

SUMMARY
Human cardiac regeneration is limited by low cardiomyocyte replicative rates and progressive polyploidization by unclear mechanisms. To study this process, we engineer a human cardiomyocyte model to track replication and polyploidization using fluorescently tagged cyclin B1 and cardiac troponin T. Using time-lapse imaging, in vitro cardiomyocyte replication patterns recapitulate the progressive mononuclear polyploidization and replicative arrest observed in vivo. Single-cell transcriptomics and chromatin state analyses reveal that polyploidization is preceded by sarcomere assembly, enhanced oxidative metabolism, a DNA damage response, and p53 activation. CRISPR knockout screening reveals p53 as a driver of cell-cycle arrest and polyploidization. Inhibiting sarcomere function, or scavenging ROS, inhibits cell-cycle arrest and polyploidization. Finally, we show that cardiomyocyte engraftment in infarcted rat hearts is enhanced 4-fold by the increased proliferation of troponin-knockout cardiomyocytes. Thus, the sarcomere inhibits cell division through a DNA damage response that can be targeted to improve cardiomyocyte replacement strategies.
Graphical Abstract

In brief
Pettinato et al. engineer human cardiomyocyte models to study replication and polyploidization using single-cell transcriptomics, chromatin-state analysis, and a CRISPR screen. This reveals how the sarcomere promotes polyploidization through enhanced oxidative metabolism, DNA damage, and p53. Exploiting this pathway improves in vivo cardiomyocyte replacement strategies.
INTRODUCTION
The adult human heart is characterized by insufficient regenerative capacity that is related to low rates of existing cardiomyocyte renewal (Bergmann et al., 2009) and lack of an appreciable progenitor pool (van Berlo et al., 2014). By contrast, the capacity for adult cardiac regeneration is well-established in other organisms, such as salamander (Becker et al., 1974) and zebrafish (Poss et al., 2002) and inversely correlates with cardiomyocyte polyploidization rates (>2n; more than two sets of homologous chromosomes). In zebrafish, it has been shown that regeneration by cardiomyocytes that are >95% diploid (2n) can be completely blocked by induction of polyploidization, supporting a causal link (González-Rosa et al., 2018). Polyploidization has also been demonstrated to regulate mammalian cardiac regeneration. For example, in a panel of 120 genetically diverse mouse strains, cardiomyocyte polyploidy rates were highly variable, genetically determined, and inversely related to cardiomyocyte proliferative responses induced by coronary artery ligation (Patterson et al., 2017). How mammalian cardiomyocyte polyploidization is regulated remains incompletely understood.
In humans, the majority of cardiomyocytes become mononuclear polyploid before the second decade of life. While both the total number of cardiomyocytes and nuclei per cardiomyocyte do not appreciably change, the average DNA content per cardiomyocyte increases 1.7-fold (Bergmann et al., 2015). This pattern of mononuclear polyploidy suggests that the human cardiomyocyte lacks the capacity to complete mitosis. If cardiomyocytes could be coaxed to complete mitosis, cardiomyocyte renewal could be enhanced to provide new treatment options for individuals who suffer from heart failure due to insufficient cardiomyocyte numbers, such as that caused by myocardial infarction (MI) or congenital heart disease. Patterns and mechanisms of human cardiomyocyte polyploidization appear to be distinct from other mammalian models such as the mouse, in which >90% of cardiomyocytes are multinuclear, likely through a block in cytokinesis (Patterson et al., 2017). Due to the lack of a model for human cardiomyocyte polyploidization and limitations in obtaining human heart samples, there is a large gap in our knowledge regarding the molecular mechanisms underlying human cardiomyocyte polyploidization (Figure 1A).
Figure 1. Engineering a human cardiomyocyte model to study polyploidization.

(A) In vivo RNA-seq analysis of adult cardiomyocyte nuclei relative to fetal (Gilsbach et al., 2018) demonstrates downregulation of G2/M cyclins CCNB1 and CCNB2 with persistence of G1/S cyclins CCND1 and CCND2.
(B) Overview of CRISPR methods to generate CCNB1-eGFP, TNNT2-T2A-NeoR, and TNNT2-mCherry iPSC lines to study polyploidization in differentiated cardiomyocytes (CMs).
(C) Representative flow cytometry plots demonstrating that iPSCs relative to CMs express more uniform CCNB1 levels that progressively increase in S-phase and peak in G2/M.
(D) Flow cytometry quantification of CCNB1-eGFP levels and Hoechst stain in CMs at differentiation day 20 to 40 shows progressive increase in DNA content paralleling inhibition of CCNB1.
(E) Representative time-lapse confocal images of live CCNB1-eGFP and cTnT-mCherry CM cell-cycle progression and cytokinesis. White arrow denotes a CM that undergoes cytokinesis into two daughter CMs. Scale bar, 10 μm.
(F) Replicative outcomes of 83 CCNB1+ CMs from time-lapse confocal imaging (green denotes CCNB1-eGFP expression and localization).
(G) CM mitosis relates to peak CCNB1-eGFP levels from time-lapse confocal imaging (n = 27 CCNB1+ CMs).
Data are n ≥ 3 and mean ± SEM; significance assessed by ANOVA with Holm-Sidak correction (D and G) and defined by *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001. See also Figure S1 and Video S1.
To address this deficit, we engineered a cellular model to study human cardiomyocyte replication and polyploidization using induced pluripotent stem cell (iPSC) technology and cyclin B1 (CCNB1) as a reporter of replicative status. We employed live-cell imaging, single-cell RNA sequencing (scRNA-seq), and chromatin-state analysis to comprehensively characterize molecular determinants of polyploidization including transcriptional signatures, signaling pathways, and gene regulatory motifs. Using a CRISPR screen adapted to study human cardiomyocytes, we identified the tumor suppressor p53 as an enhancer of cardiomyocyte polyploidization through CCNB1 inhibition. Our study implicates sarcomere assembly and function as an activator of p53 signaling through a DNA damage response secondary to oxidative stress related to metabolic reprogramming. As proof of concept, we exploited these findings to enhance human cardiomyocyte replication and promote remuscularization in an in vivo rodent model of cardiac engraftment after MI.
RESULTS
Characterizing human cardiomyocyte replication and polyploidization in vitro
To develop a model to study human cardiomyocyte replication and polyploidization, we first analyzed RNA-seq data from human cardiomyocyte nuclei isolated from fetal and adult hearts, including samples from heart failure patients (Gilsbach et al., 2018). We observed that adult cardiomyocytes maintain expression of G1/S cyclins CCND1 and CCND2 but not G2/M cyclins CCNB1 and CCNB2, which diminish with age and do not increase with stress such as heart failure (Figure 1A). While the cell cycle is regulated at complex transcriptional and post-transcriptional levels (Jensen et al., 2006), this pattern is consistent with the observation that adult human cardiomyocytes may retain some capacity for DNA synthesis but not mitosis (Bergmann et al., 2009). As GFP fused to the C terminus of CCNB1 can be exploited to track live replicating cells in vitro (Hagting et al., 1998) and in vivo (Klochendler et al., 2012) and could provide a tool to understand CCNB1 regulation, we used CRISPR-Cas9 to fuse enhanced GFP (eGFP) to endogenous CCNB1 in a human iPSC line (Figure 1B). Into this CCNB1-eGFP iPSC line, we also fused either self-cleaving T2A-NeoR or mCherry to cardiac troponin T (cTnT; encoded by TNNT2) to allow iPSC-derived cardiomyocyte (CM) purification and live-CM imaging, respectively (Figures 1B and S1A). We confirmed that selection of TNNT2-T2A-NeoR CMs with G418 resulted in improved purity (Figures S1B and S1C) compared to a well-established metabolic selection method (Tohyama et al., 2013).
After confirming that GFP fused to CCNB1 did not alter iPSC replication rates and cell-cycle status (Figures S1F–S1H), we studied CCNB1-eGFP expression in both iPSCs and CMs costained with Hoechst to analyze ploidy. We could distinguish 4n cells that are in G2/M using CCNB1-eGFP expression status (denoted as CCNB1+). iPSCs expressed uniform levels of CCNB1 in early S phase that peaked in G2/M (Figures S1D and S1E), in accord with previous studies (Clute and Pines, 1999; Klochendler et al., 2012). Relative to iPSCs, CMs expressed distinctly different CCNB1 levels. CMs expressed lower CCNB1 levels in S phase and with greater heterogeneity in G2/M (Figure S1E). We also observed 4n+ (>4n) CCNB1+ CMs, suggesting progressive polyploidization. CMs, but not iPSCs, also contained 4n and 4n+ cells that were CCNB1−, which was not due to a transient cell-cycle arrest or CM doublets, as persistence of polyploidy was documented in CMs cultured for 48 h after fluorescence-activated cell sorting (FACS) (Figure S1I). To determine how CCNB1-eGFP expression related to other cell-cycle-regulated factors, we utilized flow cytometry to analyze CMs immunostained for cell-cycle markers and anti-GFP costain (Figures S1J and S1K). 45% of CCNB1+ CMs were in S-phase as determined using a 30-min pulse with 5-ethynyl-2'-deoxyuridine (EdU). 68% and 62% of CCNB1+ CMs expressed replication markers Ki67 and Aurora, respectively. Finally, 1.7% of CCNB1+ CMs were in M-phase as determined by phospho-Histone H3 (p-H3) expression (Preuss et al., 2003). Because CCNB1− CMs did not express these makers, we conclude that CCNB1-eGFP is a marker of S and G2/M phases as observed in other cell types (Zielke et al., 2014). We also segmented CCNB1+ CMs into CCNB1+high and CCNB1+low, which demonstrated that CCNB1+low CMs were enriched in S-phase, while CCNB1+high CMs were in enriched in G2/M-phase (Figures S1L and S1M), which has also been observed in other cell types (Innocente et al., 1999; Strauss et al., 2018).
To test known effectors of cardiomyocyte replication, we varied differentiation time (Figures 1C and 1D), ambient oxygen (Figure S1N), and insulin (Figure S1O) and performed knockdown of MEIS1 (Figure S1P), a repressor of in vivo mouse cardiomyocyte replication (Mahmoud et al., 2013). From differentiation day 20 to 40, the proportion of 4n CCNB1+ CMs progressively decreased, while 4n CCNB1− CMs increased. Hypoxia (2.5% O2), insulin, and MEIS1 knockdown all increased the relative proportion of 4n CCNB1+ CMs, in accord with previous replication studies (McDevitt et al., 2005; Nakada et al., 2017). These results demonstrate that CMs recapitulate age-dependent progressive CCNB1 inhibition and polyploidization and respond similarly to previously established regulators of in vivo cardiomyocyte replication.
To understand CM replicative outcomes and potential relationships with CCNB1 expression, we performed time-lapse confocal imaging studies of CCNB1-eGFP cTnT-mCherry iPSCs and CMs (Figure 1E; Video S1). We first observed that all iPSCs expressed CCNB1 in the uniform and oscillatory pattern observed in other cell types (Clute and Pines, 1999; McDevitt et al., 2005), starting in the cytoplasm followed by nuclear entry and degradation preceding cytokinesis. Among 83 CCNB1+ CMs analyzed, 75% lost CCNB1 expression prior to completing mitosis, either in the cytoplasm or immediately after nuclear entry, which is consistent with the greater CCNB1 expression heterogeneity observed in CMs by flow cytometry. Among the 21 CMs observed to undergo nuclear entry of CCNB1 followed by mitosis, nine completed cytokinesis, five became multinuclear, and seven were inconclusive within 24 h of observation (Figure 1F). As we observed high CCNB1 heterogeneity in 4n CCNB1+ CMs relative to iPSCs, we considered whether CCNB1 expression levels and localization may relate to CM replicative outcomes. In CMs that completed mitosis, we observed the highest CCNB1 levels that were associated with CCNB1 nuclear entry (Figure 1G), with CCNB1 levels being the lowest in CMs that degraded CCNB1 prior to nuclear entry and intermediate in CMs with nuclear CCNB1 entry without mitosis, in accord with CM immunostaining results (Figure S1M). Taken together, we quantified replicative outcomes of CCNB1+ CMs and demonstrated that 75% do not complete mitosis in association with low CCNB1 levels that results in mononuclear polyploidy. Another 11% of the CCNB1+ CMs complete cytokinesis to generate two daughter CMs, while 6% result in a multinuclear polyploid CM, which did not relate to CCNB1 levels (Figure S1Q). The high occurrence of mononuclear polyploidy that we observed in CCNB1+ CMs is consistent with studies from in vivo human cardiomyocytes (Mollova et al., 2013) and illuminates a previously undescribed mitotic checkpoint related to CM-specific CCNB1 expression heterogeneity that could potentially be targeted to enhance CM replication and reduce polyploidization.
CRISPR genetic screen identifies TP53 as a regulator of CM polyploidization and CCNB1
We next sought to identify genetic regulators of CM replication and polyploidization by implementing a CRISPR knockout screen. Because pooled genetic screens to study replication have commonly utilized cell lines with high replicative rates (Hart et al., 2015; Marcotte et al., 2012), we had to adapt our strategy to account for low replication and high polyploidization in CMs. Our approach to address these inherent limitations was to utilize the CCNB1-eGFP CM model to provide a method to distinguish 4n CCNB1+ CMs from 4n CCNB1– (cell cycle arrested) and 2n (Figure 2A). Using FACS, we could collect these three populations and identify genetic modifiers of polyploidization and CCNB1 expression by tracking population-dependent sgRNA enrichment or depletion (Figure 2B).
Figure 2. CRISPR screen identifies p53 as an activator of CM polyploidy and inhibitor of CCNB1.

(A) Schematic used to conduct CRISPR screen in CMs differentiated from iPSCs transduced with SpCas9 and sgRNA library, as well as CM collection strategy by FACS.
(B) Simplified distribution of three theoretical sgRNAs, showing sgRNA B (red) enriched in 4n CCNB1+ CMs and depleted in 4n CCNB1− relative to 2n, as it targets a putative activator of polyploidization and inhibitor of CCNB1 expression.
(C) All four sgRNAs targeting TP53 are enriched in 4n CCNB1+ CMs and depleted in 4n CCNB1− relative to 2n across three biological CRISPR sub-screen replicates.
(D) Representative immunoblots and (E) quantification demonstrate increased p53 and p21 levels in FACS-collected 4n CCNB1− CM protein lysates compared to 2n.
(F) Representative immunoblots and (G) quantification of p53, p21, phospho-CCNB1, and GAPDH protein levels illustrate that p53 knockdown (KD) by TP53 sgRNA compared to non-targeting (NT) control results in p21 reduction and CCNB1 induction.
(H) p53 KD increases the proportion of CCNB1+ CMs while decreasing the proportion of CCNB1− 4n CMs, as quantified by flow cytometry.
(I) Flow cytometry analysis of immunostained p53 KD CMs demonstrates increased positivity for EdU, Ki67, Aurora A, and p-H3.
(J) Time-lapse imaging outcomes of p53 KD CCNB1+ CMs that were treated with Cas9 + NT or TP53 sgRNA 5–7 days prior to FACS collection and imaging (n = 488 CMs from 3 experiments).
(K) Ratio of diploid:polyploid outcomes from time-lapse imaging of p53 KD CCNB1+ CMs in (J).
(L) Nutlin-3 decreases the proportion of CCNB1+ CMs while increasing the proportion of CCNB1− 4n CMs, as quantified by flow cytometry.
(M) Nutlin-3 decreases the proportion of EdU+, Ki67+, Aurora A+, and p-H3+ CMs.
(N) Time-lapse imaging outcomes of FACS-collected CCNB1+ CMs (post-G1/S) treated with either DMSO or Nutlin-3 prior to imaging (n = 416 CMs from 3 experiments).
(O) Ratio of diploid:polyploid outcomes from time-lapse imaging of Nutlin-treated CCNB1+ CMs in (N).
Data are n ≥ 3 and mean ± SEM; significance assessed by FDR-adjusted p (C) or t test (D–O) and defined by *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001. See also Figure S2 and Table S2.
To perform the screen, we transduced lentiviral SpCas9 into CCNB1-eGFP TNNT2-T2A-NeoR iPSCs. The genome-wide Brunello sgRNA library (Doench et al., 2016) was then transduced at a low multiplicity of infection to optimize for one sgRNA per iPSC, followed by selection and expansion over two passages to minimize sgRNA drift. We then differentiated iPSCs to CMs, purified by G418 selection, and sorted live, Hoechst-stained CMs by FACS to collect the three desired populations (Figure 2A). We sequenced sgRNA recognition sites and performed Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout (MAGeCK) (Li et al., 2014) to identify gene-specific sgRNAs enriched in 4n CCNB1+ and depleted in 4n CCNB1− relative to 2n CMs. We used this genome-wide screen to identify candidate genes that satisfied moderate stringency cutoffs (false discovery rate [FDR] <0.25; Table S2) and designed an sgRNA sub-library for independent replication with higher stringency (FDR <0.005; Table S2). Using this strategy, we identified the tumor suppressor p53 as both a promoter of CM polyploidy and repressor of the proportion of CMs that express CCNB1, as all four sgRNAs targeting TP53 were consistently enriched in 4n CCNB1+ and depleted in 4n CCNB− relative to 2n CMs (Figure 2C).
We next tested how p53 regulates the CM cell cycle. We started by quantifying protein levels of p53 and p21, a direct p53 transcriptional target (el-Deiry et al., 1993), in lysates obtained from 4n and 2n CCNB1− CMs. 4n relative to 2n CMs exhibited increased p53 and p21 levels (Figures 2D and 2E). We then studied a p53 knockdown (KD) model by transducing CMs with lentiviral SpCas9 and an sgRNA targeting TP53, which decreased p53 and p21 levels and increased levels of active, phosphorylated CCNB1 relative to treatment with a non-targeting (NT) control sgRNA (Figures 2F and 2G; Toyoshima-Morimoto et al., 2001). In accord with our CRISPR knockout screens, p53 KD increased the proportion of CCNB1+ CMs and reduced polyploidization (Figure 2H). Additionally, immunostaining for cell-cycle markers following p53 KD revealed increased positivity for Ki67, Aurora, and p-H3 (Figure 2I), as well as enhanced EdU incorporation. This result in CMs is consistent with studies in other cell types implicating p53 as an inhibitor of G1/S and G2/M checkpoints (Agarwal et al., 1995; Bunz et al., 1998; Innocente et al., 1999). To specifically address the role of p53 in G2/M checkpoint regulation, we FACS-enriched p53 KD and NT control CMs post-G1/S checkpoints by collecting CCNB1+ CMs. We then quantified CCNB1+ CM replicative outcomes using live-cell imaging as done previously (Figure 1F). We observed that p53 KD increased the proportion of mitotic CMs (Figure 2J) and the ratio of 2n relative to 4n CMs (Figure 2K).
To assess whether p53 activation could regulate CM cell-cycle checkpoints, we treated CMs with the p53 activator Nutlin-3 versus DMSO control (Figures S2A and S2B; Vassilev et al., 2004). Nutlin-3 treatment reduced the proportion of CCNB1+ CMs and increased polyploidization (Figure 2L). Nutlin-3 treatment also reduced Ki67, Aurora, and p-H3 expression, as well as EdU incorporation (Figure 2M), which confirmed that p53 activates G1/S and G2/M checkpoints in CMs. To specifically study the G2/M checkpoint, we again FACS-collected CCNB1+ CMs and immediately treated with Nutlin-3 or DMSO. We then performed time-lapse imaging to quantify replicative outcomes, which demonstrated a decrease in the proportion of mitotic CMs (Figure 2N) and the ratio of 2n relative to 4n CMs (Figure 2O). Taken together, these results demonstrate that p53 promotes CM polyploidization through G2/M checkpoint activation and CCNB1 repression.
Chromatin-state segmentation reveals enhancers associated with polyploidy
Gene regulatory sequences and changes in chromatin state are known to be important for cardiac development and disease (Gilsbach et al., 2018). To determine epigenetic changes associated with CM polyploidization, we analyzed FACS-collected 2n and 4n CCNB1− CMs by chromatin immunoprecipitation with DNA sequencing (ChIP-seq) to assess histone modifications. Each ploidy state was imputed in conjunction with Roadmap Epigenome data and segmented using a 25-state model, as we have previously employed (VanOudenhove et al., 2020; Wilderman et al., 2018). We generated expected numbers of each chromatin state (Figures S3A and S3B) and identified activation of early heart specification genes, such as NKX2–5 (Figure S3C). To assess how our CMs compared to primary human heart samples, we performed t-Distributed Stochastic Neighbor Embedding (tSNE) analysis of H3K27ac levels, typically associated with tissue-specific enhancer activation, at segments identified as enhancer states in CMs and Roadmap segmentations (Figure 3A). Globally, 2n and 4n CCNB1− CMs were similar, as their tSNE positions were inseparable. In addition, global CM enhancer activation clustered between in vivo adult heart (HRT) and fetal heart samples but were distinct from stem cells (ESCs) and non-CM ESC derivatives.
Figure 3. Polyploidy is associated with distinct enhancer activation states.

(A) tSNE created using H3K27ac signal at enhancers called by ChromHMM segmentation across 127 tissues including 2n and 4n CCNB1− CMs, Roadmap Epigenome, and cardiomyocyte nuclei isolated from human hearts (Gilsbach et al., 2018). Samples are color coded by tissue group.
(B) Plot of differential enhancer motif enrichment. Dot size reflects the percentage of enhancers containing the given motif and color represents the directionality and magnitude of the log p value difference between 2n and 4n enhancer enrichment. Indicated motifs are significantly enriched in at least one sample and have a differential ≥2 on log scale.
(C) GO term analysis of genes predicted to be regulated by enhancers increased in 2n and 4n CMs, as determined by GREAT.
(D) UCSC Genome Browser snapshot of the locus containing CDKN1A, a cyclin-dependent kinase inhibitor with regulatory elements overlapping p53 binding sites that are more activated in 4n compared to 2n CMs. At the top are p53 binding sites, chromatin-state segmentations for 2n and 4n CMs, followed by imputed signals for five histone marks, and a track indicating vertebrate conservation. ChIP-qPCR primer locations are denoted that relate to Figure 5K.
We next sought to determine whether regulatory elements were differentially activated between 2n and 4n CCNB1− CMs. To do this, we compared H3K27ac, H3K4me2, and H3K4me3 signals at promoter and enhancer chromatin-state segmentations (Figure 3B). The majority (~96%) of regulatory sequences had similar levels of activation in both ploidy states. Among differentially activated enhancers (Table S3), we found that 2n-biased enhancer segments were enriched in binding sites for factors regulating stem cell maintenance and proliferation, including SOX6 that has been shown to regulate cardiomyocyte development in vivo (Hagiwara et al., 2000), brachyury that is necessary for cardiogenic mesoderm differentiation (Herrmann et al., 1990), and FOX family members that regulate cell proliferation and mitosis (Laoukili et al., 2005; Figure 3B). Gene Ontology (GO) analysis of genes predicted to be regulated by these 2n-biased enhancers showed enrichment of genes related to stem cell differentiation and division (Figure 3C). In contrast, 4n-biased enhancer state segments were enriched for motifs including MEF2 (Myocyte Enhancer Factor 2) family members such as MEF2A, which activates muscle-specific genes required for normal heart function (Naya et al., 2002), as well as MEIS1, a known repressor of in vivo cardiomyocyte replication (Mahmoud et al., 2013). GO analysis supported these results, as putative 4n-biased enhancer target genes were enriched in functions related to cardiac chamber development and myofibril assembly (Figure 3C). 4n-biased enhancers were also enriched in binding sites for p53 and the antioxidant NRF2. For example, CDKN1A encodes for p21, a potent cyclin-dependent kinase inhibitor, which contains multiple 4n-biased enhancers overlapping previously validated p53 binding sites (Figure 3D; el-Deiry et al., 1993; Nguyen et al., 2018). In summary, CM polyploidization is associated with distinct chromatin regulatory element usage characterized, in part, by loss of stemness and gain of myofibril assembly, oxidative stress, and p53 signaling activation.
Single-cell transcriptomics to predict determinants of CM replication and polyploidization
To determine and exploit CM heterogeneity underlying polyploidization and replication, we studied the CCNB1-eGFP CM model using scRNA-seq. We FACS-collected 2n, 4n CCNB1+, and 4n CCNB1− CMs in 384-well plates (Figure 4A) and then generated single-cell transcriptomics data using a plate-based 3' end counting method (Table S4). Using Seurat (Butler et al., 2018), we visualized cell clusters using Uniform Manifold Approximation and Projection (UMAP) (Figure 4B). We next segregated the seven clusters by CCNB1 and ploidy status (Figure 4C) and observed that CMs clustered by CCNB1 expression, as clusters 4 and 6 were non-contiguous with the other clusters and uniformly expressed CCNB1 (Figure 4D) and other G2/M markers (Figure S4A), while 4n CCNB1+ CMs that expressed lower CCNB1 transcript levels were distributed across the other clusters. 4n CCNB1− CMs were abundant in clusters 0 and 1 (containing ~66% of polyploid CMs), and 2n CMs were more abundant in clusters 2, 3, and 5 (containing ~60% of diploid CMs) (Figure 4E). Additionally, we confirmed that single-cell expression heterogeneity was not related to changes in CM differentiation subtypes such as determined by cardiac chamber-specific marker expression across clusters (Figure S4B). Transcript and protein expression data also reflected the fetal-like differentiation state of CMs based on the predominance of skeletal (TNNI1; encodes ssTnI) relative to cardiac (TNNI3; encodes cTnI) troponin I isoforms (Figures S4B and S4C; Bedada et al., 2014).
Figure 4. scRNA-seq analyses predict mechanisms of polyploidization.

(A) Representative FACS plot of CCNB1-eGFP CMs stained with Hoechst to collect 2n, 4n CCNB1+, and 4n CCNB1− single-CMs in wells of 384-well plates.
(B) UMAP clustering analysis by Seurat uncovers seven CM clusters.
(C–E) Segregation of Seurat clusters by (C) CCNB1 and ploidy status identifies clusters abundant for 4n CCNB1− (0 and 1), 2n (3 and 5), and 4n CCNB1+ CMs (4 and 6), in accord with (D) CCNB1 transcript levels and (E) proportional breakdown of Seurat clusters by sorted cell type.
(F) Gene set enrichment analysis (GSEA) using the top 100 significantly upregulated genes (adjusted p ≤ 0.05) from clusters 4 and 6 (containing ~94% CCNB1+ CMs) identified transcripts related to the cell cycle, mitosis, and cell division.
(G) GSEA using the top 100 significantly upregulated genes from clusters 0 and 1 (containing ~66% of polyploid CMs) identified transcripts related to sarcomere function and structure, oxidative metabolism, and p53 signaling.
(H) Single CMs are displayed using UMAP overlapped with Slingshot pseudotime trajectories. Trajectory 1 begins in the diploid-abundant cluster 5 and ends in the CCNB1-abundant cluster 4.
(I and J) The top 100 genes defining trajectory 1 have (I) functions related to mitosis and the cell cycle, such as (J) G2/M checkpoint (CDK1 and UBE2C) and proliferation markers (MKI67).
(K–M) Trajectory 2 (K) begins in the diploid-abundant cluster 5 and ends in the polyploid-abundant cluster 0 and is (L) defined by genes related to sarcomere function and structure, oxidation, and p53 activity, such as (M) MYH6, TPM1, and CDKN1A, suggesting a causal relationship with polyploidization.
To identify transcriptional networks related to polyploidization, we next performed gene set enrichment analysis (GSEA) using the top 100 significantly upregulated genes (adjusted p ≤ 0.05) in the CCNB1-abundant and polyploid-abundant clusters (Table S4). The CCNB1-abundant clusters exhibited high levels of genes involved in mitosis, cell-cycle regulation, and cell division (Figure 4F). In contrast, polyploid-abundant clusters exhibited higher levels of genes related to p53 signaling and myofibril assembly (Figure 4G), in accord with our chromatin segmentation analysis, as well as mitochondrial function and oxidative metabolism, which have been shown to associate with murine polyploidization in vivo (Jiang et al., 2020; Puente et al., 2014), suggesting that these processes may also promote human CM polyploidization. In summary, we exploited expression heterogeneity to identify molecular signatures of CM polyploidization and CCNB1 regulation including oxidative stress, myofibril assembly, and p53 signaling activation.
We next exploited single-cell expression heterogeneity using Slingshot pseudotime analysis (Street et al., 2018) to generate cell trajectories to computationally predict molecular determinants of CM replication and polyploidization decisions. This analysis identified two independent trajectories: trajectory 1, which starts in diploid-abundant clusters and ends in CCNB1-abundant clusters (Figure 4H) and trajectory 2, starting in diploid-abundant clusters and ending in polyploid-abundant clusters (Figure 4K). Given that trajectories resembled polyploidization decisions, we used Slingshot to study the top 100 genes whose expression related to the two trajectories. GSEA of the genes defining the G2/M-abundant trajectory 1 identified mitosis, cell-cycle checkpoint, and proliferation markers (Figure 4I), such as CDK1, MKI67, and UBE2C (Figure 4J), which are highly expressed in the late pseudotime CMs of trajectory 1. We then performed this analysis on trajectory 2 to identify genes predicted to regulate polyploidization, which included muscle contraction and sarcomere genes (Figure 4L), such as MYH6 and TPM1 (Figure 4M), which increased with pseudotime in the polyploid-abundant clusters. Moreover, GSEA also identified genes upregulated by reactive oxygen species, p53 pathway activation such as GADD45B and CDKN1A (Figure 4M), and genes related to DNA damage-induced regulation of p53 (Childs et al., 2018; Monte et al., 2003). These results additionally implicate sarcomere function, DNA damage, and p53 pathway activation in the genesis of CM polyploidy.
Sarcomere function inhibits CCNB1 through p53 activation
To study the role of sarcomere function in CM replication and polyploidization, we engineered sarcomere assembly deficient CM models based on knowledge that the troponin (Tn) complex is necessary for sarcomere assembly and contractile function (Huang et al., 1999; Nishii et al., 2008). We used CRISPR-Cas9 to generate iPSC lines containing either a homozygous frameshift in TNNT2 (denoted cTnT-KO), as we have previously employed (Pettinato et al., 2020), or frameshifts in both TNNI1 and TNNI3 (denoted TnI-DKO). The engineered CMs showed the expected absence of their respective troponin isoforms (Figures 5E and 5G), and neither KO line visibly contracted, in contrast to wild-type (WT) CMs that spontaneously contract. Correspondingly, α-actinin immunofluorescence revealed that both KO lines had impaired sarcomere formation, lacking the well-formed Z-disks that result from myofibril bundling (Figure 5A). In support of our hypothesis, we observed decreased polyploidization in sarcomere-deficient CMs compared to controls (Figures 5B and 5C), increased CCNB1+ CMs (Figure 5D), and increased positivity of EdU, Ki67, Aurora, and p-H3 (Figure S5A). We next tested whether CCNB1 overexpression could prevent the high polyploidization and low CCNB1 observed in sarcomere-containing CMs. We transduced lentivirus encoding CCNB1 fused to a nuclear localization signal (NLS-CCNB1), as we found that nuclear CCNB1 promoted mitosis (Figure 1G), to provide overexpression co-incident with sarcomere assembly in WT CMs, which reduced CM polyploidization and increased the proportion of CCNB1+ CMs (Figures S5B). Taken together, these results demonstrate that myofibrils promote polyploidization, and this can be antagonized by enhancing CCNB1 levels.
Figure 5. Cellular and molecular consequences of sarcomere assembly.
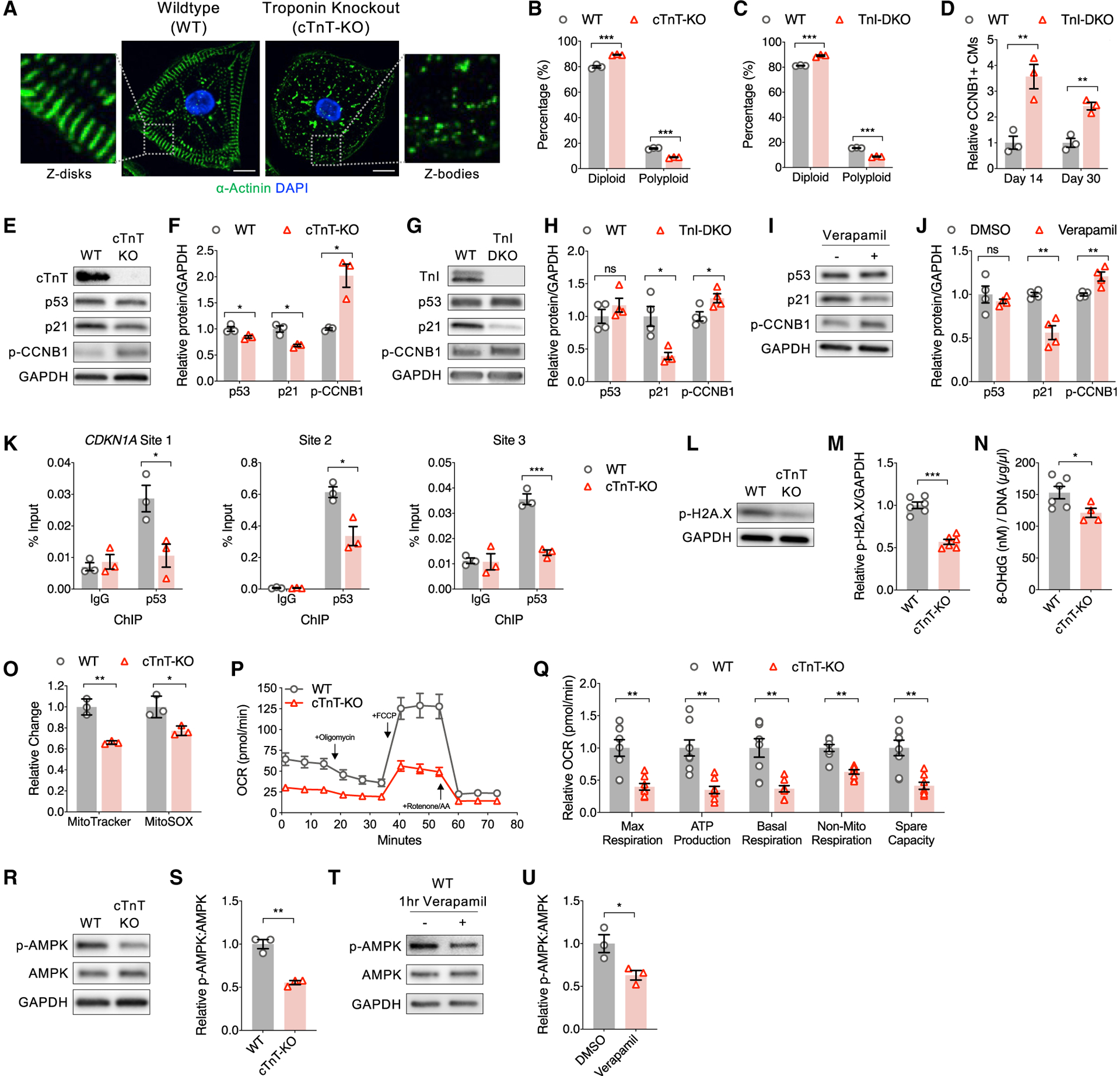
(A) Representative immunofluorescence images of wild-type (WT) control and cardiac troponin T knockout (cTnT-KO) CMs stained for α actinin (green; sarcomere Z-disk) and DAPI (blue; nuclei). KO of troponin, either cTnT-KO or double KO of skeletal and cardiac troponin I (TnI-DKO), leads to lack of striated sarcomere Z-disks. Scale bar, 10 μm.
(B and C) Flow cytometry of Hoechst-stained CMs demonstrates decreased polyploidy in (B) cTnT-KO and (C) TnI-DKO CMs relative to WT.
(D) Flow cytometry of CCNB1-stained WT and TnI-DKO CMs demonstrates increased proportion of CCNB1+ CMs with TnI-DKO, which reduces with age.
(E and F) Representative immunoblots (E) with (F) quantification of protein lysates from WT and cTnT-KO CMs probed for cTnT, p53, p21, phospho-CCNB1, and GAPDH.
(G and H) Representative immunoblots (G) with (H) quantification of protein lysates from WT and TnI-DKO CMs probed for TnI (cardiac and skeletal), p53, p21, p-CCNB1, and GAPDH.
(I and J) Representative immunoblots (I) with (J) quantification of protein lysates from day 20 WT CMs treated with verapamil from day 9 and probed for p53, p21, p-CCNB1, and GAPDH.
(K) Anti-p53 ChIP-qPCR of WT and cTnT-KO CMs targeting previously reported p53-bound ChIP-seq peaks directly upstream of CDKN1A (Nguyen et al., 2018), which demonstrates decreased p53 binding of CDKN1A in cTnT-KO CMs at three separate genomic sites (see Figure 3D). (L and M) Representative immunoblots probed for phospho-H2AX and GAPDH (L) with (M) quantification demonstrates sarcomere assembly activates a DNA damage response in WT CMs.
(N) Quantification of genomic DNA lysates from WT and cTnT-KO CMs probed via ELISA for 8-OHdG, a marker of oxidative DNA damage.
(O) Flow cytometry quantification of MitoTracker and MitoSOX dyes in WT and cTnT-KO CMs demonstrates reduced mitochondrial content and ROS in cTnT-KO CMs.
(P and Q) Seahorse Mito Stress oxygen consumption rates (OCR) (P) with (Q) quantification shows a decrease in respiration across all parameters in cTnT-KO CMs compared to WT, demonstrating that sarcomere assembly promotes oxidative metabolism.
(R and S) Representative immunoblots (R) with quantification (S) of protein lysates from WT and cTnT-KO CMs probed for p-AMPK, AMPK, and GAPDH.
(T and U) Representative immunoblots (T) with quantification (U) of protein lysates from WT CMs treated with verapamil for 1 h and probed for p-AMPK, AMPK, and GAPDH.
Data are n ≥ 3 and mean ± SEM; significance assessed by t test and defined by p > 0.05 (ns), *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001. See also Figure S5.
With multiple datasets implicating a link between the sarcomere and p53 activation in promoting polyploidization and CCNB1 inhibition, we analyzed WT, cTnT-KO, and TnI-DKO lysates for relevant protein marks. We confirmed increased p-CCNB1 and decreased p21 in both Tn-KO models (Figures 5E–5H), similar to our p53 KD studies. Unlike p53 KD, however, total p53 levels were not consistently different in Tn-KO CMs relative to WT. Additionally, treatment with verapamil, an L-type calcium channel blocker that inhibits sarcomere function (De Deyne, 2000; Lam et al., 2019), also demonstrated a similar molecular phenotype including increased p-CCNB1, decreased p21, and no change in total p53 protein levels (Figures 5I and 5J). As total p53 levels may not reflect p53 activity (Bode and Dong, 2004) and p21 can be activated by other factors (Jung et al., 2010), we additionally studied WT and cTnT-KO CMs by p53 ChIP-qPCR targeting known p53-enriched response elements on CDKN1A (Nguyen et al., 2018). Relative to WT, we found decreased enrichment of p53 at polyploidy-associated CDKN1A response elements in cTnT-KO CMs (Figure 5K), suggesting that inhibiting sarcomere assembly decreases p53 activity through a post-translational mechanism.
Direct molecular connections between sarcomere function and p53 activation have not been well established in human cardiomyocytes. We hypothesized that sarcomere function could regulate p53 through a DNA damage response, as sarcomere gene expression was related to activation of oxidative stress pathways by pseudotime (Figure 4L), and polyploid CMs exhibited increased oxidative signaling pathway activaion (Figure 4G). To test this, we measured levels of both phospho-H2AX, an epigenetic marker of DNA damage (Ayoub et al., 2008), and 8-oxo-2'-deoxyguanosine (8-OHdG), a DNA-level marker highly specific for oxidative damage (Du et al., 2016). Both markers exhibited decreased levels in the absence of sarcomere assembly (Figures 5L–5N). To assess potential sources of sarcomere-dependent oxidative damage, we quantified mitochondrial mass and superoxide production by flow cytometry analysis of MitoTracker- and MitoSOX-stained CMs, respectively (Figure 5O). We observed reductions in both parameters in the absence of sarcomere assembly, as well as reductions in oxygen consumption rates (Figures 5P and 5Q). To test whether inhibition of oxidative stress could rescue the reduced proportion of CCNB1+ CMs observed in sarcomere-containing CMs, we treated WT CMs with the antioxidant N-acetylcysteine (NAC). NAC increased the proportion of CCNB1+ CMs and reduced polyploidization (Figure S5C).
To identify potential molecular linkages between sarcomere function and oxidative metabolism, we hypothesized that sarcomere function-dependent changes in ATP hydrolysis could be sensed by AMP-activated protein kinase (AMPK), which is an activator of oxidative metabolism (Herzig and Shaw, 2018). To test this, we measured the levels of AMPK phosphorylation (p-AMPK) in cTnT-KO relative to WT CMs. We observed that cTnT-KO CMs exhibited reduced p-AMPK levels (Figures 5R and 5S), which was similarly observed after acute inhibition of sarcomere function using verapamil in WT CMs (Figures 5T and 5U). While AMPK can also be activated by calcium (Herzig and Shaw, 2018), we found no differences in calcium transients between cTnT-KO relative to WT CMs (Figure S5D), and verapamil treatment had no effect on p-AMPK levels in cTnT-KO CMs despite inhibition of calcium transients (Figures S5D–S5F). Taken together, these functional studies illuminate how the sarcomere promotes polyploidization through metabolic reprogramming in association with AMPK activation, oxidative stress, and, ultimately, p53 activation.
Inhibiting sarcomere function enhances CM engraftment and proliferation in a MI model
As sarcomere function decreased CM replication and increased polyploidization, we hypothesized that sarcomere inhibition could improve CM engraftment in a myocardial infarction (MI) rat model, since improving engraftment rates has been a long-standing obstacle for cell-therapy strategies (Laflamme et al., 2007; Zhu et al., 2018). To test this, rats underwent ischemia/reperfusion (I/R) surgery to induce MI, followed by injection of 1 × 107 human CMs (WT or TnI-DKO) 4 days later (Figure 6A), as we have previously described (Fernandes et al., 2010; Weyers et al., 2020). Bromodeoxyuridine (BrdU) was injected periodically to measure DNA synthesis, and rats were sacrificed 3 months post-transplantation for histological analysis. Sections of the left ventricle (LV) were immunohistochemically stained for β-myosin heavy chain (β-MHC), the predominant isoform expressed in human CMs, which was used to visualize the human-derived graft (Figure 6B). WT CMs produced a mean graft size of 0.11% ± 0.05% of the LV, while TnI-DKO CMs produced a size of 0.46% ± 0.10%, more than 4-fold larger (Figure 6C; p = 0.0041), demonstrating improved in vivo cardiac engraftment when transplanting TnI-DKO CMs. Additionally, immunofluorescence was performed on LV sections to assess proliferation of human CMs following in vivo engraftment using cumulative BrdU incorporation and Ki67 expression as markers. Sections were labeled for β-MHC (human CMs), Hoechst (nuclei), and either BrdU (Figure 6D) or Ki67 (Figure 6F). BrdU staining demonstrated that WT CMs were 0.40% ± 0.22% BrdU+, while TnI-DKO CMs were 1.52% ± 0.30% BrdU+ (Figure 6E; p = 0.012), and Ki67 staining demonstrated that WT CMs were 0.14% ± 0.08% Ki67+, whereas TnI-DKO CMs were 0.50% ± 0.10% Ki67+ (Figure 6G; p = 0.021). To also assess the contribution of cellular apoptosis to the engraftment phenotypes, we performed terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assays at 3 months post-engraftment, in which we observed no difference in apoptosis between WT and TnI-DKO CMs (Figure S6). We conclude that sarcomere function impairs CM engraftment and remuscularization after cell therapy, in part, through reduced replicative capacity.
Figure 6. Sarcomere-deficient CMs enhance in vivo cardiac engraftment.

(A) Overview of experimental workflow used to study cardiac engraftment of human CMs in a rat model of myocardial infarction (MI).
(B) Representative immunohistochemistry images and (C) quantification of rat left ventricle (LV) sections stained for human β-myosin heavy chain (β-MHC) to assess human CM graft size, which was increased when using TnI-DKO CMs compared to WT. Scale bar, 250 μm.
(D–G) Representative immunofluorescence images and quantification of rat LV sections probed for β-MHC (green; human CMs), Hoechst (blue; nuclei), and either (D and E) BrdU (magenta; cumulative proliferation) or (F and G) Ki67 (magenta; active proliferation), which show increased % BrdU+ and %Ki67+ human CMs when using TnI-DKO for engraftment, demonstrating enhanced proliferative capacity compared to WT CMs. Scale bars, 25 μm (D) or 50 μm (F).
Data are n ≥ 3 and mean ± SEM; significance assessed by t test and defined by *p ≤ 0.05 and **p ≤ 0.01. See also Figure S6.
DISCUSSION
The principal finding of this study is that human cardiomyocyte polyploidy and cell-cycle arrest are driven by downregulation of cyclin B1 by the tumor suppressor p53. We provide evidence that p53 is activated through a DNA damage response that is promoted by reactive oxygen species, which appear to result from mitochondrial metabolism that is enhanced by sarcomere contraction, the principal ATP-consuming process of the cardiomyocyte. We show that this pathway can be exploited to enhance cardiac remuscularization following cell transplantation in a rat model of myocardial infarction.
MIs are common events secondary to acute coronary artery blockages that result in large-scale cardiomyocyte death and progressive heart failure due to inadequate mechanical function (O’Gara et al., 2013). Low adult cardiomyocyte replication rates exacerbate this condition as yearly turnover rates have been estimated to be <1% (Bergmann et al., 2015). Human cardiomyocytes remain diploid during the first year of life, but age-dependent polyploidization and replicative arrest occur over the first 2 decades of life (Bergmann et al., 2015) by incompletely understood mechanisms. New knowledge of how to control endogenous human cardiomyocyte replication or to implant exogenous human cardiomyocytes as cell therapy could be transformative for these patients. To date, the majority of cardiomyocyte replication studies have focused on non-human model systems but have revealed that low ambient oxygen, low-pressure circulation, glycolytic metabolism, and absence of polyploidy enhance cardiac regenerative capacity (Vivien et al., 2016). The knowledge of the conservation of these replicative levers to human cardiomyocytes has lagged in part due to the lack of human model systems and the relative inaccessibility of viable human heart samples.
In this study, we utilized iPSC-derived cardiomyocytes modified to track CCNB1 as a model system to study mechanisms of human cardiomyocyte replication and polyploidization. Resembling in vivo cardiomyocytes, we observe that iPSC-derived cardiomyocytes undergo a time-dependent, progressive replicative arrest and polyploidization. The ~25% polyploidy that we observe at differentiation day 40 in this study is similar to what has been documented in ~8-year-old human hearts (Bergmann et al., 2015). Using time-lapse imaging of cardiomyocytes labeled with cTnT-mCherry and CCNB1-eGFP, we found that ~75% of CCNB1+ cardiomyocytes endocycle and become mononuclear polyploid in association with CCNB1 levels that are likely insufficient to reach the threshold required for nuclear entry and execution of mitosis, as recently described for other cell types in mouse embryos (Strauss et al., 2018). This pattern of polyploidization is distinct from rodent cardiomyocytes, which are mostly multinuclear polyploid, and further illustrates the unique replicative characteristics of human cardiomyocytes and the rationale for the establishment of a human cardiomyocyte model system.
We also uncovered molecular signatures and pathways associated with polyploidization using chromatin-state analysis and scRNA-seq. We found that, while ~96% of regulatory sequences were similarly activated between diploid and polyploid cardiomyocytes, polyploid samples exhibited increased activation of elements overlapping specific transcription factors including those involved in myofibrillogenesis such as MEF2 family members and oxidative stress such as NRF2 and TP53; polyploid CMs also showed reduced levels of early developmental factors like brachyury. In addition to providing transcriptional signatures and pathways associated with replication and polyploidization, scRNA-seq analyzed by pseudotime uncovered potentially causal relationships between the function of the sarcomere in cell-cycle regulation. While previous studies have implicated the sarcomere’s role in promoting oxidative metabolism (Ulmer et al., 2018) and cell-cycle arrest (Mills et al., 2017), our study utilized sarcomere-poor cardiomyocytes to directly link the sarcomere to metabolic reprogramming and polyploidization in human cardiomyocytes.
During time-lapse imaging studies, while we confirmed that sarcomere disassembly is a universal feature of cardiomyocytes undergoing mitosis, as has been observed in other studies (Ahuja et al., 2004), we observe that human sarcomere-containing cardiomyocytes fail to initiate the earliest stages of mitosis, as CCNB1 is degraded prior to nuclear entry and chromatin condensation does not occur. Moreover, we find that CCNB1 overexpression is sufficient to both reduce polyploidization and increase the proportion of G2/M cardiomyocytes, in accord with replication studies using non-human cardiomyocytes (Bicknell et al., 2004; Mohamed et al., 2018), though ploidy was not specifically addressed. The reduction in polyploidization that we observed indicates that the presence of sarcomere-containing myofibrils is not a total block to cytokinesis in our model, in contrast to what has been proposed in rodent models that have high rates of multinuclear polyploidy (Engel et al., 2006). Our study demonstrates that sarcomere-dependent CCNB1 inhibition appears to occur through transcriptional repression by p53 signaling, which, in turn, is activated by a DNA damage response as a consequence of increased oxidative metabolism. This is consistent with the previous finding that activation of p53 indirectly attenuates the CCNB1 promoter and induces G2/M arrest in other cell types through a p21-dependent mechanism (Fischer et al., 2016), as well as other studies that have implicated p53 activity in regulating mouse cardiomyocyte cell cycling (Nakajima et al., 2004; Pasumarthi et al., 2001) and polyploidy in mouse hepatocytes (Kurinna et al., 2013). While we could find no changes in histone methylation or acetylation at the CCNB1 promoter in polyploid cardiomyocytes relative to diploid cardiomyocytes, our data support a model whereby p53 activation results in progressive and ultimately irreversible arrest of human cardiomyocyte mitosis through CCNB1 inhibition.
To understand the functional relevance of sarcomere-dependent polyploidization and replicative arrest, we studied cardiomyocytes that were rendered non-contractile and sarcomere-poor by KO of troponin I in an in vivo cell-therapy model of MI. We found that impaired sarcomere function enhanced cardiomyocyte engraftment in the heart by 4-fold and enhanced proliferation by >3-fold. While we observed no change in apoptosis between WT and TnI-DKO conditions, we cannot exclude that differential survival may occur at earlier time points nor that other factors additionally contribute to the enhanced engraftment rate of TnI-DKO CMs. As delivery of in vitro differentiated cardiomyocytes to the injured heart is an alternative to coaxing endogenous cardiomyocyte regeneration (Chong et al., 2014), our study demonstrates that improved replicative capacity and reduced polyploidization can enhance engraftment, though future studies will need to assess the functional outcomes of the enhanced graft size produced by TnI-DKO cardiomyocytes. Additionally, inhibition of sarcomere assembly did not completely abolish polyploidization nor the age-dependent reduction in CCNB1, indicating that other mechanisms contributing to cardiomyocyte polyploidization need to be investigated, potentially such that polyploidization can be minimized while maintaining sarcomere function through the discovery of additional levers that coax cardiomyocyte replication. In summary, our study provides a comprehensive assessment of human cardiomyocyte replication, refines new sarcomere crosstalk with cell-cycle regulation through engagement with p53 signaling, and provides a list of therapeutic targets that could be exploited to coax endogenous cardiomyocyte replication or enhance cardiac cell therapy for cardiac regenerative medicine applications.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Requests for information and resources should be directed to the Lead Contact, Dr. J. Travis Hinson (travis.hinson@jax.org).
Materials availability
Materials generated in this study are available from the Lead Contact upon reasonable request.
Data and code availability
The Gene Expression Omnibus (GEO) accession numbers for the next-gen sequencing data generated in this study are as follows: GEO: GSE147417 for CRISPR screen data; GEO: GSE130285 for ChIP-seq data; and GEO: GSE147249 for scRNA-seq data.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
iPSC handling and directed differentiation
The parental human iPSC lines used for this study included PC1 for all CCNB1-eGFP studies (Hinson et al., 2016), PGP1 for all cTnT-KO studies (Coriell Institute Biorepository GM23338) (Cohn et al., 2019; Hinson et al., 2016; Hinson et al., 2015; Pettinato et al., 2020), and WTC-11 for all TnI-DKO studies (Gladstone UCSFi001-A). All iPSC lines were assessed by karyotype analysis for genomic integrity and maintained in mTeSR1 (STEMCELL Technologies 85850) on Matrigel-coated tissue culture plates (Corning 354230). Media was replenished daily, and cells were passaged at a 1:6 ratio using Accutase (BD 561527) and 10 μM ROCK inhibitor Y-27632 (Tocris 1254) once they reached 80%–90% confluency. PGP1 and PC1 iPSCs were differentiated into CMs through modulation of Wnt/β-catenin signaling as previously described (Lian et al., 2012). Briefly, Day 0 differentiation was initiated with 12 μM CHIR99021 (Tocris 4423) for 24 hours in RPMI 1640 (GIBCO 11875093) supplemented with B27 minus insulin (GIBCO A1895601) and GlutaMAX (GIBCO 35050061). On Day 3 of differentiation, cells were treated with 5 μM IWP-4 (Tocris 5214) for 48 hours. On Day 9, cells were maintained in RPMI with B27 supplement (GIBCO 17504044). On Day 13, metabolic enrichment was performed with glucose-free DMEM (GIBCO 11966025) supplemented with 4 mM lactate (Sigma 71718) for 24–48 hours, as previously described (Tohyama et al., 2013). Alternatively, the TNNT2-T2A-NeoR line (CCNB1-eGFP studies only) was enriched via antibiotic selection with 10 μg/mL Geneticin G418 sulfate (GIBCO 10131035) in RPMI-B27. Following selection, CMs were trypsinized (GIBCO 25200056) and re-plated onto fibronectin-coated tissue culture plates (GIBCO 33016015), unless noted otherwise. CMs were replenished with RPMI-B27 every other day. CM analysis was performed on Day 25–35 unless noted otherwise. For all PGP1 cTnT-KO CM experiments, PGP1 CMs were used as WT control.
WTC-11 iPSCs were maintained and differentiated similarly as above, with the following modifications. Cells were maintained in mTeSR1 and passaged with Versene (Thermo 15040066). On differentiation Day −1, media was changed to 1 μM CHIR99021(Cayman 13122) in mTeSR1. On Day 0, media was changed to 5 μM CHIR99021 in RPMI supplemented with 500 μg/mL BSA (Sigma A9418) and 213 μg/mL ascorbic acid (Sigma A8960), denoted RBA media. On Day 2, cells were washed with PBS and treated with 2 μM Wnt-C59 (Selleck S7037) in RBA media. On Day 4, media was changed to RBA without small molecules. On Day 6, media was changed to RPMI-B27, which was replenished every other day. CMs underwent metabolic selection on Day 13 with glucose-free DMEM supplemented with 4 mM lactate for 48 hours. Following lactate treatment, CMs were trypsinized and re-plated onto fibronectin-coated tissue culture plates (Fisher 33-016-015) in RPMI-B27 containing 10 μM Y-27632 + 5% FBS (BioWest S1620). RPMI-B27 was replenished every other day until analysis. For all WTC-11 TnI-DKO CM experiments, WTC-11 CMs were used as WT control.
METHOD DETAILS
Plasmid cloning
The homologous recombination (HR) targeting vector (System Biosciences HR220PA-1) was modified prior to being used in genome editing experiments. First, constitutively-expressed RFP was removed in order to prevent conflicting background fluorescence. Next, T2A-NeoR and mCherry cassettes were obtained as IDT gBlocks and were used to replace the eGFP cassette in the parent vector via HiFi DNA Assembly (NEB E2621). TNNT2 and CCNB1 ~800bp 5' and 3' HR arm gBlocks were then cloned into the appropriate backbones using the built-in multiple cloning sites. Finally, the EF-1α core-driven hygromycin-resistance cassette was replaced with a zeocin-resistance cassette to enable separate antibiotic resistance for clonal selection (hygromycin for TNNT2-T2A-NeoR and TNNT2-mCherry, and zeocin for CCNB1-eGFP). All HR vector propagation steps were performed in DH5α E.coli (NEB C2987). All CRISPR and primer sequences used for this study are provided in Table S1.
Individual lentiviral single-guide RNAs (sgRNA) were designed using https://zlab.bio/guide-design-resources, obtained from IDT, cloned into BsmBI-digested lentiGuide-Puro (Addgene 52963), and used in conjunction with lentiviral Cas9 (Dharmacon CAS10138) for gene knockdown, as previously described (Sanjana et al., 2014; Shalem et al., 2014). All lentiviral vectors were propagated in Stbl3 E.coli (Invitrogen C737303). The Brunello human genome-wide sgRNA knockout library (76,441 sgRNAs targeting 19,114 human genes and 1,000 non-targeting controls) was obtained as a pooled plasmid library in lentiGuide-Puro (Addgene 73178) and amplified according to the recommended protocol from the Broad Institute’s Genetic Perturbation Platform (Doench et al., 2016). The custom sub-library containing a 2,032 sgRNA subset from the Brunello library targeting 483 genes and 100 controls was ordered from IDT, ligated into lentiGuide-Puro with NEB HiFi DNA Assembly, and amplified in Stbl4 E.coli (Invitrogen 11635018).
CRISPR/Cas9 genome editing
Isogenic iPSC genetic modifications were engineered using a modified CRISPR/Cas9 protocol adapted from previous studies (Cohn et al., 2019; Hinson et al., 2015; Pettinato et al., 2020). 8×106 PC1 iPSCs were co-electroporated with 20 μg pCas9-GFP (Addgene 44719), 20 μg of the appropriate hU6-driven sgRNA (designed using https://zlab.bio/guide-design-resources), and 20 μg of the appropriate HR targeting vector to generate TNNT2-T2A-NeoR, CCNB1-eGFP, or TNNT2-mCherry knock-ins. Cells and plasmids were mixed in 800 μL PBS, transferred to a 4 mm cuvette (Bio-Rad 1652088), and electroporated (250V, 500uF, ∞ resistance, 4mm; Bio-Rad Gene Pulser II). Electroporated cells were transferred to a Matrigel-coated 100 mm dish containing mTeSR1 and 10 μM Y-27632. The following day, selection was started with the appropriate antibiotic (50 μg/mL Invitrogen Hygromycin B or 50 μg/mL GIBCO Zeocin) to eventually isolate single iPSC clones, which were then manually picked, expanded, and screened via Sanger sequencing. Generation of the PGP1 cTnT-KO CM model was previously described (Pettinato et al., 2020).
To generate a TnI-DKO CM model, double isogenic knockout of TNNI1 and TNNI3 in WTC-11 iPSCs was performed as follows. sgRNAs targeting TNNI1 and TNNI3 were designed using the online CRISPR design tool (https://zlab.bio/guide-design-resources) and ligated into the PX459v2 (Addgene 62988) vector (Cas9–2A-Puro). 3×105 WTC-11 iPSCs were transfected with 1 μg TNNI1-PX459v2 vector using GeneJuice (Sigma 70967) and selected with 0.5 μg/mL puromycin for 2 days beginning the day after transfection. After selection, cells were re-plated to obtain and genotype single-cell colonies. After obtaining a homozygous frameshift mutation in TNNI1, the same procedure was repeated for TNNI3.
Flow cytometry analysis
Flow cytometry analysis was performed using a BD Biosciences FACSymphony A5 or BD FACSCanto running FACSDiva software. On the day of live cell analysis, cells were dissociated and incubated for 30 min at 37°C suspended in RPMI-B27 containing 10 μM Hoechst 33342 (Thermo 62249), 200 nM MitoTracker Green (Invitrogen M7514), and/or 2.5 μM MitoSOX Red (Invitrogen M36008), where indicated. Cells were then washed in PBS, suspended in RPMI-B27 containing 500 nM TO-PRO-3 (Invitrogen T3605), and then filtered into a 35-μm cell strainer snap cap FACS tube (Corning 352235). Cells were gated on the basis of forward-scatter and side-scatter, TO-PRO-3 to identify only live cells, and Hoechst-area versus Hoechst-height for doublet discrimination. For analysis of CCNB1-eGFP cells, Hoechst and eGFP gating was used for cell cycle and ploidy analysis. For mitochondrial analysis, Hoechst was used to gate for single cells, followed by subsequent analysis of mitochondrial content (MitoTracker) and oxidative stress (MitoSOX).
For analysis by intracellular staining, suspended cells were fixed with 4% paraformaldehyde for 15 min at room temperature and then incubated for 1 hour at room temperature with 1:100 primary antibody and 10 μM Hoechst 33342 in PBS + 5% FBS + 0.75% saponin. After a spin down and wash, cells were immediately analyzed or, if using unconjugated primary antibodies, incubated for 45 minutes at room temperature with 1:200 secondary antibody, either goat anti-mouse 488 (Invitrogen A11001) or goat anti-rabbit 594 (Invitrogen A11012), in PBS + 5% FBS + 0.75% saponin. Final analysis was performed using FlowJo software. The primary antibodies used were as follows: mouse anti-cyclin B1 (Invitrogen MA5–14319), rat anti-GFP 488 (BioLegend 338008), rat anti-phospho-Histone H3 (Ser28) 647 (BioLegend 641005), mouse anti-Ki67 PE-Cy7 (BD 561283), and rabbit anti-Aurora A (Cell Signaling 14475). 5-Ethynyl-2'-deoxyuridine (EdU) incorporation was detected using the Click-iT EdU 647 flow cytometry kit (Invitrogen C10635) according to the manufacturer’s protocol following a 30 min pulse with 10 μM EdU.
Live cell sorting (FACS) and collection for CRISPR screen, ChIP-seq, scRNA-seq, and other experiments was performed using a BD Biosciences FACSAria Fusion or FACSymphony S6 CT running FACSDiva software. Samples were processed as described above prior to sorting. Cells were sorted into RPMI-B27 containing 2% FBS.
Drug treatment experiments
For analysis of the cell cycle response to disparate oxygen conditions, CCNB1-eGFP CMs were incubated in normoxia (20% O2, 5% CO2) or hypoxia (2.5% O2, 5% CO2) conditions for 48 hours, followed by flow cytometry analysis. For analysis of insulin response, CCNB1-eGFP CMs were cultured in RPMI-B27 without insulin for 48 hours, followed by culturing in RPMI-B27 with or without insulin for 48 hours before flow cytometry analysis. For Nutlin-3 treatment, CCNB1-eGFP CMs were cultured in RPMI-B27 containing either DMSO or 10 μM Nutlin-3 in DMSO for 24 hours before flow cytometry analysis. For chronic verapamil treatment, WT CMs were cultured in RPMI-B27 containing either DMSO or 200 nM verapamil in DMSO starting at Day 9 of cell differentiation and continuing to protein lysis on Day 20. For acute verapamil treatment, replated CMs were treated with either DMSO or 200 nM verapamil in DMSO for 1 hour prior to protein lysis. For N-acetyl cysteine (NAC) treatment, CCNB1-eGFP CMs were cultured in RPMI-B27 containing either DMSO or 500 nM NAC in DMSO starting at Day 9 of cell differentiation and continuing to flow cytometry analysis on Day 20.
Lentivirus production
24 hours prior to transfection, 9×106 HEK293T cells (ATCC CRL-3216) were seeded onto 150 mm plates in 20 mL DMEM (GIBCO 11965092) supplemented with 10% FBS (Gemini 100–106), GlutaMAX (GIBCO 35050061), and 1 mM sodium pyruvate (GIBCO 11360070). The next day, cells were co-transfected with 18 μg of the desired lentiviral transfer vector, 12 μg psPAX2 (Addgene 12260), and 6 μg pCMV-VSV-G (Addgene 8454). The plasmids were pre-mixed with 2 mL Opti-MEM (GIBCO 31985062) and 162 μg polyethylenimine (PEI) and incubated at room temperature for 20 minutes. Meanwhile, 293T were switched into 18 mL fresh media, followed by gentle dropwise addition of the incubated transfection mixture. Media was replenished the following day and virus-containing media was harvested at 48, 72, and 96 hours post-transfection, followed by concentration using PEG-6000 as previously described (Kutner et al., 2009).
Individual gene knockdown and overexpression experiments
Lentiviral-based CRISPR/Cas9 was used to individually knockdown genes directly in CMs. Cas9-expressing CMs were transduced with lentivirus expressing sgRNA (non-targeting, TP53, or MEIS1) at a MOI of 2 in RPMI-B27. Media was replenished the following day. CMs were analyzed 5–7 days post transduction via western blot and/or flow cytometry, as described above.
NLS-tagged CCNB1 cDNA was cloned into a lentiviral vector (derived from Addgene 17448) under the constitutive expression of the EF-1a core promoter. The construct contained a hemagglutinin (HA) tag, and an empty HA-only vector was used as a control. Lentiviral particles were produced as outlined above. For overexpression, CCNB1-eGFP CMs were transduced with control or NLS-CCNB1 lentivirus at a MOI of ~2 on Day 9 of differentiation. Flow cytometry analysis was performed on Day 20 to determine the proportion of CCNB1+ and polyploid CMs.
Time-lapse imaging
To determine CCNB1+ replicative outcomes and expression status, CCNB1-eGFP cTnT-mCherry CMs were subjected to time-lapse confocal imaging. CMs were plated on fibronectin-coated imaging dishes (MatTek P35G-1.5–14-C) and time-lapse imaging was performed the following day on an Andor Dragonfly 500 microscopy system in confocal mode (40x or 63x oil-immersion objective) using a Zyla sCMOS camera and an Okolab enclosure to control temperature (37°C) and pH (5% CO2). Images were taken every 15 minutes for 16–24 hr. Using ImageJ, CCNB1+ CMs were categorized as mononuclear polyploid if CCNB1-eGFP expression was lost either in the cytoplasm or immediately after nuclear entry. For CMs with sustained nuclear CCNB1-eGFP, mitotic status was determined by monitoring nuclear morphology for condensation and mitotic commitment by exploiting CCNB1-eGFP’s previously known localization to the mitotic apparatus (Clute and Pines, 1999). After nuclear CCNB1-eGFP was lost, cytokinesis or multinuclear polyploidization were determined by monitoring cTnT-mCherry morphology to assess successful cellular division or presence of multiple nuclei, respectively. An example CM undergoing mitosis and cytokinesis is provided in Figure 1E and Video S1. To quantify CCNB1-eGFP levels from confocal images, mean cellular CCNB1-eGFP signal was measured in ImageJ for all CCNB1+ CMs that exhibited nuclear CCNB1-eGFP entry, as well as CCNB1+ CMs without nuclear entry within the same field of view.
For comparative time-lapse imaging studies using Nutlin-3 and p53 knockdown, CCNB1-eGFP cTnT-mCherry CMs were subjected to FACS to collect live cTnT+ CCNB1+ CMs (post-G1/S), which were seeded onto fibronectin-coated imaging dishes. After waiting 2–3 hr for cell attachment, time-lapse imaging was performed on an Andor Dragonfly 500 microscopy system in widefield mode (20x air objective) using a iXon EMCCD camera and an Okolab enclosure to control temperature (37°C) and pH (5% CO2). Replicative outcomes were determined as described above, and statistical analysis was conducted using one-tailed, ratio-paired t tests. Nutlin-3 experiments were performed by treating three independent differentiation and sort batches of CMs with DMSO or 10 μM Nutlin-3 just prior to the start of imaging. For p53 knockdown, CRISPR editing (using lentiviral Cas9 + NT versus p53 sgRNA) was performed 5–7 days prior to sorting of three independent differentiation batches. Both imaging experiments included 200 nM TO-PRO-3 in order to identify and exclude dead cells. CMs with indeterminate replicative outcomes were also excluded from this analysis.
CRISPR/Cas9 screen
CCNB1-eGFP TNNT2-T2A-NeoR iPSCs (3×106) were transduced with Cas9 lentivirus at a MOI of < 0.1 and selected with 2 μg/mL blasticidin (GIBCO A1113903) in mTeSR1. After two passages, the lentiGuide-Puro sgRNA library (Brunello genome-wide or sub-library) was transduced into 1×108 Cas9-expressing iPSCs at a MOI of < 0.1 and selected with 1 μg/mL puromycin (GIBCO A1113803) in mTeSR1. The library iPSCs were passaged two times (p2) and cryopreserved in aliquots containing 2×107 iPSCs. These p2 aliquots were thawed onto 150 mm plates, grown to ~90% confluency, and plated at a seeding density of 8×106 on 100 mm plates (p3). Once confluent, p3 library iPSCs were differentiated into CMs, subjected to G418 selection, re-plated, and then processed for FACS as described above. Three populations were collected: CCNB1- diploid (2n), CCNB1- polyploid (4n), and CCNB1+. Three separate differentiation batch replicates were processed and collected. Genomic DNA (gDNA) was isolated from sorted cell pellets (QIAGEN 69506) and sgRNAs were amplified for next-generation sequencing (NGS) analysis according to the recommended protocol from the Broad Institute’s Genetic Perturbation Platform. Each 50 μL PCR reaction consisted of a maximum of 2 μg gDNA, 0.5 μL of Takara Ex Taq DNA polymerase (Takara RR001C), 5 μL of 10x Ex Taq buffer, 4 μL dNTPs, 0.25 μL of 100 μM P5 stagger-mix forward primer, 5 μL of 5 μM P7 barcode reverse primer, and water to 50 μL total. The PCR cycling conditions were: 1 minute at 95°C, followed by 30 s at 95°C, 30 s at 53°C, 30 s at 72°C, for 29 cycles; and a 10-minute extension at 72°C. PCR reactions were electrophoresed, gel extracted (QIAGEN 28706), and quantified using a Qubit dsDNA high sensitivity assay kit (Invitrogen Q32854) on a Qubit 4 Fluorometer. Samples were then sequenced on an Illumina NextSeq 550 and analyzed using MAGeCK software (Li et al., 2014). The GEO accession number for the CRISPR screen data reported in this paper is GSE147417.
Western blotting
To obtain protein lysates, plated cells were washed once in PBS and then lysed in ice-cold RIPA buffer (Cell Signaling 9806) containing protease inhibitor cocktail (Roche 11836170001), 1 mM PMSF, and phosphatase inhibitor (Pierce A32957). Lysates were centrifuged to remove cell debris, quantified and normalized via Pierce BCA (Thermo 23225), and then reduced and denatured in sample buffer (Thermo 39000). Protein lysates were separated on 4%–20% Mini-PROTEAN TGX precast gels (Bio-Rad 4561095), transferred onto PVDF membranes (Bio-Rad 1704272), washed once in TBS-T (50 mM Tris-Cl, 150 mM NaCl, 0.1% TWEEN-20), blocked for 1 hour in TBS-T with 5% BSA (Fisher BP1605) or nonfat milk, and then probed overnight with primary antibody in TBS-T with BSA or milk, depending on the antibody manufacturer’s recommendation. The following day, blots were washed three times in TBS-T for 15 minutes, probed for 1 hour at room temperature with HRP-linked secondary antibody (Cell Signaling 7076; 7074), and then washed three times in TBS-T for 15 minutes. Signal detection was performed using ECL substrate (Thermo 34580) and a Bio-Rad ChemiDoc MP imaging system. Blot images were digitally processed and analyzed in either Bio-Rad Image Lab or ImageJ. The primary antibodies used were as follows: 1:1000 mouse anti-p53 (Cell Signaling 48818), 1:1000 rabbit anti-p21 (Cell Signaling 2947), 1:1000 rabbit anti-phospho-CCNB1 (Cell Signaling 4133), 1:1000 rabbit anti-GAPDH (Cell Signaling 5174), 1:500 mouse anti-cardiac troponin T (Invitrogen MA5–12960), 1:1000 rabbit anti-troponin I (Santa Cruz sc-15368; detects both cardiac (cTnI) and skeletal (ssTnI) isoforms), 1:1000 rabbit anti-phospho-Histone H2A.X (Cell Signaling 9718), 1:1000 rabbit phospho-AMPKα (Thr172) (Cell Signaling 2535), and 1:1000 rabbit AMPKα (Cell Signaling 5832).
ChIP-seq
Fixed CM pellets were processed for ChIP as previously described (Cotney and Noonan, 2015). Briefly, samples were thawed in 1 mL of 1x Cell Lysis buffer and incubated on ice for 20 minutes. Cells were lysed with dounce homogenization and nuclei were collected by centrifugation (5 min, 2500 g, 4°C). Nuclei were resuspended in 300 μL of 1x Nuclear Lysis buffer + 0.3% SDS + 2 mM sodium buty-rate and incubated on ice for 20 minutes. Chromatin was sheared with a Qsonica Q800R1 sonicator system operating at amplitude 20 and 2°C for 30 minutes (10 s duty, 10 s rest). Samples were cleared by centrifugation (5 min, 20,000 g, 4°C) and soluble chromatin was transferred equally into seven separate tubes with 10% reserved as an input control. SDS concentration was reduced to 0.18% with ChIP-seq Dilution buffer. Protein G Dynabeads (ThermoFisher) separately preloaded with 2.5–5 μg of antibodies were added to each chromatin aliquot. Antibodies used in this study were as follows: anti-H3K27ac (C15410196, Diagenode), anti-H3K4me1 (C15410194, Diagenode), anti-H3K4me2 (AB7766, Abcam), anti-H3K4me3 (C15410003, Diagenode), anti-H3K27me3 (C16410195, Diagenode), anti-H3K9me3 (C15410193, Diagenode) and anti-H3K36me3 (C15410192, Diagenode). All Diagenode antibodies came pre-validated for ChIP, and the antibody from Abcam (H3K4me2) was validated using Absurance H3 Histone Peptide Array (16–667, Millipore). ChIP samples were incubated overnight at 4°C on a rotisserie. The chromatin was then immunoprecipitated on a magnet and the supernatant was discarded. Beads were washed 8 times with 1 mL of 500 mM LiCl ChIP-Seq Wash Buffer and once with 1 mL of TE. Chromatin was eluted from the beads twice with ChIP Elution buffer at 65°C for 10 minutes with constant agitation. Combined eluates for each ChIP were subjected to crosslink reversal overnight at 65°C. Samples were then sequentially treated with RNase A and proteinase K, purified with a PCR Purification Kit (QIAGEN), and eluted in 40 uL of EB. ChIP samples were then quantified with picoGreen (ThermoFisher) and ChIP-seq libraries were prepared (R400427, Takara), quantified by qPCR (NEB E7630L), multiplexed, and sequenced for 75 cycles across multiple flow cells on an Illumina NextSeq 500 instrument using a NextSeq 500/550 High Output v2 kit (75 cycles, Cat No. FC-404-2005).
ChIP-seq data analysis
Quality control was performed on ChIP-seq reads using FastQC (version [v.] 0.11.5) and MultiQC (v.1.1). Trimming for adapters, quality and length was performed using Trimmomatic (v.0.36) for single end data. ChIP-seq reads were aligned to the human genome (hg19) using Bowtie2 (v. 2.2.5) (Langmead and Salzberg, 2012). Fragment sizes of each library were estimated using PhantomPeak-QualTools (v.1.14) (Landt et al., 2012). We then generated P-value-based signal tracks relative to appropriate input controls based on estimated library fragment size using MACS2 (2.1.1.20160309) (Feng et al., 2012). Bedgraph files for all P-value signals from primary ChIP-Seq data were converted to 25 bp resolution and processed for model training and generation of imputed signals for all samples using ChromImpute (v1.0.3) as previously described (Ernst and Kellis, 2015). Resulting imputed signal tracks were converted to bigWig format for display in UCSC genome browser and converted for use with ChromHMM (v1.12) (Ernst and Kellis, 2012) using ChromImpute’s ExportToChromHMM. Signal files for individual chromosomes for each epigenome were binarized and segmentation was performed using the previously published 25-state chromatin models using ChromHMM as previously described (Kundaje et al., 2015). Following segmentation, annotation of states and generation of genome browser files was performed based on annotations provided by Roadmap Epigenome. The GEO accession number for the ChIP-seq signals, imputed signal files, and chromatin state segmentations reported in this paper is GSE130285.
Differential regulatory site activation and motif enrichment
To identify putative regulatory elements that are differentially utilized between mononuclear and multinucleated cardiomyocytes we compared H3K27ac, H3K4me2, and H3K4me3 signals at promoter and enhancer chromatin state segmentations independently using DiffBind (v2.10; https://bioconductor.org/packages/DiffBind in R (v3.4.1). For a specific chromatin signal, uniquely aligned reads from three replicates of each type of cardiomyocyte were quantified and normalized for input signal at enhancer segments (states 13 through 18 from 25 state model) or promoter segments (states 1 through 4 and 22) using fragment sizes determined by phantom-peakqualtools (Kharchenko et al., 2008; Landt et al., 2012) and the DBA_SCORE_TMM_MINUS_FULL_CPM function of DiffBind. Differential signals were determined by DiffBind using DESeq2 and filtered for a false discovery rate less than 0.1. Differentially enriched regions were assigned to the single nearest gene up to 1 Mb away and resulting gene lists were assessed for gene ontology (GO) enrichments using GREAT (McLean et al., 2010). All results from GREAT were retrieved programmatically using rGREAT (v1.14; https://bioconductor.org/packages/rGREAT. De novo and known motif enrichment in differentially activated regions for each histone modification were determined using HOMER with the options “-size given -len 8,10,12,14 -mask -gc” (v4.9) (Heinz et al., 2010). Resulting HOMER output files were loaded into R using homerkit (https://github.com/slowkow/homerkit) and −log10 transformed P-values for each motif were compared between regions more active in mononuclear versus multinucleated cardiomyocytes.
Global multi-tissue comparisons of H3K27ac signals
To qualitatively assess similarity of cardiomyocytes generated here to other tissues throughout the human body, we first assembled a list of all enhancer states (states 13 through 18) from 127 tissues profiled by Roadmap Epigenome, FACS-collected cardiomyocyte nuclei isolated from fetal and adult heart tissue (Gilsbach et al., 2018), and previously profiled samples from our laboratory (Wilderman et al., 2018). We next extracted imputed H3K27ac signals from all samples at all enhancer regions using the multiBigwigSummary command from DeepTools (v3.1.2) (Ramírez et al., 2016) excluding all regions blacklisted by ENCODE. The resulting signal matrix were filtered to remove regions where signal was low (> 10) across all samples (n = 146) and log10 transformed. This transformed matrix was used to calculate the Euclidean distance between each sample. The resulting distance matrix was then processed for t-Distributed Stochastic Neighbor Embedding using the Rtsne package (v0.15; https://github.com/jkrijthe/Rtsne) using options “is_distance = true, perplexity = 10, theta = 0.5, dims = 2, max_iter = 1000.” The x and y dimensions were combined with sample and group labels for plotting with ggplot2 in R.
Single-cell transcriptomics
CCNB1-eGFP and TNNT2-T2A-NeoR CMs for scRNA-Seq were processed as described above in the Flow Cytometry Analysis section. For cell sorting, 2n (n = 384 CMs), 4n (n = 384 CMs), and CCNB1+ (n = 384 CMs) were collected into individual 384-well plates using sorting strategy demonstrated in Figure S1D. We utilized an in-house 384-well plate-based approach for 3' end counting of transcripts of single cells individually sorted into each well. Each well incorporates a unique reverse transcription (RT) primer (AAGCAGTGGTATCAACGCAGAGTAC[12-bp well bar code][8-bp UMI][Tx30]VN) incorporating a well bar code and unique molecular identifier (UMI) (IDT custom order). scRNA-seq transcript metrics are summarized in Table S4. All RT reagents were dispensed using an Echo 525 acoustic liquid handler for a final total reaction volume of 1 ul. First and second strand synthesis was performed using Maxima H Minus reverse transcriptase (Thermo EP0751) and template switching at 42°C for 90 min. Reactions from each well were subsequently pooled into a single tube assisted by a Caliper liquid handler. The pooled libraries were treated with ExoI (NEB M0293L) at 37°C for 45 min to trim single-stranded ends and then cleaned up with AMPure beads (Beckman Coulter A63881). Amplification of libraries was for 12 cycles using KAPA HiFi-based (Kapa Biosystems KM2602) PCR followed by AMPure bead clean up with quantity and quality evaluated on Agilent Bioanalyzer. cDNA (1 ng) was fragmented and indexed through Nextera XT Library Prep Kit (tagmentation) (Illumina 15032354), followed by PCR with indexing primers for sequencing with a final AMPure bead clean up and bioanalyzer analysis prior to sequencing.
All plates were prepared on the same day with identical reagents, and all libraries were sequenced in a single Illumina NextSeq 500 run using a 75-cycle mid-output kit. Paired-end FASTQs were generated using BCL2Fastq v2.18.0.12 (Illumina) where the first read contains a 12bp cellular barcode and 8bp UMI and the second read contains 60bp of the mRNA 3' end. Digital expression matrices were constructed for each pair of FASTQs using Drop-seq_tools v1.13 (http://mccarrolllab.org/dropseq) in the following manner: (A) bam creation with Picard (v2.9.3) FastqToSam; (B) cell and UMI tagging, filtering, trimming with Drop-seq_tools TagBamWithRead-SequenceExtended, FilterBAM, TrimStartingSequence, PolyATrimmer; (C) alignment with STAR (v2.5.4a) to GRCh38_74 genome and Picard (v2.9.3) SortSam; (D) merging and tagging with Picard (v2.9.3) MergeBamAlignment and Drop-seq_tools (v1.13) TagReadWithGeneExon; (E) expression matrix creation with Drop-seq_tools (v1.13) DigitalExpression. Eight cells were removed due to high expression of alpha-fetoprotein (AFP), a lineage marker of endodermal differentiation.
Analysis of expression was performed with Seurat (Butler et al., 2018). Briefly, the UMI count matrix was normalized across genes using a centered log ratio transformation. Cells were then clustered using the FindNeighbors() and FindClusters() commands with a resolution of 0.8. Non-linear dimensional reduction with Uniform Manifold Approximation and Projection (UMAP) was then performed using RunUMAP() with 15 dimensions. Differential gene expression was performed using FindMarkers(). The differentially expressed genes were filtered for adjusted P-value ≤ 0.05 and then gene set enrichment analysis (GSEA) was performed using the Molecular Signature Database (Subramanian et al., 2005) to identify enriched Hallmark, Gene Ontology (GO), and Reactome gene sets. Pseudotime analysis was performed with Slingshot (Street et al., 2018) using the Seurat object as input, which identified two independent pseudotime trajectories. The top 1000 highly variable genes from Seurat were used as the input to predict pseudotime values using random forest machine learning in order to find genes deemed most important to predicting pseudotime. The top 100 genes from this analysis were used for GSEA of each trajectory. The GEO accession number for the scRNA-seq data reported in this paper is GSE147249.
Immunofluorescence
For immunofluorescence images, cells were fixed on coverslips (Fisherbrand 12–545-100) in PBS containing 4% paraformaldehyde (EMS 50–980-487), washed three times in PBS, permeabilized and blocked in PBS-T (PBS containing 0.1% Triton-X) with 1% BSA (Fisher BP1605). Cells were probed with 1:400 mouse anti-alpha actinin primary antibody (Sigma A7811) in PBS-T with BSA overnight at 4°C, washed in PBS-T, incubated for 1 hour at room temperature in PBS containing 1 μg/mL DAPI (Invitrogen D1306) and 1:400 goat anti-mouse 488 secondary antibody (Invitrogen A-11008), washed, and mounted onto slides (Corning 2948) in ProLong Diamond mountant (Invitrogen P36965). Slides were imaged on an Andor Dragonfly 500 confocal microscope system using a Zyla sCMOS camera and a Leica DMi8 63x oil immersion lens. Images were acquired using Andor Fusion and analyzed in ImageJ.
ChIP-qPCR
WT and cTnT-KO CMs were fixed and cross-linked using 1% formaldehyde for 15 min at room temperature with gentle tilt-shaking. The formaldehyde was then quenched by adding glycine to a final concentration of 125 μM and incubating for 5 min at room temperature with gentle tilt-shaking. The cells were then washed twice with ice-cold PBS and processed using the iDeal ChIP kit for Transcription Factors (Diagenode C01010055), according to the manufacturer’s protocol. For shearing, sample lysates were sonicated for 12 cycles using a Branson 250 sonicator at 30% power for 20 s with 60 s cool-down between each cycle. The ChIP-verified antibodies used for immunoprecipitation were IgG control (Cell Signaling 2729) and anti-p53 (Cell Signaling 48818). For qPCR of p53 binding sites on CDKN1A regulatory elements (Nguyen et al., 2018), input (1%) and immunoprecipitated DNA samples were diluted 1:10 and 5 μL were used per 20 μL qPCR reaction using Fast SYBR Green (Applied Biosystems 4385612) and run on a ViiA 7 Real-Time PCR System. Ct values were used to calculate the percent input (IP relative to input) using the following formula: %Input = 100*2^((Ct input - 6.644) - Ct IP). Primers used for qPCR are listed in Table S1.
Oxidative DNA damage assay
Genomic DNA was extracted (QIAGEN 69506) from WT and cTnT-KO CMs, quantified by Qubit dsDNA high sensitivity assay kit (Invitrogen Q32854) on a Qubit 4 Fluorometer, and assayed by ELISA to quantify 8-hydroxy-2'-deoxyguanosine (8-OHdG) using the HT 8-oxo-dG ELISA Kit II (Trevigen 4380–096-K), according to the manufacturer’s protocol.
Seahorse OCR assay
WT and cTnT-KO CMs were plated on Seahorse XFe96 Cell Culture Microplates (Agilent 101085–004) at a density of 3×104 CMs per well and cultured for 5 days before performing the Seahorse XF Cell Mito Stress Test (Agilent 103015–100) according to manufacturer’s protocol. Compounds were injected at the following final concentrations and assay times: 1 μM oligomycin at minute 18, 1 μM FCCP at minute 36, and 0.5 μM Antimycin A/Rotenone at minute 54. The test was conducted on an Agilent Seahorse XFe96 Analyzer running the Seahorse Wave Desktop software.
Calcium transients
Analysis of cellular calcium transients was performed using the red genetically-encoded calcium indicator RGECO, as previously described (Pettinato et al., 2020; Sparrow et al., 2019). WT and cTnT-KO CMs were seeded onto glass-bottom dishes and transduced at a MOI of ~0.3 with RGECO lentivirus two days prior to imaging. For imaging, RGECO-expressing CMs were exposed to pacing conditions of 1 Hz using a C-Pace EP stimulator (IonOptix) and C-Dish (IonOptix). Fluorescence videos were acquired using an Andor Dragonfly microscopy system equipped with an enclosed live-cell chamber and iXon EMCCD camera in 561-RFP laser widefield mode. Ten minutes prior to imaging, cells were treated with DMSO or 200 nM verapamil in DMSO. Whole cell fluorescence time series data were acquired in ImageJ using the Z axis profile function, followed by automated signal analysis using a custom Python script (github.com/TheJacksonLaboratory/hinson_excel_signal2).
Cryopreservation and cell preparation for transplantation
Cardiomyocytes used for in vivo studies were cryopreserved on Day 20–22 of differentiation and thawed immediately prior to cell injection, following our previously described protocol (Gerbin et al., 2015; Laflamme et al., 2007). One day prior to cryopreservation, cells were heat-shocked for 30 minutes at 42°C, treated with 10 μM Y-27632 for 1 hour, and dispersed with 0.25% trypsin in EDTA. Cells were spun down, resuspended in CryoStor (Sigma C2874) at 1×107 cells/mL, and frozen in cryovials. To thaw cryopreserved cells, cryovials were thawed briefly at 37°C, followed by addition of RPMI-B27 + 200 U/mL DNase. Cells were washed and resuspended in an RPMI-based pro-survival cocktail containing 50% (vol/vol) growth factor-reduced Matrigel, 100 μM ZVAD (benzylox-ycarbonyl-Val-Ala-Asp(O-methyl)-fluoro-methyl ketone, Millipore 627610), 50 nM Bcl-XL BH4 (cell-permeant TAT peptide, Millipore 197217), 200 nM cyclosporine A (Novartis), 100 ng/mL IGF-1 (Peprotech 100–11), and 50 μM pinacidil (Sigma P154).
Ischemia/reperfusion injury and cell transplantation
All animal procedures were conducted in accordance with the National Institutes of Health Policy on Humane Care and Use of Laboratory Animals and the University of Washington Institutional Animal Care and Use Committee (IACUC). Fourteen rats were enrolled per experimental group (injection of WT control versus TnI-DKO CMs). The protocol for ischemia/reperfusion surgery has been previously published (Gerbin et al., 2015; Laflamme et al., 2007). Briefly, male athymic Sprague-Dawley rats (Harlan/Envigo) were anesthetized with intraperitoneal injection of 68.2 mg/kg ketamine and 4.4 mg/kg xylazine, and were intubated and mechanically ventilated. To maintain body temperature of 37°C, the animals were placed on a heating pad with a rectal probe. A thoracotomy was performed and the left anterior descending coronary artery (LAD) was ligated for 60 minutes, re-perfused, and the chest was aseptically closed. Animals underwent a second thoracotomy 4 days after ischemia/reperfusion injury under 5% isoflurane supplemented with oxygen. Animals were randomly assigned to one of the two experimental groups. Each animal in the treatment group received 1×107 CMs resuspended in 100 μL pro-survival cocktail. Cell suspension was injected into three different areas within the infarct – one into the center of the infarct and two in the lateral infarct border zone. After cell injection, animals received subcutaneous injections of 5 mg/kg Cyclosporine A for seven consecutive days starting the day before cell transplantation. To assess proliferation in the grafts, all animals received intraperitoneal injection of 50 mg/kg BrdU (Sigma B5002) on days 1, 7, 30, 60, and 90 post-cell transplantation.
Immunohistochemical analysis
Histological stains and subsequent analysis were conducted as described previously by our group (Gerbin et al., 2015; Laflamme et al., 2007). Briefly, hearts were perfused with PBS and 150 mM KCl solution after harvesting, fixed overnight in 4% paraformaldehyde, sliced into 2 mm thick sections, processed, sectioned, and stained with appropriate primary and secondary antibodies. To visualize grafts, sections were incubated overnight with mouse β-MHC antibody (Developmental Studies Hybridoma Bank A4.951, supernatant) followed by a 1-hour incubation with biotin-SP goat anti-mouse antibody (Jackson ImmunoResearch 115–065-003, 1:500) and developed with diaminobenzadene (DAB, Vector Labs PK-6100) for brightfield images. To quantify cell proliferation in the grafts, sections were incubated overnight with β-MHC antibody followed by a 1-hour incubation with mouse anti-mouse 488 (Invitrogen A11001). Subsequently, sections were incubated overnight with either Ki67–647 antibody (BD 558615; 1:20) or peroxidase-conjugated anti-BrdU primary antibody (Roche 1585560, 1:40) followed by AF647 tyramide (Invitrogen B40916) to amplify BrdU and 10 μM Hoechst 33342. Cardiomyocyte proliferation was quantified by counting β-MHC+ and either BrdU+ or Ki67+ double-stained cells from images captured on a Nikon A1R confocal microscope.
Apoptotic cells were identified with Click-iT Plus terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay with Alexa Fluor 594 dye (Invitrogen C10618) using the manufacturer’s protocol. Briefly, paraffin-embedded tissues sections were deparaffinized initially in xylene followed by rehydration in serial dilutions of ethanol. Slides were then fixed in 4% paraformaldehyde for 15 minutes at 37°C, washed twice in PBS, and permeabilized with Proteinase K at room temperature for 15 minutes. After permeabilization, slides were fixed in 4% paraformaldehyde for 5 minutes at 37°C. Positive control slides were treated with DNase I and incubated at room temperature for 30 minutes. Terminal deoxynucleotidyl transferase reaction and Click-iT Plus reaction were performed using the components in the kit. Slides were then incubated with 10 μM Hoechst 33342 for 30 minutes in the dark. To identify grafts, serial sections were deparaffinized and incubated overnight with mouse β-MHC antibody, followed by a one hour incubation with 1:100 goat anti-mouse 488 secondary and 30 minutes incubation with 10 μM Hoechst 33342 in the dark. Apoptotic cells were quantified by counting TUNEL+ nuclei in the β-MHC+ positive graft regions from images captured on a high-resolution widefield microscope.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed and graphed using a combination of R and GraphPad Prism. Data are presented as mean ± standard error of the mean (SEM). All experiments were conducted with three or more biological replicates (n ≥ 3). Statistical comparisons were conducted via two-tailed unpaired Student’s t test or ANOVA corrected for multiple comparisons using Holm-Sidak post-test, unless noted otherwise. Statistical outcomes were defined as p > 0.05 (ns; not significant), p ≤ 0.05 (*), p ≤ 0.01 (**), and p ≤ 0.001 (***).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Phospho-Cyclin B1 (S133) | Cell Signaling | Cat. #4133; RRID:AB_2072264 |
| Cyclin B1 | ThermoFisher | Cat. #MA5-14319; RRID:AB_10987286 |
| cTnT-647 | BD Biosciences | Cat. #565744; RRID:AB_2739341 |
| cTnT | ThermoFisher | Cat. #MA5-12960; RRID:AB_11000742 |
| TnI | Santa Cruz | Cat. #sc-15368; RRID:AB_793465 |
| Alpha-actinin | Sigma | Cat. #A7811; RRID:AB_476766 |
| Alpha-actinin | Abcam | Cat. #AB9465; RRID:AB_307264 |
| p53 (ChIP-validated) | Cell Signaling | Cat. #48818; RRID:AB_2713958 |
| p21 | Cell Signaling | Cat. #2947; RRID:AB_823586 |
| GAPDH | Cell Signaling | Cat. #5174; RRID:AB_10622025 |
| Phospho-H2AX (S139) | Cell Signaling | Cat. #9718; RRID:AB_2118009 |
| Phospho-AMPK (Thr172) | Cell Signaling | Cat. #2535; RRID:AB_331250 |
| AMPK | Cell Signaling | Cat. #5832; RRID:AB_10624867 |
| GFP-488 | BioLegend | Cat. #338008; RRID:AB_2563288 |
| Phospho-Histone H3 (Ser28) 647 | BioLegend | Cat. #641005; RRID:AB_1279419 |
| Ki67-PE-Cy7 | BD | Cat. #561283; RRID:AB_10716060 |
| Aurora Kinase A | Cell Signaling | Cat. #14475; RRID:AB_2665504 |
| b-MHC | DSHB | Cat. #A4.951; RRID:AB_528385 |
| Peroxidase-conjugated anti-BrdU | Roche | Cat. #11585860001; RRID:AB_514485 |
| Ki67-647 | BD | Cat. #558615; RRID:AB_647130 |
| Goat anti-mouse PE | Jackson | Cat. #115-116-072; RRID:AB_2338627 |
| Goat anti-mouse 488 | Invitrogen | Cat. #A11001; RRID:AB_2534069 |
| Goat anti-mouse 594 | Invitrogen | Cat. #A11005; RRID:AB_2534073 |
| Goat anti-rabbit 594 | Invitrogen | Cat. #A11012; RRID:AB_2534079 |
| Rabbit anti-mouse 488 | Invitrogen | Cat. #A11059; RRID:AB_2534106 |
| Biotin-SP goat anti-mouse | Jackson ImmunoResearch | Cat. #115-065-003; RRID:AB_2338557 |
| Goat anti-rabbit IgG HRP | Cell Signaling | Cat. #7074; RRID:AB_2099233 |
| Goat anti-mouse IgG HRP | Invitrogen | Cat. #A11008; RRID:AB_143165 |
| H3K27ac (ChIP-validated) | Diagenode | Cat. #C15410196; RRID:AB_2637079 |
| H3K4me1 (ChIP-validated) | Diagenode | Cat. #C15410194; RRID:AB_2637078 |
| H3K4me2 (ChIP-validated) | Abcam | Cat. #AB7766; RRID:AB_2560996 |
| H3K4me3 (ChIP-validated) | Diagenode | Cat. #C15410003; RRID:AB_2616052 |
| H3K27me3 (ChIP-validated) | Diagenode | Cat. #C15410195; RRID:AB_2753161 |
| H3K9me3 (ChIP-validated) | Diagenode | Cat. #C15410193; RRID:AB_2616044 |
| H3K36me3 (ChIP-validated) | Diagenode | Cat. #C15410192; RRID:AB_2744515 |
| IgG control (ChIP-validated) | Cell Signaling | Cat. #2729; RRID:AB_1031062 |
| Critical commercial assays | ||
| NextSeq 500/550 v2 | Illumina | Cat. #FC4042005 |
| Absurance H3 Histone Peptide Array | Millipore | Cat. #16667 |
| SMARTer ThruPLEX DNA-seq | Takara | Cat. #R400427 |
| NEBNext Library Quant kit | New England BioLabs | Cat. #E7630 |
| Nextera XT Library Prep kit | Illumina | Cat. #15032354 |
| HiFi DNA Assembly | New England BioLabs | Cat. #E2621 |
| Seahorse XF Cell Mito Stress | Agilent | Cat. #103015-100 |
| Ex-Taq polymerase | Takara | Cat. #RR001C) |
| Qubit dsDNA kit | Invitrogen | Cat. #Q32854 |
| iDeal ChIP Kit for Transcription Factors | Diagenode | Cat. #C01010055 |
| Fast SYBR Green Master Mix | Applied Biosystems | Cat. #4385612 |
| HT 8-oxo-dG ELISA kit | Trevigen | Cat. #4380-096-K |
| Click-iT Plus EdU Alexa Fluor 647 Flow Cytometry Assay Kit |
Invitrogen | Cat. #C10635 |
| AF647 tyramide | ThermoFisher | Cat. #B40916 |
| Click-iT Plus TUNEL Alexa Fluor 594 Assay Kit |
Invitrogen | Cat. #C10618 |
| Experimental models: Cell lines and animals | ||
| PGP1 human iPSCs | Coriell | Cat. #GM23338 |
| PC1 human iPSCs | Christine Seidman, Harvard | (Hinson et al., 2016) |
| WTC-11 human iPSCs | Gladstone, UCSF | A gift from Dr. Bruce Conklin |
| Sprague-Dawley rats | Harlan/Envigo | N/A |
| HEK293T | ATCC | Cat. #CRL3216 |
| Oligonucleotides | ||
| Brunello sgRNA library | Addgene | Cat. #73178 |
| HR220PA | SBI | Cat. #HR220PA-1 |
| EditR-Cas9 | Dharmacon | Cat. #CAS10138 |
| psPAX2 | Addgene | Cat. #12260 |
| pCMV-VSV-G | Addgene | Cat. #8454 |
| LentiGuide-Puro | Addgene | Cat. #52963 |
| pCas9-GFP | Addgene | Cat. #44719 |
| pLenti CMV GFP Puro | Addgene | Cat. #17448 |
| PX459v2 | Addgene | Cat. #62988 |
| Oliognucleotides for custom sgRNA library | IDT | Custom order |
| Oligonucleotides for scRNA-seq UMIs | IDT | Custom order |
| Software and algorithms | ||
| MAGeCK | Bitbucket | https://bitbucket.org/liulab/mageck/src/master/ |
| DiffBind | Bioconductor | https://bioconductor.org/packages/DiffBind |
| rGREAT | Bioconductor | https://bioconductor.org/packages/rGREAT |
| Homerkit | GitHub | https://github.com/slowkow/homerkit |
| Rtsne | GitHub | https://github.com/jkrijthe/Rtsne |
| Seurat | GitHub | https://github.com/satijalab/seurat |
| STAR (v.2.5.4a) | GitHub | https://github.com/alexdobin/STAR |
| Molecular Signature Database | Broad | https://www.gsea-msigdb.org/gsea/msigdb/index.jsp |
| EdgeR | Bioconductor | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| CellView | GitHub | https://github.com/mohanbolisetty/CellView |
| GraphPad Prism | GraphPad Software | http://survey-smiles.com |
| FlowJo | FLOWJO | https://flowjo.com |
| Fiji | NIH | https://fiji.sc |
| Drop-seq_tools | GitHub | https://github.com/broadinstitute/Drop-seq/releases/tag/v1.13 |
| Calcium signal analysis | (Pettinato et al., 2020) | https://github.com/TheJacksonLaboratory/hinson_excel_signal2 |
| Chemicals, peptides, and recombinant proteins | ||
| IWP4 | Tocris | Cat. #5214 |
| CHIR99021 | Tocris | Cat. #4423 |
| Y-276932 | Tocris | Cat. #1254 |
| Nutlin-3 | Sigma | Cat. #444151 |
| Fibronectin (human) | Corning | Cat. #33016015 |
| Matrigel GFR | Corning | Cat. #354230 |
| Insulin (human) | Sigma | Cat. #91077C |
| Verapamil | Tocris | Cat. #0654 |
| NAC | Alfa Aesar | Cat. #A1540914 |
| GeneJuice Transfection Reagent | EMD Millipore | Cat. #709675 |
| GoTaq Flexi DNA polymerase | Promega | Cat. #M8297 |
| ROCK inhibitor Y-27632 | Tocris | Cat. #1254 |
| CHIR99021 | Cayman | Cat. #13122 |
| Wnt-C59 | Selleck | Cat. #S7037 |
| Fibronectin | Fisher | Cat. #33-016-015 |
| Benzyloxycarbonyl-Val-Ala-Asp(O-methyl)-fluoro-methyl ketone (ZVAD) | Millipore | Cat. #627610 |
| Bcl-XL BH4 | Millipore | Cat. #197217 |
| Cyclosporine A | Novartis | |
| IGF-1 | Peprotech | Cat. #100-11 |
| Pinacidil | Sigma | Cat. #P154 |
| 5-Bromo-2’-deoxyuridine (BrdU) | Sigma | Cat. #B5002 |
| Other | ||
| Accutase | BD Biosciences | Cat. #561527 |
| RPMI 1640 | ThermoFisher | Cat. #11875093 |
| DMEM (glucose-free) | GIBCO | Cat. #11966025 |
| Opti-MEM | GIBCO | Cat. #31985062 |
| Polyethylenimine (PEI) | Polysciences | Cat. #239662 |
| PEG-6000 | Millipore Sigma | Cat. #528877 |
| Trypsin-EDTA | GIBCO | Cat. #25200056 |
| Geneticin (G418) | GIBCO | Cat. #10131035 |
| Zeocin | GIBCO | Cat. #R25005 |
| Protein G Dynabeads | ThermoFisher | Cat. #10004 |
| Puromycin | GIBCO | Cat. #A1113803 |
| Blasticidin | GIBCO | Cat. #A1113903 |
| B27 supplement | GIBCO | Cat. #17504044 |
| B27 supplement (−insulin) | GIBCO | Cat. #A1895601 |
| Penicillin/Streptomycin | GIBCO | Cat. #15140122 |
| GlutaMAX | GIBCO | Cat. #35050061 |
| Sodium lactate | Sigma | Cat. #71718 |
| N-acetylcysteine (NAC) | MilliporeSigma | Cat. #1064255 |
| mTeSR1 | STEMCELL Technologies | Cat. #85850 |
| DAPI | Invitrogen | Cat. #D1306 |
| TO-PRO-3 | Invitrogen | Cat. #T3605 |
| Hoechst 33342 | ThermoFisher | Cat. #62249 |
| MitoTracker Green | Invitrogen | Cat. #M7514 |
| MitoSOX Red | Invitrogen | Cat. #M36008 |
| Fetal bovine serum (heat-inactivated) | Gemini | Cat. #100-106 |
| Protein G Dynabeads | ThermoFisher | Cat. #10014 |
| PBS | GIBCO | Cat. #10010049 |
| picoGreen | ThermoFisher | Cat. #P7589 |
| KOD Hot Start Master Mix | MilliporeSigma | Cat. #71842-4 |
| SuperSignal West Pico Chemiluminescent substrate | ThermoFisher | Cat. #34580 |
| ProLong Gold Mountant | Invitrogen | Cat. #P36965 |
| 4–20% Mini-PROTEAN TGX gels | Bio-Rad | Cat. #4561095 |
| RTA PVDF Transfer kit | Bio-Rad | Cat. #1704272 |
| BSA, Fraction V | Fisher | Cat. #BP1605 |
| Pyruvate | GIBCO | Cat. #11360070 |
| Tris-Glycine SDS 10X | Fisher | Cat. #BP13414 |
| Maxima H Minus Reverse Transcriptase | ThermoFisher | Cat. #EP0751 |
| Exo I | New England BioLabs | Cat. #M0293 |
| AMPure beads | Beckman Coulter | Cat. #A63881 |
| KAPA HiFi | Kapa Biosystems | Cat. #KM2602 |
| DH5a E.coli cells | New England BioLabs | Cat. #C2987 |
| Stbl3 E.coli cells | Invitrogen | Cat. #C737303 |
| Stbl4 E.coli cells | Invitrogen | Cat. #11635018 |
| 4mm cuvettes | Bio-Rad | Cat. #1652088 |
| RIPA buffer | Cell Signaling | Cat. #9806 |
| Protease inhibitor cocktail | Roche | Cat. #11836170001 |
| Phosphatase inhibitor | Pierce | Cat. #A32957 |
| BCA kit | ThermoFisher | Cat. #23225 |
| Protein sample buffer | ThermoFisher | Cat. #39000 |
| Puromycin | ThermoFisher | Cat. #A1113803 |
| Versene | ThermoFisher | Cat. #15040066 |
| PBS | GIBCO | Cat. #14190144 |
| RPMI | GIBCO | Cat. #11875093 |
| Bovine Serum Albumin | Sigma | Cat. #A9418 |
| Ascorbic Acid | Sigma | Cat. #A8960 |
| FBS | BioWest | Cat. #S1620 |
| 0.05% Trypsin-EDTA | GIBCO | Cat. #25300054 |
| Saponin | Sigma | Cat. #47036 |
| CryoStor | Sigma | Cat. #C2874 |
| Diaminobenzadene (DAB) | Vector Labs | Cat. #PK-6100 |
| Deposited data | ||
| scRNA-seq data | GEO | GEO: GSE147249 |
| CRISPR screen data | GEO | GEO: GSE147417 |
| ChIP-seq data | GEO | GEO: GSE130285 |
Highlights.
Replication and polyploidization are tracked using engineered human cardiomyocytes
Polyploidization is associated with transcriptomic and epigenomic signatures
The sarcomere inhibits mitosis through a p53-dependent G2/M checkpoint
Sarcomere knockout cardiomyocytes improve in vivo engraftment and replication
ACKNOWLEDGMENTS
We thank Michael Stitzel (JAX) for his suggestions, the JAX-UConn Single Cell Genomics Center for assistance with scRNA-seq, Anthony Carcio and Tiffany Prosio from the JAX Flow Core for cell sorting, and the University of Washington Garvey Imaging core for assistance with microscopy. Artwork in this study was made, in part, using BioRender. This work was supported by the NIH (J.T.H., K08HL125807, R01HL142787, and U01EB028898; C.E.M., R01HL146868, R01HL128362, R01HL128368, U54DK107979, and R01HL141570; J.C., R35GM119465; M.R., RM1GM131981 and R01HL128368; D.Y.; T32EB001650), American Heart Association (A.M.P., PRE34381021; F.A.L., PRE35110005), and UConn Health (J.T.H., institutional funds).
Footnotes
DECLARATION OF INTERESTS
C.E.M. is a scientific founder, employee, and equity holder in Sana Biotechnology. C.E.M. has multiple issued and pending patents pertaining to stem cell biology and heart regeneration.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109088.
REFERENCES
- Agarwal ML, Agarwal A, Taylor WR, and Stark GR (1995). p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc. Natl. Acad. Sci. USA 92, 8493–8497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja P, Perriard E, Perriard JC, and Ehler E (2004). Sequential myofibrillar breakdown accompanies mitotic division of mammalian cardiomyocytes. J. Cell Sci 117, 3295–3306. [DOI] [PubMed] [Google Scholar]
- Ayoub N, Jeyasekharan AD, Bernal JA, and Venkitaraman AR (2008). HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature 453, 682–686. [DOI] [PubMed] [Google Scholar]
- Becker RO, Chapin S, and Sherry R (1974). Regeneration of the ventricular myocardium in amphibians. Nature 248, 145–147. [DOI] [PubMed] [Google Scholar]
- Bedada FB, Chan SS, Metzger SK, Zhang L, Zhang J, Garry DJ, Kamp TJ, Kyba M, and Metzger JM (2014). Acquisition of a quantitative, stoichiometrically conserved ratiometric marker of maturation status in stem cell-derived cardiac myocytes. Stem Cell Reports 3, 594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, et al. (2009). Evidence for cardiomyocyte renewal in humans. Science 324, 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, Sjostrom SL, Szewczykowska M, Jackowska T, Dos Remedios C, et al. (2015). Dynamics of Cell Generation and Turnover in the Human Heart. Cell 161, 1566–1575. [DOI] [PubMed] [Google Scholar]
- Bicknell KA, Coxon CH, and Brooks G (2004). Forced expression of the cyclin B1-CDC2 complex induces proliferation in adult rat cardiomyocytes. Biochem. J 382, 411–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode AM, and Dong Z (2004). Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 4, 793–805. [DOI] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, and Vogelstein B (1998). Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282, 1497–1501. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs BG, Li H, and van Deursen JM (2018). Senescent cells: a therapeutic target for cardiovascular disease. J. Clin. Invest 128, 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong JJ, Yang X, Don CW, Minami E, Liu YW, Weyers JJ, Mahoney WM, Van Biber B, Cook SM, Palpant NJ, et al. (2014). Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature 510, 273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clute P, and Pines J (1999). Temporal and spatial control of cyclin B1 destruction in metaphase. Nat. Cell Biol 1, 82–87. [DOI] [PubMed] [Google Scholar]
- Cohn R, Thakar K, Lowe A, Ladha FA, Pettinato AM, Romano R, Meredith E, Chen YS, Atamanuk K, Huey BD, and Hinson JT (2019). A Contraction Stress Model of Hypertrophic Cardiomyopathy due to Sarcomere Mutations. Stem Cell Reports 12, 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotney JL, and Noonan JP (2015). Chromatin immunoprecipitation with fixed animal tissues and preparation for high-throughput sequencing. Cold Spring Harb. Protoc 2015, 191–199. [DOI] [PubMed] [Google Scholar]
- De Deyne PG (2000). Formation of sarcomeres in developing myotubes: role of mechanical stretch and contractile activation. Am. J. Physiol. Cell Physiol 279, C1801–C1811. [DOI] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Yamaguchi H, Wei Y, Hsu JL, Wang HL, Hsu YH, Lin WC, Yu WH, Leonard PG, Lee GR 4th., et al. (2016). Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat. Med 22, 194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, and Vogelstein B (1993). WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825. [DOI] [PubMed] [Google Scholar]
- Engel FB, Schebesta M, and Keating MT (2006). Anillin localization defect in cardiomyocyte binucleation. J. Mol. Cell. Cardiol 41, 601–612. [DOI] [PubMed] [Google Scholar]
- Ernst J, and Kellis M (2012). ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, and Kellis M (2015). Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat. Biotechnol 33, 364–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Liu T, Qin B, Zhang Y, and Liu XS (2012). Identifying ChIP-seq enrichment using MACS. Nat. Protoc 7, 1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes S, Naumova AV, Zhu WZ, Laflamme MA, Gold J, and Murry CE (2010). Human embryonic stem cell-derived cardiomyocytes engraft but do not alter cardiac remodeling after chronic infarction in rats. J. Mol. Cell. Cardiol 49, 941–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M, Quaas M, Steiner L, and Engeland K (2016). The p53-p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic Acids Res. 44, 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbin KA, Yang X, Murry CE, and Coulombe KL (2015). Enhanced Electrical Integration of Engineered Human Myocardium via Intramyocardial versus Epicardial Delivery in Infarcted Rat Hearts. PLoS ONE 10, e0131446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilsbach R, Schwaderer M, Preissl S, Grüning BA, Kranzhöfer D, Mulero-Navarro TG, Weichenhan S, Braun D, Schneider P, Nuhrenberg C, et al. (2018). Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat. Commun 9, 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Rosa JM, Sharpe M, Field D, Soonpaa MH, Field LJ, Burns CE, and Burns CG (2018). Myocardial Polyploidization Creates a Barrier to Heart Regeneration in Zebrafish. Dev. Cell 44, 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara N, Klewer SE, Samson RA, Erickson DT, Lyon MF, and Brilliant MH (2000). Sox6 is a candidate gene for p100H myopathy, heart block, and sudden neonatal death. Proc. Natl. Acad. Sci. USA 97, 4180–4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagting A, Karlsson C, Clute P, Jackman M, and Pines J (1998). MPF localization is controlled by nuclear export. EMBO J. 17, 4127–4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, et al. (2015). High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 163, 1515–1526. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann BG, Labeit S, Poustka A, King TR, and Lehrach H (1990). Cloning of the T gene required in mesoderm formation in the mouse. Nature 343, 617–622. [DOI] [PubMed] [Google Scholar]
- Herzig S, and Shaw RJ (2018). AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol 19, 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinson JT, Chopra A, Nafissi N, Polacheck WJ, Benson CC, Swist S, Gorham J, Yang L, Schafer S, Sheng CC, et al. (2015). HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 349, 982–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinson JT, Chopra A, Lowe A, Sheng CC, Gupta RM, Kuppusamy R, O’Sullivan J, Rowe G, Wakimoto H, Gorham J, et al. (2016). Integrative Analysis of PRKAG2 Cardiomyopathy iPS and Microtissue Models Identifies AMPK as a Regulator of Metabolism, Survival, and Fibrosis. Cell Rep. 17, 3292–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Pi Y, Lee KJ, Henkel AS, Gregg RG, Powers PA, and Walker JW (1999). Cardiac troponin I gene knockout: a mouse model of myocardial troponin I deficiency. Circ. Res 84, 1–8. [DOI] [PubMed] [Google Scholar]
- Innocente SA, Abrahamson JL, Cogswell JP, and Lee JM (1999). p53 regulates a G2 checkpoint through cyclin B1. Proc. Natl. Acad. Sci. USA 96, 2147–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen LJ, Jensen TS, de Lichtenberg U, Brunak S, and Bork P (2006). Co-evolution of transcriptional and post-translational cell-cycle regulation. Nature 443, 594–597. [DOI] [PubMed] [Google Scholar]
- Jiang YH, Wang HL, Peng J, Zhu Y, Zhang HG, Tang FQ, Jian Z, and Xiao YB (2020). Multinucleated polyploid cardiomyocytes undergo an enhanced adaptability to hypoxia via mitophagy. J. Mol. Cell. Cardiol 138, 115–135. [DOI] [PubMed] [Google Scholar]
- Jung YS, Qian Y, and Chen X (2010). Examination of the expanding pathways for the regulation of p21 expression and activity. Cell. Signal 22, 1003–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Tolstorukov MY, and Park PJ (2008). Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat. Biotechnol 26, 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klochendler A, Weinberg-Corem N, Moran M, Swisa A, Pochet N, Savova V, Vikeså J, Van de Peer Y, Brandeis M, Regev A, et al. (2012). A transgenic mouse marking live replicating cells reveals in vivo transcriptional program of proliferation. Dev. Cell 23, 681–690. [DOI] [PubMed] [Google Scholar]
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al. ; Roadmap Epigenomics Consortium (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurinna S, Stratton SA, Coban Z, Schumacher JM, Grompe M, Duncan AW, and Barton MC (2013). p53 regulates a mitotic transcription program and determines ploidy in normal mouse liver. Hepatology 57, 2004–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutner RH, Zhang XY, and Reiser J (2009). Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat. Protoc 4, 495–505. [DOI] [PubMed] [Google Scholar]
- Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, Reinecke H, Xu C, Hassanipour M, Police S, et al. (2007). Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat. Biotechnol 25, 1015–1024. [DOI] [PubMed] [Google Scholar]
- Lam CK, Tian L, Belbachir N, Wnorowski A, Shrestha R, Ma N, Kitani T, Rhee JW, and Wu JC (2019). Identifying the Transcriptome Signatures of Calcium Channel Blockers in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circ. Res 125, 212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. (2012). ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 22, 1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laoukili J, Kooistra MR, Brás A, Kauw J, Kerkhoven RM, Morrison A, Clevers H, and Medema RH (2005). FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat. Cell Biol 7, 126–136. [DOI] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, Raval KK, Zhang J, Kamp TJ, and Palecek SP (2012). Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA 109, E1848–E1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud AI, Kocabas F, Muralidhar SA, Kimura W, Koura AS, Thet S, Porrello ER, and Sadek HA (2013). Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 497, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte R, Brown KR, Suarez F, Sayad A, Karamboulas K, Krzyzanowski PM, Sircoulomb F, Medrano M, Fedyshyn Y, Koh JLY, et al. (2012). Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2, 172–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDevitt TC, Laflamme MA, and Murry CE (2005). Proliferation of cardiomyocytes derived from human embryonic stem cells is mediated via the IGF/PI 3-kinase/Akt signaling pathway. J. Mol. Cell. Cardiol 39, 865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RJ, Titmarsh DM, Koenig X, Parker BL, Ryall JG, Quaife-Ryan GA, Voges HK, Hodson MP, Ferguson C, Drowley L, et al. (2017). Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc. Natl. Acad. Sci. USA 114, E8372– E8381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed TMA, Ang YS, Radzinsky E, Zhou P, Huang Y, Elfenbein A, Foley A, Magnitsky S, and Srivastava D (2018). Regulation of Cell Cycle to Stimulate Adult Cardiomyocyte Proliferation and Cardiac Regeneration. Cell 173, 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollova M, Bersell K, Walsh S, Savla J, Das LT, Park SY, Silberstein LE, Dos Remedios CG, Graham D, Colan S, and Kühn B (2013). Cardiomyocyte proliferation contributes to heart growth in young humans. Proc. Natl. Acad. Sci. USA 110, 1446–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monte M, Benetti R, Buscemi G, Sandy P, Del Sal G, and Schneider C (2003). The cell cycle-regulated protein human GTSE-1 controls DNA damage-induced apoptosis by affecting p53 function. J. Biol. Chem 278, 30356–30364. [DOI] [PubMed] [Google Scholar]
- Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, Shah AM, Zhang H, Faber JE, Kinter MT, et al. (2017). Hypoxia induces heart regeneration in adult mice. Nature 541, 222–227. [DOI] [PubMed] [Google Scholar]
- Nakajima H, Nakajima HO, Tsai SC, and Field LJ (2004). Expression of mutant p193 and p53 permits cardiomyocyte cell cycle reentry after myocardial infarction in transgenic mice. Circ. Res 94, 1606–1614. [DOI] [PubMed] [Google Scholar]
- Naya FJ, Black BL, Wu H, Bassel-Duby R, Richardson JA, Hill JA, and Olson EN (2002). Mitochondrial deficiency and cardiac sudden death in mice lacking the MEF2A transcription factor. Nat. Med 8, 1303–1309. [DOI] [PubMed] [Google Scholar]
- Nguyen TT, Grimm SA, Bushel PR, Li J, Li Y, Bennett BD, Lavender CA, Ward JM, Fargo DC, Anderson CW, et al. (2018). Revealing a human p53 universe. Nucleic Acids Res. 46, 8153–8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishii K, Morimoto S, Minakami R, Miyano Y, Hashizume K, Ohta M, Zhan DY, Lu QW, and Shibata Y (2008). Targeted disruption of the cardiac troponin T gene causes sarcomere disassembly and defects in heartbeat within the early mouse embryo. Dev. Biol 322, 65–73. [DOI] [PubMed] [Google Scholar]
- O’Gara PT, Kushner FG, Ascheim DD, Casey DE Jr., Chung MK, de Lemos JA, Ettinger SM, Fang JC, Fesmire FM, Franklin BA, et al. (2013). 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol 61, e78–e140. [DOI] [PubMed] [Google Scholar]
- Pasumarthi KB, Tsai SC, and Field LJ (2001). Coexpression of mutant p53 and p193 renders embryonic stem cell-derived cardiomyocytes responsive to the growth-promoting activities of adenoviral E1A. Circ. Res 88, 1004–1011. [DOI] [PubMed] [Google Scholar]
- Patterson M, Barske L, Van Handel B, Rau CD, Gan P, Sharma A, Parikh S, Denholtz M, Huang Y, Yamaguchi Y, et al. (2017). Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat. Genet 49, 1346–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettinato AM, Ladha FA, Mellert DJ, Legere N, Cohn R, Romano R, Thakar K, Chen YS, and Hinson JT (2020). Development of a Cardiac Sarcomere Functional Genomics Platform to Enable Scalable Interrogation of Human TNNT2 Variants. Circulation 142, 2262–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poss KD, Wilson LG, and Keating MT (2002). Heart regeneration in zebrafish. Science 298, 2188–2190. [DOI] [PubMed] [Google Scholar]
- Preuss U, Landsberg G, and Scheidtmann KH (2003). Novel mitosis-specific phosphorylation of histone H3 at Thr11 mediated by Dlk/ZIP kinase. Nucleic Acids Res. 31, 878–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R, Garcia JA, et al. (2014). The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 157, 565–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44 (W1), W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, and Zhang F (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow AJ, Sievert K, Patel S, Chang YF, Broyles CN, Brook FA, Watkins H, Geeves MA, Redwood CS, Robinson P, and Daniels MJ (2019). Measurement of Myofilament-Localized Calcium Dynamics in Adult Cardiomyocytes and the Effect of Hypertrophic Cardiomyopathy Mutations. Circ. Res 124, 1228–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss B, Harrison A, Coelho PA, Yata K, Zernicka-Goetz M, and Pines J (2018). Cyclin B1 is essential for mitosis in mouse embryos, and its nuclear export sets the time for mitosis. J. Cell Biol 217, 179–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, and Dudoit S (2018). Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohyama S, Hattori F, Sano M, Hishiki T, Nagahata Y, Matsuura T, Hashimoto H, Suzuki T, Yamashita H, Satoh Y, et al. (2013). Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 12, 127–137. [DOI] [PubMed] [Google Scholar]
- Toyoshima-Morimoto F, Taniguchi E, Shinya N, Iwamatsu A, and Nishida E (2001). Polo-like kinase 1 phosphorylates cyclin B1 and targets it to the nucleus during prophase. Nature 410, 215–220. [DOI] [PubMed] [Google Scholar]
- Ulmer BM, Stoehr A, Schulze ML, Patel S, Gucek M, Mannhardt I, Funcke S, Murphy E, Eschenhagen T, and Hansen A (2018). Contractile Work Contributes to Maturation of Energy Metabolism in hiPSC-Derived Cardiomyocytes. Stem Cell Reports 10, 834–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Berlo JH, Kanisicak O, Maillet M, Vagnozzi RJ, Karch J, Lin SC, Middleton RC, Marbán E, and Molkentin JD (2014). c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature 509, 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanOudenhove J, Yankee TN, Wilderman A, and Cotney J (2020). Epigenomic and Transcriptomic Dynamics During Human Heart Organogenesis. Circ. Res 127, e184–e209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. (2004). In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848. [DOI] [PubMed] [Google Scholar]
- Vivien CJ, Hudson JE, and Porrello ER (2016). Evolution, comparative biology and ontogeny of vertebrate heart regeneration. NPJ Regen. Med 1, 16012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyers JJ, Gunaje JJ, Van Biber B, Martinson A, Reinecke H, Mahoney WM, Schwartz SM, Cox TC, and Murry CE (2020). Sonic Hedgehog upregulation does not enhance the survival and engraftment of stem cell-derived cardiomyocytes in infarcted hearts. PLoS ONE 15, e0227780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilderman A, VanOudenhove J, Kron J, Noonan JP, and Cotney J (2018). High-Resolution Epigenomic Atlas of Human Embryonic Craniofacial Development. Cell Rep. 23, 1581–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Zhao M, Mattapally S, Chen S, and Zhang J (2018). CCND2 Overexpression Enhances the Regenerative Potency of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes: Remuscularization of Injured Ventricle. Circ. Res 122, 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielke N, Korzelius J, van Straaten M, Bender K, Schuhknecht GFP, Dutta D, Xiang J, and Edgar BA (2014). Fly-FUCCI: A versatile tool for studying cell proliferation in complex tissues. Cell Rep. 7, 588–598. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Gene Expression Omnibus (GEO) accession numbers for the next-gen sequencing data generated in this study are as follows: GEO: GSE147417 for CRISPR screen data; GEO: GSE130285 for ChIP-seq data; and GEO: GSE147249 for scRNA-seq data.