

Abstract
Aims
Bone metastasis ultimately occurs due to a complex multistep process, during which the interactions between cancer cells and bone microenvironment play important roles. Prior to colonization of the bone, cancer cells must succeed through a series of steps that will allow them to gain migratory and invasive properties; epithelial-to-mesenchymal transition (EMT) is known to be integral here. The aim of this study was to determine the effects of G protein subunit alpha Q (GNAQ) on the mechanisms underlying bone metastasis through EMT pathway.
Methods
A total of 80 tissue samples from patients who were surgically treated during January 2012 to December 2014 were used in the present study. Comparative gene analysis revealed that the GNAQ was more frequently altered in metastatic bone lesions than in primary tumour sites in lung cancer patients. We investigated the effects of GNAQ on cell proliferation, migration, EMT, and stem cell transformation using lung cancer cells with GNAQ-knockdown. A xenograft mouse model tested the effect of GNAQ using micro-CT analyses and histological analyses.
Results
GNAQ-knockdown showed down-regulation of tumour growth through mitogen-activated protein kinase (MAPK) signalling in lung cancer cells, but not increased apoptosis. We found that GNAQ-knockdown induced EMT and promoted invasiveness. GNAQ-knockdown cells injected into the bone marrow of murine tibia induced tumour growth and bone-to-lung metastasis, whereas it did not in control mice. Moreover, the knockdown of GNAQ enhanced cancer stem cell-like properties in lung cancer cells, which resulted in the development of resistance to chemotherapy.
Conclusion
The present study reveals that the GNAQ-knockdown induced cancer stem cell-like properties.
Cite this article: Bone Joint Res 2021;10(5):310–320.
Keywords: GNAQ, Lung cancer, Bone metastasis
Article focus
The purpose of this study was to evaluate the relationship between G protein subunit alpha (GNAQ) and mechanisms underlying bone metastasis.
Key messages
By using human lung adenocarcinoma A549_shGNAQ cells that stably suppressed GNAQ using lentiviral transduction of short hairpin RNA (shRNA), this study confirmed the mechanisms involving GNAQ in lung cancer cell metastasis.
Knockdown of GNAQ may promote metastasis and resistance against chemotherapy by inducing epithelial-to-mesenchymal transition (EMT) and inducing cancer stem cell (CSC)-like properties.
Strengths and limitations
This study reports for the first time that GNAQ-knockdown induced CSC-like properties.
A limitation of this study is that how GNAQ affects osteogenic differentiation in vivo needs to be studied further.
Introduction
Many cancers, including breast, prostate, lung, renal, and thyroid, have a high propensity to metastasize to bone.1,2 Bone metastasis causes spinal cord compression, fractures, and intractable pain, all of which decrease patients’ quality of life. Nevertheless, an actionable mutation specific to bone metastasis has not yet been uncovered. Identification of specific genes that are responsible for metastasis is crucial to elucidate the mechanisms underlying metastasis. As cancer refers to a heterogeneous population of cells, containing different genetic alterations in a single tumour,3 a specific alteration in a given gene could help researchers to understand why a certain cancer cell migrates to the bone and survives in the bone microenvironment.4 In this study, we examined a subpopulation of lung cancer cases that involved bone metastases and compared their genetic alterations with those from lung cancers in primary lung lesions using comparative gene analysis. We found that alterations in G protein subunit α Q (GNAQ) were specific to cancer cells sampled from metastatic bone lesions.
GNAQ is a proto-oncogene closely related to the α subunit of guanine nucleotide-binding proteins (G proteins) that serve as key intermediates between membrane-bound G-protein-coupled receptors (GPCRs) and GPCR signalling nodes.5 Aberrant expression and activity of G proteins and GPCRs are frequently associated with tumorigenesis. Recent studies show that oncogenic activating mutations in GNAQ are present in 80% of the melanomas.6 GNAQ mutations are associated with poor prognosis in melanoma patients.7 However, to date, associations between GNAQ and bone metastasis have not been studied.
Epithelial-mesenchymal transition (EMT) is a reversible process of trans-differentiation in which epithelial cells lose their polarity and cell–cell interactions, and adopt a mesenchymal phenotype associated with the ability to migrate to distant body sites. EMT is driven by transcription factors, including Snail, ZEB, and Twist, which reportedly increase the invasiveness of epithelial cells.8 These processes are closely linked to a variety of other mechanisms enabling increased metastatic potential. Moreover, it has been shown that EMT induction enhances self-renewal and cancer stem cell (CSC)-like properties.9,10
The CSC theory assumes that tumours contain a subpopulation of cells with self-renewal potential, differentiation capacities, and tumorigenic properties.11,12 Moreover, several studies have demonstrated that CSCs are resistant to chemotherapy and radiation therapy, and are the main source of metastasis.13,14 However, no studies have been conducted to further validate the direct association between GNAQ alterations and CSCs.
Therefore, we sought to identify whether GNAQ alterations increase the metastatic potential of lung cancer tumours through EMT or CSC transformation. In the present study, we used a GNAQ-knockdown stable lung cancer cell line and evaluated: 1) EMT; 2) stem cell transformation and differentiation; and 3) the acquired resistance to chemotherapy compared to that in wild-type cells.
Methods
Patient samples
Patient studies were conducted with approved experimental protocols according to institutional guidelines for use. A total of 80 tissue samples from patients who were surgically treated between January 2012 and December 2014 were used in the present study. Among them, 53 samples were from primary lung tumours (not bone), and 27 were tissues from lower limb bone metastases. We first performed the gene analysis in these non-paired samples. Each collected metastatic bone sample was frozen, sectioned, and stained for cytokeratin. Samples were eligible for analysis if they contained > 50% epithelial cells (healthy bone should not contain any epithelial cells). Furthermore, samples were rejected if > 50% necrosis was observed after haematoxylin and eosin (H&E) staining. We obtained informed consent from all patients for the use of clinical specimens.
Comparative gene analysis
Non-paired samples from 80 patients were analyzed using CancerSCAN Version 2, a targeted sequencing platform designed covering 381 genes.15,16 GNAQ was also included in this gene panel. Data were further confirmed by Sanger sequencing.
Cell culture
All cells were obtained from the American Type Culture Collection (ATCC, USA). A549, NCI-H1299, NCI-H23, NCI-H1437, and HCC827 cells were cultured in RPMI 1640 medium (HyClone, USA) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic-antimycotic (Gibco, USA). NCI-H1739 cells were grown in DMEM:F12 medium containing 10% FBS and 1% antibiotic-antimycotic (Gibco). The cells were maintained in a humidified atmosphere containing 5% CO2 at 37°C, and the culture medium was replaced every third day.
Lentivirus construction and transfection
Short hairpin (shRNA) (5′-CCGGCTATGATAGACGACGAGAATACTCGAGTATTCTCGTCGTCTATCATAGTTT TTG-3′) against human GNAQ (NM_002072.4) and a control (SHC002) shRNA were purchased from Sigma-Aldrich (USA). The recombinant GNAQ silencing and control plasmid were transfected into 293 T cells using a lentiviral packaging mix (Sigma-Aldrich) and Lipofectamin2000 according to the manufacturer’s protocol. After transfection for 48 hours, the lentiviral supernatant was harvested and the lentiviral particles were used to infect the A549 cells.
Establishment of the A549_GNAQ cell line
A549_shGNAQ cell line with a stable knockdown of GNAQ, and A549_shNC cell line, which served as the control, were established in this study. A549 cells were seeded at a density of 2 × 105 cells per well and transduced using shNC or shGNAQ lentiviral particles. After 48 hours of infection, cells were maintained in a selection media containing 0.5 μg/ml puromycin (Sigma-Aldrich), and grown until colony formation. Each colony was transferred after trypsinization to 96-well plates, 24-well plates, and then to six-well plates for continued culture. For colony selection, the cells were seeded in 60 mm culture dishes. After 24 hours, the cells were harvested to assess GNAQ silencing efficiency using quantitative reverse transcription polymerase chain reaction (RT-qPCR) and western blotting.
Cell proliferation and cytotoxicity assays
Cell proliferation and cytotoxicity assays were conducted using the MTS (3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay, according to the manufacturer’s protocol (Promega, USA). Cells were seeded at a density of 5 × 103 cells/well in 96-well plates and then treated with doxorubicin for 48 hours. After adding MTS, the plates were incubated at 37°C in a CO2 incubator for one hour, and absorbance was read at 490 nm using a SPECTRA max PLUS 384 instrument (Molecular Devices, Germany).
Soft agar colony formation assay
To examine anchorage-dependent cell growth, we used a soft agar colony formation assay according to a published protocol.17 Briefly, cells were seeded for colony formation in six-well plates at a density of 1 × 103 cells per well. Each well consisted of a top layer (0.3% agarose) and bottom layer (0.5% agarose). After ten days, colonies were stained with crystal violet and counted under a light microscope. This experiment was performed in triplicate.
In vitro differentiation assay
Stable A549 cell lines were seeded in 24-well plates at a density of 5 × 103 cells/well. Cells were cultured in osteogenic medium, which was refreshed every two to three days. Osteogenic induction media comprised Dulbecco’s Modified Eagle Medium (DMEM; HyClone, USA) supplemented with 10% FBS, 1% antibiotic-antimycotic (Gibco), 0.1 mM ascorbic acid, 10 mM glycerophosphate, and 100 nM dexamethasone (Sigma-Aldrich).18 Chondrogenic differentiation media comprised DMEM supplemented with 2% FBS, 10 ng/ml transforming growth factor-β1 (TGF-β1; R&D Systems, USA), 50 μM ascorbic acid, 100 μM dexamethasone, and 6.25 μg/ml insulin (Sigma-Aldrich). Adipogenic differentiation medium comprised DMEM supplemented with 5% FBS, 1 μM dexamethasone, 10 μg/ml insulin, 0.5 mM isobutylmethylxanthine, and 200 μM indometacin (Sigma-Aldrich).19
Reverse transcription PCR and quantitative PCR analysis
Total RNA from cultured A549_shNC and A549_shGNAQ cells was reverse-transcribed into complementary DNA (cDNA) using oligo (dT) primer, and PCR was performed using AccuPower Hotstart PCR PreMix (Bioneer, South Korea) following the manufacturer’s protocol. RT-qPCR was performed on an Applied Biosystems 7900HT real-time PCR system. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and ACTB were used to normalize the expression levels in subsequent quantitative analyses. To amplify the target genes, the following primers were used: GNAQ forward, 5′-gcacaataaggctcatgcac-3′ and reverse, 5′-tggaaccaggggtatgtgat-3′; Snail (SNAI1) forward, 5′-tcaagatgcacatccgaagcca-3′ and reverse. 5′-gcttgtggagcagggacattcg-3′; GAPDH forward, 5′-tgatgacatcaagaaggtgg-3′ and reverse, 5′-tccttggaggccatgtgggc-3′; β-actin (ACTB) forward, 5′-tcatgaagtgtgacgtggac-3′ and reverse, 5′-gcagtgatctccttctgcat-3′; collagen type I (COL1A1) forward, 5′- gtgctaaaggtgccaatggt-3′ and reverse, 5′-accaggttcaccgctgttac-3′; osterix; (OSX) forward, 5′-ggcacaaagaagccgtactc-3′ and reverse 5′-caggtgaaaggagcccatta-3′; Sox9 forward, 5′-tacgactacaccgaccacca-3′ and reverse 5′-tcaaggtcgagtgagctgtg-3′; collagen type II (COL2A)1 forward, 5′-gggagtaatgcaaggaccaa-3′ and reverse 5′-atcatcaccaggctttccag-3′; Oct4 forward, 5′-agtgagaggcaacctggaga-3′ and reverse 5′-gcctcaaaatcctctcgttg-3′; Sox2 forward, 5′-accagctcgcagacctacat-3′; and reverse 5′-tggagtgggaggaagaggta-3′; NANOG; forward, 5′-ttccttcctccatggatctg-3′; and reverse 5′-tctgctggaggctgaggtat-3′; c-Met forward, 5′-aagagggcattttggttgtg-3′, reverse 5′-gatgattccctcggtcagaa-3′. PCR primers for osteopontin (OPN) and RUNX2 were designed as described in a previous report.18
Western blotting
Cells were lysed in RIPA lysis buffer (Thermo Fisher Scientific, USA), following which they were centrifuged at 14,000× g for 15 minutes at 4°C to obtain protein. Proteins were quantified using a BCA protein assay reagent (Pierce, Thermo Fisher Scientific), separated by sodium dodecyl sulphate (SDS)-polyacrylamide gel electrophoresis (PAGE), and transferred onto a polyvinylidene fluoride (PVDF) membrane (Bio-Rad, USA) for western blotting. After blocking with 5% skim milk in TBST (50 mM Tris-Cl, pH 7.5; 150 mM NaCl; and 0.1% Tween 20) for one hour, the membrane was incubated with primary antibodies against GNAQ (Abcam, UK), N-cadherin, β-actin, nuclear factor-κB (NF-κB; Santa Cruz Biotechnology, USA), E-cadherin, Snail, β-catenin, β-tubulin p-NF-κB, p-Akt, Akt, ERK1/2, p-ERK1/2, STAT3, and p-STAT3 (Cell Signalling Technology, USA) at a dilution of 1:1,000 overnight at 4°C. After the membrane was washed, it was incubated with secondary antibody conjugated to horseradish peroxidase (HRP) (Bio-Rad) in TBST containing 5% skim milk for one hour at room temperature (18°C to 25 °C).
Immunofluorescence
Cells were fixed in 4% paraformaldehyde for ten minutes at room temperature and then washed with PBS. Following permeabilization with 0.5% Triton X-100, cells were incubated with 1% bovine serum albumin (BSA, South Korea) to block non-specific binding. Subsequently, cells were incubated with anti-N-cadherin (Santa Cruz Biotechnology, USA) and firefly luciferase antibodies (Abcam), following which the cells were incubated with fluorescence-conjugated secondary antibodies (goat anti-mouse-Alexa 488 and goat anti-rabbit-Alexa 568; Invitrogen) for one hour at 37°C. The cells were mounted using a solution containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, USA), and then visualized using a Zeiss LSM780 confocal microscope (Carl Zeiss, Germany). Images were analyzed using Vectra 3 inForm version 2.3 software (PerkinElmer, USA).
Cell migration assay
Cell migration assays were performed in triplicate in 24-well Transwells. Briefly, 1 × 105 cells in 100 µl serum-free medium were seeded into the upper chamber of each well, while medium supplemented with 1% FBS (500 µl) was placed in the lower chamber. Following incubation at 37°C for 18 hours, cells adhering to the upper surface of the membrane were removed with a cotton swab. Cells which had migrated through the filter and adhered to the lower surface of the membrane were fixed in methanol at room temperature for five minutes and stained with H&E solution. The mean number of migratory cells was calculated by counting six randomly selected fields under a light microscope at 200× magnification. The experiments were repeated three times.
In vivo assay
A549_shNC and A549_shGNAQ cells (1 × 106 cells/mouse) were injected into the calvarial bones (n = 10) and tail veins (n = 10) of eight-week-old BALB/c nude mice (Orient Bio, South Korea). Female mice were used for all animal experiments. The A549_shNC cells-injected group was used as a control. The calvarial bone injection method was as follows: the injection site was the centre between the parietal and interparietal. After loading the cells in an insulin syringe, the cells were injected subcutaneously over the calvaria. The syringe was then slowly withdrawn. Tumour size was measured at two and four weeks after injection, and the tumour was fixed in 4% paraformaldehyde. Subsequently, H&E staining and immunohistochemistry (IHC) were performed according to a previously described protocol.20 Luciferase-expressing A549_shNC_luc and A549_shGNAQ_luc cells (1 × 106 cells/mouse) were injected into the left tibia (n = 8), and then lung metastases were tracked using bioluminescent imaging. Mice were administered 150 mg/kg D-luciferin via intraperitoneal injection, anaesthetized by isoflurane inhalation and imaged using the 560 nm filter.
Statistical analysis
All data were analyzed using GraphPad Prism 5 (GraphPad, USA) and are presented as means and standard deviations. Mann-Whitney U test and two-tailed independent-samples t-test were used to detect the differences between groups. All in vitro experiments were repeated three times according to the following significance thresholds: p < 0.05, p < 0.01, and p < 0.001. Our Supplementary Material includes an ARRIVE checklist to show that we have conformed to the ARRIVE guidelines.
Results
In order to understand the mechanisms involved in metastasis, it is necessary to identify the specific gene(s) responsible. We have identified genes associated with bone metastasis using CancerSCAN V2 in non-paired samples from 80 patients. We found GNAQ mutations in 40.7% (n = 11) of the lung cancer cases that involved metastasis to bone (Supplementary Table i). We also found that the GNAQ mutation rate in lung cancer is only 0.6% in the publicly accessible database from the Cancer Genome Atlas (TCGA), an open access repository.21 In this study, we aimed to identify the potential roles and underlying mechanisms involving GNAQ in lung cancer cell metastasis. Using human lung adenocarcinoma A549 cells, we established A549_shGNAQ cells stably suppressing GNAQ using lentiviral transduction of shRNA (Figure 1).
Fig. 1.
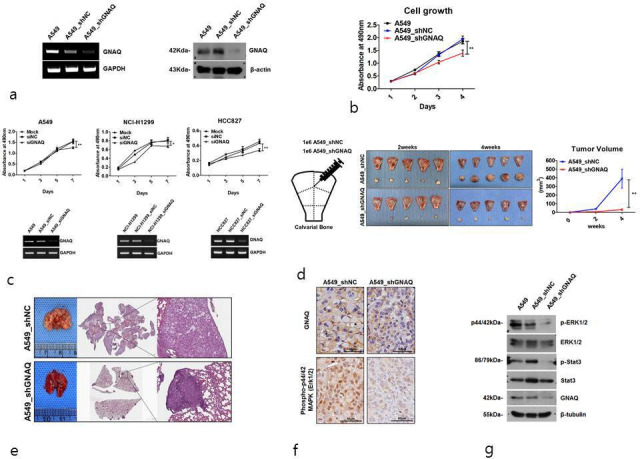
G protein subunit alpha Q (GNAQ) regulates tumour growth through mitogen-activated protein kinase (MAPK) signalling but does not increase apoptosis levels. a) GNAQ transcript levels in A549, A549_shNC, and A549_shGNAQ cells were measured by semiquantitative polymerase chain reaction (PCR) (left), and GNAQ protein expression was visualized by western blotting (right). b) Cell proliferation capacities in GNAQ-knockdown cells. c) GNAQ small interfering RNA (siRNA) was transfected into lung cancer cell line (A549, NCI-H1299, and HCC827), with negative siRNA serving as a control (upper panel). GNAQ transcript levels in cells were measured by semi-quantitative PCR (lower panel). d) Analysis of tumour growth for four weeks after injection of A549_shNC and A549_shGNAQ cells into the calvarial bone. Schematic diagram of location and the number of injected cells in mouse (left). Photographs show size comparisons of xenograft tumours in the different groups (n = 5 per group) (middle). A tumour growth curve was plotted based on the tumour volumes and the days elapsed (right). e) Representative images showing nodules on the lung surface and lung histology in mice. Mice (n = 5 per group) were injected via the tail vein with A549_shNC and A549_shGNAQ cells. Three months later, the mice were euthanized, and tumour nodules in the lungs were assessed. Two mice in the control group (A549_shNC) died before being euthanized. Lung nodules were visualized by haematoxylin and eosin (H&E) staining, magnification ×40 (Scanscope XT, Aperio, Leica Biosystems, USA). f) The expression of GNAQ and phosphylated extracellular signal-regulated kinase (p-ERK) in each group is shown by immunohistochemical staining of the xenograft tumours, magnification ×40 (Scanscope XT). g) The phosphorylation status of ERK1/2 and signal transducer and activator of transcription 3 (STAT3) was determined by western blotting. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; shNC, negative control short hairpin RNA; siNC, negative control small interfering RNA.
GNAQ regulates tumour growth through MAPK signalling but not does increase apoptosis
As GNAQ may affect the growth rate of cancer cells,22 we evaluated cell proliferation capacity using the MTS assay. The results showed that proliferation of A549 cells with GNAQ-knockdown was significantly (p < 0.01) decreased in comparison with that of control cells (Figure 1b). To determine the effects of GNAQ small interfering RNA (siRNA) on lung cancer cell growth, cells were transfected with siRNA or non-targeting siRNA (NC). As shown in Figure 1c, siNC-transfected cells presented a growth pattern similar to that of non-transfected cells, while siGNAQ reduced growth rates at two and seven days in NCI-H1299 and HCC827 cells, respectively. In various cancer cells, the knockdown of GNAQ inhibited cell proliferation (Supplementary Figure a). To determine whether GNAQ affects the growth of tumour xenografts in nude mice, A549_shNC and A549_shGNAQ cells were transplanted into the calvarias subcutaneous tissue of nude mice. We found that stable knockdown of GNAQ significantly (p = 0.008, two-tailed independent-samples t-test) inhibited the growth of A549 solid tumours compared to those in control mice (Figure 1d). In parallel, knockdown of GNAQ retarded the growth of lung tumour nodules in a xenograft mouse model of lung cancer (Figure 1e). Signal transducer and activator of transcription-3 (STAT3) and MAPK are important mediators of signal transduction during various cellular processes, including cell growth, proliferation, and apoptosis.23,24 We performed immunohistochemistry for phosphorylated extracellular signal-regulated kinase (p-ERK) to compare MAPK activity between GNAQ-knockdown tumours and control tumour tissues in the orthotropic model. Immunostaining for GNAQ revealed that GNAQ was present in the plasma membrane of control tumours, but this signal was weak in GNAQ-knockdown tumours. The levels of p-ERK in GNAQ-knockdown tumours were also suppressed compared to those in control tumours (Figure 1f). We also performed western blotting to evaluate the levels of p-ERK and STAT3, and found that the levels were significantly decreased in GNAQ-knockdown cells (Figure 1g). These results showed that the growth of tumour cells was suppressed through both the STAT3 and MAPK pathways in GNAQ-knockdown cells. However, our results further demonstrated that GNAQ-knockdown did not significantly alter the cell cycle, nor did it increase apoptosis (Figure 1h and Supplementary Figure b).
Promotion of EMT in response to knockdown of GNAQ
In lung cancer, metastasis is known to be associated with EMT.25,26 Given that GNAQ mutation is frequently associated with bone metastasis, we hypothesized that GNAQ-knockdown may promote EMT and cell migration in lung cancer. Therefore, we examined the expression of EMT markers E-cadherin, N-cadherin, vimentin, Snail, and beta-catenin in GNAQ-knockdown cells. The expression of the epithelial marker E-cadherin was downregulated in GNAQ-knockdown cells compared to that in control cells, while that of mesenchymal markers N-cadherin, vimentin, Snail, and beta-catenin was markedly upregulated in GNAQ-knockdown cells compared to that in control cells (Figure 2a). Immunofluorescence revealed that N-cadherin expression was also significantly increased in GNAQ-knockdown cells (Figure 2b). To further delineate how knockdown of GNAQ promotes EMT, we sought to assess the activity of the protein kinase B (Akt)/NF-κB signalling pathway, one of the pathways most commonly involved in EMT.27,28 We found that the levels of phosphorylated Akt and phosphorylated NF-κB were significantly increased upon the knockdown of GNAQ (Figure 2c). In order to improve our understanding of the effects of GNAQ on the metastasis of lung cancer cells, Transwell assays were performed. As shown in Figure 2d, the number of migrated cells was > two-fold higher in GNAQ-knockdown cells than that in control cells. These results demonstrated that GNAQ plays an important role in the motility of lung cancer cells via EMT.
Fig. 2.
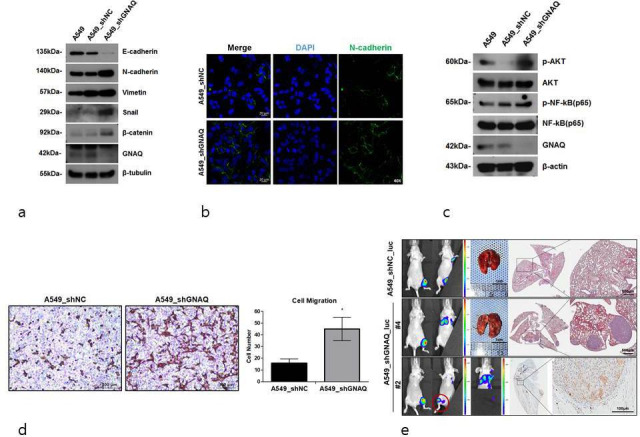
Promotion of epithelial-to-mesenchymal transition (EMT) by the knockdown of G protein subunit alpha Q (GNAQ). a) Expression of EMT markers. Epithelial and mesenchymal markers were determined by western blotting. β-tubulin is used as an internal control. b) Representative images demonstrating the expression of N-cadherin in control (A549_shNC) or GNAQ-knockdown (A549_shGNAQ) cells. c) Protein kinase B (Akt) and nuclear factor-κB (NF-κB) expression was determined by western blotting. β-tubulin served as a loading control. d) Representative microscopy images of migrating quantified by haematoxylin and eosin (H&E) staining. Transwell assays were conducted to detect cell migration. Significant differences are marked by asterisks (p = 0.014, two-tailed independent-samples t-test). e) Representative images of lung metastases monitored using bioluminescent signal imaging (left panel, n = four per group). Imaging times vary due to saturation at later acquisition time. Representative H&E-stained images of mouse lung (right panel). Arrows indicate metastatic nodules in the lungs. Scale bars, 500 μm. A549_shGNAQ_luc cells metastasized to other bone sites (bottom, left panel) (○ indicates bone metastasis site). Representative images of human mitochondria level (indicated by the arrows) by immunohistochemistry (IHC) of the bone (bottom, right panel); scale bars, 100 μm. Mice were euthanized 60 days after tibia injection with A549_shNC_luc and A549_shGNAQ_luc cells. DAPI, 4′,6-diamidino-2-phenylindole. shNC, negative control short hairpin RNA.
We next investigated the effects of GNAQ on bone to lung metastasis by injecting luciferase-labelled cells into the bone marrow of murine tibia, followed by evaluation of tumour growth and metastasis using bioluminescent imaging. Bone to lung metastasis was observed only in mice with GNAQ-knockdown tumours (three out of four cases). Consistently, H&E staining showed metastatic nodules in the lungs of mice with GNAQ-knockdown tumours. Moreover, metastasis to other bones was identified in mice with GNAQ-knockdown tumours (A549_shGNAQ_luc #2), which was not seen in control mice. We stained the mitochondria of human cells to confirm that the tumour in the right tibia of A549_shGANQ_luc #2 had metastasized (Figure 2e and Supplementary Figure c). These findings showed that knockdown of GNAQ increased the metastatic potential of lung cancer cells.
Knockdown of GNAQ enhances stem cell-like properties in lung cancer cells
The stem cell markers Oct4, Sox2, NANOG, and cMET are key cellular factors that have been used for identifying CSCs. These transcription factors are capable of reprogramming adult human somatic cells into pluripotent stem cells.29 To test whether knockdown of GNAQ results in production of characteristics associated with CSCs, stem cell markers were evaluated at the messenger RNA (mRNA) level. We found that Oct4 was significantly upregulated in response to the knockdown of GNAQ, but that the expression of other genes was not significantly affected (Figures 3a and 4). Soft agar colony formation assays indicated that knockdown of GNAQ significantly (p = 0.029, two-tailed independent-samples t-test) promoted the self-renewing potential of cancer cells (Figure 3b). CD44, a transmembrane receptor, is a CSC marker and also a critical regulator of stemness and metastatic potential.30 To investigate stem cell properties, we compared the expression of CD44 using flow cytometric analysis. Knockdown of GNAQ resulted in significant upregulation of CD44 on day 7 (Figure 3c). Previous studies have demonstrated that CSCs are the main players in the development of chemoresistance to a variety of drugs, including doxorubicin, in many cancers.31,32 To confirm whether GNAQ plays a role in doxorubicin-induced drug resistance, we assessed the effects of GNAQ on cell survival in response to doxorubicin treatment. Cells were treated with various concentrations of doxorubicin, and cell viability was measured using MTS assays. Doxorubicin treatment decreased cell viability in a concentration-dependent manner in both control and GNAQ-knockdown cells. Knockdown of GNAQ significantly (p = 0.050, two-tailed independent-samples t-test) increased cell viability in the background of high doses of doxorubicin compared to that in controls (Figure 3d). CSCs comprise a small proportion of the total number of cells in heterogeneous tumours. As conventional anticancer agents usually target rapidly growing cancer cells, slow-growing CSCs can persist and increase in proportion. This may be the main cause of relapse and resistance to chemotherapy.33,34 To mimic heterogeneous tumours, we established co-cultures of both GNAQ-knockdown cells and control cells. To identify the proportion of GNAQ-knockdown cells, we used GNAQ-knockdown cells expressing luciferase, and control cells lacking luciferase. Due to the differences in sensitivity to doxorubicin between GNAQ-knockdown cells and control cells, the proportion of GNAQ-knockdown cells remaining in the co-culture was increased in a dose-dependent manner after treatment with doxorubicin (Figures 3e and 5).
Fig. 3.

a) Knockdown of G protein subunit alpha Q (GNAQ) enhances stem cell-like properties in lung cancer cells. Real-time quantitative polymerase chain reaction (PCR) analysis of Oct4, SOX2, NANOG, and cMet messenger RNA (mRNA) expression in A549 cells. b) Control and GNAQ-knockdown cells were cultured on soft agar. The number of colonies was counted 14 days after plating. c) An overlaid histogram representing the expression of CD44 in GNAQ-knockdown cells compared to that in the control cells analyzed by flow cytometry. Cells were stained with anti-CD44-PE. d) For determination of dose-dependency, cell viability after treatment with various concentrations of doxorubicin (0, 0.25, 0.5, and 1 μM) for 48 hours was measured by MTS assays. e) Proportion of cells in response to treatment with doxorubicin. Control and stably luciferase-expressing GNAQ-knockdown cells were 1:1 co-cultured and treated with doxorubicin at concentrations ranging from 0.25 μM to 1 μM for 72 hours. The expression of luciferase (red) was analyzed by immunofluorescence. 4′,6-diamidino-2-phenylindole (DAPI)‐stained nuclei (blue) are represented in the middle panel, and the left panel shows a merged image. The images were acquired using the same exposure times. shNC, negative control short hairpin RNA.
Fig. 4.

Knockdown of G protein subunit alpha Q (GNAQ) enhances stem cell-like properties in lung cancer cells. Real-time quantitative polymerase chain reaction (PCR) analysis of Oct4, SOX2, NANOG, and cMet messenger RNA (mRNA) expression in lung cancer cell lines (NCI-H1793, NCI-H1299, NCI-H1437, NCI-H23, and HCC827). si, small interfering.
Fig. 5.
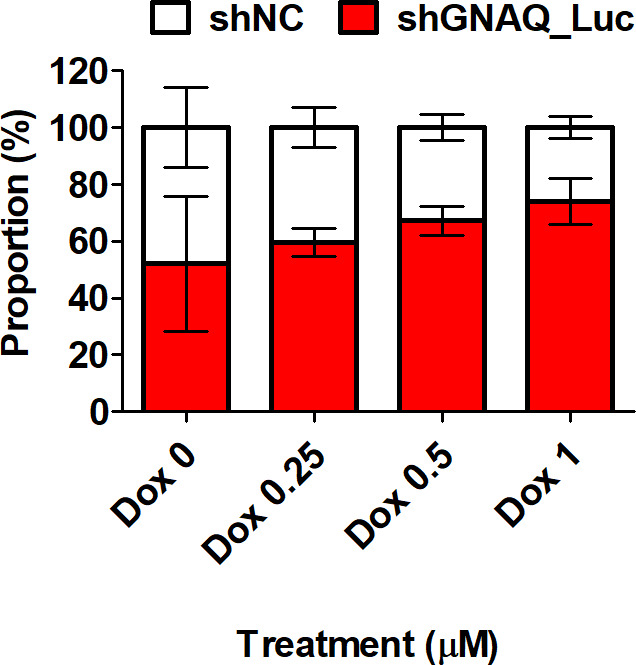
The corresponding graph for Figure 3e: proportion of cells in response to treatment with doxorubicin. Control and stably luciferase-expressing G protein subunit alpha Q (GNAQ)-knockdown cells were 1:1 co-cultured and treated with doxorubicin at concentrations ranging from 0.25 μM to 1 μM for 72 hours. shNC, negative control short hairpin RNA.
Knockdown of GNAQ promotes the further differentiation of carcinoma into mesenchymal tissue
In order to evaluate whether GNAQ regulates further mesenchymal differentiation, we attempted to assess osteogenic differentiation markers using quantitative real-time PCR. At 14 days post-osteogenic induction, GNAQ-knockdown had significantly induced the upregulation of COL1A1 (9.7-fold) and RUNX2 (2.7-fold) compared to control. The expression of OSX and OPN was upregulated to a maximal level (1.4-fold and 4.2-fold higher, respectively) relative to that in the control at day 10 after induction of osteogenic differentiation (Figure 6aA). Quantitative PCR also demonstrated that the expression of bone markers was increased significantly in GNAQ-knockdown cells compared to that in control cells. SOX9 and COL2A1, a regulator of chondrogenic differentiation, also exhibited increased mRNA levels at day 14 in chondrogenic differentiation media (Figure 6bB). However, the expression of PPARγ, C/EBPα, and adiponectin (ADIPOQ), markers of adipogenic differentiation, was not significantly increased in either GNAQ-knockdown or control cells (data not shown).
Fig. 6.
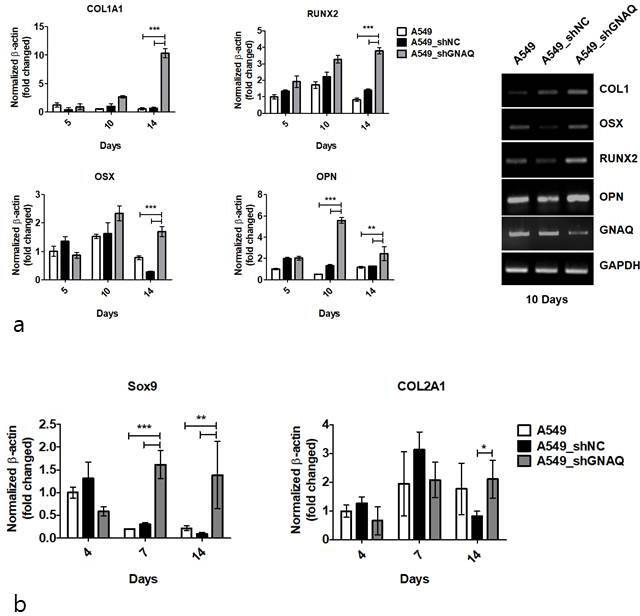
Knockdown of G protein subunit alpha Q (GNAQ) promotes the differentiation of carcinoma into mesenchymal tissue. a) COL1a1, RUNX2, OSX, and OPN messenger RNA (mRNA) expression was determined by quantitative real-time polymerase chain reaction (PCR) at five, ten, and 14 days after osteogenic differentiation (left). Osteogenic marker mRNA expression examined via semi-quantitative PCR at ten days after osteogenic differention (right). b) Quantitative real-time analysis of Sox9 and COL2A1 on days 4, 7, and 14 during chondrogenic differentiation induction. The results were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA expression. Values are means and standard deviations (n = 4). shNC, negative control short hairpin RNA.
Discussion
Little is known regarding the genetic alterations in non-small cell lung cancer (NSCLC), a malignancy associated with bone metastasis. Previous studies have described a positive correlation between mutant GNAQ and metastasis in melanomas.35 However, the role of GNAQ in the mechanisms underpinning metastasis has not been previously described. Recent studies have shown that not only specific gene mutations, but also their target organ specificity are relevant to a tumour’s potential to migrate.36,37 It is known that K-RAS mutations are often present in bone, brain, and lung metastases of colon cancer.38,39 As for breast cancer, overexpression of CXCR4 is known to cause bone metastasis via interactions with the bone microenvironment.40 Furthermore, a previous study demonstrated that high expression of RUNX2 in solid tumours such as prostate, breast, pancreatic, and lung cancer is associated with bone metastases.41 Identifying an association between mutant genes and a metastatic site will help the clinicians identify patients at a high risk of disease spread. Bone metastases are generally categorized as osteolytic, osteoblastic, and mixed lesions.42 The activation of osteoclasts is an important prerequisite of osteolytic bone metastasis.43 Our previous study demonstrated significant upregulation of receptor activator of nuclear factor-κB ligand (RANKL) and increased macrophage colony-stimulating factor (M-CSF) in GNAQ-knockdown cells. It was revealed that RANKL expression in metastatic bone cancer is regulated by GNAQ.44 In this study, we first identified GNAQ-knockdown induced mesenchymal stem cell (MSC)-like properties, and further osteogenic differentiation of lung cancer cells. The molecular mechanisms through which GNAQ regulates MSCs have not previously been reported. MSCs residing in the bone marrow differentiate into various lineages: osteogenic, chondrogenic, or adipogenic.45 COL1 is among the earliest markers expressed by the osteoblast lineage. RUNX2 plays a major role, along with OSX and OPN in driving the differentiation of mesenchymal precursor cells into osteoblasts, and is involved in skeletal morphogenesis.46,47 The current study found that GNAQ-knockdown upregulated the expression of osteogenic markers, including COL1, RUNX2, OSX, and OPN in cancer cells, which is strong evidence that GNAQ is related to osteogenesis. β‐catenin is involved in early differentiation, and is also required for osteogenesis.48 Interestingly, we noticed that knockdown of GNAQ induced the activation of β‐catenin, which is expected to affect not only the EMT but also osteogenic differentiation. In previous studies, a series of processes, including CD44 expression, were found to contribute to the metastatic homing of cancer cells in various organs.49,50 Our study suggested that CSCs with a potential for osteogenic differentiation and high CD44 expression are likely to metastasize to the bone. Activation of Akt and NK-κB signalling has been proposed to be involved in metastasis.27,28 Consistently, our data showed that knockdown of GNAQ induced hyper-phosphorylation of both Akt and NF-κB. Recent studies show that STAT3 inhibits the expression of mesenchymal markers in tumour cells.51,52 Suppression of GNAQ was shown to inhibit the activation of STAT3, which may also promote EMT. When compared with controls, GNAQ knockdown effectively inhibited the phosphorylation of ERK in vitro and in vivo, consistent with reports that GNAQ mutation negatively regulates cell growth.22 However, GNAQ suppression did not promote cancer cell apoptosis. Such GNAQ inhibition is closely related to the suppression of tumour growth. CSCs comprise a slow-growing proportion of cells in heterogeneous tumours, which are the main cause of tumour recurrence and chemoresistance, because the conventional chemotherapeutic agents usually target rapidly growing cells.53,54 The current study also showed that GNAQ-knockdown increased chemoresistance. Our tumorous heterogeneity model showed that the proportion of GNAQ-knockdown cells was higher, compared to that of the highly proliferating control cells during chemotherapy. This result indicated that GNAQ-knockdown cells acquire stem cell-like properties associated with resistance against chemotherapy and subsequent tumour relapse. Therefore, inhibiting G-protein activity may regulate cell growth and increase cellular stemness by protecting cancers from hostile environments, such as those induced by chemotherapy.
Collectively, our findings suggest that knockdown of GNAQ may promote metastasis and resistance against chemotherapy by inducing EMT and inducing CSC-like properties. This study provided an in-depth insight into GNAQ with respect to metastasis, and we hope that future studies will unravel further mechanisms involving GNAQ and metastasis.
Author contributions
J-Y. Choi: Conducted the experiments, Analyzed the data, Wrote the manuscript.
Y. S. Lee: Conducted the experiments, Drafted the manuscript.
D. M. Shim: Conducted the experiments.
Y. K. Lee: Interpreted the data.
S. W. Seo: Conceptualized and designed the study, Reviewed and revised the manuscript.
Funding statement
Although none of the authors has received or will receive benefits for personal or professional use from a commercial party related directly or indirectly to the subject of this article, benefits have been or will be received but will be directed solely to a research fund, foundation, educational institution, or other non-profit organization with which one or more of the authors are associated. This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education and Science Technology (MEST) (2016R1E1A1A0191433, 2019R1I1A1A01061544), the Grant of the Samsung Medical Center (OTC1190111), and the National Cancer Center (NCC) (No.194154).
Supplementary material
Figures showing the effect of G protein subunit alpha Q (GNAQ)-knockdown on the proliferation, cell cycle, apoptosis, and metastasis of various types of cancer cells. Table showing GNAQ status in patient-derived cancer cells from patients with primary lung cancer and bone metastasis of lung cancer.
© 2021 Author(s) et al. This is an open-access article distributed under the terms of the Creative Commons Attribution Non-Commercial No Derivatives (CC BY-NC-ND 4.0) licence, which permits the copying and redistribution of the work only, and provided the original author and source are credited. See https://creativecommons.org/licenses/by-nc-nd/4.0/.
References
- 1. Tofe AJ, Francis MD, Harvey WJ. Correlation of neoplasms with incidence and localization of skeletal metastases: an analysis of 1,355 diphosphonate bone scans. J Nucl Med. 1975;16(11):986–989. [PubMed] [Google Scholar]
- 2. Wu K, Hou S-M, Huang T-S, Yang R-S. Thyroid carcinoma with bone metastases: a prognostic factor study. Clin Med Oncol. 2008;2:CMO.S333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Badalian G, Barbai T, Rásó E, Derecskei K, Szendrôi M, Tímár J. Phenotype of bone metastases of non-small cell lung cancer: epidermal growth factor receptor expression and K-ras mutational status. Pathol Oncol Res. 2007;13(2):99–104. [DOI] [PubMed] [Google Scholar]
- 4. Krawczyk P, Nicoś M, Ramlau R, et al. The incidence of EGFR-activating mutations in bone metastases of lung adenocarcinoma. Pathol Oncol Res. 2014;20(1):107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. O'Hayre M, Vázquez-Prado J, Kufareva I, et al. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat Rev Cancer. 2013;13(6):412–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Scholz SL, Möller I, Reis H, et al. Frequent GNAQ, GNA11, and EIF1AX mutations in iris melanoma. Invest Ophthalmol Vis Sci. 2017;58(9):3464–3470. [DOI] [PubMed] [Google Scholar]
- 7. Sheng X, Kong Y, Li Y, et al. GNAQ and GNA11 mutations occur in 9.5% of mucosal melanoma and are associated with poor prognosis. Eur J Cancer. 2016;65:156–163. [DOI] [PubMed] [Google Scholar]
- 8. Jie X-. X, Zhang X-. Y, Xu C-. J. Epithelial-to-mesenchymal transition, circulating tumor cells and cancer metastasis: Mechanisms and clinical applications. Oncotarget. 2017;8(46):81558–81571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ansieau S, Bastid J, Doreau A, et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell. 2008;14(1):79–89. [DOI] [PubMed] [Google Scholar]
- 10. Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7):927–939. [DOI] [PubMed] [Google Scholar]
- 11. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–111. [DOI] [PubMed] [Google Scholar]
- 12. Vlashi E, Pajonk F. Cancer stem cells, cancer cell plasticity and radiation therapy. Semin Cancer Biol. 2015;31:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meirelles K, Benedict LA, Dombkowski D, et al. Human ovarian cancer stem/progenitor cells are stimulated by doxorubicin but inhibited by Mullerian inhibiting substance. Proc Natl Acad Sci U S A. 2012;109(7):2358–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Steinbichler TB, Dudás J, Skvortsov S, Ganswindt U, Riechelmann H, Skvortsova II. Therapy resistance mediated by cancer stem cells. Semin Cancer Biol. 2018;53:156–167. [DOI] [PubMed] [Google Scholar]
- 15. Shin H-T, Choi Y-L, Yun JW, et al. Prevalence and detection of low-allele-fraction variants in clinical cancer samples. Nat Commun. 2017;8(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Park G, Park JK, Son D-S, et al. Utility of targeted deep sequencing for detecting circulating tumor DNA in pancreatic cancer patients. Sci Rep. 2018;8(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Borowicz S, Van Scoyk M, Avasarala S, Rathinam MKK, Tauler J, Bikkavilli RK. The soft agar colony formation assay. J Vis Exp. 014;92:e51998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiawen S, Jianjun Z, Jiewen D, et al. Osteogenic differentiation of human amniotic epithelial cells and its application in alveolar defect restoration. Stem Cells Transl Med. 2014;3(12):1504–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Park J-R, Jung J-W, Lee Y-S, Kang K-S. The roles of Wnt antagonists DKK1 and sFRP4 during adipogenesis of human adipose tissue-derived mesenchymal stem cells. Cell Prolif. 2008;41(6):859–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee YS, Choi J-Y, Lee J, et al. TP53-dependence on the effect of doxorubicin and Src inhibitor combination therapy. Tumour Biol. 2018;40(8):101042831879421. [DOI] [PubMed] [Google Scholar]
- 21. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang Y, Xiao H, Wu H, et al. G protein subunit α q regulates gastric cancer growth via the p53/p21 and MEK/ERK pathways. Oncol Rep. 2017;37(4):1998–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun Y, Liu W-Z, Liu T, Feng X, Yang N, Zhou H-F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Signal Transduct Res. 2015;35(6):600–604. [DOI] [PubMed] [Google Scholar]
- 24. Jo DH, Kim JH, Cho CS, et al. Stat3 inhibition suppresses proliferation of retinoblastoma through down-regulation of positive feedback loop of STAT3/miR-17-92 clusters. Oncotarget. 2014;5(22):11513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mani SA, Guo W, Liao M-J, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Otsuki Y, Saya H, Arima Y. Prospects for new lung cancer treatments that target EMT signaling. Dev Dyn. 2018;247(3):462–472. [DOI] [PubMed] [Google Scholar]
- 27. Larue L, Bellacosa A. Epithelial-Mesenchymal transition in development and cancer: role of phosphatidylinositol 3' kinase/Akt pathways. Oncogene. 2005;24(50):7443–7454. [DOI] [PubMed] [Google Scholar]
- 28. Julien S, Puig I, Caretti E, et al. Activation of NF-kappaB by Akt upregulates snail expression and induces epithelium mesenchyme transition. Oncogene. 2007;26(53):7445–7456. [DOI] [PubMed] [Google Scholar]
- 29. Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–1920. [DOI] [PubMed] [Google Scholar]
- 30. Morath I, Hartmann TN, Orion-Rousseau V. Cd44: more than a mere stem cell marker. Int J Biochem Cell Biol. 2016;81(Pt A):166–173. [DOI] [PubMed] [Google Scholar]
- 31. Zhang N, Li R, Tao K-S, et al. Characterization of a stem-like population in hepatocellular carcinoma MHCC97 cells. Oncol Rep. 2010;23(3):827–831. [PubMed] [Google Scholar]
- 32. Yan C, Luo L, Guo C-Y, et al. Doxorubicin-Induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett. 2017;388:34–42. [DOI] [PubMed] [Google Scholar]
- 33. Dawood S, Austin L, Cristofanilli M. Cancer stem cells: implications for cancer therapy. Oncology. 2014;28(12):1101–1110. [PubMed] [Google Scholar]
- 34. Ajani JA, Song S, Hochster HS, Steinberg IB. Cancer stem cells: the promise and the potential. Semin Oncol. 2015;42 Suppl 1:S3–S17. [DOI] [PubMed] [Google Scholar]
- 35. Kim CY, Kim DW, Kim K, Curry J, Torres-Cabala C, Patel S. GNAQ mutation in a patient with metastatic mucosal melanoma. BMC Cancer. 2014;14:516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hendriks LEL, Smit EF, Vosse BAH, et al. Egfr mutated non-small cell lung cancer patients: more prone to development of bone and brain metastases? Lung Cancer. 2014;84(1):86–91. [DOI] [PubMed] [Google Scholar]
- 37. Guan J, Chen M, Xiao N, et al. Egfr mutations are associated with higher incidence of distant metastases and smaller tumor size in patients with non-small-cell lung cancer based on PET/CT scan. Med Oncol. 2016;33(1):1. [DOI] [PubMed] [Google Scholar]
- 38. Kemeny NE, Chou JF, Capanu M, et al. Kras mutation influences recurrence patterns in patients undergoing hepatic resection of colorectal metastases. Cancer. 2014;120(24):3965–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yaeger R, Cowell E, Chou JF, et al. Ras mutations affect pattern of metastatic spread and increase propensity for brain metastasis in colorectal cancer. Cancer. 2015;121(8):1195–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mukherjee D, Zhao J. The role of chemokine receptor CXCR4 in breast cancer metastasis. Am J Cancer Res. 2013;3(1):46. [PMC free article] [PubMed] [Google Scholar]
- 41. Valenti MT, Serafini P, Innamorati G, et al. Runx2 expression: a mesenchymal stem marker for cancer. Oncol Lett. 2016;12(5):4167–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Macedo F, Ladeira K, Pinho F, et al. Bone metastases: an overview. Oncol Rev. 2017;11(1):321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang M, Xia F, Wei Y, Wei X. Molecular mechanisms and clinical management of cancer bone metastasis. Bone Res. 2020;8(30):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Choi J-Y, Lee YS, Shim DM, Seo SW, Research J. Effect of GNAQ alteration on RANKL-induced osteoclastogenesis in human non-small-cell lung cancer. Bone Joint Res. 2020;9(1):29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Benayahu D, Akavia UD, Shur I. Differentiation of bone marrow stroma-derived mesenchymal cells. Curr Med Chem. 2007;14(2):173–179. [DOI] [PubMed] [Google Scholar]
- 46. Aubin JE. Bone stem cells. J Cell Biochem. 1998;72 Suppl 30-31(S30-31):73–82. [DOI] [PubMed] [Google Scholar]
- 47. Nakashima K, Zhou X, Kunkel G, et al. The novel zinc finger-containing transcription factor Osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108(1):17–29. [DOI] [PubMed] [Google Scholar]
- 48. Hu H, Hilton MJ, Tu X, Yu K, Ornitz DM, Long F. Sequential roles of hedgehog and Wnt signaling in osteoblast development. Development. 2005;132(1):49–60. [DOI] [PubMed] [Google Scholar]
- 49. Vermeulen M, Le Pesteur F, Gagnerault MC, Mary JY, Sainteny F, Lepault F. Role of adhesion molecules in the homing and mobilization of murine hematopoietic stem and progenitor cells. Blood. 1998;92(3):894–900. [PubMed] [Google Scholar]
- 50. Liesveld JL, Sharma N, Aljitawi OS. Stem cell homing: From physiology to therapeutics. Stem Cells. 2020;38(10):1241–1253. [DOI] [PubMed] [Google Scholar]
- 51. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2010;120(5):1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee J, Kim JCK, Lee S-E, et al. Signal transducer and activator of transcription 3 (STAT3) protein suppresses adenoma-to-carcinoma transition in ApcMin/+ mice via regulation of Snail-1 (SNAI) protein stability. J Biol Chem. 2012;287(22):M111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells - what challenges do they pose? Nat Rev Drug Discov. 2014;13(7):497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chang JC. Cancer stem cells. Medicine. 2016;95(Suppl 1):S20–S25. [DOI] [PMC free article] [PubMed] [Google Scholar]