

Abstract
Lysine-specific demethylase-1 (LSD1) is an epigenetic regulator of gene transcription. LSD1 risk allele in humans and LSD1 deficiency (LSD1+/−) in mice confer increasing salt-sensitivity of blood pressure (BP) with age, which evolves into salt-sensitive hypertension (HTN) in older individuals. However, the mechanism underlying the relationship between LSD1 and salt-sensitivity of BP remains elusive. Here, we show that LSD1 genotype (in humans) and LSD1 deficiency (in mice) lead to similar associations with increased BP and urine potassium levels, but with decreased aldosterone levels during a liberal salt diet. Thus, we hypothesized that LSD1 deficiency leads to an MR-dependent hypertensive state. Yet, further studies in LSD1+/−mice treated with the mineralocorticoid receptor (MR) antagonist eplerenone demonstrate that HTN, kaliuria and albuminuria are substantially improved, suggesting that the ligand-independent activation of the MR is the underlying cause of this LSD1 deficiency- mediated phenotype. Indeed, while MR and ENaC expression levels were increased in LSD1+/− mouse kidney tissues, aldosterone secretion from LSD1+/− glomerulosa cells was significantly lower. Collectively, these data establish that LSD1 deficiency leads to an inappropriate activation and increased levels of the MR during a liberal salt regimen and suggest that inhibiting the MR pathway is a useful strategy for treatment of HTN in human LSD1 risk allele carriers.
Keywords: aldosterone, hypertension, lysine-specific demethylase 1, mineralocorticoid receptor, salt-sensitive blood pressure
Graphical Abstract
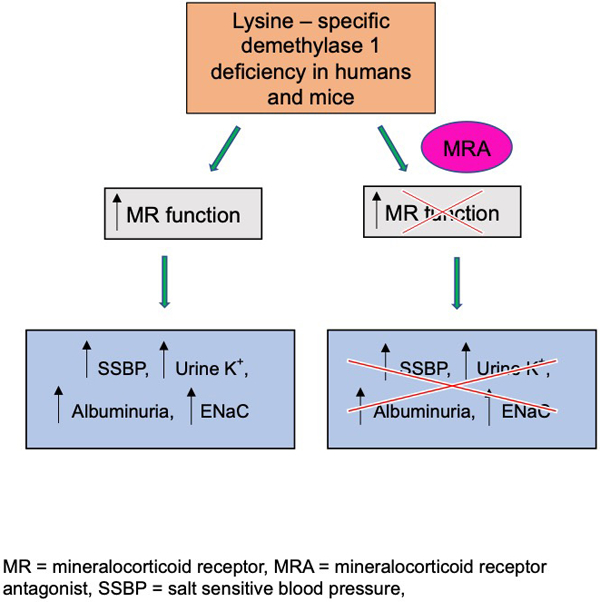
Summary
Increased BP levels and renal damage in LSD1+/− mice, and by inference in LSD1 African risk allele carriers, are mediated in part by an overactivation of MR independent of elevated ALDO levels.
Introduction
Hypertension (HTN) is a common condition that increases the risk for cardiovascular disease and stroke1, hence it is a major public health issue. Not only environmental factors such as liberal salt (LibS) intake2, but also genetic susceptibility3 lead to the development of HTN. There is now emerging evidence that epigenetic, as well as genetic factors are important in regulating and maintaining blood pressure (BP)4,5.
Lysine-specific demethylase-1 (LSD1), also known as KDM1A, is a flavin-dependent monoamine oxidase, which can demethylate mono- and di-methylated lysines, specifically histone 3, lysine 4 and 9 (H3K4 and H3K9)6. Because LSD1 modifies histone structure and is modulated by sodium intake7, we reasoned that its level may influence the BP response to dietary sodium. This was documented in both LSD1 heterozygote knockout mice (LSD1+/−) and human carriers of an LSD1 risk allele associated with decreased LSD1 expression in silico: both had hypertension with salt sensitivity of the BP (SSBP). Yet, in these older mice, SSBP was not associated with increased ALDO levels. However, young mice did have SSBP accompanied by increased ALDO levels on a LibS diet; interestingly, as they aged, the SSBP became greater but the ALDO levels were reduced in these animals. This provided entrée for the current study to test the hypothesis that LSD1 deficiency is associated with a dysregulation of ALDO’s homeostatic mechanism: initially secondary to increased ALDO levels and with time increased activity of the mineralocorticoid receptor (MR). To assess this hypothesis, we treated both WT and LSD1+/− with the MR antagonist (MRA) eplerenone (Epl) and assessed the effect of the MRA on several aldosterone/MR mediated processes.
Methods
Subjects in this study were consented well before the development of guidelines to promote openness. Therefore, requests for select deidentified study data and analytic methods will be considered on a case-by-case basis from qualified researchers with institutional review board approvals and executed institutional data transfer agreements. The dataset will be available from Dr. Gordon H. Williams, Principal Investigator for the HyperPATH consortium, by email request (gwilliams@bwh.harvard.edu).
The animal data that support the findings of this study are available from the corresponding author upon reasonable request.
Human Studies.
The subjects included in this study were part of the Genetics of Hypertension Pathophysiology (HyperPATH) cohort. The HyperPATH consortium was developed to characterize the genetic underpinnings of HTN. Its strengths include an enhanced signal-to-noise ratio through rigorous control of factors that influence BP including medication washout, body positioning, diurnal variation, and dietary salt. Three consecutive readings were averaged for analysis. Salt sensitivity of BP was recorded as a continuous variable representing the change in systolic BP (SBP) in response to dietary salt (Delta SBP)8. See additional details provided in the Online Data Supplement (Methods). We have previously reported, in hypertensive of African descent, that minor allele carriers of the LSD1 single nucleotide polymorphism (SNP) rs587168 display increased BP sensitivity to dietary salt intake7.
HyperPATH Sample Analyses.
All HyperPATH samples were analyzed at a central laboratory. Samples were collected on ice and centrifuged for 20 minutes. Serum and urine specimens were stored at −20°C without preservatives until assayed. Na+ and K+ levels were measured by flame photometry. ALDO levels were measured using Coat-A-Count Radioimmunometric Assay (RIA) kit (SIEMENS, Los Angeles, CA); PRA by RIA assay (DiaSorin, Stillwater, MN). A subset of subjects of African descent (n=63) also completed an assessment of effective renal blood flow (RBF) by para-aminohippuric acid clearance method9.
Animal Studies.
In this study, adult male WT and LSD1+/− mice aged 31–35 weeks old were used. The rationale for using male mice is that female LSD1+/− mice did not display increased SBP on a 1.6% Na+ diet10. WT and LSD1+/− mice were fed a 1.6% Na+ diet for three weeks; after one week animals were randomized to placebo or Epl treatment (100 mg/kg/day) for an additional two weeks. Data was obtained after the mice were in salt balance.
Blood Pressure Measurements in Mice.
With appropriate environmental controls, systolic blood pressure (SBP) was measured in conscious mice before randomization (day 7) and at the end of treatment period (day 21) using tail-cuff plethysmography and CODA noninvasive BP system (KENT Scientific Corporation, Torrington, CT). Animals were first trained for a week. We have previously demonstrated excellent correlation between SBPs assessed simultaneously by tail cuff and telemetry in mice11. The change in SBP (∆SBP) was calculated in each animal as SBP after treatment minus SBP before treatment.
Mouse Urine and Plasma Analyses
Mouse Urine and Plasma Analyses are described in the Online Data Supplement (Methods).
Mouse Zona Glomerulosa (ZG) Cell Stimulation.
Adrenal glands were excised during euthanasia, and ZG cells were isolated as previously reported12, 13. The details are provided in the Online Data Supplement (Methods).
Mouse Tissue Transcript Analysis.
Total mRNA was extracted from the kidneys using the RNeasy mini-kit (Qiagen Sciences, Germantown, MD) as previously described14, 15. cDNA was synthesized from 1.5µg RNA with the first-strand cDNA synthesis kit (GE healthcare, Piscataway, NJ). PCR amplification reactions were performed in duplicate using the ABI Prism 7000 sequence detection system (Thermo Fisher Scientific, Waltham, MA) and the ∆∆CT method to determine mRNA levels. PCR amplification was performed with TaqMan gene expression assays for MR, epithelial sodium channel (ENaC) subunits α-ENaC and γ-ENaC,11β-hydroxysteroid dehydrogenase 2 (11β-HSD2), and LSD1. Data are presented as fold increase relative to the measurements in WT mice.
Statistics
A detailed statistical analysis plan is provided in the Online Data Supplement (Methods).
Results
LSD1 minor allele carriers of African descent have increased salt-sensitivity of systolic blood pressure, lower ALDO levels, yet higher urine K+ excretion
Baseline characteristics of the study population are shown, by rs587168 genotype, in Table S1. The cohort consisted of 96 individuals of African descent:48% homozygous major allele (CC) carriers; 43% heterozygous (AC) carriers; and 9% homozygous minor allele (AA) carriers; the genotypes were in Hardy-Weinberg equilibrium (P=0.98). The distribution of these characteristics did not differ significantly by rs587168 genotype as shown in Online Data Supplement (Table S1). Minor allele carriers displayed a tendency to lower PAH clearance on a LibS diet.
We assessed the relationship of genotype to change in BP to dietary salt intake (Delta SBP), plasma ALDO and urine and serum K+ levels (Figure 1). There was a significant allele dose trend in the Delta SBP (P=0.024), consistent with our previously published results7. Surprisingly, on a LibS diet there was a significant negative allele dose trend in plasma ALDO levels (P=0.03), but a positive allele dose trend in urine K+ levels (P=0.036). No significant differences were observed in serum K+ and cortisol and plasma renin activity levels on LibS diet between genotypes as shown in Online Data Supplement (Table S1.)
Figure 1. Delta systolic blood pressure (SBP), Aldosterone (ALDO) and potassium (K+) levels in the HyperPATH individuals of African descent.
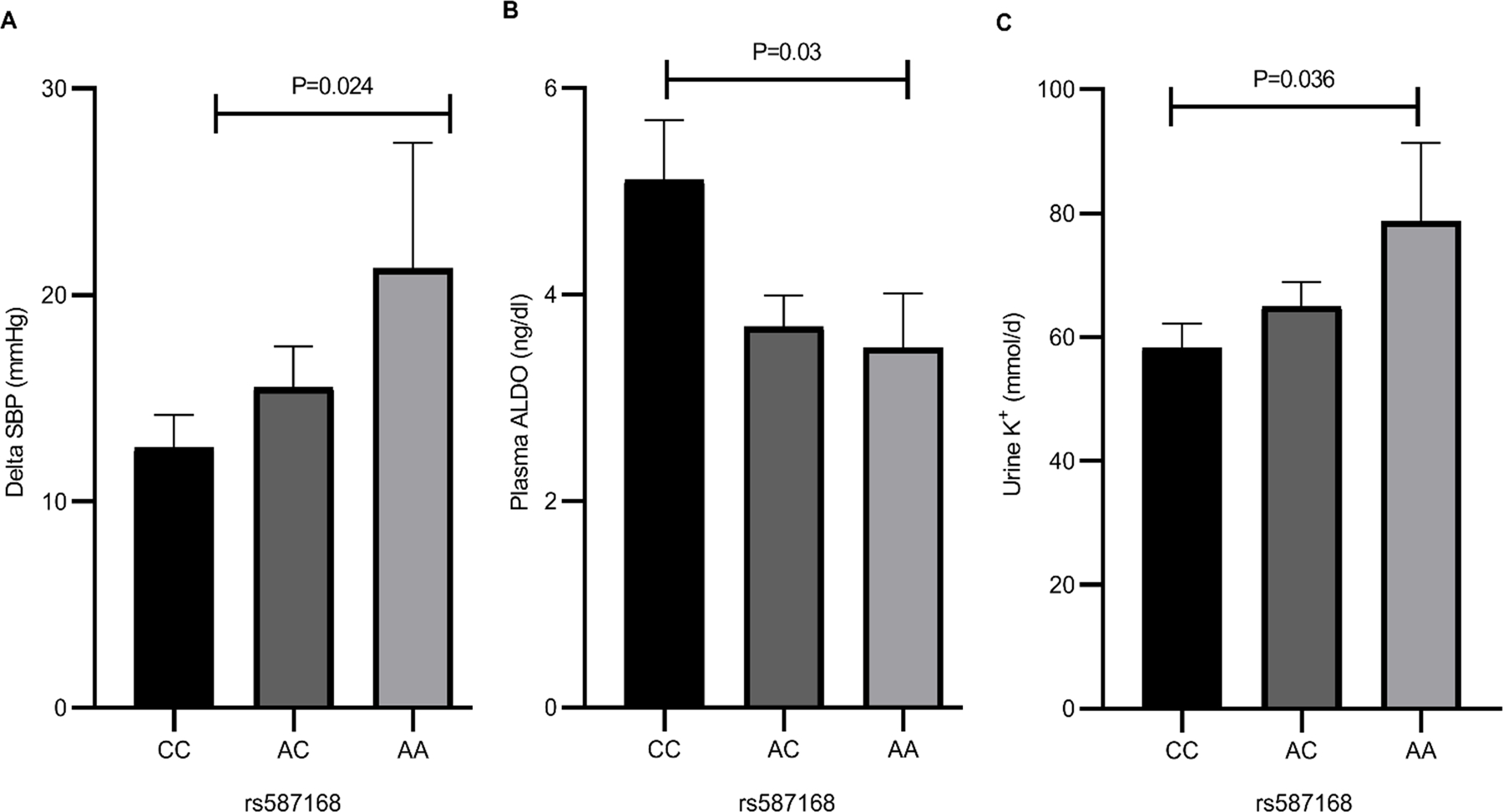
Comparison in LSD1 rs587168 CC (n=46), AC (n=41) and AA (n=9) genotypes of change in SBP in response to dietary salt (A), plasma ALDO (B), and urine K+ (C) on a LibS diet. P-value was determined from multiple linear regression models, adjusting for sex, age, BMI, disease state and study sites. Data are mean ± SEM.
Baseline characteristics in LSD1+/− vs. WT mice.
Body weights were higher in placebo-treated LSD1+/− as compared with placebo-treated WT (34.2 ± 1.8 g vs. 28.9 ± 0.7 g, P = 0.009)(Table S2); however, treatment with Epl ablated this difference between the two genotypes. Heart weights were similar in the two genotype groups, irrespective of the treatment (Table S2). In contrast, heart/body weight ratios were significantly lower in the placebo-treated LSD1+/− compared with WT (3.8 ± 0.1 mg/g vs. 4.6 ±0.2 mg/g, P=0.003) (Table S2), likely due to the higher body weights in the LSD1+/−; this difference between the genotypes was absent in the Epl treatment groups. As compared to the placebo-treated WT, kidney weigths in the placebo-treated LSD1+/− mice were significantly higher (P=0.03 and P=0.04 for left and right kidney weights, respectively). Treatment with Epl significantly decreased the kidney weights in the LSD1+/− but not in the WT. The kidney to body weight ratios were similar in the four genotype/treatment groups (Table S2).
LSD1+/− mice have increased systolic blood pressures which were partially yet significantly lowered by Epl treatment
Before the intervention, LSD1+/− mice had higher SBP than WT (132 ± 2.6 mmHg vs. 113.8 ± 2.0 mmHg, P<0.001). This difference was maintained post randomization, in the placebo-treated groups (Figure 2A). Treatment with Epl induced a significant SBP decrease in LSD1+/− ( LSD1+/−+ Epl 120.8 ± 1.6 mmHg vs. LSD1+/− + placebo : 134.1± 4.3 mmHg, P=0.02), but not in WT mice. (Figure 2A.). The change between SBP before and after treatment was significantly greater in Epl-treated vs. placebo-treated LSD1+/− mice ( LSD1+/−+ Epl: −5.1 ± 2.2 mmHg vs. LSD1+/− + placebo: 1.5 ± 1.7 mmHg, P=0.02). (Figure 2B.). However, the BP levels were still higher in the LSD1+/− + Epl vs. WT + Epl mice. These data suggest that MR overactivation may be, at least in part, the cause for the higher SBP in LSD1 deficiency.
Figure 2.
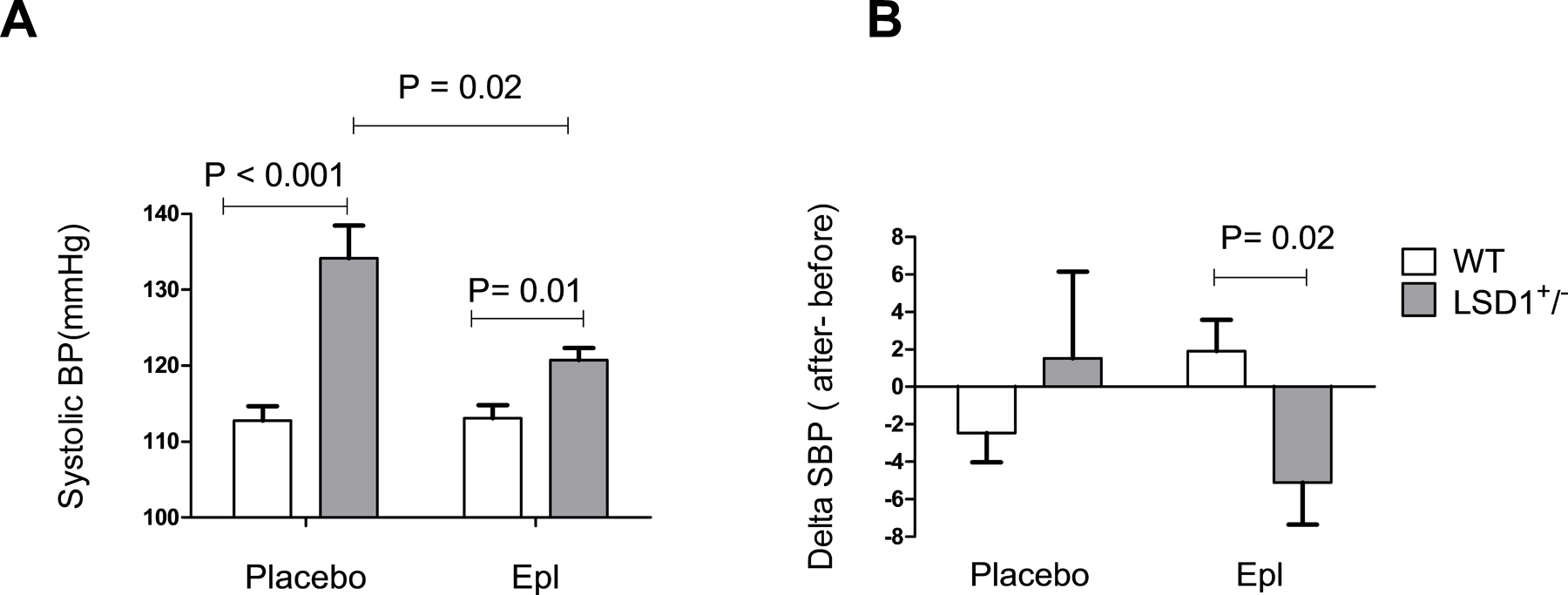
Effect of placebo and eplerenone (Epl) treatment on blood pressure in WT and LSD1+/− mice. LSD1+/− mice have higher systolic blood pressure (SBP) (A). SBP were measured at the end of the study by tail-cuff measurements. LSD1+/− mice display a greater change in SBP in response to eplerenone treatment (B). Data are Mean ± SEM. WT n=22, LSD1+/− n=18, P-value shows two-tailed Student’s t-test.
Eplerenone treatment completely reverses the kidney damage in LSD1+/− mice
Given the higher BP in the LSD1+/−mice, we next inquired on the potential damaging effects of BP on the kidney, as assessed by A/C ratios. As compared to WT, LSD1+/− mice displayed significantly higher A/C ratios (68.0 ± 23.6 µg/mg vs. 22.1 ± 1.6 µg/mg, P=0.04), and treatment with Epl reduced A/C in LSD1+/− mice to levels indistinguishable from those in the WT (LSD1+/− + Epl: 27.3 ± 2.9 µg/mg vs. WT+ Epl: 26.0 ± 1.7 µg/mg) (Figure 3A). These results suggest that kidney damage in LSD1 haploinsufficiency is completely reliant on MR overactivation. Interestingly, the A/C ratios were significantly correlated with SBP levels in the LSD1+/− (Figure 3B), but not in the WT animals (Figure 3C), suggesting that LSD1 haploinsufficiency may be a mechanism underlying BP-mediated kidney damage.
Figure 3.
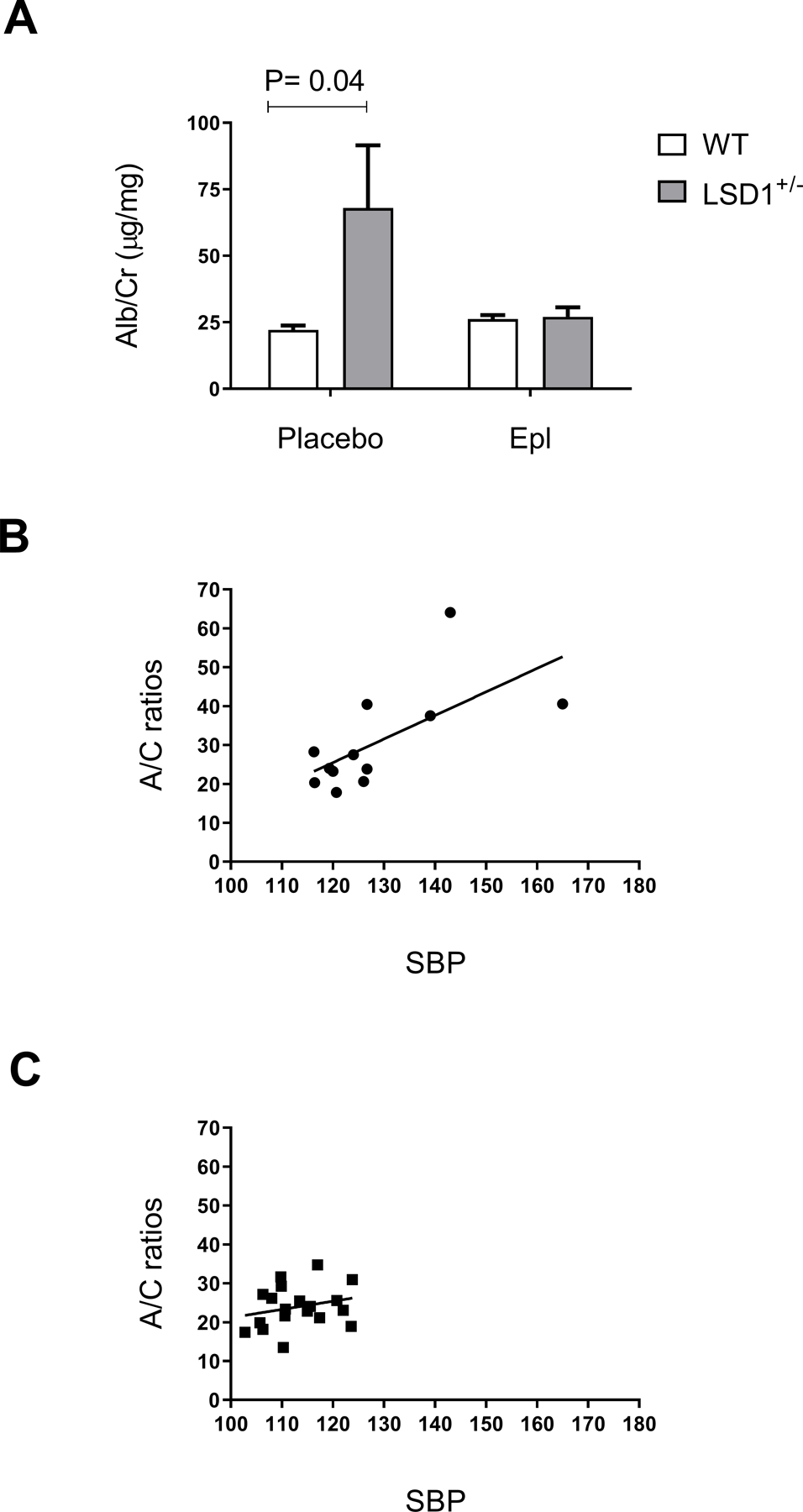
LSD1+/− mice display higher Albumin/Creatinine (A/C) ratios in the placebo-treated group (A). The A/C ratios were significantly correlated with systolic blood pressure (SBP) levels in the LSD1+/− (B, P= 0.02, R = 0.65) but not in the WT (C, P= 0.3, R = 0.25). Data are Mean ± SEM. P-value shows two-tailed Student’s t-test.
LSD1+/− mice have suppressed urine and plasma ALDO levels but higher K+ excretion
To determine the underlying cause for MR overactivation in the LSD1+/−, we next measured urine and plasma ALDO levels as well as urine K+ excretion. Consistent with our previously published results7,14, LSD1+/− mice had significantly suppressed ALDO excretion as compared to WT (26.1 ± 4.0 pg/mg vs. 39.4 ± 4.1 pg/mg, P=0.03) (Figure 4A.). A similar trend was observed for plasma ALDO levels but did not reach signficance (LSD1+/− : 21.8 ± 5.1 ng/dl vs. WT: 33.5 ± 6.1 ng/dl, P=0.17) (Figure 4B). As expected, ALDO excretion was significantly increased by treatment with Epl in both the WT (100.7 ± 21.3 pg/mg vs. 39.4 ± 4.1 pg/mg, P=0.01) and LSD1+/− mice (59.1 ± 11.5 pg/mg vs. 26.1 ± 4.0 pg/mg, P=0.01). Plasma ALDO was also increased significantly by treatment with Epl in the WT (33.5 ± 6.1 vs. 59.5 ± 9.8 ng/dl, P=0.04),but did not reach the significance level in the LSD1+/− with Epl treatment (30.3 ± 9.5 ng/dl vs. 21.8 ± 5.1 ng/dl, P = 0.43). Despite the lower ALDO levels, LSD1+/− mice had higher 24-hr urine K+ excretion as compared with WT mice (0.29 ± 0.1 mmol vs. 0.15 ± 0.02 mmol, P= 0.05) (Figure 4C.), in agreement with the effect of the LSD1 variant allele on urine K+ in humans (Figure 1). Interestingly, treatment with Epl decreased urine K+ to levels similar to those in the WT mice (LSD1+/− + Epl: 0.19 ± 0.03 mmol vs. WT+ Epl: 0.18 ± 0.02 mmol). No significant differences were observed between 24-hr urine Na+ excretion in LSD1+/− and WT mice (0.29 ± 0.1 mmol vs. 0.27 ± 0.04 mmol, P=0.72) (Table S2.). Consistent with the above, the urine ALDO/K+ was significantly decreased in LSD1+/− compared with WT mice (36.2 ± 7.4 pg/mmol vs. 69.9 ± 10.83 pg/mmol, P= 0.03), and treatment with Epl increased this ratio in both genotpye groups, to levels indistinguishable from one another (Figure 4D). Together, these findings are consistent with an ALDO-independent overactivation of the kidney MR in the LSD1+/−, leading to dysregulation in the Na+- K+ homeostasis.
Figure 4.
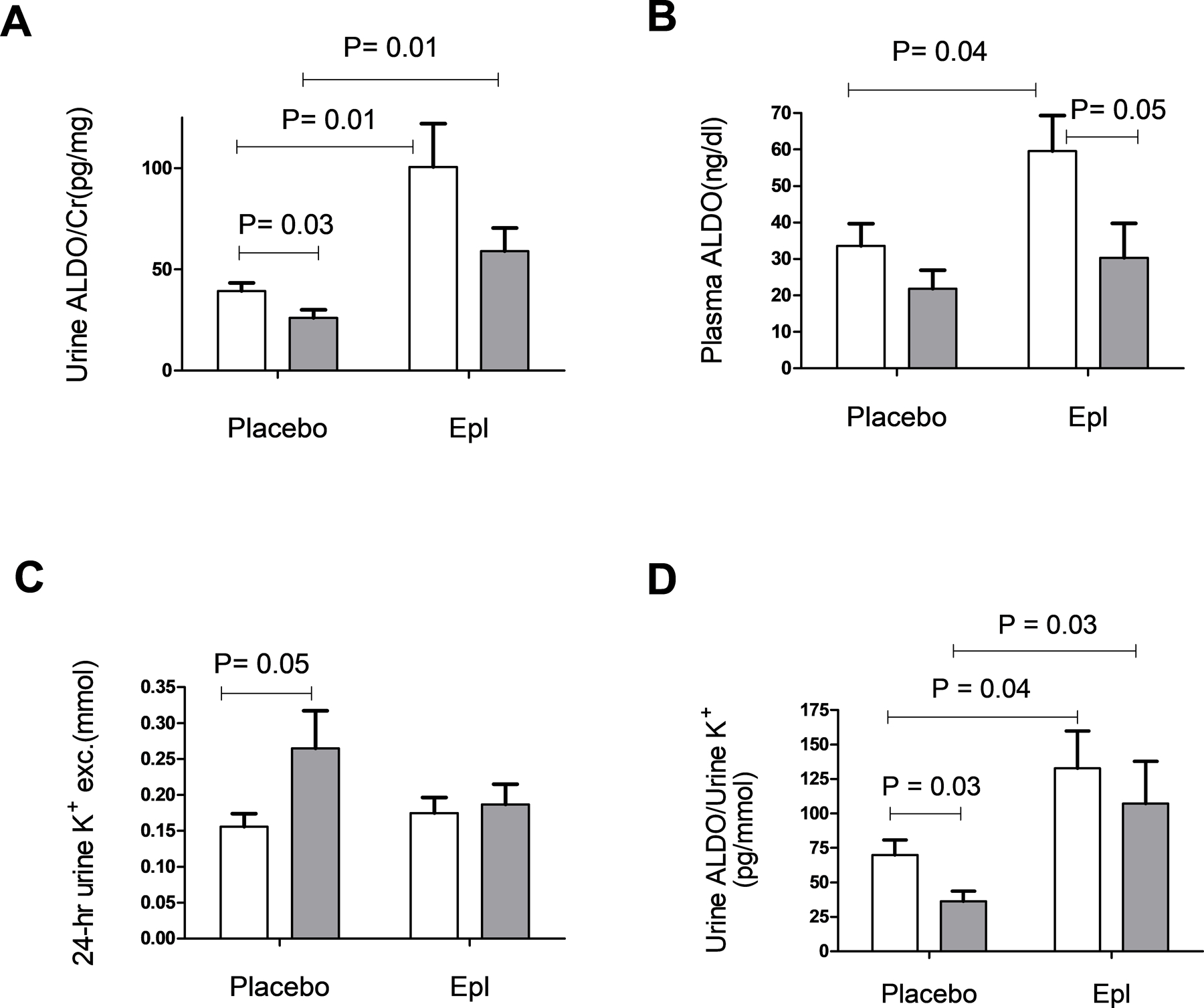
Aldosterone (ALDO) levels and 24-hr urine potassium (K+) in WT and LSD1+/− mice. LSD1+/− mice display suppressed ALDO excretion, reported as urine ALDO/Creatinine (Cr) ratio (A) and suppressed plasma ALDO (B). LSD1+/− mice display higher 24-hr urine K+ in placebo-treated group (C) and lower urine ALDO/K+ (D). Data are mean ± SEM. WT n=17–19, LSD1+/− n=13–15, P-value shows two-tailed Student’s t-test.
LSD1 haploinsufficiency associates with exaggerated MR activation in adrenal ZG cells.
To confirm that the decreased urine and plasma ALDO levels in the LSD1+/− mice are due to a suppressed ALDO secretion from the adrenal, we then measured ALDO secretion in acutely isolated ZG cells. Indeed, ALDO release was significantly lower in ZG cells from LSD1+/− vs. WT mice at either baseline (P=0.04) (Figure 5A) or in response to AngII (P=0.02) or ACTH stimulation (P=0.03) (Figure 5B–C). Treatment with Epl did not significantly modulate the ALDO secretion from the WT ZG cells, but it significantly increased ALDO release from LSD1+/− cells, both at baseline (P=0.0005) or in response to stimulation (ANG II P=0.02; ACTH P=0.04) (Figure 5A–C). These data suggest a hypersensitivity to MR blockade, and are consistent with an overactivation of the MR-mediated ultrashort feedback loop12 in the adrenal.
Figure 5.
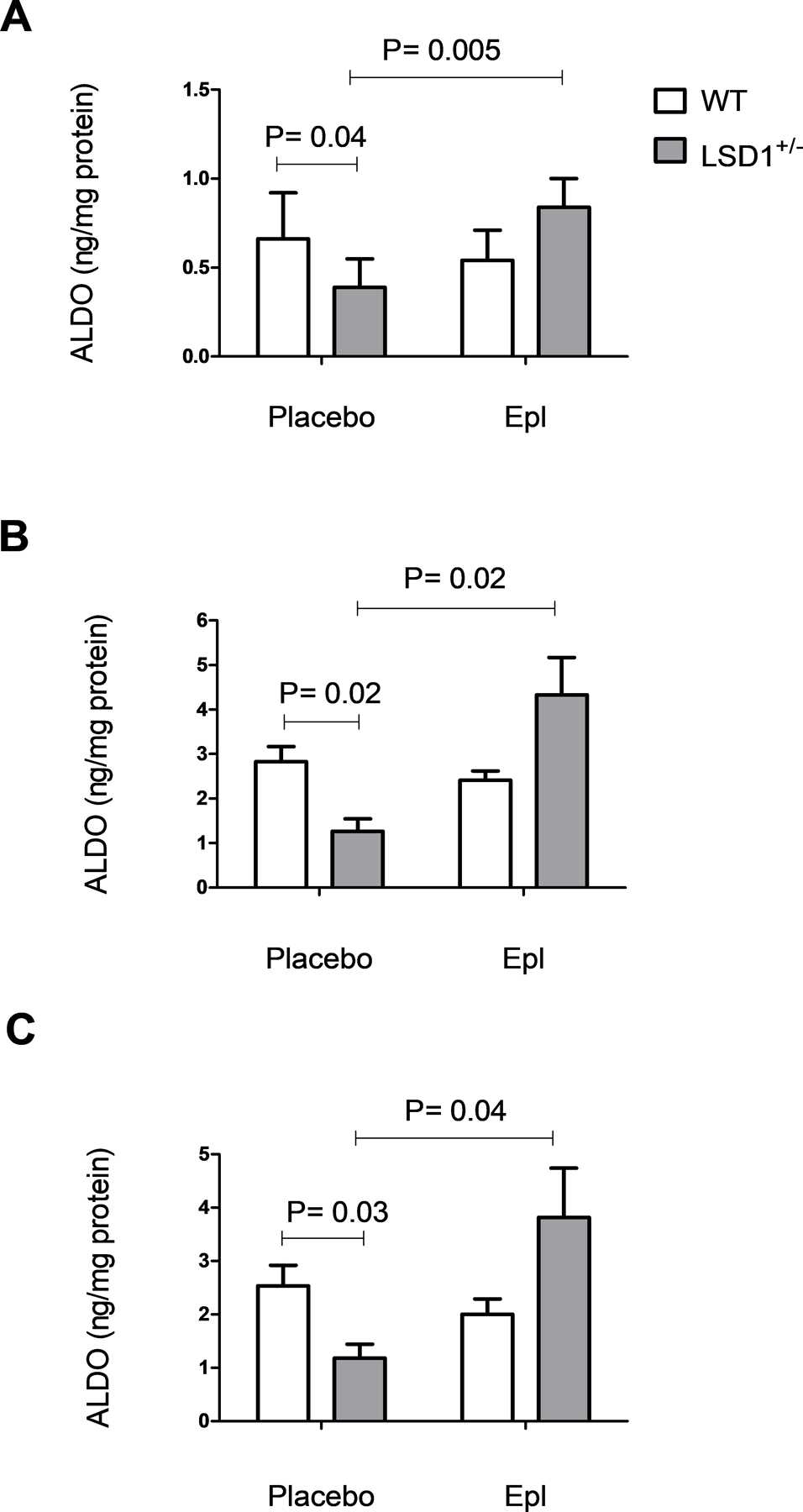
Aldosterone (ALDO) secretion from Zona Glomerulosa (ZG) cells is suppressed in LSD1 deficiency and restored by treatment with eplerenone (Epl). Acutely isolated ZG cells from placebo- or Epl-treated mice were incubated with vehicle (A), 10−8M AngII (B) or 10−10M ACTH (C) as described in the Methods section. P-value shows two-tailed Student’s t-test.
Overactivation of the MR pathway in the LSD1+/− kidney is due to increased MR levels.
To test whether the MR is overexpressed in LSD1 deficiency, we performed real time RT-PCR in renal cortex samples isolated from WT and LSD1+/− mice. Indeed, LSD1+/− mice had significant higher MR mRNA levels in kidney compared to WT (P=0.03)(Figure 6A). Furthermore, the downstream MR effectors α-ENaC and γ-ENaC were also upregulated in the LSD1+/− vs. WT samples (P< 0.05)(Figure 6B–C). As the MR specificity for ALDO in the kidney is dictated by the 11β-HSD2 enzyme (which oxidates corticosterone to 11-dehydrocorticosterone), we assessed its transcript levels in the same renal tissues. LSD1+/− mice displayed slightly lower levels of 11β-HSD2 vs WT (not shown), but this difference did not reach signficance (0.80 ± 0.20 vs. 1.00 ± 0.28, P=0.54). Furthermore, 24-hr urine corticosterone levels were also similar in the two genotype groups (WT: 38.7 ± 6.76 ng/mL; LSD1+/−: 37.9 ± 5.04 ng/mL+, P=0.94). As expected, LSD1 transcript levels were signficantly decreased in kidney samples from LSD1+/− vs. WT mice (P=0.03)(Figure 6D).
Figure 6.
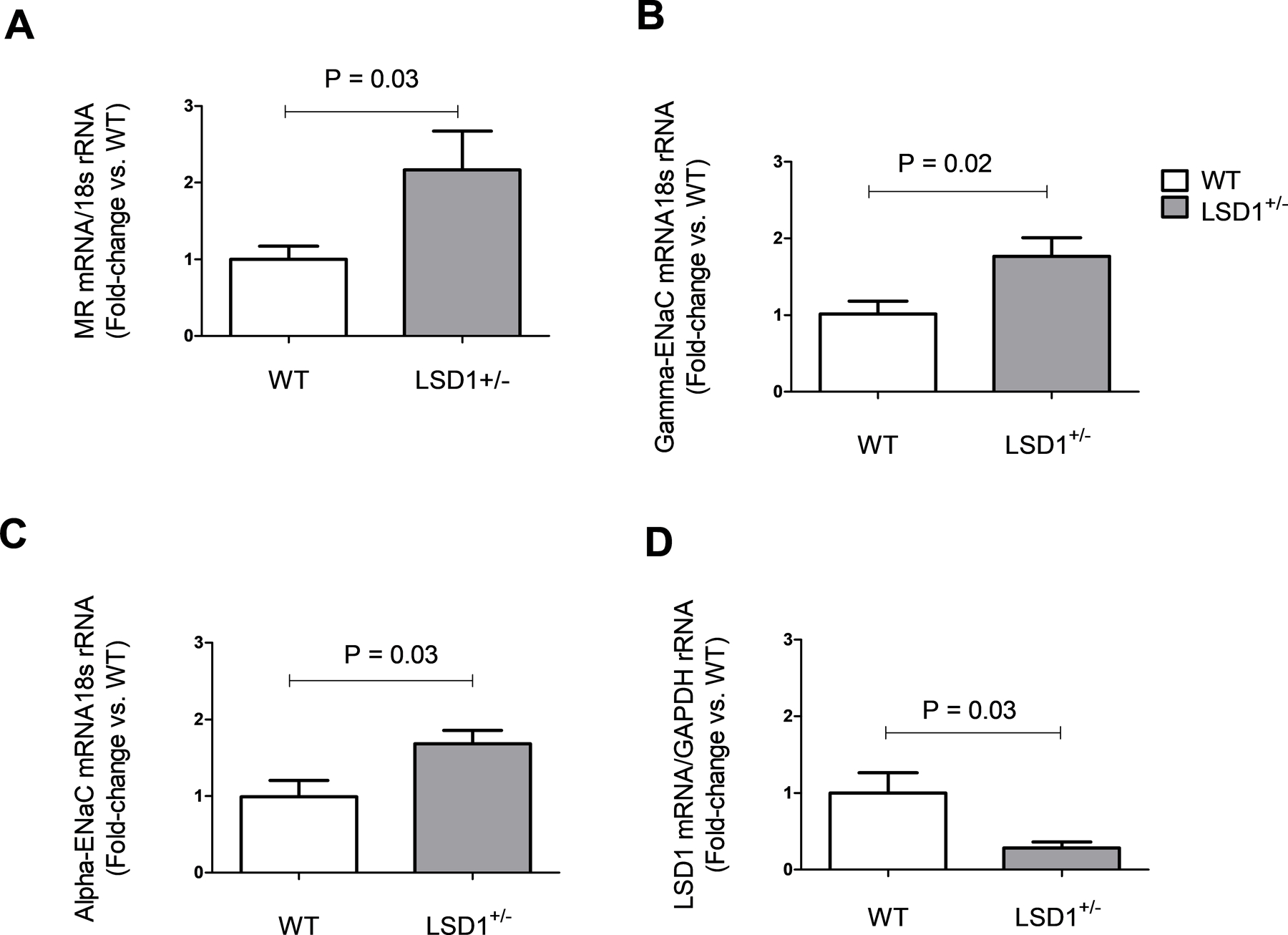
LSD1 deficiency associates with alterations in kidney transcript levels. Renal cortex mRNA expression for MR (A), gamma and alpha-ENaC (B, C) are significantly increased in LSD1+/− mice compared to WT. LSD1 levels are reduced in LSD1+/− mice compared to WT (D). Assessments were performed by RT PCR in kidney cortex tissues (WT n=8, LSD1+/− n=6). Data are mean ± SEM. P-value shows two-tailed Student’s t-test.
Discussion
Our results document a similar association between SSBP ALDO and urine K+ levels on a LibS diet and either the LSD1 risk allele in humans or LSD1 deficiency in mice, thus supporting our posit that the LSD1+/− mouse is a good model for the human disease. In addition, we documented a significant increase in albuminuria in LSD1+/− mice. To determine the mechanism(s) that may explain these features common to both mouse and human genetically defined groups, we performed additional studies in the LSD1+/− mice. The primary tool used was MR blockade. The results support our working hypothesis: MR overactivation is a critical mechanism underlying this phenotype. Compared to WT, Epl treatment in LSD1+/− mice substantially reduced SBP, urine K+ and albuminuria, suggesting that these defects were mediated by ALDO. However, this effect appears to rely on ligand-independent activation of the MR, as ALDO levels were suppressed. Support for this possibility came from two additional findings: as compared to WT, LSD1+/− mice has increased renal MR and ENaC expression, but decreased ALDO secretion from ZG cells.
We previously reported that LSD1 deficiency in mice leads to an age-depedent progression from salt-sensitivity of BP in the young, to overt salt-sensitive HTN in old animals10,14,16, suggesting a defect in BP homeostasis during Na+ loading14,16. The data herein support these previous findings. Furthermore, treatment with Epl for two weeks induced a signficant drop in BP in the LSD1+/− animals, yet the levels were still significantly elevated as compared to WT. Thus, LSD1 deficiency is responsible for the inappropriate activation of the MR during LibS diet, which in turn leads to dysregulation – at least in part – of the Na+-depedent BP homeostasis. These data are in agreement with previous studies that demonstrate a direct effect of increased MR signaling on cardiovascular readouts17, 18.
We used as a readout for ALDO-mediated renal damage, microalbuminuria. Compared to WT, LSD1+/− mice displayed heavier kidneys and increased A/C ratios, which correlated with the BP readings. Epl completely reversed these effects, suggesting that the renal phenotype in LSD1 haploinsufficiency relies on MR overactivation. Indeed, numerous clinical and preclinical studies have demonstrated that the MR pathway plays a crucial role in renal injury19. Thus, MR overexpression in mice was shown to induce renal anatomical and functional abnormalities21, while MR KO mice are protected against age-related increases in SBP and against AngII-mediated vasoconstriction20. We and others have shown that MR blockade prevents or reduces ALDO-mediated renal damage, independently of BP effects11, 19, 21,22. The current study cannot rule out this possibility, as the beneficial effect of Epl on the kidney may be mediated, in part, by its effect on BP. Indeed, our data show a strong correlation between SBP and A/C ratios. Whether independent of each other or not, the effect of MR blockade on both BP and albuminuria is strongly suggestive of an inappropriate activation of the MR signaling pathway in LSD1+/− animals. Such overactivation could be mediated by either increased levels of its cognate ligand ALDO, increased levels of alternate ligands (e.g. corticosterone), increased levels of the receptor itself, or by activation of the downstream MR signaling (e.g. the small GTPase Rac1).
The primary mechanism underlying the above mentioned phenotype is unlikely to be due to excess ALDO, as urine and plasma ALDO levels were reduced in LSD1+/− mice, consistent with the expected RAAS suppression in response to BP elevation. Consistently, we had previously reported that older LSD1+/− animals displayed suppressed ALDO levels as compared to age-matched WT10, 14. Furthermore, the increased urine K+ and decreased urine ALDO/K+ ratios in the LSD1+/− animals are consistent with the overactivation of ENaC function23, the primary effector of MR signaling in the kidney, with normalization of these levels when Epl was given. The beneficial effect of Epl in the absence of elevated ALDO levels is not unique to LSD1 deficiency; indeed, MRA have been shown to produce significant improvement even when ALDO levels are not elevated23–27.
Could the MR be activated by glucocorticoids rather than ALDO in LSD1+/− animals? Our results suggest this is unlikely. Urine corticosterone and 11β-HSD2 trascript levels were not different between LSD1+/− and WT mice. However, we speculate that the LSD1 deficiency-mediated phenotype observed relies on ligand-independent MR activation. Further, changes in 11β-HSD2 methylation pattern have been associated with changes in its activity28. Since LSD1 is an epigenetic modulator, although unlikely, we cannot exclude the possibility of corticosterone or one of its metabolites activating the MR in our study.
MR activation has been shown to occur even in the absence of ligand binding, via the Rac1 – a member of the Rho family of small GTPases, which regulates many cellular processes29. Such activation may occur in the context of oxidative stress30, salt loading31, and cardiac pressure overload32. However, further studies are needed to establish the Rac1-mediated molecular mechanisms of MR overactivity, and how LSD1 may impact those pathways.
We herein report significantly higher MR transcript levels in kidney cortex tissues from LSD1+/− vs. WT animals. Thus, even in the presence of reduced ligand concentrations, excess MR levels may be sufficient to induce cardiovascular and renal dysfunction via exaggerated activation of MR downstream signaling, as previously shown in transgenic mice overexpressing hMR18. Indeed, our results show that both α and γ-subunits of the ENaC – some of the main downstream effectors of the MR pathway in the kidney – were significantly upregulated in these animals, consistent with MR hypersensitivity withLSD1 deficiency. In normal rodents, renocortical α-ENaC and γ-ENaC is downregulated by sodium loading (low ALDO state) and upregulated by administrering ALDO, leading to increased channel expression at the cell surface, hyperkaliuria and decreased urine ALDO/K+ ratios although alteration in these relationships have been reported in genetically altered rodents.23, 33. Intriguingly, our previous report showed that in aged mice, LSD1 deficiency associated with decreased α-ENaC protein expression in the kidney7. Several factors may contribute to the apparent discrepancy relative to the data presented herein: first, the LSD1+/− mice investigated in the current work were signficantly younger than those in our earlier work7. Indeed, it has been reported that both benzamil responsiveness and ENaC subunit abundance was signficantly decreased with age34, an effect that may be enhanced/accelerated in LSD1 deficiency. Second, the aged LSD1+/− animals displayed extreme HTN7, 14, which led to the expected suppression of the RAAS and may have also induced a secondary mechanism to prevent Na+ retention: downregulation of ENaC. Third, there may be significant differences between ENaC subunit mRNA and protein levels, as well as ENaC subunit availability, as a result of cleavage. Further studies will be needed to completely address these possibilites.
We have recently shown that the MR regulates ALDO production in the ZG via a negative feedback mechanism, with acute MR stimulation suppressing ALDO secretion from the ZG cells 12. Based on the data in this report, the apparent dissociation between ALDO and MR levels can be reconciled. Our results herein show that the LSD1+/− mouse (where MR is overactivated) displays significant suppression of ALDO production by the ZG, results that are mirrored by the circulating and 24-hr urine ALDOs. Furthermore, our prior studies have shown that acute MRA positively regulates steroidogenesis12. However, no studies have assessed the effect of chronic MR blockade on the ultrashort feedback loop in the ZG. Our current report sheds additional light on this matter. Chronic treatment with Epl significantly increased ALDO release from isolated ZG cells in the LSD1+/−, at baseline and in response to secretagogues, an effect which persists even when the cells are acutely removed from the influence of circulating factors, be they endogenous (e.g. hormones) or exogenous (Epl). In contrast, ALDO secretion was not different in ZG cells from WT chronically treated with MRA or placebo, suggesting the possibility of compensatory mechanisms that are intact in the WT, but modified in the LSD1+/−adrenals. Thus, the increased MR activity would result in both MR mediated SSBP and hypertension in the kidney and potentially the cardiovascular system and suppression of ALDO production via an MR mediated ZG cell ultrashort feedback loop.
There are several potential clinically relevant facets of our findings. For example, based on our findings in humans, previously published7 or reported herein one would assume that African carriers of the LSD1 risk allele would not be candidates for MRA therapy because they had low ALDO levels. Based on the pre-clinical findings in this study, MRAs may be the most appropriate therapy for them. Secondly, the presence of an activated MR with little evidence that the mechanism is secondary to classical MR modulators, e.g., ALDO and corticosterone (cortisol in humans), provides entrée to identifying another mechanism that – if effectively targeted – may provide a treatment that would not have the side effects associated with MRAs.
Limitations of our study include the absence of plasma renin activity (an indicator of volume status), serum K+ levels measurements and the absence of a control arm using an antihypertensive agent that would not modify ALDO or MR levels. However, the LibS diet and the effect of MRAs on urine K+ levels likely minimized these limitations. Further, in the human phase of this study, both were measured and did not reveal any additional potential mechanistic possibilities. Second, we do not have data in human carriers of the LSD1 variant treated with Epl, to directly compare with our mouse model. Third, we do not know if Epl will have the same effect in LSD1+/− female mice as it did in males. However, in our previous publication, female LSD1+/− mice did not have SSBP at a time when males did. Therefore, the phenotypes are different for uncertain reasons. Fourth, there may be other mechanisms modified by LSD1 deficiency, in addition to an activated MR that could contribute to our findings. For example, LSD1 deficiency could modify renovascular function, which was not assessed in this study. Fifth, the molecular mechanism by which LSD1 deficiency induces overactivation of the MR pathway is unclear. Given the LSD1 role as a regulator of gene transcription by modulating methylation states at H3K4 and H3K935, to the extent that LSD1 directly modulates gene expression in the MR pathway, we can speculate that it does so by acting as a transcription repressor at H3K4 for the MR gene, or for a gene that can stimulate its expression (e.g. Rac1). Thus, in cases of LSD1 deficiency, this repressor mechanism may be unable to keep MR transcription under the tight control it does in the WT. Finally, due to the complexity of the effect of age on biologic readouts, we do not know if the increased ALDO in the young LSD1+/− mice10 could have induced a memory effect on the altered MR levels in older mice. Thus, future studies need to assess: 1) the potential interaction between LSD1 deficiency and MR signaling in various tissues, including the kidney and the adrenal – two tissues in which our current data postulates we may see this effect; 2) the effect of inhibiting the increased ALDO production or action in the young mice on the development of the molecular and pathophysiologic phenotype; and 3) the role salt intake, per se, plays in the development of the age-related HTN and associated target organ damage in LSD1 deficiency.
In conclusion, our results support the hypothesis that the increased BP levels and renal damage in LSD1+/− mice, and by inference in LSD1 African risk allele carriers, are mediated in part by an overactivation of MR independent of elevated ALDO levels. These findings support the concept that treatment with an MRA may result in improved cardio- and reno- vascular outcomes in African LSD1+/− risk allele carriers.
Supplementary Material
Perspectives.
The present study shows the relationship between salt-sensitive BP, ALDO levels, and K+ levels in humans by LSD1 genotype and in LSD1 deficient mice. In the present study, both humans and mice demonstrate similar associations with increased BP and urine potassium levels, but with decreased ALDO levels. In LSD1+/−mice treated with the MRA eplerenone demonstrate that HTN, kaliuria and albuminuria are substantially improved, suggesting that MR blockade may be an efficient therapeutic model for hypertensive individuals that carry the LSD1 gene variant -personalized medicine.
Novelty and Significance.
What is new?
LSD1 deficiency increases aldosterone signaling via mineralocorticoid receptor activation
Chronic treatment with eplerenone significantly increased ALDO release from isolated ZG cells in LSD1 deficiency.
What is relevant?
Individuals with LSD1 risk alleles may have benefits with mineralocorticoid receptor antagonists as a preventive or therapeutic modality
Acknowledgments
We acknowledge the helpful technical assistance of Paul Loutraris and the support staff from the HyperPATH cohort.
Sources of Funding
This work was supported by the National Institute of Health Grants R01-HL-127146 (JSW), R01-HL-104032 (LHP), DP50HL055000 (HyperPATH Cohort), T32HL007609 (GHW); the American Heart Association Grants 18POST33990336 (DLB) and GRNT20500000 (LHP) and by a postdoctoral fellowship from the Ministry of Public Health of Thailand and the College of Medicine, Rangsit University (TT).
Footnotes
Disclosures
None.
References
- 1.OMS. 2003 world health organization (WHO)/ international society of hypertension (ISH) statement of hypertension. J Hypertens 2003;21(11):1983–1992. [DOI] [PubMed] [Google Scholar]
- 2.Carvalho JJ, et al. Blood pressure in four remote populations in the INTERSALT Study. Hypertens (Dallas, Tex 1979) 1989;14(3):238–246. [DOI] [PubMed] [Google Scholar]
- 3.Manosroi W, Williams GH. Genetics of Human Primary Hypertension: Focus on Hormonal Mechanisms. Endocr Rev December 2018. doi: 10.1210/er.2018-00071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loscalzo J, Handy DE. Epigenetic modifications: basic mechanisms and role in cardiovascular disease (2013 Grover Conference series). Pulm Circ 2014;4(2):169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lorenzen JM, Martino F, Thum T. Epigenetic modifications in cardiovascular disease. Basic Res Cardiol 2012;107(2):245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi Y, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004;119(7):941–953. [DOI] [PubMed] [Google Scholar]
- 7.Williams JS, et al. Lysine-Specific Demethylase 1: An Epigenetic Regulator of Salt-Sensitive Hypertension. Am J Hypertens 2012;25(7):812–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiang AH, et al. Evidence for joint genetic control of insulin sensitivity and systolic blood pressure in hispanic families with a hypertensive proband. Circulation 2001;103(1):78–83. [DOI] [PubMed] [Google Scholar]
- 9.Hopkins PN, et al. Blunted renal vascular response to angiotensin II is associated with a common variant of the angiotensinogen gene and obesity. J Hypertens 1996;14(2):199–2 [DOI] [PubMed] [Google Scholar]
- 10.Huang Y, et al. Histone demethylase LSD1 and biological sex: impact on blood pressure and aldosterone production. J Endocrinol 2019;240(2):111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo C, et al. Mineralocorticoid receptor antagonist reduces renal injury in rodent models of types 1 and 2 diabetes mellitus. Endocrinology 2006;147(11):5363–5373. [DOI] [PubMed] [Google Scholar]
- 12.Chong C, et al. Regulation of aldosterone secretion by mineralocorticoid receptor mediated signaling. J Endocrinol 2017;232(3):525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Braley LM, Menachery AI, Yao T, Mortensen RM, Williams GH. Effect of progesterone on aldosterone secretion in rats. Endocrinology 1996;137(11):4773–4778. [DOI] [PubMed] [Google Scholar]
- 14.Pojoga LH, et al. Histone demethylase LSD1 deficiency during high-salt diet is associated with enhanced vascular contraction, altered NO-cGMP relaxation pathway, and hypertension. Am J Physiol Heart Circ Physiol 2011;301(5):H1862–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garza AE, et al. Variants in Striatin Gene Are Associated With Salt-Sensitive Blood Pressure in Mice and Humans. Hypertens 2015;65(1):211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krug AW, et al. Lysine-specific demethylase-1 modifies the age effect on blood pressure sensitivity to dietary salt intake. Age (Dordr) 2013;35(5):1809–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le Menuet D, et al. Alteration of cardiac and renal functions transgenic mice overexpressing human mineralocorticoid receptor. J Biol Chem 2001;276(42):38911–38920. [DOI] [PubMed] [Google Scholar]
- 18.DeLano FA, Schmid-Schönbein GW. Enhancement of glucocorticoid and mineralocorticoid receptor density in the microcirculation of the spontaneously hypertensive rat.Microcirculation 11(1):69–78. [DOI] [PubMed] [Google Scholar]
- 19.Shibata Hirokata & Itoh Hiroshi. Mineralocorticoid Receptor-Associated Hypertension and Its Organ Damage: Clinical Relevance for Resistant Hypertension. Am J Hypertens 2012;25(5):514–523. [DOI] [PubMed] [Google Scholar]
- 20.McCurley A, et al. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nature Medicine 2012;18(9):1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pojoga LH, et al. Cooperative Role of Mineralocorticoid Receptor and Caveolin-1 in Regulating the Vascular Response to Low Nitric Oxide-High Angiotensin II-Induced Cardiovascular Injury. J Pharmacol Exp Ther 2015;355(1):32–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kobayashi N, et al. Eplerenone shows renoprotective effect by reducing LOX 1-mediated adhesion molecule, PKCepsilon-MAPK-p90RSK, and Rho-kinase pathway. Hypertens (Dallas, Tex 1979) 2005;45(4):538–544. [DOI] [PubMed] [Google Scholar]
- 23.Ambrosius WT, et al. Genetic variants in the epithelial sodium channel in relation to aldosterone and potassium excretion and risk for hypertension. Hypertens (Dallas, Tex 1979) 1999;34(4 Pt 1):631–637. [DOI] [PubMed] [Google Scholar]
- 24.Bertocchio J-P, Warnock DG, Jaisser F. Mineralocorticoid receptor activation and blockade: an emerging paradigm in chronic kidney disease. Kidney Int 2011;79(10):1051–1060. [DOI] [PubMed] [Google Scholar]
- 25.Nagata K, et al. Mineralocorticoid receptor antagonism attenuates cardiac hypertrophy and failure in low-aldosterone hypertensive rats. Hypertens (Dallas, Tex 1979) 2006;47(4):656–664 [DOI] [PubMed] [Google Scholar]
- 26.Pitt B, et al. The Effect of Spironolactone on Morbidity and Mortality in Patients with Severe Heart Failure. N Engl J Med 1999;341(10):709–717. [DOI] [PubMed] [Google Scholar]
- 27.Baudrand R, Vaidya A. The Low-Renin Hypertension Phenotype: Genetics and the Role of the Mineralocorticoid Receptor. Int J Mol Sci 2018;19(2):546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alikhani-Koopaei R, Fouladkou F, Frey FJ, Frey BM. Epigenetic regulation of 11β hydroxysteroid dehydrogenase type 2 expression. J Clin Invest 2004;114(8):1146–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shibata S, et al. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nature medicine December 2008;14(12):1370–1376. [DOI] [PubMed] [Google Scholar]
- 30.Nagase M, et al. Oxidative stress causes mineralocorticoid receptor activation in rat cardiomyocytes: role of small GTPase Rac1. Hypertension 2012;59(2):500–506. [DOI] [PubMed] [Google Scholar]
- 31.Shibata S, et al. Rac1 GTPase in rodent kidneys is essential for salt-sensitive hypertension via a mineralocorticoid receptor-dependent pathway. J Clin Invest 2011;121(8):3233–3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ayuzawa N, et al. Rac1-Mediated Activation of Mineralocorticoid Receptor in Pressure Overload-Induced Cardiac Injury. Hypertension 2016;67(1):99–106. [DOI] [PubMed] [Google Scholar]
- 33.Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC alpha,beta,and gamma subunit proteins in rat kidney. J Clin Invest 1999; 104:R19–R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tian Y, et al. Renal ENaC subunit, Na-K-2Cl and Na-Cl cotransporter abundances in aged, water-restricted F344 x Brown Norway rats. Kidney Int January 2006;69(2):304–312. [DOI] [PubMed] [Google Scholar]
- 35.Wissmann M, et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol 2007;9(3):347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.