

Abstract
Rhinovirus (RV) infections are associated with asthma exacerbations. MicroRNA‐146a and microRNA‐146b (miR‐146a/b) are anti‐inflammatory miRNAs that suppress signaling through the nuclear factor kappa B (NF‐κB) pathway and inhibit pro‐inflammatory chemokine production in primary human bronchial epithelial cells (HBECs). In the current study, we aimed to explore whether miR‐146a/b could regulate cellular responses to RVs in HBECs and airways during RV‐induced asthma exacerbation. We demonstrated that expression of miR‐146a/b and pro‐inflammatory chemokines was increased in HBECs and mouse airways during RV infection. However, transfection with cell‐penetrating peptide (CPP)‐miR‐146a nanocomplexes before infection with RV significantly reduced the expression of the pro‐inflammatory chemokines CCL5, IL‐8 and CXCL1, increased interferon‐λ production, and attenuated infection with the green fluorescent protein (GFP)‐expressing RV‐A16 in HBECs. Concordantly, compared to wild‐type (wt) mice, Mir146a/b−/− mice exhibited more severe airway neutrophilia and increased T helper (Th)1 and Th17 cell infiltration in response to RV‐A1b infection and a stronger Th17 response with a less prominent Th2 response in house dust mite extract (HDM)‐induced allergic airway inflammation and RV‐induced exacerbation models. Interestingly, intranasal administration of CPP‐miR‐146a nanocomplexes reduced HDM‐induced allergic airway inflammation without a significant effect on the Th2/Th1/Th17 balance in wild‐type mice. In conclusion, the overexpression of miR‐146a has a strong anti‐inflammatory effect on RV infection in HBECs and a mouse model of allergic airway inflammation, while a lack of miR‐146a/b leads to attenuated type 2 cell responses in mouse models of allergic airway inflammation and RV‐induced exacerbation of allergic airway inflammation. Furthermore, our data indicate that the application of CPP‐miR‐146a nanocomplexes has therapeutic potential for targeting airway inflammation.
Keywords: asthma, bronchial epithelial cell, house dust mite, microRNA, neutrophils, noncoding RNA, viral infection
miR‐146a has anti‐inflammatory properties in human bronchial epithelial cells and mouse airways during rhinovirus infection and in case of allergic inflammation. Particular cell penetrating peptide(CPP)‐miR‐146a nanocomplexes have therapeutic potential for targeting of airway inflammation.
Abbreviations
- 3′UTRs
3′ untranslated regions
- BAL
bronchoalveolar lavage fluid
- BMDCs
bone marrow‐derived dendritic cells
- CARD10
caspase recruitment domain‐containing protein 10
- CCL5
C‐C Motif Chemokine Ligand 5
- CPP
cell‐penetrating peptide
- CXCL1
chemokine (C‐X‐C motif) ligand 1
- ELISA
enzyme‐linked immunosorbent assay
- GFP
green fluorescent protein
- HBECs
human bronchial epithelial cells
- HDM
house dust mite extract
- i.n.
intranasal
- ICAM‐1
intercellular adhesion molecule 1
- IFITM1
interferon‐induced transmembrane protein 1
- IFN‐λ
interferon‐λ
- ILC2
type 2 innate lymphoid cells
- IRAK1
interleukin‐1 receptor‐associated kinase 1
- IRF1
interferon regulatory factor 1
- LDLR
low‐density lipoprotein receptor
- miR‐146a/b
miR‐146a and miR‐146b
- miRNA
microRNA
- NF‐κB
nuclear factor kappa B
- RV
rhinovirus
- Th
T helper
- Tregs
regulatory T cells
- wt
wild‐type
1. INTRODUCTION
Asthma is the most common chronic disease of the airways and causes coughing and wheezing due to airway narrowing. 1 Asthma symptoms are often triggered by various allergic reactions; however, the disease itself is considered very heterogeneous. Accordingly, asthma can be divided into different phenotypes depending on the cell types triggering airway inflammation. 2 , 3 The type 2 phenotype includes allergic asthma, which is associated with increased T helper (Th)2 responses and IgE levels, and eosinophilic non‐allergic asthma, which is characterized by type 2 innate lymphoid cells (ILC2) and increased expression of epithelial alarmins. 4 , 5 Non‐eosinophilic asthma is characterized by airway neutrophilia and involvement of Th17 cells. 6 , 7 To provide asthma control, inhaled corticosteroids and long‐acting bronchodilators are most commonly used. 8 In addition to allergens, infections with respiratory tract viruses, including human rhinoviruses (RVs), are important triggers for worsening asthma symptoms and may lead to hospitalization or even death. 9 Among asthma exacerbations caused by viral infections, human RVs make up approximately 65% of cases. 10
RVs are positive‐strand RNA viruses that infect the upper and lower airways and cause the common cold. 11 , 12 RVs can be categorized based on the receptor they use to enter cells. 13 The majority of RV‐A and all RV‐B serotypes, including RV‐A16, use intercellular adhesion molecule‐1 (ICAM‐1) as their receptor and are classified as major group RVs. The minor group consists of only 10 RV‐A serotypes, which enter cells using the low‐density lipoprotein receptor (LDLR). 14 From the minor group, RV‐A1b has often been used in research. RV infection and replication in epithelial cells activate innate immune responses and induce expression of growth factors and chemokines, which leads to recruitment and activation of granulocytes, including neutrophils, destruction of normal airway tissue, and development of respiratory symptoms. 13 , 15 , 16
MicroRNAs (miRNAs) are short non‐coding RNAs that modulate various biological processes through posttranscriptional regulation of gene expression. 17 More specifically, miRNAs recognize and bind to the 3′ untranslated regions (3′UTRs) of multiple target mRNAs via the complementarity of 6‐ to 8‐nt‐long seed regions present in miRNAs. Once mRNA is bound by miRNA, its translation is inhibited or mRNA degradation is triggered by miRNA‐binding proteins. 18 In this way, miRNAs are also capable of regulating immune responses in various conditions. 19 , 20 , 21 The miR‐146 family consists of miR‐146a and miR‐146b (miR‐146a/b) that differ by only two nucleotides in their 3′ region, which is considered less significant for their interaction with mRNA targets. As such, miR‐146a/b has been reported to have a similar set of target genes. 22 , 23 , 24 Previous studies have demonstrated that multiple components, including interleukin‐1 receptor‐associated kinase 1 (IRAK1) and caspase recruitment domain 10 (CARD10), from the NF‐κB pathway are directly targeted by miR‐146a/b. 23 , 25 Accordingly, overexpression of miR‐146a/b has been shown to suppress endogenous levels of IRAK1 and CARD10 and of NF‐κB‐inducible chemokines IL‐8 and chemokine (C‐X‐C motif) ligand 1 (CXCL1) in multiple cell types, including human bronchial epithelial cells (HBECs). 26 An interesting example is C‐C Motif Chemokine Ligand 5 (CCL5), which has been shown to be directly suppressed by miR‐146a and through IRAK1 and CARD10. 27 Altered expression of miR‐146a has been identified in various asthma studies, 20 , 28 , 29 , 30 , 31 and we previously demonstrated that development of the neutrophilic phenotype of asthma may be among the factors associated with reduced expression of miR‐146a in human airway epithelial cells. 26 Therefore, we hypothesized that miR‐146a/b may play an important role in the regulation of immune responses to RV infections in bronchial epithelial cells and the airways, as well as in asthma exacerbations.
In this study, we examined the expression and function of miR‐146a/b during RV infection in HBECs and in mouse models of RV‐induced airway inflammation, house dust mite extract (HDM)‐induced allergic asthma, and RV‐induced exacerbation of allergic airway inflammation. We demonstrate that miR‐146a has the capacity to suppress inflammatory responses to RVs in HBECs and during RV‐ and/or HDM‐induced airway inflammation; however, mice lacking miR‐146a/b develop less prominent type 2 cell responses in mouse models of HDM‐induced allergic airway inflammation and RV‐induced exacerbation of allergic airway inflammation.
2. RESULTS
2.1. MiR‐146a inhibits the expression of pro‐inflammatory chemokines and induces interferon response genes during RV infection in HBECs
To examine the role of miR‐146a/b in the regulation of immune responses to RVs, we first used monolayer cultures of primary HBECs, which is an easy to transfect primary cell culture system that can be infected by RVs. We infected HBECs with RV‐A16 or RV‐A1b and analyzed the expression of miR‐146a/b 24 h and 48 h after infection. A two‐ to six‐fold upregulation of miR‐146a and a two‐ to three‐fold increase in miR‐146b in response to RVs were observed, with higher expression of miR‐146a in all analyzed conditions (Figure 1A). RV infection also altered the expression of miR‐146a target genes from the NF‐κB pathway, including interleukin‐1 receptor‐associated kinase 1 (IRAK1) and caspase recruitment domain‐containing protein 10 (CARD10), as well as tested pro‐inflammatory chemokines and interferon response genes in HBECs (Figure S1). Since RV infection induced both miR‐146a/b (Figure 1A) and genes that have been shown to be downregulated by miR‐146a, such as CCL5, 27 CXCL1, and IL‐8 26 (Figure S1B and S1C), we hypothesized that endogenous levels of miR‐146a/b are not sufficient to fully suppress these genes and next assessed the influence of transfected miR‐146a mimics during RV infection. As miR‐146a mimics had very similar effects as miR‐146b mimics in keratinocytes, 24 we used only miR‐146a and control mimics in these experiments (Figure S2). As a result, a significant downregulation in mRNA levels of direct miR‐146a targets, including IRAK1, CARD10, CCL5, IL‐8, and CXCL1, was observed in mock and RV‐infected cells (Figures 1B and 1C) following transfection with miR‐146a. Interestingly, miR‐146a transfection led to increased interferon‐regulated interferon regulatory factor 1 (IRF1) and a tendency for enhanced interferon‐induced transmembrane protein 1 (IFITM1) during RV‐A16 infection compared to control transfection (Figure 1D). In addition, miR‐146a overexpression significantly reduced the expression of the major group RV receptor ICAM‐1 (Figure 1E). In line with the mRNA expression results, overexpression of miR‐146a significantly inhibited IL‐8 and CXCL1 protein secretion and simultaneously led to increased interferon‐λ (IFN‐λ) production, as measured by enzyme‐linked immunosorbent assay (ELISA), in the supernatants of miR‐146a‐transfected and RV‐infected HBECs (Figure 1F). To further assess the miR‐146 effect in bronchial epithelial cells during RV infection, we performed pathway analyses of putative miR‐146a/b Targetscan 32 targets, which expression was shown to be altered during RV infection in HBECs 33 and determined interferon‐gamma response, TNF‐alpha signaling via NF‐κB, interferon‐alpha response and IL‐2/STAT5 signaling from Molecular Signatures Database (MSigDB) Hallmark Gene Set Collection 34 as pathways preferentially targeted by miR‐146a/b (Table S1). In summary, these results show that miR‐146a inhibits the expression of NF‐κB‐dependent pro‐inflammatory genes while stimulating interferon response genes during RV infection in HBECs.
FIGURE 1.
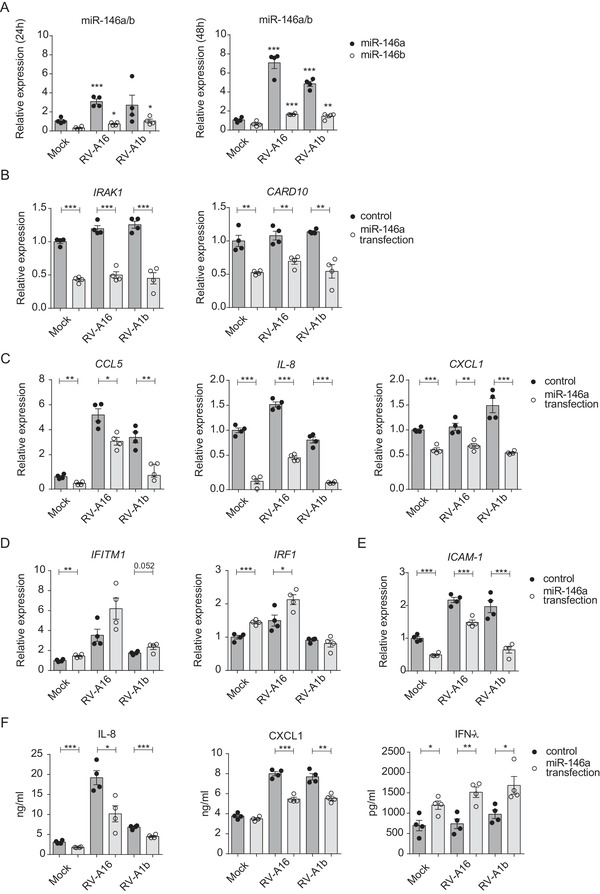
MiR‐146a/b inhibit expression of pro‐inflammatory cytokines and induce interferon response genes in HBECs during RV infection. (A) HBECs were stimulated with RVs for 24 h or 48 h. Relative expression of miR‐146a/b in HBECs was measured by RT‐qPCR and is shown in comparison to corresponding mock‐stimulated cells. (B‐F) HBECs were transfected with miR‐146a or control mimics and stimulated 24 h later with mock or the indicated RVs for 48 h. (B‐E) mRNA expression of the indicated genes in primary HBECs was measured by RT‐qPCR and compared to expression levels of the mock‐stimulated control group mean (= 1). (F) Protein expression of selected genes was measured by ELISA. (A‐F) Data represent mean ± SEM. Unpaired t‐test, *p < 0.05, **p < 0.01, ***p < 0.001. One representative of three independent experiments in HBECs from two different donors is shown
2.2. MiR‐146a reduces the number of infected cells and inhibits neutrophil migration during RV infection in HBECs
Because miR‐146a overexpression induced IRF1 and IFN‐λ expression and inhibited RV‐A16 receptor ICAM‐1 mRNA levels (Figures 1D‐1F), we next assessed whether miR‐146a influences RV infection in HBECs. HBECs were transfected with miR‐146a mimics, infected with RV‐A16‐green fluorescent protein (GFP), and then assessed by fluorescence microscopy (Figure S3B) or flow cytometry (Figures 2A and 2B). In HBECs transfected with miR‐146a mimics, significantly fewer GFP‐positive cells (3.7% ± 0.2%) were detected than in control cells (5.3% ± 0.3%) (Figures 2A and 2B). The percentage of RV‐infected HBECs was in line with previous studies indicating that only a fraction of HBECs is infected by RVs. 10 However, transfection nonspecifically enhanced the infection of HBECs with RV‐A16‐GFP. Similar to wild‐type RV‐A16, RV‐A16‐GFP had the capacity to stimulate miR‐146a/b and IL‐8, CXCL1, and ICAM‐1 mRNA expression in HBECs (Figure S3A, S3C and S3D).
FIGURE 2.
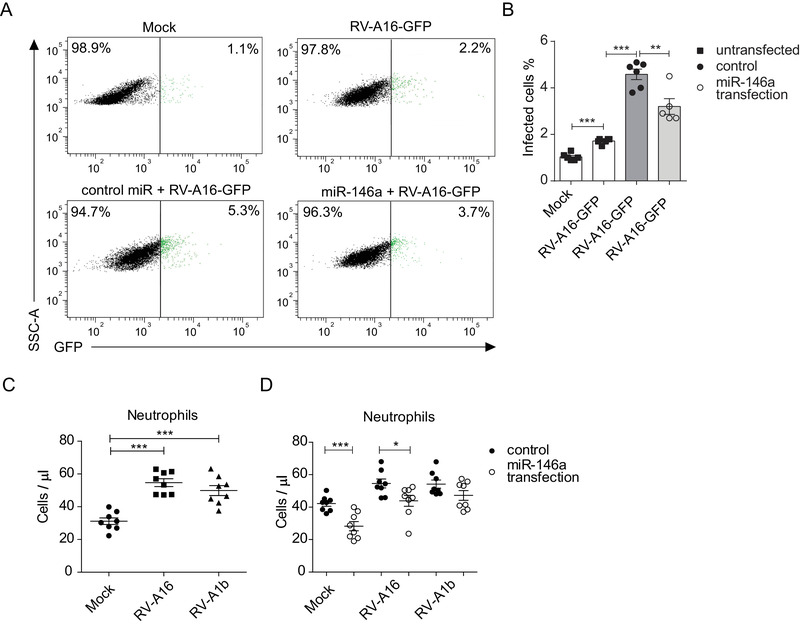
MiR‐146a inhibits RV infection and neutrophil migration. HBECs were transfected with miRNA mimics for 24 h and stimulated with the indicated RVs for 48 h or stimulated with only RV or mock. (A) Representative flow cytometry plots of HBECs infected with RV‐A16‐GFP (green) and (B) corresponding quantification graph. (C‐D) Chemotaxis assay of human neutrophils toward supernatants of primary HBECs stimulated with RVs (C) or toward supernatants of HBECs transfected with the indicated miRNA mimics and stimulated with RVs (D). (C‐D) Data from two independent experiments in HBECs from two different donors are shown. Data represent mean ± SEM. Unpaired t‐test, *p < 0.05, **p < 0.01, ***p < 0.001
As RV infection in HBECs induced neutrophil‐attracting chemokines, IL‐8 and CXCL1, and miR‐146a transfection inhibited secretion of these chemokines (Figures 1C, 1F, and S1B), we next performed a neutrophil chemotaxis assay. Indeed, increased neutrophil migration toward the supernatants of RV‐A16‐ or A1b‐infected HBECs was detected compared to mock‐infected HBECs (Figure 2C), and significantly fewer neutrophils migrated toward the supernatants of miR‐146a mimic‐transfected and RV‐A16‐infected HBECs compared to the corresponding control. There was a similar tendency with RV‐A1b as well (Figure 2D). In summary, the overexpression of miR‐146a reduces the number of infected HBECs and inhibits the secretion of neutrophil chemoattractants from HBECs during RV infection.
2.3. Mir146a/b−/− mice develop more severe airway neutrophilia in a mouse model of RV‐induced airway inflammation
To investigate whether miR‐146a/b may influence immune responses to RVs in vivo, a previously developed model that uses the capacity of minor group RVs to infect mouse airways, was applied. 35 Accordingly, RV‐A1b or PBS was applied intranasally (i.n.) to C57Bl/6J Mir146a/b−/− and wild‐type (wt) mice. Twenty‐four hours later, mice were sacrificed, and bronchoalveolar lavage fluid (BAL) and lung tissue were collected (Figure 3A). First, total BAL cells and the number of neutrophils and lymphocytes were significantly elevated in Mir146a/b−/− mice in response to RV‐A1b infection, as observed when BAL cells were differentially stained and counted by microscopy (Figure S4). Flow cytometry analysis (for sorting strategy, please see Figure S5A) revealed similarly strong increases in total cell number, neutrophils, T cells, B cells and dendritic cells (DCs) in BAL collected from mice infected with RV‐A1b, with further increases in BAL total cell count, neutrophils, T cells, and DCs in Mir146a/b−/− mice (Figures 3B and 3C). No significant differences in the numbers of B cells, macrophages, or eosinophils were found between wt and Mir146a/b−/‐ mice in response to RV infection (Figures 3B and 3C). In addition to BAL cells, we analyzed the expression of miR‐146a/b and relevant genes in lung tissue. Similar to in vitro results in HBECs (Figure 1A), RV‐A1b infection stimulated the expression of miR‐146a/b in wt mouse lungs (Figure 4A), while no miR‐146a/b was detected in Mir146a/b−/− mice. Analysis of mRNA levels revealed a significant decrease in expression of the miR‐146a/b direct target gene Irak1 during RV‐A1b infection and increased Card10 expression in the PBS treatment group in Mir146a/b−/− mouse lungs compared to wt mice (Figure 4B). Although Card10 and Irak1 mRNA was not upregulated in infected Mir146a/b−/− mice, RV infection more strongly induced Cxcl1 mRNA expression in the lungs (Figure 4C) and protein levels in the BAL fluid (Figure 4F) in Mir146a/b−/− mice than in wt mice. In addition, we observed significantly increased expression of Irf1 (Figure 4D) and neutrophil markers S100a8 and S100a9 (Figure 4E) in the lungs of Mir146a/b−/− mice compared to wt mice in response to RV infection. Differences in expression levels of Cxcl2 (Figure 4C) and Ifitm1 (Figure 4D) were not statistically significant between RV‐A1b‐stimulated Mir146a/b−/− and wt mouse lungs. Taken together, these findings reveal that the lack of miR‐146a/b in mice leads to increased numbers of neutrophils, DCs, and T cells in the airways with concomitantly enhanced expression of the neutrophil chemoattractant Cxcl1, as well as neutrophil markers S100a8 and S100a9 in the lung tissue during RV‐A1b infection.
FIGURE 3.
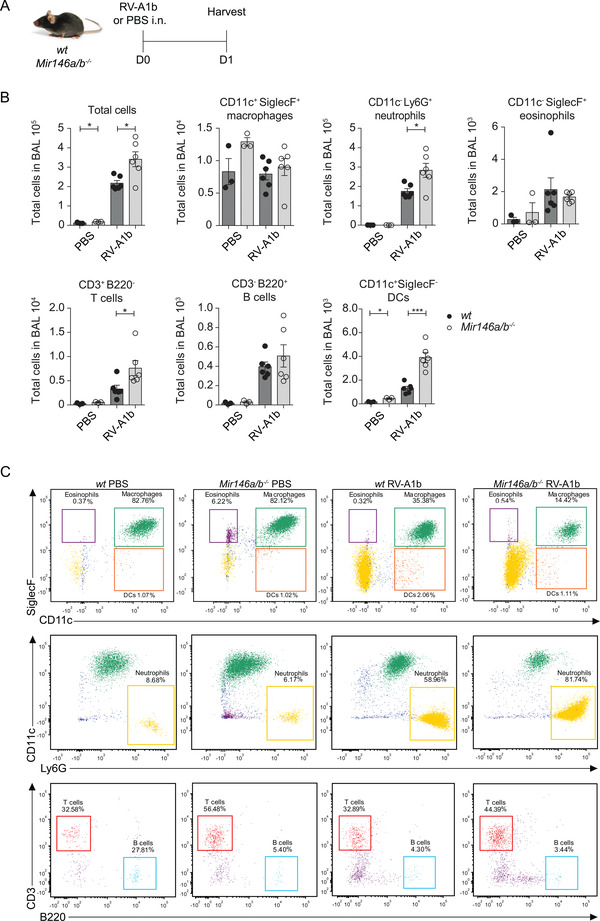
Mir146a/b−/− mice exhibit increased airway neutrophilia in a mouse model of RV‐induced airway inflammation. (A) RV‐A1b or PBS was administered intranasally (i.n.) to wt or Mir146a/b−/− mice. Twenty‐four hours after RV‐A1b infection, BAL was analyzed. (B‐C) BAL cells were counted using a hemocytometer and then subjected to FACS analysis, and according to dot plots, the total numbers of immune cells were calculated. Data represent mean ± SEM. Unpaired t‐test, *p < 0.05, ***p < 0.001. (C) One representative FACS dot plot of BAL fluid from three PBS‐treated and five RV‐A1b‐treated wt and Mir146a/b−/− mice. Eosinophils, macrophages, DCs, and neutrophils (upper and middle panel) were analyzed as a percentage of the granulocyte population, and T cells and B cells (bottom panel) were analyzed as a percentage of the lymphocyte population
FIGURE 4.
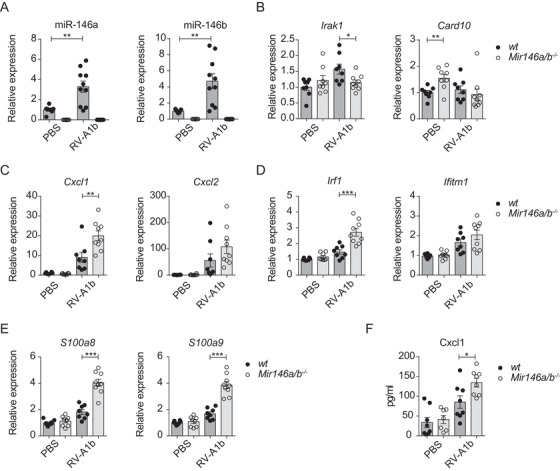
Gene expression of wt and Mir146a/b−/− mouse lungs in an RV‐induced mouse model of airway inflammation. Twenty‐four hours after i.n. administration of PBS or RV‐A1b, mouse lung lobes were collected. (A) Relative miRNA expression and (B‐E) mRNA expression in lung lobes was measured with RT‐qPCR and are shown compared to the PBS wt group mean (= 1). (F) Protein content in BAL measured with ELISA. Data from two independent experiments. Data represent mean ± SEM. Unpaired t‐test, *p < 0.05, **p < 0.01, ***p < 0.001
2.4. Mir146a/b−/− mice exhibit reduced Th2 cell responses in the airways in models of HDM‐induced allergic airway inflammation and RV‐induced exacerbation of allergic airway inflammation
We next studied the susceptibility of mice lacking miR‐146a/b to allergic airway inflammation and RV‐induced exacerbation of allergic airway inflammation. To that end, we used a previously developed mouse model involving repeated i.n. application of (HDM) extract 36 , 37 and additionally applied RV‐A1b i.n. to induce exacerbation 35 in some mice (Figure 5A). Flow cytometry analysis of BAL cells (for sorting strategy, please see Figure S5B) revealed an increased total number of cells, T cell subsets, and B cells, as well as a tendency for enhanced levels of macrophages and eosinophils in response to HDM treatment in wt and Mir146a/b−/− mice when compared to the PBS control group, with a robust increase in neutrophils when mice were additionally subjected to RV infection (Figures S6 and S7). When HDM was applied alone, significantly increased numbers of B cells and strongly reduced numbers of Th2 cells (Figures 5B, 5C, S6, and S7A) were observed in BAL fluid of miR‐146a/b‐deficient mice compared to wt mice. When RV was additionally applied, significantly elevated FOXP3+ regulatory T cells (Tregs) and a tendency for an increased number of CD44+ memory T cells were found in the BAL fluid of Mir146a/b−/− mice compared to wt mice (Figures 5B, 5C, and S7). In the same model, flow cytometry analysis of T helper cell subsets revealed significantly more Th17 cells and a trend for increased numbers of Th1 cells in Mir146a/b−/‐ mice, whereas wt mice displayed a tendency toward a higher number of Th2 cells in BAL fluid (Figures 5B‐5C, and Figure S7). Further RT‐qPCR analysis revealed increased miR‐146a/b expression in wt mouse lungs in response to HDM treatment, which was further elevated following RV infection (Figure 6A). In line with flow cytometry results, HDM treatment followed by RV infection resulted in increased expression of Cxcl1, Cxcl2, and Ccl11 in wt mouse lungs compared to mice treated with HDM alone, confirming the exacerbation of inflammation in the lungs (Figures 6B and 6C). When wt and Mir146a/b−/‐ mice were compared, a tendency for decreased Cxcl1, Cxcl2, Il‐13, Il‐4, Il‐17a, S100a8, S100a9 expression (Figures 6B, 6C, and S8A) and significantly decreased Ccl11 and increased Ifitm1 expression (Figure 6B‐C) were detected in lungs from Mir146a/b−/‐ mice following HDM and RV‐A1b application. Differences in the expression levels of Irf1 and Ifn‐γ were not statistically significant between wt and Mir146a/b−/− mice.
FIGURE 5.
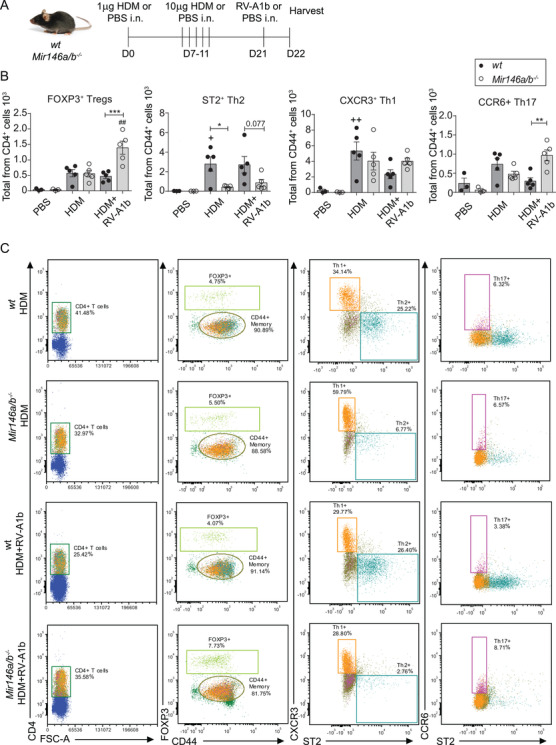
Lack of miR‐146a/b in mice results in an altered immune response in allergic airway inflammation and RV‐induced exacerbation of allergic airway inflammation models. (A) Mice were sensitized (D0) and challenged (D7‐11) with house dust mite extract (HDM) and, when indicated, infected with RV‐A1b (D21) for 24 h. Control mice received only PBS. (B) BAL cells were counted with a hemocytometer and then subjected to FACS analysis. From the dot plot percentages, the total numbers of immune cells were calculated. Data represent mean ± SEM. One‐way ANOVA with Tukey's multiple comparisons test and adjusted p‐values are shown. + p < 0.05, ++ p < 0.01 HDM compared to the same mouse line with PBS; ## p < 0.01 HDM+RV‐A1b compared to same mouse line with HDM; *p < 0.05, **p < 0.01, ***p < 0.001 wt compared to Mir146a/b−/− mice for the same treatment. (C) One representative FACS dot plot of BAL fluid from five HDM‐treated or HDM+RV‐A1b‐treated wt and Mir146a/b−/− mice. Data from one representative of three independent experiments are shown
FIGURE 6.
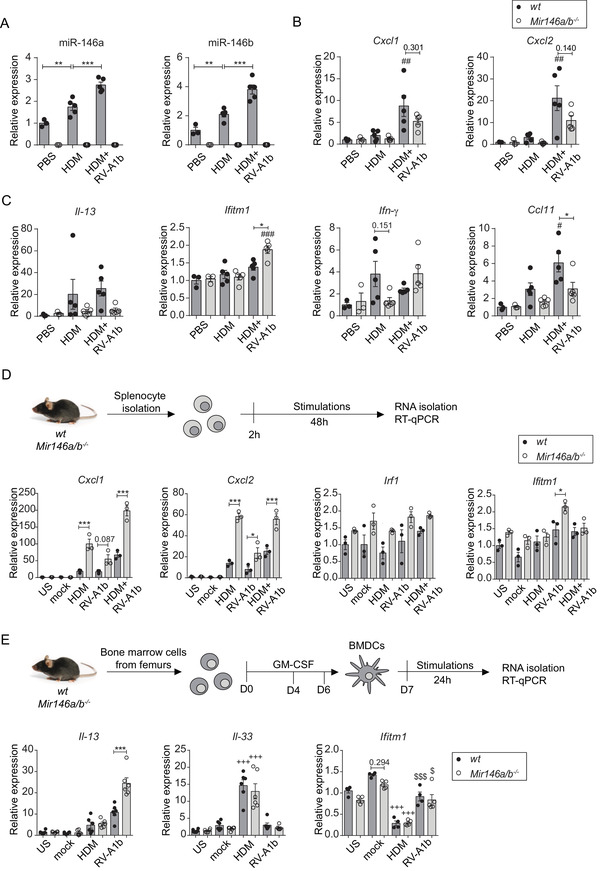
Mir146a/b−/− mice display differential gene expression in their lungs, splenocytes, and DCs in response to HDM or RV‐A1b. Relative miRNA (A) and mRNA expression in (B‐C) mouse lungs subjected to HDM‐induced airway inflammation and RV‐induced exacerbation of allergic airway inflammation models, (D) in splenocytes and (E) in BMDCs as measured by RT‐qPCR. Data are presented as the mean ± SEM and are compared to mean values of the wt PBS group (= 1) using one‐way ANOVA with Tukey's multiple comparisons test. Adjusted p‐values are shown. (A) **p < 0.01 wt PBS compared to wt HDM, ***p < 0.001 wt HDM compared to wt HDM+RV‐A1b. Data from one representative of three independent experiments are shown. (B‐C) #p < 0.05, ##0.01, ###p < 0.001 HDM+RV‐A1b compared to the same HDM mouse line; *p < 0.05 wt compared to Mir146a/b−/− mice during the same treatment. (D) Splenocytes from wt and Mir146a/b−/− mice were stimulated for 48 h; *p < 0.05, **p < 0.01, ***p < 0.001 wt compared to Mir146a/b−/− mice during the same treatment. (E) BMDCs from wt and Mir146a/b−/− mice were stimulated for 24 h; $ p < 0.05, $$$ p < 0.001 RV‐A1b compared to the same line mock; +++ p < 0.001 HDM compared to the same line US; ***p < 0.001 wt compared to Mir146a/b−/− during same treatment
To further compare the responses of the immune system between these two mouse lines, we extracted splenocytes and bone marrow‐derived DCs (BMDCs) and stimulated these cells in vitro using HDM or RV‐A1b alone or in combination. In contrast to the in vivo HDM‐induced airway inflammation and RV‐induced exacerbation of allergic airway inflammation models (Figure 6B), however consistent with the RV‐induced airway inflammation model (Figure 4C), a significantly increased expression of Cxcl1 and Cxcl2 was detected in Mir146a/b−/‐ mouse splenocytes (Figure 6D) in response to HDM alone but also in the case of HDM and RV‐A1b co‐stimulation. Mir146a/b−/‐ mouse splenocytes exhibited elevated Ifitm1 expression following the addition of RV‐A1b, while no significant changes in Irf1 were observed (Figure 6D). In addition, we detected elevated expression of Il‐33 and decreased expression of Ifitm1 in bone marrow‐derived DCs (BMDCs) from both mouse lines, with no significant differences between Mir146a/b−/‐ and wt mice in response to HDM stimulation, while significantly higher expression of Il‐13 was observed in RV‐A1b stimulated Mir146a/b−/‐ BMDCs compared to cells from wt mice (Figure 6E). No differences in expression levels of Cxcl1, Cxcl2, or Irf1 were found between Mir146a/b−/‐ and wt BMDCs (Figure S8B). Together, these results demonstrate that Mir146a/b−/‐ mice develop more dominant Th1/Th17 responses and less prominent Th2 cell responses in mouse models of HDM‐induced allergic airway inflammation and RV‐induced exacerbation of allergic airway inflammation, while BMDCs lacking miR‐146a/b did not show a gene expression pattern supporting Th1/Th17 skewing.
2.5. Intranasal administration of CPP‐miR‐146a nanocomplexes reduces HDM‐induced allergic airway inflammation
The transfection of miR‐146a mimics suppressed RV‐induced inflammation in HBECs (Figure 1, 2), and Mir146a/b−/− mice developed increased inflammation in the RV infection model (Figure 4). However, knockout mice displayed less prominent Th2 cell responses in HDM‐induced allergic airway inflammation and RV‐induced exacerbation of allergic airway inflammation models (Figures 5 and 6). Therefore, we next assessed whether in vivo application of miR‐146a has therapeutic effects in the case of allergic airway inflammation in mice. We subjected wt mice to an HDM‐induced allergic inflammation model (54, 55) and administered CPP‐miR‐146a or control nanocomplexes either 2 h before (Figure S9) or after each challenge (Figure 7A), followed by flow cytometry analysis of BAL fluid cells and RT‐qPCR of total RNA from the lung. When nanocomplexes were applied 2 h before the challenge, the treatment with HDM resulted in increase in numbers of measured cell counts without any significant differences between the HDM‐treated groups. Note that cell counts in HDM‐treated groups did not depend on whether CPP‐miRNAs were included, indicating that application of CPP‐miRNA nanocomplexes does not increase airway inflammation (Figure S9). A significantly reduced total number of eosinophils and B cells were found in BAL following administration of CPP‐miR‐146a nanocomplexes compared to CPP‐control from HDM‐treated mice (Figures 7B, 7C, S10A). A tendency for reduced numbers of regulatory T cells, memory T cells, and Th2 cells was found; however the differences were statistically not significant (Figures 7D and 7E). We also evaluated the delivery efficiency and localization of administered CPP‐miRNA nanocomplexes in mouse airways using Cy5‐labeled miR‐146a mimics and confocal microscopy. Figure 8A demonstrates that Cy5‐miR‐146a mimics localize around bronchioles in frozen mouse lung lobe sections. No Cy5 signal was detected in the kidney, liver, spleen, or MLN sections from mice transfected with CPP‐miR‐146a nanocomplexes when analyzed by confocal microscopy (data not shown). In line with this, RT‐qPCR analysis of total lung tissue RNA demonstrated that administration of CPP‐miR‐146a nanocomplexes significantly inhibited HDM‐induced expression of Il‐13, Il‐17A, Ifn‐γ, and the eosinophil attracting chemokine Ccl11 (Figure 8B). In addition, a tendency for decreased expression of Cxcl1, Cxcl2, Irf1, and Ifitm1 was observed (Figure S10B) in mouse lungs in the presence of CPP‐miR‐146a nanocomplexes. Taken together, our results demonstrate that the application of CPP‐miR‐146a nanocomplexes exerts an anti‐inflammatory effect in mouse airways during HDM‐induced allergic inflammation.
FIGURE 7.
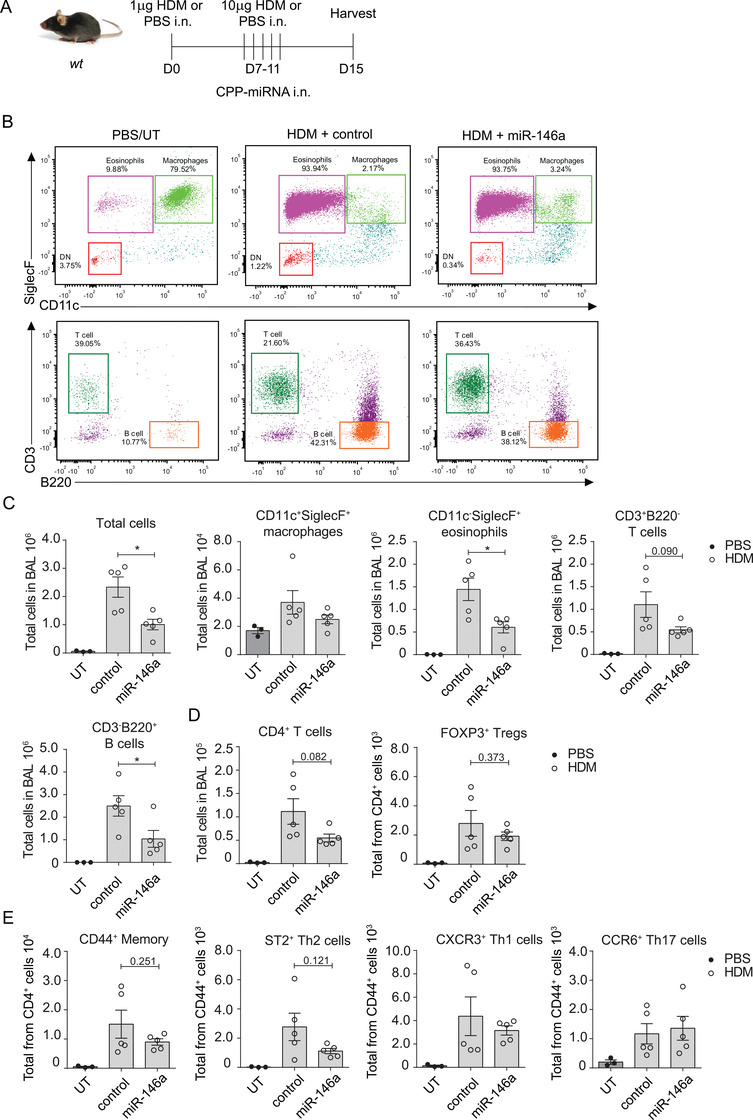
Administration of CPP‐miR‐146a nanocomplexes inhibits the accumulation of eosinophils, T cells, and B cells in an allergic airway inflammation model. Wt mice were sensitized i.n. at day 0 with 1 μg and challenged at days 7–11 daily with 10 μg of HDM. CPP‐miR‐146a (miR‐146a) and CPP‐miRNA control (control) nanocomplexes containing 60 pmol of corresponding miRNA mimic were applied 2 h after each challenge. On day 15, BAL cells and lung lobes for RNA were harvested. PBS group was left untransfected (UT). (A) Schematic of the experimental setup. (B and C) BAL fluid cells were counted with a hemocytometer, then subjected to FACS analysis. From the dot plots, the total numbers of immune cells were calculated. (B) One representative FACS dot‐plot of BAL macrophages, eosinophils, CD3+ T cells, and B220+ B cells. (C) Data are represented as mean ± SEM from five mice in study groups and three mice in UT PBS group, unpaired t‐test, *p < 0.05. Data from one representative of two independent experiments are shown. (C‐E) BAL cells were counted using a hemocytometer and then subjected to FACS analysis. From the dot plots, the total numbers of immune cells were calculated. Data are represented as the mean ± SEM, unpaired t‐test, *p < 0.05. Data from one representative of two independent experiments are shown
FIGURE 8.
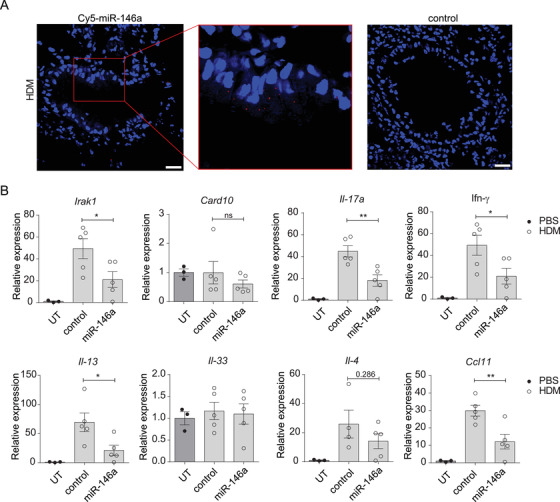
CPP‐miR‐146a nanocomplexes inhibit the expression of pro‐inflammatory genes in an HDM‐induced airway inflammation model. (A) Localization of labeled Cy5‐miR‐146a mimics (red) in lung lobes counterstained with DAPI (blue). Scale bar = 100 μm. (B) Relative mRNA expression levels of the indicated genes were compared to the PBS‐treated untransfected (UT) group (= 1) or HDM‐treated and miR‐146a mimic‐ or control‐transfected mouse lung lobes measured by RT‐qPCR. Data are represented as the mean ± SEM. Unpaired t‐test, *p < 0.05, **p < 0.01, ***p < 0.001. Data from one representative of two independent experiments are shown
3. DISCUSSION
Numerous studies have reported altered miRNA expression in asthmatic patients, 38 , 39 , 40 , 41 and accumulating data on miRNA functions from cell culture experiments 42 and animal models 43 suggest that miRNAs play important roles in the regulation of airway inflammation and the development of asthma. In the current study, we used both cell culture and mouse models to delineate miR‐146a/b functions in airway inflammation. We demonstrated that miR‐146a inhibits the pro‐inflammatory chemokines IL‐8, CXCL1, and CCL5, induces the production of IFN‐λ, and has the capacity to limit infection by RV‐A16 in HBECs. In line with the results from cell culture experiments, mice lacking miR‐146a/b displayed increased neutrophil infiltration and elevated secretion of chemokines in an RV‐induced airway inflammation model. Interestingly, while Mir146a/b−/− mice had less prominent Th2 responses in airway inflammation models involving HDM as an allergen, the application of CPP‐miR‐146a nanocomplexes led to general anti‐inflammatory effects in allergic airway inflammation, suggesting therapeutic potential for these nanocomplexes (Figure 9).
FIGURE 9.
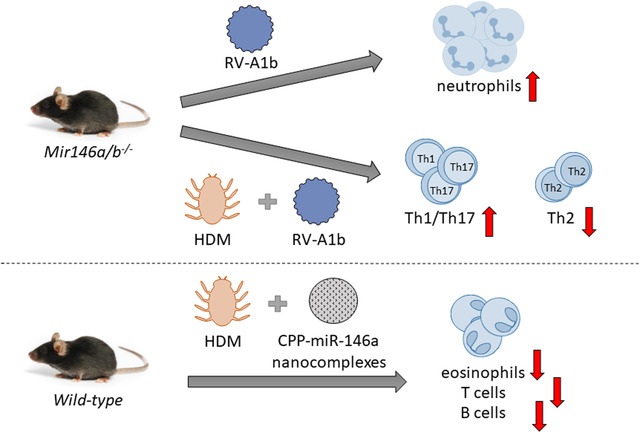
miR‐146a/b‐deficient mice develop more severe airway neutrophilia during RV infection and a less prominent Th2 cell mediated immune response in HDM‐induced allergic airway inflammation and RV‐induced exacerbation of allergic airway inflammation when compared with wild‐type mice. Application of cell‐penetrating peptide (CPP)‐miR‐146a nanocomplexes into the airways has an anti‐inflammatory effect in the HDM‐induced allergic airway inflammation model in wild‐type mice.
The first set of experiments demonstrated that miR‐146a/b expression is induced in response to RV‐A16 and RV‐A1b in HBECs along with changes in the studied pro‐inflammatory chemokines and interferon response genes. Previous publications have shown that miR‐146a and miR‐146b expression is dependent on activation of the NF‐κB and JAK‐STAT pathways, respectively, 23 , 44 , 45 which is in line with studies showing that both of these signaling pathways are induced during RV‐induced inflammation in the airways. 46 , 47 , 48 , 49
When miR‐146a was transfected into HBECs, we observed significantly reduced expression of neutrophil attracting chemokines, IL‐8 and CXCL1, 50 , 51 and fewer migrating neutrophils toward supernatants of RV‐stimulated HBECs. This finding suggests that miR‐146a may act to relieve RV‐induced inflammation mediated by neutrophils and is in line with our previous results demonstrating a link between decreased expression of miR‐146a in airway epithelial cells and the development of a neutrophilic phenotype of asthma. 26
Interestingly, we observed elevated levels of IFN‐λ in the supernatants and reduced mRNA levels of the RV‐A16 receptor ICAM‐1 in HBECs transfected with miR‐146a and infected with RVs. Moreover, a reduced infection rate of HBECs transfected with miR‐146a and infected with GFP‐tagged RV‐A16 was detected. It has been previously shown that both type I and type III interferon responses are activated during RV infection and that this activation is needed to limit viral spread. 47 , 48 Thus, our results indicate that miR‐146a may limit the viral spread of RVs through its capacity to increase IFN‐λ and additionally may inhibit the major group of RVs through suppression of ICAM‐1. Similar antiviral properties of miR‐146a in A549 52 and other bronchial epithelial cell lines 42 , 53 have been reported, however, not in primary epithelial cells.
To assess the function of the miR‐146 family during RV infection in vivo, we next performed a set of experiments using wt and miR‐146a/b knockout mice. We opted to use Mir146a/b−/− mice, as miR‐146a/b has identical seed sequences, can affect the same target genes 44 , 45 and therefore may partially compensate for each other. 54 , 55 We first generated a short mouse model of RV‐induced airway inflammation, which involved infection of mice with RV‐A1b. As major group RVs do not bind to mouse Icam‐1, 56 , 57 we only used RV‐A1b in this experiment. We found an increased total number of cells in the airways, including neutrophils, DCs, and T cells, as well as enhanced expression of Cxcl1, S100a8, and S100a9 after RV‐A1b infection in Mir146a/b−/‐ mice compared to wt mice. Interestingly, there was no difference in Ifitm1 mRNA levels, and Irf1 was even increased in Mir146a/b−/ − mice infected with RV compared to the control group, while in vitro results in HBECs showed that transfection of miR‐146a resulted in increased expression of interferon response genes. This difference might be due to the effect of other cell types present in the airways. Previously, the involvement of Irf1 in the regulation of type III interferons and its capacity to control the spread of influenza A virus have been shown. 58 , 59 In our experiments, the higher airway expression of Irf1 in RV‐infected mice lacking miR‐146a/b was accompanied by a greater pro‐inflammatory response. As suggested by previous publications, airway neutrophilia 13 and T‐cell recruitment 60 , 61 act to mediate viral clearance via Th1 cytokine production during RV infection. However, if cellular and immune responses are too strong during viral infection, a more serious health condition may develop. Our data suggest that the miR‐146 family is a part of an immune regulatory network needed for optimal immune responses in the airways during RV infection. Accordingly, we next studied how the lack of the miR‐146 family influences immune responses in RV‐induced exacerbation of allergic airway inflammation. As asthma exacerbations occur seasonally peaking from fall to winter, it has been suggested that despite the phase of an allergic reaction, viral infections play a major role in the exacerbation of asthma. 62 For example, it has been shown that RV infection occurring even 1 month after antigen exposure increases the occurrence of a late‐phase asthmatic response to the allergen by causing prolonged eosinophil infiltration to the lower airways of patients. 63 Therefore, we modified the previous protocol according to which mice are infected together with the last challenge 35 and inoculated mice with RV A1b 10 days after the last challenge. Surprisingly, in this model, flow cytometry analysis revealed no significant differences in the number of total cells in BAL fluid after HDM and RV co‐administration between the two mouse lines. However, strongly reduced numbers of Th2 cells were detected after HDM treatment, while significantly increased Th17 and FOXP3+ regulatory T cell infiltrations were found after HDM and RV application to the airways of Mir146a/b−/− mice. Similarly, we observed significantly decreased expression of eosinophils attracting chemokine Ccl11 and elevated expression of Ifitm1 in the lungs of Mir146a/b−/− mice following HDM and RV‐A1b administration. Previously, a loss of peripheral T cell tolerance, excessive amounts of Ifn‐γ expressed by CD4+ T cells, and elevated Th17 numbers have been reported for miR‐146a‐deficient mice due to the overactivation of STAT1 when mice are 6–8 months old. 54 , 64 , 65 , 66 , 67 , 68 Our results indicate that Th2 responses may already be impaired in Mir146a/b−/‐ mice at 8–10 weeks old or similarly to Mir146a−/− mice, dampened due to excessive Th17/Th1 cell‐mediated immune responses revealing in inflammatory conditions. In line with results from the RV‐induced airway inflammation model, increased mRNA expression of Cxcl1, Cxcl2, and Ifitm1 in HDM‐stimulated Mir146a/b−/− splenocytes was detected. However, the mRNA levels of other cytokines, such as Il‐17A, Il‐13, Il‐33, and Il‐4, were very low under these conditions (data not shown). As BMDCs from Mir146a/b−/− mice expressed increased Il‐13, the dominance of Th17/Th1 cell‐mediated immune responses in Mir146a/b−/− mice is likely not caused by differences in antigen‐presenting cells. As one possible scenario, T helper cells from Mir146a/b−/− mice might be more prone to Th17/Th1 type cytokine expression when activated, and that further suppresses the development of Th2 responses, eosinophilia, and inflammation during HDM treatment. Also, differences in epithelial or other tissue‐resident cell responses may lead to impaired Th2 responses in Mir146a/b−/− mice. Therefore, the precise mechanism that leads to reduced Th2 responses at the organism level in the absence of miR‐146a/b remains to be studied.
Since Mir146a/b−/− mice reacted somewhat differently in response to RV‐induced inflammation and exacerbation of allergic airway inflammation, we next assessed the effect of CPP‐miR‐146a nanocomplexes in a mouse model of HDM‐induced allergic airway inflammation, a Th2‐dependent model with a high number of eosinophils. 69 Confocal microscopy of mouse lungs revealed efficient delivery of Cy5‐labeled miR‐146a mimics into airway cells. Interestingly, wt mice receiving CPP‐miR‐146a nanocomplexes displayed a significantly reduced total number of cells, eosinophils, and B cells in their BAL following HDM administration. In addition, decreased expression of Il‐17a, Ifn‐γ, Il‐13, and Ccl11 upon HDM exposure was found in lung lobes in mice treated with CPP‐miR‐146a. Taken together, these results suggest that the administration of CPP‐miR‐146a nanocomplexes affects all immune responses, leading to decreased inflammation and attenuated airway symptoms without significant changes in the Th2/Th1/Th17 balance.
Generally, the therapeutic application of miRNA mimics is limited due to their low capacity to penetrate cell membranes due to their negative charge. 70 To date, many substances have been assessed for the delivery of nucleic acids, including CPPs, but there is still a lack of efficient, safe, and specific methods for the in vivo delivery of potentially therapeutic nucleic acids, including miRNA mimics. 71 , 72 We previously demonstrated the capacity of CPP‐miR‐146a noncovalent nanocomplexes to suppress inflammatory responses in cell cultures and a mouse model of irritant contact dermatitis. 73 To our knowledge, we report for the first time that intranasal administration of CPP‐miRNA nanocomplexes is efficient for delivering miRNA mimics into mouse airways.
One of the main concerns in the development of nucleic acid‐based therapies is off‐target effects. However, as miRNA‐s are designed by nature, it has been suggested that there are fewer off‐target effects caused by the binding of either strand of miRNA mimics to unexpected targets than artificially designed siRNAs. 74 In our study, we also assessed the presence of the Cy5 signal in frozen sections of organs collected at the end of the experiment; however, we did not detect any signal in the kidneys, spleen, liver, or MLN sections (data not shown). This suggests that most of the CPP‐miRNA nanocomplexes accumulated in the lungs, and therefore, there is less chance for off‐target effects.
It should be noted that several open questions concerning the functions of miR‐146a/b in the regulation of cellular responses of HBECs and immune responses during airway inflammation remain. For example, we previously reported reduced miR‐146a expression in bronchial brushings from asthma patients. Therefore, it would be interesting to study miR‐146 family expression and function during RV infection in HBECs from asthma patients. Another limitation of our study is that all experiments in HBECs were performed in submerged undifferentiated HBECs, and this did not enable us to assess the influence of miR‐146a/b on the differentiation of HBECs. Yet another question is whether miR‐146a/b affects all types of RVs. For example, several recent studies have revealed the importance of RV‐C in the development and exacerbation of asthma in children. 75 , 76 , 77 Our study did not include RV‐C; however, similar to the included RV species, increased secretion of CXCL10 and IFN‐λ in response to RV‐C has been reported from human primary nasal epithelial cells. 78 This indicates that the cellular responses of epithelial cells are similar in the case of RV‐A16, RV‐A1b, and RV‐C and that miR‐146a/b may also act as an anti‐inflammatory miRNA during RV‐C infection. Similar to the cell culture experiments, many questions remain to be studied regarding the influence of miR‐146a/b on immune responses in mouse models. For example, although we demonstrated increased infiltration of Th1/Th17 cells in the BAL of Mir146a/b−/− mice subjected to RV‐induced exacerbation of allergic airway inflammation, further studies are needed to better describe the individual effects of miR‐146a/b in different cell types. Accordingly, to confirm changes in Th1/Th17/Th2 balance, it would be interesting to repeat the experiment with antibodies for transcription factors GATA‐3, RORgt, and T‐bet to distinguish effector T cell subsets in flow cytometry. 79 , 80 , 81 Similarly, it would be also interesting to assess the effect of miR‐146a/b deficiency in a case when RV infection is performed simultaneously with antigen challenge. In addition, it would be interesting to investigate the suppressor capacity of Treg cells in Mir146a/b−/− mice, as previously, Treg cells from Mir146a−/− mice were shown to be defective in their capacity to suppress Th1 responses. 64 Also, since Mir146a/b−/− mice displayed impaired Th2 responses in the mouse models of HDM‐induced airway inflammation and RV‐induced exacerbation of allergic airway inflammation, it would be interesting to study whether there is an effect on type 2 ILC2, which likewise to Th2 cells, are capable of producing type 2 cytokines, including IL‐4, IL‐5, and IL‐13 82 , 83 and may therefore compensate for the reduced effect of Th2 cells.
Finally, as CPP‐miR‐146a nanocomplexes had an effect when applied after, but not before the challenge, our data suggest that these complexes penetrate to the tissue only during active inflammation and are therefore suitable for treatment, but not for prevention. Also, in the current study, the therapeutic potential of intranasal administration of CPP‐miRNA nanocomplexes was analyzed in the HDM‐induced airway inflammation model; however, the effect in the mouse model of RV‐induced exacerbation of allergic airway inflammation remains to be explored. Furthermore, it would be interesting to study whether continued treatment with CPP‐miRNA nanocomplexes would have a stronger anti‐inflammatory effect in the airways when compared with administration of nanocomplexes during the HDM challenge phase only. Therefore, although these first results are promising, additional preclinical experiments are clearly needed to assess the safety and optimize the methods of applying CPP‐miRNA nanocomplexes to target airway inflammation before any clinical trials could be initiated.
In conclusion, in the current study, we demonstrated that miR‐146a/b exerts anti‐inflammatory properties in HBECs and mouse airways during RV infection. Furthermore, even though the lack of miR‐146a/b led to reduced inflammation in murine HDM‐induced airway inflammation and RV‐induced exacerbation models, the intranasal application of CPP‐miR‐146a nanocomplexes strongly inhibited allergic inflammation in mouse airways, suggesting that the temporary overexpression of miR‐146a may exert a therapeutic effect in targeting different airway inflammation conditions.
4. MATERIALS AND METHODS
4.1. Isolation and culture of HBECs
HBECs were isolated by initial short‐term pronase (Roche, Basel, Switzerland) and DNase (Sigma‐Aldrich) digestion of bronchoscopy biopsies obtained from nonasthmatic donors from the Department of Pulmonology, Jagiellonian University Medical College (Krakow, Poland). In total, HBECs from two nonasthmatic donors were obtained (one male and one female Caucasian of age 26 and 30, respectively). Both donors underwent diagnostic bronchoscopy, and chronic airway disease was ruled out during further analyses. Frozen HBECs of passage 0 were thawed and cultured in BEGM Bulletkit (Lonza, Switzerland) media containing bovine pituitary extract, insulin, hydrocortisone, gentamicin and amphotericin‐B, retinoic acid, transferrin, triiodothyronine, epinephrine, and human epidermal growth factor as recommended by the manufacturer, and cells were transferred to a cell culture incubator (at 37°C under 5% CO2) for further experiments. This study was approved by the Ethics Committee of the Jagiellonian University Medical College.
4.2. Culture and maintenance of RV stocks
RV‐A16‐GFP was generated from the pA16‐eGFP plasmid by reverse genetics as previously described. 84 , 85 To propagate RV‐A16‐GFP and other RV serotypes used in the study, A16 and A1b RVs were cultured in Ohio HeLa cells. 35 All virus stocks from crude cell lysates were titrated by standard methods using HeLa monolayers in RPMI media and serially diluted RVs to estimate TCID50/ml by the Kremser method and stored at ‐80°C until further use.
4.3. Infection and transfection of HBECs
For experiments in HBECs, 3 × 104 cells per well were seeded into 12‐well plates (Greiner Bio‐one, Germany). Twenty‐four hours later, BEGM media from plated cells was replaced with fresh media, and cells were infected with RV‐A16, RV‐A1b, or RV‐A16‐GFP at a multiplicity of infection (MOI) of 0.1 or a negative control "mock," containing RPMI media from HeLa cells without RV particles. An MOI of 0.1 was chosen as one of the lowest concentrations previously shown to infect and activate HBECs 86 and because it was sufficient to induce cellular responses of HBECs (Figure S1). For transfection, HBECs were seeded into 12‐well plates at a density of 4 × 104 cells per well, and 24 h later, 30 nM miRIDIAN microRNA Mimic Negative Control #1 (Dharmacon, USA) or miRIDIAN microRNA hsa‐miR‐146a‐5p mimic (Dharmacon, USA) was transfected using MIRFECT (RNAexact, Estonia) according to the manufacturer's protocol. Twenty‐four hours after transfection, HBECs were infected with the indicated RVs or negative control mock. The supernatant and cells were harvested 24 h or 48 h later for further assays. For harvesting RNA, 500 μl Qiazol (Qiagen, Germany) was added to cells and stored at ‐20°C until RNA isolation.
4.4. RNA extraction, cDNA synthesis, and RT‐qPCR
To isolate RNA from HBECs and mouse lung lobes, a Total RNA Zol‐Out kit (A&A Biotechnology, Poola) and a Total RNA Zol‐Out D kit (A&A Biotechnology, Poola) were used according to the manufacturer's guidelines, respectively. To analyze relative miRNA and mRNA expression in cells and mouse lungs, RT‐qPCR and ΔΔCt calculations were used. As housekeeping genes for normalization, let7a was used for miRNA analysis, and EEF1A1 or Hprt was used for mRNA analysis. The data were compared relative to the mean value of the control group or condition, which was normalized to 1 and is indicated in each figure legend. More detailed descriptions of RNA isolation, cDNA synthesis, RT‐qPCR, and primer sequences are included in this article's Supplementary Information.
4.5. Neutrophil chemotaxis assay
Primary human neutrophils were isolated from a healthy donor's whole blood essentially as previously described 87 using gradient centrifugation on Ficoll‐Paque Plus (GE Healthcare, Chicago, IL, United States). To lyse erythrocytes, red blood cell lysis buffer (Merck, Darmstadt, Germany) was used. Neutrophils were seeded at a density of 4 × 105 on ThinCert cell culture inserts (3‐μm pore size) (Greiner Bio‐One, Kremsmünster, Austria) and placed into 24‐well plates. The outer chamber contained supernatants from HBECs transfected with miRNA mimics and stimulated with cytokines. Sixty minutes after incubation at 37°C under 5% CO2, the number of neutrophils migrating from the insert into the outer chamber supernatant was analyzed using a BD LSRFortessa (BD Biosciences, USA) cell analyzer. For each sample, the number of events was recorded for 1 min.
4.6. Mouse lines
All mice were bred and maintained under specific pathogen‐free conditions in the Laboratory Animal Center of the University of Tartu (Tartu, Estonia). The Mir146a/b−/− mouse line was created by crossing Mir146b−/− and Mir146a−/− lines in the Laboratory Animal Center of the University of Tartu (Tartu, Estonia). Mir146b−/− mice were generated as previously described. 24 C57Bl/6J wild‐type (wt) and Mir146a−/− mice were purchased from Jackson Laboratory (Bar Harbor, US). Six‐ to ten‐week‐old mice were used in all experiments. No significant behavioral or phenotypical differences between Mir146a/b‐/‐ and wt mice were observed during maintenance.
4.7. Mouse disease models
All animal studies were approved by the Animal Ethics Committee at the Ministry of Agriculture Estonian Government (01.03.2018, license 117). A mouse model of RV‐A1b‐induced airway inflammation was established as previously described. 35 Briefly, C57Bl/6J wt and Mir146a/b−/‐ mice were lightly anesthetized with isofluorane, and 40 μl RV‐A1b (7.78 × 105 TCID50) or PBS as a control was administered i.n. Twenty‐four hours after infection, mice were sacrificed, and BAL fluid and the left upper lobe of the lung were collected from each mouse.
Mouse models of HDM‐induced allergic airway inflammation and RV‐A1b‐induced exacerbation of allergic airway inflammation were developed based on previous studies. 35 , 36 , 37 In both models, 1 μg HDM extract (Greer Laboratories, Lenoir, NC) in 40 μl PBS was administered i.n. on day 0 to sensitize the C57Bl/6J wt and Mir146a/b−/− mice. On day 7–11, mice were challenged daily with 10 μg HDM extract in 40 μl of PBS (controls received PBS only) to induce allergic airway inflammation. On day 21 mice were i.n. infected with 40 μl RV‐A1b (7.78 × 105 TCID50) or PBS as a control. Mice were sacrificed on day 22, and BAL fluid and lung tissue were collected for further analysis.
The effect of CPP‐miR‐146a nanocomplexes was assessed in a mouse model of HDM‐induced allergic airway inflammation. 36 , 37 For that, C57Bl/6J wt mice were lightly anesthetized, and for sensitization, 1 μg HDM extract in 40 μl PBS was administered i.n. to mice on day 0. On day 7–11, 10 μg HDM extract in 40 μl PBS was administered daily, and 2 h later, 60 pmol CPP‐miR‐146a or CPP‐control miR mimic nanocomplexes in 40 μl PBS were administered i.n. On day 15, mice were sacrificed, and blood, BAL fluid, and lung lobes were collected. More detailed information can be found in the Supplementary Materials and Methods.
4.8. Collection and analyses of BAL fluid
BAL fluid samples were obtained by lavaging the airways of mice three times with 1 ml of PBS through a tracheal cannula. First, 1 ml of collected BAL was collected into separate tubes and centrifuged at 300 rcf for 5 min, and supernatants were used for protein quantification using ELISA. Next, erythrocytes were lysed using Red Blood Cell Lysis Buffer (Roche, Basel, Switzerland), and BAL cells were counted and stained with fluorochrome‐labeled antibodies according to the manufacturer's instructions and subjected to flow cytometry analysis using BD LSRFortessa (BD Biosciences, USA). For each sample, 50,000 events were recorded, and data were analyzed using the FCS Express 7 program. The gating strategy used to identify BAL cells is shown in Figure S5. A more precise sorting protocol appears in the Supplementary Materials and Methods section.
4.9. Isolation and stimulation of mouse splenocytes and BMDCs
Single splenocyte suspensions were prepared from wt and Mir146a/b−/− mouse spleens by homogenization through 100 μm cell strainers followed by washing in RPMI‐1640 medium (Merck) supplemented with 10% FBS and 1% Pen‐Strep. To obtain DCs, bone marrow cells from wt and Mir146a/b−/− mice were isolated and cultured in RPMI‐1640 medium supplemented with 1% FBS and 20 ng/ml GM‐CSF as previously described. 88 For stimulation, 50 μg HDM, 100 μl of RV‐A1b (TCID50 = 1.53*104) or 50 μg HDM + 100 μl of RV‐A1b (TCID50 = 1.53*104) was added to 2 × 106 splenocytes in 500 μl of RPMI for 48 h or to 5 × 105 BMDCs per well for 24 h. As a negative control, splenocytes were left unstimulated or 100 μl of "mock" medium containing RPMI medium from uninfected HeLa cells was used.
4.10. Protein quantification
Protein levels in the supernatants of HBECs or BAL fluid were analyzed by ELISA. For HBEC samples, human IL‐8 ELISA MAX Deluxe Set (BioLegend, 431504), human CXCL1/GRO alpha DuoSet ELISA (R&D Systems, DY275‐05), and human IL‐29/IL‐28B (IFN‐lambda 1/3) DuoSet ELISA kit (R&D Systems, DY1598B‐05) were used according to the manufacturers’ instructions. For BAL fluids, a mouse CXCL1/KC DuoSet ELISA kit (R&D Systems, DY453‐05) was used as instructed by the manufacturer. The data were acquired with a Ledetect 96 Microplate reader (Labexim Products, Lengau, Austria). Online analysis software Myassays.com and a four‐parameter logistic regression model were used to calculate the concentrations of the proteins.
4.11. Target and pathway analysis
One thousand eighty one unconserved targets with cumulative weighted context score < −0.1 were selected with TargetScan 7.2 32 and compared with the dataset of 4647 genes detected to be differentially expressed in response to human RVs in HBECs from non‐asthmatic individuals. 33 Three hundred fifty overlapped genes we subjected to pathway analysis using enrichr 89 and the MSigDB Hallmark Gene Set Collection. 34
4.12. Statistical analysis
GraphPad Prism 6 (GraphPad Software Inc., USA) and unpaired two‐sided Student's t‐test or one‐way ANOVA with Tukey's multiple comparisons test were used for visualization and statistical analysis, as indicated in the figure legends. The results were considered to be significant at *p < 0.05; **p < 0.01; ***p < 0.001.
CONFLICT OF INTEREST
Ana Rebane and Margus Pooga are board members of RNAexact OÜ. All other authors declare no conflict of interest regarding this manuscript.
AUTHOR CONTRIBUTIONS
Anet Laanesoo performed the experiments, analyzed the data, contributed to the design of the study, and wrote the manuscript. Egon Urgard and Jonathan Jonathan M. Coquet contributed to the design and performance of experiments in mice. Kapilraj Periyasamy performed experiments in mouse splenocytes and BMDCs. Yury A. Bochkov, James E. Gern, Alar Aab, and Sebastian L. Johnston provided GFP‐tagged and wt RV strains and contributed to the study design. Jesper Wengel designed and provided miRNA mimics for mouse experiments. Grazyna Bochenek and Bogdan Jakiela prepared HBECs and contributed to the study design. Mark P. Boldin and Nathaniel Magilnick provided knockout mouse lines. Martti Laan, Margus Pooga, and Alan Altraja contributed to the study design and review of the manuscript. Ana Rebane designed the study, supervised the project, and wrote the manuscript. All authors read and approved the final version of the manuscript.
Supporting information
Supporting information
ACKNOWLEDGMENTS
This work was supported by personal research grants PUT1669 and 1617 from the Estonian Research Council, European Union through the European Regional Development Fund (project number: 2014–2020.4.01.15‐0012; Centre of Excellence for Genomics and Translational Medicine), in part by National Institutes of Health (NIH) R01AI125615 and Research Career Development Award by the STOP CANCER Foundation to M. P. B. and by Polish National Science Center Grants UMO‐2014/15/B/NZ5/01539, UMO‐2014/13/B/NZ3/02393. Sebastian L. Johnston is the Asthma UK Clinical Chair (grant CH11SJ) and a National Institute of Health Research (NIHR) Emeritus Senior Investigator and is funded in part by European Research Council Advanced Grant 788575 and by the NIHR Imperial Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care.
Laanesoo A, Urgard E, Periyasamy K, et al. Dual role of the miR‐146 family in rhinovirus‐induced airway inflammation and allergic asthma exacerbation. Clin Transl Med. 2021;11:e427. 10.1002/ctm2.427
DATA AVAILABILITY STATEMENT
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- 1. Boonpiyathad T, Sözener ZC, Satitsuksanoa P, Akdis CA. Immunologic mechanisms in asthma. Semin Immunol. 2019;46:101333. [DOI] [PubMed] [Google Scholar]
- 2. Traister RS, Wenzel SE. Inflammatory phenotypes in asthma pathogenesis. Drug Discovery Today: Disease Mechanisms. 2012;9(3‐4):e75–e81.24976849 [Google Scholar]
- 3. Tliba O, Panettieri RA. Paucigranulocytic asthma: Uncoupling of airway obstruction from inflammation. Journal of Allergy and Clinical Immunology. 2019;143 (4):1287–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Malmhäll C, Alawieh S, Lu Y, Sjöstrand M, Bossios A, Eldh M, Rådinger M. MicroRNA‐155 is essential for TH2‐mediated allergen‐induced eosinophilic inflammation in the lung. Journal of Allergy and Clinical Immunology. 2014;133 (5):1429–1438.e7. [DOI] [PubMed] [Google Scholar]
- 5. Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, Arron JR, Koth LL, Fahy V. T‐helper Type 2–driven Inflammation Defines Major Subphenotypes of Asthma. American Journal of Respiratory and Critical Care Medicine. 2009;180(5):388–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pelaia G, Vatrella A, Busceti MT, et al. Cellular mechanisms underlying eosinophilic and neutrophilic airway inflammation in asthma. Mediat Inflamm. 2015;879783:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Priller J, Böttcher C. A role for neutrophils in asthma exacerbations. Nat Med. 2017;23:659–661.28586340 [Google Scholar]
- 8. Pavord ID, Beasley R, Agusti A, Anderson GP, Bel E, Brusselle G, Cullinan P, Custovic A, Ducharme FM, Fahy JV, Frey U, Gibson P, Heaney LG, Holt PG, Humbert M, Lloyd CM, Marks G, Martinez FD, Sly PD, von Mutius E, Wenzel S, Zar HJ, Bush A. After asthma: redefining airways diseases. The Lancet. 2018;391 (10118):350–400. [DOI] [PubMed] [Google Scholar]
- 9. Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. 2015;16:45–56. [DOI] [PubMed] [Google Scholar]
- 10. Nakagome K, Bochkov YA, Ashraf S, Brockman‐Schneider RA, Evans MD, Pasic TR, Gern JE. Effects of rhinovirus species on viral replication and cytokine production. Journal of Allergy and Clinical Immunology. 2014;134 (2):332–341.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jackson DJ, Gern JE, Lemanske RF. The contributions of allergic sensitization and respiratory pathogens to asthma inception. Journal of Allergy and Clinical Immunology. 2016;137 (3):659–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Potaczek DP, Unger SD, Zhang N, Taka S, Michel S, Akdağ N, Lan F, Helfer M, Hudemann C, Eickmann M, Skevaki C, Megremis S, Sadewasser A, Alashkar Alhamwe B, Alhamdan F, Akdis M, Edwards MR, Johnston SL, Akdis CA, Becker S, Bachert C, Papadopoulos NG, Garn H, Renz H. Development and characterization of DNAzyme candidates demonstrating significant efficiency against human rhinoviruses. Journal of Allergy and Clinical Immunology. 2019;143 (4):1403–1415. [DOI] [PubMed] [Google Scholar]
- 13. Gern JE. The ABCs of rhinoviruses, wheezing, and asthma. J Virol. 2010;84:7418–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kennedy JL, Turner RB, Braciale T, Heymann PW, Borish L. Pathogenesis of rhinovirus infection. Curr Opin Virol. 2012;2:287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Makris S, Johnston S. Recent advances in understanding rhinovirus immunity. F1000Res. 2018;7:1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sokulsky LA, Garcia‐Netto K, Nguyen TH, Girkin JLN, Collison A, Mattes J, Kaiko G, Liu C, Bartlett NW, Yang M, Foster PS. A Critical Role for the CXCL3/CXCL5/CXCR2 Neutrophilic Chemotactic Axis in the Regulation of Type 2 Responses in a Model of Rhinoviral‐Induced Asthma Exacerbation. The Journal of Immunology. 2020;205 (9):2468–2478. [DOI] [PubMed] [Google Scholar]
- 17. Rebane A, Akdis CA. MicroRNAs: Essential players in the regulation of inflammation. Journal of Allergy and Clinical Immunology. 2013;132 (1):15–26. [DOI] [PubMed] [Google Scholar]
- 18. Bartel DP. Metazoan MicroRNAs. Cell. 2018;173 (1):20–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kingsley SMK, Bhat BV. Role of microRNAs in sepsis. Inflamm Res. 2017;66:553–569. [DOI] [PubMed] [Google Scholar]
- 20. Rebane A, Akdis CA. MicroRNAs in Allergy and Asthma. Current Allergy and Asthma Reports. 2014;14 (4):. [DOI] [PubMed] [Google Scholar]
- 21. Lu TX, Rothenberg ME. Diagnostic, functional, and therapeutic roles of microRNA in allergic diseases. Journal of Allergy and Clinical Immunology. 2013;132 (1):3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vaher H, Kivihall A, Runnel T, et al. SERPINB2 and miR‐146a/b are coordinately regulated and act in the suppression of psoriasis‐associated inflammatory responses in keratinocytes. Exp Dermatol. 2019;29:51–60. [DOI] [PubMed] [Google Scholar]
- 23. Taganov K.D, Boldin MP, Chang KJ, Baltimore D. NF‐ B‐dependent induction of microRNA miR‐146, an inhibitor targeted to signaling proteins of innate immune responses. Proceedings of the National Academy of Sciences. 2006;103 (33):12481–12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hermann H, Runnel T, Aab A, et al. miR‐146b probably assists miRNA‐146a in the suppression of keratinocyte proliferation and inflammatory responses in psoriasis. J Invest Dermatol. 2017;137:1945–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Crone S, Jacobsen A, Federspiel B, Bardram L, Krogh A, Lund AH, Friis‐Hansen L. microRNA‐146a inhibits G protein‐coupled receptor‐mediated activation of NF‐κB by targeting CARD10 and COPS8 in gastric cancer. Molecular Cancer. 2012;11 (1):71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kivihall A, Aab A, Soja J, Sładek K, Sanak M, Altraja A, Jakiela B, Bochenek G, Rebane A. Reduced expression of miR‐146a in human bronchial epithelial cells alters neutrophil migration. Clinical and Translational Allergy. 2019;9 (1):. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rebane A, Runnel T, Aab A, Maslovskaja J, Rückert B, Zimmermann M, Plaas M, Kärner J, Treis A, Pihlap M, Haljasorg U, Hermann H, Nagy N, Kemeny L, Erm T, Kingo K, Li M, Boldin MP, Akdis CA. MicroRNA‐146a alleviates chronic skin inflammation in atopic dermatitis through suppression of innate immune responses in keratinocytes. Journal of Allergy and Clinical Immunology. 2014;134 (4):836–847.e11. [DOI] [PubMed] [Google Scholar]
- 28. Panganiban RP, Wang Y, Howrylak J, Chinchilli VM, Craig TJ, August A, Ishmael FT. Circulating microRNAs as biomarkers in patients with allergic rhinitis and asthma. Journal of Allergy and Clinical Immunology. 2016;137 (5):1423–1432. [DOI] [PubMed] [Google Scholar]
- 29. Feng MJ, Shi F, Qiu C, Peng WK. MicroRNA‐181a, ‐146a and ‐146b in spleen CD4+ T lymphocytes play proinflammatory roles in a murine model of asthma. International Immunopharmacology. 2012;13 (3):347–353. [DOI] [PubMed] [Google Scholar]
- 30. Collison A, Herbert C, Siegle JS, Mattes J, Foster PS, Kumar RK. Altered expression of microRNA in the airway wall in chronic asthma: miR‐126 as a potential therapeutic target. BMC Pulmonary Medicine. 2011;11 (1):. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Han S, Ma C, Bao L, Lv L, Huang M. miR‐146a Mimics Attenuate Allergic Airway Inflammation by Impacted Group 2 Innate Lymphoid Cells in an Ovalbumin‐Induced Asthma Mouse Model. International Archives of Allergy and Immunology. 2018;177 (4):302–310. [DOI] [PubMed] [Google Scholar]
- 32. Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. eLife. 2015;4 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Helling BA, Sobreira DR, Hansen GT, Sakabe NJ, Luo K, Billstrand C, Laxman B, Nicolae RI, Nicolae DL, Bochkov YA, Gern JE, Nobrega MA, White SR, Ober C. Altered transcriptional and chromatin responses to rhinovirus in bronchial epithelial cells from adults with asthma. Communications Biology. 2020;3 (1):. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Systems. 2015;1 (6):417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bartlett NW, Walton RP, Edwards MR, Johnston SL. Mouse models of rhinovirus‐induced disease and exacerbation of allergic airway inflammation. Nat Med. 2008;14:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tibbitt CA, Stark JM, Martens L, Ma J, Mold JE, Deswarte K, Oliynyk G, Feng X, Lambrecht BN, De Bleser P, Nylén S, Hammad H, Arsenian Henriksson M, Saeys Y, Coquet JM. Single‐Cell RNA Sequencing of the T Helper Cell Response to House Dust Mites Defines a Distinct Gene Expression Signature in Airway Th2 Cells. Immunity. 2019;51 (1):169–184.e5. [DOI] [PubMed] [Google Scholar]
- 37. Rodriguez‐Perez N, Schiavi E, Frei R, Ferstl R, Wawrzyniak P, Smolinska S, Sokolowska M, Sievi NA, Kohler M, Schmid‐Grendelmeier P, Michalovich D, Simpson KD, Hessel EM, Jutel M, Martin‐Fontecha M, Palomares O, Akdis CA, O'Mahony L. Altered fatty acid metabolism and reduced stearoyl‐coenzyme a desaturase activity in asthma. Allergy. 2017;72 (11):1744–1752. [DOI] [PubMed] [Google Scholar]
- 38. Jardim MJ, Dailey L, Silbajoris R, Diaz‐Sanchez D. Distinct MicroRNA Expression in Human Airway Cells of Asthmatic Donors Identifies a Novel Asthma‐Associated Gene. American Journal of Respiratory Cell and Molecular Biology. 2012;47 (4):536–542. [DOI] [PubMed] [Google Scholar]
- 39. Martinez‐Nunez RT, Rupani H, Platé M, et al. Genome‐wide posttranscriptional dysregulation by MicroRNAs in human asthma as revealed by Frac‐seq. J Immunol. 2018;201:251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bartel S, Schulz N, Alessandrini F, et al. Pulmonary microRNA profiles identify involvement of Creb1 and Sec14l3 in bronchial epithelial changes in allergic asthma. Sci Rep. 2017;7:46026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Solberg OD, Ostrin EJ, Love MI, Peng JC, Bhakta NR, Hou L, Nguyen C, Solon M, Nguyen C, Barczak AJ, Zlock LT, Blagev DP, Finkbeiner WE, Ansel KM, Arron JR, Erle DJ, Woodruff PG. Airway Epithelial miRNA Expression Is Altered in Asthma. American Journal of Respiratory and Critical Care Medicine. 2012;186 (10):965–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Perry MM, Moschos SA, Williams AE, Shepherd NJ, Larner‐Svensson HM, Lindsay MA. Rapid Changes in MicroRNA‐146a Expression Negatively Regulate the IL‐1β‐Induced Inflammatory Response in Human Lung Alveolar Epithelial Cells. The Journal of Immunology. 2008;180 (8):5689–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lu TX, Munitz A, Rothenberg ME. MicroRNA‐21 Is Up‐Regulated in Allergic Airway Inflammation and Regulates IL‐12p35 Expression. The Journal of Immunology. 2009;182 (8):4994–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Curtale G, Mirolo M, Renzi TA, Rossato M, Bazzoni F, Locati M. Negative regulation of Toll‐like receptor 4 signaling by IL‐10‐dependent microRNA‐146b. PNAS. 2013;110:11499–11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ahn J, Lee H, Jung CH, Jeon TI, Ha TY. Micro RNA ‐146b promotes adipogenesis by suppressing the SIRT 1‐ FOXO 1 cascade. EMBO Molecular Medicine. 2013;5 (10):1602–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Girkin J, Hatchwell L, Foster P, et al. CCL7 and IRF‐7 mediate hallmark inflammatory and IFN responses following rhinovirus 1B infection. J Immunol. 2015;194:4924–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xander N, Reddy Vari H, Eskandar R, Li W, Bolla S, Marchetti N, Sajjan US. Rhinovirus‐Induced SIRT‐1 via TLR2 Regulates Subsequent Type I and Type III IFN Responses in Airway Epithelial Cells. The Journal of Immunology. 2019;203 (9):2508–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bartlett NW, Slater L, Glanville N, Haas JJ, Caramori G, Casolari P, Clarke DL, Message SD, Aniscenko J, Kebadze T, Zhu J, Mallia P, Mizgerd JP, Belvisi M, Papi A, Kotenko SV, Johnston SL, Edwards MR. Defining critical roles for NF‐κB p65 and type I interferon in innate immunity to rhinovirus. EMBO Molecular Medicine. 2012;4 (12):1244–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim J, Sanders SP, Siekierski ES, Casolaro V, Proud D. Role of NF‐κB in Cytokine Production Induced from Human Airway Epithelial Cells by Rhinovirus Infection. The Journal of Immunology. 2000;165 (6):3384–3392. [DOI] [PubMed] [Google Scholar]
- 50. Sawant KV, Poluri KM, Dutta AK, et al. Chemokine CXCL1 mediated neutrophil recruitment: role of glycosaminoglycan interactions. Sci Rep. 2016;6:33123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Laan M, Chu Z‐H, Hiroshi H, Lötvall J, Sjöstrand M, Gruenert DC, Skoogh B‐E, Linden A, et al. Neutrophil recruitment by human IL‐17 via C‐X‐C chemokine release in the airways. J Immunol. 1999;162(4):2347–2352. [PubMed] [Google Scholar]
- 52. Terrier O, Textoris J, Carron C, Marcel V, Bourdon J‐C, Rosa‐Calatrava M. Host microRNA molecular signatures associated with human H1N1 and H3N2 influenza A viruses reveal an unanticipated antiviral activity for miR‐146a. Journal of General Virology. 2013;94 (5):985–995. [DOI] [PubMed] [Google Scholar]
- 53. Tsai M‐J, Tsai Y‐C, Chang W‐A, Lin Y‐S, Tsai P‐H, Sheu C‐C, Kuo P‐L, Hsu Y‐L. Deducting MicroRNA‐Mediated Changes Common in Bronchial Epithelial Cells of Asthma and Chronic Obstructive Pulmonary Disease—A Next‐Generation Sequencing‐Guided Bioinformatic Approach. International Journal of Molecular Sciences. 2019;20 (3):553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, Garcia‐Flores Y, Luong M, Devrekanli A, Xu J, Sun G, Tay J, Linsley PS, Baltimore D. miR‐146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. Journal of Experimental Medicine. 2011;208 (6):1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhao JL, Rao DS, Boldin MP, Taganov KD, O'connell RM, Baltimore D. NF‐kappa B dysregulation in microRNA‐146a‐deficient mice drives the development of myeloid malignancies. PNAS. 2011;108:9184–9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Register RB, Uncapher CR, Naylor AM, Lineberger DW, Colonno RJ. Human‐murine chimeras of ICAM‐1 identify amino acid residues critical for rhinovirus and antibody binding. Journal of Virology. 1991;65 (12):6589–6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Staunton DE, Gaur A, Chan PY, Springer TA. Internalization of a major group human rhinovirus does not require cytoplasmic or transmembrane domains of Icam‐1. J Immunol. 1992;148(10):3271–3274. [PubMed] [Google Scholar]
- 58. Odendall C, Kagan JC. The unique regulation and functions of type III interferons in antiviral immunity. Curr Opin Virol. 2015;12:47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ueki IF, Min‐Oo G, Kalinowski A, Ballon‐Landa E, Lanier LL, Nadel JA, Koff JL. Respiratory virus–induced EGFR activation suppresses IRF1‐dependent interferon λ and antiviral defense in airway epithelium. Journal of Experimental Medicine. 2013;210 (10):1929–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kelly JT, Busse WW. Host immune responses to rhinovirus: Mechanisms in asthma. Journal of Allergy and Clinical Immunology. 2008;122 (4):671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Triantafilou K, Vakakis E, Richer EAJ, Evans GL, Villiers JP, Triantafilou M. Human rhinovirus recognition in non‐immune cells is mediated byToll‐like receptors and MDA‐5, which trigger a synergetic pro‐inflammatory immune response. Virulence. 2011;2 (1):22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ortega H, Nickle D, Carter L. Rhinovirus and asthma: challenges and opportunities. Rev Med Virol. 2020; e2193. 10.1002/rmv.2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Oliver BGG, Robinson P, Peters M, Black J. Viral infections and asthma: an inflammatory interface?. Eur Respir J. 2014;44:1666–1681. [DOI] [PubMed] [Google Scholar]
- 64. Lu LF, Boldin MP, Chaudhry A, Lin L‐L, Taganov KD, Hanada T, Yoshimura A, Baltimore D, Rudensky AY. Function of miR‐146a in Controlling Treg Cell‐Mediated Regulation of Th1 Responses. Cell. 2010;142 (6):914–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yang L, Boldin MP, Yu Y, Liu CS, Ea C‐K, Ramakrishnan P, Taganov KD, Zhao JL, Baltimore D. miR‐146a controls the resolution of T cell responses in mice. Journal of Experimental Medicine. 2012;209 (9):1655–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li B, Wang X, Choi IY, Wang Y‐C, Liu S, Pham AT, Moon H, Smith DJ, Rao DS, Boldin MP, Yang L. miR‐146a modulates autoreactive Th17 cell differentiation and regulates organ‐specific autoimmunity. Journal of Clinical Investigation. 2017;127 (10):3702–3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Maitra U, Davis S, Reilly CM, Li L. Differential Regulation of Foxp3 and IL‐17 Expression in CD4 T Helper Cells by IRAK‐1. The Journal of Immunology. 2009;182 (9):5763–5769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Okoye IS, Czieso S, Ktistaki E, et al. Transcriptomics identified a critical role for Th2 cell‐intrinsic miR‐155 in mediating allergy and antihelminth immunity. PNAS. 2014;111:E3081–E3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Coquet JM, Schuijs MJ, Smyth MJ, Deswarte K, Beyaert R, Braun H, Boon L, Hedestam GBK, Nutt SL, Hammad H, Lambrecht BN. Interleukin‐21‐Producing CD4+ T Cells Promote Type 2 Immunity to House Dust Mites. Immunity. 2015;43 (2):318–330. [DOI] [PubMed] [Google Scholar]
- 70. Chen B, Yoo K, Xu W, Pan R, Han XX, Chen P. Characterization and evaluation of a peptide‐based siRNA delivery system in vitro. Drug Delivery and Translational Research. 2017;7 (4):507–515. [DOI] [PubMed] [Google Scholar]
- 71. Wittrup A, Lieberman J. Knocking down disease: a progress report on siRNA therapeutics. Nat Rev Genet. 2015;16:543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dua K, Hansbro NG, Foster PS, Hansbro PM. Targeting MicroRNAs: Promising Future Therapeutics in the Treatment of Allergic Airway Disease. Critical Reviews in Eukaryotic Gene Expression. 2018;28 (2):125–127. [DOI] [PubMed] [Google Scholar]
- 73. Urgard E, Lorents A, Klaas M, Padari K, Viil J, Runnel T, Langel K, Kingo K, Tkaczyk E, Langel Ü, Maimets T, Jaks V, Pooga M, Rebane A. Pre‐administration of PepFect6‐microRNA‐146a nanocomplexes inhibits inflammatory responses in keratinocytes and in a mouse model of irritant contact dermatitis. Journal of Controlled Release. 2016;235 195–204. [DOI] [PubMed] [Google Scholar]
- 74. Kanasty RL, Whitehead KA, Vegas AJ, Anderson DG. Action and reaction: the biological response to sirna and its delivery vehicles. Mol Ther. 2012;20:513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bizzintino J, Lee W‐M, Laing IA, Vang F, Pappas T, Zhang G, Martin AC, Khoo S‐K, Cox DW, Geelhoed GC, McMinn PC, Goldblatt J, Gern JE, Le Souef PN. Association between human rhinovirus C and severity of acute asthma in children. European Respiratory Journal. 2011;37 (5):1037–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cox DW, Khoo S‐K, Zhang G, Lindsay K, Keil AD, Knight G, Gern JE, Laing IA, Bizzintino J, Le Souëf PN. Rhinovirus is the most common virus and rhinovirus‐C is the most common species in paediatric intensive care respiratory admissions. European Respiratory Journal. 2018;52 (2):1800207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bergroth E, Aakula M, Elenius V, Remes S, Piippo‐Savolainen E, Korppi M, Piedra PA, Bochkov YA, Gern JE., Camargo CA, Jartti T. Rhinovirus Type in Severe Bronchiolitis and the Development of Asthma. The Journal of Allergy and Clinical Immunology: In Practice. 2020;8 (2):588–595.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Basnet S, Bochkov YA, Brockman‐Schneider RA, Kuipers I, Aesif SW, Jackson DJ, Lemanske RF, Ober C, Palmenberg AC, Gern JE. CDHR3 Asthma‐Risk Genotype Affects Susceptibility of Airway Epithelium to Rhinovirus C Infections. American Journal of Respiratory Cell and Molecular Biology. 2019;61 (4):450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman C.G, Glimcher LH. A Novel Transcription Factor, T‐bet, Directs Th1 Lineage Commitment. Cell. 2000;100 (6):655–669. [DOI] [PubMed] [Google Scholar]
- 80. Zheng W‐P, Flavell RA. The Transcription Factor GATA‐3 Is Necessary and Sufficient for Th2 Cytokine Gene Expression in CD4 T Cells. Cell. 1997;89 (4):587–596. [DOI] [PubMed] [Google Scholar]
- 81. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The Orphan Nuclear Receptor RORγt Directs the Differentiation Program of Proinflammatory IL‐17+ T Helper Cells. Cell. 2006;126 (6):1121–1133. [DOI] [PubMed] [Google Scholar]
- 82. Helou DG, Shafiei‐Jahani P, Lo R, et al. PD‐1 pathway regulates ILC2 metabolism and PD‐1 agonist treatment ameliorates airway hyperreactivity. Nat Commun. 2020;11:3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Boberg E, Johansson K, Malmhäll C, Calvén J, Weidner J, Rådinger M. Interplay between the IL‐33/ST2 axis and bone marrow ILC2s in protease allergen‐induced IL‐5‐dependent eosinophilia. Front Immunol. 2020;11:1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Burrill CP, Strings VR, Andino R. Poliovirus: generation, quantification, propagation, purification, and storage. Curr Protoc Microbiol. 2013;29:15H.11.11–15H.11.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bochkov YA, Watters K, Ashraf S, et al. Cadherin‐related family member 3, a childhood asthma susceptibility gene product, mediates rhinovirus C binding and replication. PNAS. 2015;112:5485–5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Calvén J, Yudina Y, Uller L. Rhinovirus and dsRNA Induce RIG‐I‐Like Receptors and Expression of Interferon β and λ1 in Human Bronchial Smooth Muscle Cells. PLoS ONE. 2013;8 (4):e62718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Oh H, Siano B, Diamond S. Neutrophil isolation protocol. J Vis Exp. 2008;745. 10.3791/745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Madaan A, Verma R, Singh AT, Jain SK, Jaggi M. A stepwise procedure for isolation of murine bone marrow and generation of dendritic cells. Journal of Biological Methods. 2014;1 (1):e1. [Google Scholar]
- 89. Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma'ayan A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research. 2016;44 (W1):W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.