

Abstract
Chromatin undergoes extensive reprogramming during immune cell differentiation. Here we report repression of controlled histone H3 amino-terminus proteolytic cleavage (H3ΔN) during monocyte-to-macrophage development. This abundant histone mark in human peripheral blood monocytes is catalyzed by neutrophil serine proteases (NSPs) cathepsin G, neutrophil elastase, and proteinase 3. NSPs are repressed as monocytes mature into macrophages. Integrative epigenomic analysis reveals widespread H3ΔN distribution across the genome in a monocytic cell line and primary monocytes, which becomes largely undetectable in fully differentiated macrophages. H3ΔN is enriched at permissive chromatin and actively transcribed genes. Simultaneous NSP depletion in monocytic cells results in H3ΔN loss and further increase in chromatin accessibility, which likely primes the chromatin for gene expression reprogramming. Importantly, H3ΔN is reduced in monocytes from patients with systemic juvenile idiopathic arthritis (sJIA), an autoinflammatory disease with prominent macrophage involvement. Together, we uncover an epigenetic mechanism that primes the chromatin to facilitate macrophage development.
Introduction
Controlled histone proteolysis is evolutionarily conserved from unicellular eukaryotes to humans1. Multiple classes of proteases show catalytic activities toward the histone H3 tail, including serine2, 3, cysteine4, aspartyl5, and metallo-proteases6. Truncation of the highly basic and heavily modified H3 N-terminal tail can alter its electrostatic interaction with DNA7 and impact chemical modifications with pivotal cell regulatory functions8. Proteolytic processing of H3 is frequently elevated during cellular differentiation, such as embryonic stem cell development4, myogenesis9 and osteoclastogenesis6, suggesting its crucial roles in regulating cell fate determination and development.
We report that neutrophil serine proteases (NSPs) cathepsin G (CTSG), neutrophil elastase (ELANE), and proteinase 3 (PRTN3) jointly catalyze histone H3 amino-terminus proteolytic cleavage (H3ΔN) in monocytes. NSPs are localized to the chromatin, generating H3ΔN that is integrated into nucleosome arrays. During monocyte-to-macrophage differentiation, NSPs and H3ΔN are repressed. Integrative epigenomic analyses reveal H3ΔN association with permissive chromatin and active transcription. Simultaneous ablation of NSPs (ΔNSPs) abolishes H3ΔN but only modestly impacts the transcriptome in the basal condition. Upon PMA-induced differentiation, ΔNSPs cells show a greater response in gene expression. Our data support a model in which chromatin reconfiguration by H3ΔN repression may prime the chromatin to facilitate gene expression reprogramming during cellular differentiation. Further, H3ΔN is repressed in monocytes from patients with sJIA, and the repression is mediated in part by ferritin, a serum protein frequently elevated in sJIA patients10, 11. Together, we identify a non-canonical nuclear function of NSPs in epigenetic gene regulation.
Results
CTSG, ELANE, and PRTN3-Mediated H3ΔN in Monocytes
Epigenetic landscape profiling using cytometry by Time-Of-Flight (EpiTOF)12 analysis revealed cell type-specific enrichment of proteolytically processed histone H3 cleaved between amino acids alanine 21 and threonine 22 (H3ΔNThr22) in HLA-DR+CD33+CD14+ monocytes (Fig. 1a). H3ΔNThr22 was not associated with cell death (Extended Data Fig. 1a), and the enrichment was specific in CD14+CD16- classical monocytes (referred to as “monocytes” below) but not in CD14+CD16+ intermediate or CD14-CD16+ nonclassical subsets13 (Extended Data Fig. 1b). At the single-cell level, H3ΔNThr22 abundance is negatively correlated with class II-MHC level (Extended Data Fig. 1c). We validated the cell type-specific enrichment of H3ΔNThr22 in purified monocytes (Fig. 1b). This molecular signature was preserved in monocytic cell lines U937 and THP-1 (Fig. 1c). Biochemical fractionation analysis demonstrated H3ΔN chromatin localization (Fig. 1d and Extended Data Fig. 1d), suggesting its physiological significance in regulating DNA-templated biological processes.
Fig. 1. CTSG, ELANE, and PRTN3 Catalyze H3ΔN in Monocytes.
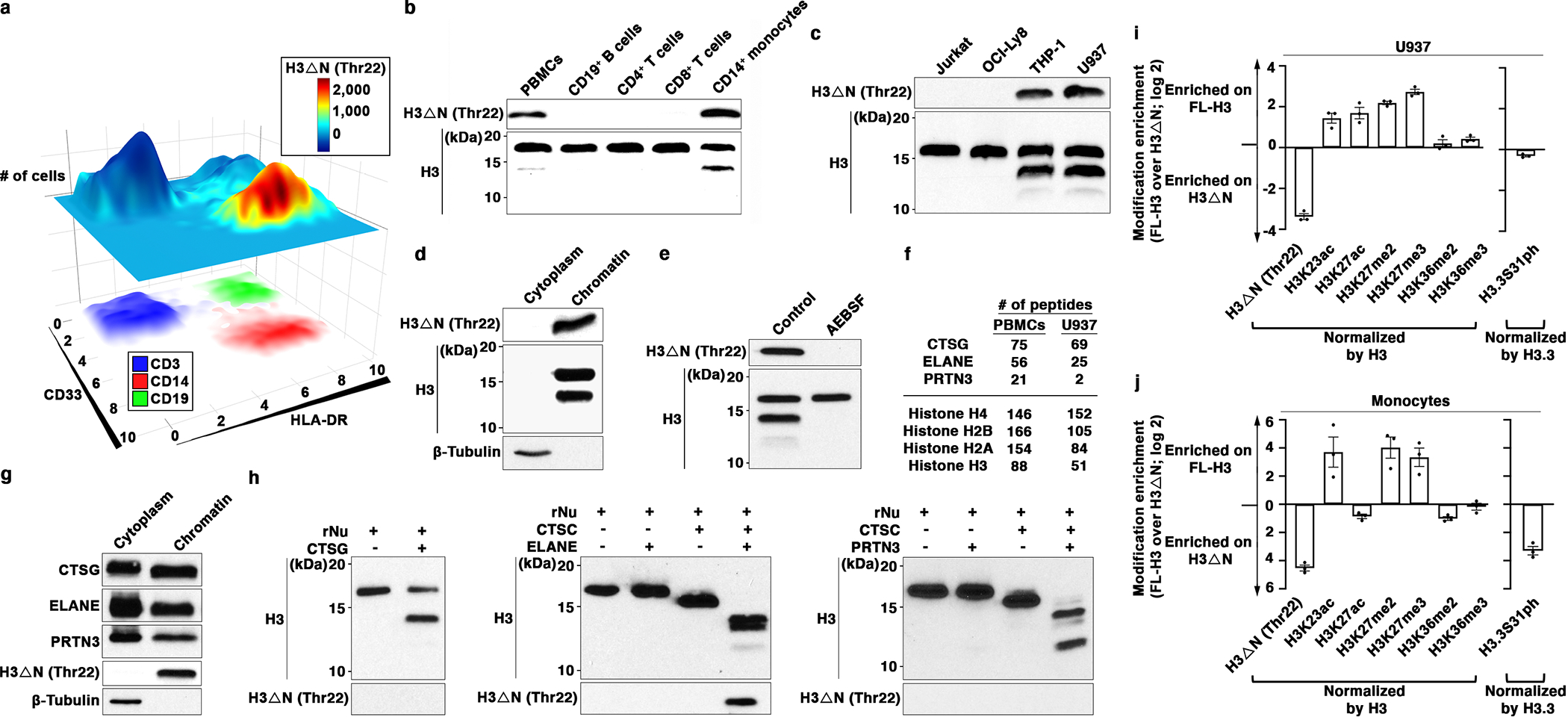
(a) H3ΔNThr22 distinguishes CD14+ monocytes. EpiTOF analysis of PBMCs from 20 healthy volunteers as described previously12. X- and y-axes, HLA-DR and CD33 levels, respectively. Top plane: color, normalized H3ΔNThr22 level; height, cell count. Bottom plane: color, CD3 (blue), CD14 (red) and CD19 (green) levels.
(b) Monocyte-specific H3ΔN enrichment relative to lymphocytes. Western blot analysis of whole cell extract (WCE) from purified primary human immune cells.
(c) Monocytic cell lines retain high H3ΔN levels in comparison with other cell lines of lymphoid origins. Western blot analysis of WCE from the indicated cell lines.
(d) H3ΔN native chromatin incorporation. Western blot analysis of U937 cells biochemically separated into cytoplasmic and chromatin fractions.
(e) Serine protease-mediated H3ΔN. Western blot analysis of WCE from AEBSF-treated U937 cells. Control, PBS-treated.
(f and g) Chromatin localization of CTSG, ELANE, and PRTN3. Protein identification of purified chromatin from PBMCs (left) or U937 cells (right) using mass spectrometry. The numbers of peptides mapped to the indicated histone proteins and serine proteases are shown (f). Western blot analysis of cytoplasmic and chromatin fractions purified from U937 cells (g). Molecular weight markers, see Source Data.
(h) CTSG, ELANE, and PRTN3 catalyze controlled H3 proteolytic cleavage in vitro. Immunoblotting analysis of protease assays using recombinant CTSG (left), ELANE (middle), or PRTN3 (right) and recombinant nucleosomes as substrate. ELANE and PRTN3 activities require pre-activation by CTSC51.
(i and j) Differential histone modifications between FL- and truncated H3. Quantification of the indicated histone marks between FL-H3 and H3ΔN in U937 cells (i) or primary monocytes (j). Y-axis, FL-H3-over-H3ΔN ratio of the indicated histone marks normalized against the ratio of H3 or H3.3 (for H3.3S31ph). Data represent mean ± S.E.M. (three biological replicates).
To identify the protease(s) catalyzing H3ΔN in monocytes, we first tested proteases with known H3-cleaving activities. Depletion of cathepsin L, which processes H3 during mouse embryonic stem cell development4, did not impact H3ΔN in monocytes (Extended Data Fig. 1e). H3ΔN both in U937 and THP-1 cells was unaffected by MMP9 inhibition, which targets H3 during osteoclastogenesis (Extended Data Fig. 1f)6, or E64d, a cell-permeable nonselective cysteine protease inhibitor (Extended Data Fig. 1g)14. Strikingly, a broad-spectrum serine protease inhibitor 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF) abolished H3ΔN in monocytic cells (Fig. 1e and Extended Data Fig. 1h). Thus, we limited our search scope to serine proteases present at the chromatin. Protein identification using mass spectrometry found abundant CTSG, ELANE, and PRTN3 at the native chromatin isolated from PBMCs and U937 cells (Fig. 1f). Chromatin localization of individual NSPs was validated by immunoblotting (Fig. 1g and Extended Data Fig. 1i). NSPs remained chromatin-bound after extensive washes of chromatin pellet and were solubilized together with histones and HP-1 in a high-salt solution15 (Extended Data Fig. 1j). Next, in vitro protease assay using NSPs and recombinant nucleosomes16 showed that all three NSPs demonstrated regulated proteolytic activities towards H3 on the physiological substrate (Fig. 1h). Notably, H3ΔNThr22 was only detected in the protease assay using ELANE but not in others with CTSG or PRTN3, indicating that ELANE is the only NSP generating H3ΔNThr22 in vitro. Tandem mass spectrometry analysis of protease assay revealed that CTSG preferentially cleaved at Leu20, and PRTN3 primarily catalyzed Lys23 cleavage (Extended Data Fig. 1k). Both ELANE and PRTN3 efficiently catalyzed the cleavage at Thr32. Together, CTSG, ELANE, and PRTN3 generate multiple truncated H3 products.
Immunoblotting revealed distinct chemical modification profiles between FL-H3 and H3ΔN in U937 cells (Fig. 1i and Extended Data Fig. 1l) and in primary monocytes (Figure 1j and Extended Data Fig. 1m). Di- and tri-methylation of H3 at lysine 27 (H3K27me2 and H3K27me3, respectively) were present almost exclusively on FL-H3. In contrast, di- and tri- methylation of H3 at lysine 36 (H3K36me2 and H3K36me3 respectively) were indiscriminately distributed between both H3 forms. In U937 cells, acetylation at lysine 27 (H3K27ac) was also enriched on FL-H3. However, H3K27ac was near evenly distributed between FL-H3 and H3ΔN in primary monocytes. Proteolytic processing also occurred on histone variant H3.3. Phosphorylation at serine 31 (H3.3S31ph) was enriched on H3ΔN in primary monocytes but not in U937 cells. The difference may be due to active cell proliferation of U937 cells17. Together, these data suggest crosstalk between H3ΔN and other chemical modifications and its functional significance.
Altered Cell Morphology and Functions Upon NSP Depletion
CTSG, ELANE and PRTN3 arose from a common ancestor through gene duplication during evolution and remain highly homologous in primary sequence18. Genetic ablation of individual NSPs did not affect H3ΔN (Extended Data Fig. 2a–c), consistent with our in vitro findings. Notably, in agreement with the in vitro data (Fig. 1h), ELANE depletion abrogated H3ΔNThr22 (Extended Data Fig. 2b), indicating that H3ΔNThr22 is generated exclusively by ELANE both in vitro (Fig. 1h, middle) and in cells (Extended Data Fig. 2b). The H3ΔNThr22 antibody used in this study was highly specific, showing negligible cross-reactivity with other H3ΔN species (Extended Data Fig. 2d).
Strikingly, H3ΔN was abolished in three U937 cell clones depleted of CTSG, ELANE, and PRTN3 simultaneously (ΔNSPs) (Fig. 2a). Control cells underwent the same viral transduction processing as ΔNSPs clones and were cultured in the presence of an identical set of antibiotics. Clonal selection of control cells did not alter H3ΔN level (Extended Data Fig. 2e), cell morphology (Extended Data Fig. 2f), or CD11b and CD11c expression (Extended Data Fig. 2g). Thus, we used control cells without clonal selection for downstream analyses to preserve potential cellular heterogeneity.
Fig. 2. Morphologic and Functional Alterations in Monocytic Cells Ablated of CTSG, ELANE, and PRTN3.
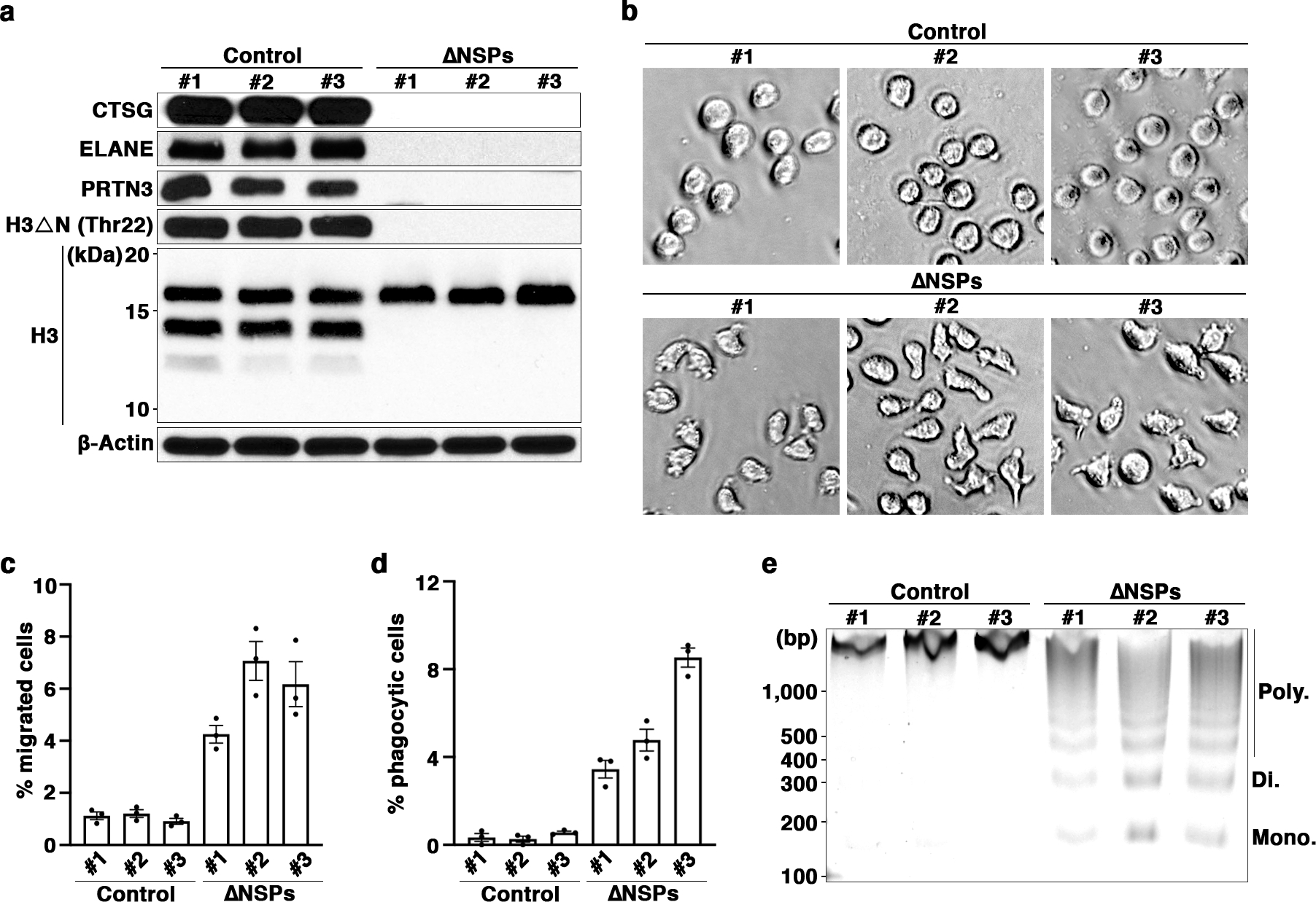
(a) CTSG, ELANE, and PRTN3 jointly catalyze H3ΔN in U937 cells. Western blot analysis of WCE from monoclonal U937 cells stably expressing CRISPR-Cas9 and sgRNAs targeting CTSG, ELANE, and PRTN3 simultaneously. Control cells express CRISPR-Cas9 without sgRNA.
(b) Morphological alterations of ΔNSPs cells. Representative light microscopy images of ΔNSPs cells as in (a).
(c and d) Functional alterations of ΔNSPs cells. Transwell cell migration (c) and phagocytosis (d) analyses of ΔNSPs cells described in (a). Data represent mean ± S.E.M. (three technical replicates).
(e) Increased sensitivity to MNase digestion upon NSP depletion. MNase sensitivity analysis of the indicated cells. DNA marker and the expected sizes of mono-, di-, and poly-nucleosomes are shown.
ΔNSPs cells exhibited an irregular shape (Fig. 2b), increased migration ability (Fig. 2c), and phagocytic capability (Fig. 2d). Increased sensitivity to micrococcal nuclease (MNase) digestion in ΔNSPs cells indicated altered global chromatin dynamics in response to NSP depletion (Figure 2e). Importantly, NSP depletion minimally impacted cell viability (Extended Data Fig. 2h). To determine if the molecular, morphological and functional alterations in ΔNSPs cells were dependent upon H3ΔN loss at chromatin, we established an experimental system to reintroduce H3ΔN into ΔNSPs cells. An epitope-tagged exogenous H3 with a Tobacco Etch Virus (TEV) protease substrate sequence inserted between Ala21 and Thr22 (H3ENLYFQS-FLAG) was introduced along with a tetracycline-inducible TEV protease (iTEV) into ΔNSPs cells (Extended Data Fig. 2i). H3ENLYFQS-FLAG can be electrophoretically resolved from endogenous H3 (Extended Data Fig. 2j). Biochemical fractionation analysis showed H3ENLYFQS-FLAG chromatin integration (Extended Data Fig. 2k). Upon doxycycline treatment, all cells remained over 93% viable (Extended Data Fig. 2l). We observed an electrophoretically fast-migrating band that matched the molecular weight of N-terminally cleaved H3ENLYFQS-FLAG (Extended Data Fig. 2m), with the cleavage product representing approximately one third of exogenous H3. ΔNSPs cells containing cleaved H3ENLYFQS-FLAG returned to wild-type cell morphology (Extended Data Fig. 2n) and showed reduced migration ability (Fig. 2o) and phagocytosis capability (Fig. 2p). Further, reintroducing H3ΔN into ΔNSPs cells resulted in reduced MNase sensitivity (Extended Data Fig. 2q), suggesting a direct role of H3ΔN in regulating chromatin architecture. While exogenous H3ENLYFQS-FLAG represents a small fraction of bulk H3, and iTEV protease-mediated proteolysis unlikely recapitulates the endogenous H3ΔN genomic distribution, these data suggest that the morphological and functional phenotypes mimicking macrophages in ΔNSPs cells are regulated, at least in part, by H3ΔN. Moreover, these data also provide direct evidence that H3ΔN affects global chromatin structure.
NSP and H3ΔN Repression During Macrophage Development
Compelling evidence from monocytic cell lines led us to extend our investigation to primary monocytes and monocyte-derived macrophages19. Mass cytometry analysis of monocytes differentiating into macrophages ex vivo revealed time-dependent H3ΔNThr22 loss and elevated CD68, CD163, and HLA-DR expression indicative of macrophage maturation20, 21 (Fig. 3a and Extended Data Fig. 3a). At day five after induction for differentiation, H3ΔNThr22 was undetectable in over 70% of cells by mass cytometry. H3ΔNThr22 repression was not associated with cell death (Extended Data Fig. 3b). At day seven, all H3ΔN species were largely undetectable by immunoblotting in fully mature macrophages (Fig. 3b and Extended Data Fig. 3c), indicating that all species of truncated H3 were repressed during macrophage development. Functionally, mature macrophages efficiently engulfed osmium-labelled E. coli particles in mass cytometry analysis22 (Fig. 3c and Extended Data Fig. 3d). Cells with undetectable H3ΔNThr22 showed the greatest degree of phagocytosis, suggesting that the loss of H3ΔNThr22 is indicative of full maturation of macrophages. H3ΔN depletion was maintained in macrophages classically or alternatively activated by LPS and IFN-γ or IL-4, respectively23 (Fig. 3d and Extended Data Fig. 3e). Moreover, we found that NSPs were repressed during macrophage differentiation (Fig. 3e and Extended Data Fig. 3f), explaining H3ΔN depletion in mature macrophages. In U937 cells stimulated with PMA to induce macrophage-like phenotypes, NSP transcripts (Extended Data Fig. 3g) and proteins (Extended Data Fig. 3h) were both suppressed. Together, our data demonstrate that NSPs and H3ΔN are repressed as monocytes differentiate into macrophages.
Fig. 3. NSP and H3ΔN Repression During Monocyte-to-Macrophage Differentiation.
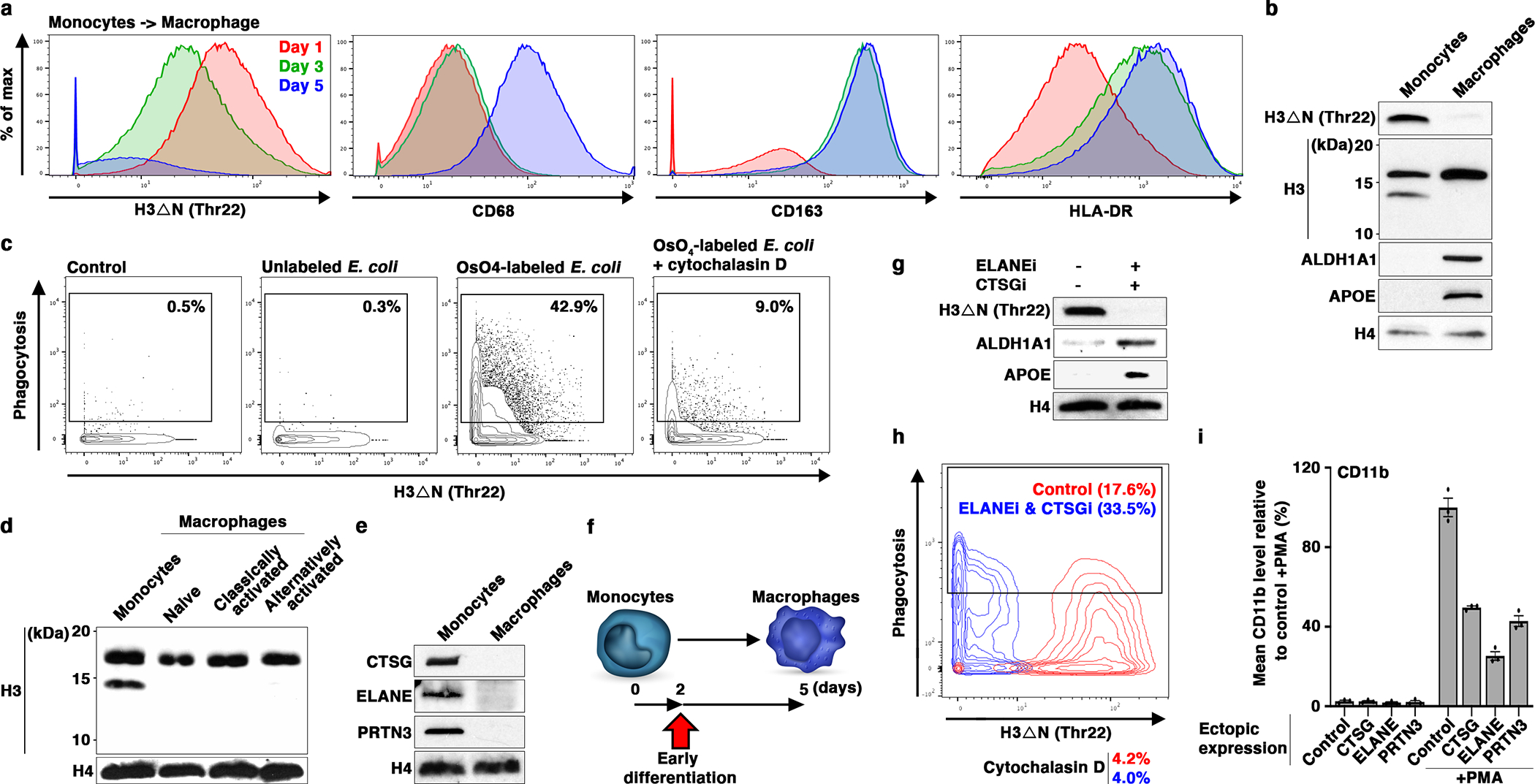
(a and b) H3ΔN repression as monocytes differentiate into macrophages. Mass cytometry (a) and western blot (b) analyses of primary monocytes and the matching monocyte-derived macrophages. Cells collected at days one (red), three (green), and five (blue) are analyzed by mass cytometry (a). Day one and seven samples are subject to immunoblotting analysis (b). Molecular weight markers, see Source Data.
(c) Increased phagocytosis capability of differentiating monocytes with repressed H3ΔN. Mass cytometry analysis of cells as in (a) (day five) incubated with osmium-labelled E. coli to assess phagocytosis capability22. Control, no E coli particle is added; cytochalasin D, control to show the specificity of phagocytosis measurement. H3ΔNThr22 (x-axis) and osmium (y-axis) levels measured by mass cytometry are shown.
(d) H3ΔN repression is maintained in polarized macrophages. Western blot analysis of peripheral blood monocytes, naïve macrophages generated as in (b), classically activated macrophages polarized by LPS and IFN-γ, and alternatively activated macrophages polarized by IL-4.
(e) CTSG, ELANE, and PRTN3 repression during monocyte-to-macrophage differentiation. Western blot analysis of the samples as in (b) using the indicated antibodies. Molecular weight markers, see Source Data.
(f-h) Accelerated macrophage development by pharmacological inhibition of NSPs. Overview of the chronological timeline for this experiment (f). Western blot analysis of WCE from peripheral blood monocytes cultured in the presence or absence of ELANE inhibitor GW311616 in combination with CTSG inhibitor CAS 429676–93-7 for two days (g). Molecular weight markers, see Source Data. Phagocytosis analysis of the cells as in (g) using mass cytometry and osmium-labeled E. coli (h).
(i) Constitutive NSP expression suppresses monocytic cell differentiation. U937 cells stably expressing the indicated NSPs (x-axis) under the control of cytomegalovirus promoter are induced for differentiation by PMA. CD11b expression is determined by mass cytometry. Data represent mean ± S.E.M. (three technical replicates).
Treatment of primary monocytes with selective CTSG and ELANE inhibitors promoted macrophage development. At day two after differentiation was initiated (Fig. 3f), differentiating cells cultured in the presence of CTSG and ELANE inhibitors expressed higher macrophage markers APOE and ALDH1A1 (Fig. 3g and Extended Data Fig. 3i) and demonstrated elevated phagocytosis capability (Fig. 3h and Extended Data Fig. 3j), suggesting accelerated macrophage development. Next, we transduced U937 cells with lentiviral vectors encoding individual NSPs under cytomegalovirus promoter control. Exogenous NSP expression was unaffected by PMA-induced NSP repression during cellular differentiation (Extended Data Fig. 3k). H3ΔN levels were largely maintained in these modified cell lines where a single NSP remained highly expressed. Ectopic overexpression of individual NSPs suppressed CD11b (Fig. 3i) and CD11c (Extended Data Fig. 3l) expression by as much as 70% upon PMA-induced differentiation. Together, these data suggest critical roles for NSPs in regulating monocyte-to-macrophage differentiation.
Widespread H3ΔN Genomic Distribution at Accessible Chromatin
To characterize the molecular functions of H3ΔN in monocytes, we adopted an integrative epigenomic approach, performing ChromatinImmunoPrecipitation followed by high-throughput sequencing (ChIP-seq) on H3ΔNThr22 and bulk H3 in wild-type U937 cells, with paired Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) and RNA sequencing (RNA-seq) on ΔNSPs and control cells. Affinity reagents were validated for ChIP-seq application (Extended Data Fig. 4a). H3ΔNThr22 immunoprecipitation of sheared chromatin containing predominantly mono-nucleosomes (Extended Data Fig. 4b) modestly enriched for the fast-migrating band observed in immunoblotting (Extended Data Fig. 4a), suggesting that a large proportion of immunoprecipitated nucleosomes may be asymmetrically cleaved at H3. Under a 5% Irreproducible Discovery Rate (IDR)24 cutoff, we identified 15,504 peaks enriched with H3ΔNThr22 consistent between both biological replicates (Fig. 4a). H3ΔNThr22 peaks were dispersed across the genome (Extended Data Fig. 4c), with preferential enrichment in genic over intergenic regions (Fig. 4b). 9,540 genes were found to have one or more H3ΔNThr22 peaks, mostly in promoters, exons, and untranslated regions (UTRs). Quantitative PCR (qPCR) analysis validated ChIP-seq findings (Fig. 4c, 4d, Extended Data Fig. 4d, and 4e). The signals were specific as they were nearly undetectable in ΔNSPs cells. Surprisingly, at the loci where H3ΔNThr22 ChIP-seq signals were low, we also detected considerable H3ΔNThr22 enrichment in control cells. These data indicate that while H3ΔNThr22 is enriched at specific genomic sites, particular in genic regions, it is also widely distributed at a low level across the genome. Moreover, antibodies against NSPs co-immunoprecipitated nucleosomal H3 under the stringent ChIP condition, further supporting NSP chromatin localization (Extended Data Fig. 4f). ChIP analysis using an antibody against ELANE, which is the sole NSP catalyzing H3ΔNThr22 in cells (Extended Data Fig. 2b), revealed its presence at loci with both high and low H3ΔNThr22 enrichment (Fig. 4d and Extended Data Fig. 4e). The signals were specific as they were largely undetectable in ΔNSPs cells. These data suggest widespread ELANE occupancy across the genome and that site-specific H3ΔNThr22 enrichment is controlled by mechanisms other than ELANE genomic localization.
Fig. 4. Widespread H3ΔN Genomic Distribution and Its Permissive Chromatin Association in Monocytic Cells.
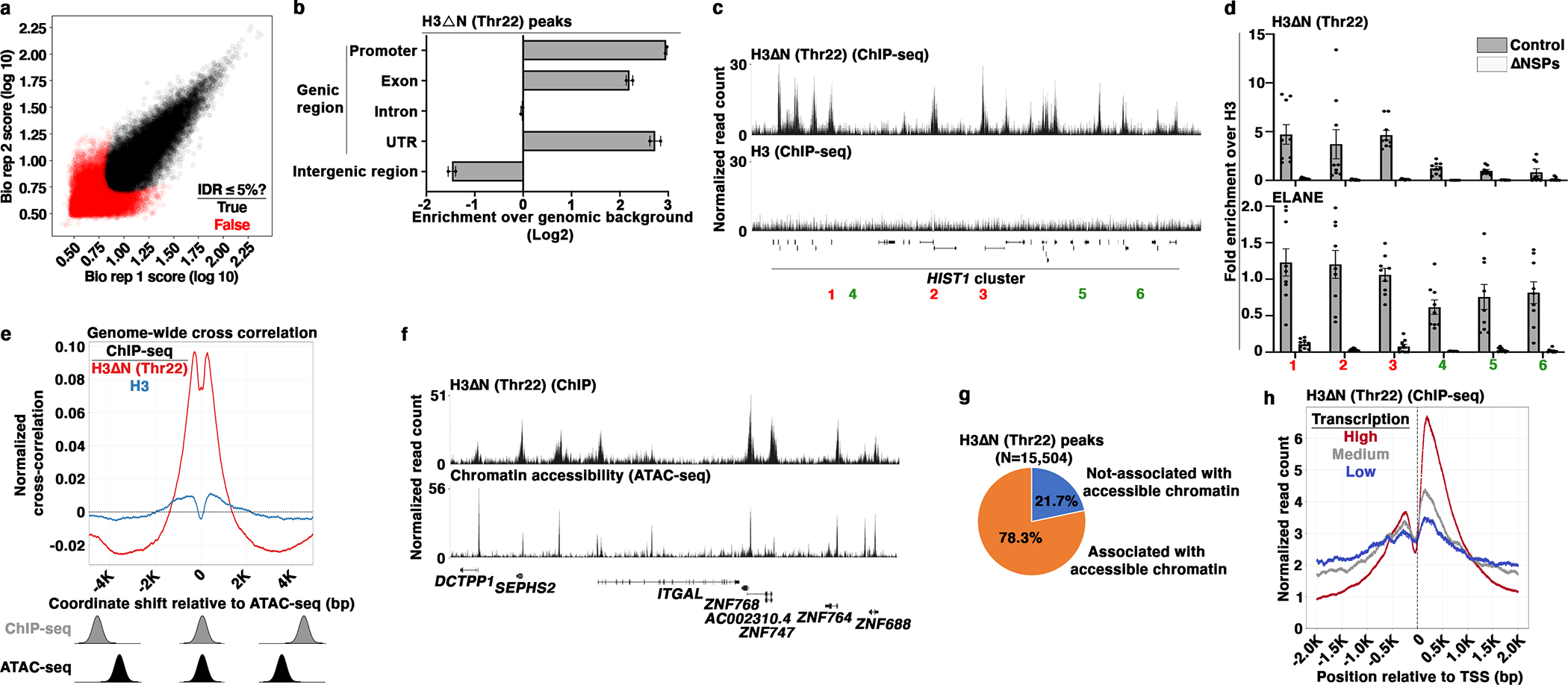
(a) Identification of H3ΔNThr22-enriched peaks in wild-type U937 cells. Scaled signal intensities of H3ΔNThr22 ChIP-seq peaks in biological replicate one (x-axis) and two (y-axis). Peaks that pass 5% IDR24 threshold are labeled (black). Red, peaks with IDR > 5%.
(b) Genic region enrichment of H3ΔNThr22. X-axis, the enrichment of H3ΔNThr22 peaks in the indicated genomic regions. Center line, genomic distribution with no enrichment. Data represent mean ± S.D. (two biological replicates).
(c and d) H3ΔNThr22 enrichment and ELANE occupancy at the HIST1 locus. Representative genomic tracks of H3ΔNThr22 (top) and bulk H3 (bottom) ChIP-seq data. Regions with high (red) or low (green) H3ΔNThr22 are tested (c). qPCR analysis of H3ΔNThr22 (top) or ELANE (bottom) ChIP DNA from control (gray) or ΔNSPs (white) cells using the indicated primer pairs. Y-axes, fold enrichment over signals from bulk H3. Data represent mean ± S.E.M. (three biological replicates (three control cell lines or three ΔNSPs clones) with three technical replicates (N=9)) (d).
(e) Association between H3ΔNThr22 enrichment and permissive chromatin at the nucleotide level. Cross correlation analysis that overlays the H3ΔNThr22 (red) or bulk H3 (blue) ChIP-seq datasets over the ATAC-seq dataset. Aggregate data from 1,000 randomly selected start points are shown. Correlations are computed as the ChIP-seq datasets are shifted with a distance in base pair depicted at the x-axis relative to the ATAC-seq dataset. Y-axis, normalized correlation score.
(f and g) Association between H3ΔNThr22 enrichment and permissive chromatin at the peak level. Representative genomic tracks of H3ΔNThr22 ChIP-seq (top) and ATAC-seq (bottom) datasets (f). Pie chart depicts the percentage of H3ΔNThr22 ChIP-seq peaks that are associated with ATAC-seq peaks (g).
(h) Association between H3ΔNThr22 enrichment and active transcription. H3ΔNThr22 levels at the TSS-proximal regions of genes with high (red), medium (gray), or low (blue) transcription activities (GSE10756625). X-axis, base pair relative to TSS; y-axis, H3ΔNThr22 ChIP-seq signal intensity.
To investigate the relationship between H3ΔNThr22 enrichment and chromatin accessibility, we overlaid the H3ΔNThr22 ChIP-seq genomic track over the ATAC-seq datasets from control cells. We performed a cross correlation analysis at the nucleotide level. We discovered a positive correlation between H3ΔNThr22 ChIP-seq and ATAC-seq signals at individual nucleotides (Fig. 4e), with the highest correlation observed as the overlay was shifted by approximately 250 bp up- or downstream. The offset suggests accessible sequences surrounding H3ΔNThr22-containing nucleosomes. With a 5% FDR cutoff, we identified 94,615 ATAC-seq peaks in control cells. H3ΔNThr22 ChIP-seq and ATAC-seq peaks were highly concordant (Fig. 4f and Extended Data Fig. 4g). 78.3% of H3ΔNThr22 peaks (Fig. 4a) were associated with one or more of these accessible sites (Fig. 4g). Moreover, H3ΔNThr22 was strongly enriched at the transcriptional start site (TSS) of actively transcribed genes relative to those with medium or low expression (GSE10756625) (Fig. 4h and Extended Data Fig. 4h). Together, these data demonstrate H3ΔNThr22 enrichment at permissive chromatin and its positive correlation with gene expression.
Next, we extended the epigenomic analysis to primary monocytes and the matching monocyte-derived macrophages. We isolated peripheral blood monocytes from three healthy volunteers (Extended Data Fig. 5a) and generated monocyte-derived macrophages19. The differentiation was confirmed by increased CD68 expression (Extended Data Fig. 5b) and other macrophage markers (Fig. 3a and Extended Data Fig. 3a). Three sets of paired monocytes and macrophages were subject to ChIP-seq analysis of H3ΔNThr22 and bulk H3. Peak-calling algorithm26 identified 4,851, 8,617, and 11,807 peaks in the three monocyte samples using a FDR 5% cutoff (Fig. 5a). 6,150 H3ΔNThr22 peaks were shared between two or three donors, which we used for downstream analyses. Strikingly, under the same 5% FDR threshold, 0, 282, and 1 H3ΔNThr22 peaks were significant in macrophages from the three donors, respectively. H3ΔNThr22 distribution was widespread in monocytes (Extended Data Fig. 5c) with genic region enrichment (Fig. 5b). 5,011 genes contained one or more H3ΔNThr22 peaks, including transcription factor PU.1 (SPI1) (Fig. 5c) and the p65 (RELA) subunit of NF-κB (Extended Data Fig. 5d). qPCR analysis validated H3ΔNThr22 enrichment at ChIP-seq peaks (Fig. 5d and Extended Data Fig. 5e). At loci with low H3ΔNThr22 ChIP-seq signals in monocytes, we detected considerable H3ΔNThr22, suggesting H3ΔNThr22 presence at a low level across the genome. The ChIP signals were specific as nearly no H3ΔNThr22 was detected in mature macrophages. ELANE occupancy was observed at loci with both high and low H3ΔNThr22, indicating mechanisms other than ELANE localization in controlling H3ΔNThr22 genomic distribution. Overall, the H3ΔNThr22 genomic distribution in primary monocytes is highly similar to that in U937 cells.
Fig. 5. Permissive Chromatin and Active Transcription Association of H3ΔN in Primary Monocytes.
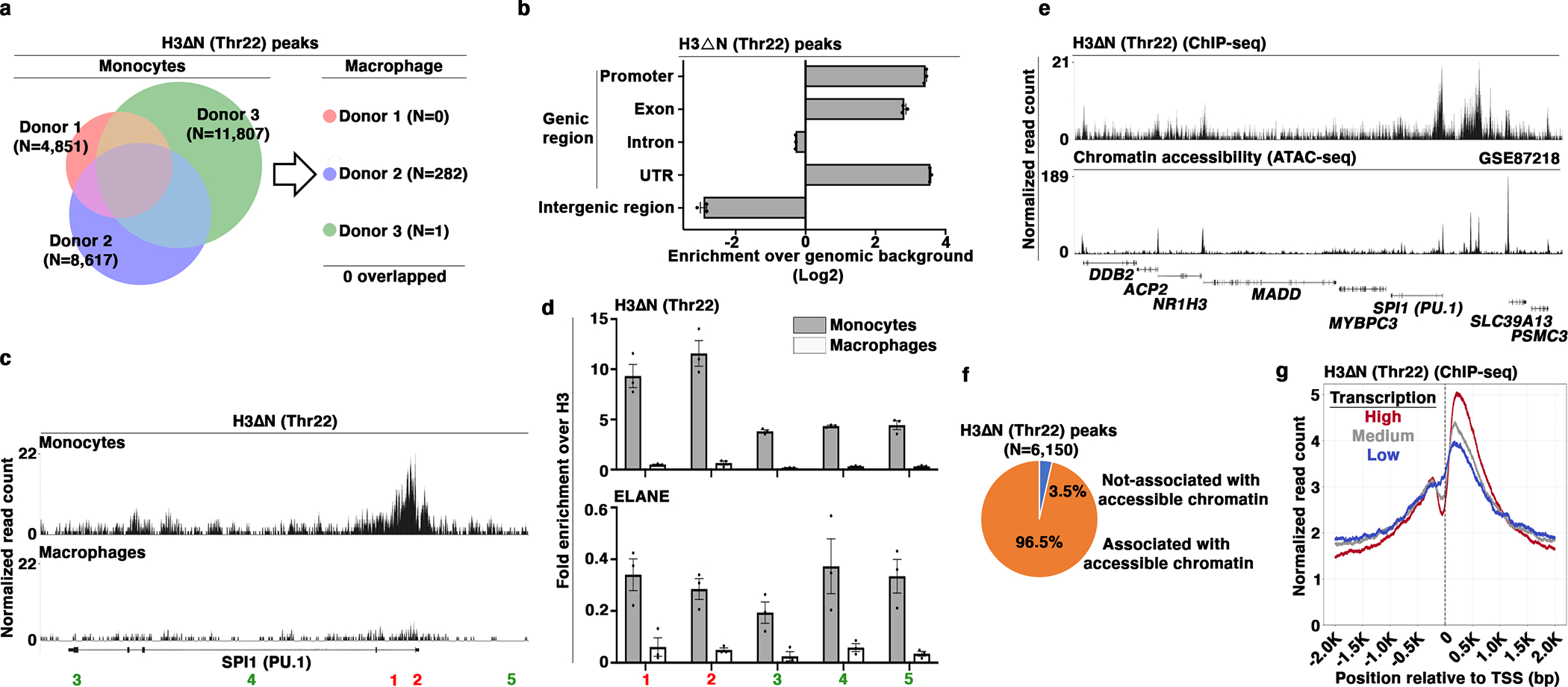
(a) Identification of H3ΔNThr22-enriched peaks in primary monocytes and paired monocyte-derived macrophages. Venn diagram depicts the number of H3ΔNThr22-enriched peaks in monocytes (left). The numbers of peaks identified in macrophages from the same donors are shown (right).
(b) Genic region enrichment of H3ΔNThr22 in primary monocytes. X-axis, H3ΔNThr22 peak enrichment in the indicated genomic regions. Center line, genomic distribution with no enrichment. Data represent mean ± S.D. (three biological replicates).
(c and d) H3ΔNThr22 enrichment and ELANE occupancy at the PU.1 (SPI1) locus in primary monocytes. Representative genomic tracks of H3ΔNThr22 ChIP-seq data from primary monocytes (top) or macrophages (bottom) (c). Genomic regions with high (red) or low (green) H3ΔNThr22 ChIP-seq signals are tested. qPCR analysis of H3ΔNThr22 enrichment (top) or ELANE occupancy (bottom) in primary monocytes (gray) or in the matching monocyte-derived macrophages (white) using the indicated primer pairs. Y-axes, fold enrichment over bulk H3. Data represent mean ± S.E.M. (three technical replicates) (d).
(e and f) Association between H3ΔNThr22 enrichment and permissive chromatin at the peak level in primary monocytes. Representative genomic tracks of H3ΔNThr22 ChIP-seq (top) and ATAC-seq (GSE8721827) (bottom) datasets from primary monocytes (e). Pie chart depicts the percentage of H3ΔNThr22 ChIP-seq peaks that are associated with ATAC-seq peaks (f).
(g) Association between H3ΔNThr22 enrichment and active transcription in primary monocytes. H3ΔNThr22 levels at the TSS-proximal regions for genes with high (red), medium (gray), or low (blue) transcription activities (GSE509919).
To investigate the relationship between H3ΔNThr22 enrichment and chromatin accessibility in primary cells, we leveraged a publicly available ATAC-seq dataset from purified monocytes (GSE8721827). Using a 5% FDR cutoff, we found a high degree of peak concordance between both datasets in the greater genomic regions surrounding PU.1 and RELA (Fig. 5e and Extended Data Fig. 5f). Strikingly, 96.5% of H3ΔNThr22 ChIP-seq peaks were associated with one or more ATAC-seq peaks (Fig. 5f). Actively transcribed genes showed the highest H3ΔNThr22 level in their TSS-proximal regions (GSE509919) (Fig. 5g). These data provide strong support for H3ΔNThr22 enrichment at open chromatin and actively transcribed genes in primary monocytes. Together, integrative analyses of ChIP-seq and ATAC-seq datasets from primary monocytes reveal H3ΔNThr22 association with permissive chromatin and active transcription, a pattern similar to that observed in U937 cells.
Chromatin Priming Mediated by NSP and H3ΔN Repression
Transcriptomic analysis by RNA-seq discovered modest gene expression changes between ΔNSPs and control cells (Fig. 6a), with 170 and 220 genes up- or down-regulated in ΔNSPs relative to control cells under a 5% FDR cutoff, respectively. Integrative analysis with the ATAC-seq dataset found increased chromatin accessibility at genes upregulated in ΔNSPs cells (Extended Data Fig. 6a), whereas the chromatin at downregulated genes became inaccessible. Gene ontology analysis revealed that genes upregulated upon NSP and H3ΔN depletion were involved in immune regulation (GO: 0002376, p<6.8E-06; GO: 0002684, 5.3E-04) (Extended Data Fig. 6b), including AIF1, CXCL8/IL8, GPR183/EBI2, ID2, JUN, and JUNB that have been linked to macrophage development and polarization. Upregulation of these genes is in agreement with the morphological and functional alterations in ΔNSPs cells (Fig. 2b–d). Importantly, reintroduction of H3ΔN into ΔNSPs cells repressed the expression of these genes (Fig. 6b). These data support a direct role of H3ΔN in transcriptional regulation and that H3ΔN repression affects genes regulating macrophage-associated phenotypes.
Fig. 6. Global Chromatin Accessibility Increase Upon NSP and H3ΔNThr22 Depletion.

(a) Differential gene expression in response to NSP depletion. Heatmap representation of differentially expressed genes between ΔNSPs and control cells under a FDR 5% cutoff. Z-score transformed gene expression levels (color) of differential genes (y-axis) in the indicated cell lines (x-axis). Dendrogram, unsupervised clustering.
(b) Reintroducing H3ΔN into ΔNSPs cells attenuates the expression of genes upregulated in ΔNSPs cells. Gene expression analysis of ΔNSPs cells with (white) or without (gray) H3ΔN reintroduction as in Extended Data Fig. 2m. Data represent mean ± S.E.M. (three technical replicates and two biological replicates (independent ΔNSPs clones) (N=6)). Statistical significance is determined by two-tailed Student’s t-test. **, P≤0.01; ***, P≤0.001.
(c) Increased chromatin accessibility in ΔNSPs cells. ATAC-seq analysis of three controls and three ΔNSPs cell lines. The numbers of differentially accessible peaks are shown. Top, differentially accessible peaks in ΔNSPs clones over the average of controls; bottom, differentially accessible peaks in controls over the average of ΔNSPs clones.
(d-f) Increased chromatin accessibility at H3ΔNThr22-enriched peaks upon NSP and H3ΔNThr22 depletion. Venn diagram depicts the number of H3ΔNThr22 peaks identified in wild-type U937 cells associated with differentially accessible peaks between ΔNSPs and control cells (d). Representative genomic tracks of the H3ΔNThr22 ChIP-seq dataset from wild-type U937 cells (top) and ATAC-seq datasets from control (middle) or ΔNSPs (bottom) cells (e). Pie chart depicts the percentage of overlapped peaks in (d) (N=9,654) that are existing accessible chromatin peaks in control cells (f).
Across the genome, differential analysis of ATAC-seq data between ΔNSPs and control cells identified 25,900 peaks with increased chromatin accessibility in ΔNSPs over control cells (Fig. 6c). In contrast, under the same FDR 5% threshold, only 16 peaks showed decreased accessibility in ΔNSPs cells relative to controls. Differentially accessible peaks in ΔNSPs cells were dispersed across the genome (Extended Data Fig. 6c) with enrichment in genic regions (Extended Data Fig. 6d). Specifically, among the 15,504 H3ΔNThr22-enriched peaks in wild-type U937 cells (Fig. 4a), 9,654 were associated with differential ATAC-seq peaks in ΔNSPs cells (Fig. 6d), suggesting increased chromatin accessibility upon H3ΔNThr22 depletion. 85.8% of these 9,654 peaks were already maintained at a permissive chromatin state in control cells (Fig. 6e, 6f, and Extended Data Fig. 6e). In summary, NSP depletion greatly impacts chromatin dynamics, resulting in a global increase in chromatin accessibility. At H3ΔNThr22-enriched loci in wild-type cells, we observed further increase in accessibility upon NSP and H3ΔNThr22 depletion.
However, dramatic alterations in chromatin accessibility were only associated with modest gene expression changes (Fig. 6a). We hypothesize that the increased chromatin accessibility in response to H3ΔN repression may represent a potentiated chromatin state, which facilitates gene expression reprogramming in the presence of external cues for differentiation. Permissive chromatin state was found in ΔNSPs cells at genes encoding proinflammatory cytokines and key immune modulators, such as IL-1β, TNF-α, MIP-1α, MIP-1β, IL-18, lymphotoxin-α, IL-10, and TNFR2 (Extended Data Fig. 7a). Loci encoding cell adhesion molecules CD11b and CD11c also showed increased chromatin accessibility in cells depleted of NSPs (Extended Data Fig. 7b). While the transcription activities of these genes were not significantly affected by the increased chromatin accessibility in ΔNSPs cells in the basal condition (Fig. 6a), upon PMA-induced differentiation, ΔNSPs cells responded with a higher magnitude of proinflammatory cytokine and immune modulator secretion (Fig. 7a) and expressed higher CD11b and CD11c relative to control cells (Fig. 7b). In addition to genes upregulated upon PMA stimulation, ΔNSPs cells also responded to PMA with a greater degree of transcriptional repression for genes downregulated during differentiation (GSE10756625) (Fig. 7c). These genes were also enriched with H3ΔNThr22 in wild-type U937 cells and showed elevated chromatin accessibility in ΔNSPs relative to control cells (Extended Data Fig. 7c).
Fig. 7. Chromatin Priming Mediated by NSP and H3ΔN Repression Facilitates Gene Expression Reprogramming.
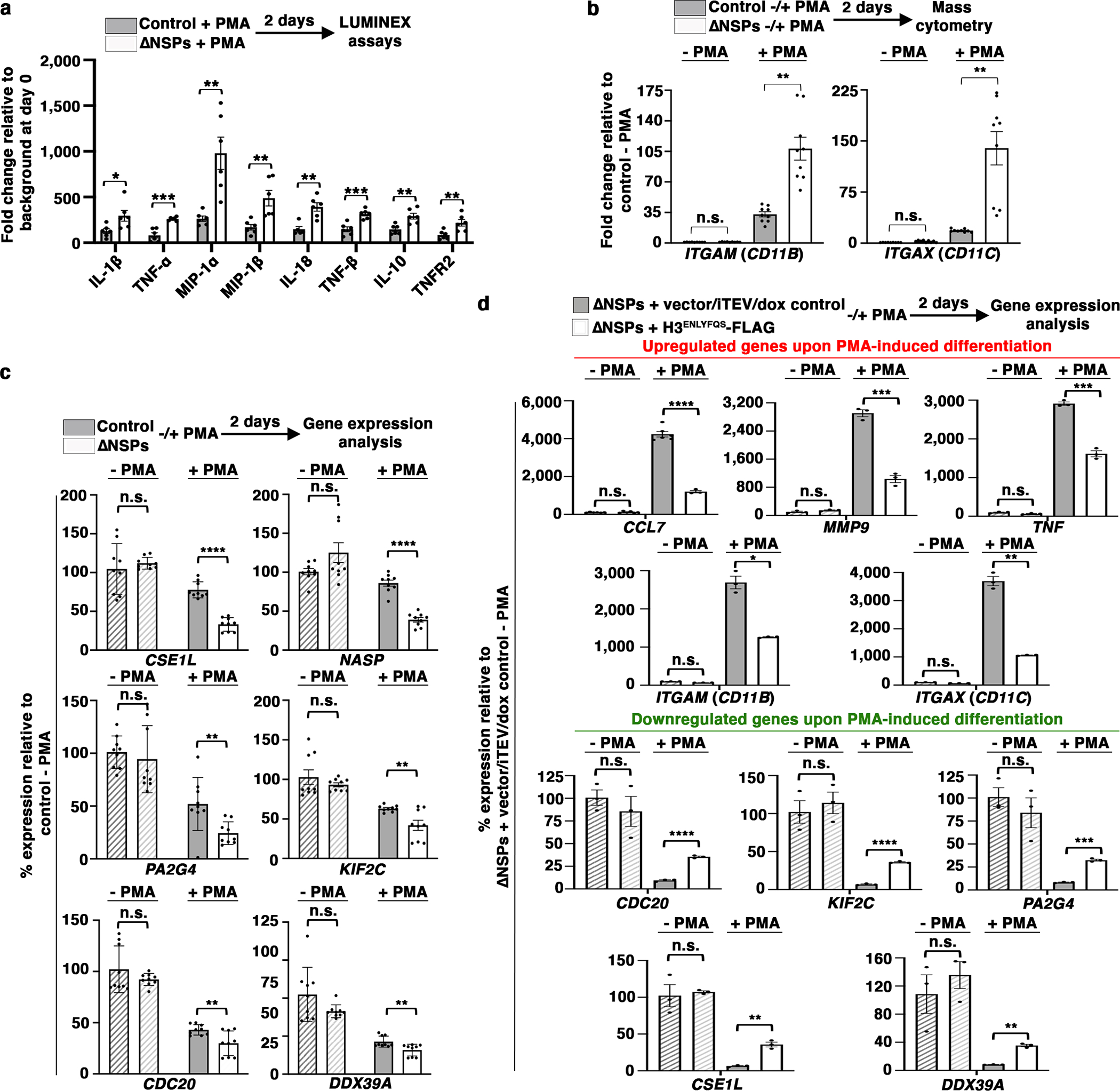
(a to c) ΔNSPs cells respond to PMA-induced differentiation with a greater magnitude of gene expression changes. Bead-based immunoassay analysis of the indicated cytokines. White, ΔNSPs cells; gray, control cells; y-axis, fold change relative to the background at day 0 (a). Mass cytometry analysis of the cells as in (a) using antibodies targeting CD11b (left) or CD11c (right). Y-axis, fold change relative to the level in control cells without PMA stimulation (b). Gene expression analysis of the indicated genes. Y-axis, expression normalized against the level in control cells without PMA stimulation (c). Data represent mean ± S.E.M. (three (two in (a)) technical replicates from three ΔNSPs clones (white) or three control cell lines (gray) (N=9 each). Statistical significance is determined by two-tailed Student’s t-test. *, P≤0.05; **, P≤0.01; ***, P≤0.001.
(d) Reintroducing H3ΔN into ΔNSPs cells attenuates cellular response to PMA-induced differentiation. Gene expression analysis of the cells as in Extended Data Fig. 2m treated with PMA to induce cellular differentiation. Y-axis, expression normalized against the level in control cells without PMA stimulation. Data represent mean ± S.E.M. (three technical replicates). Statistical significance is determined by two-tailed Student’s t-test. *, P≤0.05; **, P≤0.01; ***, P≤0.001; ****, P≤0.0001.
To determine H3ΔN involvement in these transcriptional effects, we treated ΔNSPs cells into which H3ΔN was reintroduced (Extended Data Fig. 2m) with PMA to induce cellular differentiation mimicking macrophage development. The presence of H3ΔN in ΔNSPs cells attenuated CCL7, MMP9, TNF-α, CD11B, and CD11C upregulation (Fig. 7d, top) upon PMA-induced cell differentiation, in addition to the repression of genes downregulated during this process (Fig. 7d, bottom). Together, these findings provide direct evidence that H3ΔN regulates gene expression reprogramming during cellular differentiation. NSP repression and the consequent H3ΔN depletion during monocyte-to-macrophage differentiation likely create a potentiated chromatin state that facilitates transcriptional alterations and promotes macrophage development.
Repressed H3ΔN in Monocytes from Patients with sJIA
We next investigated H3ΔN in sJIA, a pathologic condition where dysregulation of the monocyte and macrophage compartment has been implicated in disease pathophysiology28, 29. We performed EpiTOF analysis of PBMCs from 16 sJIA patients separated into two biological replicates with equivalent disease activity, in addition to 10 age- and sex-matched healthy controls (HCs) (Fig. 8a and Supplementary Table 1). We measured the global levels of 40 histone marks in 16 major immune cell subtypes. Effect size comparison of the 640 parameters showed marked epigenetic alterations between HCs and sJIA patients consistently captured in two independent biological replicates (Extended Data Fig. 8a) with negligible batch effect (Extended Data Fig. 8b). The epigenetic landscape between sJIA patients and HCs was substantially distinct (Extended Data Fig. 8c). Monocyte-specific PCA separated sJIA patients from HCs (Fig. 8b). H3ΔNThr22 in monocytes is lower in most sJIA patients (Fig. 8c), with a median z-score 0.393 lower than that in HCs (Extended Data Fig. 8d). However, significant heterogeneity was observed (p = 2.3E-02, Bartlett’s test on S.D.). sJIA patients receiving therapies targeting IL-130, 31 (N=5), neutralizing TNF-α or on no biologic treatment (N=6) showed lower H3ΔNThr22 in monocytes (Extended Data Fig. 8d). Patients treated with the IL-6 inhibitor, tocilizumab32, did not show reduced H3ΔNThr22 in monocytes. To validate these findings, we performed an expanded EpiTOF analysis on an independent cohort of 14 sJIA patients and 4 HCs (Supplementary Table 2). Differential analysis found repressed H3ΔNThr22 in monocytes from sJIA patients (Extended Data Fig. 8e). Notably, the only patient on tocilizumab therapy in this cohort showed the highest H3ΔNThr22 in monocytes among all patients, comparable to the HC level. Together, our data show reduced H3ΔNThr22 in sJIA-derived monocytes, suggesting that their chromatin state is primed for macrophage differentiation.
Fig. 8. Repressed H3ΔN in sJIA Patients.
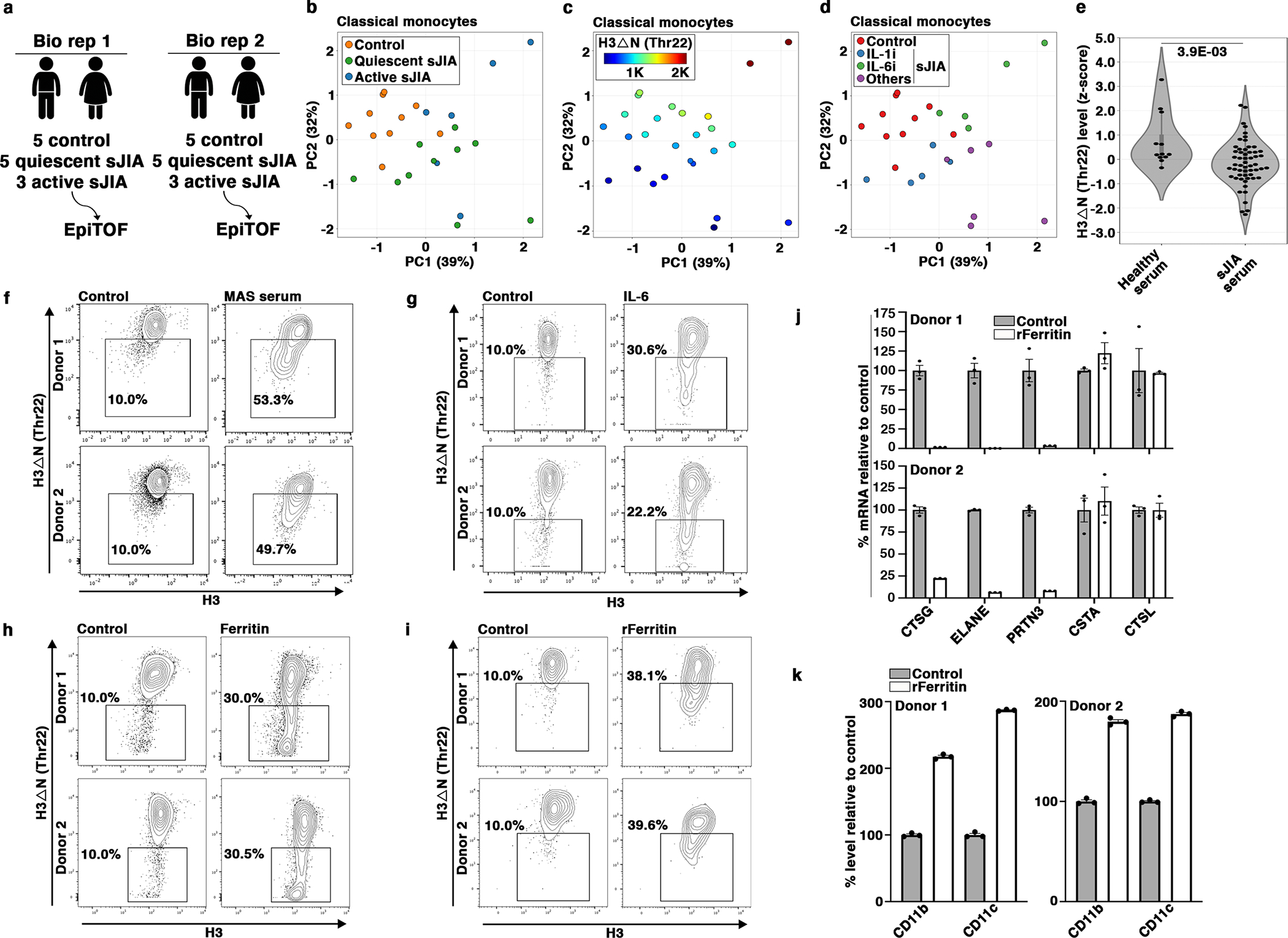
(a) EpiTOF analysis.
(b) Separation of monocytes from sJIA patients or from healthy volunteers by histone modification profiles. PCA of EpiTOF data. Each dot, monocyte from a sJIA patient; green, quiescent disease; blue, active disease; orange, healthy volunteer; %, the fraction of variance explained by each principal component.
(c) Reduced H3ΔN in monocytes from patients with sJIA. PCA as in (b). Color, H3ΔNThr22 level.
(d) sJIA patients receiving IL-6 blockade therapy do not show reduced H3ΔNThr22 in monocytes. PCA as in (b). Blue, biologics targeting IL-1; green, biologics targeting IL-6; purple, others (see Results).
(e) Sera from sJIA patients induce H3ΔN repression in monocytes from healthy volunteers. Mass cytometry analysis of H3ΔNThr22 levels in monocytes cultured in the presence of sera from 14 sJIA patients (right, 56 data points) or 3 healthy donors (left, 12 data points) for 24 hours. Statistical significance is determined by mixed effect linear model with P value depicted.
(f) Sera from sJIA patients with MAS induce marked H3ΔN reduction. Mass cytometry analysis of monocytes from two healthy donors (top and bottom) cultured in the presence of sera from a healthy donor (left) or a sJIA patient with MAS (right) for 24 hours. X-axis, bulk H3 level; y-axis, H3ΔNThr22 level.
(g) IL-6 induces H3ΔN reduction. In vitro stimulation assay as in (f) using IL-6.
(h) Native ferritin induces H3ΔNThr22 repression in monocytes. In vitro stimulation assay as in (f) using native ferritin.
(i) Recombinant ferritin induces H3ΔNThr22 repression in monocytes. In vitro stimulation assay as in (f) using recombinant ferritin.
(j) Ferritin treatment suppresses NSP expression. qPCR analysis of the indicated genes (x-axis). White, with recombinant ferritin; gray, without recombinant ferritin; y-axis, expression relative to that in control cells; top, donor 1; bottom, donor 2. Data represent mean ± S.E.M. (three technical replicates).
(k) Ferritin promotes monocyte-to-macrophage differentiation. Mass cytometry analysis. White, with recombinant ferritin; gray, without recombinant ferritin. CD11b (left) and CD11c (right) expression is analyzed. Top, donor 1; bottom, donor 2; y-axis, expression relative to control. Data represent mean ± S.E.M. (three technical replicates).
Ferritin-Induced H3ΔNThr22 Repression in Monocytes
We next asked if soluble serum factors contribute to H3ΔNThr22 repression in monocytes from sJIA28. In vitro stimulation of PBMCs from four HCs treated ex vivo with sera from 14 sJIA patients, 8 with active and 6 with quiescent disease (Supplementary Table 3), showed reduced H3ΔNThr22 in monocytes relative to cells treated with control sera (Fig. 8e). Moreover, sera from sJIA patients with macrophage activation syndrome (MAS)33 induced a greater magnitude of H3ΔNThr22 reduction in HC monocytes (Fig. 8f and Extended Data Fig. 8f). The data argue that the dysregulated monocyte and macrophage compartment in sJIA patients may be driven in part by signaling molecules present in the circulation. In vitro stimulations of HC monocytes with several cytokines and acute-phase reactants frequently elevated in sJIA sera28 showed H3ΔNThr22 repression induced by IL-6 (Fig. 8g)34. In contrast, stimulations with IL-1β and IL-18 did not significantly impact H3ΔNThr22 in monocytes (Extended Data Fig. 8g and 8h).
IL-6 blockade therapy reduces serum ferritin35. We found that ferritin purified from human liver induced H3ΔNThr22 repression in monocytes (Fig. 8h). To exclude the effects of possible contaminants carried over from primary human tissues, we validated the finding using ferritin purified from a recombinant source36 (Fig. 8i and Extended Data Fig. 8i). Ferritin treatment in vitro suppressed NSP expression in HC monocytes (Fig. 8j) and promoted macrophage development (Fig. 8k). Together, our data support that both IL-6 and ferritin, two clinically important serum factors implicated in sJIA pathophysiology, may promote monocyte-to-macrophage differentiation through a chromatin-based mechanism involving regulated histone proteolysis.
Discussion
In this work, we identify a noncanonical nuclear function of NSPs in monocytes and their epigenetic role in governing cellular differentiation. An important open question is how NSPs are translocated to the nucleus in resting monocytes. Whether the same mechanisms controlling apoptosis or “NETosis” are utilized but at an attenuated level, or there is a specialized transport system shuttling NSPs into the nuclei of resting monocytes requires further investigation.
NSPs share a similar promoter architecture37. During monocyte-to-macrophage differentiation, NSP repression likely involves trans-factors binding to the promoters of all three NSPs simultaneously. These regulators may be activated by signaling pathways that promote macrophage differentiation, such as M-CSF and IL-6. Additionally, an open question is whether NSP and H3ΔN repression is specific to macrophage development or also occurs as monocyte differentiation is skewed to dendritic cells (DCs). Cytokines that promote DC development, such as GM-CSF, IL-4, and TNF-α, may also regulate NSP expression.
The experiments where H3ΔN is reintroduced into ΔNSPs cells provide direct evidence that H3ΔN can induce chromatin architectural changes (Extended Data Fig. 2q) and affect cell morphology and functions (Extended Data Fig. 2n, 2o, and 2p). However, future proteomic analysis will almost certainly find additional NSP substrates in the nucleus, whose involvements in transcription regulation and macrophage development require further investigation.
Asymmetric H3ΔN in monocytes is suggested in our work. First, H3ΔNThr22 ChIP using sheared chromatin containing predominantly mono-nucleosomes (Extended Data Fig. 4b) results in modest H3ΔN enrichment (Extended Data Fig. 4a). Second, the TSS-proximal H3ΔNThr22 enrichment at actively transcribed genes (Fig. 4h and 5g) is similar to that of H3K4me338. H3ΔNThr22 and H3K4me3 are mutually exclusive. However, both in U937 cells and primary monocytes, we observe concurrent H3ΔNThr22 and H3K4me3 peaks using publicly available ChIP-seq datasets. Modification symmetry represents an important yet poorly understood aspect of chromatin regulation39. Future studies using sequential ChIP or nucleosome-resolution mass spectrometry40 may provide additional insights into H3ΔN asymmetry.
Our data also raise an important question about how H3ΔN genomic distribution is determined in monocytes. First, NSP enzymatic activities may be modulated by MNEI/SERPINB141, whose nucleocytoplasmic distribution has been well-documented42. It is possible that a significant proportion of ELANE molecules immunoprecipitated during our ChIP analysis is in complex with MNEI, which renders ELANE catalytically inactive. Second, crosstalk between H3ΔN and other histone modifications may affect its genomic distribution. NSP catalytic activities may be affected by pre-existing histone marks43, and H3ΔN may promote or prevent subsequent modifications44.
Our findings are of particular significance to the training and tolerance of innate immunity45, 46. Several histone marks known to regulate innate immune memory, including H3K4me1, H3K4me3, H3K9me2, H3K27me3, and H3K27ac, are directly affected by H3ΔN. An integrative analysis of H3ΔN and these histone marks will allow us to better understand epigenetic regulation of innate immune memory.
EpiTOF analysis can be expanded to other diseases where monocytes and macrophages play key pathologic roles, such as atherosclerosis, hemophagocytic lymphohistiocytosis (HLH), and other autoinflammatory conditions47, 48. Our data link IL-6 to a chromatin-based mechanism promoting macrophage development34. IL-6 blockade is under clinical evaluation for giant cell arteritis, Takayasu arteritis, and polymyalgia rheumatica. EpiTOF can be employed to investigate the relationships between H3ΔN in monocytes, tocilizumab therapy, and disease activity in these vasculitides.
Identifying the receptor mediating ferritin uptake by monocytes49, 50 and characterizing the dependence of ferritin-induced chromatin alterations on its iron loading will provide important mechanistic insights. Elevated ferritin is found in sepsis and antiphospholipid syndrome, in which NSPs and H3ΔN may play key pathologic roles. Similarly, NSPs have been implicated in the pathophysiology of chronic obstructive pulmonary disease (COPD) and acute lung injuries, for which targeting excessive NSP activities represents a promising therapeutic strategy18. Selective inhibition of pathologically relevant NSP functions will be key to the development of an efficacious and safe NSP inhibitor for therapeutic applications.
Methods
Cell culture and transfection
Jurkat, OCI-Lys3, U937, THP1 cells were cultured in RPMI 1640 media (Gibco) supplemented with 10% fetal bovine serum (FBS) (ATCC), glutamine (Gibco), and penicillin/streptomycin (Gibco) (referred to as complete media below unless otherwise specified). 293T cells were cultured in complete advanced DMEM media (ThermoFisher). Transfection of 293T cells with plasmid DNA was performed using TransIT-293 (Mirus) following the manufacturer’s protocol.
Small-scale biochemical fractionation to isolate native chromatin
1×107 to 2×107 U937, THP1 cells or PBMCs were collected, washed with PBS and resuspended in buffer A (10 mM HEPES, pH=7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM dithiothreitol (DTT), complete protease inhibitor (Roche) and 1 mM PMSF)52. Triton X-100 was added to a final concentration of 0.1%. After incubating the cells for 8 mins on ice, cells were centrifuged at 1300 g for 5 mins at 4°C. The supernatant containing the cytoplasm was clarified by centrifugation at 20,000 g for 5 mins at 4°C. The nuclei were washed once with buffer A and lysed with buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, complete protease inhibitor and 1 mM PMSF) for 30 mins. Chromatin was collected by centrifugation at 1,700 g for 5 mins at 4°C. Chromatin was washed once with buffer B, resuspended in 5X SDS sample buffer (250 mM Tris-HCl pH=6.8, 10% SDS, 30% glycerol, 5% β-mercaptoethanol, 0.02% bromophenol blue), sonicate briefly to shear the DNA, and boiled at 90°C for 10 mins prior to electrophoresis analysis.
Biochemical fractionation to isolate chromatin at different salt conditions
1×107 to 2×107 U937 were collected, washed with PBS and swelled in 5 packed cell volume (PCV) of buffer A (10 mM Tris-HCl pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 1 mM PMSF, 0.5 mM DTT) for 10 min on ice. Cells were collected by centrifugation at 420 g for 5 mins and homogenized using a type B (loose) pestle in 2 PCV of buffer A. Nuclei were collected by centrifugation at 10,000 g for 20 mins, resuspended in 3 ml buffer C (20 mM Tris-HCl pH 7.9, 0.42M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM PMSF, 0.5 mM DTT, 25 % glycerol) per 109 cells and homogenized using a type B pestle. The suspension is then gently rotated for 30 mins at 4 °C. The nuclear extract was collected by centrifugation at 20,000 g for 5 mins. The chromatin pellet was washed with low salt buffer (20 mM Tris-HCl pH 7.9, 1.5 mM MgCl2, 0.2 mM EDTA, 0.15 M NaCl) three times. To extract NSPs from chromatin, chromatin was resuspended in high salt buffer (20 mM Tris-HCl pH 7.9, 1.5 mM MgCl2, 0.2 mM EDTA, 0.65 M NaCl).
Protein identification by mass spectrometry
To profile associated proteins in the purified chromatin fractions descried above, the isolated native chromatin was subject to mass spectrometry analysis (MSBioworks). Briefly, sample were loaded onto a 10% Bis-Tris SDS-PAGE gel (Novex, Invitrogen) and separated for approximately 1cm. After Coomassie staining, the excised gel slice was washed with 25 mM ammonium bicarbonate followed by acetonitrile, reduced with 10 mM DTT at 60°C followed by alkylation with 50 mM iodoacetamide at room temperature (RT), and digested with trypsin (Promega) at 37°C for 4 hours. The digested peptides were analyzed by nano LC/MS/MS with a Waters NanoAcquity HPLC system interfaced to a ThermoFisher Q Exactive. Peptides were loaded on a trapping column and eluted over a 75 μm analytical column at 350 nL/min. Both columns were packed with Luna C18 resin (Phenomenex). The mass spectrometer was operated in data-dependent mode, with MS and MS/MS performed in the Orbitrap at 70,000 FWHM and 17,500 FWHM resolution, respectively. The fifteen most abundant ions were selected for MS/MS. Data were searched using a local copy of Mascot and parsed into the Scaffold software for validation, filtering and to create a non-redundant list of identified proteins per sample.
Crispr-mediated knock out strategy
sgRNA targeting CTSL, CTSG, ELANE, and PRTN3 were synthesized and cloned into plentiCRISPR v2 to generate lentiviral particles for stable transduction. The sgRNA sequences are: 5’-ACTGGAAAGCATAATC CATT-3’ (CTSL#1); 5’-CAGTATGTTCAGGATA-3’ (CTSL#2); 5’-GGTCGTAGGAACCGAAGATG-3’ (CTSG#1); 5’-GATCTGAAGATACGCCATGT-3’ (CTSG#2); 5’-GGAAAAGACACGCGAGTCGG-3’ (ELANE#1); 5’- GAGTCGGCGGCCGAGGGTCA-3’ (ELANE#2); 5’-CACTTTCGTCCCTCGCCGCA-3’(PRTN3#1); and 5’-TGCTCGGAGCCCACAACGTG (PRTN3#2). In order to generate a plasmid expressing sgRNA targeting CTSG, ELANE and PRTN3 simultaneously, oligos CTSG#2, ELANE #1 and PRTN#2 were cloned into pMule ENTR U6 stuffer sgRNA scafford/plentiNeo using the multiple lentiviral expression (MuLE) system (Addgene). Transduced U937 cells were selected with 500 μg/mL geneticin.
In vitro protease assay
5 μg of recombinant cathepsin C (R&D systems) was first incubated with 5 μg of recombinant ELANE (R&D systems) or PRTN3 (Novoprotein) for 2 hours at 37°C in PBS (Gibco) to activate these enzymes. 10 μg of recombinant nucleosomes16 were subsequently added and incubated for another 2 hours at 37°C. A sample with only cathepsin C was used as a control to exclude cleavage mediated by cathepsin C. For CTSG protease assay, 5 μg of recombinant CTSG (Sigma) was incubated with 10 μg of recombinant nucleosomes for 2 hours at 37°C. Reactions were terminated with 5X SDS sample buffer and run on a polyacrylamide gel followed by Coomassie staining. Excised gel slices were subject to mass spectrometry analysis.
Mass spectrometry to identify H3 cleavage sites
Protein bands corresponding to cleaved H3 products were cut and digested with Glu-C endoproteinase in 50 mM NH4HCO3 buffer. Samples were reconstituted in 0.1% formic acid and analyzed on a Fusion Lumos mass spectrometer (Thermo Fisher Scientific, San Jose, USA) equipped with a Dionex Ultimate 3000 LC-system. Peptides were separated by capillary reverse phase chromatography on a 24 cm reversed phase column (100 μm inner diameter, packed in-house with ReproSil-Pur C18-AQ 3.0 m resin (Dr. Maisch GmbH)). Full MS scans, data dependent HCD (higher-energy collisional dissociation) and EThcD (electron transfer dissociation with 25% of supplemental collision energy) MS/MS scans were all acquired in the Orbitrap mass analyzer. H3 cleavage products with cleavage sites range from H3K14 to H3R42 were analyzed. Peptide sequences were confirmed based on the EThcD spectra manually. In cases where multiple cleavage products were found, peak area of the precursor peptides were used to compare the relative abundance of each cleavage product.
Cell sorting
FACS was performed as previously described12. Briefly, PBMCs were resuspended in PBS containing Zombie Aqua reagents (Biolegend) for 15 mins at RT and quenched with CyTOF buffer (PBS, 1% BSA, 2 mM EDTA, and 0.05% sodium azide). Cells were centrifuged at 400 g for 8 mins and resuspended in CyTOF buffer containing antibodies against immunophenotypic markers for 30 mins. Markers for sorting: APC anti-CD19 (Biolegend) for B cells, PE-Cy7 anti-CD3 (Biolegend) for T cells, PE anti-CD14 (Biolegend) for monocytes, and FITC anti-CD45 (BD) for total PBMCs. Cells were washed once in CyTOF buffer and twice in sorting buffer (PBS with 0.1% BSA). FACS was performed on sorters in Stanford Shared FACS Facility. Sorted cells collected in CyTOF buffer were centrifuged at 400 g for 8 mins and resuspended in 5X SDS sample buffer at 1×106 cells per 100 μL. Samples were sonicated using Bioruptor (Diagenode) for 10 mins prior to western blot analysis. To isolate clonal ΔNSPs U937 cells, single transduced cells were sorted into 96-well plate filled with media based on forward and side scatters.
Reconstitution of H3ΔN in ΔNSPs U937 cells
Full-length H3 cDNA with TEV protease recognition site inserted between Ala21 and Thr22 and 3X-FLAG epitope tag was synthesized and cloned into pENTR3C (Invitrogen) and recombined into pLenti CMV Blast DEST (Addgene). TEV protease S219V mutant cDNA with added nuclear localization sequences was cloned into pCW57.1 (Addgene). For lentiviral transduction, viral particles were prepared with packaging plasmids pCMVΔ8.91 and pMD.G. Transduced ΔNSPs U937 cells were selected with 2 μg/mL puromycin and 10 μg/mL blasticidin. Cell lines were cultured in RPMI 1640 media (Gibco) supplemented with 10% fetal bovine serum-TET tested (FBS-TET) (R&D Systems), glutamine (Gibco), and penicillin/streptomycin (Gibco). To induce the expression of TEV protease, cells were incubated with 5 μg/mL of doxycycline (Sigma) for 7 days.
In vitro cell migration assay
Control or ΔNSPs U937 cells were cultured in serum-free RPMI 1640 media overnight. 1×106 cells were then plated in the upper chamber of the 5 μm transwell and incubated for 5 hours. Lower chambers were filled with RPMI 1640 media with FBS. Cells were collected from the lower chamber, centrifuged at 300 g for 5 mins and resuspended in 100 μL of PBS for counting.
Phagocytosis assay
1×105 cells were plated in 96-well plates and incubated with 18 ug protein A Dynabeads (Invitrogen) for 4 hours. The number of cells that contained the Dynabeads and the total number of cells were counted under the microscope. Each cell line was counted in three technical replicates. Phagocytosis of primary cells was measured using osmium-labeled E.coli and mass cytometry22. DH5-α E.coli was washed with PBS and fixed in 1.6% PFA (Electron Microscopy Sciences) for 10 mins at RT at 1 × 109 cells/mL. Cells were washed twice with PBS and stained at a final concentration of 0.00008% OsO4 for 7 mins at RT. After staining, cells were washed twice with PBS, filtered, and stored at −20 o C. For experiments, cytochalasin D was added to a concentration of 5 uM and incubated for 10 mins. Unlabeled or osmium labeled E.coli were added to primary cells at a 1:100 ratio and incubated for 30 mins. Cells were then washed and processed for mass cytometry.
MNase sensitivity assay
1×106 cells were centrifuged at 300g for 5 mins and washed once with PBS. Cells were then treated with 1 mL NP-40 buffer (10 mM Tris-HCl pH=7.4, 10 mM NaCl, 3 mM MgCl 2, 0.5% NP-40) and incubated on ice for 5 mins. Nuclei were pelleted at 120g for 10 mins and supernatant was removed by pipetting. MNase buffer (10 mM Tris-HCl ph=7.4, 15 mM NaCl, 60 mM KCl) was used to wash nuclei twice. Nuclei were resuspended in 200 uL MNase buffer and treated with 0.015 U MNase for 12 mins at 37 °C. Reaction was stopped with 100 uL 0.5 M EDTA. 3 uL proteinase K and SDS to a final concentration of 2% were added and samples were incubated at 37 °C overnight. The next day, equal volume of phenol/chloroform was added and samples were mixed and centrifuged at 16,000g for 10 mins. The aqueous layer was pipetted off and transferred into a new tube. Samples were then incubated with 3 uL RNase A at 37 o C for at least 2 hours. Equal volume of chloroform was added and samples were mixed and centrifuged at 16,000 g for 10 mins. The aqueous layer was pipetted off and transferred into a new tube. 1/7 volume of 3 M Na Acetate and 2–2.5 volume of ice-cold ethanol were added to each sample. Samples were incubated at −80 °C for at least 30 mins. Samples were then centrifuged at 16,000 g for 10 mins at 4 °C. Supernatant was poured off, 1 mL 70% ethanol was added, and samples were centrifuged at 16,000 g for 10 mins. Ethanol was poured off and pellets were dried. DNA was resuspended in 50 uL TE buffer at 55 °C and run on a 5% TBE gel.
Primary monocyte differentiation and macrophage polarization
Differentiation of monocytes to macrophages was performed following a standard protocol19. Mononuclear cells purified from buffy coat were plated and incubated overnight in complete RPMI 1640 media. The plates were washed 3 times with PBS (Gibco) to remove nonadherent cells, and the adherent cells were cultured for 7 days to allow for differentiation into macrophages. Alternatively, monocytes were isolated using the Pan Monocyte Isolation kit, a magnetic bead-based negative selection kit (Miltenyl Biotec, 130–096-537) and plated in RPMI media with 30 ng/mL M-CSF. Plates were incubated for 5 days before collection of differentiated cells. To test the effects of NSP inhibition on differentiation, cells were cultured in media containing 100 μM AEBSF (Sigma) or 1 μM GW311616 hydrochloride ELANE inhibitor (APExBIO) and 1 μM cathepsin G inhibitor (APExBIO) for 2 days. To obtain polarized macrophages, monocyte-derived macrophages were first generated by the protocol described above. Naïve macrophages were then cultured in media containing 100 ng/mL LPS and 20 ng/mL IFN-γ (classically activated) or 20 ng/mL IL-4 (alternatively activated) for 18 hours before collecting for western blot analysis.
Generation of U937 cells expressing exogenous NSPs
Full-length CTSG, ELANE, or PRTN3 cDNA was cloned into pENTR3C (Invitrogen)/plentiCMV Blast Dest (Addgene) for overexpression in U937 cells. For lentiviral transduction, viral particles were prepared with packaging plasmids pCMVΔ8.91 and pMD.G. Transduced U937 cells were selected with 10 μg/mL blasticidin. To induce differentiation of these transduced cells, cells were treated with 25 ng/mL PMA for one day and cultured in complete media for 2 days. Cells were then collected for CyTOF and western blot analysis.
Cell preparation for Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) analysis
Three independently generated control cell lines and three ΔNSPs clones were pelleted, resuspended in fresh media with 1:100 volume of 100x DNase buffer (250 mM MgCl2 and 50 mM CaCl2) and 1:100 volume of DNase solution (20,000 units/mL DNase in HBSS) (Invitrogen) and incubated at 37°C for 30 mins. Cells were then washed once with PBS and cryopreserved in media containing 5% DMSO prior to ATAC-seq analysis (Active Motif) following the standard protocol53.
Transcriptomic profiling by RNA-sequencing
RNA was extracted from control and ΔNSPs U937 cells using the RNeasy Plus mini kit (Qiagen) following the manufacturer’s protocol. Purified RNA was used for library construction and analyzed by high-throughput sequencing (Novogene).
Chromatin immunoprecipitation sequencing (ChIP-seq) analysis
ChIP analysis was performed following a published protocol with minor changes54. Briefly, U937 cells were cross-linked with 1% formaldehyde (Sigma) for 8 mins at RT with gentle mixing. Cross-linking reaction was stopped by adding final concentration of 125 mM glycine and incubating for 5 mins at RT. Cells were then centrifuged at 300 g for 5 mins and washed with PBS (Gibco) two times. Cells were resuspended in ChIP lysis buffer (50 mM HEPES-KOH pH=7.5, 140 mM NaCl, 1 mM EDTA pH=8.0, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, and protease inhibitors) and incubated for 10 mins on ice. Cell lysate was sonicated with Bioruptor (Diagenode) and diluted with RIPA buffer 10-fold before incubating overnight with antibody/protein-A magnetic bead complexes. Antibodies used were as follows: H3 (abcam, #1791), H3ΔNThr22 (Cell Signaling Technology, #12576), CTSG (abcam, #49854), ELANE (abcam 21595), PRTN3 (abcam, #133613). Beads were washed two times with ChIP wash buffer I (20 mM Tris pH=8.0, 0.1% SDS, 1% Triton, 0.1% sodium deoxycholate, 2 mM EDTA, and 150 mM NaCl) and two times with ChIP wash buffer II (20 mM Tris-HCl pH=8.0, 0.1% SDS, 1% Triton, 0.1% sodium deoxycholate, 2 mM EDTA, 500 mM NaCl). IP’ed DNA was eluted from beads, de-crosslinked at 68°C and purified using the MinElute PCR Purification Kit (Qiagen). Purified-DNA was subjected for library preparation using the NEBNext Ultra II DNA library prep kit (NEB) following the manufacturer’s protocol. DNA library was then submitted for sequencing at the Stanford Genome Sequencing Service Center.
Multiplex protein quantitation using the Luminex instrument platform
Control or ΔNSPs U937 cells were treated with 25 ng/mL PMA for one day. Cells were washed with PBS and cultured in complete RPMI 1640 media for 2 days. Supernatant was collected, filtered through 0.22 μM filter and stored at −80°C for downstream analysis. Multiplex protein quantitation was performed using the Human Immune Monitoring Panel of ProcartaPlex Multiplex Immunoassay (Invitrogen) following the manufacturer’s protocol. In short, supernatant samples were diluted 1:10 and incubated with magnetic beads in a 384-well plate for 1 hour at RT. Antibody/streptavidin-PE conjugates were added for detection. Data were acquired using Luminex FlexMap 3D system (Luminex).
Mass cytometry sample processing, staining, barcoding and data acquisition
EpiTOF was performed following the protocol described previously12. Briefly, cryopreserved PBMCs were thawed and incubated in complete RPMI 1640 media at 37°C for 1 hour prior to processing. Cisplatin (ENZO Life Sciences) was added to 10 μM final concentration for 5 mins before quenching with CyTOF Buffer (PBS with 1% BSA, 2 mM EDTA, and 0.05% sodium azide). Cells were centrifuged at 400 g for 8 mins and stained with lanthanide-labeled antibodies against immunophenotypic markers in CyTOF buffer containing Fc receptor blocker (BioLegend) for 30 mins at RT. Following extracellular marker staining, cells were washed 3 times with CyTOF buffer and fixed in 1.6% PFA (Electron Microscopy Sciences) at 1×106 cells/mL for 15 mins at RT. Cells were centrifuged at 600 g for 5 mins and permeabilized with 1 mL ice-cold methanol (Fisher Scientific) for 20 mins at 4°C. 4 mL of CyTOF buffer was added to stop permeabilization followed by 2 PBS washes. Mass-tag sample barcoding was performed following the manufacturer’s protocol (Fluidigm). Individual samples were then combined and stained with intracellular antibodies in CyTOF buffer containing Fc receptor blocker (BioLegend) overnight at 4°C. The following day, cells were washed twice in CyTOF buffer and stained with 250 nM 191/193Ir DNA intercalator (Fluidigm) in PBS with 1.6% PFA for 30 mins at RT. Cells were washed twice with CyTOF buffer and once with double-deionized water (ddH2O) (ThermoFisher) followed by filtering through a 35 μm strainer. Cells were resuspended in ddH2O containing four element calibration beads (Fluidigm) and analyzed on CyTOF (Fluidigm). Raw data were concatenated and normalized using calibration beads following the manufacturer’s protocol for downstream processing.
PBMC isolation for in vitro stimulation assays
Mononuclear cells were purified from buffy coat by density gradient centrifugation using Ficoll-Paque Plus (GE Healthcare) in SepMate tubes (STEMCELL Technology). Crude PBMCs were treated with RBC lysis buffer (BioLegend) for 5 mins to remove residual red blood cells followed by 3 PBS washes. 2.5×105 PBMCs were resuspended in 250 μL of RPMI with 20 μL of serum from healthy donors or sJIA patients. Cells were processed for CyTOF analysis after overnight incubation. For in vitro PBMC stimulations, 1×106 PBMCs were resuspended in 1 mL RPMI and treated with 50 ng/mL IL-6 (R&D), 25 ng/mL IL-1B (R&D), 10 ng/mL IL-18, 5 μg/mL native ferritin (abcam), or 5 μg/mL recombinant ferritin. All human subject research was approved by Stanford Institutional Review Board (IRB).
Expression and purification of recombinant human ferritin
Full-length ferritin heavy chain and light chain cDNA was cloned into pET-21a (+) and pET-28a (+) vectors (Millipore), respectively. pET-21 ferritin heavy chain construct and pET-28 ferritin light chain construct were transformed into BL21-CodonPlus (DE3)-RILP Competent Cells (Agilent) sequentially. For protein expression, positive transformants were grown in LB medium with carbenicillin and kanamycin overnight. The saturated culture was diluted in the following day to obtain an exponential-phase culture, to which 1.0 mM IPTG was added to induce ferritin expression for 2.5 hours at 37°C. Cells were pelleted and sonicated, and supernatant was heated at 70°C for 15 mins. Ferritin was isolated from the supernatant by centrifugation. Ferritin was purified by fast protein liquid chromatography (FPLC) using a HiPrep 26/60 Sephacryl S-300 HR gel filtration column (Sigms) filled with 50 mM Tris pH=7.5, 50 mM NaCl buffer followed by a HiTrap Q HP ion exchange column filled with 50 mM Tris pH 7.5 buffer with a 50–300 mM NaCl gradient. Purified protein was dialyzed to the buffer containing 50 mM Tris pH 7.5 and 50 mM NaCl for storage.
PCR primers
| Fig. 4d #1-F | tagcgctcttcttcggagtt |
| Fig. 4d #1-R | gggcggaaccaaacctaa |
| Fig. 4d #2-F | ggcgtccttcctaacatcc |
| Fig. 4d #2-R | tgtcaaatcacttgcccttg |
| Fig. 4d #3-F | gtcgggttccttcaaactca |
| Fig. 4d #3-R | gctcctgctggcttcttg |
| Fig. 4d #4-F | ttccacctcctggtttcaag |
| Fig. 4d #4-R | cgcttgtactcccagctacc |
| Fig. 4d #5-F | gcaggaaaatcgctggaa |
| Fig. 4d #5-R | cagtgctgccgtcttagctt |
| Fig. 4d #6-F | caattggcaggacagaacatc |
| Fig. 4d #6-R | gcataggcttgagggacaaa |
| Extended Data Fig. 4e #1-F | atacacagctacgggatacgg |
| Extended Data Fig. 4e #1-R | gctcggtttcaggagtttgt |
| Extended Data Fig. 4e #2-F | ccccttggaggaggacag |
| Extended Data Fig. 4e #2-R | cgaggaccctcgacttagaga |
| Extended Data Fig. 4e #3-F | ccaggctctgctgtctcg |
| Extended Data Fig. 4e #3-R | ggagacctcggctatcctg |
| Extended Data Fig. 4e #4-F | gctgtggctattatgacacataca |
| Extended Data Fig. 4e #4-R | tggatggttgactggaagg |
| Extended Data Fig. 4e #5-F | gcaggaggatcctttgaacc |
| Extended Data Fig. 4e #5-R | gaaacgagtttcattgtgttgc |
| Extended Data Fig. 4e #6-F | gagggatcccacctgacc |
| Extended Data Fig. 4e #6-R | cctggcctcacacctagact |
| Fig. 4l #1-F | caccctaccaggagacactca |
| Fig. 4l #1-R | ctccttgggccatcccta |
| Fig. 4l #2-F | gaggggaaacccttccatt |
| Fig. 4l #2-R | caggggatctgaccgactc |
| Fig. 4l #3-F | tctctcatttcctctctctaaacca |
| Fig. 4l #3-R | gactagggatgtgtgggtagga |
| Fig. 4l #4-F | ttttgatacaggcataaattgtgtaat |
| Fig. 4l #4-R | tgttggtaacacaaggaaatgaa |
| Fig. 4l #5-F | gtctgccctggctgagtg |
| Fig. 4l #5-R | gcagcagtggggtctttg |
| Extended Data Fig. 4m #1-F | tgagtcaaggttccctctgc |
| Extended Data Fig. 4m #1-R | tgactcagtttcccctctgg |
| Extended Data Fig. 4m #2-F | cggtgtggagggaaactg |
| Extended Data Fig. 4m #2-R | tggcccttgactcagcat |
| Extended Data Fig. 4m #3-F | ctctacaggggcttcgtgag |
| Extended Data Fig. 4m #3-R | catcttccctccctccact |
| Extended Data Fig. 4m #4-F | tgaaacgttagtcctttcaaataatc |
| Extended Data Fig. 4m #4-R | ccaaagcactggcattacaa |
| Extended Data Fig. 4m #5-F | tccctctgtttgcattgct |
| Extended Data Fig. 4m #5-R | agggaaggagaggtgaatgg |
| Extended Data Fig. 4m #6-F | gaaggtacagccactgtggaa |
| Extended Data Fig. 4m #6-R | ttgatcataggatagttccgtgttt |
| Extended Data Fig. 4m #7-F | ttctctgctttgttgtgttatgact |
| Extended Data Fig. 4m #7-R | cagagtggcacttgtctgtga |
| CSE1L-F | agattctgctaacaaaccttttcaa |
| CSE1L-R | ggagagaaaaacttctcatgatagc |
| NASP-F | cagatgaaagagggtgaagaaac |
| NASP-R | tttggcatttcttcggtctta |
| PA2G4-F | cagggcctggcttaggag |
| PA2G4-R | cagactctagccgctctcg |
| KIF2C-F | cctcagttgtcgccctca |
| KIF2C-R | ggaaagaagggcaacacaaa |
| CDC20-F | ctgtctgagtgccgtggat |
| CDC20-R | tccttgtaatggggagacca |
| DDX39A-F | ggtttgaagttaatgtggcagaa |
| DDX39A-R | gttaccggctctgctcgat |
| CCL7-F | atgaagcacctggacaagaaa |
| CCL7-R | gaaccactctgagaaaggacag |
| MMP9-F | gaactttgacagcgacaagaag |
| MMP9-R | cggcactgaggaatgatctaa |
| TNF-F | agagggagagaagcaactaca |
| TNF-R | gggtcagtatgtgagaggaaga |
| ITGAM-F | tcagcatcaccttcagtttca |
| ITGAM-R | ggagtcctcaccatcatttctc |
| ITGAX-F | actcagatcggctcctactt |
| ITGAX-R | gttagcagccacgaacaattc |
Computational Methods
RNA-seq analysis
FASTQ files were assessed for quality using FastQC55 (available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and aligned to the human genome (GRCh38 assembly) using HISAT256. Following alignment, HTSeq-counts57 was used to produce gene-level counts (using gene features from ENSEMBL) which were supplied to DESeq258 to determine differentially expressed genes between control U937 cells and ΔNSP U937 cells at an FDR cutoff of 5%. To account for transduction batch effects, transduction batch was included as a covariate in the DESeq2 analysis. DAVID59 was used to determine enriched Gene Ontology (GO) terms using the list of differentially expressed genes. To generate the heatmap of differentially expressed genes, genes where all counts were 0 were removed and log2-transformed transcripts per million (log-TPM) were calculated for the remaining genes. A linear model including a covariate for transduction batch was then used to correct for batch effects. Genes were then filtered, keeping genes with FDR <5% (as determined by DESeq2) and then plotted using Seaborn Clustermap60. Samples and genes were clustered using hierarchical clustering with the Euclidean distance metric and Ward’s minimum variance method for linkage. The values plotted are z-score transformed across each gene.
ATAC-seq analysis
FASTQ files for the ATAC-seq datasets were analyzed using PEPATAC (http://code.databio.org/PEPATAC/) with parameters as described in Corces et al.61. Control U937 cell line was used as the control/background population for MACS226. Briefly, adapters were trimmed using Skewer62, followed by removal of reads aligning to the Revised Cambridge Reference Sequence (rCRS) of human mitochondrial DNA (http://www.ncbi.nlm.nih.gov/nuccore/251831106) and human repeats (https://github.com/databio/ref_decoy) references. The resulting reads were then aligned to the human genome (GRCh38) using bowtie263 and duplicates were removed using Picard Tools (https://broadinstitute.github.io/picard/). MACS2 callpeak was then used to discover peaks in the pooled ΔNSP reads with a comparison to the pooled control U937 cell line alignments as background (or discovering peaks in pooled control U937 without a background for comparison) at a q-value of 5%, using options ‘nomodel’ and ‘keep-dup all’.
As described in Corces et al, 2018, peak summits were extended by 250 bp in both directions and removed if they extended beyond the chromosome ends or if they were within blacklisted regions from the ENCODE project64. To assess the quality of the data, read counts per million at each base in a range of −1000 to +1000 from all annotated transcriptional start sites (TSS) were plotted. Peaks were classified as being either in promoters, exons, introns, UTRs, or intergenic regions by determining if the peak overlapped with either a UTR (‘five_prime_utr’ or ‘three_prime_utr’), exon, intron (peak within a gene but not in a promoter, exon, or UTR) as annotated by Ensembl (https://uswest.ensembl.org/info/data/ftp/index.html). As promoters are not annotated, a window of 2000 bp was used from the TSS. All other peaks were classified as intergenic. The fraction of peaks in each of these features were then compared to the genomic background, i.e. the expected distribution if the peaks were uniformly distributed. Enrichment statistics were calculated using the normal approximation of the hypergeometic distribution for large N. The number of reads across selected genes (as determined by the Luminex cytokine assay) were calculated as pileups generated by `samtools mpileup` and then plotted.
ChIP-seq analysis
PEPATAC was used to perform the alignment and preprocessing of the FASTQ files from three biological replicates for ChIP-seq for both macrophages and monocytes. The resulting aligned, sorted, and deduplicated BAM files were then supplied to MACS2 using a q-value of 5% to call peaks with a comparison against H3. Peaks were annotated by genomic feature as described for ATAC-seq data. Few peaks were found in macrophages and they did no overlap across biological replicates. To select consistent peaks for the monocytes, peaks were first merged across the three biological replicates (i.e., if any part of a peak overlapped with any part of another, no matter which replicate it came from, the peaks were merged into a single peak). Peaks with at least two biological replicates represented were used for downstream analysis. Normalized pileups for each gene were plotted with ATAC-seq pileups. The same analysis was then completed for all TSSs to plot normalized reads relative to TSSs.
To compare H3ΔNThr22 ChIP-seq to the monocyte transcriptional profile from GSE5099, mean expression from that dataset was used to bin genes into low, medium, and highly expressed genes by taking the 1st, 2nd, and 3rd terciles respectively. ChIP-seq signals (pileups) were calculated for TSSs for genes in these groups with a bandwidth of 4 kbp. The aggregate signal across the three biological replicates was included in the plot.
For the U937 cell lines, two biological replicates were processed using PEPATAC and MACS2 as described above (with a lenient q-value of 25% as recommended for the following filtering steps). The resulting peaks from the two biological replicates were sorted and then filtered using the irreproducible discovery rate (IDR) method (https://github.com/nboley/idr), ranking by signal value. The IDR cutoff of 5% was then used to filter for significant peaks.
To compare H3ΔNThr22 ChIP-seq in U937 cells to the U937 transcriptional profile from GSE107566, mean expression from that dataset was used to bin genes into low, medium, and highly expressed genes by taking the 1st, 2nd, and 3rd terciles respectively. ChIP-seq signals (pileups) were calculated for TSSs for genes in these groups with a bandwidth of 4 kbp. The aggregate signal across the three biological replicates was included in the plot.
ATAC-Seq vs. ChIP-Seq analysis
To calculate the cross correlation between ATAC-seq and U937 H3ΔNThr22 ChIP-seq data, a representative bigwig file generated by PEPATAC for both the ATAC-seq and ChIP-seq data were processed by the numpy (v.1.18.1) “correlate” function over a region of 10 kbp from a randomly selected starting nucleotide. The correlations were then normalized analogously to Pearson correlation by dividing the raw correlation values by the square root of the product of each signal taken as a dot product with itself. This process was completed 1000 times and the results averaged for each shift/offset.
To find overlapping H3ΔNThr22 ChIP-seq peaks with ATAC-seq peaks, in both U937 and primary monocytes, ATAC-seq peaks found in ΔNSP U937s and not U937 controls were first merged and compared to H3ΔNThr22 ChIP-seq peaks. Any portion of a peak overlapping another was counted as an overlap.
EpiTOF analysis
For both experiments involving EpiTOF (PBMCs derived from sJIA patients and healthy PBMCs exposed to sJIA sera), CSVs representing hand-gated populations of monocytes were exported from FlowJo and pre-processed as described previously12. To summarize, histone modification marker levels were normalized to total H3/H4 by fitting a linear model with H3 and H4 values as covariates and the marker as the response variable. The residuals were then used for downstream analyses. Cell population-level means for each marker were then calculated and used in principal components analysis (PCA). The sample correlation matrix rather than the sample covariance matrix was used for each PCA.
Statistics and Reproducibility
Statistical analysis for nonsequencing data was performed using GraphPad Prism software. Unpaired two-tailed Student’s t-test was used unless otherwise stated. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P<0.0001. For the EpiTOF analyses related to sJIA samples, a preliminary unpublished dataset on H3ΔThr22 levels in sJIA patients suggested marked decrease in H3ΔThr22 level in sJIA relative to that in healthy subjects with effect sizes <−1. Using this estimate, a power analysis assuming an unpaired one-tailed t-test at 80% power, effect size −1, and significance level 0.05 was performed. The analysis suggested that a sample size greater than 13 is required. We were able to obtain clinical samples from 16 patients with sJIA. Samples from patients with quiescent or active sJIA were randomly assigned and analyzed in two biological replicates. Samples from healthy volunteers were obtained from Stanford Blood Center with age being the only selection criterion (between 17 and 18). All samples were randomly assigned to different experimental groups. For analyses related to the sJIA serum, no statistical method was used to predetermine sample size. The design was based on the number of samples that can be barcoded for combined mass cytometry processing and analysis to minimize technical variability. All experiments described in the manuscripts were repeated at least 2 times. Statistical analyses for sequencing data are described above. No statistical method was used to predetermine sample size. For ChIP-seq, we analyzed wild-type U937 cells in two biological replicates. ChIP-seq analysis of H3ΔThr22 was performed on three primary monocyte samples and their matching monocyte-derived macrophages. ATAC-seq and RNA-seq were employed to analyze three ΔNSPs clones and control cell lines. The sequencing experiments were not randomized. For all analyses, no data were excluded. The Investigators were not blinded to allocation during experiments and outcome assessment. All replicates were successful. All measurements were objectively quantifiable (e.g. instrument output). Thus, blinding was not applicable.
Extended Data
Extended Data Fig. 1. CTSG, ELANE, and PRTN3-Mediated H3ΔN in Monocytes.

(a) H3ΔNThr22 enrichment in monocytes is not associated with cell death. Viability of the cells shown in Fig. 1a measured by cisplatin staining. Data represent mean ± S.E.M. (N = 20).(b) CD14+CD16- classical monocyte-specific H3ΔNThr22 enrichment. EpiTOF analysis of the indicated monocyte subsets. Y-axis, normalized H3ΔNThr22 level; center line, median; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range; points, outliers. Statistical significance is determined by two-tailed Welch’s t-test with P values depicted.
(c) Inverse relationship between class II MHC expression and H3ΔNThr22 in monocytes. Single-cell analysis of EpiTOF data as in (a). Each dot represents a single monocyte. X-axis, HLA-DR; y-axis, H3ΔNThr22 (top) or bulk H3 levels (bottom).
(d) Chromatin localization of H3ΔN in monocytic cells. Western blot analysis of THP-1 cells biochemically separated into cytoplasmic and insoluble chromatin fractions.
(e) H3ΔN in monocytes is not catalyzed by cathepsin L. Western blot analysis of WCE from THP-1 cells expressing CRISPR-Cas9 and two independent sgRNAs targeting CTSL. Control cells express CRISPR-Cas9 but lack sgRNA.
(f) H3ΔN in monocytes is not catalyzed by matrix metalloproteases (MMPs). Western blot analysis of WCE from U937 (top) or THP-1 (bottom) cells treated with MMP9-specific or broad-spectrum MMP inhibitors. Control, DMSO treated.
(g) H3ΔN in monocytes is not catalyzed by cysteine proteases. Western blot analysis of WCE from U937 (left) or THP-1 (right) cells cultured in the presence of a cell-permeable broad-spectrum cysteine protease inhibitor E-64d. Control, DMSO treated.
(h) Serine proteases generate H3ΔN in monocytic THP-1 cells. Western blot analysis of WCE from THP-1 cells treated with nonselective serine protease inhibitor AEBSF. Control, PBS treated.
(i) Chromatin localization of CTSG, ELANE, and PRTN3. Western blot analysis of the cytoplasmic and chromatin-enriched fractions purified from THP-1 cells.
(j) Release of chromatin-bound NSPs in high-salt solution. Chromatin pellet as in Fig. 1d is washed extensively with a buffer containing physiological ionic strength and is subsequently treated with a high-salt solution to solubilize chromatin proteins. Supernatant (left) and pellet (right) fractions are subject to immunoblotting analysis. HP1α serves as a chromatin protein control.
(k) Controlled proteolytic activities of CTSG, ELANE, and PRTN3 on nucleosomal H3 in vitro. Tandem mass spectrometry analysis of protease assays as in Fig. 1h using individual NSPs and recombinant nucleosomes. Primary cleavage sites accounting for greater than 20% of proteolytic products are labeled. CTSG (red); ELANE (green); PRTN3 (blue).
(l and m) Distinct histone modification profiles between FL- and truncated H3. Immunoblotting analysis of WCE from U937 cells (l) or primary monocytes (m) using the indicated antibodies. Three biological replicates are shown. These results are used for the quantitative analyses shown in Fig. 1i and 1j.
Extended Data Fig. 2. Reintroduction of H3ΔN into ΔNSPs Cells Reverses the Morphologic and Functional Alterations Associated with NSP Depletion.
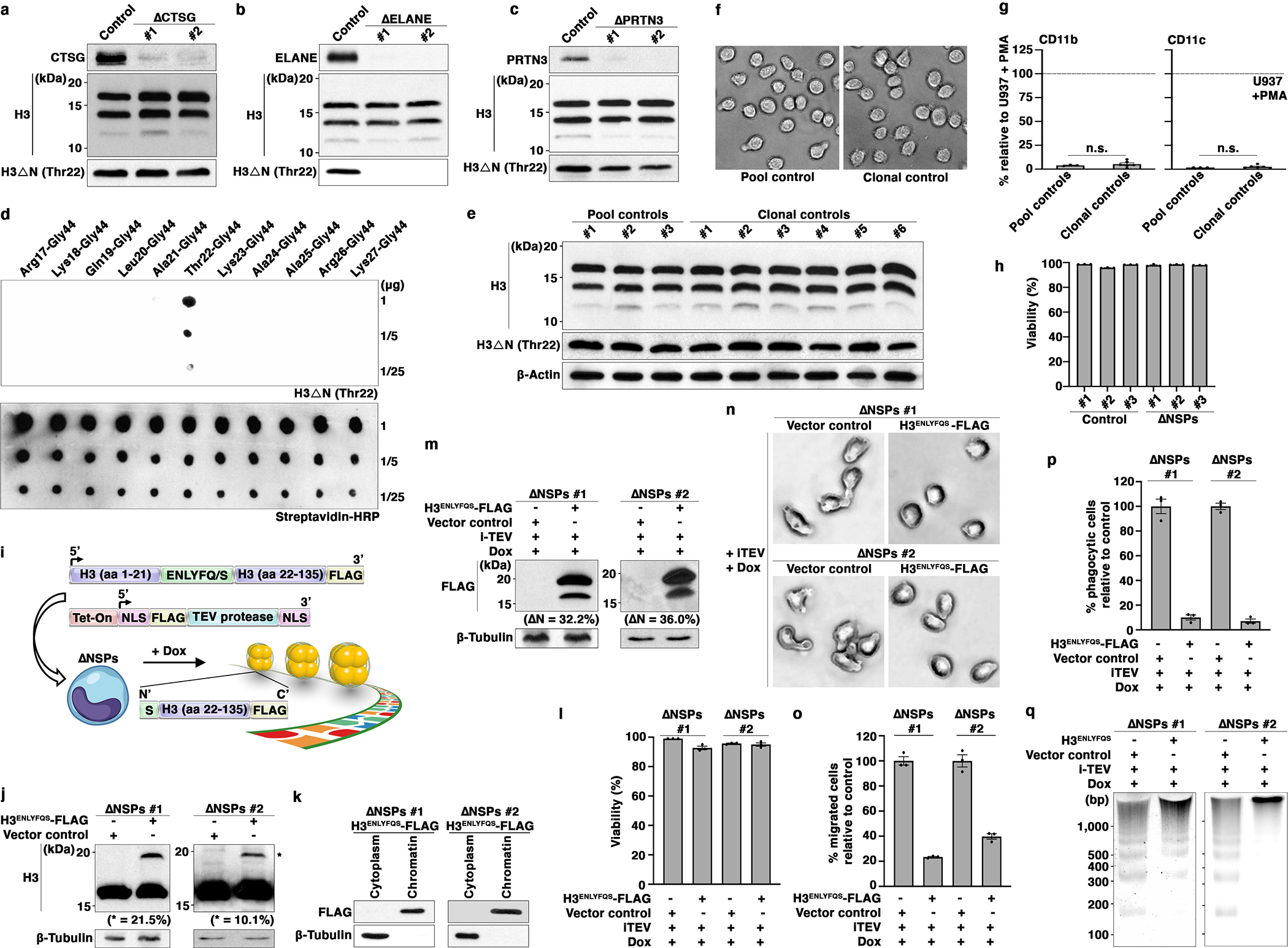
(a, b, c) Depletion of individual NSPs does not affect global H3ΔN level in monocytes. Western blot analysis of WCE from U937 cells depleted of CTSG (a), ELANE (b), or PRTN3 (c). Control cells express CRISPR-Cas9 but lack sgRNA.
(d) Highly specific anti-H3ΔNThr22 antibody used in this study. Dot blot analysis of the anti-H3ΔNThr22 antibody using the indicated synthetic peptides. This affinity reagent is employed throughout the entire study.
(e, f, g) Clonal selection by FACS does not alter H3ΔN level and pattern, cell morphology, or activation markers CD11b and CD11c expression. Western analysis of WCE from control cells as in Fig. 2a with (six samples on the right) or without (three samples on the left) clonal selection by FACS (e). Representative light microscopy images of the control cells as in (e) (f). Mass cytometry analysis of the cells as in (e) (g). Y-axis, mean signal intensities of CD11b (left) or CD11c (right) from the indicated cells relative to the signals from PMA-activated U937 cells as a positive control. Data represent mean ± S.E.M. (three pool controls (left) or six clonal controls (right)). Clonal controls are randomly selected from approximately 400 sorted clones. Statistical significance is determined by two-tailed Student’s t-test.
(h) Simultaneous NSP depletion does not affect cell viability. Mass cytometry analysis of ΔNSPs and control cells. The percentage of cells negative for cisplatin staining for each cell line is shown. Data represent mean ± S.E.M. (three technical replicates).
(i) Strategy to reintroduce H3ΔN into ΔNSPs cells.
(j) Expression of epitope-tagged exogenous H3 (H3ENLYFQS-FLAG) in ΔNSPs cells. Western blot analysis of WCE from ΔNSPs cells transduced with H3ENLYFQS-FLAG (right lane) or empty vector (left lane). The relative abundance of H3ENLYFQS-FLAG to endogenous H3 determined by ImageJ software is shown.
(k) Chromatin localization of exogenous H3. Western blot analysis of cytoplasmic and chromatin fractions from ΔNSPs cells expressing H3ENLYFQS-FLAG.
(l) Doxycycline treatment to induce TEV protease expression and H3ENLYFQS-FLAG cleavage does not affect cell viability. Viability of the indicated cells is determined by trypan blue staining and an automatic cell counter. Data represent mean ± S.E.M. (three technical replicates).
(m) H3ENLYFQS-FLAG cleavage in response to doxycycline-induced TEV protease expression. Western analysis of WCE from the indicated cells. The abundance of cleavage product relative to total H3ENLYFQS-FLAG is determined by ImageJ software.
(n, o, p) Reintroduction of H3ΔN into ΔNSPs cells reverses morphological and functional alterations associated with NSP depletion. Light microscopy (n), transwell cell migration (o), and phagocytosis (p) analyses of the cells as in (m). Data represent mean ± S.E.M. (three technical replicates).
(q) Reintroduction of H3ΔN into ΔNSPs cells alters global chromatin architecture. MNase sensitivity analysis of the cells as in (m).
Extended Data Fig. 3. NSP and H3ΔN Repression as Primary Monocytes Mature into Macrophages.

(a) H3ΔNThr22 repression during monocyte-to-macrophage differentiation. Mass cytometry analysis of monocytes and monocyte-derived macrophage. Cells collected at days one (red), three (green), and five (blue) in culture for differentiation are analyzed. Independent biological replicate of Fig. 3a from a different donor.
(b) H3ΔNThr22 repression during monocyte-to-macrophage differentiation is not associated with cell death. Viability of the cells as in Fig. 3a (donor 1) and (a) (donor 2) assessed by cisplatin staining and mass cytometry.
(c) Repression of all species of H3ΔN as monocytes differentiate into macrophages. Western blot analysis of WCE from primary monocytes and monocyte-derived macrophages. Macrophages, cells collected at day seven in culture. Molecular weight markers, see Source Data. Independent biological replicate of Fig. 3b from a different donor.
(d) Macrophages depleted of H3ΔNThr22 show robust phagocytosis capability. Mass cytometry analysis of mature macrophages as in (a) (five-day differentiation in vitro) incubated with osmium-labelled E. coli to measure phagocytosis capability. Control, no E coli particle. Cytochalasin D is used to demonstrate the specificity of phagocytosis measurement. H3ΔNThr22 (x-axis) and osmium (y-axis) levels measured by mass cytometry are shown. Independent biological replicate of Fig. 3c from a different donor.
(e) H3ΔN repression is maintained in polarized macrophages. Western blot analysis of freshly isolated monocytes, naïve, classically activated, and alternatively activated macrophages from a healthy volunteer. Independent biological replicate of Fig. 3d.
(f) CTSG, ELANE, and PRTN3 repression during monocyte-to-macrophage differentiation. Western blot analysis of the samples as in (c) using the indicated antibodies. Molecular weight markers, see Source Data. Independent biological replicate of Fig. 3e from a different donor.
(g) Transcriptional repression of CTSG, ELANE, and PRTN3 in U937 cells treated with PMA. Differential gene expression analysis of CTSG, ELANE, and PRTN3 (x-axis) in undifferentiated (green) and differentiated (purple) U937 cells using the publicly available dataset GSE107566. Each dot represents an independent sample. Y-axis, reads per kilobase per million mapped reads (RPKM).
(h) Repression of CTSG, ELANE, and PRTN3 proteins in U937 cells treated with PMA. Western blot analysis of WCE from U937 cells treated with or without PMA using the indicated antibodies.
(i and j) Pharmacological inhibition of NSPs accelerates macrophage development. Western blot analysis of WCE from peripheral blood monocytes cultured in the absence or presence of ELANE inhibitor GW311616 in combination with CTSG inhibitor CAS 429676–93-7 (i). Phagocytosis analysis of the cells as in (i) using mass cytometry and osmium-labeled E. coli (j). Independent biological replicate of Fig. 3g and h from a different donor.
(k) Exogenous NSP expression is unaffected by NSP repression during cellular differentiation. Western blot analysis of WCE from U937 cells stably expressing exogenous CTSG (left), ELANE (middle), or PRTN3 (right) under the control of a cytomegalovirus promoter treated with or without PMA to induce differentiation. Control, cells transduced with lentiviral vector only without a transgene.
(l) Constitutive NSP expression suppresses cellular differentiation. Ectopic overexpression of the indicated NSPs (x-axis) in U937 cells treated with PMA. CD11c expression is determined by mass cytometry. Data represent mean ± S.E.M. (three technical replicates).
Extended Data Fig. 4. Widespread H3ΔN Genomic Enrichment in Monocytic Cells is Associated with Permissive Chromatin and Active Transcription.

(a) Validation of affinity reagents for ChIP-seq. Immunoprecipitation under the stringent ChIP condition using formaldehyde-crosslinked chromatin and the indicated antibodies. ChIP samples are subject to immunoblotting analysis using an antibody raised against H3. Control, bare magnetic beads.
(b) Mono-nucleosome-enriched sheared chromatin for ChIP-seq analysis. Gel electrophoresis analysis of chromatin input for ChIP-seq analysis. DNA ladder in base pairs is shown.
(c) Widespread H3ΔNThr22 distribution across the genome. Percentages of H3ΔNThr22 peaks at the indicated 22 pairs of autosomes.
(d and e) Widespread H3ΔNThr22 enrichment and ELANE occupancy across the genome. Representative genomic tracks of H3ΔNThr22 (top) and bulk H3 (bottom) ChIP-seq data. Genomic regions with high (red) or low (green) H3ΔNThr22 ChIP-seq signals are tested (d). qPCR analysis of H3ΔNThr22 (top) or ELANE (bottom) ChIP DNA from control (gray) or ΔNSPs (white) cells using the indicated primer pairs. Y-axes, fold enrichment over bulk H3. Data represent mean ± S.E.M. (three biological replicates (three control cell lines or thee ΔNSPs clones) and three technical replicates (N=9)) (e).
(f) Validation of affinity reagents for ChIP analysis of NSP occupancy. Immunoprecipitation as in (a) using antibodies recognizing the indicated NSPs. Immunoprecipitation samples are subsequently analyzed by immunoblotting using an antibody recognizing bulk H3. Control, bare magnetic beads.
(g) Peak association between H3ΔNThr22 ChIP-seq and ATAC-seq datasets. Representative genomic tracks of H3ΔNThr22 ChIP-seq data from wild-type U937 cells (top) and ATAC-seq data from control cells (bottom).
(h) Positive correlation between transcriptional activity and chromatin accessibility. TSS-proximal ATAC-seq signals for the three groups of genes with high (red), medium (gray), or low (blue) gene expression activity (GSE107566).
Extended Data Fig. 5. Widespread H3ΔN Genomic Enrichment in Primary Monocytes is Associated with Permissive Chromatin and Active Transcription.
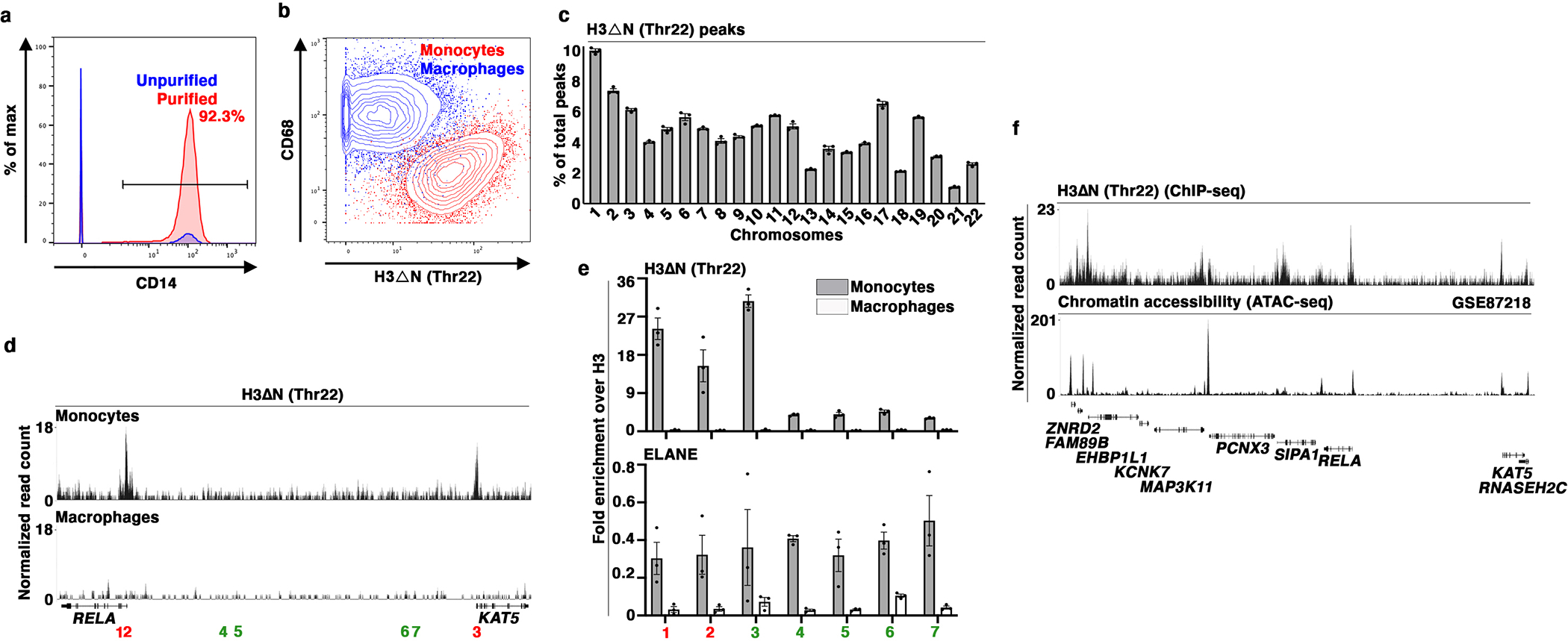
(a) Purity of primary monocytes used for ChIP-seq analysis. Representative mass cytometry data showing the percentages of CD14+ monocytes in bulk PBMC (blue) or purified monocyte samples (red) used for ChIP-seq analysis. X-axis, CD14 level; y-axis, percentage of the maximal count.
(b) Fully differentiated macrophages used for ChIP-seq analysis. Representative mass cytometry data showing H3ΔNThr22 (x-axis) level and CD68 expression (y-axis) in macrophages (blue) relative to those in monocytes (red) for the cells used for ChIP-seq analysis.
(c) Widespread H3ΔNThr22 distribution across the genome in primary monocytes. Percentages of H3ΔNThr22 peaks at the indicated 22 pairs of autosomes. Data represent mean ± S.E.M. (three biological replicates).
(d and e) H3ΔNThr22 enrichment and ELANE occupancy at the p65 subunit (RELA) of NF-κB locus in primary monocytes. Representative genomic tracks of H3ΔNThr22 ChIP-seq data from primary monocyte (top) or the matching monocyte-derived macrophages (bottom). Genomic regions with high (red) or low (green) H3ΔNThr22 are tested (d). qPCR analysis of H3ΔNThr22 enrichment (top) or ELANE occupancy (bottom) in primary monocytes (gray) or the matching macrophages (white) using the indicated primer pairs. Y-axes, fold enrichment over bulk H3. Data represent mean ± S.E.M. (three technical replicates) (e).
(f) H3ΔNThr22 is associated with permissive chromatin in primary monocytes. Representative genomic tracks of H3ΔNThr22 ChIP-seq (top) and ATAC-seq (GSE87218) (bottom) datasets from primary monocytes.
Extended Data Fig. 6. NSP and H3ΔN Depletion Is Associated with Increased Chromatin Accessibility.

(a) Differential gene expression in ΔNSPs cells is associated with chromatin accessibility changes. Integrative analysis of ATAC-seq and RNA-seq datasets focusing on differentially expressed genes in Fig. 6a. Y-axis, relative chromatin accessibility determined by ATAC-seq; left, genes downregulated in ΔNSPs cells relative to controls (N=220); right, genes upregulated in ΔNSPs cells relative to controls (N=170). Statistical significance is determined by two-tailed Welch’s t test with P value depicted.
(b) Differential gene expression in U937 cells depleted of NSPs affects selective biological processes. Gene ontology analysis of the differentially expressed genes in Fig. 6a. Gene ontology terms in which differential genes are enriched are ranked by FDR.
(c) Increased chromatin accessibility across the genome in U937 cells depleted of NSPs and H3ΔN. The distribution of differentially accessible peaks across 22 pairs of autosomes (x-axis) in ΔNSPs cells. Color, three independent ΔNSPs clones; y-axis, percentages of peaks at specific chromosomes.
(d) Genic region enrichment of differentially accessible peaks in ΔNSPs cells. X-axis, the presence of differentially accessible peaks in ΔNSPs cells in the indicated genomic regions over their relative proportions in the genome. Center line, genomic distribution with no enrichment. Data represent mean ± S.D. (three biological replicates).
(e) Further increase in chromatin accessibility upon NSP and H3ΔNThr22 depletion. Representative genomic tracks of the H3ΔNThr22 ChIP-seq dataset from wild-type U937 cells (top) and the ATAC-seq datasets from control (middle) or ΔNSPs (bottom) cells at the STAT5A-STAT3 locus.
Extended Data Fig. 7. NSP and H3ΔN Repression Primes the Chromatin to Facilitate Transcription Reprogramming During Cellular Differentiation.
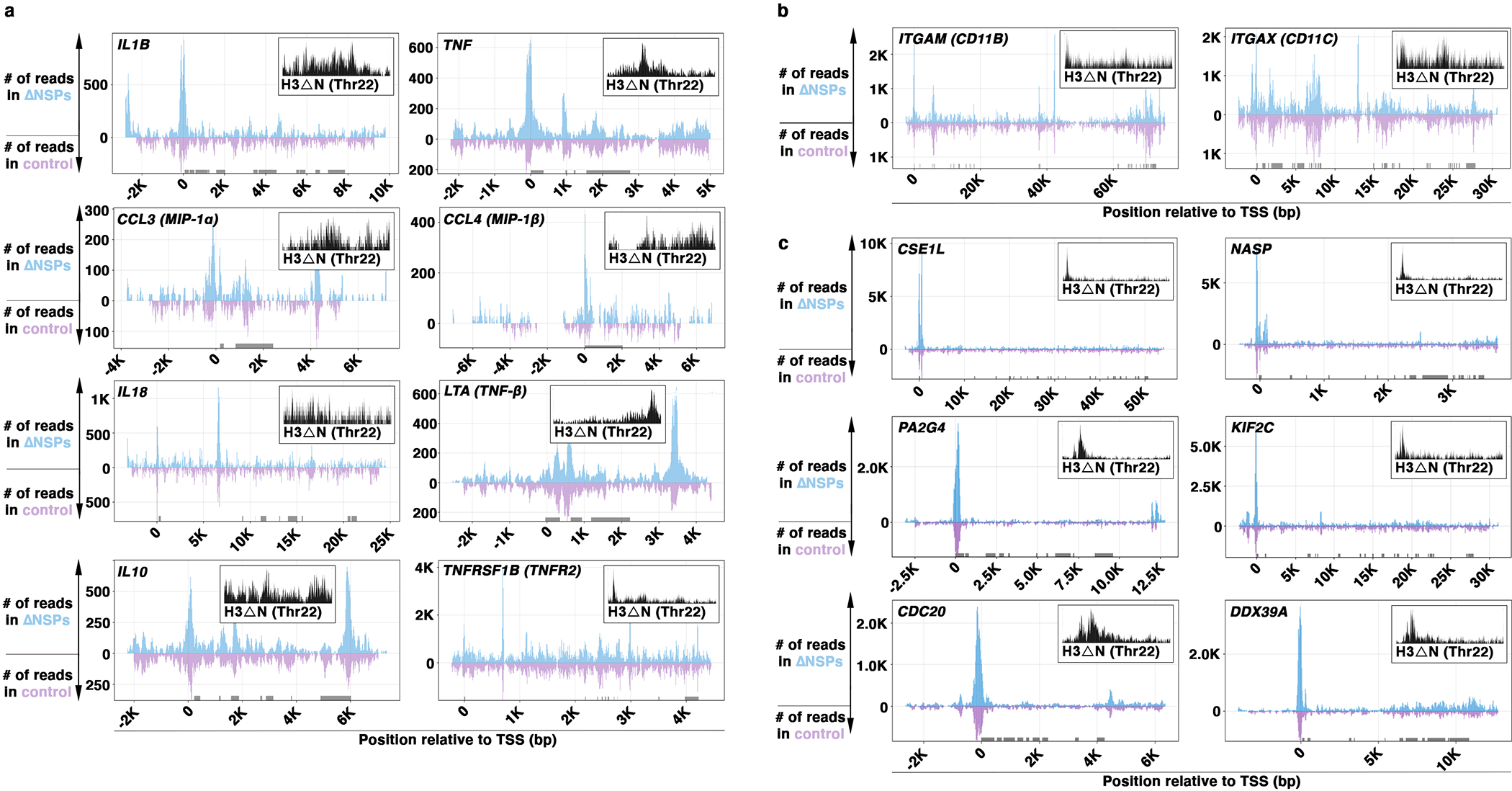
(a-c) Increased chromatin accessibility at genes with important immune regulatory functions in ΔNSPs cells. ATAC-seq sequencing tracks of the loci encoding proinflammatory cytokines, immune regulators (a), differentiation markers (b), or differential genes upon differentiation (c) in ΔNSPs (blue) or control (purple) cells. X-axis, relative position to TSS in base pairs; y-axis, normalized read count in ΔNSPs (above zero) or control (below zero) cells; gray block, exon. H3ΔNThr22 genomic track in wild-type U937 cells is shown.
Extended Data Fig. 8. IL-6 and Ferritin Contribute to H3ΔN Repression in Monocytes from sJIA patients.
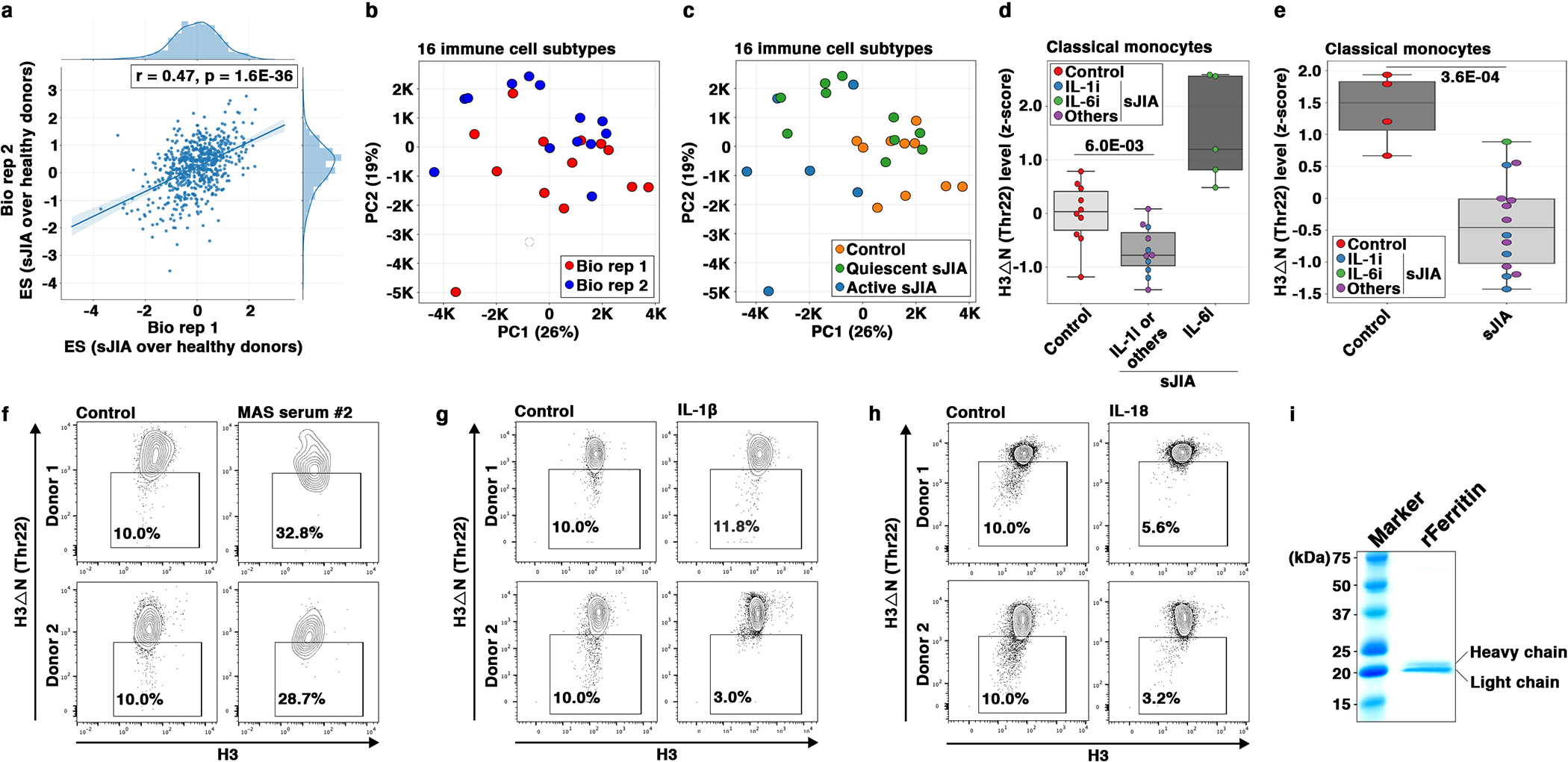
(a) Robust epigenetic alterations in sJIA patients captured by independent replicates. EpiTOF analysis of the global levels of 40 histone marks in 16 major immune cell subtypes from sJIA patients and healthy volunteers. Effect size comparisons of 560 data points are computed using the biological replicates 1 (x-axis) or 2 (y-axis) datasets. The trendline and Pearson’s correlation coefficient are shown. Statistical significance is determined by Student’s t-test for Pearson correlation with P value depicted.
(b) Insignificant batch effect between the two biological replicates. PCA of the EpiTOF data as in (a) where each dot represents a single subject from biological replicate 1 (red) or 2 (blue) using the variance of 40 histone marks in 16 major immune cell subtypes (560 data points). The proportion of the variance explained by each principal component is shown.
(c) Separation of sJIA patients from healthy volunteers by epigenetic landscape. PCA as in (b). Orange, healthy donors; green, sJIA patients with quiescent disease; blue, sJIA patients with active disease.
(d) Repressed H3ΔNThr22 in sJIA patients with the exception of those on IL-6-blocking therapy. Box plot representation of the H3ΔNThr22 levels in monocytes from healthy volunteers, sJIA patients on IL-1 (blue) or IL-6 targeting therapies (green), or on no or other biologic treatments (purple). Center line, median; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range; points, outliers. Statistical significance is determined by two tailed Student’s t-test with P value depicted.
(e) Reduced H3ΔNThr22 in monocytes from sJIA patients. EpiTOF analysis of PBMCs from 14 sJIA patients and 4 healthy volunteers focusing on H3ΔNThr22 in monocytes. An independent validation cohort in addition to the one described in Fig. 8a. Each dot represents the H3ΔNThr22 level in monocytes from a subject. sJIA patients are colored based on treatments as in (d). Center line, median; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range; points, outliers. Statistical significance is determined by two tailed Student’s t-test with P value depicted.
(f) Sera from sJIA patients with MAS induce H3ΔNThr22 repression in monocytes from healthy volunteers. Mass cytometry analysis of monocytes from two healthy volunteers (top and bottom) cultured in vitro with sera from a healthy donor (left) or a sJIA patient with MAS (right) for 24 hours. Biological replicate of the experiment described in Fig. 8f using independent serum sample from an sJIA patient with MAS and independent PBMCs from two healthy volunteers.
(g and h) Clinically important cytokines IL-1β and IL-18 do not induce H3ΔNThr22 repression in monocytes. Mass cytometry analysis of PBMCs from two healthy volunteers (top and bottom) treated with (right) or without (left) IL-1β (g) or IL-18 (h) for 24 hours. X-axis, bulk H3; y-axis, H3ΔNThr22 level; each dot represents a single monocyte.
(i) Purity of recombinant ferritin for ex vivo monocyte stimulation. Coomassie staining of ferritin complex purified from E. coli ectopically co-expressing the heavy and light subunits of ferritin. Molecular weight marker is shown.
Supplementary Material
Acknowledgements
We thank our colleagues at the Oklahoma Medical Research Foundation for helpful discussion. We also thank Bhupinder Nahal and the Division of Pediatric Rheumatology at University of California San Francisco, led by Dr. Emily von Scheven, for collection of several serum samples and associated clinical data. This work was supported in part by the Donald E. and Delia B. Baxter Foundation (to P.J.U.), Elizabeth F. Adler (to P.J.U.), the Henry Gustav Floren Trust (to P.J.U.), the Bill & Melinda Gates Foundation (OPP1113682 to P.J.U. and P.K.), EMD Serono (to P.J.U. and P.K.), the Department of Defense contracts W81XWH-18–1-0253 and W81XWH1910235 (to P.K.), the Ralph & Marian Falk Medical Research Trust (to P.K.), the sJIA Foundation (to E.D.M. and G.S.S.), the Lucile Packard Foundation for Children’s Health (to E.D.M.), the Fundación Bechara (to P.A.N.), the Arbuckle Family Foundation for Arthritis Research (to P.A.N.), Cincinnati Children’s Research Foundation ARC Grant (to G.S.S. and A.A.G.), and the NIH the Autoimmunity Center of Excellence U19 AI110491 (to P.J.U.), R01 AI125197 (to P.K. and P.J.U.), U19 AI109662 (to P.K.), U19 AI057229 (to P.K.), R01AR061297 (to E.D.M.), R35 GM139569 (to O.G.), P30 AR070253 (to P.A.N.), R01 AR073201 (to P.A.N.), K08 AR073339 (to L.A.H.), R01 AR059049 (to A.A.G.), P30 AR070549 (to G.S.S. and A.A.G.), and K08 AR072075 (to G.S.S.).
Acknowledgements
Competing Interests
sJIA-related consultation or research support: E.D.M., Novartis; P.A.N., Novartis; A.A.G., Juno, Novartis, NovImmune and AB2Bio; G.S.S, Novartis. The remaining authors declare no competing interests.
Data Availability
ChIP-seq, ATAC-seq, and RNA-seq datasets have been deposited in the Gene Expression Omnibus (GEO) with accession numbers GSE142661, GSE142660, and GSE142662, respectively. Other data generated during and/or analyzed during the current study, and all reagents, including cell lines and plasmid DNA, described in this work are available from the corresponding author on reasonable request.
Footnotes
Figures associated with individual datasets are listed below.
ATAC-seq, GSE142660 (Fig. 4e, 4f, 4g, 6c, 6d, 6e, 6f, Extended Data Fig. 4g, 6a, 6c, 6d, 6e, 7a, 7b, and 7c)
ChIP-seq, GSE142661 (Fig. 4a, 4b, 4c, 4e, 4f, 4g, 4h, 5a, 5b, 5c, 5e, 5f, 5g, 6d, 6e, 6f, Extended Data Fig. 4c, 4d, 4g, 5c, 5d, 5f, and 6e)
RNA-seq, GSE142662 (Fig. 6a and Extended Data Fig. 6a, and 6b)
Code Availability
Custom code and mathematical algorithm to analyze ChIP-seq, ATAC-seq, RNA-seq, and EpiTOF datasets are available from the corresponding author on reasonable request.
References
- 1.Dhaenens M, Glibert P, Meert P, Vossaert L & Deforce D Histone proteolysis: a proposal for categorization into ‘clipping’ and ‘degradation’. Bioessays 37, 70–79 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Santos-Rosa H et al. Histone H3 tail clipping regulates gene expression. Nat Struct Mol Biol 16, 17–22 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xue Y, Vashisht AA, Tan Y, Su T & Wohlschlegel JA PRB1 is required for clipping of the histone H3 N terminal tail in Saccharomyces cerevisiae. PLoS One 9, e90496 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duncan EM et al. Cathepsin L proteolytically processes histone H3 during mouse embryonic stem cell differentiation. Cell 135, 284–294 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khalkhali-Ellis Z, Goossens W, Margaryan NV & Hendrix MJ Cleavage of Histone 3 by Cathepsin D in the involuting mammary gland. PLoS One 9, e103230 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim K et al. MMP-9 facilitates selective proteolysis of the histone H3 tail at genes necessary for proficient osteoclastogenesis. Genes Dev 30, 208–219 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iwasaki W et al. Contribution of histone N-terminal tails to the structure and stability of nucleosomes. FEBS Open Bio 3, 363–369 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kouzarides T Chromatin modifications and their function. Cell 128, 693–705 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Asp P et al. Genome-wide remodeling of the epigenetic landscape during myogenic differentiation. Proc Natl Acad Sci U S A 108, E149–158 (2011).21551099 [Google Scholar]
- 10.Fall N et al. Gene expression profiling of peripheral blood from patients with untreated new-onset systemic juvenile idiopathic arthritis reveals molecular heterogeneity that may predict macrophage activation syndrome. Arthritis Rheum 56, 3793–3804 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Ravelli A et al. 2016 Classification Criteria for Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopathic Arthritis: A European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Ann Rheum Dis 75, 481–489 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Cheung P et al. Single-Cell Chromatin Modification Profiling Reveals Increased Epigenetic Variations with Aging. Cell 173, 1385–1397 e1314 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ziegler-Heitbrock L et al. Nomenclature of monocytes and dendritic cells in blood. Blood 116, e74–80 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Wilcox D & Mason RW Inhibition of cysteine proteinases in lysosomes and whole cells. Biochem J 285 (Pt 2), 495–502 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herrmann C, Avgousti DC & Weitzman MD Differential Salt Fractionation of Nuclei to Analyze Chromatin-associated Proteins from Cultured Mammalian Cells. Bio Protoc 7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luger K, Rechsteiner TJ & Richmond TJ Preparation of nucleosome core particle from recombinant histones. Methods Enzymol 304, 3–19 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Hake SB et al. Serine 31 phosphorylation of histone variant H3.3 is specific to regions bordering centromeres in metaphase chromosomes. Proc Natl Acad Sci U S A 102, 6344–6349 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korkmaz B, Horwitz MS, Jenne DE & Gauthier F Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev 62, 726–759 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinez FO, Gordon S, Locati M & Mantovani A Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol 177, 7303–7311 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Holness CL & Simmons DL Molecular cloning of CD68, a human macrophage marker related to lysosomal glycoproteins. Blood 81, 1607–1613 (1993). [PubMed] [Google Scholar]
- 21.Fabriek BO, Dijkstra CD & van den Berg TK The macrophage scavenger receptor CD163. Immunobiology 210, 153–160 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Schulz D, Severin Y, Zanotelli VRT & Bodenmiller B In-Depth Characterization of Monocyte-Derived Macrophages using a Mass Cytometry-Based Phagocytosis Assay. Sci Rep 9, 1925 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mantovani A, Sozzani S, Locati M, Allavena P & Sica A Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23, 549–555 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Li Q, Brown JB, Huang H & Bickel PJ Measuring reproducibility of high-throughput experiments. The annals of applied statistics 5, 1752–1779 (2011). [Google Scholar]
- 25.Haney MS et al. Identification of phagocytosis regulators using magnetic genome-wide CRISPR screens. Nat Genet 50, 1716–1727 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Novakovic B et al. beta-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell 167, 1354–1368 e1314 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mellins ED, Macaubas C & Grom AA Pathogenesis of systemic juvenile idiopathic arthritis: some answers, more questions. Nat Rev Rheumatol 7, 416–426 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schneider R, Canny SP & Mellins ED Cytokine Storm Syndrome Associated with Systemic Juvenile Idiopathic Arthritis. In: Cron RQ & Behrens EM (eds). Cytokine Storm Syndrome. Springer International Publishing: Cham, 2019, pp 349–379. [Google Scholar]
- 30.Ruperto N et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med 367, 2396–2406 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Quartier P et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis 70, 747–754 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Benedetti F et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med 367, 2385–2395 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Grom AA & Mellins ED Macrophage activation syndrome: advances towards understanding pathogenesis. Curr Opin Rheumatol 22, 561–566 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chomarat P, Banchereau J, Davoust J & Palucka AK IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat Immunol 1, 510–514 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Schulert GS et al. Effect of Biologic Therapy on Clinical and Laboratory Features of Macrophage Activation Syndrome Associated With Systemic Juvenile Idiopathic Arthritis. Arthritis Care Res (Hoboken) 70, 409–419 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Santambrogio P et al. Production and characterization of recombinant heteropolymers of human ferritin H and L chains. J Biol Chem 268, 12744–12748 (1993). [PubMed] [Google Scholar]
- 37.Lennartsson A, Garwicz D, Lindmark A & Gullberg U The proximal promoter of the human cathepsin G gene conferring myeloid-specific expression includes C/EBP, c-myb and PU.1 binding sites. Gene 356, 193–202 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Barski A et al. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Xie J, Wooten M, Tran V & Chen X Breaking Symmetry - Asymmetric Histone Inheritance in Stem Cells. Trends Cell Biol 27, 527–540 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Voigt P et al. Asymmetrically modified nucleosomes. Cell 151, 181–193 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cooley J, Takayama TK, Shapiro SD, Schechter NM & Remold-O’Donnell E The serpin MNEI inhibits elastase-like and chymotrypsin-like serine proteases through efficient reactions at two active sites. Biochemistry 40, 15762–15770 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Bird CH et al. Nucleocytoplasmic distribution of the ovalbumin serpin PI-9 requires a nonconventional nuclear import pathway and the export factor Crm1. Mol Cell Biol 21, 5396–5407 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simon MD et al. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell 128, 1003–1012 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmitges FW et al. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol Cell 42, 330–341 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Netea MG et al. Trained immunity: A program of innate immune memory in health and disease. Science 352, aaf1098 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saeed S et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 345, 1251086 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheung P, Khatri P, Utz PJ & Kuo AJ Single-cell technologies - studying rheumatic diseases one cell at a time. Nat Rev Rheumatol 15, 340–354 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheung P et al. Single-cell epigenetics - Chromatin modification atlas unveiled by mass cytometry. Clin Immunol 196, 40–48 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li L et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc Natl Acad Sci U S A 107, 3505–3510 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen TT et al. TIM-2 is expressed on B cells and in liver and kidney and is a receptor for H-ferritin endocytosis. J Exp Med 202, 955–965 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McGuire MJ, Lipsky PE & Thiele DL Generation of active myeloid and lymphoid granule serine proteases requires processing by the granule thiol protease dipeptidyl peptidase I. J Biol Chem 268, 2458–2467 (1993). [PubMed] [Google Scholar]
- 52.Mendez J & Stillman B Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol 20, 8602–8612 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buenrostro JD, Wu B, Chang HY & Greenleaf WJ ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 21–21 29 29 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dahl JA & Collas P Q2ChIP, a quick and quantitative chromatin immunoprecipitation assay, unravels epigenetic dynamics of developmentally regulated genes in human carcinoma cells. Stem Cells 25, 1037–1046 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Andrews S FastQC: a quality control tool for high throughput sequence data. Babraham Bioinformatics, Babraham Institute, Cambridge, United Kingdom; 2010. [Google Scholar]
- 56.Kim D, Langmead B & Salzberg SL HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Anders S, Pyl PT & Huber W HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang da W, Sherman BT & Lempicki RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 60.Waskom M et al. mwaskom/seaborn: v0. 9.0 (July 2018). DOI: 10.5281/zenodo1313201 (2018). [DOI] [Google Scholar]
- 61.Corces MR et al. The chromatin accessibility landscape of primary human cancers. Science 362 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jiang H, Lei R, Ding SW & Zhu S Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics 15, 182 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Consortium EP An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ChIP-seq, ATAC-seq, and RNA-seq datasets have been deposited in the Gene Expression Omnibus (GEO) with accession numbers GSE142661, GSE142660, and GSE142662, respectively. Other data generated during and/or analyzed during the current study, and all reagents, including cell lines and plasmid DNA, described in this work are available from the corresponding author on reasonable request.