

Abstract
The proteasome mediates most selective protein degradation. Proteolysis occurs within the 20S core particle (CP), a barrel-shaped chamber with an α7β7β7α7 configuration. CP biogenesis proceeds through an ordered multistep pathway requiring five chaperones, Pba1–4 and Ump1. Using S. cerevisiae, we report high-resolution structures of CP assembly intermediates by cryogenic-electron microscopy. The first structure corresponds to the 13S particle which consists of a complete α-ring, partial β-ring (β2–4), Ump1, and Pba1/2. The second structure contains two additional subunits (β5–6) and represents a later pre-15S intermediate. These structures reveal the architecture and positions of Ump1 and β2/β5 propeptides, with important implications for their functions. Unexpectedly, Pba1’s N-terminus extends through an open CP pore, accessing the CP interior to contact Ump1 and the β5 propeptide. These results reveal how the coordinated activity of Ump1, Pba1, and the active site propeptides orchestrate key aspects of CP assembly.
Keywords: proteasome, core particle, Ump1, 13S, assembly, propeptide
The proteasome is responsible for most selective protein degradation and preferentially recognizes substrates that have been covalently modified by the small protein ubiquitin. The 2.5 MDa 26S proteasome holoenzyme consists of the central core particle (CP), a barrel shaped chamber, capped at either axial end by the regulatory particle (RP). The RP recognizes the ubiquitin tag, unfolds the substrate, and threads it through the central pore of the CP, while also releasing ubiquitin for reuse. The CP has three types of active site--chymotryptic, tryptic, and post-acidic--which endow the proteasome with the capacity to render most proteins into small peptides1,2.
Like other large multisubunit protein complexes, the proteasome cannot assemble spontaneously. Rather, it is built through defined pathways orchestrated by dedicated chaperones that mediate assembly but which are themselves excluded from the final proteasome product. There are ten such chaperones known to contribute to assembly of the full 26S proteasome1,2. Given the essential nature of the proteasome for viability, defects in proteasome assembly are typically associated with cellular dysfunction and a reduced capacity to respond to proteotoxic stress. In some cases, loss of chaperone proteins is the direct cause of human disease3.
The CP is assembled first and is a 700 kDa 28-member complex with an α7β7β7α7 configuration. Its assembly requires 5 dedicated chaperones: the heterodimeric pairs Pba1/2 and Pba3/4, and Ump1. Current models of CP assembly1,2, based mainly on biochemical and genetic analyses, suggest that a complete α-ring is formed first with the assistance of Pba1/2 and Pba3/4, followed by sequential incorporation of β-subunits (Fig. 1a). The first β-subunit-containing intermediate that has been identified is the 13S complex, which contains the alpha ring (α1–7), three beta subunits (β2–4), Pba1/2, and Ump14–6 (Fig. 1a). Pba3/4 are released prior to 13S formation, which has been thought to reflect steric clash between Pba4 and β47,8. β1, β5, and β6 join next, resulting in the 15S complex9. β7 enters last, creating the half-CP, two of which then fuse in a poorly understood Ump1-dependent fashion to create the pre-holo-proteasome1. Five β-subunits, including the three proteolytic subunits (β1, β2, β5), are synthesized as precursor proteins with N-terminal propeptides. These propeptides prevent premature activation of the proteasome, but also play critical but still unexplained roles in CP assembly6,10. Final activation of the proteasome results in cleavage of these propeptides, degradation of Ump1, and release of Pba1/2, generating mature 20S proteasome.
Fig. 1 |. Biochemical characterization of CP mutants.
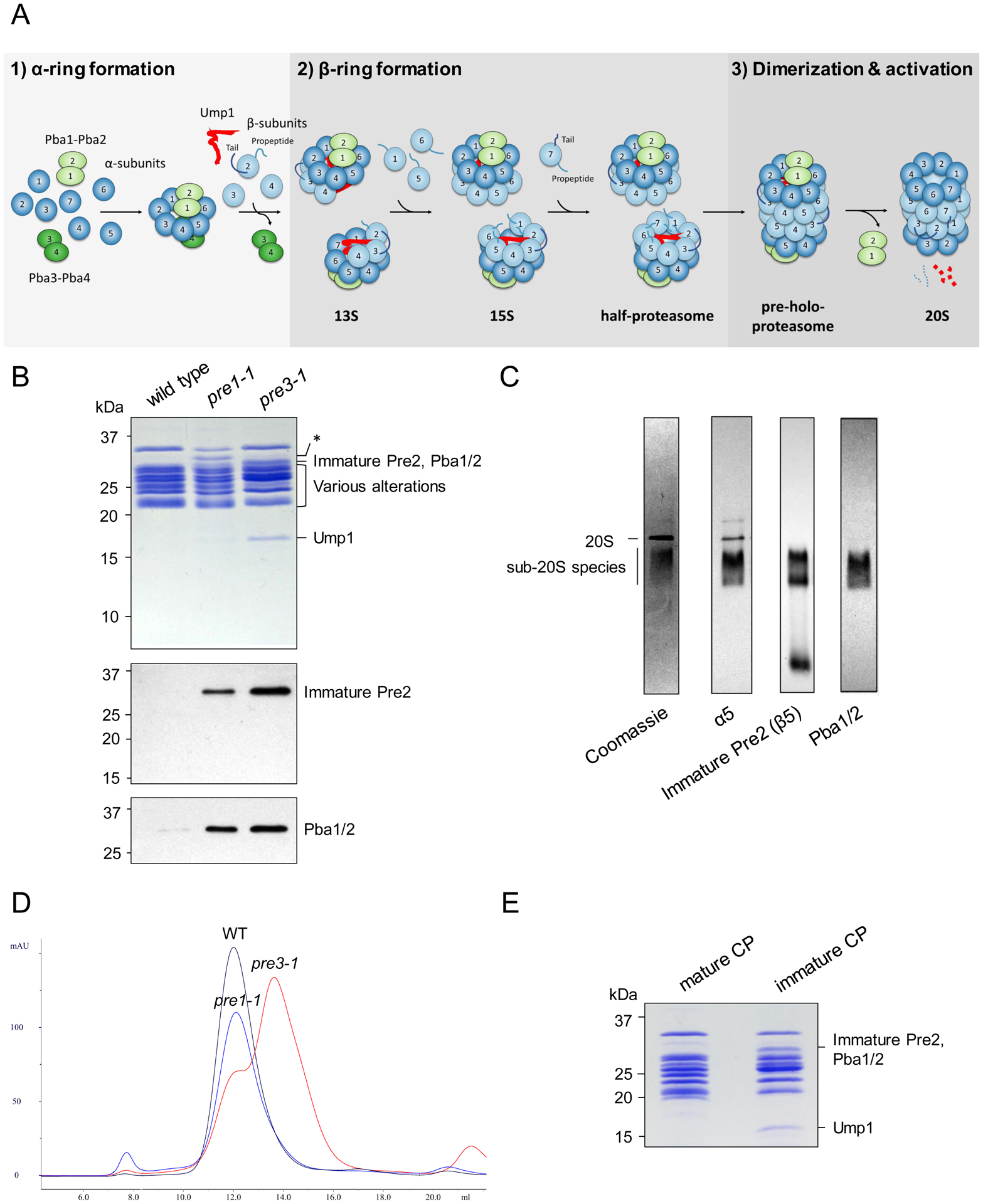
a, Schematic of CP assembly. b, Electrophoretic profiles of wild-type and mutant CP visualized by Coomassie staining (upper panel). Immature Pre2, Pba1, and Pba2 are of similar size and migrate together. Asterisk, an apparent breakdown product of α7 seen in pre1-1 CP, the significance of which is uncertain. Relative abundance of immature Pre2 (β5) and Pba1/2, as determined by immunoblot (lower panels). Similar results were obtained in five independent experiments. c, Non-denaturing gel electrophoresis of pre3-1 CP followed by Coomassie staining or immunoblot with the indicated antibodies. In addition to the 20S CP, there is a population of sub-20S particles that is not seen in wild-type CP. Immature Pre2 is detected within these sub-20S species, confirming that they contain post-13S species. Pba1/2 were only identified within sub-20S species, consistent with their preferential binding to immature CP. Similar results were obtained in five independent experiments. d, Size exclusion chromatography of the purified CP species shown in panel b. e, The heavier and lighter peaks from pre3-1 were pooled and analyzed by SDS-PAGE and Coomassie staining. Similar results were obtained in two independent experiments. Uncropped images for panels b, c and e are available online as source data.
While the structure of mature CP has been known for decades11, there has been little structural analysis of CP assembly, largely due to an inability to generate intermediates in sufficient quantities for analysis. In particular, the structure, localization, and function of Ump1 and the active site propeptides remain poorly understood. We hypothesized that specific CP mutants might stall in the assembly process, leading to enrichment of intermediates. We report here the first high-resolution structures of CP assembly intermediates, which explain key aspects of the process, identify new aspects of the assembly pathway, and establish a robust framework for mechanistic analysis of CP biogenesis.
Results
Enrichment of Assembly Intermediates in Defined CP Mutants
We selected the pre1-1 (β4; S142F)12 and pre3-1 mutants (β1, the post-acidic active site; G34D)13 mutants for initial analysis. Pre1-1 was previously known to accumulate CP assembly intermediates14. Pre3-1 not previously known to accumulate intermediates, but it showed a strong phenotype12 and so we considered it worth investigating. We purified CP from both mutants using a C-terminal Pre1-TEV-ProA affinity tag inserted at the endogenous locus. CP from both mutants showed an altered electrophoretic profile compared to wild-type (Fig. 1b). Additional bands present around 31 kD (Fig. 1b) represent Pba1/2 and immature, unprocessed β5 (Pre2), the latter specifically identified using a novel antibody directed against the 75-residue β5-propeptide (Fig. 1b,c). A band at 16 kD, more prominent in pre3-1 than pre1-1, was identified as Ump1 by mass spectrometry (Supplementary Table 1, Extended Data Set 1). Thus, both pre1-1 and pre3-1 preparations are enriched in immature CP species.
We analyzed the purified species by size exclusion chromatography using a Superose 6 column. Most pre1-1 species co-migrated with wild-type 20S CP (Fig. 1d), suggesting a late block in assembly. Consistent with this, a prior study reported a low resolution (>20 Å) cryo-EM structure of pre-holo-proteasome from this mutant14. In contrast, pre3-1 showed a prominent second peak of lower molecular weight (Fig. 1d,e), indicating that sub-20S particles were enriched in this mutant. These findings confirm that individual CP mutants may arrest at different stages of assembly. We chose to focus on the pre3-1 mutant given its strong accumulation of sub-20S species.
Structure and Function of Ump1
Cryo-electron microscopy of the pre3-1 material identified a mixture of 20S and sub-20S particles (Extended Data Fig. 1–3 and Table 1). We focused on two abundant sub-20S species, the first of which precisely corresponded to the 13S particle (3.6 Å) and consisted of Pba1/2, α1–7, β2–4, and Ump1 (Fig. 2a). The second (3.2 Å) consisted of the 13S and two additional subunits, β5 (the chymotryptic active site) and β6 (Fig. 2b). Consistent with this, native gel analysis of the pre3-1 sample detected immature β5 within sub-20S particles, confirming the presence of post-13S species (Fig. 1c). We refer to this particle as pre-15S. These structures reveal four key aspects of CP maturation: the overall architecture and position of Ump1, the structures of the β2 and β5 active site propeptides, and the relationship of Pba1/2 to the maturing CP.
Table 1 |.
Statistics for cryo-EM analysis.
| 13S (EMD-23508, PDB 7LSX) | Pre-15S (EMD-2350, PDB 7LS6) | Pre3-1 20S (EMD-23502, PDB 7LS5) | |
|---|---|---|---|
| Data collection and processing | |||
| Magnification | 47,169 | ||
| Voltage (kV) | 300 | ||
| Electron exposure (e−/Å−2) | 55.94 | ||
| Defocus range (μm) | 0.8, 2.2 | ||
| Pixel size (Å) | 1.06 | ||
| Symmetry imposed | C1 | C1 | C2 |
| Initial particle images (no.) | 1,633,892 | ||
| Final particle images (no.) | 76,731 | 95,288 | 216,361 |
| Map Resolution (Å) | 3.61 | 3.17 | 2.74 |
| FSC threshold | 0.143 | ||
| Map resolution range (Å) | 3.25–7.0 | 3.0–7.0 | 2.4–3.9 |
| Refinement | |||
| Initial model used | 7LS6 | 4G4S | 1RYP |
| Model resolution (Å) | 3.60 | 3.20 | 2.70 |
| FSC threshold | 0.143 | 0.143 | 0.143 |
| Map-sharpening B-factor (Å2) | −83.7 | −80 | −91.7 |
| Model composition | |||
| Non-hydrogen atoms | 22,220 | 25,100 | 49,153 |
| Protein residues | 2,838 | 3,210 | 6,315 |
| B factors (Å2) | |||
| Protein | 109.00 | 66.79 | 59.80 |
| R.M.S.D. deviations | |||
| Bond lengths (Å) | 0.003 | 0.003 | 0.004 |
| Bond angles (°) | 0.557 | 0.555 | 0.529 |
| Validation | |||
| MolProbity score | 1.3 | 1.32 | 1.17 |
| Clashscore | 4.98 | 5.51 | 3.81 |
| Poor rotamers (%) | 0 | 0.00 | 0.00 |
| Ramachandran plot | |||
| Favored (%) | 97.82 | 97.90 | 98.29 |
| Allowed (%) | 2.18 | 2.10 | 1.71 |
| Disallowed (%) | 0 | 0 | 0 |
Fig. 2 |. Structures of the 13S and pre-15S CP Assembly Intermediates.

a, Cryo-EM structure of a CP intermediate corresponding to the 13S particle (3.6 Å). b, Cryo-EM structure of the pre-15S particle (3.2 Å), which additionally contains β5 and β6. c, Rotated view of pre-15S providing a clearer view of the architecture and position of Ump1. β5/6 have been omitted for clarity. d, Overall architecture of Ump1. The first and last resolved residues are labeled. e, Position of Ump1 within the α-ring making contacts with α7 and α1–4, as shown in the fitted atomic model. f, Ump1’s major hinge region stretches between α7 and the incomplete β-ring.
In solution, free Ump1 is largely disordered15. In contrast, Ump1 formed a well-defined density within the 13S and pre-15S with similar local resolution to the rest of the structures (Fig. 2c,d and Extended Data Fig. 4). Ump1 was nestled into the undersurface of the α-ring, making multiple contacts with α7 and α1–α4 (Fig. 2e, Extended Data Fig. 5, and Supplementary Table 2). Interestingly, Ump1 was situated opposite from the α4–6 side, which is the site occupied by Pba3/4 at an earlier stage of assembly8. Nevertheless, there would be significant steric clash between Ump1 and Pba4 in the vicinity of α4 (Extended Data Fig. 6), suggesting that Pba3/4 exit the complex prior to Ump1’s incorporation. This could explain prior observations that co-immunoprecipitation with Ump1 was much stronger for Pba1/2 than for Pba3/416.
Ump1’s overall structure consists of seven α-helices with intervening loops of variable length (Fig. 2d). Its N-terminus is situated near β5/6 and winds around the β-ring towards β2–4. Its central portion then forms a hinge or open safety pin-like structure that crosses from the β–ring into the α-ring (Fig. 2f). This region begins near the highly conserved HPLE sequence at residue 51 with a short helix (H2) that is immediately followed by a longer helix (H3) that runs from the β3–β4 interface alongside β2 to end near α7, where it is cradled between two adjacent α7 helices (Fig. 2f). After a short turn, there is a second long helix (H4) that traverses deeper into the α-ring. A series of loops and smaller helices then wind around the CP cavity, ultimately terminating near the α3/α4 interface. Ump1 is fully enclosed within the CP cavity (i.e. not extended beyond the β-ring), although Ump1’s first 26 residues are not resolved. The overall position of Ump1 is supported by its concordance with prior cross-linking studies (Supplementary Table 3)14.
The β2 and β5 Propeptides are Coordinately Positioned with Ump1
The propeptides of active site subunits prevent premature proteolysis, but also play enigmatic roles in CP maturation6,10,17,18. The 29-residue β2 propeptide was resolved and ran across the inner surface of the β-ring from β2’s active site threonine along the entire width of β3 (Fig. 3a). The propeptide then turned sharply, forming a helix that coursed along the length of β3 toward the α-ring where, surprisingly, the propeptide terminated in contact with Ump1, sandwiched in between two segments of Ump1, β3, and β4 (Fig. 3b). Additional contacts were present between Ump1 and two adjacent helices within the main body of β2 (Fig. 3c and Supplementary Table 2), implicating Ump1 in the overall positioning of β2.
Fig. 3 |. Structures of the β2 and β5 propeptides.

a, Partial view of the pre-15S with the β-ring oriented upwards; β6 has been omitted for clarity. The β2 propeptide runs in front of β3 before turning sharply towards the α-ring. Only an N-terminal segment of the β5 propeptide is resolved, and it terminates in contact with Ump1 deep within the CP cavity. b, Close-up view of the β2 propeptide’s N-terminus, which terminates sandwiched in-between Ump1, β3, and β4 (top panel). Close-up view of the extended interaction between the β2 propeptide and both β3 and β4 (bottom panel). c, Additional contacts between Ump1 and the main body of β2, with close-up views shown in side panels. d, Interactions between Ump1 and β5, with close-up views shown in side panels. In c and d, helices are numbered to facilitate orientation of the close-up views.
β2 possesses a C-terminal extension that wraps around the exterior surface of β3 and is thought to promote proper placement of β3 during maturation (Extended Data Fig. 7)19. The propeptide’s position, running across the inner surface of β3, suggests that the propeptide may further promote β3 incorporation. Interestingly, in the mature CP β2’s C-terminal extension also makes contacts with the other half of the CP barrel11 and the C-terminal extension’s central portion was largely unresolved in the 13S and pre-15S complexes (Extended Data Fig. 7), suggesting that it may be fully stabilized only upon joining of CP half-mers. If the C-terminal extension is not structurally stable at early stages of maturation, this might create a need for further stabilization of β3 by the β2 propeptide.
In the pre-15S, a small segment of the β5 propeptide was resolved and corresponded to its N-terminal ten residues (Fig. 3a, d, and Extended Data Fig. 8). This density was in direct contact with Ump1’s helix #4 and α7, situated near the point where the hinge area of Ump1 is cradled by α7. This position is compatible with a previously detected cross-link between Lys-16 in β5 and α614.
Pba1 Accesses the CP Interior through a Novel Open Pore Configuration
Pba1 and Pba2 are thought to contribute to the generation of properly configured α-rings but may also function to prevent α-α ring dimerization and premature association of the RP with CP20,21. Both proteins contain C-terminal HbYX motifs which are found in proteasome activators like the RP that open the CP gate. Interestingly, Pba1/2 show higher affinity for immature versus mature CP22 and are released from the 20S upon final CP activation. A prior crystal structure visualized Pba1/2 bound to CP, but since this structure was generated by combining recombinant bacterially produced Pba1/2 with wild-type mature yeast 20S21, it did not visualize Pba1/2 within its physiologic context of CP assembly, and could not explain why Pba1/2 preferentially bind immature CP (Fig. 1c) or how Pba1/2 are released upon completion of assembly. Of note, Pba1’s N-terminus, which is discussed in detail below, was also not resolved in that crystal structure21.
In the 13S and pre-15S, Pba1/2 cap the outer surface of the α-ring with Pba1 located mainly at the α5–α6 interface and Pba2 positioned more over the central pore with its C-terminus extending into the α6–α7 interface (Fig. 4a). Both proteins’ C-terminal HbYX motifs are inserted into their respective α-ring pockets (Fig. 4a), consistent with the prior study21. The CP pore is open and, unexpectedly, the N-terminus of Pba1 extends through the pore into the CP interior (Fig. 4b,c). The pore displays a conformation distinct from previously described open and closed states11,23. The N-termini of α5–6 are rotated dramatically upward, contacting Pba1 and running in parallel for a significant distance (Fig. 4d,e). In contrast, the N-terminus of α2 is rotated downward into the CP cavity and runs in parallel with Pba1. Remarkably, Pba1’s N-terminus contacts not only α2, but also Ump1, the β5 propeptide, and the other 6 α-subunits (Fig. 4d). This unique pore/α-ring conformation is dependent on interactions between Pba1, Ump1, and the β5 propeptide, and therefore likely exists only during CP maturation.
Fig. 4 |. Pba1 Transits Through the Open CP Pore to Contact Ump1 and the β5 propeptide.
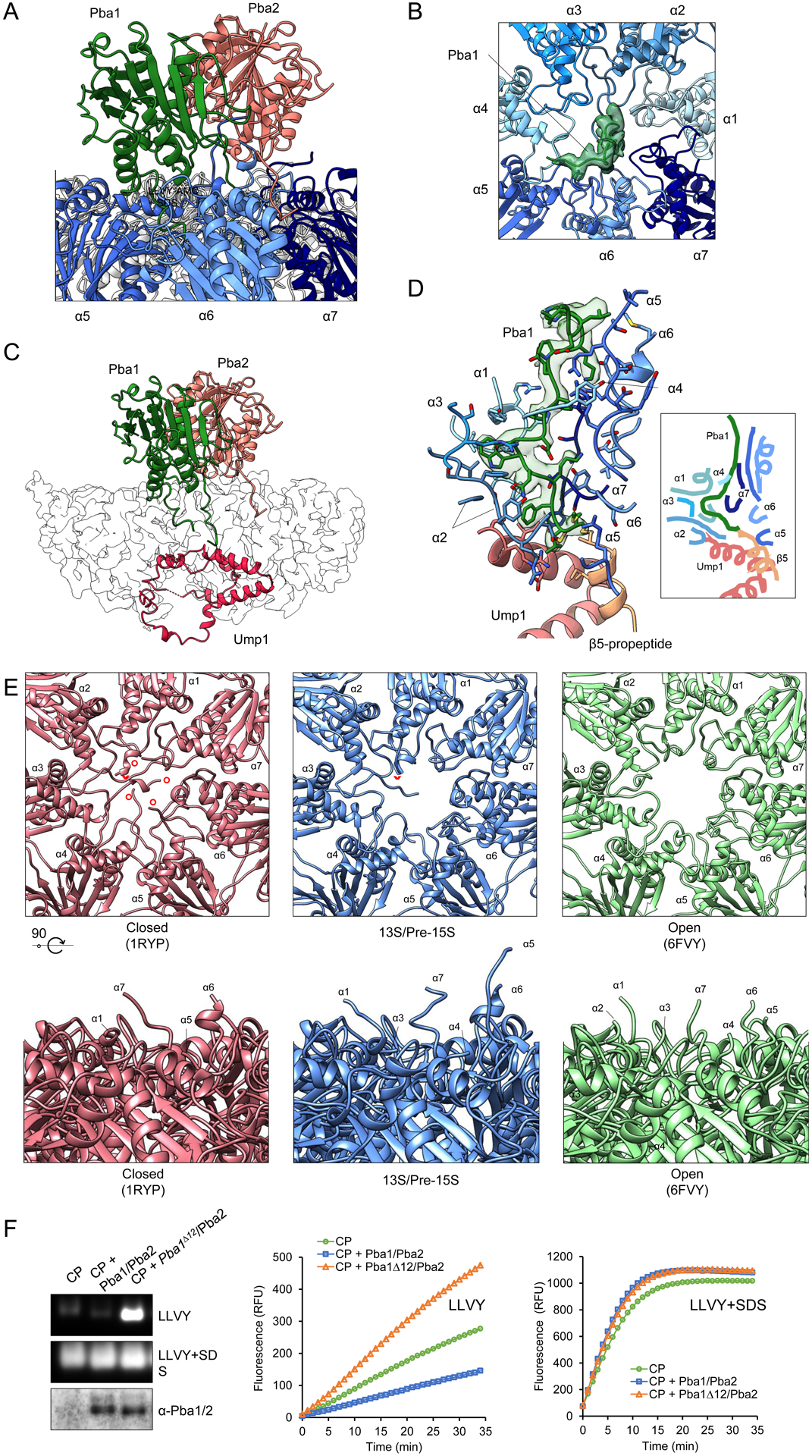
a, Overall position of Pba1 and Pba2 with their HbYX motifs inserted into the α5/6 and α6/7 interfaces, respectively. b, The open gate of the CP with Pba1 filling the pore (top view). c, Passage of Pba1’s N-terminus through the pore and into CP cavity where it terminates in contact with Ump1. d, Pba1’s N-terminus contacts Ump1, the β5 propeptide, and all 7 α-subunits. Right box, simplified cartoon rendering of the interactions. e, Comparison of the CP pore in closed, 13S/pre-15S and open states. Upper panels show the CP pore, as viewed from the exterior surface of the α-ring. The N-terminus of α2 is indicated by a red arrowhead; the N-termini of α1 and α3–5 are indicated by red circles. Bottom panels show side views of the CP pore. Note that Pba1 has been omitted from these images for clarity, but that Pba1’s N-terminus extends through and largely fills the pore (see panel b), contributing to a conformation that is distinct from the open and closed states. f, CP enzymatic activity in the presence of Pba1wt/Pba2 or Pba1Δ12/Pba2, as determined by native in-gel activity assay (left panels) or liquid assay (right panels). Pba1/2 immunoblot confirms binding of Pba1Δ12/Pba2 to CP. Similar results were obtained in two independent experiments. Uncropped images and data for graphs in panel f are available online as source data.
Despite possessing HbYX motifs, prior work found that Pba1/2 do not activate the CP in vitro21. This finding may be explained by occlusion of the pore by Pba1’s N-terminus (Fig. 4b). To test this model, we deleted Pba1’s N-terminal 12 residues, purified Pba1/2 from bacteria, and added them to purified CP. Pba1Δ12/Pba2 stimulated hydrolysis of the peptide substrate LLVY-AMC, while Pba1wt/Pba2 did not (Fig. 4f).
Discussion
Several lines of evidence support the physiologic relevance of the structures reported here. First, the composition of one of the two structures precisely recapitulates the biochemically defined 13S, a bona fide assembly intermediate that has been detected even in wild-type cells4–6. Second, the positions of Ump1 and the β5 propeptide are fully consistent with previously determined cross-links to other CP subunits (Supplementary Table 3)14. Third, an arrest at the 13S or pre-15S stage is consistent with a molecular defect in pre3-1 since maturation of 13S to 15S requires the addition of β1 (Pre3), β5, and β6, although the exact order of addition remains unsettled9. An important, but unanswered question, is why the specific mutation in pre3-1 results in such a strong assembly defect.
By providing the first high-resolution structures of maturing CP, the results presented here allow us to reassess important aspects of CP biogenesis. Ump1 was identified over 20 years ago24, but little was known about its structure. It is intrinsically disordered outside of the CP15 and two low resolution (~20 Å) structures of CP assembly intermediates could not resolve its structure14. Furthermore, most models assumed that Ump1 projected out of the β-ring, an orientation that could have explained its role in promoting CP half-mer dimerization/fusion24. In contrast, we show that Ump1 is highly structured in the context of maturing CP and no part of it extends beyond the β-ring (although residues 1–26 are not resolved). Ump1 appears to coordinate multiple aspects of CP biogenesis, interacting with the β2/5 propeptides and Pba1, as well as 9 different α/β subunits (Extended Data Fig. 5 and Supplementary Table 2). A major aspect of Ump1 function appears to be proper β-ring configuration, and this central organizing role is facilitated by its extended α-helical nature which allows it to wind around the CP interior. It has been proposed that conformational changes in Ump1 drive CP half-mer dimerization/fusion24. It is hard to envision how that might occur from these structures, although we can’t exclude that such conformational changes might occur later in assembly. An alternate model is that Ump1 facilitates proper configuration of the β-ring, leading to half-CP which may have the inherent capacity to dimerize. Ump1’s position also clashes strongly with Pba4, suggesting that it is the entry of Ump1 that evicts Pba3/4, and not β4 as previously thought7,8. Thus, Pba3/4 likely exit the maturing CP soon after completion of the α-ring.
The β2 and β5 propeptides have been known to play key roles in CP assembly for more than 20 years10,17, but their structures were unknown and no precise function has been assigned to either of them. Our data indicate that the β2 propeptide facilitates incorporation of β3, and likely also β4, suggesting that propeptides may play an important role in β-ring assembly. Ump1 helps position the β2 propeptide in a manner that was not previously anticipated. The β5 propeptide is the longest and most important of the propeptides, and previous models have suggested that it might also project out of the half-CP to promote dimerization6. In contrast, our structures indicate that the β5 propeptide is directed toward the α-ring, consistent with prior cross-linking data14. This makes the prior models extremely unlikely, although again we cannot exclude conformational changes later in assembly. An interaction between Ump1 and the β5 propeptide was first proposed more than two decades ago6,24 and our structures identify this interaction for the first time. It is interesting that both the β2 and β5 propeptides contact Ump1, and it is possible that the different lengths of the active site propeptides (19, 29, or 75 residues) may primarily be determined by the distance between each subunit and Ump1.
A major conundrum in the field has been the role of the C-terminal HbYX motifs in Pba1/2, which are found in proteasome activators like the RP which open the CP gate. There was no prior rationale for an open gate during CP assembly and experimental evidence indicated Pba1/2 did not stimulate CP activity in vitro21. Our structures explain these findings: Pba1/2 do open the CP gate but then Pba1’s N-terminus enters the pore and fills it, preventing access of substrates to the CP interior. This highly unanticipated structural arrangement clarifies two other major unexplained aspects of Pba1/2: their preferential association with immature CP and their release from the CP upon completion of assembly. By accessing the CP interior, Pba1 makes contacts with immature aspects of the CP, including Ump1 and the β5 propeptide. Upon completion of assembly, the β5 propeptide is cleaved and Ump1 is destroyed; disruption of these interactions with Pba1 could reduce its affinity for mature CP.
A final conundrum of CP assembly relates to the observation that while Pba1/2 and Pba3/4 mediate α-ring assembly, all 4 proteins sit on the same side of the ring (α4–7, with minor contacts to α1)7,21. Thus, it has been unclear how Pba1–4 can ensure proper ordering of the subunits of the α-ring. Our structures indicate that Pba1’s N-terminus contacts all 7 α-subunits, with major contacts to α2 and α1, providing a mechanistic basis for understanding the proper configuration of complete α-rings.
Proteasome assembly provides one of the most dramatic examples of dedicated molecular chaperone action in nature. Our approach establishes a framework for detailed mechanistic investigation of CP biogenesis, and suggests a general experimental approach to elucidate the structures of additional assembly intermediates. With enough informative mutants, it may be possible to generate a comprehensive structural atlas of the pathway of CP assembly.
Methods
Strains and Antibodies
The isogenic wild-type (YHI29W; MATa ura3 leu2–3,112 his3–11,15 Cans Gal+), pre1-1 (YHI29W/1), and pre3-1 (YRG11) strains have been previously described12. A C-terminal HIS3-marked TEV-ProA tag was inserted by homologous recombination into each strain to allow for CP purification, yielding sMB163 (WT), sMB164 (pre1-1), and sMB186 (pre3-1). Yeast (S. cerevisiae) were cultured in YPD (1% yeast extract, 2% Bactopeptone, and 2% dextrose) at 30°C.
The β5 (Pre2) propeptide (residues 1–75) was cloned into pET45 which provides for an N-terminal 6xHis-tag (pMB62). This construct was expressed in E. coli and purified by Nickel chromatography to near homogeneity. The purified protein was used to generate a rabbit polyclonal antibody. This antibody recognizes immature, unprocessed Pre2 and does not recognize mature Pre2. Full-length alpha5 was cloned into pGEX-4T-1 which provides for an N-terminal GST tag followed by a thrombin cleavage site (pMB45). This construct was expressed in E. coli, purified by glutathione-sepharose chromatography to near homogeneity, and cleaved with thrombin. The purified protein was used to generate a rabbit polyclonal antibody. The Pba1/2 antibody has been previously described22. The following antibody dilutions were used: anti-alpha5 (1:5000), anti-Pre2 propeptide (1:5000), and Pba1/2 (1:4000).
CP Purification & Analysis
CP was affinity purified using IgG resin (MP Biomedicals; ICN55961) as previously described25. Analysis was by standard SDS-polyacrylamide gel electrophoresis followed by Coomassie staining or immunoblotting. For native gel analysis, 3.6 μg of purified pre3-1 material was analyzed using a 3–8% Tris-Acetate gel (Invitrogen), followed by Coomassie staining or immunoblotting.
Cryo-EM Sample Preparation
Purified CP was concentrated and injected over a Superose 6 10/300 GL column (GE Healthcare) equilibrated in SEC buffer (50mM Tris, pH 7.5, 1mM EDTA, and 100 mM NaCl). The CP-containing fractions were pooled and concentrated to ~4.5 mg/mL. Immediately prior to disposition onto 400 mesh Quantifoil Cu 1.2/1.3 grids that had been glow discharged in a PELCO easiGLOW (Ted Pella) at 0.39mBar, 15mA for 30s, 3 μL of sample was mixed with 0.5 μL of a 7 mM Fos-Choline, fluorinated solution (Anatrace). Fos-Choline, fluorinated solution (at a final concertation of 1 mM) was used to diversify the particle orientations in ice. Samples were vitrified in 100% liquid ethane using a Vitrobot Mark IV (Thermo Fisher Scientific), with a wait time of 15 s, blot time of 8 s, and a blot force of 20 at 100% humidity.
Cryo-EM Data Collection and Processing
Cryo-EM data were collected on a 300 kV Titan Krios G3i Microscope (Thermo Fisher Scientific) equipped with a K3 direct electron detector (Gatan) and a GIF quantum energy filter (25 eV) (Gatan) using counted mode. Data were acquired utilizing image shift and real-time comma correction by beamtilt using the automated data collection software SerialEM26; nine holes were visited per stage position acquiring a single movie per hole. Details of the data collection and dataset parameters are summarized in Table 1. Dose-fractionated images were gain normalized, aligned, dose-weighted, and summed using MotionCor227. Contrast transfer function (CTF) and defocus value estimation were performed using CTFFIND428. Details of the data processing strategy are shown in Extended Data Fig.1. In short, particle picking was carried out using crYOLO29 followed by initial 2D classification within Relion30 to give 1,633,892 particles. Heterogeneous classification in cryoSPARC31, using initial models generated ab initio from prior small cryo datasets, was used to sort particles into 20S-like or 13S-like groups based on presence of presumed Ump1 density. The “13S-like” particles were subjected to further heterogeneous refinement within cryoSPARC leading to the identification of 259,892 13S particles. Following Bayesian particle polishing within Relion 3.1 and further 2D/3D classification, 76,731 13S particles were identified ultimately leading to a 3.6Å reconstruction following CTF refinement and non-uniform refinement within cryoSPARC. Additionally, a “pre-15S” class was identified which following further polishing and CTF refinement resulted in a 3.17Å reconstruction from 95,288 particles. 17,927 particles corresponding to 13S + β5 were identified via subtraction of the β5–6 domains followed by alignment free classification within Relion resulting in a 4.6Å reconstruction following non-uniform refinement within cryoSPARC.
The 20S particles underwent a similar polishing, 2D/3D classification, CTF refinement procedure to produce a final reconstruction at 2.7Å with C2 symmetry imposed (2.8 Å C1). Structural biology applications used in this project were compiled and configured by SBGRid32.
Model Building and Refinement
The higher resolution pre-15S model was built first. A starting model for Ump1 was generated using the robetta server (https://robetta.bakerlab.org/)33.Relevant chains from 4G4S (α1–α7, β2–β6, Pba1, Pba2) were used as a starting model for the remainder of the pre-15S complex. Components were first rigid body fit into the electron density using UCSF Chimera34. This was followed by iterative cycles of manual building and refinement using Coot35 and ISOLDE36 for manual building and Phenix37 for real space refinement. The pre-15S complex and 1RYP were used as initial models for the 13S complex and the 20S pre3-1 complex respectively. The 13S complex and the 20S pre3-1 complex were subjected to the same iterative process of manual building and refinement. Protein-protein interactions were annotated using PDBePISA.
Figures were prepared using both UCSF Chimera and Chimera X.
Mass Spectrometry
Gel bands were excised, fragmented, and destained with 50 mM HEPES pH 8.2/acetonitrile (70/30 v/v). Proteins were reduced with 5 mM TCEP for 25 min and alkylated with 14 mM iodoacetamide for 30 min, and the reaction quenched with 10 mM DTT for 15 min. Dried gel fragments were rehydrated with 10 ng/μl of trypsin and digested overnight at 37°C. The resulting peptides were desalted and analyzed using a Q-Exactive mass spectrometer (Thermo Scientific).
Expression and Purification of Pba1/2
Plasmids for the expression of Pba1 or Pba1Δ12 from E. coli were derived from pJR58622. Primers pRL1000 (GGGCCCCTGGAACAGAAC) and pRL1001 (CTTTTTAAACAATGGAATGAC) were used to generate plasmid pJR924, which allowed for the expression of GST-Pba1; after PreScission cleavage, two amino acids (Gly Pro) remain N-terminal of Pba1. To create an N-terminal truncation of the first 12 amino acids of Pba1, primers pRL1000 (GGGCCCCTGGAACAGAAC) and pRL1002 (AAACATCTGCTAGATCTCCCAG) were used to create plasmid pJR931.
Pba1 or Pba1Δ12 was co-expressed with Pba2 (pJR585) in E. coli rosetta II cells and the dimer was purified using glutathione resin as described previously22. Following purification, the GST moiety was removed using GST PreScission protease. The protein complex was dialyzed against 50 mM Tris-HCl pH 6.8, 150 mM NaCl, 1 mM EDTA, 1 mM DTT, and 0.01% Triton X-100 overnight at 4°C or further purified in this buffer using size exclusion chromatography over a Superdex 200 6 10/300 GL column on an Äkta™ go and concentrated using 10 kDa MW cutoff concentrator (PALL Life Sciences).
Reconstitution assays of CP with Pba1-Pba2
CP was purified as described previously38 3.25 μM purified CP was reconstituted with a 10-fold molar excess of Pba1-Pba2 dimer, either Pba1 or Pba1Δ12, in reconstitution buffer (50mM Tris-HCl (pH-7.5), 5mM MgCl2, 1mM EDTA, and 10% glycerol) for a total volume of 5 uL and incubated for 30 min at 30 °C. Samples were mixed with 10x native gel loading buffer (50 mM Tris‐HCl (pH 7.4), 50% glycerol and 60 ng ml−1 xylene cyanol) and loaded onto a native gel. Electrophoresis was performed for 2 hours at 90 V, 4°C followed by in-gel CP activity (LLVY-AMC hydrolysis) assay using LLVY-AMC as fluorogenic substrate39. The images were captured using G-Box imaging system from SynGene with GeneSnap software. Next, gels were transferred onto polyvinylidene fluoride membrane for immunoblotting with polyclonal antibody against Pba1-Pba2. For in-solution assay, the CP was reconstituted with Pba1-Pba2 dimers as described above, but in a 1:20 molar ratio. The addition of 0.02% SDS to LLVY-AMC assays (in solution or in gel) resulted in activated CP through opening of the CP gate. The increase in fluorescence was monitored using Synergy 2 plate reader (BioTek Instruments Inc.).
Reporting Summary statement
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Cryo-EM maps and atomic model coordinates have been deposited in the EMDB and RCSB respectively: 13S (EMD-23508, PDB 7LSX), Pre-15S (EMD-2350, PDB 7LS6), Pre3-1 20S (EMD-23502, PDB 7LS5). Additional structures referenced here include: PDB 4G4S, PDB 1RYP, PDB 2Z5C, and PDB 6FVY. Source data are available with the paper online.
Extended Data
Extended Data Fig. 1. Cryo-EM classification of CP species.

Processing scheme for classification and refinement of proteasome species. “Junk” classes throughout colored grey – identifiable species colored by species. All 3D classification steps other than the subtracted β5–6 classification were carried out in cryoSPARC.
Extended Data Fig. 2. Cryo-EM data analysis for CP species.

a, Representative micrograph of proteasome particles embedded in vitreous ice (scale bar = 500Å). A total of 21,000 micrographs were collected from a single multi day experiment. b, Selected 2D class averages of 20S and 13S particles (scale bar = 200Å). c, Proteasome reconstructions filtered and colored by local resolution (left), gold-standard Fourier shell correlation (FSC) curves from cryoSPARC (center) and viewing direction distribution plots (right). Resolution determined at FSC = 0.143.
Extended Data Fig. 3. Structure of 20S CP from pre3-1 mutant.
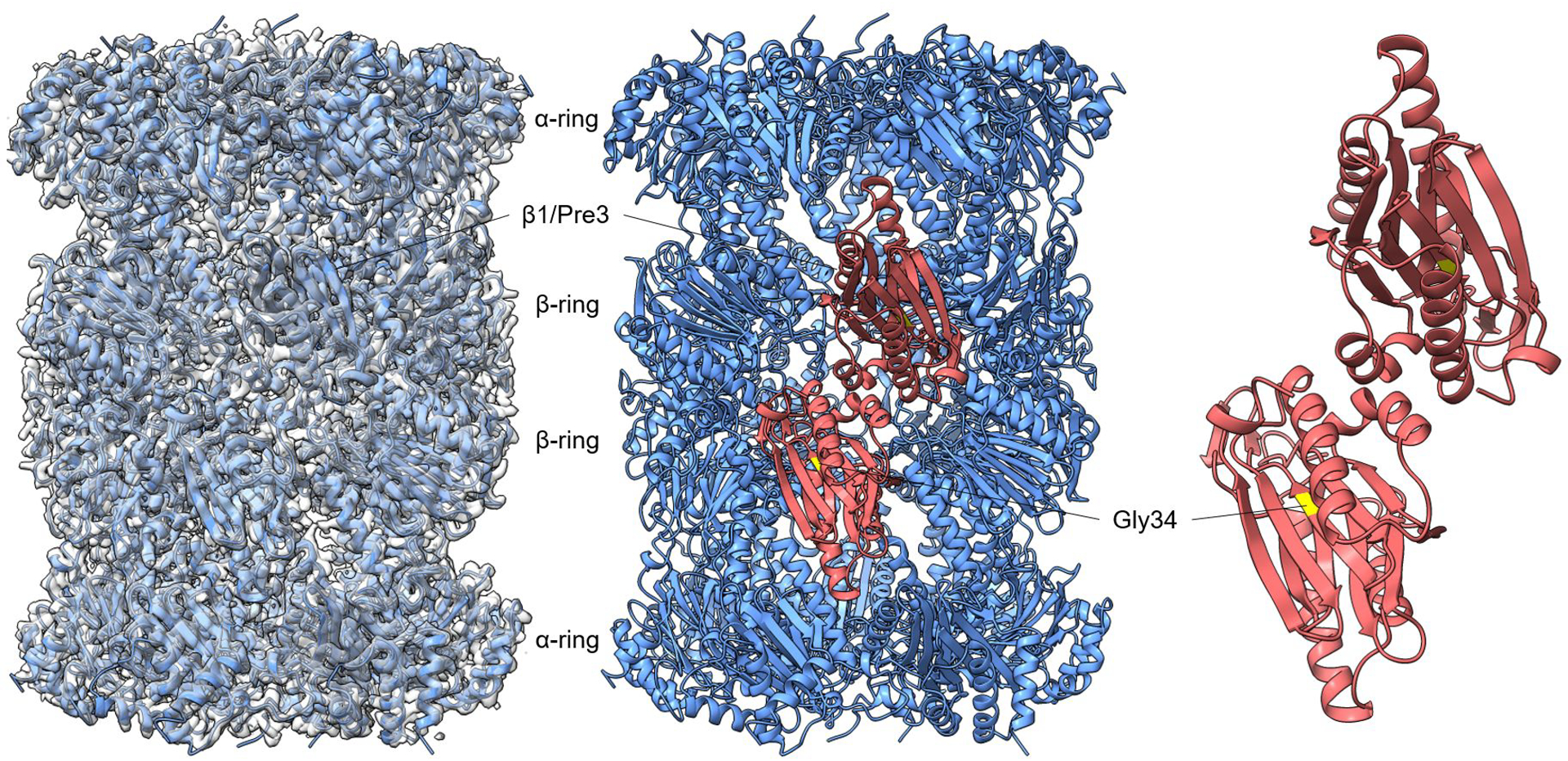
Cryo-EM density of the pre3-1 20S species (2.7 Å) modeled onto the crystal structure of wild-type mature 20S (PDB: 1RYP). The position of Pre3 (β1) is indicated in red in the middle panel. Pre3-1 harbors a G34D mutation13 and the position of G34 in 1RYP is shown in yellow in the right panel.
Extended Data Fig. 4. Confirmation of the assignment of Ump1 to the novel central density within 13S and pre-15S structures.

The Ump1 model is shown overlaid onto the primary cryo-EM map density. The four boxed panels show close-up views confirming that the density precisely matches the modeled amino acid side chains of Ump1.
Extended Data Fig. 5. Extensive contacts between Ump1 and the CP.

a, Multiple views of Ump1’s contacts with α-subunits and Pba1. b, Multiple views of β-subunits. In both panels, contacts were determined using PDBePISA (see Supplementary Table 1 for details).
Extended Data Fig. 6. Potential steric clash between Ump1 and Pba4.

Surface of the α-ring with the associated Ump1 density. Pba3 and Pba4 (PDB: 2Z5C) have been modeled onto this structure, and Pba4 (yellow) shows extensive clash with Ump1 (red) in the vicinity of α4.
Extended Data Fig. 7. Comparison of β2’s N-terminal propeptide and C-terminal loop in mature CP and pre-15S structures.

Relationship between β2 and β3 in the wild-type mature 20S (purple; PDB: 1RYP) and the pre-15S structure (green). Multiple views are shown. The propeptide is absent in mature 20S, while the C-terminal loop is largely unresolved in the maturing CP.
Extended Data Fig. 8. Identification of N-terminal β5 propeptide helix.

a, Ump1 hinge region showing clear density assigned to β5 propeptide (orange) in pre-15S reconstruction. b, Corresponding region of the 13S reconstruction shows no density. c, Low resolution map of 13S + β5 reconstruction showing density is restored. Surrounding density in all panels hidden for clarity using a 2–3Å carve radius.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
Cryo-EM data were collected at the Harvard Cryo-Electron Microscopy Center for Structural Biology at Harvard Medical School. This work was supported by NIH grants DP5-OD019800 (to J.H.), R01-GM043601 (to D.F), R01-GM67945 (to S.G.), R01-GM132129 (to J.A.P.), P20-GM103418 (to J.R.), and R01-GM118660 (to J.R.).
Footnotes
Competing Interests Statement
The authors declare that they have no conflict of interest.
References
- 1.Budenholzer L, Cheng CL, Li Y & Hochstrasser M Proteasome Structure and Assembly. J. Mol. Biol 429, 3500–3524 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rousseau A & Bertolotti A Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol 19, 697–712 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Dahlqvist J et al. A Single-Nucleotide Deletion in the POMP 5′ UTR Causes a Transcriptional Switch and Altered Epidermal Proteasome Distribution in KLICK Genodermatosis. Am. J. Hum. Genet 86, 596–603 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frentzel S, Pesold-Hurt B & Seelig A 20 S Proteasomes are Assembled via Distinct Precursor Complexes Processing of LMP2 and LMP7 Proproteins Takes Place in 13–16 S Preproteasome Complexes. J. Mol. Biol 236–975 (1991). [DOI] [PubMed] [Google Scholar]
- 5.Schmidtke G, Schmidt M & Kloetzel P-M Maturation of Mammalian 20 S Proteasome: Purification and Characterization of 13 S and 16 S Proteasome Precursor Complexes. J. Mol. Biol 268, 95–106 (1997). [DOI] [PubMed] [Google Scholar]
- 6.Li X, Kusmierczyk AR, Wong P, Emili A & Hochstrasser M β-Subunit appendages promote 20S proteasome assembly by overcoming an Ump1-dependent checkpoint. EMBO J 26, 2339–2349 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yashiroda H et al. Crystal structure of a chaperone complex that contributes to the assembly of yeast 20S proteasomes. Nat. Struct. Mol. Biol 15, 228–236 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Takagi K et al. Pba3-Pba4 heterodimer acts as a molecular matchmaker in proteasome α-ring formation. Biochem. Biophys. Res. Commun 450, 1110–1114 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Hirano Y et al. Dissecting β-ring assembly pathway of the mammalian 20S proteasome. EMBO J 27, 2204–2213 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaeger S, Groll M, Huber R, Wolf DH & Heinemeyer W Proteasome β-type Subunits: Unequal Roles of Propeptides in Core Particle Maturation and a Hierarchy of Active Site Function. J. Mol. Biol 291, 997–1013 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Groll M et al. Structure of 20S proteasome from yeast at 2.4A resolution. Nature 386, 463–471 (1997). [DOI] [PubMed] [Google Scholar]
- 12.Gerlinger UM, Gückel R, Hoffmann M, Wolf DH & Hilt W Yeast cycloheximide-resistant crl mutants are proteasome mutants defective in protein degradation. Mol. Biol. Cell 8, 2487–2499 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gueckel R, Enenkel C, Wolf DH & Hilt W Mutations in the Yeast Proteasome-Type Subunit Pre3 Uncover Position-dependent Effects on Proteasomal Peptidase Activity and in Vivo Function. J. Biol. Chem 273, 19443–19452 (1998). [DOI] [PubMed] [Google Scholar]
- 14.Kock M et al. Proteasome assembly from 15S precursors involves major conformational changes and recycling of the Pba1-Pba2 chaperone. Nat. Commun 6:6123, (2015). [DOI] [PubMed] [Google Scholar]
- 15.Sá-Moura B et al. Biochemical and biophysical characterization of recombinant yeast proteasome maturation factor Ump1. Comput. Struct. Biotechnol. J 7, e201304006 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.le Tallec B et al. 20S Proteasome Assembly Is Orchestrated by Two Distinct Pairs of Chaperones in Yeast and in Mammals. Mol. Cell 27, 660–674 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Chen P & Hochstrasser M Autocatalytic Subunit Processing Couples Active Site Formation in the 20S Proteasome to Completion of Assembly. Cell 86, 961–972 (1996). [DOI] [PubMed] [Google Scholar]
- 18.Arendt CS & Hochstrasser M Eukaryotic 20S proteasome catalytic subunit propeptides prevent active site inactivation by N-terminal acetylation and promote particle assembly. EMBO J 18, 3575–3585 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramos PC, Marques AJ, London MK & Dohmen RJ Role of C-terminal Extensions of Subunits β2 and β7 in Assembly and Activity of Eukaryotic Proteasomes. J. Biol. Chem 279, 14323–14330 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Hirano Y et al. A heterodimeric complex that promotes the assembly of mammalian 20S proteasomes. Nature 437, 1381–1385 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Stadtmueller BM et al. Structure of a proteasome Pba1-Pba2 complex implications for proteasome assembly, activation, and biological function. J. Biol. Chem 287, 37371–37382 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wani PS, Rowland MA, Ondracek A, Deeds EJ & Roelofs J Maturation of the proteasome core particle induces an affinity switch that controls regulatory particle association. Nat. Commun 6:6123, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eisele MR et al. Expanded Coverage of the 26S Proteasome Conformational Landscape Reveals Mechanisms of Peptidase Gating. Cell Reports 24, 1301–1315.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramos PC, Hoeckendorff J, Johnson ES, Varshavsky A & Dohmen JR Ump1p Is Required for Proper Maturation of the 20S Proteasome and Becomes Its Substrate upon Completion of the Assembly. Cell 92, 489–499 (1998). [DOI] [PubMed] [Google Scholar]
- 25.Leggett DS et al. Multiple Associated Proteins Regulate Proteasome Structure and Function. Mol. Cell 10, 495–507 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Mastronarde DN Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Zheng SQ et al. MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rohou A & Grigorieff N CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wagner T et al. SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun. Biol 2:218, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scheres SHW RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA CryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Morin A et al. Collaboration gets the most out of software. eLife 2:e01456, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raman S et al. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins 77, 89–99 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pettersen EF et al. UCSF Chimera - A visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Cryst D66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Croll TI ISOLDE: A physically realistic environment for model building into low-resolution electron-density maps. Acta Cryst D74, 519–530 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liebschner D et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Cryst D75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kleijnen MF et al. Stability of the proteasome can be regulated allosterically through engagement of its proteolytic active sites. Nat. Struct. Mol. Biol 14, 1180–1188 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Roelofs J, Suppahia A, Waite KA, Park S (2018) Native Gel Approaches in Studying Proteasome Assembly and Chaperones. In: Mayor T, Kleiger G (eds) The Ubiquitin Proteasome System. Methods in Molecular Biology, vol 1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cryo-EM maps and atomic model coordinates have been deposited in the EMDB and RCSB respectively: 13S (EMD-23508, PDB 7LSX), Pre-15S (EMD-2350, PDB 7LS6), Pre3-1 20S (EMD-23502, PDB 7LS5). Additional structures referenced here include: PDB 4G4S, PDB 1RYP, PDB 2Z5C, and PDB 6FVY. Source data are available with the paper online.