

Abstract
Approximately 30% of patients with newly diagnosed acute myeloid leukemia (AML) harbor mutations in the fms-like tyrosine kinase 3 (FLT3) gene. While the adverse prognostic impact of FLT3-ITDmut in AML has been clearly proven, the prognostic significance of FLT3-TKDmut remains speculative. Current guidelines recommend rapid molecular testing for FLT3mut at diagnosis and earlier incorporation of targeted agents to achieve deeper remissions and early consideration for allogeneic stem cell transplant (ASCT). Mounting evidence suggests that FLT3mut can emerge at any timepoint in the disease spectrum emphasizing the need for repetitive mutational testing not only at diagnosis but also at each relapse. The approval of multi-kinase FLT3 inhibitor (FLT3i) midostaurin with induction therapy for newly diagnosed FLT3mut AML, and a more specific, potent FLT3i, gilteritinib as monotherapy for relapsed/refractory (R/R) FLT3mut AML have improved outcomes in patients with FLT3mut AML. Nevertheless, the short duration of remission with single-agent FLT3i’s in R/R FLT3mut AML in the absence of ASCT, limited options in patients refractory to gilteritinib therapy, and diverse primary and secondary mechanisms of resistance to different FLT3i’s remain ongoing challenges that compel the development and rapid implementation of multi-agent combinatorial or sequential therapies for FLT3mut AML.
Subject terms: Acute myeloid leukaemia, Targeted therapies
Overview
Fms-like tyrosine kinase 3 (FLT3) is a recurrent genetic abnormality in AML (~30%)1–3. FLT3 activating mutations (FLT3mut) may involve either the juxta membrane domain [internal tandem duplication mutations (FLT3-ITD)]4 or the tyrosine kinase domain (FLT3-TKD)5,6. In patients with newly diagnosed AML, FLT3-ITDmut is a poor prognostic factor in terms of relapse-free (RFS) and overall survival (OS)7–10. FLT3-TKD activating mutations also constitutively activate FLT311; however, they have not been associated with a consistent prognostic impact12.
Prognostic impact of FLT3 mutations
Not all FLT3-ITDmut are equal; the prognostic impact is influenced by the allele ratio (AR), insertion site, ITD length, co-mutations (NPM1), and karyotype. AR is defined as the ratio of ITD-mutated alleles to wild-type allele (FLT3‐ITD/FLT3 wild-type)13. Variant allele frequency (VAF) is the ratio of ITD-mutated alleles to ITD-mutated + wild-type alleles (FLT3‐ITD/FLT3‐ITD + FLT3 wild-type)14. Schlenk et al. evaluated the impact of AR in 323 patients with newly diagnosed FLT3-ITDmut AML. After post-remission therapy with either consolidation (high-dose cytarabine-based) or allogeneic stem cell transplant (ASCT), AR ≥0.51 and FLT3-ITD insertion site in TKD1 were associated with an unfavorable RFS (P = 0.0008) and OS (P = 0.004)15. In fact, every quartile increase in FLT3-ITD AR (from 0.01 to 0.20, 0.20 to 0.53, 0.53 to 0.80, 0.80 to 1.19) was associated with worsening complete remission (CR) rates, RFS, and OS, highlighting the prognostic value of AR. It is important to note that none of these patients received a FLT3 inhibitor (FLT3i) during induction, consolidation, or post-ASCT.
The UKMRC group evaluated the presence of NPM1 co-mutations in young adult patients with AML. Favorable relapse risk and OS were seen in NPM1mut with FLT3 wild-type; intermediate prognosis in FLT3-ITDmut with concurrent NPM1mut, and adverse prognosis in FLT3-ITDmut with NPM1 wild-type patients16. The Spanish group evaluated intermediate-risk AML patients treated with intensive chemotherapy. In patients with concurrent NPM1mut, the OS and relapse risk were comparable between FLT3 wild-type and FLT3-ITDmut AR <0.5, but worse when AR ≥0.5. Among those with NPM1 wild-type, all FLT3-ITDmut patients had an increased risk of relapse and inferior OS, regardless of the AR17. The current European Leukemia Net (ELN) guidelines categorize FLT3 -ITDmut AML as favorable (NPM1mut with FLT3 wild-type Or NPM1mut with FLT3-ITD AR<0.5), intermediate (NPM1mut with FLT3-ITD AR>0.5 Or NPM1WT with FLT3-ITD AR<0.5), or adverse (NPM1WT with FLT3-ITD AR>0.5)18. However, a subsequent UKMRC study of 1600 patients with cytogenetic intermediate-risk AML showed that relapse risk did not differ based on the FLT3-ITDmut AR, and that the cumulative incidence of relapse in patients with NPM1mut was increased with a concurrent FLT3-ITDmut irrespective of the AR19. Oran et al. recently showed that ASCT in CR1 improved RFS and OS independent of the FLT3-ITDmut AR or NPM1mut status in patients with FLT3-ITDmut AML20. Collectively, NPM1mut even with FLT3-ITDmut AR <0.5 are likely higher risk than truly “favorable risk” AML and we continue to consider them for ASCT in CR1.
Minetto and colleagues retrospectively evaluated the efficacy of fludarabine, high-dose cytarabine, and idarubicin (FAI) in 149 newly diagnosed FLT3-ITDmut and/or NPM1mut AML (only FLT3-ITDmut = 29; FLT3-ITDmut NPM1mut = 59, only NPM1mut = 61). In patients ≤55 years, this regimen appeared to overcome the negative impact of FLT3-ITDmut in NPM1 co-mutated patients, regardless of the FLT3 AR, with comparable 3-year OS rates of 64% and 68% in FLT3-ITDmut NPM1mut and FLT3-ITDWT NPM1mut patients, respectively (P > 0.05). Moreover, ASCT in CR1 only benefitted patients with isolated FLT3-ITDmut (without NPM1mut) irrespective of AR (P < 0.05)21.
Taken together, utilizing baseline FLT3-ITDmut AR to guide the post-remission therapy remains controversial. We currently recommend the incorporation of FLT3i’s and ASCT in CR1 in all ASCT eligible patients with a FLT3-ITDmut AML, irrespective of the AR and/or NPM1 co-mutation status. However, emerging data does suggest that patients with FLT3-ITDmut AR<0.5 and NPM1 co-mutation without concurrent high-risk mutations such as DNMT3A, TP53, TET2, or high-risk cytogenetics may be a more favorable subset, who may be considered for induction, consolidation followed by maintenance therapy without ASCT on a case by case basis if they achieve early MRD negativity using a highly sensitive MRD assay.
First-generation FLT3 inhibitors
Type I FLT3i’s are active against both the FLT3-ITD or TKD, type II inhibitors are only active against FLT3-ITD, not TKD. The first-generation FLT3i’s lack specificity for FLT3 and inhibit multiple downstream RTKs that may result in more off-target toxicities. Second-generation FLT3i’s potently and specifically target FLT3 with fewer off-target effects.
Midostaurin is a type I FLT3i active against PDGFR, KIT, SRC, and other RTKs22,23. In the randomized phase III RATIFY trial of midostaurin combined with cytarabine and daunorubicin (3 + 7) induction and consolidation, midostaurin improved OS compared to placebo in patients <60 years of age with newly diagnosed FLT3 (ITD and/or TKD) AML24, regardless of AR (≤0.7 or ≥0.7) or the type of mutation (ITD or TKD). Patients treated with midostaurin had higher rates of anemia and skin rash compared to placebo and these were generally manageable with supportive care without necessitating dose reductions or interruptions in the majority of cases. Pulmonary infiltration and acute pneumonitis-like picture are rare (<1%) but noted side effects of midostaurin that treating physicians should be aware of. Midostaurin has been approved and widely used in combination with induction and consolidation therapy in patients with newly diagnosed FLT3mut AML25.
Sorafenib is a first generation, type II multi-kinase FLT3i26 that demonstrated safety and efficacy (14/15 CR) in combination with the standard anthracycline/cytarabine induction therapy in newly diagnosed FLT3mut AML27. SORAML, a randomized placebo-controlled trial evaluated the efficacy and tolerability of 3 + 7 induction-consolidation with or without sorafenib in patients ≤60 years with newly diagnosed AML, irrespective of a FLT3mut (only 34% had FLT3mut). The addition of sorafenib significantly improved the event-free survival (EFS; 21 months vs 9 months; P = 0.013) and RFS (56% vs 38%), but not OS28, although a recent update suggested an emerging trend toward improved OS29. The sorafenib treatment arm had increased rates of adverse events, particularly diarrhea, bleeding, cardiac events, hand-foot-skin reaction, and rash but with no significant increase in the 30- or 60-day mortality between the two treatment arms. Intriguingly, this was the first large study to show that the FLT3i may also benefit FLT3 wild-type patients, perhaps through multi-kinase blockade or prevention of emergent FLT3 clones at relapse28. Recently, a double-blind placebo-controlled study reported a trend toward improved OS but not EFS with sorafenib combined with intensive chemotherapy in the frontline setting, especially among those with high FLT3-ITDmut AR >0.730. Sorafenib with azacitidine combination reported an overall response rate (ORR) of 78% (n = 27) in the frontline patients not eligible for intensive induction31 and an ORR of 46% with an acceptable safety profile in R/R FLT3-ITDmut 32 which led to the inclusion of sorafenib with azacitidine combination as a 2B guideline in National Comprehensive Cancer Network (NCCN) for R/R FLT3-ITDmut AML33.
Second-generation FLT3 inhibitors
Quizartinib, a second-generation, type I FLT3i is active against FLT3, KIT, CSF1R, PDGFR, and RET kinase34. Unlike midostaurin, quizartinib monotherapy, even at lower doses demonstrated significant marrow remissions in R/R FLT3mut AML35–37. In a single-arm phase II trial of quizartinib (90 or 135 mg), the CRc rates were between 46 and 56% in ~250 R/R FLT3-ITDmut patients treated across two cohorts. QTc prolongation >500 ms emerged as a significant adverse event36. A subsequent randomized phase IIb trial evaluated lower doses, 30 or 60 mg of quizartinib daily, in patients with R/R FLT3-ITDmut AML. The CRc rates (47%) were similar between both doses, and the frequency of QTcF >500 ms was significantly reduced (3–5%) with these lower doses of quizartinib35.
QuANTUM-R, a phase 3 randomized controlled trial, evaluated quizartinib monotherapy vs investigator choice salvage chemotherapy in R/R FLT3-ITDmut AML. Quizartinib demonstrated an OS of 6.2 months compared with 4.7 months with salvage chemotherapy (hazard ratio 0.76 and P = 0.02). The CRc rates with quizartinib were similar to prior studies (48.2%), and 32% patients on the quizartinib arm underwent ASCT compared with 11% with salvage chemotherapy. Given the magnitude of OS benefit and concerns over therapeutic equipoise and potential cardiac safety signals, quizartinib was not approved in the US and Europe, but approved in Japan as a monotherapy in R/R FLT3-ITDmut AML. In general, quizartinib is well tolerated with minimal skin, gastrointestinal, or pulmonary side effects. However, in addition to QTcF prolongation, quizartinib is also more myelosuppressive than many other FLT3 inhibitors likely due to the inhibition of KIT.
Gilteritinib, a second-generation type I FLT3i demonstrated tolerability with CRc rates of 45–55% in patients with R/R FLT3 (ITD or TKD)mut AML38,39. The randomized phase III ADMIRAL trial evaluated gilteritinib vs investigator choice salvage chemotherapy in patients with R/R FLT3mut AML. Gilteritinib decreased the risk of death by 36% compared with salvage chemotherapy, with a median OS of 9.3 months vs 5.6 months (P < 0.001), and a superior CR+CRh rate (34% vs 15.3%). Gilteritinib was generally well tolerated but was associated with increased incidence of gastrointestinal side effects, most frequently diarrhea although nausea has been occasionally observed. Increase in bilirubin and transaminase can be seen with giltertiinib but are usually self-resolving and transient. Posterior reversible encephalopathy and pancreatitis are rare (<1–2%) but important side effects to be aware of. These results led to the approval of gilteritinib monotherapy in the US and Europe in patients with R/R FLT3mut AML40.
Post-ASCT maintenance with FLT3 inhibitors
In patients with FLT3mut AML who relapsed after first ASCT, sorafenib was found to be tolerable with long-lasting remissions in 7 of 29 patients treated, suggesting a potential synergy with post-ASCT alloimmune effects41. SORMAIN, a placebo-controlled randomized phase II trial evaluated post-transplant sorafenib maintenance in patients with FLT3-ITDmut AML with RFS post-ASCT as the primary endpoint. At a median follow-up of 42 months, sorafenib demonstrated a 2-year estimated RFS of 85% and OS of 90.5% compared with 53.3% (P = 0.002), and 66.2% with placebo, respectively (P = 0.007). Although the toxicity-related discontinuation rate was low (22%), sorafenib-treated patients did experience higher rates of graft-versus-host disease (GVHD) and skin toxicity42. In another randomized phase III study comparing post-ASCT sorafenib maintenance (n = 100) to non-maintenance (n = 102), sorafenib demonstrated an improved 1-year OS (82.1% vs 68%, P = 0.012) and a decreased 1-year cumulative incidence of relapse (7% vs 24.5%, P = 0.001) in FLT3-ITDmut AML patients undergoing ASCT in CR143. We currently recommend post-transplant maintenance with a FLT3i for at least 2 years (potentially indefinitely as there is limited data on the incidence of possible late relapses) in all FLT3mut AML. The MORPHO phase III placebo-controlled trial evaluating post-transplant maintenance with gilteritinib in FLT3mut AML recently completed accrual and results are eagerly awaited (NCT02997202).
Moving forward to maximize benefit: FLT3 inhibitors combination therapy
Frontline FLT3i’s with anthracycline/cytarabine induction or hypomethylating agents (HMAs)
A phase I study evaluating gilteritinib with 7 + 3 induction and high-dose cytarabine consolidation chemotherapy, followed by single-agent maintenance therapy, in patients with newly diagnosed AML showed that gilteritinib 120 mg daily was well tolerated. Among 38 patients with FLT3mut AML who received gilteritinib 120 mg daily, the CRc rate was 81.6% (n = 31) including 39.5% CR and median OS was not reached at a median follow-up of 35.8 months. Two randomized trials are evaluating the addition of gilteritinib vs midostaurin to induction and consolidation therapy in patients with newly diagnosed FLT3mut AML44 (NCT04027309, NCT03836209).
The LACEWING phase III randomized trial evaluated gilteritinib with azacitidine vs azacitidine monotherapy (NCT02752035) in patients with newly diagnosed FLT3mut AML not eligible for intensive induction chemotherapy. The CRc rate was 67% (n = 10/15) in the combination arm in the safety cohort prior to commencement of randomization45. However, in a recently released planned interim analysis, the study did not meet its primary endpoint of overall survival and may be terminated for futility46.
Strati et al. evaluated midostaurin with azacitidine in patients with both newly diagnosed and R/R AML regardless of FLT3 mutational status. Among the FLT3mut patients, response rates were numerically higher (33%) and remission duration was longer (31 versus 16 weeks, P = 0.09) in those who were naive to treatment with FLT3 inhibitors compared with those who had been exposed to prior FLT3 inhibitors. Although activity was seen, the response rates were overall modest with this combination and the combination of HMA with midostaurin is not one that we routinely use or recommend for frontline FLT3-mutated AML47.
Swaminathan et al. evaluated quizartinib (60 mg daily) combined with either azacitidine or low-dose cytarabine in patients with newly diagnosed or R/R FLT3mut AML not eligible for intensive chemotherapy. Among patients treated with azacitidine and quizartinib in the frontline setting, the CRc rate was 78% (n = 7/9) with a median OS of 21.1 months. In the R/R setting, the CRc rate was 64% (n = 18/28) with a median OS of 12.0 months, with responses observed even in prior FLT3i exposed patients48. A randomized, placebo-controlled phase III study of 3 + 7 with quizartinib (QuANTUM-First; NCT02668653) in patients with newly diagnosed FLT3-ITDmut AML eligible for induction therapy recently completed accrual. Quizartinib is also being evaluated in combination with CPX-351 (NCT04209725) and with CLIA (NCT04047641) in treatment naive and R/R FLT3mut AML.
Combinations with venetoclax with or without HMA
Based on the strong preclinical synergy and synthetic lethality with venetoclax and FLT3i combination49–51, and the fact that BCL2 upregulation may confer resistance to FLT3 inhibition52, evaluation of several doublet and triplet combinations of venetoclax and FLT3i are ongoing. Gilteritinib with venetoclax (NCT03625505) was evaluated in 41 patients with heavily pretreated R/R FLT3mut AML (median salvage 2, 65% previously exposed to FLT3i)40,53. Using the same response criteria, the CRc rate was 85.4% (n = 35/41) which compared favorably to 52% with gilteritinib alone in the ADMIRAL study. However, the true CR/CRi rate was only 34%. Molecular clearance of FLT3 was noted in 50% of all evaluable patients. Encouragingly, the response rate was maintained among patients previously exposed to other FLT3 TKIs. The combination continues to enroll.
Maiti et al. recently presented the first triplet combination of venetoclax, FLT3i (mainly gilteritinib or sorafenib), and decitabine from the FLT3mut subset of the prospective decitabine 10 days with venetoclax study (NCT03404193)54. Among 16 patients with newly diagnosed FLT3mut AML not eligible for intensive induction, the CRc rate was 88% with FLT3-PCR negativity in 100% of responders and a projected 2-year OS of >80%. Among 14 R/R FLT3mut AML patients, the CRc rate was 64% with FLT3-PCR negativity in 88% of responders. In the treatment-naive setting, the median time to neutrophil and platelet recovery among responders was 45 and 30 days, respectively, suggesting cumulative myelosuppression is to be expected and further optimization of triplets schedules is ongoing55.
Yilmaz et al. prospectively evaluated decitabine and quizartinib (doublet) with or without venetoclax (triplet) in patients with newly diagnosed and R/R FLT3-ITDmut AML. While the seven patients treated with the doublet had a CRc rate of 57% (n = 4/7) and a median OS of 5.7 months, the fifteen R/R FLT3mut AML patients treated with the triplet had a CRc rate of 81% (n = 11) with a projected 1-year OS of 60%. In the frontline setting (n = 4), the CRc rate with the triplet was 100% with FLT3-PCR negativity in all four patients56.
These data highlight the potent anti-leukemic activity of the triplet approach in FLT3mut AML. We believe that triplets may be the optimal way to use FLT3i to improve long-term survival and “cure rates” in older patients, able to tolerate this approach. Further evaluation and optimization of triplets is a major area of clinical research focus in FLT3mut AML.
Treatment algorithm of FLT3-mutated AML
Clinical trial enrollment (if available) is always the first option, in both frontline and R/R FLT3mut AML. The choice of treatment backbone depends on the patient’s ability to successfully tolerate intensive chemotherapy. Accumulating evidence have shown improved outcomes in FLT3-ITDmut patients receiving induction with higher dose anthracyclines57, cladribine58, or fludarabine added to induction backbone21, and incorporating FLT3i with induction (either first or second generation) in FLT3mut AML24,44,59,60 (Fig. 1A). Our treatment approach for FLT3mut AML in MD Anderson Cancer Center is as follows: in newly diagnosed patients who are eligible to receive intensive chemotherapy (Fig. 1B) we add a second generation FLT3i to the intensive induction backbone of cladribine or fludarabine with cytarabine and idarubicin (CLIA or FIA, respectively) as published previously by our group61,62. Addition of venetoclax to this backbone may be associated with prolonged and potentially prohibitive myelosuppression; we have not routinely added and do not at this time recommend adding venetoclax to the backbone of CLIA/FIA with FLT3i63. We prefer a second-generation FLT3i (ideally gilteritinib) in the newly diagnosed setting, and administer the FLT3i D1-D14 during induction, and continuously starting Cycle 2 Day 1 through consolidation.
Fig. 1. Treatment algorithm of FLT3-ITD-mutated AML in patients eligible for intensive chemotherapy.

A Conventional approach. B MD Anderson Cancer Center Approach. 7+3—7 days of cytarabine and 3 days of daunorubicin. F fludarabine, I idarubicin, CL cladribine, A cytarabine 1.5–2 g/m2, HMA hypomethylating agent, CR complete remission, ECOG PS Eastern Cooperative Oncology Group Performance Status, CG cytogenetics, MRD measurable residual disease, SCT stem cell transplant, HiDAC high-dose cytarabine, CBC complete blood count.
Upon achieving CR, the decision for ASCT is based on the risk-benefit assessment for ASCT. All eligible intermediate or high-risk patients (defined as patients with FLT3-ITDmut AR>0.50 irrespective of NPM1mut status, or FLT3-ITDmut AR<0.50 without NPM1mut) are equivocally recommended to proceed to ASCT in CR1 followed by post-ASCT FLT3i maintenance for at least 2 years (although we often continue indefinite FLT3i maintenance until long-term maintenance data becomes available). For post-ASCT maintenance, our agent of choice has been gilteritinib 80–120 mg day either as a single agent or combined with low-dose azacitidine. The role of ASCT in patients with FLT3-ITDmut AR<0.50 with concomitant NPM1mut in the absence of concomitant high-risk features such as DNMT3A, TP53, or RUNX1 co-mutations, adverse cytogenetics, therapy-related or secondary AML, who achieve MRD negativity by high-sensitivity PCR (ideally for NPM1mut), or patients with FLT3-TKDmut is an area of ongoing debate. We evaluate these patients on a case by case basis and may consider maintenance with 4–5 consolidation cycles of CLIA or FAI with FLT3i followed by FLT3i +/− HMA maintenance for two years vs ASCT based on donor availability, age, performance status, MRD negativity, and patient preference.
In patients with FLT3mut AML unsuitable for intensive chemotherapy, azacitidine with venetoclax demonstrated encouraging CR/CRi rates (55–70%) and a median OS of 13.3 months64 which prompted the inclusion of this combination approach as part of NCCN AML guidelines (Fig. 2A). However, the median OS was 19.2 months in FLT3-TKDmut AML (19.2 months), but only 11.5 months in FLT3-ITDmut patients65. This is in line with the preclinical data49 and molecular profiling of pre- and post-treatment samples66 identifying FLT3-ITDmut as a putative mechanism of resistance to venetoclax based therapies67, suggesting that FLT3-ITDmut patients may need a FLT3i incorporated into the HMA with venetoclax therapy either in a triplet or sequential approach to improve OS. Therefore, in patients not eligible for intensive chemotherapy at MDACC, we prefer a combination of HMA with venetoclax and FLT3i (gilteritinib) over an HMA with venetoclax doublet (Fig. 2B). Administration of the triplet is associated with prolonged cytopenias, requiring close monitoring and experience with venetoclax based combinations.
Fig. 2. Treatment algorithm of FLT3-mutated AML in patients not eligible for intensive chemotherapy.

A Conventional approach. B MD Anderson Cancer Center Approach. FLT3i FLT3 inhibitor, HMA hypomethylating agent, VEN venetoclax, CR complete remission, ECOG PS Eastern Cooperative Oncology Group Perfromance Status, CG cytogenetics, MRD measurable residual disease, SCT stem cell transplant, CBC complete blood count.
We administer a second-generation FLT3i (ideally gilteritinib) continuously with HMA from cycle 1 Day 1. We introduce venetoclax with a ramp-up when the WBC is <10,000/µL to decrease the risk of tumor lysis syndrome. To mitigate prolonged myelosuppression with the triplet and avoid over-treatment, we perform an early bone marrow assessment on Cycle 1 Day 14 (Fig. 3). We stop the venetoclax and the FLT3i after Day 14 in patients who achieve marrow remission (<5% blasts) and/or marrow aplasia/hypoplasia/insufficiency (<5% cellularity). We continue the venetoclax and FLT3i until Day 21 if the Day 14 bone marrow shows >5% blasts with >/=5% cellularity. In patients with ongoing cytopenias (ANC</=0.5 and/or platelets </=50K) on Day 28, we repeat a bone marrow on Day 28 to confirm marrow remission and once confirmed recommend administering growth factors starting Day 28 to boost recovery. In subsequent cycles: FLT3i is continued for the entire duration of the cycle and the venetoclax duration is reduced to 14 days or lower to mitigate cumulative prolonged cytopenias. Although the triplet approaches are still in development, emerging data with the triplets as discussed previously, suggest rapid and high potency, deep molecular remissions, and encouraging survival. An alternate option would be to consider sequencing with alternate cycles of HMA with venetoclax and HMA with FLT3i. Such sequential approaches need to be formally evaluated in the context of prospective clinical trials.
Fig. 3. Initial response assessment with the triplet regimen.

*C1 D14: Perform bone marrow biopsy; if bone marrow shows <5% blasts and/or <5% cellularity/insufficient sample → Stop venetoclax and FLT3i on D14. **If the C1 D14 bone marrow show >5% blasts → continue venetoclax, FLT3i till D21. @Repeat a C1 D28 bone marrow on all patients to confirm remission. If C1 D28 marrow confirms remission and ANC<0.5 and/or platelet < 50K consider interrupting FLT3i and using neupogen to enhance count recovery.
In patients with relapsed or refractory FLT3mut AML (Fig. 4), diligent effort must be made to refer the patient to large academic centers with clinical trial options as the outcomes remain dismal with a median OS < 10 months with almost any approach. In the absence of clinical trial options: among patients eligible for intensive chemotherapy who had a prior remission >10–12 months, we would prefer a regimen incorporating intensive therapy (FLAG-Ida, CLAG-M, CLIA, MEC) in combination with a FLT3 inhibitor with an intent to achieve a rapid and hopefully deep remission and transition patients to ASCT followed by post-ASCT maintenance. In older patients not eligible for intensive therapy, patients with primary refractory disease or early relapse with a persistent FLT3 mutation we would suggest gilteritinib based therapy. Although the label indication for gilteritinib is as a single agent we have never used it as a single agent but always in combination with either HMA alone, venetoclax alone or as a triplet with HMA and venetoclax. These combinations appear to improve the efficacy over single agent gilteritinib and could be considered if there is expertise in using such an approach, For patients relapsing while on gilteritinib or soon after gilteritinib based therapy a combination of azacitidine with sorafenib or azacitinde with venetoclax or gemtuzumab based approaches may be considered as salvage options (with clinical trials being clearly the best option if available).
Fig. 4. Treatment algorithm of relapsed or refractory FLT3mut AML.
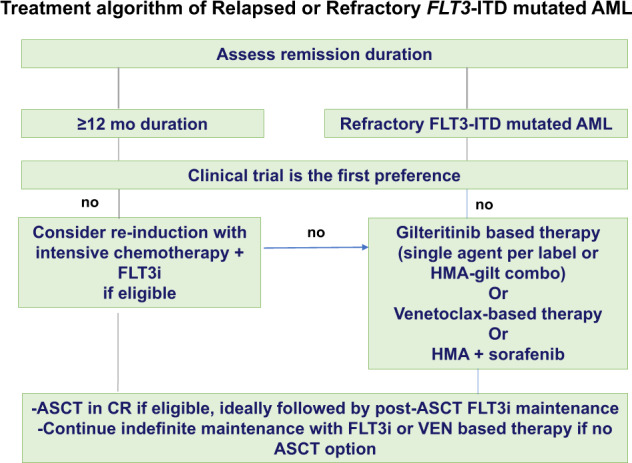
SCT stem cell transplant.
Mechanisms of resistance to FLT3 inhibitors
Despite the encouraging development of FLT3i, resistance to FLT3i is not uncommon and it can be either primary or secondary. The primary resistance mechanisms include specific FLT3-TKD mutations (either single TKD mutations or compound mutations within the FLT3-ITD allele), mutations in genes other than FLT3, activation of alternative signaling pathways in leukemic cells or the bone marrow microenvironment that confer resistance to FLT3i68.
Secondary resistance to FLT3i could be either on-target (changes in the FLT3) or off-target (constitutive activation of non-FLT3-dependent oncogenic pathways). The on-target mechanism of resistance includes emergence of secondary TKD mutations in patients treated with type II inhibitors like quizartinib or sorafenib69,70. Type I FLT3i’s like gilteritinib are less prone to develop secondary mutations in the TKD, although the gatekeeper F691M can confer resistance to gilteritinib71. Off-target resistance includes clonal evolution during FLT3i therapy even when FLT3-ITDmut clone is lost70. In a study that identified molecular mechanisms of resistance to gilteritinib, 32% of patients had emergent mutations in the RAS/MAPK pathway (K/NRAS), and 5% had emergent BCR/ABL1 fusions71. More recently, the emergence of BCR-ABL1-positive clone was shown as a resistance mechanism to multiple FLT3i’s72. It is important to acknowledge the diverse mechanisms of FLT3i resistance after different FLT3i’s, and it is essential to proactively evaluate for these mechanisms at the time of FLT3i failure to optimize subsequent therapy.
Future direction
In the QuANTUM-R and ADMIRAL trials, only 4% and 12% of patients had received prior FLT3i therapy with induction, making it difficult to draw conclusions regarding the outcomes of contemporary patients, most of whom will have received a FLT3i (commonly midostaurin) with induction36,40. Yilmaz et al. evaluated the outcomes of sequential FLT3i-based therapies in FLT3mut AML. In the frontline setting, there was a sequential decrease in CRc rates (77%→31%→25%) and OS (16.7→6.0→1.4 months). A comparable decrease in CRc rates (45%→21%→10%) and OS (7.9→4.0→4.1 months) was observed with sequential FLT3i-based therapies in the R/R AML setting73. Perl and colleagues investigated whether prior FLT3i therapy influenced outcomes in patients treated with gilteritinib. Regardless of prior FLT3i therapy, gilteritinib-treated patients had CRc rates >40%, however, the median OS with single-agent gilteritinib was 6.5 vs 9.6 months in prior FLT3i exposed (n = 31) vs naive patients (n = 216) with FLT3mut R/R AML74. These data suggests that although responses may still be achieved with gilteritinib in patients refractory to prior first-generation FLT3i-based therapies, optimization with doublet or triplet combinations with second-generation FLT3i is likely needed to significantly improve OS with prior TKI exposure.
Acknowledgments
Funding
This work was supported in part by the MD Anderson Cancer Centre Support Grant (CCSG) CA016672, the MD Anderson Cancer Center Leukemia SPORE CA100632, the Charif. Souki Cancer Research Fund and generous philanthropic contributions to the MD Anderson Moon Shots Program.
Conflict of interest
N.D. has received research funding from Daiichi-Sankyo, Bristol-Myers Squibb, Pfizer, Gilead, Sevier, Genentech, Astellas, Daiichi-Sankyo, Abbvie, Hanmi, Trovagene, FATE, Amgen, Novimmune, Glycomimetics, and ImmunoGen and has served in a consulting or advisory role for Daiichi-Sankyo, Bristol-Myers Squibb, Pfizer, Novartis, Celgene, AbbVie, Astellas, Genentech, Immunogen, Servier, Syndax, Trillium, Gilead, Amgen, and Agios. F.R. has received research funding from Astellas, and Novartis and has served as a member of advisory board in Astellas and Novartis. S.V. has nothing to disclose.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Naval Daver, Sangeetha Venugopal
References
- 1.Rosnet O, et al. Expression and signal transduction of the FLT3 tyrosine kinase receptor. Acta Haematol. 1996;95:218–223. doi: 10.1159/000203881. [DOI] [PubMed] [Google Scholar]
- 2.Timothy J, Ley C, Network CGAR. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Welch John S, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakao M, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10:1911–1918. [PubMed] [Google Scholar]
- 5.Yamamoto Y, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97:2434–2439. doi: 10.1182/blood.V97.8.2434. [DOI] [PubMed] [Google Scholar]
- 6.Abu‐Duhier F, et al. Identification of novel FLT‐3 Asp835 mutations in adult acute myeloid leukaemia. Br. J. Haematol. 2001;113:983–988. doi: 10.1046/j.1365-2141.2001.02850.x. [DOI] [PubMed] [Google Scholar]
- 7.Kiyoi H, et al. Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood. 1999;93:3074–3080. [PubMed] [Google Scholar]
- 8.Kottaridis PD, et al. Studies of FLT3 mutations in paired presentation and relapse samples from patients with acute myeloid leukemia: implications for the role of FLT3 mutations in leukemogenesis, minimal residual disease detection, and possible therapy with FLT3 inhibitors. Blood. 2002;100:2393–2398. doi: 10.1182/blood-2002-02-0420. [DOI] [PubMed] [Google Scholar]
- 9.Thiede C, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Presented in part at the 42nd Annual Meeting of the American Society of Hematology, December 1–5, 2000, San Francisco, CA (abstract 2334) Blood. 2002;99:4326–4335. doi: 10.1182/blood.V99.12.4326. [DOI] [PubMed] [Google Scholar]
- 10.Fröhling S, et al. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood. 2002;100:4372–4380. doi: 10.1182/blood-2002-05-1440. [DOI] [PubMed] [Google Scholar]
- 11.Kiyoi H, Ohno R, Ueda R, Saito H, Naoe T. Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene. 2002;21:2555–2563. doi: 10.1038/sj.onc.1205332. [DOI] [PubMed] [Google Scholar]
- 12.Mead AJ, et al. FLT3 tyrosine kinase domain mutations are biologically distinct from and have a significantly more favorable prognosis than FLT3 internal tandem duplications in patients with acute myeloid leukemia. Blood. 2007;110:1262–1270. doi: 10.1182/blood-2006-04-015826. [DOI] [PubMed] [Google Scholar]
- 13.Whitman SP, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a cancer and leukemia group B study. Cancer Res. 2001;61:7233–7239. [PubMed] [Google Scholar]
- 14.Yalniz F, et al. Prognostic significance of baseline FLT3-ITD mutant allele level in acute myeloid leukemia treated with intensive chemotherapy with/without sorafenib. Am. J. Hematol. 2019;94:984–991. doi: 10.1002/ajh.25553. [DOI] [PubMed] [Google Scholar]
- 15.Schlenk RF, et al. Differential impact of allelic ratio and insertion site in FLT3-ITD–positive AML with respect to allogeneic transplantation. Blood. 2014;124:3441–3449. doi: 10.1182/blood-2014-05-578070. [DOI] [PubMed] [Google Scholar]
- 16.Gale RE, et al. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood. 2008;111:2776–2784. doi: 10.1182/blood-2007-08-109090. [DOI] [PubMed] [Google Scholar]
- 17.Pratcorona M, et al. Favorable outcome of patients with acute myeloid leukemia harboring a low-allelic burden FLT3-ITD mutation and concomitant NPM1 mutation: relevance to post-remission therapy. Blood. 2013;121:2734–2738. doi: 10.1182/blood-2012-06-431122. [DOI] [PubMed] [Google Scholar]
- 18.Döhner H, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–447. doi: 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Linch DC, Hills RK, Burnett AK, Khwaja A, Gale RE. Impact of FLT3ITD mutant allele level on relapse risk in intermediate-risk acute myeloid leukemia. Blood. 2014;124:273–276. doi: 10.1182/blood-2014-02-554667. [DOI] [PubMed] [Google Scholar]
- 20.Oran B, et al. Allogeneic transplantation in first remission improves outcomes irrespective of FLT3-ITD allelic ratio in FLT3-ITD-positive acute myelogenous leukemia. Biol. Blood Marrow Transplant. 2016;22:1218–1226. doi: 10.1016/j.bbmt.2016.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minetto P, et al. Intensive fludarabine, high dose cytarabine and idarubicin-based induction for younger NPM1-mutated AML patient: overcoming the negative prognosis of FLT3-ITD mutation. Blood. 2020;136:32–33. doi: 10.1182/blood-2020-142887. [DOI] [Google Scholar]
- 22.Tamaoki T, et al. Staurosporine, a potent inhibitor of phospholipid Ca++ dependent protein kinase. Biochem. Biophys. Res. Commun. 1986;135:397–402. doi: 10.1016/0006-291X(86)90008-2. [DOI] [PubMed] [Google Scholar]
- 23.Weisberg E, et al. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell. 2002;1:433–443. doi: 10.1016/S1535-6108(02)00069-7. [DOI] [PubMed] [Google Scholar]
- 24.Stone RM, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N. Engl. J. Med. 2017;377:454–464. doi: 10.1056/NEJMoa1614359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rydapt Prescribing Information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/207997s000lbl.pdf2017 (2017).
- 26.Zhang W, et al. Mutant FLT3: a direct target of sorafenib in acute myelogenous leukemia. J. Natl Cancer Inst. 2008;100:184–198. doi: 10.1093/jnci/djm328. [DOI] [PubMed] [Google Scholar]
- 27.Ravandi F, et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J. Clin. Oncol. 2010;28:1856. doi: 10.1200/JCO.2009.25.4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Röllig C, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015;16:1691–1699. doi: 10.1016/S1470-2045(15)00362-9. [DOI] [PubMed] [Google Scholar]
- 29.Rollig C, et al. The addition of sorafenib to standard AML treatment results in a substantial reduction in relapse risk and improved survival. Updated results from long-term follow-up of the randomized-controlled SORAML trial. Blood. 2017;130:721. doi: 10.1182/blood.V130.Suppl_1.721.721. [DOI] [Google Scholar]
- 30.Andrew, H. et al. Results of a Phase 2, Randomized, Double-Blind Study of Sorafenib Versus Placebo in Combination with Intensive Chemotherapy in Previously Untreated Patients with FLT3-ITD Acute Myeloid Leukemia (ALLG AMLM16) (ASH, 2020).
- 31.Ohanian M, et al. Sorafenib combined with 5-azacytidine in older patients with untreated FLT3-ITD mutated acute myeloid leukemia. Am. J. Hematol. 2018;93:1136–1141. doi: 10.1002/ajh.25198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ravandi F, et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood. 2013;121:4655–4662. doi: 10.1182/blood-2013-01-480228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tallman MS, et al. Acute myeloid leukemia, Version 3.2019, NCCN clinical practice guidelines in oncology. J. Natl Compr. Canc. Netw. 2019;17:721–749. doi: 10.6004/jnccn.2019.0028. [DOI] [PubMed] [Google Scholar]
- 34.Zarrinkar PP, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML) Blood. 2009;114:2984–2992. doi: 10.1182/blood-2009-05-222034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cortes JE, et al. Phase 2b study of 2 dosing regimens of quizartinib monotherapy in FLT3-ITD–mutated, relapsed or refractory AML. Blood. 2018;132:598–607. doi: 10.1182/blood-2018-01-821629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cortes J, et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: an open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 2018;19:889–903. doi: 10.1016/S1470-2045(18)30240-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cortes, J. E. et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3—internal tandem duplication status. J. Clinl. Oncol.31, 3681 (2013). [DOI] [PMC free article] [PubMed]
- 38.Perl AE, et al. Final results of the chrysalis trial: a first-in-human phase 1/2 dose-escalation, dose-expansion study of gilteritinib (ASP2215) in patients with relapsed/refractory acute myeloid leukemia (R/R AML) Blood. 2016;128:1069. doi: 10.1182/blood.V128.22.1069.1069. [DOI] [Google Scholar]
- 39.Perl AE, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1-2 study. Lancet Oncol. 2017;18:1061–1075. doi: 10.1016/S1470-2045(17)30416-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perl AE, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N. Engl. J. Med. 2019;381:1728–1740. doi: 10.1056/NEJMoa1902688. [DOI] [PubMed] [Google Scholar]
- 41.Metzelder S, et al. High activity of sorafenib in FLT3-ITD-positive acute myeloid leukemia synergizes with allo-immune effects to induce sustained responses. Leukemia. 2012;26:2353–2359. doi: 10.1038/leu.2012.105. [DOI] [PubMed] [Google Scholar]
- 42.Burchert A, et al. Sorafenib maintenance after allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia with FLT3–internal tandem duplication mutation (SORMAIN) J. Clin. Oncol. 2020;38:2993–3002. doi: 10.1200/JCO.19.03345. [DOI] [PubMed] [Google Scholar]
- 43.Xuan L, et al. Sorafenib maintenance in patients with FLT3-ITD acute myeloid leukaemia undergoing allogeneic haematopoietic stem-cell transplantation: an open-label, multicentre, randomised phase 3 trial. Lancet Oncol. 2020;21:1201–1212. doi: 10.1016/S1470-2045(20)30455-1. [DOI] [PubMed] [Google Scholar]
- 44.Pratz KW, et al. A phase 1 study of gilteritinib in combination with induction and consolidation chemotherapy in patients with newly diagnosed AML: final results. Blood. 2020;136:16–17. doi: 10.1182/blood-2020-137685. [DOI] [Google Scholar]
- 45.Wang, E. S. et al. Phase 3, Multicenter, Open-label Study of Gilteritinib, Gilteritinib plus Azacitidine, or Azacitidine Alone in Newly Diagnosed FLT3-Mutated (FLT3mut+) Acute Myeloid Leukemia (AML) Patients Ineligible for Intensive Induction Chemotherapy (ASH, 2020).
- 46.Astellas Reports XOSPATA® (gilteritinib) in combination with azacitidine did not meet endpoint of overall survival in newly diagnosed flt3 mutation-positive acute myeloid leukemia patients ineligible for intensive induction chemotherapy (2020).
- 47.Strati P, et al. Phase I/II trial of the combination of midostaurin (PKC412) and 5-azacytidine for patients with acute myeloid leukemia and myelodysplastic syndrome. Am. J. Hematol. 2015;90:276–281. doi: 10.1002/ajh.23924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Swaminathan M, et al. The combination of quizartinib with azacitidine or low dose cytarabine is highly active in patients (Pts) with FLT3-ITD mutated myeloid leukemias: interim report of a phase I/II trial. Blood. 2017;130:723. doi: 10.1182/blood.V130.Suppl_1.723.723. [DOI] [Google Scholar]
- 49.Mali, R. S. et al. Venetoclax combines synergistically with FLT3 inhibition to effectively target leukemic cells in FLT3-ITD+ acute myeloid leukemia models. Haematologica106, 1034 (2020). [DOI] [PMC free article] [PubMed]
- 50.Brinton LT, et al. Synergistic effect of BCL2 and FLT3 co-inhibition in acute myeloid leukemia. J. Hematol. Oncol. 2020;13:139. doi: 10.1186/s13045-020-00973-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu R, Li L, Nguyen B, Duffield AS, Small D. Gilteritinib and venetoclax synergize to eliminate FLT3/ITD+ leukemia cells through BIM. Blood. 2019;134:2564. doi: 10.1182/blood-2019-131635. [DOI] [Google Scholar]
- 52.Yamatani K, et al. Upregulation of Bcl-2 confers resistance to FLT3 inhibition in FLT3-ITD AML with secondary acquired mutations. Blood. 2018;132:3944. doi: 10.1182/blood-2018-99-109957. [DOI] [Google Scholar]
- 53.Naval Daver, M. D. et al. Efficacy and Safety of Venetoclax in Combination with Gilteritinib for Relapsed/refractory FLT3-mutated Acute Myeloid Leukemia in the Expansion Cohort of a Phase 1b Study (ASH, 2020).
- 54.DiNardo CD, et al. 10-day decitabine with venetoclax for newly diagnosed intensive chemotherapy ineligible, and relapsed or refractory acute myeloid leukaemia: a single-centre, phase 2 trial. Lancet Haematol. 2020;7:e724–e736. doi: 10.1016/S2352-3026(20)30210-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abhishek Maiti, M. D. et al. Venetoclax, FLT3 Inhibitor and Decitabine in FLT3mut Acute Myeloid Leukemia: Subgroup Analysis of a Phase II Trial (ASH, 2020).
- 56.Musa Yilmaz, M. et al. Quizartinib with Decitabine +/− Venetoclax is Highly Active in Patients (Pts) with FLT3-ITD Mutated (mut) Acute Myeloid Leukemia (AML): Clinical Report and Signaling Cytof Profiling from a Phase IB/II Trial (ASH, 2020).
- 57.Burnett AK, Russell NH, Hills RK. Group obotUKNCRIAMLS. Higher daunorubicin exposure benefits FLT3 mutated acute myeloid leukemia. Blood. 2016;128:449–452. doi: 10.1182/blood-2016-04-712091. [DOI] [PubMed] [Google Scholar]
- 58.Libura M, et al. Cladribine added to daunorubicin-cytarabine induction prolongs survival of FLT3-ITD+ normal karyotype AML patients. Blood. 2016;127:360–362. doi: 10.1182/blood-2015-08-662130. [DOI] [PubMed] [Google Scholar]
- 59.Altman JK, et al. Phase 1 study of quizartinib in combination with induction and consolidation chemotherapy in patients with newly diagnosed acute myeloid leukemia. Am. J. Hematol. 2018;93:213–221. doi: 10.1002/ajh.24974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang ES, et al. Low relapse rate in younger patients ≤60 years old with newly diagnosed FLT3-mutated acute myeloid leukemia (AML) treated with crenolanib and cytarabine/anthracycline chemotherapy. Blood. 2017;130:566. [Google Scholar]
- 61.Jain P, et al. Cladribine combined with idarubicin and Ara-C (CLIA) as a frontline and salvage treatment for young patients (≤65 yrs) with acute myeloid leukemia. Blood. 2016;128:1639. doi: 10.1182/blood.V128.22.1639.1639. [DOI] [Google Scholar]
- 62.Sasaki K, et al. Sorafenib plus intensive chemotherapy improves survival in patients with newly diagnosed, FLT3-internal tandem duplication mutation-positive acute myeloid leukemia. Cancer. 2019;125:3755–3766. doi: 10.1002/cncr.32387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kadia, T. et al. Venetoclax Added to Cladribine, Idarubicin, and Cytarabine with or without a FLT3 Inhibitor in Newly Diagnosed Acute Myeloid Leukemia (EHA, 2020).
- 64.DiNardo CD, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N. Engl. J. Med. 2020;383:617–629. doi: 10.1056/NEJMoa2012971. [DOI] [PubMed] [Google Scholar]
- 65.Konopleva M, et al. Results of venetoclax and azacitidine combination in chemotherapy ineligible untreated patients with acute myeloid leukemia with FLT3 mutations. Blood. 2020;136:8–10. doi: 10.1182/blood-2020-134100. [DOI] [Google Scholar]
- 66.Chyla B, et al. Genetic biomarkers of sensitivity and resistance to venetoclax monotherapy in patients with relapsed acute myeloid leukemia. Am. J. Hematol. 2018;93:E202–E205. doi: 10.1002/ajh.25146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DiNardo CD, et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood. 2020;135:791–803. doi: 10.1182/blood.2019003988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Daver N, et al. Secondary mutations as mediators of resistance to targeted therapy in leukemia. Blood. 2015;125:3236–3245. doi: 10.1182/blood-2014-10-605808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smith CC, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485:260–263. doi: 10.1038/nature11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Alotaibi AS, et al. Patterns of resistance differ in patients with acute myeloid leukemia treated with type I versus type II FLT3-inhibitors. Blood Cancer Discov. 2020;2:125. doi: 10.1158/2643-3230.BCD-20-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McMahon CM, et al. Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov. 2019;9:1050–1063. doi: 10.1158/2159-8290.CD-18-1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Alotaibi AS, et al. Emergence of BCR-ABL1 fusion in AML post-FLT3 inhibitor-based therapy: a potentially targetable mechanism of resistance—a case series. Front. Oncol. 2020;10:588876-. doi: 10.3389/fonc.2020.588876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yilmaz M, et al. Outcomes with sequential FLT3-inhibitor-based therapies in patients with AML. J. Hematol. Oncol. 2020;13:132. doi: 10.1186/s13045-020-00964-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Perl AE, et al. Clinical outcomes in patients with relapsed/refractory acute myeloid leukemia treated with gilteritinib who received prior midostaurin or sorafenib. Blood. 2020;136:22–23. doi: 10.1182/blood-2020-136395. [DOI] [PMC free article] [PubMed] [Google Scholar]