

Abstract
Objective:
Humans and mice with loss-of-function variants of genes in the transforming growth factor beta (TGF-β) signaling pathway develop aortic aneurysms. These aneurysms could be caused by decreased aortic smooth muscle cell (SMC) contractile-protein levels and impaired aortic SMC contractile-unit function. Accordingly, we investigated whether loss of SMC TGF-β signaling in mice alters aortic contractile-protein levels and aortic contractility.
Approach and Results:
We used immunoblotting, wire myography, histologic analyses, and measurements of aortic nitric oxide and superoxide levels to assess aortic contractile-protein levels and vasomotor function in mice with SMC-specific deletion of the type 2 TGF-β receptor (TBR2SMΔ mice). Aortic contractile-protein levels were not altered in TBR2SMΔ mice. Surprisingly, TBR2SMΔ mice had increased aortic contractility and severe endothelial dysfunction. Endothelial dysfunction was manifested as decreased relaxation to acetylcholine (Emax 37% versus 97%; P<0.0001), decreased aortic nitric oxide (50%; P=0.005), decreased endothelial nitric oxide synthase activation (31%; P=0.002), and lower aortic levels of phosphorylated vasodilator-stimulated phosphoprotein (an indicator of nitric oxide bioavailability: 65%; P<0.0001). Aortic hypercontractility was reduced by mechanical denudation of endothelium and was eliminated by pretreatment of TBR2SMΔ and control aortas with a nitric oxide synthase inhibitor, revealing a significant positive interaction between aortic hypercontractility and absence of endothelium-derived nitric oxide (P<0.05 for both denudation and nitric oxide inhibition).
Conclusions:
Aortic aneurysms that develop in TBR2SMΔ mice are not caused by decreased SMC contractility. Loss of physiologic SMC TGF-β signaling causes endothelial dysfunction leading to aortic hypercontractility. Endothelial dysfunction may contribute to vascular pathologies associated with abnormal TGF-β signaling.
Keywords: endothelial dysfunction, myography, transforming growth factor beta, vasomotor function
Subject Codes: Aneurysm, Endothelium/Vascular Type/Nitric Oxide, Genetically Altered and Transgenic Models, Growth Factors/Cytokines, Vascular Biology
Loss of transforming growth factor beta (TGF-β) signaling is associated with aortic aneurysms in humans and in mice.1–6 However, the mechanisms through which reduced TGF-β signaling causes aneurysms are poorly understood. Because TGF-β signaling in cultured smooth muscle cells (SMC) increases expression of contractile proteins,7–12 loss of SMC TGF-β signaling is expected to decrease levels of SMC contractile proteins and impair vascular contractility. Decreased functionality of the SMC contractile unit and associated impairment of vascular contractility are widely considered to be critical precursors to aortic aneurysm formation.13–17 Accordingly, it seems intuitive that genetic variants (in humans) and experimental manipulations (in mice) that decrease SMC TGF-β signaling would decrease aortic SMC contractile protein abundance, lessen aortic contractility, and lead to aneurysm formation. However, despite generation of several mouse models of impaired SMC TGF-β signaling and aortic aneurysms,2–4, 6 the combination of impaired SMC TGF-β signaling, reduced SMC contractile protein levels, and decreased aortic contractility has not been reported in any of these mouse models.
We and others have generated mice with postnatal SMC-specific deletion of the type 1 or type 2 TGF-β receptors (Tgfbr1 or Tgfbr2).2–4 All of these mice have defective SMC TGF-β signaling and develop aortic aneurysms. However, SMC contractile protein abundance and aortic contractility were not fully investigated in any of these studies. Here we report experiments that test whether young mice with homozygous deletion of Tgfbr2 in SMC (hereafter termed TBR2SMΔ mice) have decreased SMC contractile protein abundance and impaired aortic contractility. Surprisingly, aortas of TBR2SMΔ mice have unaltered levels of SMC contractile proteins and are hypercontractile to both potassium chloride (KCl) and phenylephrine (PE). The increased contractility of TBR2SMΔ aortas is due to endothelial dysfunction, manifested as decreased nitric oxide NO bioavailability and severely impaired endothelium-dependent relaxation.
Materials and Methods
The data that support the findings of this study are available from the corresponding author on reasonable request.
Animals
All mice in this study were homozygous for a floxed Tgfbr2 allele (exon 4;Tgfbr2f/f).18 We generated male mice with deleted Tgfbr2 alleles in SMC (TBR2SMΔ) by mating male Tgfbr2f/f mice carrying a Y chromosome-integrated Myh11-CreERT2 allele19 with female Myh11-CreERT2 0/0 Tgfbr2f/f mice. Because the Myh11-CreERT2 allele is integrated in the Y chromosome, only female—not male—littermate controls (Myh11-CreERT2 0/0 Tgfbr2f/f) are generated by these matings. In a small number of initial experiments (wire myography studies performed 3 weeks after tamoxifen injection, including ex vivo testing of pharmacologic inhibitors), we used these littermate females as controls. In the remaining experiments—to avoid leaving sex as an uncontrolled variable—we used non-littermate male Myh11-CreERT2 0/0 Tgfbr2f/f mice as controls. The male Myh11-CreERT2 0/0 Tgfbr2f/f controls were generated in our facility by mating male and female Myh11-CreERT2 0/0 Tgfbr2f/f mice.
We consider female and male Myh11-CreERT2 0/0 Tgfbr2f/f mice (hereafter termed TBR2f/f) to be equivalent controls because vasomotor properties of male and female TBR2f/f isolated aortic rings—measured 3 weeks after tamoxifen injections—are nearly identical (Figure I and Tables I and II in the Data Supplement). The small male-female difference in ACh sensitivity (–logEC50) may be a chance finding, and it is far less than the differences in −logEC50 for ACh between TBR2SMΔ and TBR2f/f aortas at all time points (Table II in the Data Supplement). The validity of tamoxifen-treated non-littermate male Myh11-CreERT2 0/0 Tgfbr2f/f as controls is further supported by an independent set of experiments in which we bred the autosomal Acta2-CreERT2 allele into the Tgfbr2f/f background. Compared to male tamoxifen-injected Acta2-CreERT2 0/0 Tgfbr2f/f littermates, male tamoxifen-injected Acta2-CreERT2 +/0 Tgfbr2f/f mice have the same hypercontractile aortic phenotype as tamoxifen-injected Myh11-CreERT2 +/0 Tgfbr2f/f mice (unpublished data).
For all experimental mice, genotypes were confirmed using allele-specific primers (Table III in the Data Supplement).3 We reported previously that the Myh11-CreERT2 allele reliably recombines the Tgfbr2flox allele after tamoxifen administration, resulting in near-complete loss of both TGFBR2 and phosphorylated SMAD2 protein in aortic medial SMC as well as loss of SMC responsiveness to TGF-β1 ligand ex vivo.3, 20 All lines were extensively backcrossed (>10 generations) into the C57BL/6J background. Mice were maintained in a specific-pathogen-free facility and fed a normal laboratory diet (PicoLab Rodent Diet 20; LabDiet.com). Experimental and control mice were enrolled at 6 weeks of age, at which time they were injected intraperitoneally with 1 mg tamoxifen for 5 days. All animal protocols were approved by the University of Washington Office of Animal Welfare.
Harvests of Aortas for Myography and Protein Extraction
Mice were anesthetized with intraperitoneal ketamine (100 mg/kg) and xylazine (10 mg/kg) and exsanguinated by cold-saline perfusion via the left ventricle. With aid of a dissecting microscope, a segment of ascending aorta extending from the aortic root to the brachiocephalic artery was dissected free and explanted. The remainder of the aorta, extending to the bifurcation, was explanted and stripped of adventitial fat by dissection. The proximal aortic segment was used immediately for myography and the distal segment was snap-frozen in liquid nitrogen for later protein extraction.
Wire Myography
Isolated aortic rings were placed in Krebs-Ringer bicarbonate solution of the following composition (mM): sodium chloride 130, potassium chloride 4.7, magnesium sulfate heptahydrate 1.2, potassium phosphate 1.2, calcium chloride 3.3, sodium bicarbonate 15, EDTA 0.03, and dextrose 6). This solution (prepared within 48 hours of each myography experiment) was used both to immerse aortic tissues during dissection and for myography. Ascending aortas were cleaned of fat and other adherent tissue, cut into rings (~2.5 mm-long), and mounted in a multichamber myograph system (Model 610M, Danish Myo Technology). Two stainless-steel wires (40-μm diameter) were introduced into the lumen of the aortic rings and then fixed to the mounting devices of a force transducer and a micrometer. The aortas were equilibrated with a baseline tension of 20 milliNewtons (mN) for 1 hour in Krebs-Ringer bicarbonate solution at 37 °C and gassed with 95% O2 and 5% CO2 (pH 7.4; Praxair BI OXCD5ZC-K). After the equilibration period, in almost all cases the rings were contracted twice with KCl (40 mM; Fisher Scientific #P217) at a 20-minute interval, to confirm viability and provide standardization of the rings. For 4 of 8 Myh11-CreERT2 0/0 Tgfbr2f/f rings and 6 of 10 Myh11-CreERT2 +/0 Tgfbr2f/f rings removed 1 week after Tgfbr2 deletion, these contractions were performed with phenylephrine (PE) instead of KCl. For this reason, normalization to KCl in rings removed at 1 week could be done for only 4 aortas in each group. The 10 PE-contracted rings responded to subsequent vasomotor stimuli identically to the 8 KCl-contracted rings, so the subsequent myography data from both groups were pooled. Thereafter, we determined concentration-response curves for PE (10–9–10–4 M; Sigma Aldrich #P6126). Endothelium-dependent and smooth muscle cell (SMC) relaxation functions of the rings were then measured by addition of acetylcholine (ACh; 10–9–10–5 M; Sigma #A6625), or the nitrovasodilator sodium nitroprusside (SNP; 10–9–10–4 M or 10–10–10–5 M; Sigma #71778), respectively. To identify the underlying mechanisms mediating altered vascular reactivity, rings were incubated for 30 min with either the endothelin-1 receptor antagonist bosentan (10−5 M; Cayman Chemical #11731), the thromboxane A2 receptor antagonist SQ-25948 (10−5 M; Cayman #19025), the nitric oxide synthase inhibitor N(ω)-nitro-L-arginine methyl ester (L-NAME; 10−4 M; Cayman #80210) or 150 U/mL superoxide dismutase (Sigma #S5395) + 200 U/mL catalase (Sigma #C1345). PE concentration-response curves or ACh-relaxation curves were then performed in the presence of these agents and with parallel vehicle-only controls.
We also performed myography on endothelium-denuded aortic rings. To remove the endothelium, the trimmed rings were first mounted on the myograph, using the two parallel steel wires. Once mounted, an additional segment of 40-μm diameter steel wire was threaded through the lumen of the ring and the ring was placed under tension to provide gentle traction. Using a dissecting microscope for better visualization, the steel wire segment was then manipulated to circumferentially scrape the ring luminal surface. The ability of an operator to reliably use this technique to remove luminal endothelium was confirmed by wire myography showing impaired (<25%) relaxation to ACh along with preservation of contraction to PE and relaxation to SNP. Denudation was further confirmed by absence of luminal CD31 immunostaining on multiple sections of denuded rings.
Western Blot Analysis
Most of the western blots were performed using extracts of whole aortic tissue distal to the innominate artery (Figure II in the Data Supplement). We selected this segment of aorta based on 4 considerations: 1) To maximize use of aortic tissue from each mouse; 2) To ensure that SMC contractile proteins and vasomotor function are measured in the same mice; 3) Because aneurysmal dilation and a consistent histopathology are present throughout the aortas of TBR2SMΔ mice,3 it is unlikely that exclusion of the ascending aorta would yield unrepresentative results; and 4) An important precedent study used immunoblots of whole aortic extracts of TBR2SMΔ and control mice to measure SMC contractile proteins.2
Snap-frozen aortas (brachiocephalic artery to bifurcation) were ground in liquid nitrogen using a mortar and pestle and resuspended in 100 μL of cOmplete™ Lysis-M buffer with protease (Roche Diagnostics) and phosphatase inhibitors (Thermo Fisher #78420). The suspension was vortexed, incubated on ice for 30 minutes, then centrifuged at 14,000 g for 10 minutes at 4 °C. The supernatant was collected, and protein concentration was measured with the Pierce BCA assay (Thermo #23227). For each western blot, equal quantities of protein from each sample (12–20 μg) were separated by SDS-PAGE and transferred to a PVDF membrane. The blots were probed with primary antibodies that detect: smooth muscle myosin heavy chain (MYH11; Santa Cruz #6956), smooth muscle alpha actin (ACTA2; Abcam #32575), phosphorylated myosin light chain (p-MLC; Abcam #2480), myosin light chain (MLC; Cell Signaling Technologies #3672), phosphorylated myosin phosphatase (p-MYPT; Cell Signaling Technologies #4563 and #5163), total endothelial nitric oxide synthase (eNOS; Cell Signaling Technologies #32027); eNOS phosphorylated on threonine 495 (p-eNOS Thr495; Cell Signaling Technologies #9574), eNOS phosphorylated on serine 1177 (p-eNOS Ser1177; Cell Signaling Technologies #9570), vasodilator-stimulated phosphoprotein phosphorylated on serine 239 (p-VASP Ser239; Cell Signaling Technologies #3114), caveolin 1 (CAV1; Cell Signaling Technologies #12506), or glyceraldehyde phosphate dehydrogenase (GAPDH; Santa Cruz #20357). All primary antibodies were produced in rabbits except for anti-GAPDH (produced in goats). We used HRP-conjugated goat anti-rabbit IgG to detect bound rabbit antibodies (initially Santa Cruz #2030, then Cell Signaling Technologies #7074 after #2030 was unavailable). To detect anti-GAPDH antibody, we used HRP-conjugated donkey anti-goat IgG (Santa Cruz #2033). Image J was used to quantify band densities. To normalize for variations in loading, band densities for all of the proteins except p-MLC and p-eNOS Ser1177 were divided by the signal for GAPDH in the same lane. Band densities for p-MLC and p-eNOS Ser1177 were divided by the density signals in the same lanes for total MLC and total eNOS, respectively.
We also performed western blot analysis of aortic endothelial lysates. Whole aortas were excised after cold saline perfusion, placed in a silicone-bottom dissection dish containing cold saline, divided axially, and pinned with the lumen up. The saline was removed, and the lumen gently scraped with a cotton swab soaked in RIPA Lysis Buffer System (Santa Cruz Biotechnology, #SC-24948) including protease and phosphatase inhibitors per the manufacturer’s instructions. The swab was then dipped repeatedly in an ice-cooled microcentrifuge tube containing 100 μL of RIPA Lysis Buffer. The aorta was transferred to an ice-cooled microcentrifuge tube containing 250 μL of RIPA Lysis Buffer. Both samples were incubated on ice with the lysis buffer for 20–30 minutes, with brief vortexing and centrifugation every 5 minutes followed by centrifugation (16,000 g at 4 °C for 15 minutes). The supernatant was collected, protein measured as described above, and stored at −20 °C.
Luminal EC lysate (5–6 μg) and of EC-denuded aortic lysate (10–11 μg) from C57BL/6J Tgfbr2WT/WT mice (n=3; not treated with tamoxifen) were separated by SDS-PAGE and blotted as described above. The membranes were incubated with primary antibodies including cadherin-5 (CDH5; Cell Signaling Technologies #2500), platelet-endothelial cell adhesion molecule 1 (PECAM1; R&D Systems #AF3628), smooth muscle actin (ACTA2; see above), vimentin (VIM; Cell Signaling Technology #5741), and beta actin (ACTB; Cell Signaling Technology #8457). Bound antibodies were detected with the secondary antibodies described above, which were visualized and measured with ECL Solution (Bio Rad, #170561) or GE ECL Select Western Blotting detection reagent, GE Healthcare, RPN2235), a BioRad ChemiDoc XRS+ imager, and ImageJ. For serial detection of antigens of different molecular weights, blots were probed sequentially without stripping. Before re-probing a blot to detect a similar-molecular-weight antigen, membranes were incubated with stripping buffer (20 mL; Thermo Scientific, #21059) for 30 minutes at 37 °C, re-blocked for 30 minutes, washed, incubated with the last-used secondary antibody, and re-imaged to confirm complete stripping.
Tissue Processing, Sectioning, and Staining
Previously published protocols3 for harvesting, processing, and staining tissues were followed with minor adjustments. Mice were exsanguinated by saline perfusion via the left ventricle. Aortas were then fixed in situ by perfusion with 10% formalin at physiological pressure. A segment of ascending aorta was harvested by dividing the aorta first at the aortic root and then just proximal to the brachiocephalic artery. The segment between these 2 landmarks was dissected free of periaortic fat and tissue, placed in 10% formalin overnight, then in 30% sucrose in PBS for 12 hours, then stored in 70% ethanol at 4 °C. The segment was then embedded in optimal cutting temperature compound (4583, Sakura Finetek USA, Torrance, CA) with the caudal end positioned towards the block face. Aortic segments with macroscopically visible intramural hematomas were excluded from analysis. For each aorta, we cut 8 serial 8-μm-thick sections beginning at each of five 114-μm steps, for a total of 40 sections per aorta, covering 520 μm from the first to the last section. For each stain, 5 sections per aorta (1 at each 114-μm step) were stained. Measurements from the 5 sections were used to calculate a single mean value for that mouse. Aortic sections were stained with hematoxylin and eosin (H&E), Prussian blue (to detect hemosiderin from past aortic bleeds), and antibodies to CD31 (R&D Systems #AF3628) or Mac2 (Cedarlane Laboratories #CL8942AP). For Prussian blue staining, an aortic specimen from a previous study,3 with positive Prussian blue staining, was used as a positive control. For both CD31 and Mac2 antibody staining, secondary-antibody-only stains were used as negative controls. Stained sections were photographed using a Leica DM4000B microscope and digital camera (model DFC295; Leica Microsystems). Planimetry [measurement of internal elastic lamina (IEL) and external elastic lamina (EEL) length, calculation of medial thickness and area] was performed on H&E-stained sections, using ImagePro Plus (Media Cybernetics). IEL and EEL lengths were measured by 2 blinded observers, and the mean of their measurements was used for data analysis. Inter-observer agreement was verified using Bland-Altman analysis.21 Medial thickness was calculated assuming circular geometry of the IEL and EEL. Medial area was calculated by subtracting the area bound by the IEL from the area bound by the EEL (both areas were measured with ImagePro software). Medial elastin damage was quantified by counting elastin breaks on H&E-stained sections imaged after illumination with a fluorescein filter, to exploit the autofluorescence of elastic laminae. Elastin breaks (defined by the presence of two free ends of what otherwise seemed to be a continuous elastin fiber) were counted by two blinded observers, and inter-observer agreement was verified using Bland-Altman analysis.21 The extent of CD31 staining of luminal endothelium was quantified by measuring the luminal length that stained with CD31, dividing this length by the total luminal circumference, and multiplying the result by 100. Color thresholding was used to measure the medial area stained by the Mac2 antibody. This value was divided by total medial area (calculated from an adjacent H&E-stained section) and multiplied by 100 to give the percentage of medial area occupied by macrophages.
Vascular Permeability Assay
To measure vascular permeability using intravenous Evans blue dye, we modified methods from published protocols.22 Mice were anesthetized with ketamine and xylazine as described above, then injected via tail vein with 200 μL of 0.5% Evans blue dye (Sigma #E2129) in sterile-filtered PBS (1 mg of Evans blue injected). After 30 minutes to allow the dye to circulate, mice were exsanguinated by saline perfusion via the left ventricle. Remaining intraluminal Evans blue dye was then removed by cardiac perfusion with 10 mL of sterile PBS. Aortas were dissected free and explanted from the aortic root to the diaphragmatic hiatus and cleaned of periaortic fat. Aortas were photographed through a dissecting microscope, then placed in 1.5 mL microcentrifuge tubes and dried overnight at 55 °C. The desiccated aortas were weighed, then incubated for 24 hours in 200 μL of formamide to extract the Evans blue dye. Tubes containing the aortas were centrifuged at 14,000 g for 20 minutes to pellet aortic tissue fragments. The supernatant was removed, and Evans blue dye quantified by measuring absorbance at 620 nm. Total formamide-extracted dye was normalized to the dry weight of each aortic segment.
Blood Pressure Measurements
Non-invasive blood pressure measurements were obtained in awake, unsedated mice using the CODA Non-Invasive Blood Pressure System (Kent Scientific).23 Mice were placed in a tube holder and allowed to acclimate on a warming platform for at least 15 minutes. When the surface temperature of the tail was between 32–35 °C, the occlusion cuff and volume pressure recorder were placed on the tail and measurements were obtained on 20 consecutive cycles with 30 seconds between cycles. Blood pressures were measured 1, 3, 6, and 9 weeks after completing tamoxifen injections.
Measurement of NO and Superoxide Levels in Aortic Extracts
For measurement of aortic NO levels, mice were anesthetized as described above and exsanguinated by perfusion via the left ventricle with Krebs-HEPES buffer (Alfa Aesar #J67795). With aid of a dissecting microscope, the thoracic aorta was cleaned of periaortic fat and transected both at 1.5 cm distal to the top of the aortic arch and at the aortic root. This segment of thoracic aorta was explanted and divided into 6 segments. After ex vivo contraction, each of these 6 segments was approximately 2.5 mm in length. The 6 segments were placed together in a single well of an ice-cooled 24-well plate, containing 1 mL cold Krebs-HEPES buffer. The segments were then treated for 15 minutes with the calcium ionophore A23187 (final concentration 5 × 10−3 M; Enzo Life Sciences #BML-CA100). During this treatment, a colloidal Fe(DETC)2 solution was prepared by combining equal volumes of 3.2 × 10−5 M of sodium diethyldithiocarbamate (Sigma Aldrich #D3506) and 1.9 × 10−5 M of ferrous sulfate (Sigma Aldrich #12354), each dissolved separately in 0.9% deoxygenated saline. The freshly prepared Fe(DETC)2 solution was immediately added to each well, in dropwise fashion. The segments were incubated at 37 °C for 60 minutes, snap frozen in fresh Krebs-HEPES buffer in a pre-cut 1 mL syringe, then stored at −80 °C. Samples were removed from syringes as described,24 and placed in a quartz Dewar (Corning, NY) filled with liquid nitrogen. Electron spin resonance spectra were recorded using an EMX electron spin resonance spectrometer (Bruker Biospin Corp., Billerica, MA) and a super high Q microwave cavity. The electron spin resonance settings were: field sweep 160 Gauss, microwave frequency 9.42 GHz, microwave power 10 milliwatts, modulation amplitude 3 Gauss, scan time 150 msec, time constant 5.2 sec, and receiver gain 60 dB (n = 4 scans).
For measurement of aortic superoxide levels, mice were anesthetized, perfused, and their thoracic aortas removed and divided as described above. The 6 aortic segments were added to a single ice-cooled well in 1 mL of Krebs-HEPES buffer, and the superoxide probe dihydroethidium (Cayman Chemical #104821-25-2) was added at a final concentration of 50 μM. After 30 minutes in the dark at 37 °C, all 6 segments were placed in a single light-insulated tube containing 300 μL of dry-ice cooled methanol, stored at −80 °C, shipped on dry ice, and again stored at −80 °C. After removal from −80 °C, each group of segments (in 300 μL of methanol) was homogenized with a glass pestle. The homogenate was passed through a 0.22 μm syringe filter and the methanol filtrates were analyzed by HPLC according to a previously published protocol.25 Dihydroethidium oxidation products (2-hydroxyethidium and ethidium), were separated using a C-18 reverse-phase column (Nucleosil 250 to 4.5 mm) and a mobile phase containing 0.1% trifluoroacetic acid and an acetonitrile gradient (from 37% to 47%) at a flow rate of 0.5 mL/min. Ethidium and 2-hydroxyethidium were detected with a fluorescence detector using an emission wavelength of 580 nm and an excitation of 480 nm. Production of cellular superoxide was measured as accumulation of 2-hydroxyethidium and normalized by protein concentration.
Statistics
All data are presented as mean±S.E.M., with “n” referring to the number of mice. Statistical analyses were performed using either Prism (GraphPad) or SigmaStat. For comparison of continuous data between two groups (e.g. band densities on western blots and blood pressures), the two-tailed Student’s t-test was applied after verifying normal distribution and equal variances. If the variances were unequal, we used the two-tailed Welch’s t-test. The contractile responses to KCl and PE are expressed as mN or % relative to KCl (40 mM); whereas relaxation to ACh and SNP are indicated as the % decreases from the precontracted tone. Sensitivities of the arteries (pD2) to PE, ACh, and SNP were calculated as the effective concentration that elicits 50% of the maximal response, using non-linear regression curve-fit, and are expressed as −logEC50. Values of −logEC50 were compared by either two-tailed Student’s t-test or Mann-Whitney rank-sum test when conditions of equal variances and normal distribution were not met. The responses of aortas from knockout and wild-type mice to multiple doses of vasoactive agents were analyzed using 2-way ANOVA with Bonferroni correction for multiple comparisons (to test the hypothesis that genotype has a significant effect on contractile force). Myography data that compared aortas from knockout and wild-type mice—with a subset of aortas of each group subjected to an additional intervention (i.e., mechanical denudation of endothelium or addition of L-NAME)—were analyzed by 2-way ANOVA at each dose of vasoactive agent. Here we tested the hypothesis that—at each dose—there is a significant interaction between genotype and either denudation or L-NAME in determining the outcome variable of force.
Results
Design of Experiments Comparing Aortas of TBR2SMΔ and TBR2f/f Mice
Six-week-old Myh11-CreERT2 +/0 Tgfbr2f/f (experimental: TBR2SMΔ) and Myh11-CreERT2 0/0 Tgfbr2f/f (control: TBR2f/f) mice were injected with tamoxifen for 5 consecutive days. One, three, or fourteen weeks after the last tamoxifen injection, mice were euthanized and aortas removed (Figure II in the Data Supplement). We and others reported that TBR2SMΔ mice have significantly reduced aortic medial TBR2 protein, aortic SMC that are unresponsive to TGF-β1 ligand, aortic aneurysmal dilation, elastolysis, and aortitis.2–4 Because these observations are already well established, we did not repeat the biochemical and histologic studies on which they are based.
Because the Myh11-CreERT2 allele is integrated into the Y chromosome,19 all TBR2SMΔ mice are male and our breeding strategy (see Materials and Methods) does not generate male littermate controls. Therefore, for most experiments, we used tamoxifen-injected non-littermate CreERT2 0/0 Tgfbr2f/f males (bred in our facility) as the TBR2f/f controls. However, for the initial 3-week myography experiments, we used tamoxifen-injected littermate CreERT2 0/0 Tgfbr2f/f females as the TBR2f/f controls. We consider female and male TBR2f/f to be equivalent controls in experiments that measure vasomotor function (see Materials and Methods, and Tables I and II in the Data Supplement).
Aortas of TBR2SMΔ Mice Have Unaltered Levels of SMC Contractile Proteins
We previously reported that postnatal deletion of SMC Tgfbr2 increased SMC expression of mRNA encoding contractile proteins, including ACTA2 and MYH11.3 However, immunoblotting of extracts of aortas removed 3 weeks after Tgfbr2 deletion showed no significant differences in levels of MYH11, ACTA2, p-MLC, MLC, or p-MYPT proteins in TBR2SMΔ aortas compared to TBR2f/f aortas (Figure 1A and Figures III and IV in the Data Supplement).
Figure 1. Aortas of TBR2SMΔ mice have unaltered expression of contractile-apparatus proteins but are hypercontractile to phenylephrine (PE).
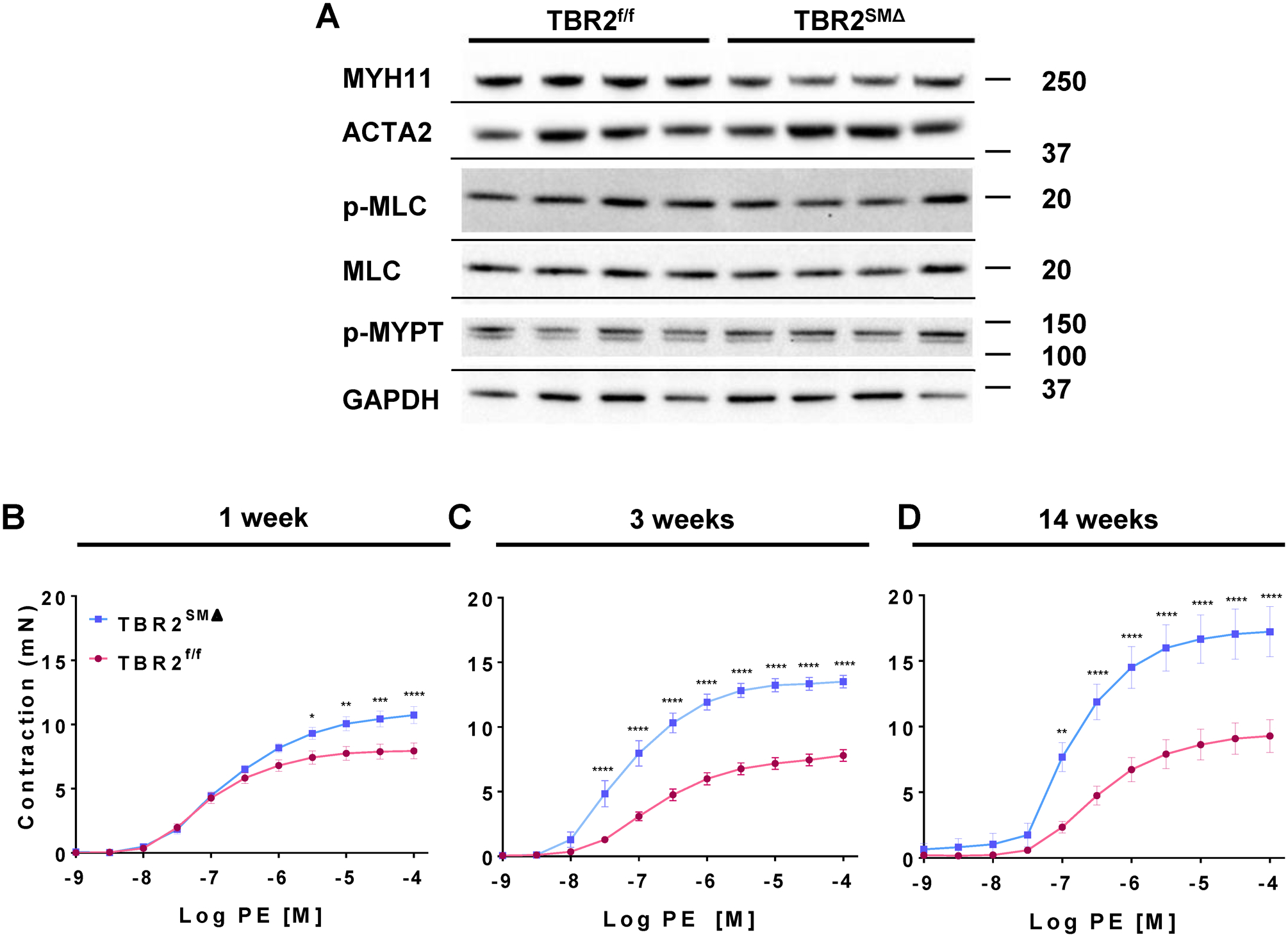
(A) Representative immunoblots of aortic extracts for contractile-apparatus proteins (n=7–15 per group, with aortas isolated 3 weeks after tamoxifen-induced Tgfbr2 deletion; see Online Figures II and III for additional blots and quantitation). Size markers are in kDa. (B–D) Force in milliNewtons (mN) developed by TBR2f/f and TBR2SMΔ aortic segments in response to increasing concentrations of PE. (B–D) Data are mean±SEM. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
Aortas of TBR2SMΔ Mice Are Hypercontractile and Have Impaired Endothelium-Dependent Relaxation
We used wire myography to measure vasomotor properties of ascending aortas of TBR2SMΔ and TBR2f/f mice. TBR2SMΔ mice often develop aortic hematomas.2, 3 To eliminate potential effects of aortic hematoma on vasomotor function, we excluded aortic segments with grossly visible hematomas (18% of TBR2SMΔ aortas). Three weeks after Tgfbr2 deletion, TBR2SMΔ ascending aortas contracted with greater force in response to both 40 mM KCl (7.6±0.3 mN versus 5.7±0.3 mN; 33% increase; P<0.0001; Figure V B in the Data Supplement) and phenylephrine (PE; Emax=14±0.5 mN versus 7.8±0.5 mN; 79% increase; P<0.0001; Figure 1C and Table I in the Data Supplement). When PE-induced contractile force was normalized to KCl-induced contractile force for each segment, the difference between TBR2SMΔ and TBR2f/f aortas remained significant (31% increase; P<0.0001; Figure V E in the Data Supplement), consistent with a receptor-dependent increase in contractility. TBR2SMΔ aortas were also more sensitive to PE (–logEC50=7.2±0.12 versus 6.7±0.056; P=0.01; Table II in the Data Supplement).
Hypercontractility of TBR2SMΔ aortas was unexpected.2 In our earlier work we observed that TBR2SMΔ aortas removed at a later time point (14 weeks after Tgfbr2 deletion) had relative medial thinning, potentially decreasing contractility.3 Therefore, we measured PE-induced contraction in aortas isolated 14 weeks after Tgfbr2 deletion. TBR2SMΔ aortas remained hypercontractile to both KCl (Figure V C in the Data Supplement) and PE (Emax=17±1.9 mN versus 9.3±1.3 mN; 83% increase; P<0.0001; Figure 1D and Table I in the Data Supplement), and were also hypersensitive to PE (–logEC50=7.0±0.21 versus 6.5±0.066; P=0.007; Table II in the Data Supplement). TBR2SMΔ aortic segments removed as early as 1 week after Tgfbr2 deletion were hypercontractile to PE (Emax=11±0.7 mN versus 7.9±0.6 mN; 40% increase; P<0.0001; Figure 1B and Table I in the Data Supplement), but their sensitivity to PE was unaltered (Table III in the Data Supplement) and they were not hypercontractile to KCl (Figure V A in the Data Supplement). When normalized to KCl-induced contraction, contractility of TBR2SMΔ aortic segments was significantly higher at 14 weeks, with a trend towards increased contractility at 1 week (Figure V D and V F in the Data Supplement).
We also tested whether Tgfbr2 deletion altered the responses of aortic rings to ACh (endothelium-dependent relaxation) or SNP (endothelium-independent relaxation). One week after Tgfbr2 deletion, TBR2SMΔ aortas already had significantly impaired relaxation and sensitivity to ACh (Emax=70±5.7% versus 104±2.9% for TBR2f/f segments; P<0.0001 Figure 2A and Table I in the Data Supplement; and −logEC50=6.5±0.17 versus 7.1±0.15; P=0.01; Table II in the Data Supplement). Relaxation and sensitivity to ACh were further impaired 3 weeks after SMC Tgfbr2 deletion (Emax=37±3.6% versus 97±1.5%; P<0.0001; Figure 2B and Table I in the Data Supplement; and −logEC50=6.6±0.13 versus 7.5±0.038; P<0.001; Table II in the Data Supplement). ACh-stimulated relaxation and ACh sensitivity were even more severely impaired in aortas removed 14 weeks after Tgfbr2 deletion (Emax=15±3.9% versus 93±1.5% for TBR2f/f; P<0.0001; Figure 2C and Table I in the Data Supplement; −logEC50=6.0±0.13 versus 7.5±0.086; P<0.001; Table II in the Data Supplement). In contrast, maximal relaxation to SNP was unaltered in TBR2SMΔ aortas removed at 1, 3, or 14 weeks after SMC Tgfbr2 deletion (Emax=102±1.8% versus 108±3.9%; 106±3.6% versus 100±0.28% and 104±1.2% versus 100±0.73%; P=0.2 at 1 week and P>0.9 at 3 and 14 weeks, respectively; Figure 2D–2F and Table I in the Data Supplement). TBR2SMΔ aortic segments isolated 3 weeks after SMC Tgfbr2 deletion were mildly less sensitive to SNP than TBR2f/f segments (–logEC50=7.4±0.09 versus 7.7±0.081; P=0.01; Table II in the Data Supplement), but segments isolated 1 and 14 weeks after SMC Tgfbr2 deletion did not differ significantly from TBR2f/f segments in their sensitivity to SNP (Table II in the Data Supplement). Therefore, SMC Tgfbr2 deletion in 6-week-old mice causes progressive hypercontractility and impaired endothelium-dependent relaxation but does not affect maximal relaxation to SNP and alters sensitivity to SNP only mildly and variably.
Figure 2. Ascending aortas from TBR2SMΔ mice have severely impaired endothelium-dependent relaxation and mildly impaired sensitivity to sodium nitroprusside.
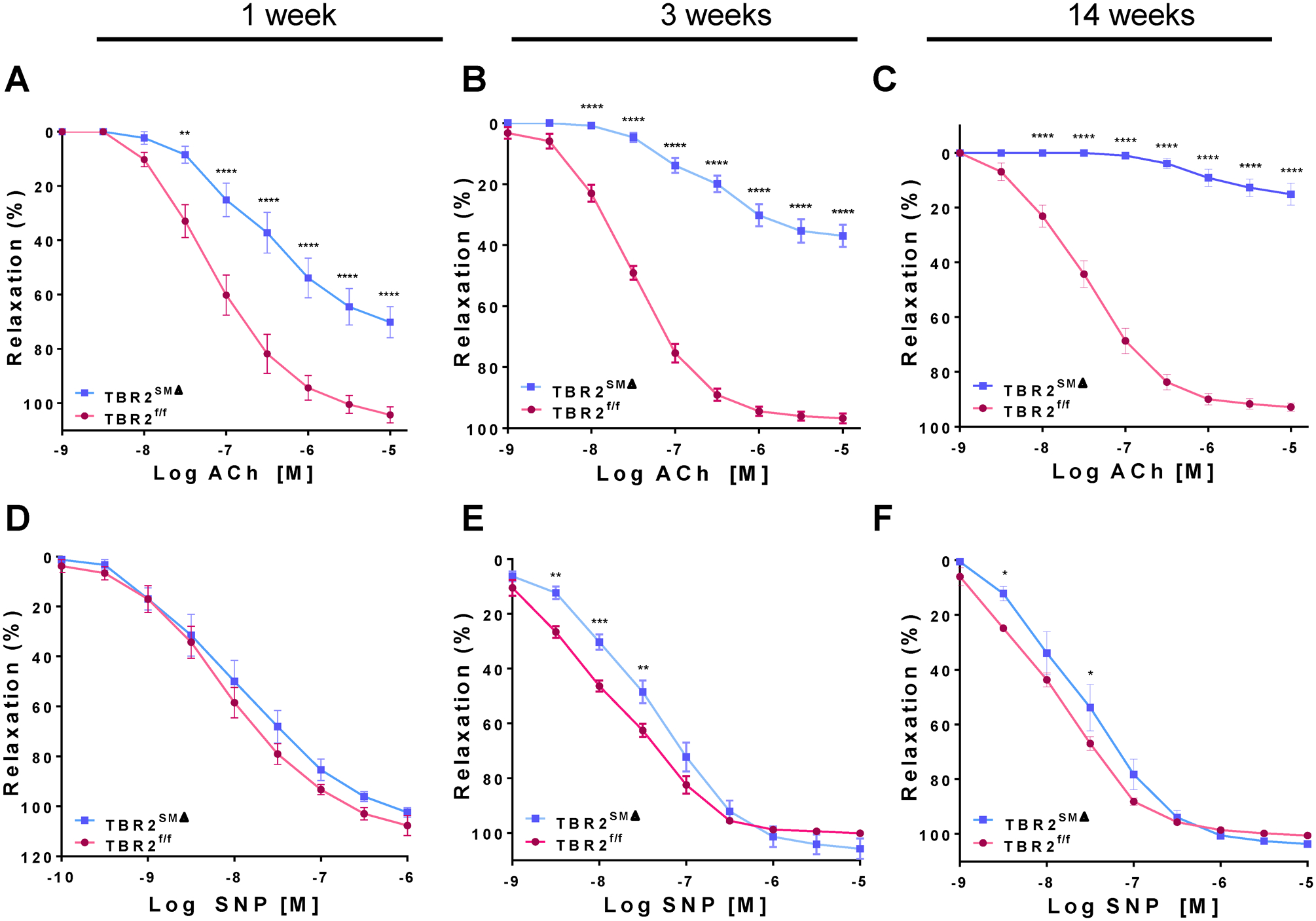
Segments of ascending aortas of TBR2SMΔ and TBR2f/f mice were mounted in a wire myograph, precontracted, and treated with acetylcholine (ACh; A–C) or sodium nitroprusside (SNP; D–F). Segments were removed 1 week (A, D), 3 weeks (B, E), or 14 weeks (C, F) after tamoxifen-induced Tgfbr2 deletion. 1 and 14 weeks: n=8–10; 3 weeks: n=19–20. Data are mean±SEM. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
Pharmacologic Inhibition of Endothelin and Thromboxane Receptors, or Reactive Oxygen Species Does Not Normalize Vasomotor Function of TBR2SMΔ Aortas
To investigate the underlying mechanisms of vasomotor abnormalities in TBR2SMΔ aortas, we treated TBR2SMΔ aortas ex vivo with the endothelin-1-receptor antagonist bosentan, the thromboxane receptor antagonist SQ29548, and a combination of superoxide dismutase and catalase. Neither bosentan nor SQ29548 reversed PE-induced hypercontractility of TBR2SMΔ aortas (Figure VIA–B and Table I in the Data Supplement), although SQ29548 increased sensitivity of the aortas to PE (–logEC50=7.5±0.13 versus 6.9±0.19; P=0.03; Table II in the Data Supplement). The combination of superoxide dismutase and catalase did not restore responsiveness of TBR2SMΔ aortas to ACh or change sensitivity to ACh (Figure VIC and Tables I and II in the Data Supplement).
Hypercontractility of TBR2SMΔ Aortas is Caused by Endothelial Dysfunction
We considered that hypercontractility of TBR2SMΔ aortas could be caused by EC loss or EC dysfunction (potentially including endothelial-to-mesenchymal transition (EndMT).26, 27 We investigated EC loss by staining sections of aortic rings used in the 3-week myography studies with an antibody to PECAM1 (an EC marker). Even after myography, the luminal endothelium in both TBR2SMΔ and TBR2f/f rings was largely intact (Figure 3A and 3B), with no difference between the two groups in luminal surface coverage by PECAM1-expressing cells (83% for both; P=0.8; Figure 3C; absence of aortic dilation at this time point is expected3).
Figure 3. Luminal endothelium is intact in TBR2SMΔ ascending aortic rings.
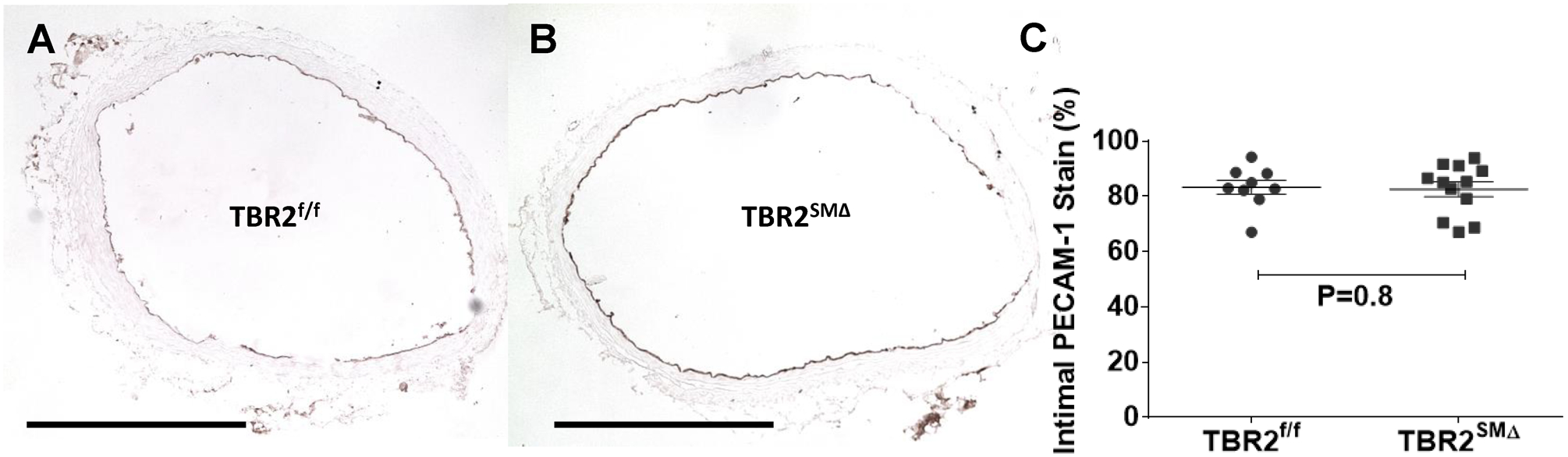
Rings were removed from the wire myograph after vasomotor studies, embedded, sectioned, and stained for expression of the endothelial marker PECAM1. (A, B), Representative PECAM1-stained sections. (C), Percentage of luminal surface of aortic sections that stained positive for PECAM1 expression. Each data point represents a single mouse; group means and SEM are indicated (n=9–12). (A, B), size bar is 0.5 mm.
We used luminal EC-specific western blotting (Figure VIIA in the Data Supplement) to investigate EndMT in aortic luminal EC of TBR2SMΔ mice. These blots showed no reduction of PECAM1 and CDH5 (EC markers) and no increase in VIM (a mesenchymal marker; Figures VII B–VII E in the Data Supplement), consistent with absence of EndMT.27, 28 To investigate a role for endothelial dysfunction independent of EndMT, we mechanically denuded luminal endothelium from segments of TBR2SMΔ and TBR2f/f aortas isolated 3 weeks after tamoxifen injection, measured responses of aortic rings to PE, and compared these responses to the PE-contraction-response curves of (previously studied) nondenuded aortic rings. Removal of endothelium was confirmed by near-absence of PECAM1 immunostaining (not shown) and by near-complete loss of responsiveness to ACh (Figure VIII in the Data Supplement). Endothelial denudation substantially increased PE-induced contractility of TBR2f/f segments but did not alter contractility of TBR2SMΔ segments (Figure 4A and Table I in the Data Supplement). Endothelial denudation also eliminated the difference in PE sensitivity between TBR2f/f and TBR2SMΔ segments (Table II in the Data Supplement). That endothelial denudation did not affect contractility of TBR2SMΔ segments could be due to saturation of effect; however, at nearly all of the higher PE concentrations, 2-way ANOVA revealed a statistically significant interaction between SMC TBR2 genotype and endothelial denudation in determining PE-induced force (Figure 4A). The significant interaction between SMC TBR2 genotype and loss of endothelium in determining contractile force supports a hypothesis that an EC-derived factor contributes to the PE-induced hypercontractility of nondenuded TBR2SMΔ aortas. We explored this hypothesis in a series of experiments that tested whether NO was this EC-derived factor.
Figure 4. Hypercontractility of TBR2SMΔ aortas is associated with decreased bioavailability of endothelium-derived nitric oxide (NO).
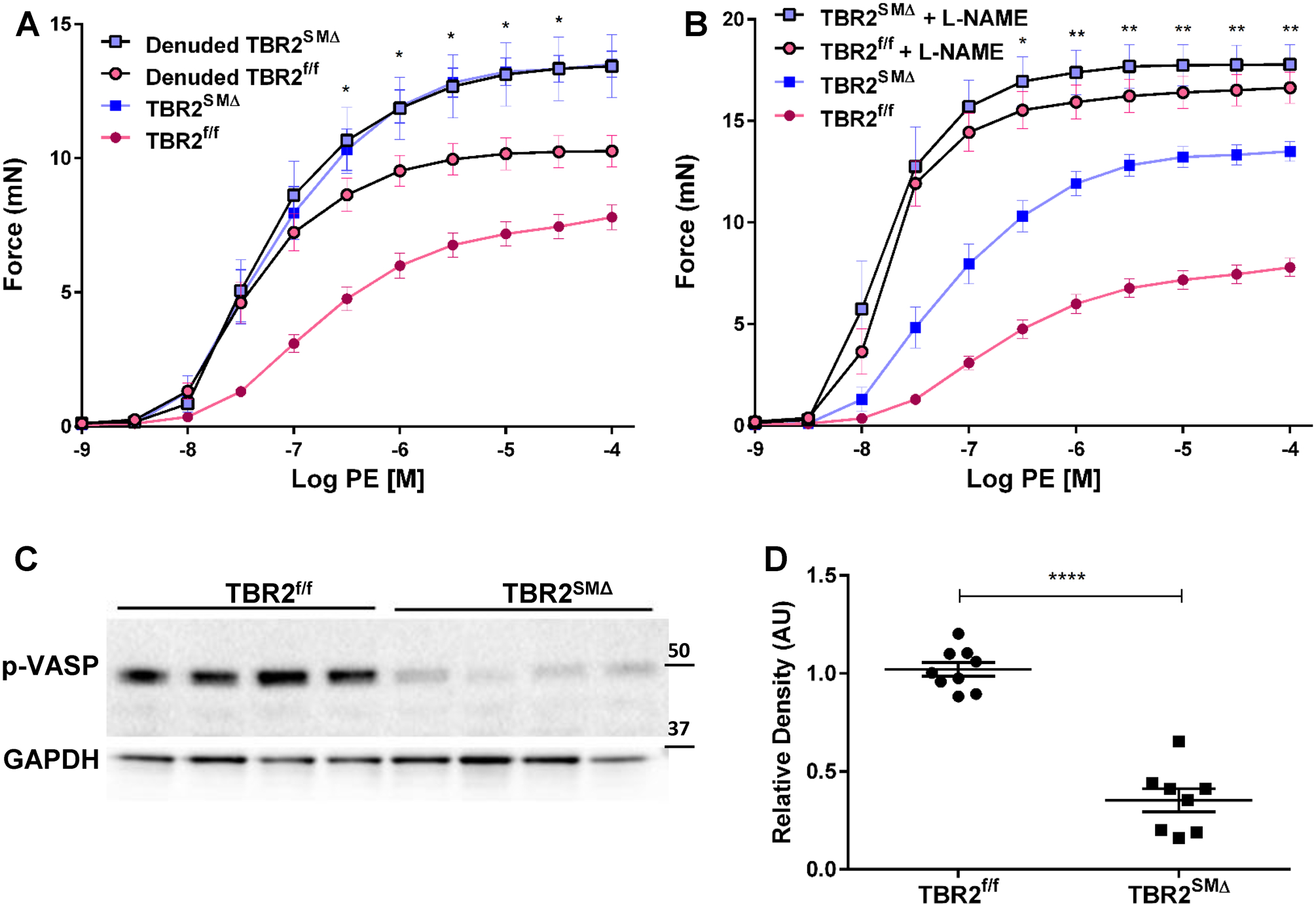
(A, B), Ascending aortic segments were removed from TBR2SMΔ and TBR2f/f mice, mounted in a wire myograph, and exposed to increasing concentrations of phenylephrine (PE). (A), Force is measured either with (n=9–10) or without (n=19–20) endothelial denudation. (B), Force is measured either in the presence (n=8) or absence (n=19–20) of N(⍵)-nitro-L-arginine methyl ester (L-NAME). (C), Immunoblot of aortic extracts for a phosphorylated form of vasodilator-stimulated phosphoprotein (p-VASP Ser239). Glyceraldehyde phosphate dehydrogenase (GAPDH) is included as a loading control. (D), Quantitation of levels of pVASP Ser239 based on densitometry of immunoblot shown in (C) and others. Each data point represents one mouse (n=8–9; ****P<0.0001). (A, B, and D), data are mean±SEM. (A, B), *P<0.05; **P<0.01 by 2-way ANOVA measuring interaction of SMC TBR2 genotype and either endothelial denudation (A) or L-NAME (B). (A, B), mN=milliNewtons; data for non-denuded segments are reproduced from Figure 2, for comparison. (C), Size markers are in kDa. (D), AU = arbitrary units.
To test the hypothesis that deficiency of endothelium-derived NO was responsible for hypercontractility to PE of TBR2SMΔ aortas, we isolated aortas 3 weeks after SMC Tgfbr2 deletion, treated them with the NOS inhibitor N(ω)-nitro-L-arginine methyl ester (L-NAME), and assessed PE concentration-response curves in the presence of L-NAME. After treatment with L-NAME, TBR2SMΔ and TBR2f/f segments had equivalent PE-induced contractility (Figure 4B; P=0.2–1.0 for comparison of L-NAME-treated TBR2SMΔ and TBR2f/f segments at all PE doses; and Table I in the Data Supplement) and equivalent sensitivity to PE (–logEC50=7.8±0.15 vs. 7.8±0.082; P=0.4; Table II in the Data Supplement). Moreover, at all of the higher PE concentrations, 2-way ANOVA revealed a statistically significant interaction between SMC TBR2 genotype and treatment with L-NAME in determining PE-induced force (Figure 4B). This interaction is profound because in the absence of NO (i.e., in L-NAME-treated segments), deletion of SMC Tgfbr2 has no effect on contractile responses to PE. Therefore, increased PE-responsiveness of TBR2SMΔ segments (Figure 1C) is most likely caused by a reduction in bioavailable NO. Decreased contractility of EC-denuded rings (Figure 4A) versus L-NAME-treated rings (Figure 4B) could reflect incomplete EC denudation or presence (in EC-denuded rings) of NO derived from a non-EC source.
To look for additional evidence of decreased NO bioavailability in TBR2SMΔ aortas, we measured levels of the Ser239-phosphorylated form of vasodilator-stimulated phosphoprotein (p-VASP). P-VASP is increased in vascular SMC via NO-stimulated increases in guanylate cyclase, cGMP, and protein kinase G,29 leading to its use as a readout of NO bioavailability.30 Western blots showed substantially lower levels of p-VASP Ser239 in TBR2SMΔ versus TBR2f/f aortas (65% decrease; P<0.0001; Figure 4C and 4D), consistent with decreased NO bioavailability in TBR2SMΔ aortas.
Endothelial Dysfunction in TBR2SMΔ Aortas Does Not Increase Blood Pressure or Vascular Permeability
Homozygous eNOS knockout mice are moderately hypertensive.31, 32 However, there were no significant differences in systolic or diastolic blood pressure between TBR2f/f and TBR2SMΔ mice, measured 1, 3, 6, and 9 weeks after SMC Tgfbr2 deletion (Figure IX in the Data Supplement). TBR2SMΔ mice have aortic dissections,2, 3 and others have hypothesized that dissections are caused by intramural edema.33 Therefore, we measured vascular permeability, which might increase along with EC dysfunction and could cause intramural edema. However, intravenous injection of Evans blue dye 3 weeks after SMC Tgfbr2 deletion did not reveal increased permeability of TBR2SMΔ aortas, either on gross examination or by measurement of Evans blue dye in aortic extracts (Figure 5A–5C; absence of aortic dilation at this time point is expected3).
Figure 5. Aortas of TBR2SMΔ mice have normal permeability 3 weeks after SMC Tgfbr2 deletion and no increase in macrophage infiltration 1 week after SMC Tgfbr2 deletion.
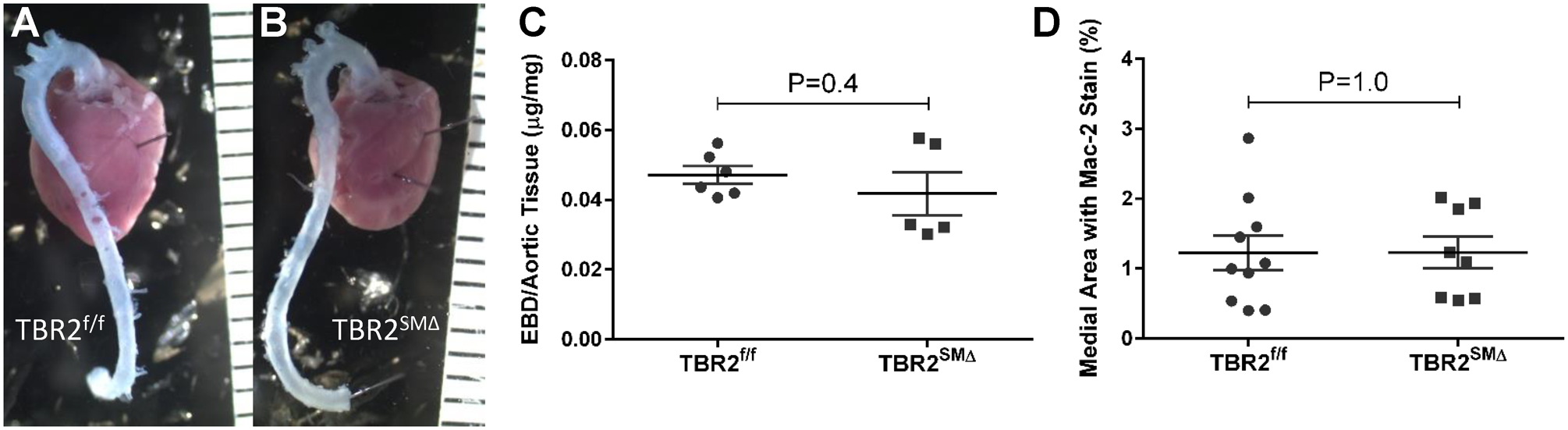
(A–C), Three weeks after completing tamoxifen injections, TBR2f/f and TBR2SMΔ mice were injected intravenously with Evans blue dye and aortas removed 30 minutes later. (A, B), Representative images. (C), Quantitation of Evans blue dye (EBD) in aortic extracts (n=5–6). (D), One week after completing tamoxifen injections, aortas of TBR2f/f and TBR2SMΔ mice (n=8–10) were removed, sectioned, and stained for expression of the macrophage antigen Mac-2. (A–B), Rulers are in mm. (C–D), Data points are individual mice; mean±SEM are indicated.
Endothelial Dysfunction in TBR2SMΔ Aortas is Independent of Inflammatory Cell Infiltration, Intramural Hemorrhage, and Elastolysis
We considered that endothelial dysfunction in TBR2SMΔ aortas might be caused by infiltrating inflammatory cells or by vasoactive agents derived from medial hemorrhage or elastolysis (all of which are present in TBR2SMΔ aortas removed 4 weeks after Tgfbr2 deletion).3 Accordingly, we removed aortas 1 week after SMC Tgfbr2 deletion (endothelial dysfunction is already present at this time; Figure 2A), stained aortic sections with hematoxylin and eosin, for detection of macrophages (Mac-2), and for past hemorrhage (Prussian blue; detects iron released from hemoglobin), and we counted elastin breaks. We excluded aortas with hematomas (3 of 12 TBR2SMΔ aortas) from these histological analyses because vasomotor function studies were performed only on aortas without grossly evident hematoma. Sections of TBR2f/f and TBR2SMΔ aortas had only rare staining for the Mac-2 antigen (~1% of total medial area; Figure 5D). Close examination of every section revealed no free red blood cells in the media, no Prussian blue staining (Figure X in the Data Supplement), and a similar number of elastin breaks in TBR2f/f and TBR2SMΔ aortas (3–4/section; P=1.0; Figure 6A). Planimetry revealed that the internal elastic lamina lengths also did not differ significantly between the 2 groups (2,822±57 μm versus 2,975±68 μm for TBR2f/f and TBR2SMΔ aortas, respectively, P=0.1; Figure 6B; absence of aortic lumen dilation at this time point is expected3). However, the external elastic lamina length was mildly increased in TBR2SMΔ aortas (7% increase; P=0.02; Figure 6B). TBR2SMΔ aortas also had thicker medias (30% increase; P=0.0001; Figure 6C), and larger medial areas (39% increase; P<0.0001; Figure 6D). Therefore, endothelial dysfunction in TBR2SMΔ aortas precedes macrophage infiltration, medial hemorrhage, elastolysis, and aneurysmal dilation.
Figure 6. Deletion of SMC Tgfbr2 increases medial area and thickness at 1 week, without increased elastolysis.
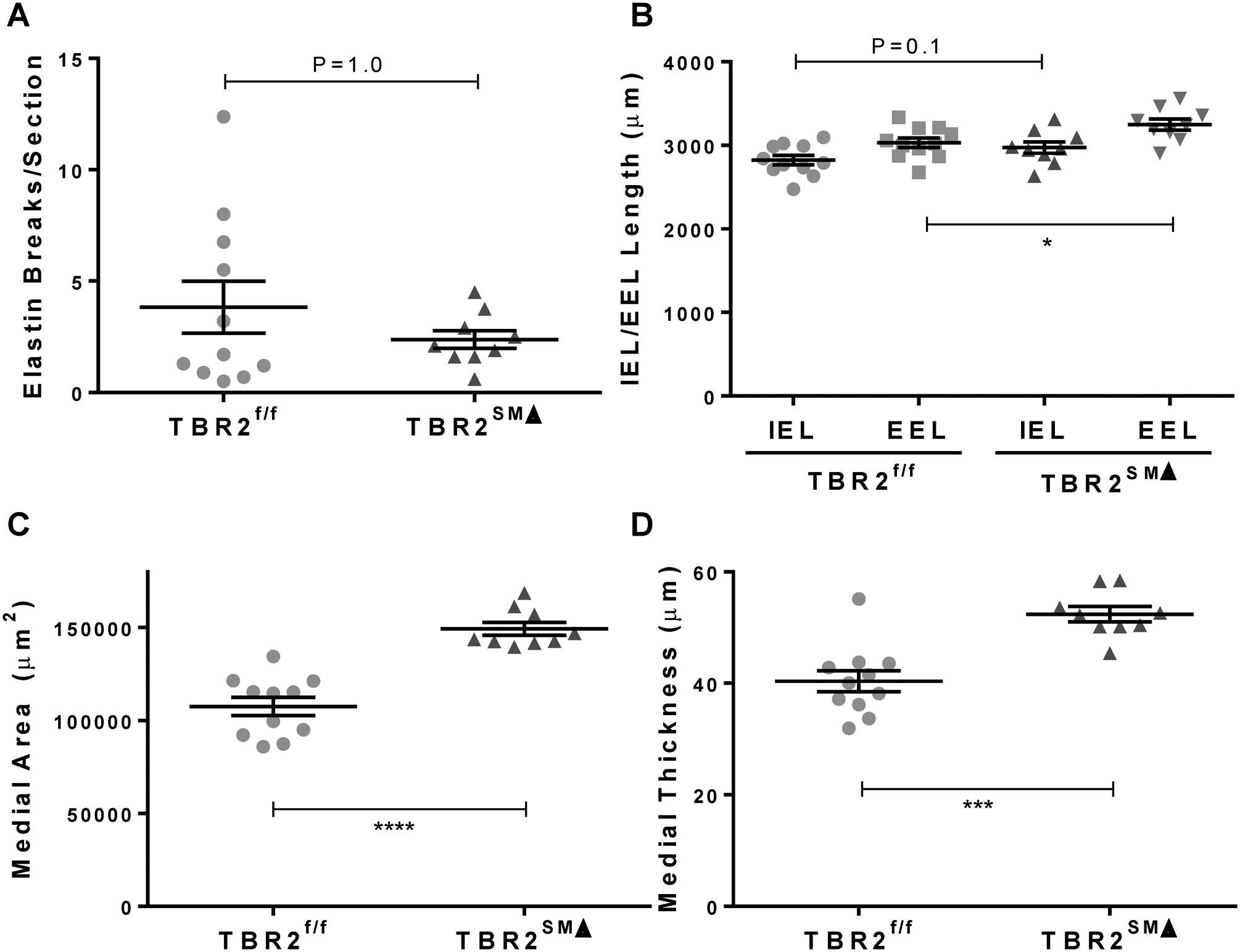
One week after completing tamoxifen injections, ascending aortas were removed from TBR2SMΔ and TBR2f/f mice, sectioned, and stained. (A), Elastin breaks were counted manually. (B), Lengths of the internal and external elastic laminae (IEL and EEL) were measured. (C–D), Medial area and thickness were calculated. (A–D), Data points are individual mice; n=9–11; mean±SEM are indicated. *P<0.05; ***P<0.001; ****P<0.0001.
eNOS Activation and NO Levels Are Decreased in TBR2SMΔ Aortas
To further investigate the mechanisms underlying endothelial dysfunction in TBR2SMΔ aortas, we measured total eNOS protein, phosphorylation of key eNOS residues, and levels of NO in extracts of TBR2SMΔ and TBR2f/f aortas isolated 3 weeks after deletion of SMC Tgfbr2. We also measured levels of caveolin and superoxide in aortic extracts because both molecules can inhibit NO-mediated endothelial function.34, 35 Total eNOS protein did not differ between the 2 groups (Figure 7A and 7B); however, eNOS phosphorylation at Ser1177 (associated with eNOS activation),36 was decreased in TBR2SMΔ segments (31%; P=0.002; Figure 7A and 7C). We could not detect p-eNOS Thr495 (associated with eNOS de-activation)37 in any of the extracts, and levels of caveolin-1 were not increased in TBR2SMΔ aortas (Figure 7D). Consistent with decreased eNOS-Ser1177, NO levels were lower in TBR2SMΔ aortas (50%; P=0.005; Figure 7E). There was no difference in superoxide levels between TBR2SMΔ and TBR2f/f aortas (Figure 7F).
Figure 7. Decreased endothelial nitric oxide synthase (eNOS) activation with unchanged levels of total eNOS and caveolin-1 in aortas of TBR2SMΔ mice.
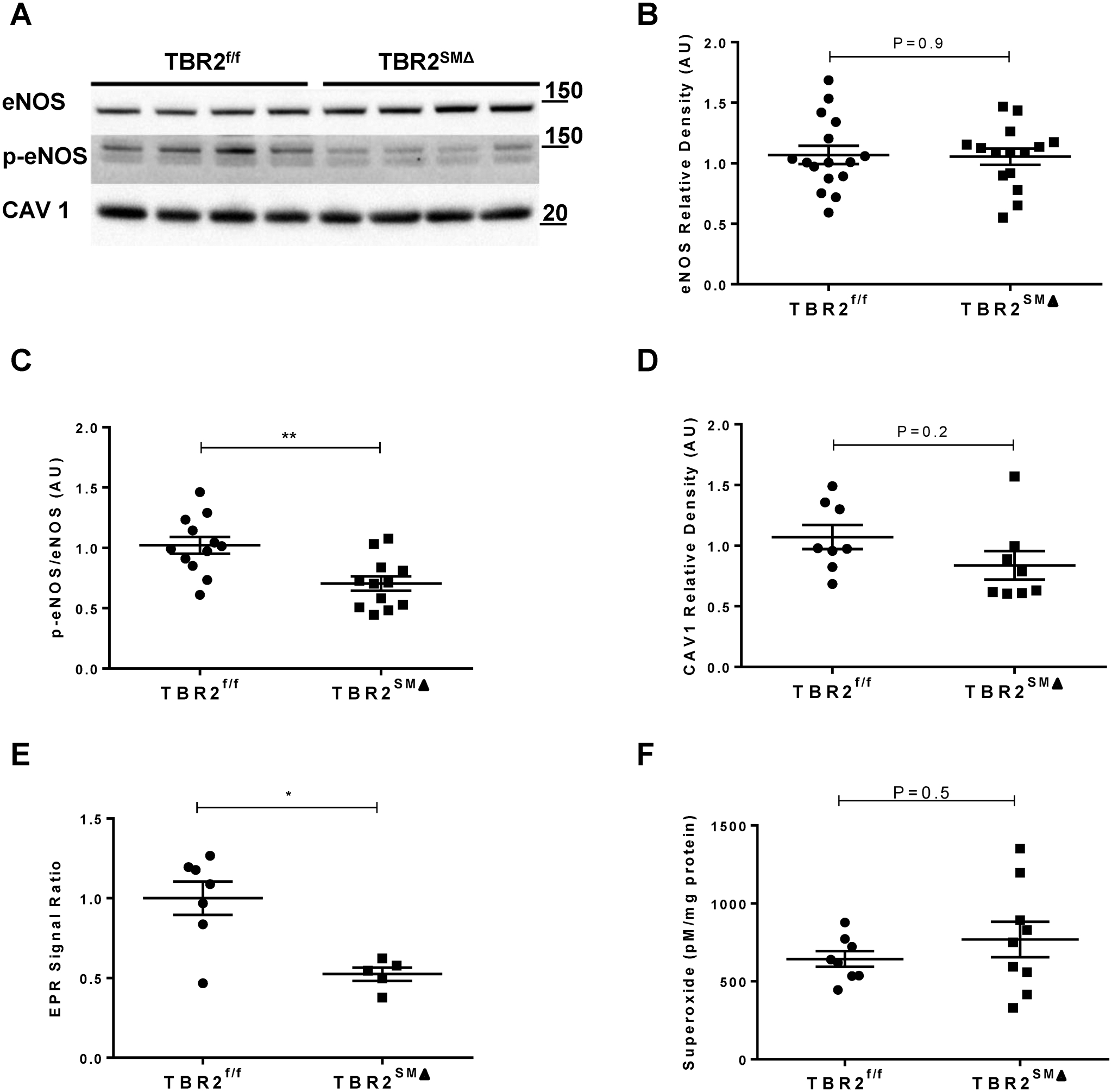
(A), Representative western blots of aortic extracts to detect: (B), total eNOS; (C), phospho-eNOS Ser1177 (p-eNOS); and (D), caveolin-1 (CAV1). (B–D), Quantitation of band densities in blots in (A) and others; AU=arbitrary units. (E), EPR=electron paramagnetic resonance. (A), Size markers are in kDa. (B), n=15–16; (C), n=12; (D), n=8; (E), n=5–7; (F), n=8–9. (B–E), two-sample Student’s t-test; (F), Rank-sum test.
Discussion
We tested the hypothesis that postnatal SMC-specific deletion of Tgfbr2 (TBR2SMΔ) would decrease SMC contractile protein expression and impair aortic contractility. Our major findings are: (1) TBR2SMΔ aortas have unaltered levels of several SMC contractile proteins; (2) TBR2SMΔ aortas are hypercontractile to both KCl and PE and have impaired endothelium-dependent relaxation; (3) hypercontractility is caused by endothelial dysfunction, manifested as decreased NO bioavailability; (4) decreased NO bioavailability is likely caused by decreased endothelial NO production; (5) the mechanism through which loss of SMC TGF-β signaling decreases EC NO production is yet to be elucidated.
At the time this study was initiated, it was unclear whether Tgfbr2 deletion in postnatal SMC would alter contractile protein abundance and aortic contractility. Several studies in cultured SMC and embryos show that increased TGF-β signaling promotes contractile protein synthesis. For example, addition of TGF-β to cultured SMC and their precursors increases expression of contractile proteins.7, 9, 38–41 Consistent with these data, in vivo deletion of Tgfbr2 in murine embryonic SMC or their precursors impairs cellular differentiation towards a contractile phenotype.42, 43 In an earlier study, we also found that Tgfbr2 deletion in mouse embryonic SMC impaired differentiation towards a contractile phenotype, based on morphologic criteria.44 However, others reported that Tgfbr2 deletion in embryonic SMC precursors did not alter contractile protein expression or differentiation.45, 46
Investigations of the impact of postnatal SMC Tgfbr2 deletion on contractile protein expression and SMC contractile function have also yielded inconsistent results. Li et al. reported that TBR2SMΔ mice had modestly decreased levels of total aortic Acta2, Tagln, and Myh11 mRNA, measured 2 weeks after Tgfbr2 deletion.2 However, they were unable to consistently detect loss of these proteins in aortic extracts, and total MLC (which is upregulated in SMC by TGF-β1)47 appeared to increase in TBR2SMΔ aortas. Explanted TBR2SMΔ SMC had a diminished ability to contract collagen gels,2 but aortic contractility was not examined. In contrast, our earlier study showed that aortic medias of TBR2SMΔ mice (analyzed 4 weeks after Tgfbr2 deletion) had consistently higher levels of mRNAs encoding several SMC contractile-unit proteins, including ACTA2, MYH11, TAGLN, and CNN1.3 These increases were substantial (2–3-fold), highly significant (typically P<0.001), and reproducible in independent experiments using both Myh11-CreERT2 and Acta2-CreERT2 drivers. A third study of TBR2SMΔ mice did not measure aortic contractile protein mRNA or protein, and did not assess SMC or aortic contractility.4
In the present study, we did not detect significant changes in MYH11, ACTA2, or MLC proteins in aortas removed 3 weeks after Tgfbr2 deletion. This result, largely congruent with the data of Li et al,2 suggests that acute postnatal loss of SMC TGF-β signaling does not alter contractile protein abundance and that studies in cultured cells and embryos showing a high dependence of SMC contractile protein abundance on cell-autonomous TGF-β signaling7, 9, 38, 40–43 may not apply to postnatal aortic SMC. We are unsure why Li et al found lower levels of aortic Acta2, Tagln, and Myh11 mRNA in TBR2SMΔ aortas; whereas, all of these mRNA were increased in our earlier study.3 A potential explanation is that Li et al measured total aortic mRNA (diluting SMC-derived mRNA) whereas we measured only aortic medial mRNA.3
Although we did not find altered levels of SMC contractile proteins and their active phosphorylated forms in TBR2SMΔ aortas, TBR2SMΔ aortas were hypercontractile to both KCl and PE and this hypercontractility was entirely attributable to endothelial dysfunction. We did not anticipate that use of the SMC-targeted Myh11-CreERT2 allele to delete Tgfbr2 would cause an EC-driven phenotype. Therefore, we considered whether Myh11-CreERT2 might also delete Tgfbr2 in EC. Although we cannot exclude this possibility based only on data from the present study, for several reasons we view this as highly unlikely. First, Myh11 is well established as a SMC-specific gene.48 Second, the Myh11-CreERT2 allele is widely used for generating SMC-specific knockouts.19, 49–52 Third, we and others have used reporter alleles to verify SMC-specificity of Myh11-CreERT2.3, 49, 53 Fourth, when compared directly, using the same floxed allele as a target, Myh11-CreERT2 does not duplicate phenotypes generated with EC-specific Cre drivers.54, 55 Fifth, we reproduced the hypercontractile vascular phenotype of the TBR2SMΔ mice using Tgfbr2f/f mice and an independent SMC-targeted Cre driver (Acta2-CreERT2;56 unpublished data). Finally, a comprehensive review of SMC-targeted Cre drivers does not identify any reports of Myh11-CreERT2-driven recombination in EC.57
Our finding that SMC Tgfbr2 deletion causes EC dysfunction is novel. EC dysfunction was evident within 1 week of Tgfbr2 deletion, before medial elastolysis and macrophage infiltration, and was independent of gross or microscopic intramural hemorrhage. Accordingly, we hypothesize that EC dysfunction is mediated by altered communication between Tgfbr2-null SMC and neighboring EC. This hypothesis is supported by several reports of SMC-EC communications that impact EC gene expression and function. SMC can communicate with EC via direct cell-cell contact, secreted protein ligands, transfer of miRNA via exosomes or tunneling nanotubes, or indirectly via altered SMC matrix-synthesis that impacts EC function.58–62 Of particular relevance, EC NO release can be altered by SMC-EC transfer of ions or small signaling molecules (e.g., Ca2+ or inositol tris phosphate).63, 64
Based on these and other studies, we considered several mechanisms through which SMC Tgfbr2 deletion could decrease EC NO release. First, SMC Tgfbr2 deletion could disrupt the positive feedback loop through which TGF-β can induce its own expression,65 lowering aortic TGF-β levels, reducing EC TGF-β signaling, and lowering eNOS phosphorylation and NO release.66–68 Lower aortic TGF-β levels—or loss of SMC TGF-β signaling alone—could also decrease expression and phosphorylation of SMC connexin 43,69, 70 decreasing gap junction-mediated SMC-EC communication,69–71 and potentially disrupting physiologic pathways through which SMC regulate EC function.72 These pathways include transfer of PE-induced/SMC-derived inositol tris phosphate to EC, causing calcium release, activation of eNOS, and NO release. Notably, if gap junction-mediated SMC-EC communication is experimentally impaired, PE-induced arterial contraction is significantly increased,72 exactly as in TBR2SMΔ aortas.
Increased contractility of TBR2SMΔ aortas contrasts with the conclusion of Li et al that postnatal SMC-specific Tgfbr2 deletion “impairs the contractile apparatus” of SMC.2 However, data that support this conclusion are limited to biochemical measurements (mostly in explanted subcultured SMC) and a cell-based collagen gel contraction assay.2 The gel-contraction assay may detect abnormalities in cellular mechanosensing and mechanoregulation that rely on relatively low SMC-generated stresses (~5 kPa). In contrast, our wire-myography assay measures the far-higher SMC-generated stresses that control vessel caliber (~100 kPa; see Figure XI in the Data Supplement). As formulated by Humphrey et al.,15 these SMC functions are likely distinct. Other variables that complicate comparison of the 2 studies include differences in: Tgfbr2flox alleles, age at time of tamoxifen injection/Cre activation, genotype of controls, injection of control mice with tamoxifen, and use by Li et al., of a lineage-tracing allele that is activated only in the experimental tamoxifen-treated mice. One or more of these variables might also explain why Li et al found a modest impact of SMC-specific Tgfbr2 deletion on systemic blood pressure, while we found no effect.
Finally, we were surprised to find aortic hypercontractility in an experimental model (TBR2SMΔ mice) that consistently manifests aneurysmal dilation.2–4 Although we did not measure aneurysmal dilation in the present study, this anatomic finding is 100% penetrant in TBR2SMΔ mice injected with tamoxifen at 4–6 weeks of age.2, 3 Importantly, however, the onset of hypercontractility of TBR2SMΔ aortas precedes dilation.3 While endothelial dysfunction leading to hypercontractility may play a role in aneurysm development in this model, it may not be the primary mechanism by which loss of SMC TGF-β signaling leads to aneurysm development. Notably, hypercontractility of TBR2SMΔ aortas seems to conflict with data and expert opinion that associate decreased SMC contractile-unit function with aneurysm formation.13–17, 73, 74 However, the TBR2SMΔ model is not unique in dissociating SMC contractile-unit function from aneurysm formation. For example, mice homozygous for a mutant SMMHC allele have impaired aortic contractility but do not develop aortic aneurysms.75 Moreover, mice haploinsufficient for elastin (a key component of the SMC contractile unit)14 develop arterial narrowing (not aneurysms) and have hypercontractile aortas.76, 77 These studies suggest a complex relationship between SMC contractile unit function and aneurysm formation.
In conclusion, postnatal SMC-specific deletion of Tgfbr2 causes profound endothelial dysfunction and aortic hypercontractility. Future studies will be aimed at identifying perturbations in SMC-EC communication through which loss of SMC TGF-β signaling causes EC dysfunction and determining whether EC dysfunction contributes to other vascular pathologies in TBR2SMΔ mice.
Supplementary Material
Highlights.
SMC-specific deletion of Tgfbr2, which causes aortic aneurysmal dilation, does not alter levels of several aortic SMC contractile proteins.
SMC-specific deletion of Tgfbr2 causes aortic hypercontractility and impaired endothelium-dependent vasodilation.
Aortic hypercontractility in mice with SMC-specific deletion of Tgfbr2 is caused by endothelial dysfunction.
Aortas of mice with SMC-specific deletion of Tgfbr2 have biochemical evidence of decreased eNOS activity and NO synthesis.
Acknowledgments
We thank Dr. Sergey Dikalov (Vanderbilt University Medical Center, Nashville, TN) for performing the nitric oxide and superoxide measurements and for helpful discussions. We thank Ms. Donya Derakshani for excellent technical assistance. We are also grateful to Dr. R. William Caldwell for perceptive comments and suggestions.
Sources of Funding
This work was supported by the National Heart Lung and Blood Institute grant (R01HL116612, to David A. Dichek) and by the John L. Locke Jr. Charitable Trust. Dr. Jay Zhu was supported by the National Heart Lung and Blood Institute (T32HL007828). Dr. Mark Majesky was supported by R01HL121877.
Nonstandard Abbreviations and Acronyms
- ACh
acetylcholine
- CreERT2
cre recombinase with tamoxifen-activated nucleus-localization signal
- EC
endothelial cell
- L-NAME
N(ω)-nitro-L-arginine methyl ester
- NO
nitric oxide
- PE
phenylephrine
- SMC
smooth muscle cell
- SNP
sodium nitroprusside
- TGFBR2
type 2 receptor for transforming growth factor-beta
- TBR2f/f
mice homozygous for a floxed allele for the type II transforming growth factor-beta receptor
- TBR2SMΔ
mice lacking type II transforming growth factor-beta receptor in smooth muscle cells
- TGF-β
transforming growth factor beta
Footnotes
Disclosures
None
References
- 1.Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in tgfbr1 or tgfbr2. Nat. Genet 2005;37:275–281 [DOI] [PubMed] [Google Scholar]
- 2.Li W, Li Q, Jiao Y, Qin L, Ali R, Zhou J, Ferruzzi J, Kim RW, Geirsson A, Dietz HC, Offermanns S, Humphrey JD, Tellides G. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J. Clin. Invest 2014;124:755-–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu JH, Wei H, Jaffe M, Airhart N, Du L, Angelov SN, Yan J, Allen JK, Kang I, Wight TN, Fox K, Smith A, Enstrom R, Dichek DA. Postnatal deletion of the type ii transforming growth factor-beta receptor in smooth muscle cells causes severe aortopathy in mice. Arterioscler. Thromb. Vasc. Biol 2015;35:2647–2656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang P, Schmit BM, Fu C, DeSart K, Oh SP, Berceli SA, Jiang Z. Smooth muscle cell-specific tgfbr1 deficiency promotes aortic aneurysm formation by stimulating multiple signaling events. Sci Rep. 2016;6:35444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boileau C, Guo DC, Hanna N, et al. Tgfb2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of marfan syndrome. Nat. Genet 2012;44:916–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dai X, Shen J, Annam NP, Jiang H, Levi E, Schworer CM, Tromp G, Arora A, Higgins M, Wang XF, Yang M, Li HJ, Zhang K, Kuivaniemi H, Li L. Smad3 deficiency promotes vessel wall remodeling, collagen fiber reorganization and leukocyte infiltration in an inflammatory abdominal aortic aneurysm mouse model. Sci Rep. 2015;5:10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen S, Lechleider RJ. Transforming growth factor-beta-induced differentiation of smooth muscle from a neural crest stem cell line. Circ. Res 2004;94:1195–1202 [DOI] [PubMed] [Google Scholar]
- 8.Hirschi KK, Lai L, Belaguli NS, Dean DA, Schwartz RJ, Zimmer WE. Transforming growth factor-beta induction of smooth muscle cell phenotpye requires transcriptional and post-transcriptional control of serum response factor. J. Biol. Chem 2002;277:6287–6295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hautmann MB, Madsen CS, Owens GK. A transforming growth factor beta (tgfbeta) control element drives tgfbeta-induced stimulation of smooth muscle alpha-actin gene expression in concert with two carg elements. J. Biol. Chem 1997;272:10948–10956 [DOI] [PubMed] [Google Scholar]
- 10.Schlumberger W, Thie M, Rauterberg J, Robenek H. Collagen synthesis in cultured aortic smooth muscle cells. Modulation by collagen lattice culture, transforming growth factor–b1, and epidermal growth factor. Arterioscler. Thromb 1991;11:1660–1666 [DOI] [PubMed] [Google Scholar]
- 11.Schönherr E, Järveläinen HT, Kinsella MG, Sandell LJ, Wight TN. Platelet-derived growth factor and transforming growth factor-b1 differentially affect the synthesis of biglycan and decorin by monkey arterial smooth muscle cells. Arterioscler. Thromb 1993;13:1026–1036 [DOI] [PubMed] [Google Scholar]
- 12.Lawrence R, Hartmann DJ, Sonenshein GE. Transforming growth factor b1 stimulates type v collagen expression in bovine vascular smooth muscle cells. J. Biol. Chem 1994;269:9603–9609 [PubMed] [Google Scholar]
- 13.Milewicz DM, Guo DC, Tran-Fadulu V, Lafont AL, Papke CL, Inamoto S, Kwartler CS, Pannu H. Genetic basis of thoracic aortic aneurysms and dissections: Focus on smooth muscle cell contractile dysfunction. Annu Rev Genomics Hum Genet. 2008;9:283–302 [DOI] [PubMed] [Google Scholar]
- 14.Karimi A, Milewicz DM. Structure of the elastin-contractile units in the thoracic aorta and how genes that cause thoracic aortic aneurysms and dissections disrupt this structure. Can. J. Cardiol 2016;32:26–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Humphrey JD, Schwartz MA, Tellides G, Milewicz DM. Role of mechanotransduction in vascular biology: Focus on thoracic aortic aneurysms and dissections. Circ. Res 2015;116:1448–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Milewicz DM, Trybus KM, Guo DC, Sweeney HL, Regalado E, Kamm K, Stull JT. Altered smooth muscle cell force generation as a driver of thoracic aortic aneurysms and dissections. Arterioscler. Thromb. Vasc. Biol 2017;37:26–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gillis E, Van Laer L, Loeys BL. Genetics of thoracic aortic aneurysm: At the crossroad of transforming growth factor-beta signaling and vascular smooth muscle cell contractility. Circ. Res 2013;113:327–340 [DOI] [PubMed] [Google Scholar]
- 18.Leveen P, Larsson J, Ehinger M, Cilio CM, Sundler M, Sjostrand LJ, Holmdahl R, Karlsson S. Induced disruption of the transforming growth factor beta type ii receptor gene in mice causes a lethal inflammatory disorder that is transplantable. Blood. 2002;100:560–568 [DOI] [PubMed] [Google Scholar]
- 19.Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, Lemmer B, Schutz G, Gutkind JS, Offermanns S. G12-g13-larg-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat. Med 2008;14:64–68 [DOI] [PubMed] [Google Scholar]
- 20.Wei H, Hu JH, Angelov SN, Fox K, Yan J, Enstrom R, Smith A, Dichek DA. Aortopathy in a mouse model of marfan syndrome is not mediated by altered transforming growth factor beta signaling. J Am Heart Assoc. 2017;6:pii: e004968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bland JM, Altman DG. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet. 1986;1:307–310 [PubMed] [Google Scholar]
- 22.Radu M, Chernoff J. An in vivo assay to test blood vessel permeability. J Vis Exp. 2013:e50062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.King VL, Lin AY, Kristo F, Anderson TJ, Ahluwalia N, Hardy GJ, Owens AP 3rd, Howatt DA, Shen D, Tager AM, Luster AD, Daugherty A, Gerszten RE. Interferon-gamma and the interferon-inducible chemokine cxcl10 protect against aneurysm formation and rupture. Circulation. 2009;119:426–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dikalov S, Fink B. Esr techniques for the detection of nitric oxide in vivo and in tissues. Methods Enzymol. 2005;396:597–610 [DOI] [PubMed] [Google Scholar]
- 25.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res 2010;107:106–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gimbrone MA Jr., Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res 2016;118:620–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kovacic JC, Dimmeler S, Harvey RP, Finkel T, Aikawa E, Krenning G, Baker AH. Endothelial to mesenchymal transition in cardiovascular disease: Jacc state-of-the-art review. J. Am. Coll. Cardiol 2019;73:190–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sánchez-Duffhues G, García de Vinuesa A, Ten Dijke P. Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev. Dyn 2018;247:492–508 [DOI] [PubMed] [Google Scholar]
- 29.Butt E, Abel K, Krieger M, Palm D, Hoppe V, Hoppe J, Walter U. Camp- and cgmp-dependent protein kinase phosphorylation sites of the focal adhesion vasodilator-stimulated phosphoprotein (vasp) in vitro and in intact human platelets. J. Biol. Chem 1994;269:14509–14517 [PubMed] [Google Scholar]
- 30.Chen L, Daum G, Chitaley K, Coats SA, Bowen-Pope DF, Eigenthaler M, Thumati NR, Walter U, Clowes AW. Vasodilator-stimulated phosphoprotein regulates proliferation and growth inhibition by nitric oxide in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol 2004;24:1403–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242 [DOI] [PubMed] [Google Scholar]
- 32.Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, Smithies O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. U. S. A 1996;93:13176–13181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mallat Z, Tedgui A, Henrion D. Role of microvascular tone and extracellular matrix contraction in the regulation of interstitial fluid: Implications for aortic dissection. Arterioscler. Thromb. Vasc. Biol 2016;36:1742–1747 [DOI] [PubMed] [Google Scholar]
- 34.Michel JB, Feron O, Sacks D, Michel T. Reciprocal regulation of endothelial nitric-oxide synthase by ca2+-calmodulin and caveolin. J. Biol. Chem 1997;272:15583–15586 [DOI] [PubMed] [Google Scholar]
- 35.Tesfamariam B, Cohen RA. Role of superoxide anion and endothelium in vasoconstrictor action of prostaglandin endoperoxide. Am. J. Physiol 1992;262:H1915–1919 [DOI] [PubMed] [Google Scholar]
- 36.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by akt-dependent phosphorylation. Nature. 1999;399:601–605 [DOI] [PubMed] [Google Scholar]
- 37.Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of thr(495) regulates ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circ. Res 2001;88:E68–75 [DOI] [PubMed] [Google Scholar]
- 38.Bjorkerud S Effects of transforming growth factor-beta 1 on human arterial smooth muscle cells in vitro. Arterioscler. Thromb 1991;11:892–902 [PubMed] [Google Scholar]
- 39.Owens GK, Geisterfer AAT, Yang YW-H, Komoriya A. Transforming growth factor-b–induced growth inhibition and cellular hypertrophy in cultured vascular smooth muscle cells. J. Cell Biol 1988;107:771–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirschi KK, Rohovsky SA, D’Amore PA. Pdgf, tgf-beta, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10t1/2 cells and their differentiation to a smooth muscle fate. J. Cell Biol 1998;141:805–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hirschi KK, Burt JM, Hirschi KD, Dai C. Gap junction communication mediates transforming growth factor-beta activation and endothelial-induced mural cell differentiation. Circ. Res 2003;93:429–437 [DOI] [PubMed] [Google Scholar]
- 42.Langlois D, Hneino M, Bouazza L, Parlakian A, Sasaki T, Bricca G, Li JY. Conditional inactivation of tgf-beta type ii receptor in smooth muscle cells and epicardium causes lethal aortic and cardiac defects. Transgenic Res. 2010;19:1069–1082 [DOI] [PubMed] [Google Scholar]
- 43.Wurdak H, Ittner LM, Lang KS, Leveen P, Suter U, Fischer JA, Karlsson S, Born W, Sommer L. Inactivation of tgfbeta signaling in neural crest stem cells leads to multiple defects reminiscent of digeorge syndrome. Genes Dev. 2005;19:530–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jaffe M, Sesti C, Washington I, Du L, Dronadula N, Chin MT, Stolz DB, Davis EC, Dichek DA. Transforming growth factor beta signaling in myogenic cells regulates vascular morphogenesis, differentiation, and matrix synthesis. Arterioscler. Thromb. Vasc. Biol 2012;32:e1–e11 [DOI] [PubMed] [Google Scholar]
- 45.Choudhary B, Ito Y, Makita T, Sasaki T, Chai Y, Sucov HM. Cardiovascular malformations with normal smooth muscle differentiation in neural crest-specific type ii tgfbeta receptor (tgfbr2) mutant mice. Dev. Biol 2006;289:420–429 [DOI] [PubMed] [Google Scholar]
- 46.Choudhary B, Zhou J, Li P, Thomas S, Kaartinen V, Sucov HM. Absence of tgfbeta signaling in embryonic vascular smooth muscle leads to reduced lysyl oxidase expression, impaired elastogenesis, and aneurysm. Genesis. 2009;47:115–121 [DOI] [PubMed] [Google Scholar]
- 47.Ramachandran A, Gangopadhyay SS, Krishnan R, Ranpura SA, Rajendran K, Ram-Mohan S, Mulone M, Gong EM, Adam RM. Junb mediates basal- and tgfbeta1-induced smooth muscle cell contractility. PLoS One. 2013;8:e53430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev 2004;84:767–801 [DOI] [PubMed] [Google Scholar]
- 49.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK. Klf4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med 2015;21:628–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cherepanova OA, Gomez D, Shankman LS, et al. Activation of the pluripotency factor oct4 in smooth muscle cells is atheroprotective. Nat. Med 2016;22:657–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xie Y, Ostriker AC, Jin Y, Hu H, Sizer AJ, Peng G, Morris AH, Ryu C, Herzog EL, Kyriakides T, Zhao H, Dardik A, Yu J, Hwa J, Martin KA. Lmo7 is a negative feedback regulator of transforming growth factor beta signaling and fibrosis. Circulation. 2019;139:679–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chang AN, Gao N, Liu Z, Huang J, Nairn AC, Kamm KE, Stull JT. The dominant protein phosphatase pp1c isoform in smooth muscle cells, pp1cbeta, is essential for smooth muscle contraction. J. Biol. Chem 2018;293:16677–16686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herring BP, Hoggatt AM, Burlak C, Offermanns S. Previously differentiated medial vascular smooth muscle cells contribute to neointima formation following vascular injury. Vasc Cell. 2014;6:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garrido-Martin EM, Nguyen HL, Cunningham TA, Choe SW, Jiang Z, Arthur HM, Lee YJ, Oh SP. Common and distinctive pathogenetic features of arteriovenous malformations in hereditary hemorrhagic telangiectasia 1 and hereditary hemorrhagic telangiectasia 2 animal models--brief report. Arterioscler. Thromb. Vasc. Biol 2014;34:2232–2236 [DOI] [PubMed] [Google Scholar]
- 55.Tang H, Babicheva A, McDermott KM, et al. Endothelial hif-2alpha contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am J Physiol Lung Cell Mol Physiol. 2018;314:L256–L275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wendling O, Bornert JM, Chambon P, Metzger D. Efficient temporally-controlled targeted mutagenesis in smooth muscle cells of the adult mouse. Genesis. 2009;47:14–18 [DOI] [PubMed] [Google Scholar]
- 57.Chakraborty R, Saddouk FZ, Carrao AC, Krause DS, Greif DM, Martin KA. Promoters to study vascular smooth muscle. Arterioscler. Thromb. Vasc. Biol 2019;39:603–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deng L, Blanco FJ, Stevens H, et al. Microrna-143 activation regulates smooth muscle and endothelial cell crosstalk in pulmonary arterial hypertension. Circ. Res 2015;117:870–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Climent M, Quintavalle M, Miragoli M, Chen J, Condorelli G, Elia L. Tgfbeta triggers mir-143/145 transfer from smooth muscle cells to endothelial cells, thereby modulating vessel stabilization. Circ. Res 2015;116:1753–1764 [DOI] [PubMed] [Google Scholar]
- 60.Li M, Qian M, Kyler K, Xu J. Endothelial-vascular smooth muscle cells interactions in atherosclerosis. Front Cardiovasc Med. 2018;5:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tang R, Zhang G, Chen SY. Smooth muscle cell proangiogenic phenotype induced by cyclopentenyl cytosine promotes endothelial cell proliferation and migration. J. Biol. Chem 2016;291:26913–26921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ren J, Zhou T, Pilli VSS, Phan N, Wang Q, Gupta K, Liu Z, Sheibani N, Liu B. Novel paracrine functions of smooth muscle cells in supporting endothelial regeneration following arterial injury. Circ. Res 2019;124:1253–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gao Y, Chen T, Raj JU. Endothelial and smooth muscle cell interactions in the pathobiology of pulmonary hypertension. Am. J. Respir. Cell Mol. Biol 2016;54:451–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Makino A, Firth AL, Yuan JX. Endothelial and smooth muscle cell ion channels in pulmonary vasoconstriction and vascular remodeling. Compr Physiol. 2011;1:1555–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Van Obberghen-Schilling E, Roche NS, Flanders KC, Sporn MB, Roberts AB. Transforming growth factor b1 positively regulates its own expression in normal and transformed cells. J. Biol. Chem 1988;263:7741–7746 [PubMed] [Google Scholar]
- 66.Saura M, Zaragoza C, Cao W, Bao C, Rodriguez-Puyol M, Rodriguez-Puyol D, Lowenstein CJ. Smad2 mediates transforming growth factor-beta induction of endothelial nitric oxide synthase expression. Circ. Res 2002;91:806–813 [DOI] [PubMed] [Google Scholar]
- 67.Vasquez R, Farias M, Vega JL, Martin RS, Vecchiola A, Casanello P, Sobrevia L. D-glucose stimulation of l-arginine transport and nitric oxide synthesis results from activation of mitogen-activated protein kinases p42/44 and smad2 requiring functional type ii tgf-beta receptors in human umbilical vein endothelium. J. Cell. Physiol 2007;212:626–632 [DOI] [PubMed] [Google Scholar]
- 68.Walshe TE, dela Paz NG, D’Amore PA. The role of shear-induced transforming growth factor-beta signaling in the endothelium. Arterioscler. Thromb. Vasc. Biol 2013;33:2608–2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kabir N, Chaturvedi K, Liu LS, Sarkar DK. Transforming growth factor-beta3 increases gap-junctional communication among folliculostellate cells to release basic fibroblast growth factor. Endocrinology. 2005;146:4054–4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tacheau C, Fontaine J, Loy J, Mauviel A, Verrecchia F. Tgf-beta induces connexin43 gene expression in normal murine mammary gland epithelial cells via activation of p38 and pi3k/akt signaling pathways. J. Cell. Physiol 2008;217:759–768 [DOI] [PubMed] [Google Scholar]
- 71.van Zoelen EJ, Tertoolen LG. Transforming growth factor-beta enhances the extent of intercellular communication between normal rat kidney cells. J. Biol. Chem 1991;266:12075–12081 [PubMed] [Google Scholar]
- 72.Straub AC, Billaud M, Johnstone SR, Best AK, Yemen S, Dwyer ST, Looft-Wilson R, Lysiak JJ, Gaston B, Palmer L, Isakson BE. Compartmentalized connexin 43 s-nitrosylation/denitrosylation regulates heterocellular communication in the vessel wall. Arterioscler. Thromb. Vasc. Biol 2011;31:399–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chung AW, Au Yeung K, Sandor GG, Judge DP, Dietz HC, van Breemen C. Loss of elastic fiber integrity and reduction of vascular smooth muscle contraction resulting from the upregulated activities of matrix metalloproteinase-2 and −9 in the thoracic aortic aneurysm in marfan syndrome. Circ. Res 2007;101:512–522 [DOI] [PubMed] [Google Scholar]
- 74.Inamoto S, Kwartler CS, Lafont AL, et al. Tgfbr2 mutations alter smooth muscle cell phenotype and predispose to thoracic aortic aneurysms and dissections. Cardiovasc. Res 2010;88:520–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kuang SQ, Kwartler CS, Byanova KL, Pham J, Gong L, Prakash SK, Huang J, Kamm KE, Stull JT, Sweeney HL, Milewicz DM. Rare, nonsynonymous variant in the smooth muscle-specific isoform of myosin heavy chain, myh11, r247c, alters force generation in the aorta and phenotype of smooth muscle cells. Circ. Res 2012;110:1411–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiao Y, Li G, Korneva A, Caulk AW, Qin L, Bersi MR, Li Q, Li W, Mecham RP, Humphrey JD, Tellides G. Deficient circumferential growth is the primary determinant of aortic obstruction attributable to partial elastin deficiency. Arterioscler. Thromb. Vasc. Biol 2017;37:930–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Osei-Owusu P, Knutsen RH, Kozel BA, Dietrich HH, Blumer KJ, Mecham RP. Altered reactivity of resistance vasculature contributes to hypertension in elastin insufficiency. Am J Physiol Heart Circ Physiol. 2014;306:H654–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.