

Abstract
Herein, we describe the development of a deconstructive strategy for the first asymmetric synthesis of (−)-thebainone A, capitalized on an enantioselective C−C bond activation and a C−O bond cleavage reaction. The Rh-catalyzed asymmetric ‘cut-and-sew’ transformation between sterically hindered trisubstituted alkenes and benzocyclobutenones allows efficient construction of the fused A/B/C rings and the all-carbon quaternary center of the natural product. The newly optimized conditions show a broad substrate scope and excellent enantioselectivity (up to 99.5:0.5 er). Taking advantage of the boron-mediated ether bond cleavage, synthesis of the morphine-alkaloid (−)-thebainone A has been completed with two complementary routes.
Keywords: C−C activation, Total synthesis, Alkaloids, Asymmetric synthesis, Fused rings
Graphical Abstract

Asymmetric total synthesis of morphine alkaloid (−)-thebainone A has been accomplished for the first time in 13 (and 14) steps from commercially available starting materials. The synthesis features a deconstructive strategy through cleaving relatively inert C‒C and C‒O bonds to build more complex scaffolds.
Introduction
Isolated from the opium poppy latex, morphine (1) and its congeners are among the oldest and most extensively studied alkaloid natural products known to date (Figure 1).[1] Given their potent neurological and immunological activity, morphine alkaloids and the unnatural semi-synthetic opioids, such as ketorfanol,[2a] oxycodone,[2b] and naltrexone,[2c] have found various important therapeutic applications, including acting as pain killers or treating opioid abuse and alcohol dependence. Besides the biological importance, these compounds also exhibit very intriguing chemical structures, such as a unique polybridged/fused ring system, an all-carbon quaternary center, a basic tertiary amine moiety, and a 1,2,3,4-tetrasubstituted arene, which have posted significant challenges for their preparation. Consequently, morphine alkaloids have continuously attracted significant attentions from the synthetic community for more than 70 years. Since the first synthesis of morphine by Gates in 1952,[3] over 40 synthesis routes of morphine alkaloids have been reported and more than 30 prominent synthetic research groups have been involved.[4,5,6] Beyond just a target of synthesis, the morphine scaffold has more or less become a touchstone for examining efficiency of new synthetic strategies or robustness of new synthetic methods.
Figure 1.

Morphine and related Alkaloids.
Among various morphine alkaloids, thebainone A (4) with a unique enone-containing C-ring has served as a precursor to access morphine (1) and codeine (2) in the seminal work of Gates.[3] The Tius group in 1992 developed the second total synthesis of racemic thebainone A in 24 steps taking advantage of a chemo- and regiospecific Diels–Alder reaction.[7] Very recently, another elegant racemic synthesis of this natural product was reported by Metz in 22 steps, which features an intramolecular nitrone cycloaddition and a Heck cyclization as the key steps.[6a] To the best of our knowledge, enantioselective synthesis of thebainone A is not known yet. Herein, we describe the development of a deconstructive strategy for the rapid and asymmetric preparation of (−)-thebainone A, capitalizing on a catalytic enantioselective C−C bond activation[8] and a C−O bond cleavage[9] reaction.
Deconstructive synthesis, namely building new (and often more challenging) structures through bond cleavage of easily accessible moieties, has emerged as a useful design principle in synthesizing bioactive natural products and other target molecules (Scheme 1a).[10] For example, from readily available cyclic ketones and ethers, more complex fused and bridged rings could be prepared rapidly through cleavage and subsequent functionalization of a C−C or C−O bond. Such deconstructive strategies could enhance the overall efficiency and provide a concise synthetic route. As one of ongoing research efforts, our laboratory has been engaged in developing catalytic “cut-and-sew” transformations for preparing bridged and fused rings through activation of relatively inert C−C bonds in cyclic ketones.[11] Here, we anticipated that such a deconstructive strategy could be applied to the first enantioselective total synthesis of (−)-thebainone A and formal syntheses of codeine and morphine (Scheme 1b).
Scheme 1.

Deconstructive Synthesis of (−)-Thebainone A.
From the retrosynthetic viewpoint (Scheme 2), the enone in the C ring of thebainone A could be introduced via a late-stage dehydrogenation of the saturated intermediate (5). The amine and the phenol functional groups (FGs) in 5 is expected to be installed via the deconstructive C−O bond cleavage strategy from intermediate 6 that contains a dihydropyran moiety. A Rh-catalyzed enantioselective“cut-and-sew” transformation with benzocyclobutenones appears to be well suited to access tetracycle 6 that contains the fused A/B/C rings along with the all-carbon quaternary center.[12] This step not only sets the important stereochemistry at the C13 and C14 positions, but also provides all the C−C bonds present in the natural product. The benzocyclobutenone precursor (7) could be readily prepared in a convergent manner from alcohol 8 and phenol 9 by the Mitsunobu reaction. Finally, fragments 8 and 9 could be straightforwardly produced from commercially available compounds 10 and 11, respectively.
Scheme 2.

Retrosynthetic Analysis
Results and Discussion
The key C−C activation reaction:
To explore the proposed synthetic strategy, the key “cut-and-sew” reaction was investigated first. While a number of alkene-tethered benzocyclobutenones have previously proved to be viable substrates for the Rh-catalyzed asymmetric carboacylation,[11d–f] compound 7 that contains an acid-sensitive ketal, a sterically hindered trisubstituted olefin and a relatively long linker nevertheless represents a new challenge (Scheme 3). Unlike mono- and di-substituted alkenes, trisubstituted alkenes have not been used in the asymmetric “cut-and-sew” reaction as they typically exhibit much lower reactivity due to the steric hindrance. In addition, almost all the previous intramolecular carboacylations with benzocyclobutenones use a five-membered-ring-forming tether,[11] which is kinetically favorable; the extended linkage in substrate 7 is expected to increase the kinetic barrier for the 2π-insertion. Moreover, the more flexible linker could also add difficulty for the enantioselective control. Therefore, the primary difficulty arises from the challenging enantio-determining migratory insertion step.
Scheme 3.
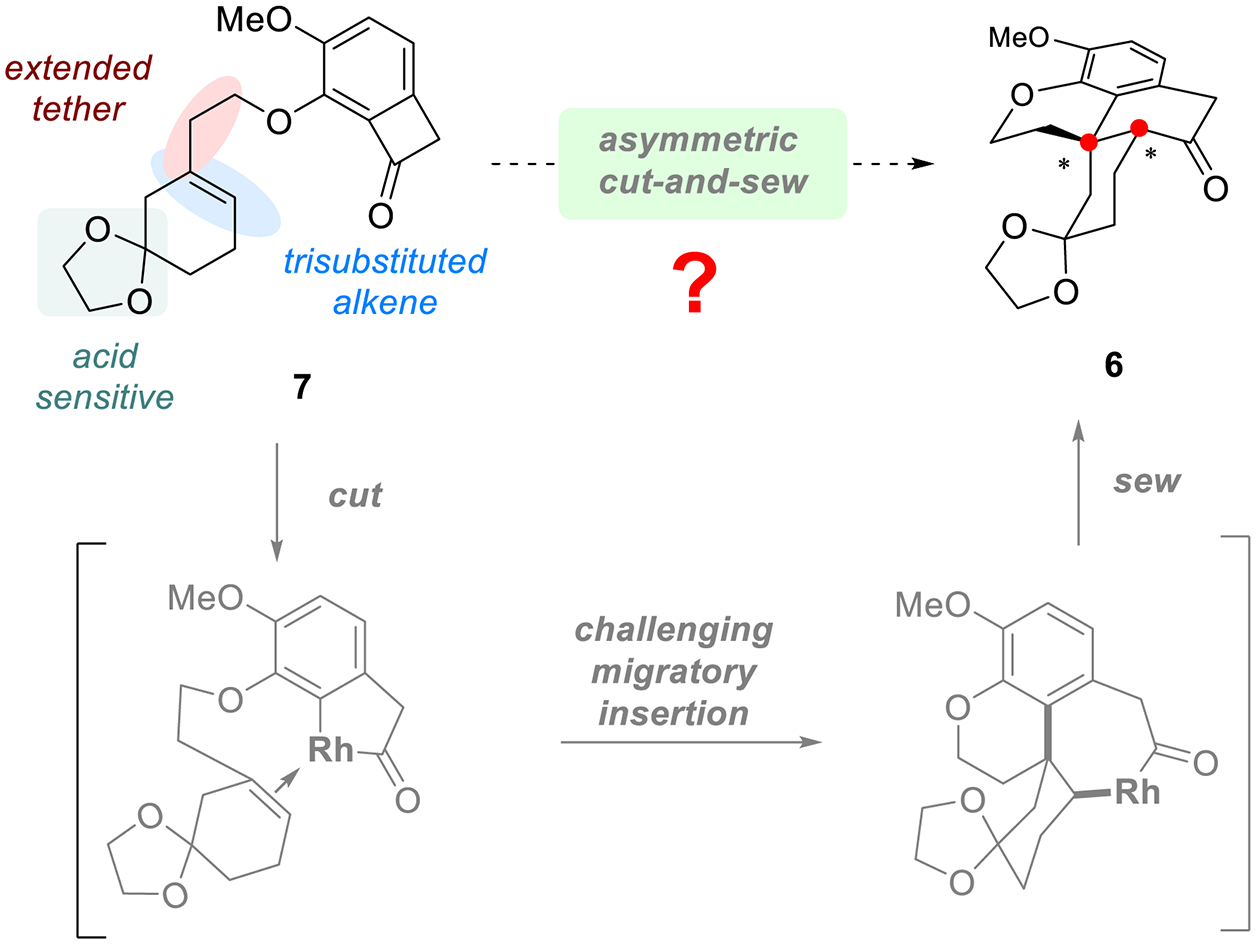
The Key Enantioselective “Cut-and-sew” Step
To explore the feasibility of the key enantioselective “cut-and-sew” step, a simplified model substrate (7a) was prepared from commercially available aryl bromide 11 through a sequence of benzocyclobutenone synthesis using our previously developed protocol,[13a] followed by the Mitsunobu coupling with a known alcohol (8a)[13b] (Scheme 4). Notably, phenol 9 can be prepared on decagram scales in 70% overall yield with only one column chromatography purification needed.
Scheme 4.
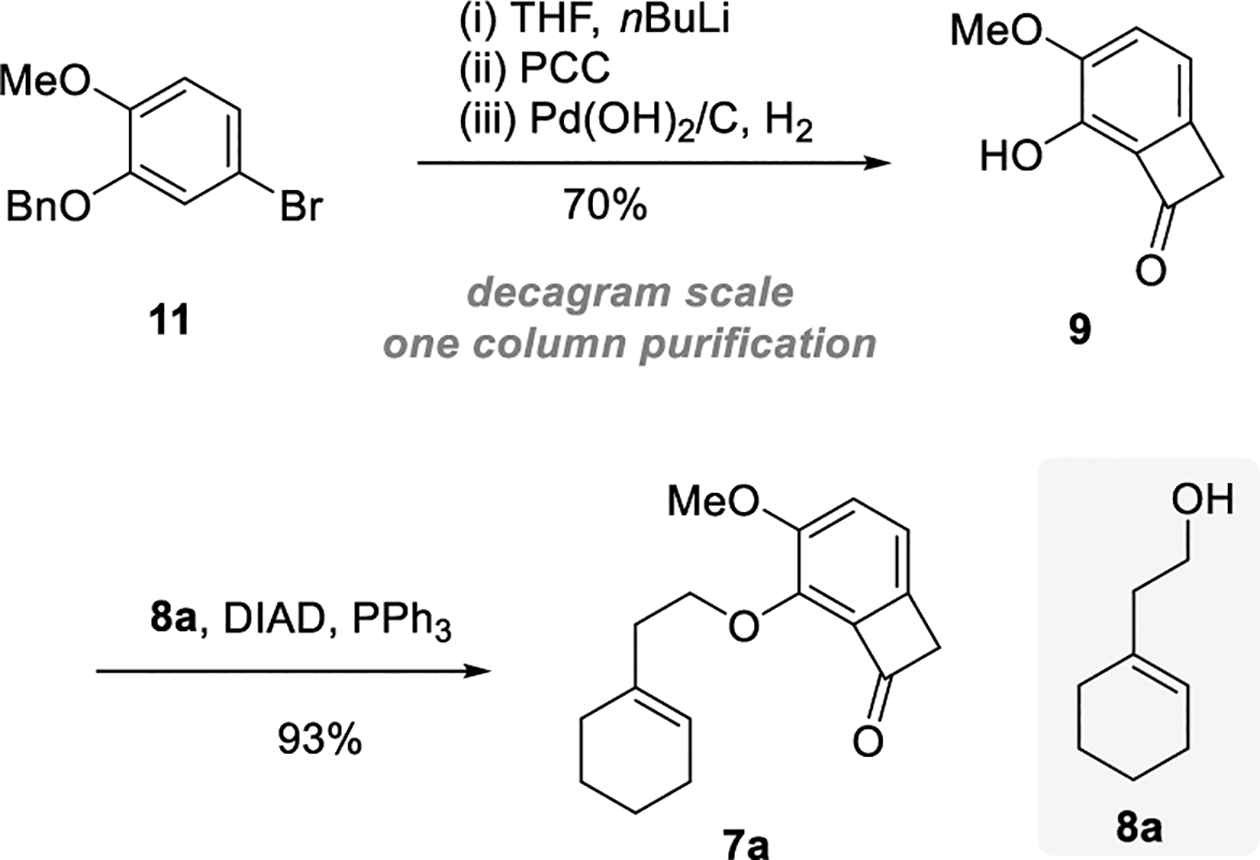
Preparation of Substrate 7a. PCC: pyridinium chlorochromate; Me: methyl; DIAD: diisopropyl azodicarboxylate.
With model substrate 7a in hand, the previously reported conditions were found to be ineffective for the desired “cut-and-sew” reaction (entries 1 and 2, Table 1).[11e] Interestingly, when using the neutral Rh/dppb/ZnCl2 system, changing the solvent from THF to 1,4-dioxane afforded the desired tetracyclic product (6a) in 37% yield (entry 3), though the exact reason is still unclear. After surveying different rhodium pre-catalysts (entries 4–8), [Rh(COD)2]BF4 proved to be optimal, giving 72% isolated yield (entry 6). With the cationic catalyst, the addition of ZnCl2 was found unnecessary (entry 9). In addition, up to 89% yield was obtained using a precoordinated [Rh(COD)dppb]BF4 catalyst (entry 10). Finally, promising enantioselectivity was obtained when switching the ligand from dppb to chiral ligand (R)-(−)DTBM-segphos (L1) (entry 11), giving 6a in 83% yield and 88.5: 11.5 er with only 5 mol% Rh loading (entry 12).
Table 1.
Selected Optimization of the C−C Activation Reaction with Substrate 7a.[a]
 | |||
|---|---|---|---|
| Entry | Catalytic conditions | 7a Conv. [%] |
6a Yield [%][b] |
| 1 | [Rh(CO)2Cl]2 (5 mol%), P(C6F5)3 (40 mol%), THF (0.04 M), 140°C, 5 days; | 50 | n.d. |
| 2[c] | [Rh(COD)Cl]2 (5 mol%), dppb (12 mol%), ZnCl2 (20 mol%), THF (0.04 M), 130°C, 12h | 10 | 5 |
| 3[c] | [Rh(COD)Cl]2 (5 mol%), dppb (12 mol%), ZnCl2 (20 mol%), 1,4-dioxane (0.04 M), 130°C, 12h | 57 | 37 |
| 4 | [Rh(COD)Cl]2 (5 mol%), dppb (12 mol%), ZnCl2 (20 mol%), 1,4-dioxane (0.1 M), 140°C, 24h | 10 | 7 |
| 5[d] | [Rh(C2H4)2Cl]2 (5 mol%), dppb (12 mol%), ZnCl2 (20 mol%), 1,4-dioxane (0.1 M), 140°C, 24h | 30 | 10 |
| 6 | [Rh(COD)2]BF4 (10 mol%), dppb (12 mol%), ZnCl2 (20 mol%), 1,4-dioxane (0.1 M), 140°C, 24h | 93 | 72 |
| 7 | [Rh(NBD)2]BF4 (10 mol%), dppb (12 mol%), ZnCl2 (20 mol%), 1,4-dioxane (0.1 M), 140°C, 24h | 10 | 5 |
| 8 | [Rh(COD)MeCN]BF4 (10 mol%), dppb (12 mol%), ZnCl2 (20 mol%), 1,4-dioxane (0.1 M), 140°C, 24h | 55 | 45 |
| 9 | [Rh(COD)2]BF4 (10 mol%), dppb (12 mol%), 1,4-dioxane (0.1 M), 140°C, 24h | 100 | 73 |
| 10 | [Rh(COD)dppb]BF4(10 mol%), 1,4-dioxane (0.1 M), 140°C, 24h | 100 | 89 |
| 11 | [Rh(COD)2]BF4 (10 mol%), L1 (12 mol%), 1,4-dioxane (0.1 M), 140°C, 24h | 100 | 60 (er 89:11) |
| 12 | [Rh(COD)2]BF4(5 mol%), L1(6 mol%), 1,4-dioxane (0.1 M), 140°C, 24h | 100 | 83 (er 88.5:11.5) |
All reactions were run on a 0.1 mmol scale.
Yields are isolated yields; numbers in parentheses are enantiomeric ratio (er) values determined by chiral HPLC.
A second portion of the same catalysts was added after 12 h.
Conv. = conversion; n.d.= not detected; COD: 1,5-cyclooctadiene; dppb: 1,4-bis(diphenylphosphino)butane; L1: (R)-(−)-DTBM-segphos.
Encouraged by the model study, the ketal-containing substrate (7) was first prepared in 93% yield through a Mitsunobu coupling between phenol 9 and alcohol 8 that was easily accessed through Birch reduction and ketal formation from anisole 10 (Scheme 5). Surprisingly, subjecting substrate 7 to the above optimized racemic conditions (entries 1 and 2, Table 2) gave only trace desired product with most of 7 decomposed, suggesting the sensitivity of the ketal moiety. To our delight, under the asymmetric conditions, the desired tetracycle 6 was obtained in 59% yield with 91.5:8.5 er (entry 3). It is likely that the enhanced reaction efficiency with the chiral ligand outcompeted the decomposition pathway.[11a] Reducing the reaction temperature to 130°C further enhanced the yield and enantioselectivity (83% yield, 93:7 er) (entry 4). After examining various chiral bidentate phosphine ligands, solvents, and counter ions (entries 5–13), product 6 was ultimately isolated in 80% yield with 97:3 er by switching [Rh(COD)2]BF4 to [Rh(COD)2]NTf2 as the catalyst and changing 1,4-dioxane to 1,2-difluorobenzene (DFB) as the solvent (entry 13). Notably, the asymmetric “cut-and-sew” reaction can be run on a two-gram scale with a reduced catalyst loading without loss of enantioselectivity (entry 14). The structure and relative configuration of compound 6 were confirmed by X-Ray crystallography;[14] its absolute stereochemistry was later confirmed by X-Ray crystallography of its derivative 16 (vide infra, Scheme 6).[14]
Scheme 5.

Preparation of Substrate 7. PPTS: pyridinium p-toluenesulfonate; glycol: ethylene glycol; DEAD: diethyl azodicarboxylate.
Table 2.
Optimization of the C−C Activation of Substance 7.[a]
 | ||||
|---|---|---|---|---|
| Entry | Catalytic conditions | 7a Conv. [%] |
6 Yield [%][b |
er[b] |
| 1 | [Rh(COD)dppb]BF4 (10 mol%), 1,4-dioxane (0.1 M), 140°C | 95 | n.d. | -- |
| 2 | [Rh(COD)2]BF4 (10 mol%), dppb (12 mol%), 1,4-dioxane (0.1 M), 140°C | 95 | 5 | -- |
| 3 | [Rh(COD)2]BF4 (5 mol%), L1 (6 mol%), 1,4-dioxane (0.1 M), 140°C | 100 | 59 | 91.5:8.5 |
| 4 | [Rh(COD)2]BF4 (5 mol%), L1 (6 mol%), 1,4-dioxane (0.1 M), 130°C | 100 | 83 | 93:7 |
| 5 | [Rh(COD)2]BF4 (5 mol%), L2 (6 mol%), 1,4-dioxane (0.1 M), 130°C | 90 | 60 | 37.5:62.5 |
| 6 | [Rh(COD)2]BF4 (5 mol%), L3 (6 mol%), 1,4-dioxane (0.1 M), 130°C | 97 | 41 | 64:36 |
| 7 | [Rh(COD)2]BF4 (5 mol%), L4 (6 mol%), 1,4-dioxane (0.1 M), 130°C | 30 | n.d. | -- |
| 8 | [Rh(COD)2]BF4 (5 mol%), L5 (6 mol%), 1,4-dioxane (0.1 M), 130°C | 95 | 28 | 72:28 |
| 9 | [Rh(COD)2]PF6 (5 mol%), L1 (6 mol%), 1,4-dioxane (0.1 M), 130°C | 53 | 6 | -- |
| 10 | [Rh(COD)2]BF4 (5 mol%), L1 (6 mol%), 1,3-DFB (0.1 M), 130°C | 100 | 60 | 94.5:5.5 |
| 11 | [Rh(COD)2]BF4 (5 mol%), L1 (6 mol%), toluene (0.1 M), 130°C | 100 | 52 | 85.5:14.5 |
| 12 | [Rh(COD)2]BF4 (5 mol%), L1 (6 mol%), 1,2-DFB (0.1 M), 130°C | 100 | 55 | 96.5:3.5 |
| 13[c, d] | [Rh(COD)2]NTf2(5 mol%), L1(6 mol%), 1,2-DFB (0.1 M), 130°C | 100 | 80 | 97:3 |
| 14[c, d] | [Rh(COD)2]NTf2(4 mol%), L1(4.8 mol%), 1,2-DFB (0.4 M), 130°C (two-gram scale) | 100 | 76 | 97:3 |

All reactions were run on a 0.1 mmol scale for 24 h.
Yields are isolated yields; the enantiomeric ratio (er) was determined by chiral HPLC.
The reaction was run for 48 h.
The reaction was run on a two-gram scale. Conv.: conversion; Tf: triflate; n.d.: not detected; DFB: difluorobenzene.
Scheme 6.

The First-generation Route to access (−)-Thebainone A (4). TMS: trimethylsilyl; OTf: trifluoromethanesulfonate; TFA: trifluoroacetic acid; DCM: dichloromethane; DMSO: dimethyl sulfoxide.
Scope of the enantioselective “cut-and-sew” reaction:
The high efficiency and selectivity enabled by the new catalytic conditions motivated us to examine the scope of the enantioselective “cut-and-sew” transformation with different cyclic trisubstiuted alkenes (Table 3). First, as expected, the model susbtrate (7a) afforded even higher yield and higher enantioselectivity under the optimal conditions (entry 1). Cyclic olefins with different ring sizes all provided the desired tetracyclic products in good to excellent yields. The 5- and 6-membered ring substrates (7a-c) generally offered excellent enantioselectivity, while the 7- and 8-membered ring substrates (7e-f) exhibited reduced enantioselectivity. In addition, the benzofused olefins (7b and 7d) gave somewhat lower yields and er than their non-conjugated counterpartners. Gratifyingly, excellent enantioselectivity was observed with these containing heterocyclic alkenes (7g-h). The more sterically demanding β-dihydronaphthyl substrate (7i) still proved to be competent, albeit in lower yield and er under the current conditions. In the absence of the OMe substituent on the arene, the 6- and 5-membered ring substrates (7j-k) gave slightly lower yield but still excellent enantioselectivity. Besides using an oxygen linker, the nitrogen-tethered substrates (7l-m) worked even better, which afforded good yield and excellent enantioselectivity (up to 99.5: 0.5 e.r.) likely benefited from the rigidity of the linkers. Finally, the new catalytic conditions are also suitable for the one-methylene-short substrate (7n), giving good yield but poor er. The exact reason for the significantly better enantioselectivity with the 6-membered-ring-forming substrates remains unclear for this specific catalytic system.
Table 3.
Substrate Scope of the Enantioselective Carboacylation with Cyclic Trisubstituted Olefins.[a]

|
The reaction was carried out on a 0.1 mmol scale of substrate with [Rh(COD)2]NTf2 (5 mol %), (R)-DTBM-segphos (6 mol%) in 1,2-DFB (1 mL) at 130°C for 48 h.
All yields are isolated yields; the enantiomeric ratio (er) value was determined by chiral HPLC.
The reaction was carried out on a 1 mmol scale of substrate with [Rh(COD)2]NTf2 (4 mol %), (R)-DTBM-segphos (4.8 mol%) in 1,2-DFB (0.5 mL) at 130°C for 48 h. Ts: p-toluenesulfonyl; Ac: acetate.
Total synthesis of (−)-thebainone A:
With a scalable asymmetric route to access key intermediate 6, the stage was set for the next deconstructive transformation through cleavage of the C−O bond and installation of the nitrogen moiety (Scheme 6). First, reduction of the ketone in compound 6 proceeded stereoselectivity with LiAlH4; the subsequent acetate protection followed by in situ ketal removal afforded ketone 12[14] in 94% yield over two steps on gram scales. There are substantial challenges to cleave the C−O bond of the dihydropyran moiety in intermediate 12: (1) the ether bond lacks sufficient ring strain for opening; (2) the presence of other oxygen functional groups, including ketones and esters, in the substrate can inhibit Lewis acid coordination; and (3) due to the rigid molecular scaffold, the C−O bond reformation was found to be facile, which became a significant difficulty for handling the ring-opened product. Ultimately, we found that treatment of 12 with BBr3 (5 equiv) in DCM at −30°C for 72 h delivered the diphenol intermediate (13) in good yield. For comparison, running the reaction at a higher temperature led to notable decomposition. Protection of the free phenol moieties proved to be necessary; otherwise, any basic conditions (tested so far) resulted in reforming the dihydropyran C−O bond. After systematic evaluation of various reaction conditions, the use of CH2N2 in methanol and ether provided an extremely mild methylation protocol, giving the desired dimethoxy product in 50% yield on a 79 mg scale (see Table S1 for details). On a gram scale, the yield of 14 dropped to 28% plus 32% of the partially protected product (13a) and 28% of the prior substrate (12).[14] However, 13a can be further transformed to 14 in 68% yield, along with forming 18% of 12 that can be cycled, combined, and reused. With a sufficient amount of 14 in hand, the subsequent ketone protection and amination via a SN2 reaction, followed by removal of the Ac protecting group, occurred smoothly in one-pot to provide 15 with 76% yield. Upon elimination of the alcohol moiety with Martin’s sulfurane, a formal hydroamination process took place using sodium naphthalenide.[15] Under this condition, the Ts-N bond was first cleaved to generate a nitrogen-centered radical that then adds to the olefin; the resulting carbon-centered radical was further reduced to a carbanion that be protonated during the acidic workup. This step completes the construction of the piperidine ring and reveals the ketone moiety in the C ring.
In the end game, the more sterically hindered methyl ether can be selectively cleaved under the nucleophilic condition, i.e. using NaSEt, in 87% yield on a 120 mg scale.[16] Such a high selectivity is likely due to a more available σ* orbital of the middle ether moiety, as the bulkiness of the structure disfavors a “flat” conformation (Figure 2). Notably, the mono-deprotected product 5 is a known precursor for the total syntheses of codeine (2) and morphine (1).[17] Finally, (−)-thebainone A (4) was prepared through the α,β-desaturation of ketone 5 using Stahl’s Pd(TFA)2/DMSO system (see Table S2 for details).[18, 5c] The spectroscopic data of synthetic (−)-thebainone A (4) match those reported previously.[6a] Thus, using this route, the asymmetric total synthesis of (−)-thebainone A was accomplished from compound 6 in 9 steps and 13% overall yield.
Figure 2.
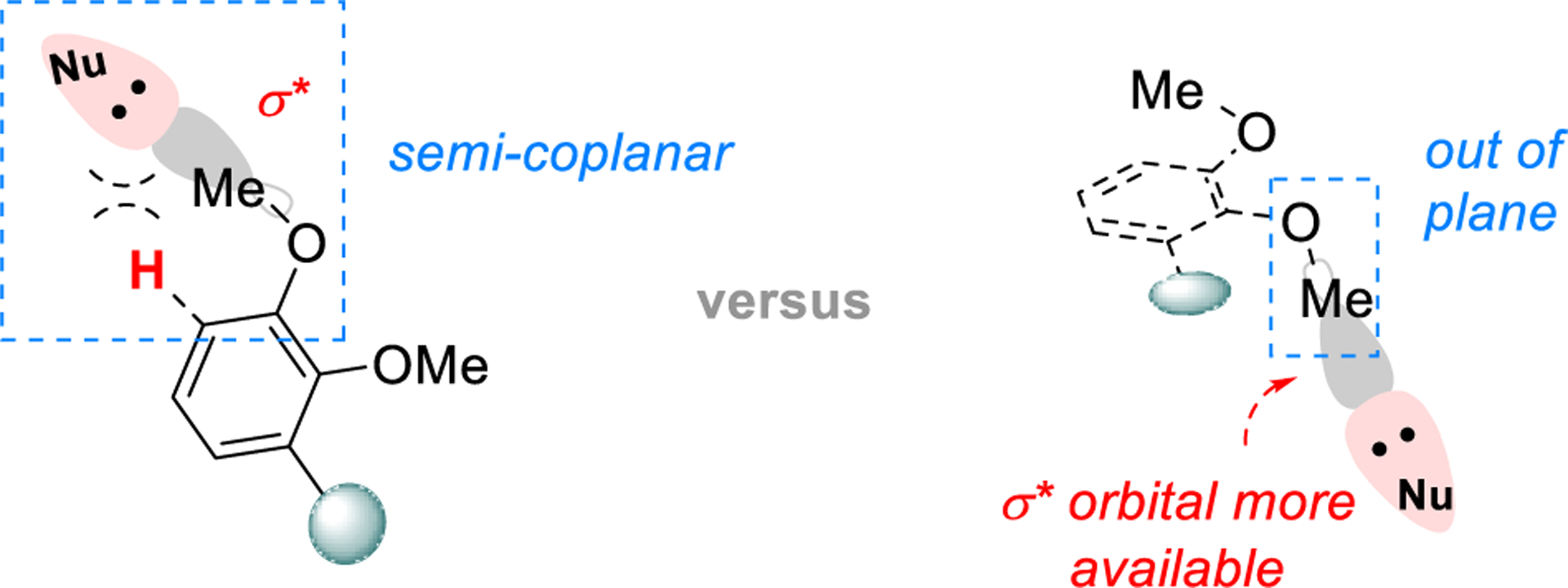
Selective Demethylation of Vicinal Ethers.
To avoid the acetate protection of the secondary C9 alcohol, an alternative approach to construct intermediate 16 has been developed (Scheme 7). From the “cut-and-sew” product 6, ketone reduction followed by dehydration provided alkene 18. The subsequent cleavage of the C−O bond with BBr3 still delivered the desired diphenol intermediate, which was then subjected to the doubly methylation conditions with CH2N2 to deliver 19 in moderate yield. Finally, a one-pot sequence of ketal installation and SN2 amination smoothly delivered sulfonamide 16 (the same one present in the first-generation route) in good yield. Overall, the second-generation route saves one step in the longest linear sequence of the synthesis.
Scheme 7.

The Second-generation Route: Avoiding the Ac Protection.
Conclusion
In summary, the first enantioselective total synthesis of (−)-thebainone A (4) has been achieved in 13 (and 14) steps with 4.7% (and 7.2%) overall yield from commercially available starting material 11. The efficiency of this synthetic strategy is primarily highlighted by an asymmetric Rh-catalyzed ‘cut-and-sew’ transformation to access the all-carbon fused-rings structure and the quaternary stereocenter. Notably, an improved catalytic system has been identified for the asymmetric ‘cut-and-sew’ transformation with sterically hindered olefins, which allows for a broad substrate scopef with high enantioselectivity (up to 99.5:0.5 er). The deconstructive approach illustrated in this synthesis, i.e. through cleaving C−C and C−O bonds in readily availble substrates to build more complex structures, could have broader implications beyond this work.
Supplementary Material
Acknowledgements
NIGMS (2R01GM109054) and University of Chicago are acknowledged for research support. S.-H. H. acknowledges the International Postdoctoral Exchange Fellowship Program 2017 from the Office of China Postdoctoral Council. We thank Mr. Shusuke Ochi and Dr. Ki-Young Yoon for X-ray crystallography. Umicore AG & Co. KG and Chiral Technology are acknowledged for their generous donation of Rh salts and chiral HPLC columns, respectively.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Blakemore PR, White JD, Chem. Commun 2002, 38, 1159–1168. [DOI] [PubMed] [Google Scholar]
- [2].a) Manmade A, Dalzell HC, Howes JF and Razdan RK, J. Med. Chem, 1981, 24, 1437–1440; [DOI] [PubMed] [Google Scholar]; b) Falk E Muench. Med. Wochenschr, 1917, 20, 38; [Google Scholar]; c) Blumberg H, Pachter IJ and Matossian Z, US Pat, 1967, vol. 3, pp. 332–950. [Google Scholar]
- [3].a) Gates M, Tschudi G, Am GJ. Chem. Soc 1952, 74, 1109–1110; [Google Scholar]; b) Gates M, Tschudi G, J. Am. Chem. Soc 1956, 78, 1380–1393. [Google Scholar]
- [4].For selected reviews on total synthesis of morphine alkaloids, see:; a) Chambers SA, DeSousa JM, Huseman ED, Townsend SD, ACS Chem. Neurosci 2018, 9, 2307–2330; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Haider S, Chittiboyina AG, Khan IA, Curr. Org. Chem 2018, 22, 148–164; [Google Scholar]; c) Li Q, Zhang H, Chin. J. Org. Chem 2017, 37, 1629–1652; [Google Scholar]; d) Reed JW, Hudlicky T, Acc. Chem. Res 2015, 48, 674–687; [DOI] [PubMed] [Google Scholar]; e) Rinner U, Hudlicky T, Top. Curr. Chem 2012, 309, 33–66; [DOI] [PubMed] [Google Scholar]; f) Chida N, Top. Curr. Chem 2011, 299, 1–28; [DOI] [PubMed] [Google Scholar]; g) Zezula J, Hudlicky T, Synlett 2005, 388–405; [Google Scholar]; h) Novak BH, Hudlicky T, Reed JW, Mulzer J, Trauner D, Curr. Org. Chem 2000, 4, 343–362. [Google Scholar]
- [5].For recent selected work on asymmetric total synthesis of morphine alkaloids, see:; a) Makarova M, Endoma-Arias MAA, Dela Paz HE, Simionescu R, Hudlicky T, J. Am. Chem. Soc 2019, 141, 10883–10904; [DOI] [PubMed] [Google Scholar]; b) Zhang Q, Zhang F-M, Zhang C-S, Liu S-Z, Tian J-M, Wang SH, Zhang X-M, Tu Y-Q, Nature Comm. 2019, 10, 2507; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sun DB, Pedersen B, Ellman JA, Chem. Sci 2019, 10, 535–541; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lipp A, Selt M, Ferenc D, Schollmeyer D, Waldvogel SR, Opatz T, Org. Lett 2019, 21, 1828–1831; [DOI] [PubMed] [Google Scholar]; e) Lipp A, Ferenc D, Gutz C, Geffe M, Vierengel N, Schollmeyer D, Schafer HJ, Waldvogel SR, Opatz T, Angew. Chem. Int. Ed 2018, 57, 11055–11059; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2018,130, 11221–11225; [Google Scholar]; f) Park KH, Chen R, Chen DYK, Chem. Sci 2017, 8, 7031–7037; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Umihara H, Yokoshima S, Inoue M, Fukuyama T, Chem. Eur. J 2017, 23, 6993–6995. [DOI] [PubMed] [Google Scholar]; For asymmetric formal synthesis of morphine alkaloids, see:; h) Rautschek J, Jäger A, Metz P, Org. Lett 2018, 20, 832–835. [DOI] [PubMed] [Google Scholar]
- [6].For recent selected work on racemic synthesis of morphine alkaloids, see:; a) Wang YZ, Hennig A, Kuttler T, Hahn C, Jager A, Metz P, Org. Lett 2020, 22, 3145–3148; [DOI] [PubMed] [Google Scholar]; b) Brousseau J, Xolin A, Barriault L, Org. Lett 2019, 21, 1347–1349; [DOI] [PubMed] [Google Scholar]; c) Chu S, Munster N, Balan T, Smith MD, Angew. Chem. Int. Ed 2016, 55, 14306–14309; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2016,128, 14518–14521. [Google Scholar]
- [7].Tius MA, Kerr MA, J. Am. Chem. Soc 1992, 114, 5959–5966. [Google Scholar]
- [8].For selected books and reviews on catalytic C-C bond activation, see:; a) Murakami M, Ito Y, Cleavage of Carbon–Carbon Single Bonds by Transition Metals. Top. Organomet. Chem 1999; 3, 97–130; [Google Scholar]; b) Dong G, C−C Bond Activation. Topics in Current Chemistry, (Ed.: Dong G), Springer: New York, 2014; Vol. 346; [DOI] [PubMed] [Google Scholar]; c) Murakami M, Cleavage of carbon-carbon single bonds by transition metals, (Ed.: Murakami M), Wiley-VCH Verlag GmbH & Co. KGaA: New York, 2015; [Google Scholar]; d) van der Boom ME, Milstein D, Chem. Rev 2003, 103, 1759–1792; [DOI] [PubMed] [Google Scholar]; e) Seiser T, Saget T, Tran DN, Cramer N, Angew. Chem. Int. Ed 2011, 50, 7740–7752; [DOI] [PubMed] [Google Scholar]; Angew. Chem 123, 2011, 7884–7896; [Google Scholar]; f) Ruhland K, Eur. J. Org. Chem 2012, 2012, 2683–2706; [Google Scholar]; g) Chen F, Wang T, Jiao N, Chem. Rev 2014, 114, 8613–8661; [DOI] [PubMed] [Google Scholar]; h) Souillart L, Cramer N, Chem. Rev 2015, 115, 9410–9464; [DOI] [PubMed] [Google Scholar]; i) Kim D-S, Park W-J, Jun C-H, Chem. Rev 2017, 117, 8977–9015; [DOI] [PubMed] [Google Scholar]; j) Murakami M, Ishida N, Chem. Rev 2021, 121, 264–299. [DOI] [PubMed] [Google Scholar]
- [9].For a review on C−O cleavage of alkyl ethers, see:; Ranu BC, Bhar S, Dealkylation of ethers. a review. Org. Prep. Proc. Int 1996, 28, 371–409. [Google Scholar]
- [10].a) Wang B, Perea MA, Sarpong R, Angew. Chem. Int. Ed 2020, 59, 18898–18919; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2020,132, 19058–19080; [Google Scholar]; b) Kerschgens I, Rovira AR, Sarpong R, J. Am. Chem. Soc 2018, 140, 9810–9813; [DOI] [PubMed] [Google Scholar]; c) Leger PR, Kuroda Y, Chang S, Jurczyk J, Sarpong R, J. Am. Chem. Soc 2020, 142, 15536–15547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Deng L, Fu Y, Lee SY, Wang C, Liu P, Dong G, J. Am. Chem. Soc 2019, 141, 16260–16265; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xia Y, Ochi S, Dong G, J. Am. Chem. Soc 2019, 141, 13038–13042; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Deng L, Chen M, Dong G, J. Am. Chem. Soc 2018, 140, 9652–9658; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Xu T, Dong G, Angew. Chem. Int. Ed 2014, 53, 10733–10736; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2014,126, 10909–10912; [Google Scholar]; e) Xu T, Dong G, Angew. Chem. Int. Ed 2012, 51, 7567–7571; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2012, 124, 7685–7689; [Google Scholar]; f) Xu T, Ko HM, Savage NA, Dong G, J. Am. Chem. Soc 2012, 134, 20005–20008. [DOI] [PubMed] [Google Scholar]
- [12].a) Zhang Y, Shen S, fang H, Xu T, Org. Lett 2020, 22, 1244–1248; [DOI] [PubMed] [Google Scholar]; b) Qin Y, Zhan J-L, Shan T-T, Xu T, Tetrahedron Lett. 2019, 60, 925–927; [Google Scholar]; c) Sun T, Zhang Y, Qiu B, Wang Y, Qin Y, Dong G, Xu T, Angew. Chem. Int. Ed 2018, 57, 2859–2863; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2018, 130, 2909–2913; [Google Scholar]; d) Qiu B, Li X-T, Zhang J-Y, Zhan J-L, Huang S-P, Xu T, Org. Lett 2018, 20, 7689–7693. [DOI] [PubMed] [Google Scholar]
- [13] <j/>(a).Chen P, Savage NA, Dong G, Tetrahedron 2014, 70, 4135–4146; [DOI] [PMC free article] [PubMed] [Google Scholar]; For an efficient synthesis of alcohol 8a, see:; b) Green SA, Vásquez-Céspedes S, Shenvi RA, J. Am. Chem. Soc 2018, 140, 11317–11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14]. Deposition Numbers 2003653 (6), 2003652 (12), and 2067511 (16) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallo-graphic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures. For more details, see Supporting Information.
- [15].For the cyclization under the Birch reductive conditions, see:; a) Parker KA, Fokas D, J. Am. Chem. Soc 1992, 114, 9688–9689; [Google Scholar]; for using a similar substrate under the same conditions, see:; b) Yamada O, Ogasawara K, Org. Lett 2000, 2, 2785–2788. [DOI] [PubMed] [Google Scholar]
- [16].Moos WH, Gless RD, Rapoport H, J. Org. Chem 1983, 48, 227–238. [Google Scholar]
- [17].a) Weller DD, Rapoport H, J. Med. Chem 1976, 19, 1171–1175; [DOI] [PubMed] [Google Scholar]; b) Rice KC, J. Org. Chem 1980,45, 3137–3139; [Google Scholar]; c) Rice KC, J. Med. Chem 1977, 20, 164–165. [DOI] [PubMed] [Google Scholar]
- [18].a) Diao T, Wadzinski TJ, Stahl SS Chem. Sci, 2012, 3, 887–891; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Diao T, Stahl SS, J. Am. Chem. Soc 2011, 133, 14566–14569. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.