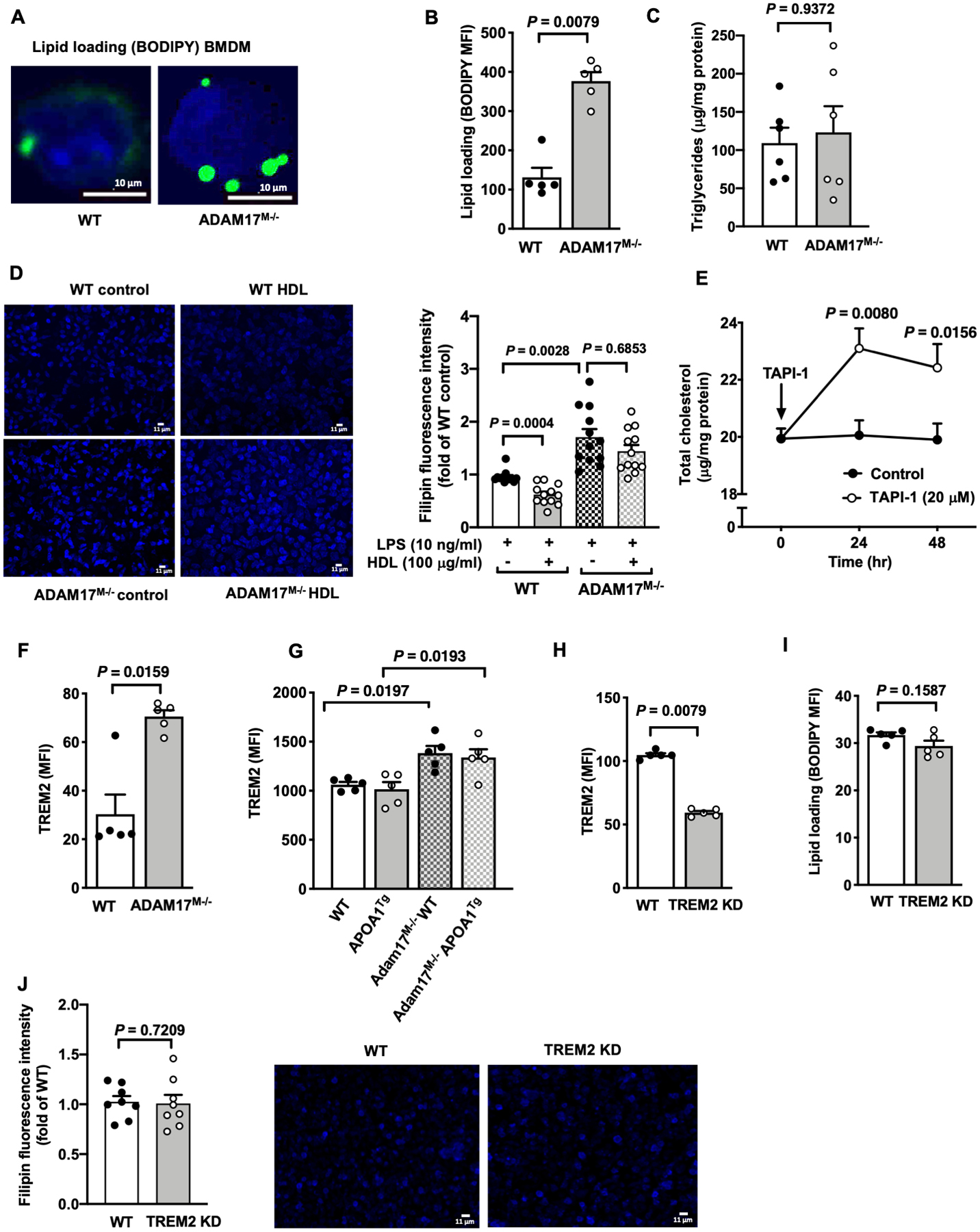
A. Representative images of neutral lipid staining with BODIPY® 493/503 (bright green) of BMDMs from male C57BL/6J WT and ADAM17-deficient mice (n=4). Representative cells are shown, the nuclei stained blue by DAPI. B. BMDMs from male WT and ADAM17-deficient mice were stained with BODIPY® 493/503 and mean fluorescent intensity (MFI) was determined by flow cytometry (n=5). C. Cellular triglyceride levels in BMDMs from male WT and ADAM17-deficient mice were analyzed (n=6). D. BMDMs from male WT and ADAM17-deficient mice were pre-treated with HDL (100 μg/ml). After 18 hr, free cholesterol was visualized by filipin staining (50 μg/ml), using fluorescence microscopy. Positive filipin staining was quantified as mean fluorescence intensity per cell and expressed as fold change of BMDMs from WT mice (n=12). E. BMDMs from female C57BL/6J mice were treated with TAPI-1 (20 μM) for indicated times. At the end of stimulation, the cells were used to analyze total cholesterol content (n=4–6). F. BMDMs from male WT and ADAM17M−/− mice were analyzed for cell surface expression of TREM2 by flow cytometry (n=5). G. Thioglycollate-elicited peritoneal macrophages from male Ldlr−/− or APOA1Tg; Ldlr−/− recipient mice transplanted with bone marrow from WT or ADAM17M−/− mice were stained for TREM2 and were analyzed by flow cytometry (n=5). H-J. TREM2 was knocked down (KD) in BMDM populations from male C57BL/6J mice by transfection of Cas9 complexed with Trem2-specific guide RNAs using lipofectamine 3000. 48 hr after transfection, cell surface expression of TREM2 (H) and (I) neutral lipid loading (BODIPY) were determined by flow cytometry. J. Free cholesterol was visualized by filipin (50 μg/ml) stain with fluorescence microscopy and quantified as mean fluorescence intensity in control and Trem2 KD BMDMs (n=8; experiment performed twice with similar results). Data are shown as mean ± SEM. Tests for normality (Shapiro-Wilk) and equal variance (Brown-Forsythe) were performed for each of the data sets. P values were determined accordingly by two-tailed Mann-Whitney test (B, C, F, H, I, J), Brown-Forsythe ANOVA followed by Dunnett’s multiple comparisons tests (D), or two-way ANOVA and Sidak’s multiple comparison tests (E). Unless otherwise stated, data are representative of at least three independent experiments performed in replicates in BMDMs from male mice.
To understand the mechanism behind the higher cholesterol loading status in ADAM17-deficient macrophages, we first determined the expression of cholesterol biosynthesis genes. ADAM17-deficiency did not affect gene expression of Srebf2 or Hmgcr, but reduced Sqle mRNA levels under baseline conditions, as compared with macrophages from WT mice (Figure VA in the online-only Data Supplement). Consistently, there was no increase in lipogenesis in ADAM17-deficient macrophages, measured as incorporation of 3H-acetate into cellular lipids (Figure VB in the online-only Data Supplement). Furthermore, measurements of uptake of DiI-labeled AcLDL in BMDMs from WT and ADAM17M−/− mice revealed no significant differences in lipoprotein uptake (Figure VC in the online-only Data Supplement), nor was there a difference in Ldlr mRNA levels (Figure VA in the online-only Data Supplement). In addition, there was no increase in the accumulation of cholesteryl ester in ADAM17-deficient macrophages loaded with AcLDL as compared with macrophages from WT mice, but rather a decrease (Figure VD in the online-only Data Supplement). These results indicate that the higher cholesterol content in ADAM17-deficient macrophages is not due to increased cholesterol biosynthesis or cholesterol uptake.
Next, we determined by flow cytometry cell surface expression of receptors known to be involved in lipid metabolism. Although CD36 and MerTK can be substrates of ADAM17,30, 46, 47 we did not observe significant changes in cell surface levels in ADAM17-deficient macrophages (Figure VE–F in the online-only Data Supplement). In addition, consistent with previous literature, cell surface levels of the VLDL receptor were low in mouse macrophages48 and were not significantly altered by ADAM17-deficiency (Figure VG in the online-only Data Supplement).
Recently, TREM2 (triggering receptor expressed on myeloid cells 2), which can bind APOE,49 was shown to act as lipid sensor and to play a role in phagocytosis and cytokine release.50 Furthermore, TREM2’s ectodomain shedding is mediated in part by ADAM17.50–52 Consistently, cell surface levels of TREM2 were significantly higher in BMDMs from ADAM17M−/− mice (Figure 4F). Similarly, ADAM17-deficient macrophages exhibited significantly increased TREM2 cell surface levels in both APOA1Tg Ldlr−/− and Ldlr−/− mice (Figure 4G), indicating that TREM2 is indeed an important ADAM17 substrate. Because adipose tissue macrophages from TREM2-deficient mice have reduced neutral lipid accumulation,53 to investigate if ADAM17 acts to lower macrophage cholesterol levels by shedding of TREM2, we next silenced TREM2 using a CRISPR/Cas9 strategy (Figure VH in the online-only Data Supplement). As shown in Figure 4H, transfection of Cas9 complexed with Trem2-specific guide RNAs reduced cell surface levels of TREM2 in BMDMs. However, the reduction in TREM2 did not alter the intracellular content of BODIPY-positive neutral lipid (Figure 4I) or free cholesterol measured by filipin staining (Figure 4J), suggesting that the increased lipid levels in ADAM17-deficient macrophages is likely to be independent of TREM2 shedding. Overall, our results show that ADAM17 is required for maintaining cholesterol levels low in macrophages and that lack of ADAM17 increases cholesterol content in these cells through a mechanism that is not dependent on increased cholesterol synthesis or uptake.
ADAM17 maintains low cholesterol levels in macrophages by enhancing ABCA1-mediated cholesterol efflux
Given that ADAM17-deficiency does not appear to increase cholesterol uptake or synthesis, we next investigated whether cholesterol efflux is impaired by ADAM17-deficiency. Compared to WT macrophages, ADAM17-deficient macrophages displayed reduced cholesterol efflux into the media under baseline conditions (Figure 5A). Following cAMP-stimulation, which increases expression of ABCA1 and ABCG1,54 cholesterol efflux was markedly reduced in ADAM17-deficient macrophages, as compared with WT macrophages (Figure 5A). Furthermore, consistent with the reduced cholesterol efflux, HDL was unable to reduce the free cholesterol level (Figure 4D) and lipid raft content (Figure 5B) in ADAM17-deficient macrophages.
Figure 5. Reduced cholesterol efflux capacity in ADAM17-deficient macrophages reflects inhibition of TNFα-induced ABCA1 expression.
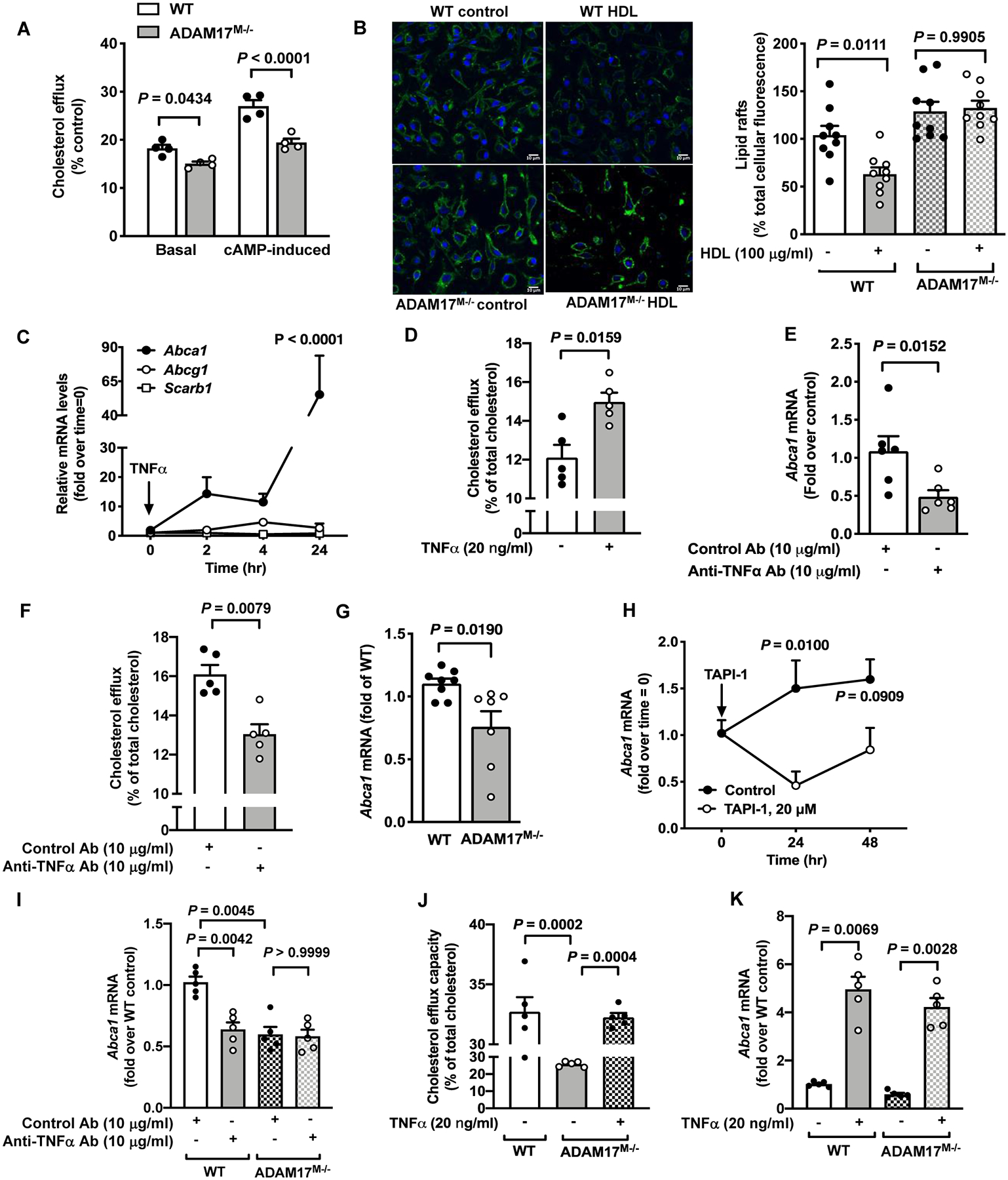
A. BMDMs from male C57BL/6J WT and ADAM17-deficient mice were loaded with radiolabeled [3H]-cholesterol and then treated with HDL (100 μg/ml) for 4 hr. Cholesterol efflux capacity was determined in cAMP-stimulated or unstimulated cells treated with an acyl–coenzyme A: cholesterol acyltransferase inhibitor (n=4). B. BMDMs from male WT and ADAM17-deficient mice treated with HDL (100 μg/ml) for 18 hr. At the end of the treatment, the cells were fixed and stained for lipid rafts with cholera toxin B. Representative pictures and quantification of lipid raft staining are shown (n=9). C. BMDMs from female C57BL/6J mice were treated with TNFα (20 ng/ml) for indicated periods of time. RNA extracts were prepared, and mRNA levels of Abca1, Abcg1 and Scrab1 were analyzed by real-time PCR (n=4; experiment performed twice with similar results). P-value represents changes compared to time point 0. D. BMDMs from male C57BL/6J mice were treated with TNFα (20 ng/ml) and cholesterol efflux was determined as in A (n=5). E. A neutralizing anti-TNFα antibody or a control antibody were used to treat BMDMs from male C57BL/6J mice for 4 hr. RNA extracts were used to measure Abca1 gene expression (n=6). F. Cholesterol efflux was measured in cells from male C57BL/6J mice treated with a neutralizing anti-TNFα antibody or a control antibody (n=5). G. BMDMs from male WT and ADAM17M−/− mice were analyzed for Abca1 gene expression after 7 days of differentiation (n=7–8). H. BMDMs from female C57BL/6J mice were treated with TAPI-1 (20 μM) for 24 and 48 hr. At the end of stimulation, cells were analyzed for Abca1 gene expression (n=3–5; experiment performed twice with similar results). I. BMDMs from male WT and ADAM17M−/− mice were treated with a neutralizing anti-TNFα antibody or a control antibody, and Abca1 mRNA levels were measured by real-time PCR (n=5). J. WT and ADAM17-deficient BMDMs from male mice were incubated with or without TNFα, and cholesterol efflux capacity was then measured as in A (n=5). K. BMDMs from WT and ADAM17M−/− male mice were treated with and without TNFα (20 ng/ml) for analysis of Abca1 mRNA levels (n=5; experiment performed twice with similar results). Data are shown as mean ± SEM. Tests for normality (Shapiro-Wilk) and equal variance (Brown-Forsythe) were performed for each of the data sets. P values were determined accordingly by two-way ANOVA followed by Sidak’s multiple comparison tests (A, H) or Tukey’s multiple comparison tests (C, J), unpaired two-tailed Mann-Whitney test (D, E, F, G) or by one-way ANOVA followed by Tukey’s multiple comparison tests (B) or Brown-Forsythe ANOVA followed by Dunnett’s multiple comparisons tests (I, K). Unless otherwise stated, data are representative of at least three independent experiments performed in replicates in BMDMs from male mice.
TNFα, the best known ADAM17 substrate, is known to enhance cholesterol efflux by upregulating ABCA155 and accordingly, a reduction in cholesterol efflux capacity was reported in TNF receptor-deficient macrophages.22 Consistently, TNFα-treatment markedly upregulated Abca1 mRNA in BMDMs without inducing a significant change in Abcg1 or Scarb1 (SRB1) gene expression (Figure 5C). In addition, TNFα pretreatment significantly increased HDL’s cholesterol efflux capacity in BMDMs (Figure 5D). Contrary, blocking the response of constitutively released TNFα from BMDMs under basal conditions by using a neutralizing anti-TNFα antibody significantly reduced Abca1 mRNA levels (Figure 5E) and reduced the cholesterol efflux capacity of HDL (Figure 5F). Importantly, ADAM17-deficient BMDMs also showed a reduction in Abca1 gene expression (Figure 5G), consistent with the lower release of biologically active soluble TNFα due to lack of ADAM17 mediated ectodomain shedding. Indeed, inhibition of ADAM17’s activity produced a similarly marked reduction in Abca1 mRNA levels (Figure 5H). Moreover, blocking the activity of released TNFα in wildtype BMDMs by the TNFα blocking antibody reduced Abca1 mRNA levels to those found in ADAM17-deficient BMDMs, in which the TNFα blocking antibody had no effect (Figure 5I), and treatment with exogenous TNFα rescued the impaired cholesterol efflux in ADAM17-deficient macrophages (Figure 5J), and increased Abca1 gene expression (Figure 5K).
Finally, if APOA1 acts through ABCA1 to suppress inflammatory effects of LPS, one would expect that ADAM17-deficient macrophages, with their lower levels of ABCA1 expression, would be less responsive to APOA1. This was indeed the case: the ability of APOA1 to suppress LPS-induced Tnfa gene expression was significantly impaired in ADAM17-deficient BMDMs, as compared with macrophages from WT mice (Figure IIIF in the online-only Data Supplement).
Together, these findings strongly suggest that the ability of HDL to efflux cholesterol is significantly reduced in ADAM17-deficient macrophages due to the absence of autocrine signaling from TNFα needed to maintain ABCA1 expression.
Myeloid cell ADAM17 is required to maintain large HDL particle concentrations in vivo
Finally, to investigate the potential physiological relevance of the reduced cholesterol efflux in response to myeloid cell ADAM17-deficiency in vivo, we quantified concentrations of the different HDL particle populations in APOA1Tg Ldlr−/− mice transplanted with bone marrow from ADAM17M−/− mice by calibrated IMA. As shown in Figures 6A–B, APOA1Tg Ldlr−/− mice transplanted with WT bone marrow exhibited 2.2-fold higher total HDL particle concentrations than Ldlr−/− mice not expressing the APOA1 transgene. APOA1Tg Ldlr−/− mice also displayed the human-like HDL particle distribution of small-, medium- and large-HDL, whereas Ldlr−/− mice had only one HDL population, consistent with previous reports.45
Figure 6. Myeloid cell-targeted ADAM17-deficiency selectively reduces the concentration of large HDL particles in APOA1Tg; Ldlr−/− mice.
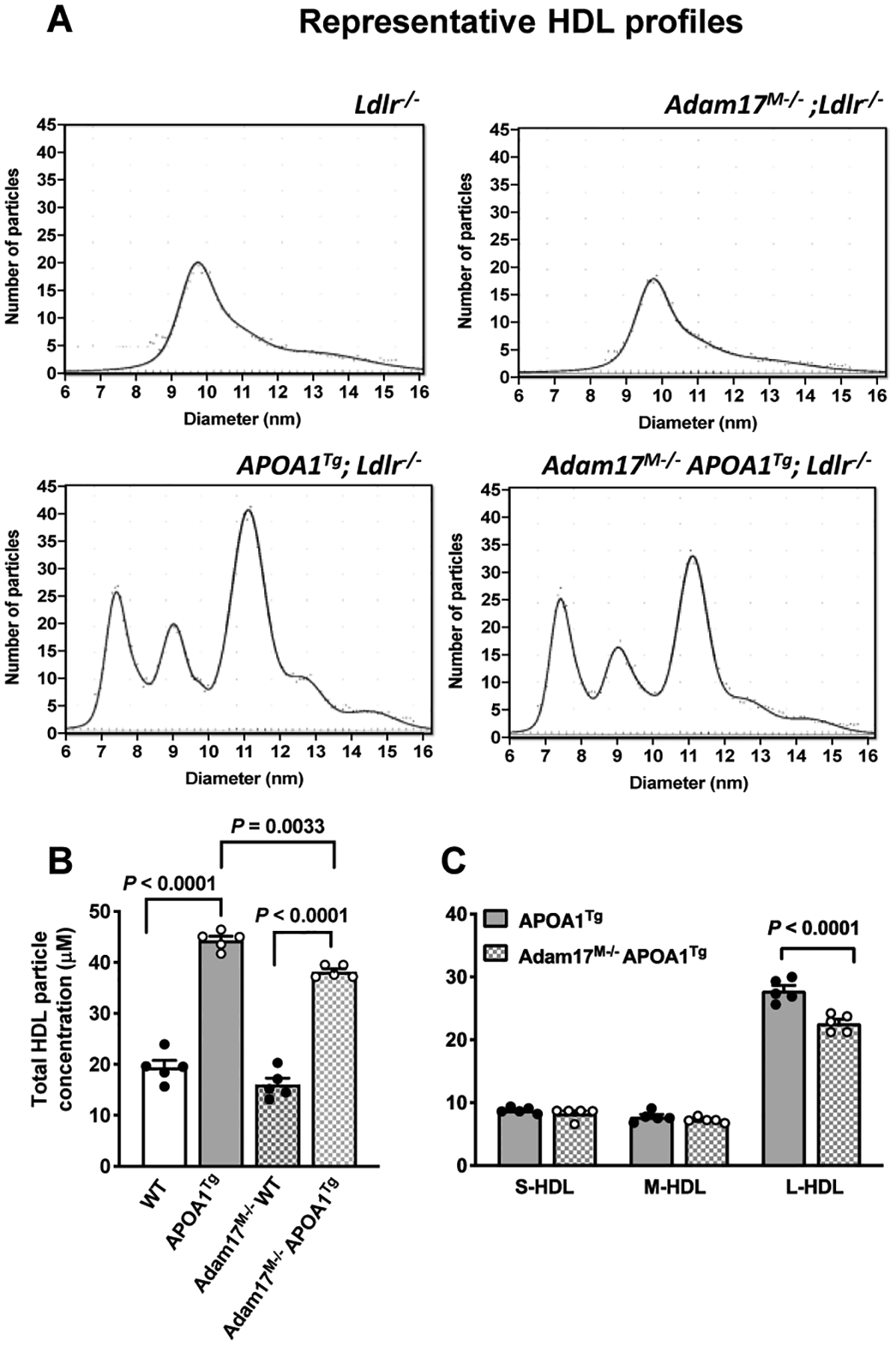
Bone marrow from myeloid cell-targeted ADAM17-deficient mice or C57BL/6J WT controls was transplanted into lethally irradiated (10 Gy) male Ldlr−/− or APOA1Tg; Ldlr−/− recipient littermates. After 8 weeks of recovery, collected plasma samples were used to measure HDL particle concentration and size by calibrated ion mobility analysis. A. Representative size distributions of small, medium, and large HDL from Ldlr−/− and APOA1Tg; Ldlr−/− recipients. B. Total HDL particle concentration (n=5). C. Small (S-HDL), medium (M-HDL) and large HDL (L-HDL) particle concentration (n=5). Data are shown as mean ± SEM. Tests for normality (Shapiro-Wilk) and equal variance (Brown-Forsythe) were performed for each of the data sets. P values were determined accordingly by one-way ANOVA followed by Tukey’s multiple comparison tests (B) and two-way ANOVA followed by Sidak’s multiple comparison tests (C).
Transplantation of bone marrow from ADAM17M−/− mice resulted in a modest reduction in the total HDL particle concentrations, as compared with transplants from control littermates in APOA1Tg Ldlr−/− mice (Figure 6B). HDL particles are heterogeneous and can be fractioned into subclasses defined by density or size, reflecting differences in the relative content of proteins and lipids. Hence, the HDL subclasses vary in composition and also differ in their capacity of contributing to reverse cholesterol transport. Small HDL particles exert the highest cholesterol efflux capacity by a pathway involving ABCA156 while impairment of cholesterol efflux due to reduced interaction of nascent HDL with ABCA1 is associated with defective maturation into large HDL.57 Indeed, measurement of particle concentrations of the different HDL subclasses revealed that APOA1Tg Ldlr−/− mice transplanted with bone marrow from ADAM17M−/− mice exhibited reduced plasma concentrations specifically of large HDL particles, compared with APOA1Tg Ldlr−/− mice transplanted with WT bone marrow (Figure 6C). These results support a role for myeloid cell ADAM17 in facilitating cholesterol efflux and maintaining large HDL particle concentrations in vivo.
Discussion
Our experiments show that ADAM17 is induced and activated by HDL in macrophages and that ADAM17 plays a causal role in maintaining macrophage cholesterol homeostasis through cholesterol efflux. Cholesterol depletion by HDL has previously been shown to activate ADAM17 in other cell types, including endothelial cells32, 33 and human monocytes.58 The mechanism whereby cholesterol depletion increases ADAM17’s sheddase activity has been suggested to be related to movement of ADAM17 from plasma membrane lipid rafts, where it is normally located32 to other membrane domains, where it can access membrane-bound protein substrates.34 Membrane disturbance and appearance of phosphatidylserine in the outer leaflet of the plasma membrane also activate ADAM17.34 The finding that HDL markedly induces ADAM17 expression and activity suggests that ADAM17 induction is part of the cellular response orchestrated by HDL in macrophages.
An important function of HDL and APOA1, its main structural protein, are to induce cholesterol efflux from macrophages, thereby mediating cholesterol removal from lesions of atherosclerosis.8 While the cholesterol depletion also mediates HDL’s pro- and anti-inflammatory effects in macrophages, we showed that the anti-inflammatory effects of HDL predominate over pro-inflammatory effects in lesions of atherosclerosis.25 Importantly, we now show a critical role for ADAM17 in HDL-mediated cholesterol efflux as well as the downstream immune-modulating activity of HDL in macrophages.
Moreover, we demonstrate that free APOA1 only elicits anti-inflammatory responses, without any pro-inflammatory effects in macrophages. The anti-inflammatory effect of APOA1 is dependent on ABCA1 and ADAM17. There are at least three possible explanations for the differential effect of HDL and APOA1 on macrophage inflammatory phenotypes. First, HDL might induce a more generalized passive diffusion of cholesterol from the plasma membrane, while APOA1 is more likely to mediate cholesterol efflux from specific membrane pools through ABCA1. The pro-inflammatory phenotype of ABCA1-deficient macrophages would be in line with this possibility.59 Second, free APOA1 might induce anti-inflammatory signaling via ABCA1 through activation of STAT3-SOCS signaling independent of cholesterol efflux.60 However, we found no evidence for increased Socs3 mRNA levels in LPS-stimulated macrophages exposed to APOA1 (data not shown), which makes this possibility less likely. Third, it is possible that HDL’s protein, -lipid or -RNA cargo influences pro-inflammatory responses directly or by affecting the cholesterol depletion status of the cell. Because HDL’s anti-inflammatory effects predominate over pro-inflammatory effects in lesions of atherosclerosis and because APOA1 shows only anti-inflammatory responses, it is likely that the induction of ADAM17 by HDL facilitates the resolution of inflammation in lesions and represents a beneficial response in the context of atherosclerosis. However, the overall contribution of HDL and APOA1 to the effects of ADAM17 in atherogenesis remains to be established in future experiments.
ABCA1 is critical in mediating cholesterol efflux to APOA1 and small HDL. ABCA1 is markedly induced by LXR, a cholesterol sensor sensitive to oxysterols, when cholesterol levels are high61 and by cyclic AMP.54 However, TNFα can also increase ABCA1 expression in macrophages. Thus, TNFα upregulates ABCA1 in macrophages through intracellular signaling (NF-κB and p38 MAPK pathways) and increases cholesterol efflux,55 and TNF receptor-deficiency impairs macrophage cholesterol efflux.22 Notably, our results strongly suggest that ADAM17 promotes HDL-induced cholesterol efflux at least in part through promoting ABCA1 upregulation by shedding membrane-bound TNFα to generate its bioactive soluble form (Figure 7). By contrast, ADAM17M−/− macrophages display impaired cholesterol efflux due to reduced expression of ABCA1 because of the lack of ADAM17-dependent shedding of soluble and active TNFα. While the evidence supports a role for shed TNFα in maintaining ABCA1 expression and low cellular cholesterol levels, we cannot rule out additional consequence of ADAM17-deficiency that could affect cholesterol efflux.
Figure 7. Schematic representation of the role of ADAM17 in mediating HDL’s pro-inflammatory and anti-inflammatory effects in macrophages.
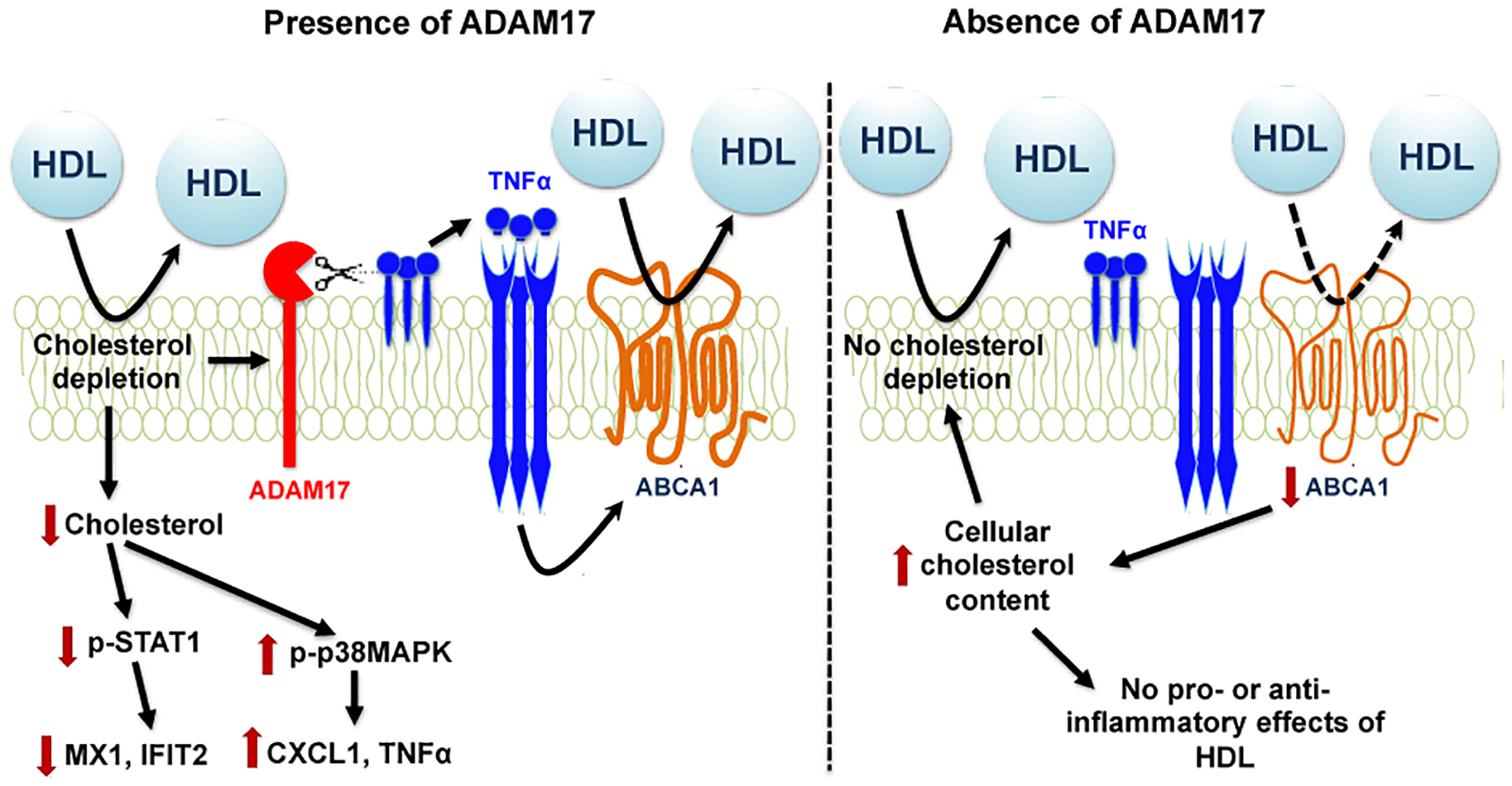
Cholesterol depletion in non-lipid-loaded macrophages by HDL increases ADAM17 expression and its activity. ADAM17 supports a lipid depleted status by shedding TNFα, allowing the cell to maintain ABCA1 expression and cholesterol efflux. Cholesterol depletion drives both the pro-inflammatory and anti-inflammatory responses to HDL in the presence of ADAM17 (left) through STAT1 and p38 MAPK signaling. In the absence of ADAM17 (right), HDL’s ability to deplete cellular cholesterol is impaired due to the low levels of bioactive soluble TNFα and ABCA1, which in turn lead to elevated cellular cholesterol. The elevated cholesterol prevents both the pro-inflammatory and anti-inflammatory effects of HDL. In vivo, myeloid cell-targeted ADAM17-deficiency lowers the concentration of large HDL particles in circulation.
The induction of ADAM17 in macrophages by HDL may be relevant to atherosclerotic plaques in which efflux of cholesterol from macrophage foam cells is thought to be central to HDL’s anti-atherogenic mechanism. It is known that efflux of cholesterol through ABCA1 is mediated most effectively to small HDL particles, and much less so to large HDL.56 Moreover, loss-of-function mutations in ABCA1 in patients with Tangier disease are associated with impaired cholesterol efflux, reduced expansion of HDL particle size, and in reduced levels of large HDL particles.57 Similar results have been obtained in cell culture systems.62 Increased ABCA1 expression in the presence of a constant amount of APOA1 results in more large HDL particles (and vice versa), demonstrating that ABCA1 abundance can affect HDL size. Consistent with these results, our data show that myeloid cell-targeted ADAM17-deficiency results in reduced concentrations specifically of large HDL particles in APOA1Tg mice. Although HDL turnover and particle size distribution are largely determined by the liver and HDL remodeling in circulation, our observations raise the possibility that ADAM17-dependent cholesterol efflux from myeloid cells contributes to plasma levels of large HDL particles.
The insertion of the human APOA1 transgene in mice causes three major size populations of HDL, like in human plasma, and as opposed to the monodispersed HDL size peak normally found in mice. However, one of the limitations of APOA1Tg Ldlr−/− mice is that they have higher HDL levels than humans and that the large HDL population predominates. Furthermore, cholesterol levels are higher in Ldlr−/− mice than in humans, even when fed a low-fat diet, like in the present study. Nevertheless, this is the best studied mouse model available to mimic the three HDL peaks together with elevated levels of APOB-containing lipoproteins. Previous studies on the effects of ADAM17-deficiency have not used mouse models with the human-like HDL particle distribution, which might explain why effects of ADAM17 on HDL levels have not been detected.27 Conversely, it is not possible to investigate the effects of myeloid cell-targeted ADAM17-deletion in humans.
Although TNFα and its receptors are the best known ADAM17 substrates in macrophages,63 ADAM17 has a plethora of effects in macrophages, likely depending on which of its many substrates is highly expressed at a given time and macrophage population, the cellular and membrane localization of ADAM17, trafficking to the cell surface,64 expression levels of ADAM17, activation of intracellular signaling leading to increased sheddase activity,65 and levels of ADAM17 inhibitors.66 Myeloid cell-targeted deletion of ADAM17 has been shown to increase atherosclerosis lesion size in Apoe−/− mice, while reducing relative lesional macrophage content and increasing collagen and smooth muscle cell content, suggesting a more fibrotic, less inflammatory lesion phenotype.28 Consistently, ADAM17-deficiency results in reduced proliferation of lesion macrophages, likely because of the reduced shedding of cell surface CSF1, an important myeloid cell growth factor.31 Moreover, MerTK cleavage by ADAM17 impairs synthesis of proresolving mediators involved in resolution of inflammation.29 Furthermore, MerTK and CD36, another ADAM17 substrate, mediate efferocytosis, and ADAM17 reduces efferocytosis by shedding these substrates, thereby increasing tissue inflammation.46, 67 ADAM17 also promotes inflammatory tissue processes in other ways, including enhancing diapedesis during transendothelial cell migration, likely by shedding the integrins CD11b/CD18 in a controlled temporal fashion.68
The present study supports a role for ADAM17 in modulating inflammatory responses in macrophages, and demonstrates a novel role for ADAM17 in maintaining cholesterol homeostasis by promoting cholesterol efflux through ABCA1 in vitro and in vivo.
Supplementary Material
Highlights.
HDL induces expression and activity of the sheddase ADAM17 in macrophages.
ADAM17 is required for HDL’s pro-inflammatory and anti-inflammatory effects in macrophages.
Myeloid cell-targeted ADAM17-deficiency prevents cholesterol efflux due to the absence of bioactive TNFα and TNFα-mediated ABCA1 upregulation.
Myeloid cell ADAM17 is required to maintain large HDL particle concentrations in vivo in APOA1Tg mice.
Acknowledgments:
We thank Jake Wimberger, Angela Irwin and Carissa Thornock (all at the University of Washington) for technical assistance. We are grateful to Dr. Alan Tall (Columbia University) for fruitful discussions and Dr. W. Sean Davidson (University of Cincinnati) for providing the wildtype APOA1 and APOA1 C-terminal deletion mutant and advice.
Sources of funding
This work was supported by grants from the National Institutes of Health P01HL151328, P01HL092969, R01HL149685, and R35HL150754 (to K.E.B), a postdoctoral fellowship award from the American Diabetes Association #9-18-CVD1-002 (to V.K) and the Vector and Transgenic Mouse Core of the Diabetes Research Center at the University of Washington (P30DK017047). This research was also supported by the Cell Analysis Facility Flow Cytometry and Imaging Core in the Department of Immunology at the University of Washington. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosures
Dr. Bornfeldt reports receiving research support from Novo Nordisk A/S on an unrelated project. The other authors report no conflicts.
Abbreviations
- ABCA1
ATP-binding cassette transporter A1
- AcLDL
Acetylated low-density lipoprotein
- LDLR
Low-density lipoprotein receptor
- ADAM17
ADAM metallopeptidase domain 17
- APOA1
Apolipoprotein A1
- ASK1
Apoptosis signal-regulating kinase 1
- BMDM
Bone marrow-derived macrophages
- CVD
Cardiovascular disease
- HDL
High-density lipoprotein
- HDL-C
HDL-cholesterol
- HDL-P
HDL particle concentration
- IFNβ
Interferon-β
- IMA
Ion mobility analysis
- LDL
Low-density lipoprotein
- LPS
Lipopolysaccharide
- MβCD
Methyl-β-cyclodextrin
- p38MAPK
p38 mitogen-activated protein kinase
- STAT1
Signal transducer and activator of transcription 1
- TLR
Toll-like receptor
- TNFα
Tumor necrosis factor-α
References
- 1.Kannel WB, Dawber TR, Friedman GD, Glennon WE, McNamara PM. Risk factors in coronary heart disease. An evaluation of several serum lipids as predictors of coronary heart disease; the framingham study. Ann Intern Med. 1964;61:888–899 [DOI] [PubMed] [Google Scholar]
- 2.Castelli WP, Garrison RJ, Wilson PW, Abbott RD, Kalousdian S, Kannel WB. Incidence of coronary heart disease and lipoprotein cholesterol levels. The framingham study. JAMA. 1986;256:2835–2838 [PubMed] [Google Scholar]
- 3.Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, Hindy G, Holm H, Ding EL, Johnson T, et al. Plasma hdl cholesterol and risk of myocardial infarction: A mendelian randomisation study. Lancet. 2012;380:572–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45:1345–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tall AR, Rader DJ. Trials and tribulations of cetp inhibitors. Circ Res. 2018;122:106–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Investigators A-H, Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low hdl cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–2267 [DOI] [PubMed] [Google Scholar]
- 7.Madsen CM, Varbo A, Nordestgaard BG. Extreme high high-density lipoprotein cholesterol is paradoxically associated with high mortality in men and women: Two prospective cohort studies. Eur Heart J. 2017;38:2478–2486 [DOI] [PubMed] [Google Scholar]
- 8.Tall AR, Yvan-Charvet L, Terasaka N, Pagler T, Wang N. Hdl, abc transporters, and cholesterol efflux: Implications for the treatment of atherosclerosis. Cell Metab. 2008;7:365–375 [DOI] [PubMed] [Google Scholar]
- 9.Oram JF. Hdl apolipoproteins and abca1: Partners in the removal of excess cellular cholesterol. Arterioscler Thromb Vasc Biol. 2003;23:720–727 [DOI] [PubMed] [Google Scholar]
- 10.Phillips MC. Molecular mechanisms of cellular cholesterol efflux. J Biol Chem. 2014;289:24020–24029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ronsein GE, Heinecke JW. Time to ditch hdl-c as a measure of hdl function? Curr Opin Lipidol. 2017;28:414–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barnett KC, Kagan JC. Lipids that directly regulate innate immune signal transduction. Innate Immun. 2020;26:4–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Westerterp M, Bochem AE, Yvan-Charvet L, Murphy AJ, Wang N, Tall AR. Atp-binding cassette transporters, atherosclerosis, and inflammation. Circ Res. 2014;114:157–170 [DOI] [PubMed] [Google Scholar]
- 15.Sorci-Thomas MG, Thomas MJ. High density lipoprotein biogenesis, cholesterol efflux, and immune cell function. Arterioscler Thromb Vasc Biol. 2012;32:2561–2565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tall AR. Plasma high density lipoproteins: Therapeutic targeting and links to atherogenic inflammation. Atherosclerosis. 2018;276:39–43 [DOI] [PubMed] [Google Scholar]
- 17.Suzuki M, Pritchard DK, Becker L, Hoofnagle AN, Tanimura N, Bammler TK, Beyer RP, Bumgarner R, Vaisar T, de Beer MC, et al. High-density lipoprotein suppresses the type i interferon response, a family of potent antiviral immunoregulators, in macrophages challenged with lipopolysaccharide. Circulation. 2010;122:1919–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song GJ, Kim SM, Park KH, Kim J, Choi I, Cho KH. Sr-bi mediates high density lipoprotein (hdl)-induced anti-inflammatory effect in macrophages. Biochem Biophys Res Commun. 2015;457:112–118 [DOI] [PubMed] [Google Scholar]
- 19.De Nardo D, Labzin LI, Kono H, Seki R, Schmidt SV, Beyer M, Xu D, Zimmer S, Lahrmann C, Schildberg FA, et al. High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator atf3. Nat Immunol. 2014;15:152–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanson M, Distel E, Fisher EA. Hdl induces the expression of the m2 macrophage markers arginase 1 and fizz-1 in a stat6-dependent process. PLoS One. 2013;8:e74676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van der Vorst EPC, Theodorou K, Wu Y, Hoeksema MA, Goossens P, Bursill CA, Aliyev T, Huitema LFA, Tas SW, Wolfs IMJ, et al. High-density lipoproteins exert pro-inflammatory effects on macrophages via passive cholesterol depletion and pkc-nf-kappab/stat1-irf1 signaling. Cell Metab. 2017;25:197–207 [DOI] [PubMed] [Google Scholar]
- 22.Smoak KA, Aloor JJ, Madenspacher J, Merrick BA, Collins JB, Zhu X, Cavigiolio G, Oda MN, Parks JS, Fessler MB. Myeloid differentiation primary response protein 88 couples reverse cholesterol transport to inflammation. Cell Metab. 2010;11:493–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ito A, Hong C, Rong X, Zhu X, Tarling EJ, Hedde PN, Gratton E, Parks J, Tontonoz P. Lxrs link metabolism to inflammation through abca1-dependent regulation of membrane composition and tlr signaling. Elife. 2015;4:e08009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fotakis P, Kothari V, Bornfeldt KE, Tall AR. Response by fotakis et al to letter regarding article, “anti-inflammatory effects of hdl (high-density lipoprotein) in macrophages predominate over proinflammatory effects in atherosclerotic plaques”. Arterioscler Thromb Vasc Biol. 2020;40:e33–e34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fotakis P, Kothari V, Thomas DG, Westerterp M, Molusky MM, Altin E, Abramowicz S, Wang N, He Y, Heinecke JW, et al. Anti-inflammatory effects of hdl (high-density lipoprotein) in macrophages predominate over proinflammatory effects in atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2019;39:e253–e272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zunke F, Rose-John S. The shedding protease adam17: Physiology and pathophysiology. Biochim Biophys Acta Mol Cell Res. 2017;1864:2059–2070 [DOI] [PubMed] [Google Scholar]
- 27.Nicolaou A, Zhao Z, Northoff BH, Sass K, Herbst A, Kohlmaier A, Chalaris A, Wolfrum C, Weber C, Steffens S, et al. Adam17 deficiency promotes atherosclerosis by enhanced tnfr2 signaling in mice. Arterioscler Thromb Vasc Biol. 2017;37:247–257 [DOI] [PubMed] [Google Scholar]
- 28.van der Vorst EP, Zhao Z, Rami M, Holdt LM, Teupser D, Steffens S, Weber C. Contrasting effects of myeloid and endothelial adam17 on atherosclerosis development. Thromb Haemost. 2017;117:644–646 [DOI] [PubMed] [Google Scholar]
- 29.Cai B, Thorp EB, Doran AC, Subramanian M, Sansbury BE, Lin CS, Spite M, Fredman G, Tabas I. Mertk cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc Natl Acad Sci U S A. 2016;113:6526–6531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thorp E, Vaisar T, Subramanian M, Mautner L, Blobel C, Tabas I. Shedding of the mer tyrosine kinase receptor is mediated by adam17 protein through a pathway involving reactive oxygen species, protein kinase cdelta, and p38 mitogen-activated protein kinase (mapk). J Biol Chem. 2011;286:33335–33344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang J, Frey JM, Wilson CL, Moncada-Pazos A, Levet C, Freeman M, Rosenfeld ME, Stanley ER, Raines EW, Bornfeldt KE. Neutrophil and macrophage cell surface colony-stimulating factor 1 shed by adam17 drives mouse macrophage proliferation in acute and chronic inflammation. Mol Cell Biol. 2018;38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tellier E, Canault M, Rebsomen L, Bonardo B, Juhan-Vague I, Nalbone G, Peiretti F. The shedding activity of adam17 is sequestered in lipid rafts. Exp Cell Res. 2006;312:3969–3980 [DOI] [PubMed] [Google Scholar]
- 33.Tellier E, Canault M, Poggi M, Bonardo B, Nicolay A, Alessi MC, Nalbone G, Peiretti F. Hdls activate adam17-dependent shedding. J Cell Physiol. 2008;214:687–693 [DOI] [PubMed] [Google Scholar]
- 34.Reiss K, Bhakdi S. The plasma membrane: Penultimate regulator of adam sheddase function. Biochim Biophys Acta Mol Cell Res. 2017;1864:2082–2087 [DOI] [PubMed] [Google Scholar]
- 35.Sommer A, Kordowski F, Buch J, Maretzky T, Evers A, Andra J, Dusterhoft S, Michalek M, Lorenzen I, Somasundaram P, et al. Phosphatidylserine exposure is required for adam17 sheddase function. Nat Commun. 2016;7:11523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis KE, Kirk EA, McDonald TO, Wang S, Wight TN, O’Brien KD, Chait A. Increase in serum amyloid a evoked by dietary cholesterol is associated with increased atherosclerosis in mice. Circulation. 2004;110:540–545 [DOI] [PubMed] [Google Scholar]
- 37.Wall VZ, Barnhart S, Kramer F, Kanter JE, Vivekanandan-Giri A, Pennathur S, Bolego C, Ellis JM, Gijon MA, Wolfgang MJ, et al. Inflammatory stimuli induce acyl-coa thioesterase 7 and remodeling of phospholipids containing unsaturated long (>/=c20)-acyl chains in macrophages. J Lipid Res. 2017;58:1174–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang L, Mei X, Atkinson D, Small DM. Surface behavior of apolipoprotein a-i and its deletion mutants at model lipoprotein interfaces. J Lipid Res. 2014;55:478–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Panagotopulos SE, Witting SR, Horace EM, Hui DY, Maiorano JN, Davidson WS. The role of apolipoprotein a-i helix 10 in apolipoprotein-mediated cholesterol efflux via the atp-binding cassette transporter abca1. J Biol Chem. 2002;277:39477–39484 [DOI] [PubMed] [Google Scholar]
- 40.Kanter JE, Shao B, Kramer F, Barnhart S, Shimizu-Albergine M, Vaisar T, Graham MJ, Crooke RM, Manuel CR, Haeusler RA, et al. Increased apolipoprotein c3 drives cardiovascular risk in type 1 diabetes. J Clin Invest. 2019;130:4165–4179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schlondorff J, Becherer JD, Blobel CP. Intracellular maturation and localization of the tumour necrosis factor alpha convertase (tace). Biochem J. 2000;347 Pt 1:131–138 [PMC free article] [PubMed] [Google Scholar]
- 42.Henderson CM, Vaisar T, Hoofnagle AN. Isolating and quantifying plasma hdl proteins by sequential density gradient ultracentrifugation and targeted proteomics. Methods Mol Biol. 2016;1410:105–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hutchins PM, Ronsein GE, Monette JS, Pamir N, Wimberger J, He Y, Anantharamaiah GM, Kim DS, Ranchalis JE, Jarvik GP, et al. Quantification of hdl particle concentration by calibrated ion mobility analysis. Clin Chem. 2014;60:1393–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pamir N, Hutchins PM, Ronsein GE, Wei H, Tang C, Das R, Vaisar T, Plow E, Schuster V, Koschinsky ML, et al. Plasminogen promotes cholesterol efflux by the abca1 pathway. JCI Insight. 2017;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rubin EM, Ishida BY, Clift SM, Krauss RM. Expression of human apolipoprotein a-i in transgenic mice results in reduced plasma levels of murine apolipoprotein a-i and the appearance of two new high density lipoprotein size subclasses. Proc Natl Acad Sci U S A. 1991;88:434–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Driscoll WS, Vaisar T, Tang J, Wilson CL, Raines EW. Macrophage adam17 deficiency augments cd36-dependent apoptotic cell uptake and the linked anti-inflammatory phenotype. Circ Res. 2013;113:52–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Couto G, Jaghatspanyan E, DeBerge M, Liu W, Luther K, Wang Y, Tang J, Thorp EB, Marban E. Mechanism of enhanced mertk-dependent macrophage efferocytosis by extracellular vesicles. Arterioscler Thromb Vasc Biol. 2019;39:2082–2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takahashi S, Ito T, Zenimaru Y, Suzuki J, Miyamori I, Takahashi M, Takahashi M, Ishida T, Ishida T, Hirata K, et al. Species differences of macrophage very low-density-lipoprotein (vldl) receptor protein expression. Biochem Biophys Res Commun. 2011;407:656–662 [DOI] [PubMed] [Google Scholar]
- 49.Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, et al. The trem2-apoe pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47:566–581 e569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kosack L, Gawish R, Lercher A, Vilagos B, Hladik A, Lakovits K, Bhattacharya A, Schliehe C, Mesteri I, Knapp S, et al. The lipid-sensor trem2 aggravates disease in a model of lcmv-induced hepatitis. Sci Rep. 2017;7:11289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feuerbach D, Schindler P, Barske C, Joller S, Beng-Louka E, Worringer KA, Kommineni S, Kaykas A, Ho DJ, Ye C, et al. Adam17 is the main sheddase for the generation of human triggering receptor expressed in myeloid cells (htrem2) ectodomain and cleaves trem2 after histidine 157. Neurosci Lett. 2017;660:109–114 [DOI] [PubMed] [Google Scholar]
- 52.Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, Llapashtica C, Wang J, Kim DJ, Xia D, et al. Trem2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron. 2020;105:837–854 e839 [DOI] [PubMed] [Google Scholar]
- 53.Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, Lundgren P, Bleriot C, Liu Z, Deczkowska A, et al. Lipid-associated macrophages control metabolic homeostasis in a trem2-dependent manner. Cell. 2019;178:686–698 e614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oram JF, Lawn RM, Garvin MR, Wade DP. Abca1 is the camp-inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J Biol Chem. 2000;275:34508–34511 [DOI] [PubMed] [Google Scholar]
- 55.Gerbod-Giannone MC, Li Y, Holleboom A, Han S, Hsu LC, Tabas I, Tall AR. Tnfalpha induces abca1 through nf-kappab in macrophages and in phagocytes ingesting apoptotic cells. Proc Natl Acad Sci U S A. 2006;103:3112–3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Du XM, Kim MJ, Hou L, Le Goff W, Chapman MJ, Van Eck M, Curtiss LK, Burnett JR, Cartland SP, Quinn CM, et al. Hdl particle size is a critical determinant of abca1-mediated macrophage cellular cholesterol export. Circ Res. 2015;116:1133–1142 [DOI] [PubMed] [Google Scholar]
- 57.Brousseau ME, Eberhart GP, Dupuis J, Asztalos BF, Goldkamp AL, Schaefer EJ, Freeman MW. Cellular cholesterol efflux in heterozygotes for tangier disease is markedly reduced and correlates with high density lipoprotein cholesterol concentration and particle size. J Lipid Res. 2000;41:1125–1135 [PubMed] [Google Scholar]
- 58.Matthews V, Schuster B, Schutze S, Bussmeyer I, Ludwig A, Hundhausen C, Sadowski T, Saftig P, Hartmann D, Kallen KJ, et al. Cellular cholesterol depletion triggers shedding of the human interleukin-6 receptor by adam10 and adam17 (tace). J Biol Chem. 2003;278:38829–38839 [DOI] [PubMed] [Google Scholar]
- 59.Zhu X, Lee JY, Timmins JM, Brown JM, Boudyguina E, Mulya A, Gebre AK, Willingham MC, Hiltbold EM, Mishra N, et al. Increased cellular free cholesterol in macrophage-specific abca1 knock-out mice enhances pro-inflammatory response of macrophages. J Biol Chem. 2008;283:22930–22941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang C, Houston BA, Storey C, LeBoeuf RC. Both stat3 activation and cholesterol efflux contribute to the anti-inflammatory effect of apoa-i/abca1 interaction in macrophages. J Lipid Res. 2016;57:848–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tontonoz P, Mangelsdorf DJ. Liver x receptor signaling pathways in cardiovascular disease. Mol Endocrinol. 2003;17:985–993 [DOI] [PubMed] [Google Scholar]
- 62.Lyssenko NN, Nickel M, Tang C, Phillips MC. Factors controlling nascent high-density lipoprotein particle heterogeneity: Atp-binding cassette transporter a1 activity and cell lipid and apolipoprotein ai availability. FASEB J. 2013;27:2880–2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bell JH, Herrera AH, Li Y, Walcheck B. Role of adam17 in the ectodomain shedding of tnf-alpha and its receptors by neutrophils and macrophages. J Leukoc Biol. 2007;82:173–176 [DOI] [PubMed] [Google Scholar]
- 64.Oikonomidi I, Burbridge E, Cavadas M, Sullivan G, Collis B, Naegele H, Clancy D, Brezinova J, Hu T, Bileck A, et al. Itap, a novel irhom interactor, controls tnf secretion by policing the stability of irhom/tace. Elife. 2018;7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grieve AG, Xu H, Kunzel U, Bambrough P, Sieber B, Freeman M. Phosphorylation of irhom2 at the plasma membrane controls mammalian tace-dependent inflammatory and growth factor signalling. Elife. 2017;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schubert K, Collins LE, Green P, Nagase H, Troeberg L. Lrp1 controls tnf release via the timp-3/adam17 axis in endotoxin-activated macrophages. J Immunol. 2019;202:1501–1509 [DOI] [PubMed] [Google Scholar]
- 67.Cai B, Thorp EB, Doran AC, Sansbury BE, Daemen MJ, Dorweiler B, Spite M, Fredman G, Tabas I. Mertk receptor cleavage promotes plaque necrosis and defective resolution in atherosclerosis. J Clin Invest. 2017;127:564–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tsubota Y, Frey JM, Tai PW, Welikson RE, Raines EW. Monocyte adam17 promotes diapedesis during transendothelial migration: Identification of steps and substrates targeted by metalloproteinases. J Immunol. 2013;190:4236–4244 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.