

Abstract
We report a nanoparticle formulation of the SHH-pathway inhibitor vismodegib that improves efficacy for medulloblastoma, while reducing toxicity. Limited blood–brain barrier (BBB) penetration and dose-limiting extraneural toxicities complicate systemic therapies for brain tumors. Vismodegib is FDA-approved for SHH-driven basal cell carcinoma, but implementation for medulloblastoma has been limited by inadequate efficacy and excessive bone toxicity. To address these issues through optimized drug delivery, we formulated vismodegib in polyoxazoline block copolymer micelles (POx-vismo). We then evaluated POx-vismo in transgenic mice that develop SHH-driven medulloblastomas with native vasculature and tumor microenvironment. POx-vismo improved CNS pharmacokinetics and reduced bone toxicity. Mechanistically, the nanoparticle carrier did not enter the CNS, and acted within the vascular compartment to improve drug delivery. Unlike conventional vismodegib, POx-vismo extended survival in medulloblastoma-bearing mice. Our results show the broad potential for non-targeted nanoparticle formulation to improve systemic brain tumor therapy, and specifically to improve vismodegib therapy for SHH-driven cancers.
Keywords: Medulloblastoma, Vismodegib, Sonic-hedgehog pathway, Polymeric micelles, Nanomedicines
Graphical Abstract
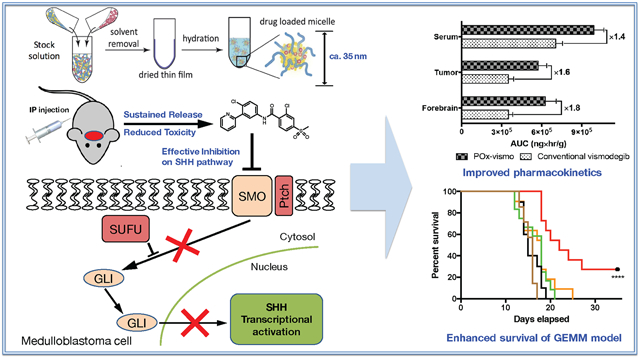
New, targeted approaches are needed for medulloblastoma. Current, non-targeted treatment extends survival for most medulloblastoma patients, but causes long-term neuro-cognitive injury. About one-third of medulloblastomas show SHH pathway hyper-activation, and for these patients, targeted inhibition of SHH signaling may improve therapy.1 Vismodegib, a small molecule inhibitor of SHH receptor component Smoothened (SMO), is FDA-approved for the treatment of basal cell carcinoma and is in clinical trials for other SHH-driven cancers.2 However, medulloblastoma treatment with vismodegib is limited by developmental bone toxicity and produces initial responses that are consistently followed by recurrence.3,4
The physicochemical properties of vismodegib present challenges. With extremely low aqueous solubility (0.1 μg/ml in at pH 7.0) and oral bioavailability of 31.8%, the drug is formulated for oral dosage.5 However, oral administration is complicated in the pediatric population most at risk for medulloblastoma.6 Furthermore, vismodegib has high affinity to serum proteins, including acid-glycoprotein and albumin, resulting in >99% of the drug circulating in protein bound form.5 This protein-binding affects the central nervous system (CNS) delivery as protein-bound vismodegib is unable to penetrate the BBB. A prior study of vismodegib pharmacokinetics in children found that drug levels in the CSF were 0.26% of total plasma vismodegib, but 26-78% of the concentration of unbound vismodegib.7 These data highlight the potential for optimized delivery to improve vismodegib therapy for medulloblastoma.
Several nanoparticle-based therapeutic products have been approved for clinical use in cancers outside the CNS, and more are currently under development.8,9 Nano-formulation can enhance the solubility of hydrophobic agents, extend their systemic circulation, provide sustained drug release, enhance drug accumulation in target tissues and reduce off-site effects.10,11
Polymeric micelles are formed by self-assembly of diblock (A-B) or triblock (A-B-A) amphiphilic block polymers. Hydrophobic segments of block copolymers play an essential role in solubilizing and encapsulating poorly soluble drugs in the core of the micelles, while hydrophilic segment forms protective corona to external environment. Commonly employed hydrophobic blocks are polyethers12 (polypropylene oxide and polybutylene oxide), polyesters13 (poly(lactic acid), poly(lactic-co-glycolic acid) and polycaprolactone) and hydrophobized poly(amino acid)s14 (poly(β-benzyl-l-aspartate and poly(γ-benzyl l-glutamate) while the hydrophilic segment is usually polyethylene glycol.15 Polymeric micelles were shown to solubilize hydrophobic drugs and have been employed as delivery carriers in clinical development.16 Triblock A-B-A copolymer of POx consists of hydrophobic poly(2-n-butyl-2-oxazoline) (PBuOx) segment with flanking hydrophilic poly(2-methyl-2-oxazoline) (PMeOx) segments, PMeOx-b-PBuOx-b-PMeOx, formed micelles with hydrophobic but highly polar core.17 Also, POx micelles have been shown to have an increased biocompatibility and decreased immunoactivation in vivo.18 Furthermore, the unique environment of POx micelles core results in unprecedentedly high loading for many hydrophobic polar compounds further reducing the excipient associated risks. It should be noted that excipient-derived toxicities have often occurred in clinical development, which are a dose limiting factor and hinder further development for human use.19 Thus, POx micelle holds an importance for improving drug delivery capacities while sparing drug-derived toxicities by virtue of polymeric micelle formulation.
We have previously shown that POx micelles can solubilize the investigational ATR inhibitor VE-821, allowing delivery into the CNS in vivo.20 To test the ability of POx delivery to optimize an FDA-approved agent with known safety profile but sub-optimal performance in prior brain tumor trials, we encapsulated vismodegib in POx micelles to generate POx-vismo.
Rodent xenograft models are frequently curable and have the potential to lead to overestimation of efficacy in preclinical studies. We tested POx-vismo in mice genetically engineered to develop endogenous medulloblastoma by breeding SmoM2 mice, which carry a Cre-conditional, oncogenic allele of Smo with Gfap-Cre mice that express Cre recombinase in CNS stem cells early in development, generating Gfap-Cre/SmoM2 (G-Smo) mouse pups. 100% of G-Smo mice develop medulloblastoma by P10, and, if untreated, die of tumor progression by P20.21 This transgenic model, which is highly aggressive and incurable with radiation therapy,22 forms tumors with native vasculature and intact blood brain barrier.23 We treated medulloblastoma-bearing G-Smo mice with either POx-vismo or conventional vismodegib either as oral suspension or parenteral formulation, and compared pharmacokinetics, toxicity, pharmacodynamics, and efficacy.
Methods
Preparation of POx-vismo micelles
The amphiphilic triblock copolymer (P(MeOx39-b-PBuOx25-b-PMeOx39)), Mn = 8.2 kg/mol, PDI = 1.11)) was synthesized and characterized as previously described.17 Vismodegib-loaded polymeric micelle formulation (POx-vismo) was prepared by the thin film hydration method.24 Briefly, stock solutions of the polymer and vismodegib (10 mg/ml in acetone) were mixed together at the pre-determined ratios (2:10-8:10 drug to polymer w/w ratios). The organic solvent was evaporated at 60 °C under a stream of nitrogen gas to form a thin-film of drug-polymer homogenous mixture. To obtain well-dried thin film, the films were dried in the vacuum chamber (approx. 0.2 mbar) overnight. Next, the thin films were rehydrated with saline and then incubated at 60 °C for 10 min to self-assembly into drug-loaded polymeric micelles solution. The formed POx-vismo micelles were centrifuged at 10,000 rpm for 3 min (Sorvall Legend Micro 21R Centrifuge, Thermo Scientific) to remove non-loaded vismodegib.
Characterization of POx-vismo micelles
The Zave and the polydispersity index (PDI) of POx-vismo were determined using a Nano-ZS (Malvern Instruments Inc., UK) dynamic light scattering (DLS). Each sample was diluted with saline to yield 1 mg/mL final polymer concentration. Zave and the polydispersity index (PDI) of POx-vismo were determined by cumulate analysis. Results are the average of three independent micelle samples measurements.
The morphology of POx-vismo micelles was determined using a LEO EM910 TEM operating at 80 kV (Carl Zeiss SMT Inc., Peabody, MA). The micelles were stained with negative staining (1% uranyl acetate) before imaging. Digital images were obtained using a Gatan Orius SC1000 CCD Digital Camera in combination with Digital Micrograph 3.11.0 software (Gatan Inc., Pleasanton, CA).
The final concentration of vismodegib in POx-vismo micelles was determined by HPLC (Agilent Technologies 1200 series) using Agilent eclipse plus C18 3.5 μm column (4.6 mm × 150 mm) with a mixture of acetonitrile/water (30%/70% v/v, 0.01% trifluoroacetic acid mobile phase. The samples were diluted with mobile phase to a final concentration of 100 μg/mL of vismodegib (and injected (10 μL) into the HPLC system. The flow rate was 1.0 mL/min, and column temperature was 40 °C. Detection wavelength was 245 nm. Vismodegib concentration was quantified against free vismodegib analytical standards.
Loading efficiency (LE) and loading capacity (LC) calculations. The following equations were used to calculate LE and LC of vismodegib in POx-vismo micelles:
| (1) |
| (2) |
Where Mdrug and Mexcipient are the mass of the solubilized drug and polymer excipient in the solution, while Mdrug added is the weight amount of the drug added to the dispersion during the preparation of the micelle formulation.
Additional methods
Materials, micelle stability, mouse breeding, analytical approaches (nuclear magnetic resonance (NMR) spectroscopy, liquid chromatography–mass spectrometry (LC–MS), infrared matrix assisted laser desorption electrospray ionization (IR-MALDESI), albumin binding analysis), in vivo toxicity, pharmacodynamics, tumor pathology and in vivo efficacy are described in the Supplementary Materials.
Results
Self-forming POx micelles encapsulate vismodegib in nanoparticles
We prepared POx-vismo polymeric micelles using POx polymer P(MeOx39-b-BuOx25-b-MeOx39) (Mn = 8.2 kg/mol, Ð (Mw/Mn) = 1.11). We generated micelles with vismodegib:POx polymer ratios of 2:10, 4:10, 6:10 and 8:10 w/w (drug to polymer) using the thin film method as previously described.17 At all tested ratios, the loading efficiency (LE, %) of vismodegib was nearly 90% and the loading capacity was 13.5-42.4% w/w depending on the drug:polymer ratio (Table 1). POx-vismo micelles had an intensity-mean z-averaged particle size (Zave) of 25-40 nm, narrow size distribution (PDI < 0.1) as determined by DLS (Figure 1, A). The POx-vismo micelles had a slightly positive surface charge of 3.7 mV (Figure 1, B); this low magnitude approximates neutral charge.25 The particle size and spherical morphology of POx-vismo micelles were further confirmed by transmission electron microscopy (Figure 1, C). The POx-vismo micelles were stable during storage at 4 °C as confirmed by HPLC and size analysis (DLS) (Figure 1, D). The POx-vismo micelles were also stable after lyophilization; lyophilized POx-vismo micelles could be redispersed in water, keeping their drug loading and particles size unchanged (Figure 1, E). Vismodegib was continuously released from the micelles in vitro under the perfect sink conditions with approximately 25% of the drug released in 2 h and 90% released in 12 h (Figure 1, F).
Table 1.
Actual vismodegib concentration, LE (%), LC (%), nanoparticles size and size distribution of POx-vismo micelles prepared at indicated drug:polymer ratios. (n = 3 ± SD).
| Ratio of drug:polymer by weight |
Theoretical drug concentration (mg/ml) |
Actual drug concentration (mg/ml) |
LE (%)a | LC (%)b | z-ave diameter (nm) |
PDI | z-potential (mV) |
|---|---|---|---|---|---|---|---|
| 2:10 | 2.00 | 1.81 ± 0.27 | 90.39 ± 13.50 | 15.31 ± 2.29 | 26.6 ± 2.4 | 0.07 ± 0.01 | 0.72 ± 3.14 |
| 4:10 | 4.00 | 3.64 ± 0.33 | 91.09 ± 8.16 | 26.71 ± 2.39 | 34.5 ± 5.3 | 0.03 ± 0.01 | 3.34 ± 0.32 |
| 6:10 | 6.00 | 5.90 ± 0.33 | 98.36 ± 5.46 | 37.11 ± 2.06 | 41.5 ± 5.9 | 0.03 ± 0.01 | 3.81 ± 0.44 |
| 8:10 | 8.00 | 7.45 ± 0.67 | 93.11 ± 8.36 | 42.69 ± 3.83 | 38.3 ± 0.3 | 0.08 ± 0.01 | 3.77 ± 0.52 |
LE (%) = Mdrug/ (Mdrug added) × 100%.
LC (%) = Mdrug / (Mdrug + Mexcipient) × 100%.
Figure 1.

POx-vismo micelles form stable, nanometer-scale spheres. (A) Particle size distribution measured by DLS (z-average, Dz). (B) Zeta potential and (C) morphology, shown by TEM. Scale bar = 200 nm (left), 50 nm (right). (D) Stability of the POx-vismo micelles at 4 °C as determined by actual drug measurements (left) and size distribution (right) overtime. (E) Size of particles after reconstitution of lyophilized POx-vismo. (F) Vismodegib release from POx-vismo incubated in 10% fetal bovine serum (FBS) solution at 37 °C over time.
POx micelles improve the brain and tumor delivery of vismodegib without penetrating CNS
We evaluated the effect of nanoparticle delivery on the biodistribution of vismodegib in medulloblastoma-bearing G-Smo mice. We administered vismodegib to G-Smo mice (Figure S1), formulated as POx-vismo injected IP or conventional vismodegib administered by either IP injection or oral gavage. We then euthanized mice after 4 h and harvested blood, tumor and forebrain samples. We prepared extracts of these samples and analyzed them using NMR spectroscopy to measure vismodegib and POx concentrations simultaneously. We identified signature peaks for vismodegib and POx that could be measured in serum and brain samples and verified that in serum samples, vismodegib and POx peaks increased in proportion to dose (Figure 2, A insets).
Figure 2.

Differential distribution of vismodegib and POx components of POx-vismo in the vascular and CNS compartments. (A) NMR spectra with the signature peaks for vismodegib and POx highlighted. Insets show dose-dependent changes in regions that correspond to vismodegib or POx in representative spectra from serum samples. Note that POx peak is a broad peak that spans several, more narrow peaks for unrelated molecules. To maximize specificity, we integrated an area within the broad POx peak that falls between unrelated peaks. The area under this portion of the curve was proportional to POx concentration over the range of tested standards. (B) Vismodegib concentrations in serum, tumor and forebrain after administration of the indicated formulation and dose. (C) POx concentration in serum, tumor and forebrain following IP injection of the indicated doses of POx-vismo. *P < 0.05, **P < 0.005, ***P < 0.0005. For B and C, dots indicate data from individual replicates and error bars are the SEM.
We compared vismodegib in blood, tumor and forebrain samples from mice treated with each formulation (Figure 2, B). Escalating POx-vismo doses of 50 mg/kg, 100 mg/kg and 150 mg/kg produced a linear increase in vismodegib concentration in blood, tumor and forebrain, and IP conventional vismodegib showed a similar pattern. Systemic administration of conventional vismodegib resulted to comparable levels to POx-vismo in serum at the same dose of 100 mg/kg. Oral vismodegib produced significantly lower vismodegib concentrations in blood, tumor and forebrain compared to POx-vismo and showed a similar trend toward lower concentrations compared to systemic vismodegib that was statistically significant in the forebrain compartment. These data show that oral administration produced similar or inferior concentrations compared to parenteral administration.
NMR spectroscopy showed that the POx polymer component of POx-vismo, unlike the vismodegib component, did not penetrate the BBB. While escalating doses of POx-vismo produced increasing concentrations of POx in the blood, POx concentrations in the tumor and forebrain remained below the threshold of detection at all tested dose (Figure 2, C). Based on these data, we conclude that the nanoparticle carrier dissociates from the vismodegib payload prior to the entry of vismodegib into the CNS.
To determine the effect of POx-vismo delivery on pharmacokinetics, we compared vismodegib concentrations in the blood, medulloblastoma/cerebellum and forebrain of tumor-bearing G-Smo mice at different intervals after parenteral administration of either POx-vismo or conventional vismodegib. We injected G-Smo mice on P10 with a single IP injection of either POx-vismo or conventional vismodegib, both dosed at 100 mg/kg vismodegib. We then harvested serum, tumor and brain tissues at 5 min, 2 h, 4 h, 8 h or 24 h after administration and measured vismodegib concentrations by LC–MS. In sagittal brain sections from replicate mice harvested 4 or 24 h after administration, we used IR-MALDESI imaging to determine the spatial distribution of vismodegib across tissue slices containing both medulloblastoma and brain.
POx-vismo induced significantly higher vismodegib concentrations in serum, medulloblastoma and forebrain at 2, 4, and 8 h, compared to conventional vismodegib (Figure 3, A-C). At 24 h, vismodegib was below the level of detection in both tumor and brain, likely due to extraction limiting the sensitivity of the assay at low tissue concentrations. Consistent with higher peaks and more sustained concentrations, the total tumor vismodegib exposure, measured as AUCinfinite was significantly higher in tumor, adjacent brain and serum following administration of POx-vismo compared to conventional vismodegib (Figure 3, D). The AUCtumor/AUCplasma ratio was much higher in the POx Vismo formulation when compared to the control formulation (0.569 compared 0.439), which indicates increased tumor exposure in the POx formulation relative to systemic exposure. The POx formulation thus improved distribution across the blood brain barrier (Figure 3, E, F), resulting in more of the drug in circulation distributing to the tumor for longer periods of time.
Figure 3.
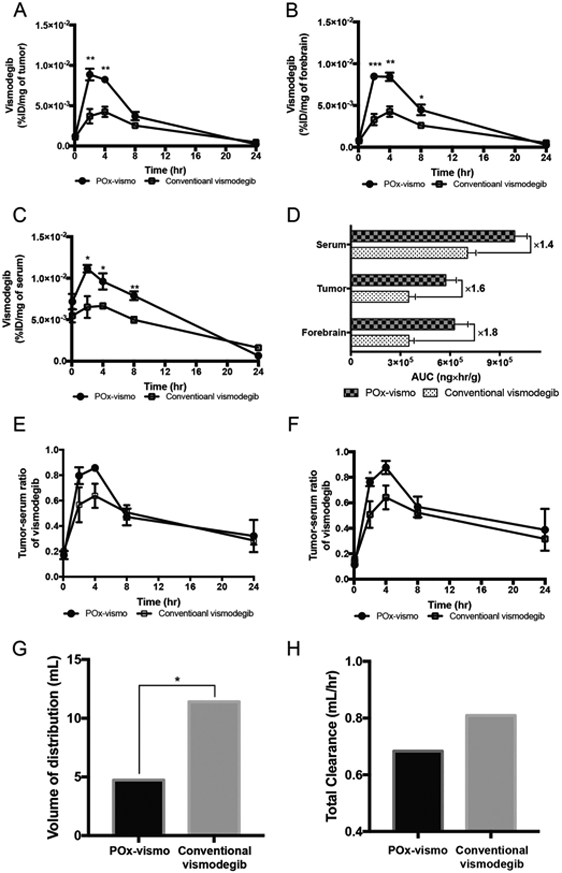
Pharmacokinetic profile of POx-vismo and conventional vismodegib in medulloblastoma-bearing G-Smo mice. Dot plots of vismodegib concentration in (A) tumor, (B) forebrain and (C) serum. (D) AUC comparison of serum, tumor, and forebrain, (E) tumor-serum ratio of vismodegib, (F) forebrain-serum ratio of vismodegib, (G) volume of distribution and (H) total clearance of vismodegib following administration of 100 mg/kg of POx-vismo or conventional vismodegib in tumor bearing mice. *P < 0.05, **P < 0.005, ***P < 0.0005. For A-C, data are expressed as dot plots with column (means ± SEM); the number of each group is expressed as the number of dots (n ≥ 3).
We examined how 1 compartment and 2 compartment models fit the data, and with both formulations a 1 compartment model was the best fit as determined by Akaike information criterion and residual plot analysis. The subsequent modeling showed that along with increased tumor drug exposure, the nanoparticle formulation decreased the vismodegib volume of distribution (Vd) (Figure 3, G) and clearance (CL) (Figure 3, H). The reduced volume of distribution indicates a lower distribution to peripheral tissues which could perhaps mitigate some systemic toxicity. Analysis of the vismodegib distribution in non-tumor mice showed similar results (Figure S2), indicating that the improved CNS penetration of vismodegib administered as POx-vismo, and reduced systemic distribution, were not due to tumor-specific changes in the BBB.
We analyzed the spatial distribution of vismodegib across sections of tumor and brain using IR-MALDESI.26 This technique uses infrared laser scanning across frozen tissue sections to excite the overlying matrix of ice, causing desorption of endogenous and exogenous ions that can be detected mass spectrometry. IR-MALDESI quantifies the concentrations of species at all detected molecular weights with spatial resolution, generating ion-specific heatmaps. We validated this technique by imaging endogenous cholesterol, which we found to be at relatively high concentration in normal brain compared to tumor (Figure 4, A, B). MALDESI detected vismodegib evenly distributed in tumor and forebrain 4 h after IP injection of either POx-vismo or conventional vismodegib (P = 0.630; Figure 4, B, C). Importantly, IR-MALDESI, in contrast to LC–MS, was able to measure vismodegib concentrations at 24 h; we attribute this increased sensitivity to the lack of an extraction step which increased sensitivity at low tissue concentrations. Prior studies using the antiretroviral EFV in brain tissue show that our IR-MALDESI system can detect drug concentrations as low as 123 fg/voxel.27
Figure 4.

Widespread vismodegib distribution in the brain, with increased retention after POx-vismo administration, demonstrated by IR-MALDESI. (A) Left panel, contour of brain sample showing forebrain and medulloblastoma (MB) and right panel, IR-MALDESI analysis of cholesterol in a control brain (m/z:369). (B) IR-MALDESI analysis of vismodegib (m/z: 421) in representative sagittal brain sections from G-Smo mice following a single dose or conventional vismodegib at 4 h (left) and 24 h (right). (C) IR-MALDESI MSI analysis of vismodegib (m/z: 421) as in B following a single dose of POx-vismo at 4 h (left) and 24 h (right).
IR-MALDESI demonstrated higher vismodegib signal at both 4 h and 24 h post injection in POx-vismo-treated mice (Figure 4, B, C). Using IR-MALDESI, we measured the distribution of vismodegib between the vascular compartment, detected by co-localization with heme, and the brain parenchyma, where heme was not detected (Figure S3). At all time points, most of the drug in each section localized to the brain parenchyma, but at 4 h, a greater fraction of drug localized to the vascular compartment in mice treated conventional vismodegib, while localization was similar at 24 h. These data show that the intravascular vismodegib was a minimal contribution to tissue extracts and suggest that at earlier time points POx-vismo penetrates better into brain tissue.
Nanoparticle carrier reduces vismodegib binding to serum albumin and improves bioavailability
The exclusion of the nanoparticle carrier from the CNS suggested that the mechanism of nanoparticle effect may occur in the blood. As the penetration of vismodegib into the brain is known to be affected by its binding to serum protein, we determined if POx formulation changes the serum protein binding of vismodegib. We compared albumin binding of vismodegib in POx-vismo and conventional formulations by measuring the quenching of the fluorescence of albumin tryptophan.28 The fluorescence quenching was lower for samples incubated with POx-vismo versus conventional vismodegib, indicating that POx formulation reduced albumin binding (Figure 5, A, B). Conversely, the intensity ratio at 340 nm was consistently higher for POx-vismo at each step in the increase of drug (Figure 5, C). Next, we evaluated protein binding over time. After 30 min, over 30% of total vismodegib remained unbound, decreasing to 10% over 4 h. In contrast, the unbound fraction of conventional vismodegib was marginal at 30 min. This is consistent with the improved tumor:serum and forebrain: serum ratio measured at early time points (up to 4 h) following administration of either POx-vismo or conventional vismodegib (Figure 3, E, F).
Figure 5.

Reduced vismodegib:protein binding with POx-vismo. (A) Tryptophan fluorescence quenching assay by addition of either POx-vismo or conventional vismodegib at given concentration of vismodegib (right); (B) the fraction of unbound vismodegib by addition of either Pox-vismo or conventional vismodegib in diluted FBS solution. (C) Unbound vismodegib after the incubation in FBS; (D) unbound versus protein bound vismodegib in serum 4 h post administration of POx-vismo or conventional vismo. *P < 0.05, **P < 0.005, ***P < 0.0005. Each point is mean ± SEM.
To assess the fraction of protein bound versus micelle-encapsulated drug in serum, we processed the samples using solid phase extraction (SPE) columns. This method was previously shown to separate albumin-bound and micelle-encapsulated paclitaxel.18 We found a 2-fold increase in unbound drug in POx-vismo treated mice, with 20% of vismodegib unbound and 80% of the drug albumin-bound compared to 9% unbound and 90% albumin-bound in mice treated with conventional vismodegib (Figure 5, D). NMR analysis on eluted fractions showed POx peaks in the fraction corresponding to micellar POx-vismo, confirming the co-loading of the polymer and the drug (Figure S4). These findings support the hypothesis that improved CNS penetration of vismodegib in POx-vismo is due to POx micelles reducing the protein binding of the drug.
POx-vismo reduces systemic toxicity of vismodegib
To determine whether nanoparticle delivery altered the systemic toxicity of vismodegib, we compared healthy P10 mouse pups treated with either PO-vismo or conventional vismodegib. To evaluate systemic toxicity, we compared growth of pups on extended regimens of POx-vismo or conventional vismodegib to that of untreated age-matched littermate controls (Figure 6, A). POx-vismo was injected IP and conventional vismodegib was administered either IP or by oral gavage. Drug was administered on postnatal days 10-12 and then every other day until P35. All mice including littermate controls were weighed daily. Mice treated with 100 mg/kg POx-vismo gained weight similar to untreated controls. In contrast mice on oral or IP conventional vismodegib at 100 mg/kg showed significantly decreased weight gain that required a 50% increased dose of 150 mg/kg POx-vismo to replicate. These studies show that POx formulation overall reduced systemic toxicity.
Figure 6.

Reduced toxicity in POx-vismo treated mice. (A) The weights of mice treated with the indicated formulations are graphed over time. The gray range indicates the mean weights ± SEM of littermate controls. (B) Whole body CT scan of mice treated as indicated. Red lines show the femur length definition used for measurements. (C) Femur growth, normalized to the mean for saline-treated controls, as determined by CT scan, or by direct measurement. *P < 0.05, **P < 0.005, ***P < 0.0005. Each point is mean ± SEM.
Vismodegib is known to impair bone growth, and we compared the bone toxicity in mice treated with POx-vismo or conventional vismodegib.29,30 We treated P10 mice with 4 daily doses of 100 mg/kg of either formulation, administered IP, then performed CT scans and measured femur length, as in prior vismodegib toxicity studies (Figure 6, B). We also isolated and directly measured the length of the dissected femurs. These studies showed that both POx-vismo and conventional vismodegib reduced femur length, as in prior studies, but that POx-vismo caused significantly less growth reduction (Figure 6, C).
POx-vismo and conventional vismodegib show similar pharmacodynamic effects
Next, we examined whether nanoparticle formulation altered the pharmacodynamic effect within the CNS. We treated G-Smo mice with a single 100 mg/kg dose of POx-vismo administered IP or conventional vismodegib, administered either IP or orally. We then harvested tumors after 24 h and compared the fractions of cells in tumors that expressed the proliferation marker phosphorylated RB (pRB; Figure 7, A). POx-vismo and conventional vismodegib IP similarly suppressed pRB (P = 0.9987), while oral vismodegib showed less suppression, approximating the effect of IP vismodegib at 50% lower dose (Figure 7, B). To compare pharmacodynamic effects of repeated doses, we examined tumor pathology after 3 daily IP doses of either POx-vismo or conventional vismodegib. We again found similar suppression of proliferation, with reduced tumor size and expression of both pRB and Proliferating Cell Nuclear Antigen (PCNA) (Figure 7, B). These data show that POx-vismo suppressed SHH-driven proliferation more effectively than the oral formulation and at least as effective as systemically administered conventional vismodegib.
Figure 7.

Pharmacodynamic response to POx-vismo and conventional vismodegib. (A) Flow cytometry quantification of pRB+ cells in untreated tumors and in tumors 24 h after a single IP administration of POx-vismo, or conventional vismodegib, administered IP or orally, compared to POx vehicle-injected controls. Cells of representative replicates treated as indicated are plotted according to pRB intensity versus DNA content. The dotted line indicates the threshold of detection for pRB. The pRB+ fractions for all replicates in each treatment group are graphed to the right, with columns indicating the means and error bars indicating the SEM. (B) PCNA immunofluorescence staining of sagittal medulloblastoma sections from representative P13 G-Smo mice, 24 h after three daily IP injections of the indicated formulation. Nuclei are counterstained with DAPI.
POx-vismo micelles prolong survival in GEMM model of SHH medulloblastoma
To determine whether improved pharmacokinetics and toxicity profile of POx-vismo improved the therapeutic efficacy of vismodegib, we compared the survival of G-Smo mice on a regimen of either POx-vismo or conventional vismodegib administered IP or orally. We randomized G-Smo mice to 5 groups: POx-vismo at 100 mg/kg IP, conventional vismodegib 100 mg/kg oral, conventional vismodegib 100 mg/kg IP, conventional vismodegib 50 mg/kg IP and saline-injected controls. The survival time to the humane endpoint was considered the event free survival (EFS). Conventional vismodegib at 50 mg/kg was included to evaluate the possibility of increased toxicity obscuring a treatment effect and was considered a reasonable dose as pharmacodynamics studies showed that 50 mg/kg was sufficient to suppress pRB (Figure 7, A). All G-Smo mice in the saline-injected control group survived less than 20 days, consistent with our prior data.31,32 Treatment with treatment with 100 mg/kg POx-vismo significantly improved the EFS (P < 0.001 log-rank test) with 30% of mice surviving to 35 days (Figure 8, A). In contrast, all conventional vismodegib regimens, including 100 mg/kg IP or orally, or with 50 mg/kg IP failed to extend the EFS. These data show that POx vismo produced superior efficacy compared to conventional vismodegib at a range of conventional vismodegib doses.
Figure 8.

Increased efficacy of POx-vismo compared to conventional vismodegib. (A) Kaplan–Meier survival curves for G-Smo mice treated with the indicated regimen, compared to no treatment; *** indicates P < 0.001 (vs. no treatment). All other curves were not significantly different from no treatment. (B) PCNA immunofluorescent staining (red), in a sagittal medulloblastoma section from a representative P35 G-Smo mouse, treated with 100 mg/kg of POx-vismo (P35). Nuclei are counterstained with DAPI. Red arrowhead indicates a region of proliferative tumor. Blue arrowhead indicates a differentiated, non-proliferative region.
We evaluated the tumor pathology in POx-vismo-treated mice that survived beyond the 20-day maximum EFS observed in the other groups. In each of these mice, we found regions of highly proliferative cells within the cerebellum, interspersed with regions of differentiated, non-proliferative cells (Figure 8, B). The non-proliferative regions indicate that vismodegib effectively interrupted SHH-driven proliferation and restricted tumor growth, while the proliferative regions show that even in the longer-term survivors, residual disease persisted. While POx-vismo was not curative, no known drug is curative as a single agent for medulloblastoma. POx-vismo was singularly effective in extending the life of G-Smo mice, while conventional vismodegib, oral or parenteral, failed to provide a survival benefit.
Discussion
Vismodegib has shown clinical efficacy for treatment of SHH-driven cancer outside the CNS, but has not been as effective for SHH subtype medulloblastoma either in animal models26 or in patients.3,33 We now show that optimizing vismodegib delivery using nanoparticles markedly reduces toxicity and improves efficacy, providing strong support for the use of the POx nanoparticle platform. POx-vismo formed uniform nanometer-scale particles and nearly neutral surface charge with high loading capacity that were stable in aqueous media and could be lyophilized and reconstituted with water, facilitating clinical implementation.
Our analyses show that POx formulation increased drug exposure in the CNS, in both medulloblastoma and forebrain and reduced systemic exposure, providing an explanation for both improved anti-tumor effect and reduced toxicity. POx-vismo produced higher, more sustained drug concentration in the target tumor tissues, with lower toxicity, resulting in improved antitumor efficacy compared to conventional vismodegib.
Our NMR studies provide important insight into the mechanism through which POx nanoparticle encapsulation improves pharmacokinetics. NMR simultaneously detected the polymer carrier and the drug, without labeling modifications that might affect biodistribution.34 We found that both vismodegib and polymer are absorbed into the circulation, with the % dose ratio of drug:polymer 1:13, suggesting that some of the drug was released from the micelles prior to absorption to systemic circulation. However, NMR and LC–MS analysis data account for total drug extracted from the biological matrix, including free drug, albumin-bound drug and carrier encapsulated-drug). In vitro analysis of vismodegib binding to serum proteins showed that encapsulation into POx micelles reduced the fraction of protein bound drug. The introducing vivo analysis is complicated because albumin-bound drug and encapsulated drug frequently have similar physicochemical properties and because polymeric micelles are dynamic and can disassemble and release the drug under standard analytical conditions. However, solid phase extraction (SPE) was shown to separate POx-micelles encapsulated and protein bound paclitaxel.18 SPE analysis of mice serum confirmed that the encapsulation into POx micelles reduced the fraction of protein bound drug. We conclude that nanoparticle encapsulation reduced vismodegib serum protein binding, increasing the availability of free vismodegib, which is known to pass through the BBB. Notably, our results are consistent with the previously observed data for POx-paclitaxel micelles following iv administration, where a significantly higher fraction of paclitaxel accumulated in brain compared to free drug.18 We propose that a POx similarly protects vismodegib from protein binding following IP administration of POx-vismo.
NMR analysis also showed that the vismodegib component of POx-vismo enters the brain and brain tumor, while the POx component does not. The absence of POx in the brain tissue shows that the nanoparticles release the payload outside the BBB, underscoring the importance of nanoparticle effects in the blood compartment. Importantly, since the nanoparticle does not enter into the CNS, there is reduced potential that it will cause untoward effects on the normal brain.
Diverse nanoparticles have shown efficacy against medulloblastoma cell lines either in vitro or in xenografts,35 including poly(lactic-co-glycolic acid) conjugated to polyethylene glycol-based nanoparticles delivering the SHH pathway inhibitor HPI-1.36 A key advance in our studies is testing nanoparticle drug delivery for brain tumors in a primary, genetic model in which tumors grow in the intracranial space, with an endogenous BBB. Several approaches to target brain and improve delivery across BBB were reported. There are several parameters that affect the efficiency of NP systemic circulation, BBB passage and cellular delivery. For example, smaller particles were shown to passively penetrate BBB.37 The surface charge of the nanoparticles was also shown to contribute to ability to penetrate BBB, with charged particles penetrating the BBB more effectively. Hydrophobicity of the carrier also contributes to BBB penetration38 as more hydrophobic particles can better interact with the BBB. However, while the POx-micelles in our study were moderately positively charged, the charge didn't contribute to the ability of the polymer to penetrate the BBB, as it was likely counterbalanced by the high hydrophilicity of the POx block copolymer, which was shown to be more hydrophilic than PEG.39 Another approach to improve BBB penetration and brain delivery is targeting BBB receptors or transporters. One nanoparticle system tested in an endogenous preclinical brain tumor model, iron oxide nanoparticles-coated with chitosan and polyethyleneimine-PEG copolymer, coated with the tumor-targeting chlorotoxin peptide, effectively delivered siRNA in a primary mouse glioma model.40,41 Our study opens new potential for brain tumor nanoparticle-drug delivery by demonstrating that a drug that is FDA-approved and in clinical use can be made more effective and less toxic. Moreover, POx micelles are a versatile system that can efficiently load diverse small molecules, and the non-targeted nature of POx micelles may be advantageous by not requiring a specific receptor that cancer cells can down-regulate to become resistant.
Our data clearly show that in mice POx-vismo is less a toxic and more effective, parenteral alternative to conventional vismodegib. Phase 1 trials to test the safety of POx-micelles loaded with other agents are currently on-going and early results of one phase 1 trial showed that poly(2-oxazoline) nanoparticles similar to POx micelles in this study were well tolerated.42 The improved efficacy of POx-vismo and the need for more effective treatments for SHH-driven cancers support the testing of POx-vismo in humans.
Resistance to vismodegib develops rapidly during therapy and was identified as main reason for treatment failure.43,44 We observed development of resistance as persistent rests of proliferating medulloblastoma cells in mice treated with POx-vismo through P35. Exposure to sub-optimal concentrations of cytotoxic drugs is associated with development of resistance and failure to respond to treatment.45,46 Alternative administration pathways such as intra CSF, interstitial or intra-arterial with or without blood brain barrier disruption delivery which had shown benefit in treatment of leptomeningeal malignancies can be explored to improve delivery and efficacy.47 However, Vismodegib-resistant cells may have different, specific sensitivities that may be defined in future studies of up-regulated signaling pathways in recurrent tumors.48 The versatility of the POx system as a potential carrier for diverse agents allows for delivery of vismodegib into the CNS through alternative delivery routes, either as single drug or combined with other specific inhibitors or chemotherapeutic agents, with the potential to improve survival and quality-of-life for brain tumor patients.
Data and materials availability
All data associated with this study are available in the main text or the supplementary materials.
Supplementary Material
Acknowledgments
We thank the UNC CGBID Histology Core supported by P30 DK 034987, the UNC Tissue Pathology Laboratory Core supported by NCI CA016086 and UNC UCRF and Ms. Yuling Zhao and Dr. Hedi Liu for assistance with histopathology, and the Chapel Hill Analytical and Nanofabrication Laboratory, supported by the National Science Foundation Grant ECCS-1542015, for help with electron microscopy.
Funding: This work was supported by the NCI Alliance for Nanotechnology in Cancer (U54CA198999, Carolina Center of Cancer Nanotechnology Excellence), by NINDS (R01NS088219, R01NS102627, R01NS106227) and by the St. Baldrick's Foundation.
Abbreviations:
- Ka
absorption rate constant
- AUC
area under the curve
- BBB
blood–brain barrier
- BSA
bovine serum albumin
- CL
clearance
- CNS
the central nervous system
- DLS
dynamic light scattering
- EFS
event-free survival
- FBS
fetal bovine serum
- G-Smo
Gfap-Cre/SmoM2
- IR-MALDESI
infrared matrix assisted laser desorption electrospray ionization
- IP
intraperitoneally
- Cmax
maximum concentration
- NMR
Nuclear magnetic resonance
- LC–MS
liquid chromatography–mass spectrometry
- LC
loading capacity
- LE
loading efficiency
- PK
pharmacokinetic
- PMeOx
poly(2-methyl-2-oxazoline)
- PBuOx
poly(2-n-butyl-2-oxazoline)
- PDI
polydispersity index
- POx-vismo
Polyoxazoline-vismodegib
- SMO
Smoothened
- SPE
solid phase extraction
- Vd
volume of distribution
Footnotes
Conflict of interest: AVK is listed as inventor on patents pertinent to the subject matter of the present contribution and co-founder of DelAqua Pharmaceuticals Inc. having intent of commercial development of POx based drug formulations. Both AVK and MSP have interest in the commercial success of DelAQUA. The other authors have no competing interests to report.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nano.2020.102345.
References
- 1.Rimkus TK, Carpenter RL, Qasem S, Chan M, Lo HW. Targeting the sonic hedgehog signaling pathway: review of smoothened and GLI inhibitors. Cancers (Basel) 2016;8(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vismodegib granted FDA approval for treatment of basal cell carcinomaOncology (Williston Park) 2012;26(2):174–213. [PubMed] [Google Scholar]
- 3.Robinson GW, Orr BA, Wu G, Gururangan S, Lin T, Qaddoumi I, et al. Vismodegib exerts targeted efficacy against recurrent sonic hedgehog-subgroup medulloblastoma: results from phase II Pediatric Brain Tumor Consortium studies PBTC-025B and PBTC-032. J Clin Oncol 2015;33(24):2646–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009;326(5952):572–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graham RA, Hop CE, Borin MT, Lum BL, Colburn D, Chang I, et al. Single and multiple dose intravenous and oral pharmacokinetics of the hedgehog pathway inhibitor vismodegib in healthy female subjects. Br J Clin Pharmacol 2012;74(5):788–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Batchelor HK, Marriott JF. Formulations for children: problems and solutions. Br J Clin Pharmacol 2015;79(3):405–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gajjar A, Stewart CF, Ellison DW, Kaste S, Kun LE, Packer RJ, et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: a pediatric brain tumor consortium study. Clin Cancer Res 2013;19(22):6305–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barenholz Y Doxil(R)—the first FDA-approved nano-drug: lessons learned. J Control Release 2012;160(2):117–34. [DOI] [PubMed] [Google Scholar]
- 9.Yamamoto Y, Kawano I, Iwase H. Nab-paclitaxel for the treatment of breast cancer: efficacy, safety, and approval. Onco Targets Ther 2011;4:123–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Misra R, Acharya S, Sahoo SK. Cancer nanotechnology: application of nanotechnology in cancer therapy. Drug Discov Today 2010;15(19–20):842–50. [DOI] [PubMed] [Google Scholar]
- 11.Ferrari M Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer 2005;5(3):161–71. [DOI] [PubMed] [Google Scholar]
- 12.Gupta S, Tyagi R, Parmar VS, Sharma SK, Haag R. Polyether based amphiphiles for delivery of active components. Polymer 2012;53(15):3053–78. [Google Scholar]
- 13.Gaucher G, Marchessault RH, Leroux J-C. Polyester-based micelles and nanoparticles for the parenteral delivery of taxanes. J Control Release 2010;143(1):2–12. [DOI] [PubMed] [Google Scholar]
- 14.Lavasanifar A, Samuel J, Kwon GS. Poly (ethylene oxide)-block-poly (L-amino acid) micelles for drug delivery. Adv Drug Deliv Rev 2002;54(2):169–90. [DOI] [PubMed] [Google Scholar]
- 15.Knop K, Hoogenboom R, Fischer D, Schubert US. Poly (ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew Chem Int Ed 2010;49(36):6288–308. [DOI] [PubMed] [Google Scholar]
- 16.Hwang D, Ramsey JD, Kabanov AV. Polymeric micelles for the delivery of poorly soluble drugs: from nanoformulation to clinical approval. Adv Drug Deliv Rev 2020;156:80–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luxenhofer R, Schulz A, Roques C, Li S, Bronich TK, Batrakova EV, et al. Doubly amphiphilic poly(2-oxazoline)s as high-capacity delivery systems for hydrophobic drugs. Biomaterials 2010;31(18):4972–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He Z, Wan X, Schulz A, Bludau H, Dobrovolskaia MA, Stern ST, et al. A high capacity polymeric micelle of paclitaxel: implication of high dose drug therapy to safety and in vivo anti-cancer activity. Biomaterials 2016;101:296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campos FC, Victorino VJ, Martins-Pinge MC, Cecchini AL, Panis C, Cecchini R. Systemic toxicity induced by paclitaxel in vivo is associated with the solvent cremophor EL through oxidative stress-driven mechanisms. Food Chem Toxicol 2014;68:78–86. [DOI] [PubMed] [Google Scholar]
- 20.Lang PY, Nanjangud GJ, Sokolsky-Papkov M, Shaw C, Hwang D, Parker JS, et al. ATR maintains chromosomal integrity during postnatal cerebellar neurogenesis and is required for medulloblastoma formation. Development 2016;143(21):4038–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell 2008;14(2):123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malawsky DS, Weir S, Ocasio J, Babcock B, Dismuke T, Wilhelmsen K, Gershon TR. Cryptic developmental events determine medulloblastoma radiosensitivity and cellular heterogeneity without altering transcriptomic profile., 08 August 2020, PREPRINT (version 1) available at Research Square 10.21203/rs.3.rs-50396/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phoenix TN, Patmore DM, Boop S, Boulos N, Jacus MO, Patel YT, et al. Medulloblastoma genotype dictates blood brain barrier phenotype. Cancer Cell 2016;29(4):508–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wan X, Beaudoin JJ, Vinod N, Min Y, Makita N, Bludau H, et al. Co-delivery of paclitaxel and cisplatin in poly(2-oxazoline) polymeric micelles: implications for drug loading, release, pharmacokinetics and outcome of ovarian and breast cancer treatments. Biomaterials 2019;192:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clogston JD, Patri AK. Zeta potential measurement. Characterization of nanoparticles intended for drug delivery: Springer; 2011. p. 63–70. [DOI] [PubMed] [Google Scholar]
- 26.Ocasio J, Babcock B, Malawsky D, Weir SJ, Loo L, Simon JM, et al. scRNA-seq in medulloblastoma shows cellular heterogeneity and lineage expansion support resistance to SHH inhibitor therapy. Nat Commun 2019;10(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Srinivas N, Rosen EP, Gilliland WM Jr, Kovarova M, Remling-Mulder L, De La Cruz G, et al. Antiretroviral concentrations and surrogate measures of efficacy in the brain tissue and CSF of preclinical species. Xenobiotica 2019;49(10):1192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sułkowska A Interaction of drugs with bovine and human serum albumin. J Mol 2002;614(1–3):227–32. [Google Scholar]
- 29.Kimura H, Ng JM, Curran T. Transient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell 2008;13(3):249–60. [DOI] [PubMed] [Google Scholar]
- 30.Robinson GW, Kaste SC, Chemaitilly W, Bowers DC, Laughton S, Smith A, et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 2017;8(41):69295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williams SE, Garcia I, Crowther AJ, Li S, Stewart A, Liu H, et al. Aspm sustains postnatal cerebellar neurogenesis and medulloblastoma growth in mice. Development 2015;142(22):3921–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gershon TR, Crowther AJ, Tikunov A, Garcia I, Annis R, Yuan H, et al. Hexokinase-2-mediated aerobic glycolysis is integral to cerebellar neurogenesis and pathogenesis of medulloblastoma. Cancer Metab 2013;1(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang JC, Hanke CW, Caro I. Vismodegib and the Hedgehog pathway inhibitors: a historical perspective to current clinical application. J Drugs Dermatol 2018;17(5):506–8. [PubMed] [Google Scholar]
- 34.Fonge H, Huang H, Scollard D, Reilly RM, Allen C. Influence of formulation variables on the biodistribution of multifunctional block copolymer micelles. J Control Release 2012;157(3):366–74. [DOI] [PubMed] [Google Scholar]
- 35.Catanzaro G, Curcio M, Cirillo G, Spizzirri UG, Besharat ZM, Abballe L, et al. Albumin nanoparticles for glutathione-responsive release of cisplatin: new opportunities for medulloblastoma. Int J Pharm 2017;517(1–2):168–74. [DOI] [PubMed] [Google Scholar]
- 36.Chenna V, Hu C, Pramanik D, Aftab BT, Karikari C, Campbell NR, et al. A polymeric nanoparticle encapsulated small-molecule inhibitor of Hedgehog signaling (NanoHHI) bypasses secondary mutational resistance to Smoothened antagonists. Mol Cancer Ther 2012;11(1):165–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Betzer O, Shilo M, Opochinsky R, Barnoy E, Motiei M, Okun E, et al. The effect of nanoparticle size on the ability to cross the blood–brain barrier: an in vivo study. Nanomedicine 2017;12(13):1533–46. [DOI] [PubMed] [Google Scholar]
- 38.Li X, Tsibouklis J, Weng T, Zhang B, Yin G, Feng G, et al. Nano carriers for drug transport across the blood–brain barrier. J Drug Target 2017;25(1):17–28. [DOI] [PubMed] [Google Scholar]
- 39.Lorson T, Lubtow MM, Wegener E, Haider MS, Borova S, Nahm D, et al. Poly(2-oxazoline)s based biomaterials: a comprehensive and critical update. Biomaterials 2018;178:204–80. [DOI] [PubMed] [Google Scholar]
- 40.Kievit FM, Stephen ZR, Wang K, Dayringer CJ, Sham JG, Ellenbogen RG, et al. Nanoparticle mediated silencing of DNA repair sensitizes pediatric brain tumor cells to gamma-irradiation. Mol Oncol 2015;9(6):1071–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kievit FM, Wang K, Ozawa T, Tarudji AW, Silber JR, Holland EC, et al. Nanoparticle-mediated knockdown of DNA repair sensitizes cells to radiotherapy and extends survival in a genetic mouse model of glioblastoma. Nanomedicine 2017;13(7):2131–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moreadith RW, Viegas TX, Bentley MD, Harris JM, Fang Z, Yoon K, et al. Clinical development of a poly (2-oxazoline)(POZ) polymer therapeutic for the treatment of Parkinson's disease—proof of concept of POZ as a versatile polymer platform for drug development in multiple therapeutic indications. Eur Polym 2017;88:524–52. [Google Scholar]
- 43.Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med 2009;361(12):1173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Metcalfe C, de Sauvage FJ. Hedgehog fights back: mechanisms of acquired resistance against smoothened antagonists. Cancer Res 2011;71(15):5057–61. [DOI] [PubMed] [Google Scholar]
- 45.Liu WM, Oakley PR, Joel SP. Exposure to low concentrations of etoposide reduces the apoptotic capability of leukaemic cell lines. Leukemia 2002;16(9):1705–12. [DOI] [PubMed] [Google Scholar]
- 46.Wang EC, Sinnott R, Werner ME, Sethi M, Whitehurst AW, Wang AZ. Differential cell responses to nanoparticle docetaxel and small molecule docetaxel at a sub-therapeutic dose range. Nanomedicine 2014;10(2):321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walker DA, Meijer L, Coyle B, Halsey C. Leptomeningeal malignancy of childhood: sharing learning between childhood leukaemia and brain tumour trials. Lancet Child Adolesc Health 2020;4(3):242–50. [DOI] [PubMed] [Google Scholar]
- 48.Bertrand KC, Faria CC, Skowron P, Luck A, Garzia L, Wu X, et al. A functional genomics approach to identify pathways of drug resistance in medulloblastoma. Acta Neuropathol Commun 2018;6(1):146. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.