

Dear Editor,
In this study, we reported the clinical and genetic characteristics of 10 Epstein‐Barr virus (EBV)‐triggered late‐onset primary hemophagocytic lymphohistiocytosis (HLH) patients. As HLH is a rare and devastating disorder characterized by uncontrolled immune activation resulting from the impaired function of natural killer and cytotoxic T cells. 1 And HLH can be further classified as either primary or secondary based on the predisposing genetic deficiency. 2 , 3 , 4 Although primary HLH usually arises in infants and children, the late‐onset primary HLH is increasingly reported in the literature. 5 It has been suggested that synergetic effects of the atypical heterozygous HLH‐associated mutations and environmental triggers, including EBV infection, were associated with the pathogenesis of late‐onset primary HLH. 6 However, the role of EBV‐infected cell subpopulation in primary HLH is unknown.
Therefore, we retrospectively investigated 10 patients with EBV‐triggered late‐onset primary HLH (occurring at over 12 years of age) from June 2013 and June 2018 in our department. According to the Diagnostic Guidelines for HLH, all of the cases met at least five of eight criteria and thereby were diagnosed as HLH, and detailed clinical parameters are listed in Table 1. Pathological studies of bone marrow, lymph node, and pleural fluids samples demonstrated that abnormal T lymphocytes, which were characterized by CD2bri+CD4–CD5–CD7dim+CD8+CD45RA–CD45RO+Ki67str+, existed in the bone marrow of case 1 and 2. In contrast, abnormal NK cell phenotype, which was marked by CD7dim+CD8–CD11bdim/–CD16dim/+CD56dim/+/str+CD57–, existed in the bone marrow for cases 7, 9, 10, and pleural effusion of case 4. No malignant clone was found in any of these patients. Despite all the patients were treated with regimens based on HLH‐2004 protocol and supportive therapy, seven of 10 patients died of disease progression or complications, and the median survival time was 3.4 months. Three patients who received allogeneic hematopoietic stem cell transplantation (allo‐HSCT) from unrelated donors acquired longer survival time compared with patients who did not receive allo‐HSCT (Figure 1A).
TABLE 1.
Clinical and laboratory characteristics of 10 EBV‐triggered late‐onset primary HLH cases
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 | Case 8 | Case 9 | Case 10 | Median (range) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gender | Female | Female | Female | Female | Female | Male | Male | Female | Male | Female | – |
| Age at onset (y), HLH | 14 | 18 | 25 | 45 | 12 | 25 | 21 | 18 | 32 | 13 | 22.3 (12–45) |
| Fever (℃) | 40.0 | 41.0 | 39.2 | 39.8 | 39.0 | 39.5 | 39.6 | 39.2 | 39.4 | 38.6 | 39.5 (39–41) |
| Neutrophils, 109/L | 0.3 | 0.2 | 0.8 | 0.1 | 0.6 | 0.1 | 0.7 | 0.9 | 0.6 | 0.8 | 0.5 (0.1–0.9) |
| Hemoglobin, g/L | 64.0 | 88.0 | 88 | 122.0 | 96.0 | 98.0 | 99.0 | 86.0 | 64.0 | 76.0 | 88.1 (64–122) |
| Platelets, 109/L | 35.0 | 14.0 | 32 | 29.0 | 90.0 | 25.0 | 44.0 | 64.0 | 9.0 | 218.0 | 56.0 (14–90) |
| ALT index * | 6.8 | 11.5 | 5.1 | 1.2 | 1.6 | 2.2 | 2.3 | 5.5 | 3.2 | 10.0 | 5.0 (1.2–11.5) |
| AST index ** | 28.0 | 24.0 | 6.2 | 4.2 | 1.5 | 2.2 | 1.2 | 1.8 | 0.9 | 9.3 | 7.7 (0.9–28) |
| LDH index # | 21.6 | 16.7 | 6.0 | 8.3 | 0.7 | 6.8 | 1.4 | 1.3 | 2.1 | 4.0 | 6.9 (0.7–21.6) |
| Triglycerides, mM | 4.5 | 3.6 | 4.1 | 13.4 | 2.7 | 2.3 | 2.3 | 3.5 | 2.6 | 2.6 | 4.2 (2.3–13.4) |
| Fibrinogen, g/L | 0.8 | 0.5 | 1.0 | 0.6 | 1.5 | 1.2 | 1.4 | 0.9 | 0.6 | 1.4 | 1.0 (0.5–1.5) |
| Ferritin, μg/L | 8000 | 50000 | 3481 | 8000 | 1500 | 8000 | 5052 | 1228 | 18403 | 923 | 8787 (1228–50000) |
| sCD25 index & | 6.1 | 6.6 | 2.1 | 7.6 | 4.8 | 2.9 | 8.3 | 3.6 | 5.2 | 4.8 | 5.2 (2.1–8. |
| Lymphadenopathy | Yes | No | Yes | Yes | No | Yes | Yes | Yes | Yes | Yes | – |
| Splenomegaly (thickness, cm) | 5.9 | 4.2 | 5.1 | 6.8 | 2.8 | 3 | 6.7 | 4.3 | 6.4 | 6.5 | 5.2 (2.8–6.8) |
| Hemophagocytosis | BM | No | BM, spleen | No | BM | BM | BM | No | BM | No | – |
| NK‐cell activity (%) | 5.6 0.6 | 8.9 0.7 | 1.2 0.3 | 7.5 1.3 | 4.0 0.5 | 16.8 2.6 | 16.0 3.1 | 4.6 0.8 | 15.8 3.2 | 4.3 0.7 | – |
| Perforin (%) | 61.2 4.7 | 50.8 3.8 | 0.9 0.2 | 27.2 2.6 | 31.2 2.4 | 76.6 5.4 | 11.9 1.2 | 52.3 5.0 | 17.6 1.8 | 26.6 3.8 | – |
| Degranulation of resting NK cells (%) | 1.1 0.2 | 6.7 0.8 | 12.6 1.8 | 2.3 0.6 | 13.2 1.6 | 1.3 0.3 | 14.5 3.0 | 27.7 4.7 | 17.2 1.7 | 0.4 0.1 | – |
| Degranulation of IL‐2 stimulated NK cells (%) | 1.4 0.1 | 42.8 2.7 | 42.1 3.6 | 3.1 0.2 | 67.9 5.7 | 2.8 0.3 | 72.4 6.6 | 49.3 3.9 | 56.9 4.6 | 73.1 6.3 | – |
| Sorting‐PCR | NK | NK | NK | NK | NK and T | NK | NK | NK | NK | NK | – |
| Sorting‐FISH | ND | ND | ND | ND | ND | ND | ND | NK | ND | NK | – |
| Treatment | HLH‐2004 | HLH‐2004 | HLH‐2004 | HLH‐2004; MDL |
HLH‐2004; BR |
HLH‐2004; P‐Gemox; Allo‐HSCT |
RCHOP; HLH‐2004; P‐Gemox |
HLH‐2004; Mp; Allo‐HSCT | HLH‐2004 |
HLH‐2004 Allo‐HSCT |
– |
| Disease reactivation | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | – |
| Outcome | Death | Death | Death | Death | Death | Alive | Death | Alive | Death | Alive | – |
| Cause of death | ICH | PD | PD | PD | Respiratory failure | – | PD | – | PD | – | – |
| Survival time (days) | 35 | 27 | 112 | 95 | 199 | 906 | 180 | 907 | 66 | 575 | – |
Abbreviations: Allo‐HSCT, allogeneic hematopoietic stem cell transplantation; BM, bone marrow; BR, brentuximab vedotin and rituximab; EBV, Epstein‐Barr virus; HLH, hemophagocytic lymphohistiocytosis; MDL, methotrexate, dexamethasone, and L‐asparaginase; ICH, intracerebral hemorrhage; Mp, methylprednisolone; ND, not done; PD, progressive disease; P‐Gemox, pegaspargase, gemcitabine, and oxaliplatin; RCHOP, rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; sCD25: soluble CD25; sorting‐FISH: immunobead sorting followed by fluorescence in situ hybridization; sorting‐PCR, immunobead sorting followed by quantitative PCR; y: year.
ALT index, patient's ALT/upper normal limit of ALT;
AST index, patient's AST/upper normal limit of AST;
LDH, patient's LDH/upper normal limit of LDH;
sCD25 index, patient's sCD25/upper normal limit of sCD25.
FIGURE 1.
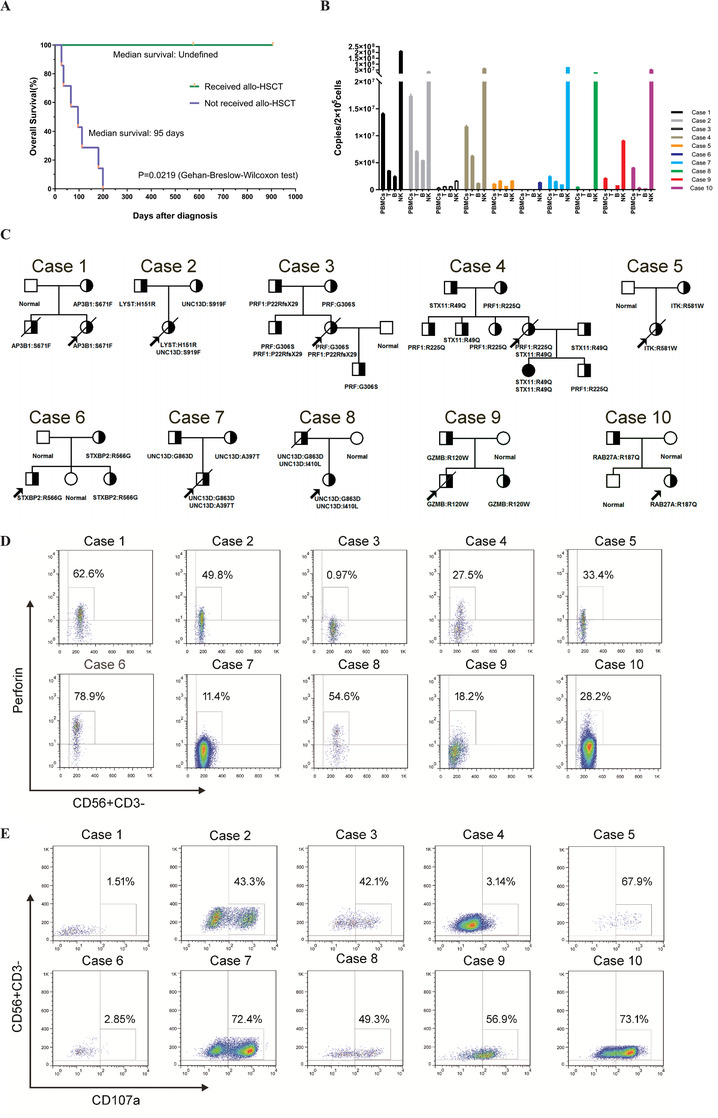
(A) Kaplan‐Meier survival curves of late‐onset primary HLH patients who received or not received allo‐HSCT. p values calculated by the Gehan‐Breslow‐Wilcoxon test. (B) Target cell identification by sorting‐PCR in the late‐onset primary HLH patients. EBV‐DNA copies quantification of different cell types (PBMCs, T, B, and NK) by qualitative PCR demonstrated that EBV primarily infected NK cells in 100% (10/10) of them, and concomitant infection in T cells in 10% of patients (1/10). (C) The pedigrees of the 10 families affected by hemophagocytic lymphohistiocytosis were included in the present study. Squares, males; circles, females; slash, deceased; half‐filled, heterozygous; darkened, homozygous; arrows, probands of the families. The affected genes and amino acid substitution caused by mutations in each family are indicated below the corresponding pedigree. (D) The expression of perforin protein on NK cells of case 2, case3, case 4, case 5, case 7, case 9, and case 10 was decreased, while relative regular perforin expression on CD56+CD3–NK cells was observed in case 1, case 6, and case 8. Numbers indicate the percentage of perforin expression. (E) The degranulation of rIL‐2 stimulated NK cells (CD56+CD3–) was determined by the expression of CD107a. The results showed that stimulated degranulation of NK cells was significantly decreased in case 1, case 4, and case 6; however, relatively healthy stimulated degranulation of NK cells was observed in case 5, case 7, case 9, and case 10. Numbers indicate the percentage of CD107a expression
Abbreviation: PBMCs, peripheral blood mononuclear cells.
Firstly, to investigate the cell subpopulation pattern of EBV infection, immunobead sorting followed by quantitative PCR (sorting‐PCR) and fluorescence in situ hybridization (sorting‐FISH) assay was performed. Peripheral blood mononuclear cells (PBMCs) were isolated and fractionated into CD3+, CD19+, and CD56+ cells using an immunobead method, purities of which were confirmed by flow cytometry to be 97%–99% for B and T cells and 91%–95% for NK cells. 7 Then purified cells were analyzed by quantitative PCR assay and fluorescence in situ hybridization with the EBER probe (green) as previously described. 7 The results demonstrated that T cells, B cells, and NK cells were all shown to be infected with EBV, while EBV significantly infected NK cells in nine patients (cases 1–4, 6–10). EBV predominantly infected both NK cells and T cells in one patient (case 5) (Figure 1B). Sorting‐FISH was performed in two cases (cases 8 and 10). EBV was found predominantly in NK cells, consistent with the results of sorting‐PCR.
Secondly, to understand the pathogenesis of EBV‐triggered late‐onset primary HLH, we carried out next‐generation sequencing (NGS) using a custom design for the HLH panel under the Ion AmpliSeq Ready‐to‐Use custom designer platform following the guide (https://www.ampliseq.com/protected/dashboard.action), and detailed NGS procedure has been reported in previous literature. 8 As described in Table 2, a total of five cases had one single gene mutation at heterozygous status (cases 1, 5, 6, 9, and 10), two cases had gene mutations in two primary HLH‐related genes at heterozygous state (cases 2 and 4). Case 3 had a compound heterozygous mutation in PRF1 (this case had been reported in the previous article 8 ), case 7 had a compound heterozygous mutation in UNC13D, while case 8 had two sites of heterozygous mutations in UNC13D, which were descended from her father. Most of the mutations were predicted to be damaging by software SIFT (Sorting Intolerant From Tolerant). All the details of 10 pedigrees are shown in Figure 1C and Table 2.
TABLE 2.
The EBV‐DNA load and genetic characteristics of 10 late‐onset primary HLH cases and their families
| Case number | Relationship | Age (y) | EBV (copies/μg) | Gene | Protein | 1000 genome | SIFT (<0.05) |
|---|---|---|---|---|---|---|---|
| Case 1 | Proband | 14 | (1.2 0.1) × 107 | AP3B1 | S671F | – | 0.049 (Damaging) |
| Father | 40 | <5 × 102 | Normal | – | |||
| Mother | 39 | <5 × 102 | AP3B1 | S671F | |||
| Brother | 0.75 | <5 × 102 | AP3B1 | S671F | |||
| Case 2 | Proband | 18 | (3.3 0.5) × 107 | LYST | H151R | – | 0.042 (Damaging) |
| UNC13D | S919F | – | 0.699 (Tolerated) | ||||
| Father | 50 | <5 × 102 | LYST | H151R | |||
| Mother | 49 | <5 × 102 | UNC13D | S919F | |||
| Case 3 | Proband | 25 | (5.4 1.5) × 105 | PRF1 | G306S | – | 0.010 (Damaging) |
| PRF1 | P22RfsX29 | – | – | ||||
| Father | 48 | (4.1 1.1) × 103 | PRF1 | P22RfsX29 | |||
| Mother | 47 | <5 × 102 | PRF1 | G306S | |||
| Brother | 23 | <5 × 102 | PRF1 | G306S | |||
| PRF1 | P22RfsX29 | ||||||
| Son | 1 | (3.2 0.8) × 103 | PRF1 | G306S | |||
| Case 4 | Proband | 45 | (1.90.3) × 107 | STX11 | R49Q | 0.02 | 1.000 (Tolerated) |
| PRF1 | R225Q | – | 0.366 (Tolerated) | ||||
| Father | 70 | <5 × 102 | STX11 | R49Q | |||
| Mother | 69 | <5 × 102 | PRF1 | R225Q | |||
| Brother | 47 | <5 × 102 | STX11 | R49Q | |||
| Brother | 43 | <5 × 102 | PRF1 | R225Q | |||
| Sister | 40 | <5 × 102 | PRF1 | R225Q | |||
| Son | 16 | <5 × 102 | PRF1 | R225Q | |||
| Daughter | 18 | <5 × 102 | STX11(hom) | R49Q | |||
| Husband | 48 | <5 × 102 | STX11 | R49Q | |||
| Case 5 | Proband | 12 | (1.3 0.4) × 107 | ITK | R581W | 0.001 | 0.007 (Damaging) |
| Father | 39 | <5 × 102 | Normal | ||||
| Mother | 37 | <5 × 102 | ITK | R581W | |||
| Case 6 | Proband | 25 | (7.7 1.9) × 105 | STXBP2 | R566G | 0.012 | 0.035 (Damaging) |
| Father | 50 | <5 × 102 | Normal | ||||
| Mother | 49 | <5 × 102 | STXBP2 | R566G | |||
| Elder Sister | 27 | <5 × 102 | STXBP2 | R566G | |||
| Younger Sister | 23 | <5 × 102 | Normal | ||||
| Case 7 | Proband | 21 | (1.30.7) × 107 |
UNC13D UNC13D |
G863D A397T |
0.001 ‐ |
0.000 (Damaging) 0.262 (Tolerated) |
| Father | 45 | <5 × 102 | UNC13D | G863D | |||
| Mother | 44 | <5 × 102 | UNC13D | A397T | |||
| Case 8 | Proband | 18 | (6.51.3) × 107 |
UNC13D UNC13D |
G863D I410L |
0.001 0.003 |
0.000 (Damaging) 1.000 (Tolerated) |
| Father | 43 | <5 × 102 |
UNC13D UNC13D |
G863D I410L |
|||
| Mother | 42 | <5 × 102 | Normal | ||||
| Case 9 | Proband | 32 | (4.0 1.1) × 106 | GZMB | R120W | – | 0.031 (Damaging) |
| Father | 54 | (1.2 0.3) × 103 | GZMB | R120W | |||
| Mother | 53 | <5 × 102 | Normal | – | |||
| Sister | 35 | <5 × 102 | GZMB | R120W | |||
| Case 10 | Proband | 13 | (1.20.4) × 107 | RAB27A | R187Q | 0.009 | 0.372 (Tolerated) |
| Father | 38 | <5 × 102 | RAB27A | R187Q | |||
| Mother | 36 | (1.20.6) × 104 | Normal | – | |||
| Brother | 8 | <5× 102 | Normal | – |
1000 genome, also known as 1000 genome project, is a map of human genome variation from population‐scale sequencing, the number stands for the frequency of amino acid substitution in the database. "‐" means amino acid substitution can't be detected in the database. SIFT predicts whether an amino acid substitution affects protein function. The lower the score, the more likely to be harmful.
Abbreviations: hom, homozygote; y, year.
Thirdly, to evaluate the impact of genetic mutations on their NK cell function, NK‐cell activity assay, NK‐cell degranulation assay, and perforin staining assay were subsequently performed on whole blood samples from these pedigrees. NK‐cell activity assay was performed as previously described. 8 Perforin staining assay was performed by staining PBMC samples for NK cell markers before fixation and permeabilization. Then anti‐perforin or phycoerythrin‐conjugated mouse immunoglobulin G2b (BD Pharmingen) was stained. NK cells were first gated and subsequently analyzed for the expression of the PRF1 protein. As for the NK‐degranulation assay, PBMCs were incubated with or without rIL‐2 to investigate activated or resting NK‐cell degranulation, respectively. For analysis, lymphocytes were gated based on forward scatter, then CD3–CD56+ NK cells were gated and assessed for surface expression of CD107a. Results demonstrated that reduced NK‐cell activity was observed in all EBV‐triggered late‐onset primary HLH patients (Table 1), while NK‐cell activity defect was found in four patients, which was consistent with previous research. 9 Substantially reduced perforin expression on CD3–CD56+ NK cells was detected in seven patients (Figure 1D). One case had abnormal resting NK‐cell degranulation, and four cases had defective resting NK‐cell degranulation. Abnormal activated NK‐cell degranulation was recorded in three cases (Figure 1E). Nine cases had either reduced perforin expression or decreased activated NK‐cell degranulation.
Based on the above findings, our study demonstrated that, unlike sporadic HLH, EBV‐triggered late‐onset primary HLH usually has one or two pathologic mutations of primary HLH‐associated genes, reduced perforin expression, NK activity, or degranulation. Most patients have high load EBV infection, and the target cells of EBV are usually NK cells, sometimes with T cells, which was typical for FHL (familial hemophagocytic lymphohistiocytosis) patients. 10 Through the pedigree investigation, we also noticed that although some family members of the patient had the same mutations, they did not develop the disease, possibly attributing to their lack of EBV infection or other "second‐hit" factors such as additional genetic mutations. Therefore, genetic mutations, as well as EBV‐infected lymphocyte subtypes, may collectively be involved in the pathogenesis of late‐onset primary HLH. HLH gene sequencing and pedigree investigation, combined with EBV‐infected cell type identification, are valuable in the differential diagnosis of late‐onset primary HLH. Timely allo‐HSCT is recommended to improve the poor prognosis of late‐onset primary HLH.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
AUTHOR CONTRIBUTIONS
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Liang Huang, Yi Xiao, Jinniu Deng, Miao Zheng, Hui Luo, and Lijun Jiang. The first draft of the manuscript was written by Lili Gao with the help of Li Yang and revised by Chunrui Li, Min Xiao, and Jianfeng Zhou. All authors commented on previous versions of the manuscript and agreed on the final manuscript.
ACKNOWLEDGMENTS
The authors would like to acknowledge laboratory colleagues for data mining and analysis of next‐generation sequencing, and we also acknowledge all the patients and their family members who participated in the study.
Gao L, Yang Li, Huang L, et al. Clinical and genetic features of Epstein‐Barr virus‐triggered late‐onset primary hemophagocytic lymphohistiocytosis: Ten pedigrees study. Clin Transl Med. 2021;11:e393. 10.1002/ctm2.393
Funding information
This work was supported by the funding from the National Natural Science Foundation of China (81873452, to Dr. Chunrui Li; 81700126, to Dr. Lili Gao; 81770211, to Dr. Min Xiao), the Key Program of the National Natural Science Foundation of China (81830008 to Dr. Jianfeng Zhou), and the Huanghe Talents Plan of Wuhan City (HHYC‐2015002, to Dr. Jianfeng Zhou).
Contributor Information
Min Xiao, Email: lfxm2000@126.com.
Chunrui Li, Email: cunrui5650@hust.edu.cn.
REFERENCES
- 1. Chandrakasan S, Filipovich AH. Hemophagocytic lymphohistiocytosis: advances in pathophysiology, diagnosis, and treatment. J Pediatr. 2013;163:1253–1259. [DOI] [PubMed] [Google Scholar]
- 2. Parikh SA, Kapoor P, Letendre L, et al. Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis. Mayo Clin Proc. 2014;89:484–492. [DOI] [PubMed] [Google Scholar]
- 3. Ramos‐Casals M, Brito‐Zerón P, López‐Guillermo A, et al. Adult haemophagocytic syndrome. Lancet. 2014;383:1503–1516. [DOI] [PubMed] [Google Scholar]
- 4. Rivière S, Galicier L, Coppo P, et al. Reactive hemophagocytic syndrome in adults: a retrospective analysis of 162 patients. Am J Med. 2014;127:1118–1125. [DOI] [PubMed] [Google Scholar]
- 5. Wang Y, Wang Z, Zhang J, et al. Genetic features of late onset primary hemophagocytic lymphohistiocytosis in adolescence or adulthood. PLoS One. 2014;9:e107386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cetica V, Sieni E, Pende D, et al. Genetic predisposition to hemophagocytic lymphohistiocytosis: report on 500 patients from the Italian registry. J Allergy Clin Immunol. 2016;137:188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang P, Zeng C, Cheng J, et al. Determination of Epstein‐Barr virus‐infected lymphocyte cell types in peripheral blood mononuclear cells as a valuable diagnostic tool in hematological diseases. Open forum Infect Dis. 2019;6:ofz171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gao L, Dang X, Huang L, et al. Search for the potential “second‐hit” mechanism underlying the onset of familial hemophagocytic lymphohistiocytosis type 2 by whole‐exome sequencing analysis. Transl Res. 2016;170:26–39. [DOI] [PubMed] [Google Scholar]
- 9. Marcenaro S, Gallo F, Martini S, et al. Analysis of natural killer‐cell function in familial hemophagocytic lymphohistiocytosis (FHL): defective CD107a surface expression heralds Munc13‐4 defect and discriminates between genetic subtypes of the disease. Blood. 2006;108:2316–2323. [DOI] [PubMed] [Google Scholar]
- 10. Fox CP, Shannon‐Lowe C, Gothard P, et al. Epstein‐Barr virus‐associated hemophagocytic lymphohistiocytosis in adults characterized by high viral genome load within circulating natural killer cells. Clin Infect Dis. 2010;51:66–69. [DOI] [PubMed] [Google Scholar]