

Abstract
M-type (KCNQ2/3) K+ channels play dominant roles in regulation of active and passive neuronal discharge properties such as resting membrane potential, spike-frequency adaptation, and hyper-excitatory states. However, plasticity of M-channel expression and function in nongenetic forms of epileptogenesis are still not well understood. Using transgenic mice with an EGFP reporter to detect expression maps of KCNQ2 mRNA, we assayed hyperexcitability-induced alterations in KCNQ2 transcription across subregions of the hippocampus. Pilocarpine and pentylenetetrazol chemoconvulsant models of seizure induction were used, and brain tissue examined 48 hr later. We observed increases in KCNQ2 mRNA in CA1 and CA3 pyramidal neurons after chemoconvulsant-induced hyperexcitability at 48 hr, but no significant change was observed in dentate gyrus (DG) granule cells. Using chromogenic in situ hybridization assays, changes to KCNQ3 transcription were not detected after hyper-excitation challenge, but the results for KCNQ2 paralleled those using the KCNQ2-mRNA reporter mice. In mice 7 days after pilocarpine challenge, levels of KCNQ2 mRNA were similar in all regions to those from control mice. In brain-slice electrophysiology recordings, CA1 pyramidal neurons demonstrated increased M-current amplitudes 48 hr after hyperexcitability; however, there were no significant changes to DG granule cell M-current amplitude. Traumatic brain injury induced significantly greater KCNQ2 expression in the hippocampal hemisphere that was ipsilateral to the trauma. In vivo, after a secondary challenge with subconvulsant dose of pentylenetetrazole, control mice were susceptible to tonic-clonic seizures, whereas mice administered the M-channel opener retigabine were protected from such seizures. This study demonstrates that increased excitatory activity promotes KCNQ2 upregulation in the hippocampus in a cell-type specific manner. Such novel ion channel expressional plasticity may serve as a compensatory mechanism after a hyperexcitable event, at least in the short term. The upregulation described could be potentially leveraged in anticonvulsant enhancement of KCNQ2 channels as therapeutic target for preventing onset of epileptogenic seizures.
Keywords: epileptogenesis, hippocampus, hyperexcitability, KCNQ channels, plasticity
1 |. INTRODUCTION
The Kv7/KCNQ family of voltage-gated K+ channels plays significant roles in control over neuronal signaling and human disease states in brain and peripheral nerves. There are five isoforms of the KCNQ family, of which KCNQ2-5 are widely expressed throughout the nervous system. KCNQ2, KCNQ3, and KCNQ5 underlie neuronal “M-type” currents (IM) (Schroeder et al., 2000; Hadley et al., 2000), either as homomers or more commonly as heteromers (Shapiro et al., 2000; Wang et al., 1998). KCNQ channels were first named as “M-type” channels due to their inhibition by stimulation of Gq/11-coupled muscarinic acetylcholine receptors (Brown and Adams, 1980). M channels strongly contribute to control of neuronal excitability via regulation of resting membrane potential, spike-frequency adaptation, bursting, and hyper-excitatory states (Brown & Passmore, 2009; Gamper and Shapiro, 2015; Greene and Hoshi, 2017). Mutations in KCNQ2 and KCNQ3 subunits are responsible for the generation of some inherited forms of epileptic encephalopathies (Biervert et al., 1998; Miceli et al., 2015; Singh et al., 1998). Similarly, transgenic suppression of M channels in mice results in epileptic phenotypes (Greene, Kosenko, & Hoshi, 2018; Niday et al., 2017; Peters, Hu, Pongs, Storm, & Isbrandt, 2005; Singh et al., 2008; Soh, Pant, LoTurco, & Tzingounis, 2014). Whereas the role of M channels in acquired epileptogenesis has not been well characterized, evidence suggests that M-channel openers can serve a protective role in preventing seizures and other insults that can progress to epilepsy, such as stroke and traumatic brain injury (Bierbower, Choveau, Lechleiter, & Shapiro, 2015; Sampath, Valdez, White, & Raol, 2017; Vigil et al., 2019).
Hippocampal excitability is critically altered in temporal lobe epilepsies and epileptogenesis (Dengler, Yue, Takano, & Coulter, 2017; Poolos and Johnston, 2012; Takano and Coulter, 2012). In hippocampal principal neurons, M channels are localized to the axon initial segment (in structural interactions with Ankyrin-G) where their activity strongly regulates responses to excitatory and inhibitory inputs (Klinger, Gould, Boehm, & Shapiro, 2011; Lezmy et al., 2017; Soh et al., 2014). Both CA1 stratum oriens interneurons (Lawrence et al., 2006) and parvalbumin+ (PV) interneurons in the dentate gyrus (DG) (Nieto-Gonzalez & Jensen, 2013) exhibit somatodendritic localization of KCNQ2/3 in control of interspike interval. Previously, our lab reported very significant upregulation of KCNQ2 and KCNQ3 transcription in sympathetic neurons 8-24 hr after one 15 min burst of hyper-activity (Zhang & Shapiro, 2012). Furthermore, strong increases in KCNQ2 and KCNQ3 mRNA were observed in mouse hippocampus 24 hr after a single episode of pilocarpine-induced or kainic acid-induced seizures (Zhang & Shapiro, 2012). Those experiments measured RNA levels only in lysates of whole hippocampus, thus lacking information as to which types of neurons or cells were responsible for transcriptional alterations. Given that neuron-selective upregulation of M-channel transcription may occur among principal neurons and inhibitory interneurons, the particular types of neurons responding to intense activity would greatly determine the net result on hippocampal circuit behavior. Therefore, we investigated cell-type specific affects of such plasticity in DG granule cells (DGGCs), CA3 pyramidal neurons, and CA1 pyramdial neurons.
The glutamatergic principal cells involved in the tri-synaptic pathway of the hippocampus are stratified by regional, morphological, and developmental differences; however, each type of neuron is also distinct in how they respond to external stimuli and handling cognitive tasks (Alkadhi, 2019; Glibert et al., 2001; Lavenex & Banta Lavenex, 2013). Furthermore, compared to DGGCs, the CA1/CA3 pyramidal neurons demonstrate greater susceptibility and vulnerability to pathophysiological conditions such as Alzhemier’s disease, cerebral ischemia, and epilepsy (Borges et al., 2003; Dao, Zagaar, Levine, & Alkadhi, 2016; Hsu et al., 1998; Steve, Jirsch, & Gross, 2014; Turski et al., 1984; Xiong & Stringer, 2000). The differential signal processing at the synapse and axons and variation in channel expression of each neuron type significantly contribute to how they respond to deleterious insults and injury (Carver and Shapiro et al., 2019; Coultrap, Nixon, Alvestad, Valenzuela, & Browning, 2005; Trieu et al., 2015; Wang & Mattson, 2014). Therefore, mechanisms in which different neuronal subtypes respond to hyperexciatory stimuli are of great interest in understanding how epileptogenesis progresses.
Here, we hypothesized that KCNQ2-containing channels are upregulated in conditions of low-level hyperexcitability prior to the onset of seizures and the development of epilepsy. We used transgenic mice that express EGFP as a reporter of KCNQ2 mRNA to assess neuron-specific changes in KCNQ2 mRNA expression after chemoconvulsant-induced hyperexcitability in the hippocampus. Upon initial observation of hippocampal subfield changes in KCNQ2 expression, we discovered significant neuronal-specific differences by individual cell quantification. Whereas CA1 and CA3 pyramidal neurons exhibited increased KCNQ2 mRNA, expression in DGGCs was not significantly altered after a single hyperexcitable, sub-threshold event. However, an increase in seizure severity induced by status epilepticus (SE) robustly augmented KCNQ2 expression in DGGCs. We also report that KCNQ2 expression increases in the hippocampus after traumatic brain injury (TBI), which is a known causative precursor to epileptic seizures (Hunt, Scheff, & Smith, 2009; Vigil et al., 2019). Using chromogenic in situ hybridization (CISH), we saw no changes in KCNQ3 transcription, and the KCNQ2 CISH results paralleled those from the transgenic mice. Our findings demonstrate cell-specific, compensatory upregulation of KCNQ2-containing M channels. Based on this work, we posit that such endogenous KCNQ2 plasticity could be leveraged to prevent further secondary hyperexcitatory cascades that promote temporal lobe epileptogenesis.
2 |. MATERIALS AND METHODS
2.1 |. Animals
KCNQ2-EGFP reporter mice (KCNQ2-EGFP/FW221Gsat/Mmucd, stock number 015412-UCD) were acquired from the GENSAT Project (Rockefeller University), in which mice are transgenic for a bacterial artificial chromosome (RP23-247P15) containing the KCNQ2 locus with EGFP inserted immediately upstream of the first exoncoding sequence gene. The reporter mice were established on a mixed background of FVB/N and Swiss Webster mice and maintained on a Swiss Webster background by mating hemizygous transgenic males with wildtype Swiss Webster females. Hemizygous male and female progeny of 2-3 months age were used for experiments. In cases of no significant sex differences of EGFP immunofluorescent intensities, group data were pooled from both sexes. To control for ovarian cycle-dependent plasticity of GABAA receptors in dentate gyrus granule cells, which may confound measures of excitability (Carver, Wu, Gangisetty, & Reddy, 2014), only male mice were used for electrophysiology experiments. In seizure susceptibility assays on KCNQ2 knockdown mice, hemizygous Cre-POMC+ mice (Tg[Pomc1-cre] 16Lowl/J, The Jackson Laboratory, RRID:IMSR_JAX: 010714) were crossed with singly (KCNQ2+/flox) or doubly (KCNQ2flox/flox) floxed mice to yield knockdown exclusive to DG granule cells (DGGCs). KCNQ2 deficient mice were established and maintained on a C57BL6 background (Jackson Laboratory), so as to be compatible with previous experiments conducted on mice with knocked-down KCNQ2 (Carver & Shapiro, 2019; Soh et al., 2014), and for the pentylenetetrazole seizure susceptibility assays. All mice were housed in an environmentally controlled animal facility with a 12 hr light/dark cycle and had access to food and water ad libitum. Animals were cared for in compliance with the guidelines in the National Institutes of Health Guide for Care and Use of Laboratory Animals. All animal procedures were performed in a protocol approved by the Institutional Animal Care and Use Committee of UT Health San Antonio.
2.2 |. Chemoconvulsant-induced hyperexcitability and seizures
The nonselective muscarinic agonist pilocarpine is widely used to generate temporal lobe epilepsy in rodents as an experimental model (Reddy & Kuruba, 2013; Turski et al., 1984). We first used subthreshold doses of pilocarpine that do not result in status epilepticus (SE) to investigate early changes in hippocampal KCNQ2 or KCNQ3 channel plasticity as a result of sub-seizure hyperexcitability. Scopolamine methyl nitrate (1 mg/kg, i.p.) was injected to block peripheral muscarinic receptor stimulation, and 30 min later, mice were treated with pilocarpine (200-350 mg/kg, i.p.) or control vehicle (0.9% saline). Video records were acquired with the entire cage in the field of view, ~15° above the surface, and seizure activity monitored by a blinded reviewer. Behavioral seizures were rated according to the Racine scale (Racine, 1972), as modified for mice: Stage 0, no response or behavioral arrest; Stage 1, chewing or facial twitches; Stage 2, chewing and head nodding; Stage 3, forelimb clonus and Straub tail; Stage 4, bilateral forelimb clonus and rearing; Stage 5, rearing and falling or jumping. At 230 mg/kg pilocarpine, animals achieved forelimb clonus and Racine Stage 2/3 behavior, but did not reach SE, and convulsions terminated after 2 hr without intervention. Therefore, this dose was used for pre-epileptiform hyperexcitability in comparison to control mice. Forty-eight hours or 7 days after pilocarpine challenge, brains were fixed in 4% paraformaldehyde (PFA), and hippocampi immunostained for EGFP and microtubule-associated protein 2 (MAP2). A separate group of mice were challenged with 300 mg/kg pilocarpine to produce Stage 5 tonic-clonic seizures and SE. The SE behavior was characterized as a persistent state of seizure that did not terminate without anticonvulsant intervention. Mice experiencing SE were treated with 5 mg/kg diazepam (Patterson Veterinary) 2 hr after initiation of seizures. Guidelines were followed in order to ensure the welfare of the mice to minimize any adverse outcomes (Lidster et al., 2016).
Mice were alternatively administered pentylenetetrazole (PTZ, 60 mg/kg, s.c.), a GABAA receptor antagonist, to induce mild, clonic convulsions in which progression of motor seizure activity could be classified (Ferraro et al., 1999). Four stages of seizure behavioral response to subcutaneous PTZ injection were defined as follows: (a) hypoactivity characterized by a progressive decrease in motor activity until the animal came to rest in a crouched or prone position, (b) myoclonic and jerking spasms characterized by brief focal seizures lasting 1 s or less, (c) generalized clonus characterized by sudden loss of upright posture, all four limb and tail clonus, rearing, and autonomic signs, and (d) tonic-clonic seizure characterized by generalized seizure followed by tonic hindlimb extension. Latencies to focal (partial clonic), generalized (generalized clonic), and maximal (tonic-clonic) behavioral seizures were recorded. Mice were monitored for 1 hr; however, they were considered protected if they did not display forelimb clonus behavior within 30 min of injection.
2.3 |. Controlled cortical impact injury (CCI) model
The controlled cortical impact injury (CCI) traumatic brain injury (TBI) model uses a pneumatic impact device, as previously described by our group (Vigil et al., 2019). The CCI model causes a TBI but the skull remains intact with minimal hematoma (Hunt et al., 2009). This type of model most closely simulates the trauma experienced during blunt-force head injuries from vehicular accidents or falls. Mice were anesthetized with isoflurane (3% induction, 1% maintenance) in 100% O2. Body temperature of 37°C was maintained using a temperature-controlled heated surgical table. A small midline incision was made on the scalp using aseptic surgical techniques. The mouse was then positioned on a stage directly under the pneumatic impact tip of a cylindrical probe of 5 mm diameter. A calibrated impact was delivered at 4.5 m/s to a depth of 1 mm over the parietal lobe of the cortex, which generates a moderate head injury to the mouse. Apnea episodes after injury and righting reflex after removal of the mouse from anesthesia were timed and recorded. Scalp incisions were sutured closed and mice were placed in a Thermo-Intensive Care Unit (Braintree Scientific model FV-1; 37°C; 27% O2) and monitored until fully awake and moving freely.
2.4 |. Western immunoblot of protein quantification
Brain samples were collected 48 hr after treatment and left and right hemispheres snap frozen in dry ice. Samples were homogenized on ice in a solution of RIPA buffer (Thermo Fisher Scientific), protease inhibitor tablet (Roche), phosphatase inhibitor cocktail 2 (Sigma) and phosphatase inhibitor cocktail 3 (Sigma), using an Ultra EZgrind tissue homogenizer (Denville Scientific Inc.) at the lowest speed setting. Protein concentrations of each sample were determined using a Pierce BCA protein assay kit (Thermo Fisher Scientific). Protein samples (25 μg of total protein) were run in 4-20% precast polyacrylamide gels (Bio-rad) at 130 V for 1 hr. Proteins were then transferred to a nitrocellulose membrane (Bio-rad) at 100 V for 1 hr. Membranes were stained with specific KCNQ2 antibody (Aviva Systems Biology #ARP35459_T100, RRID:AB_842293, 1:1000) for semi-quantification of protein expression (Zhang et al., 2013). Immunoblot bands obtained were revealed via horseradish peroxidase-conjugated secondary antibodies and enhanced chemilumiscence western blotting detection reagents (GE Healthcare). Quantification of bands was performed with ImageJ software (National Institutes of Health). For normalization, the results of every protein of interest were divided by the results of glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:25,000, Abcam #ab125247, RRID:AB_11129118) at 36 kDa. To confirm the results obtained a second independent normalization was also performed using Ponceau S solution (Sigma) and the band intensity quantified.
2.5 |. Immunohistochemistry
Forty-eight hours after treatment, mice were anesthetized with isoflurane, and brains rapidly excised and fixed in paraformaldehyde (4% wt/vol in PBS) for 2 hr. Alternatively, brain tissue was also acquired from mice monitored for 7 days post vehicle or pilocarpine treatments. Five to seven mice were imaged and analyzed for each experimental condition. Fixed brains were transferred to sucrose solution (20% wt/vol in PBS) overnight at 4°C and 40 μm coronal sections cut in a cryostat at −20°C the next day. Dorsal hippocampus brain sections containing EGFP were detected using a goat anti-GFP antibody (1:2000 dilution, Rockland #600-101-215, RRID:AB_218182) and FITC donkey anti-goat IgG (1:150, Jackson ImmunoResearch#705-005-147, RRID:AB_2340385). MAP2 was used to normalize the relative level of EGFP and detected with chicken anti-MAP2 (1:5000, Abcam #ab5392, RRID:AB_2138153) and rhodamine red rabbit anti-chicken IgG (1:400, Jackson ImmunoResearch #303-295-008, RRID:AB_2339316). In other preparations, anti-cFos (1:250, ThermoFisher Scientific#MA5-15055, RRID:AB_10984728), anti-PROX1 (1:100, R&D Systems#AF2727, RRID:AB_2170716), anti-NeuN clone A60 (1:200, Millipore #MAB377, RRID:AB_2298772), or anti-ZnT3 (1:250, Synaptic Systems #197 003) primary antibodies were used. After washing with PBS, sections were mounted in anti-fading reagent and imaged on a Nikon Eclipse FN1 upright microscope with ×4 Plan Fluor, ×20 Plan Apo, and ×40 objectives and swept-field confocal imaging for EGFP and MAP2 in the dentate gyrus, CA3, and CA1 subregions of the hippocampus. To avoid variability in signal intensity, imaging was completed from a fixed depth of 20 μm from the tissue surface using Z-axis control of the microscope. Intensity ratios for EGFP/MAP2 were obtained for each image by alternating laser excitation light sources (488 and 561 nm, respectively) at a fixed exposure time of 500 ms in 10 MHz/14-bit readout mode. Initially, each hippocampal subregion was independently quantified with the experimenter blinded to the image source derived from either vehicle-treated control or pilocarpine-treated animals. Subsequently for ×40 magnification images, individual two-plane cell ROIs were drawn and quantified using ImageJ software. From each mouse, eight to ten single slice images were analyzed according to each DG, CA3, and CA1 principal neuron region. An intensity value was derived for each cell in the image, and a group average was tabulated for each mouse.
2.6 |. In situ hybridization
Forty-eight hours after delivery of vehicle or pilocarpine challenge (230 mg/kg), mice were anesthetized with isoflurane, then transcardially perfused with PBS followed by 4% PFA. Brains were extracted and postfixed in 4% PFA in PBS overnight at 4°C. Brains were then transferred to 30% sucrose in PBS for 72 hr at 4°C. Brains were embedded in Optimal Cutting Temperature media and 16 μm coronal sections cut in a cryostat at −80°C. For chromogenic in situ hybridization (cISH), RNAscope® 2.0 Assay (Wang et al., 2012; Advanced Cell Diagnostics, Inc.) was used to label KCNQ2 and KCNQ3 mRNA of each section. Specificity of the hybridization was evaluated by the use of a negative control probe. Bright field microscopy images were taken of the chromogenically-labeled hippocampus. For semi-quantitative analysis, densitometric measurements of each hippocampal area were assessed for each mRNA probe using ImageJ with using color deconvoluion (Ruifrok & Johnston, 2001), with the analyzer blinded to the experimental group of the sections.
2.7 |. Brain slice electrophysiology
Transverse slices (300 μm) of hippocampus were cut with a vibratome (Thermo Scientific Microm HM650V) from mice using standard techniques, as reported previously (Carver et al., 2014). Mice were anesthetized with isoflurane, and brains excised and placed in artificial cerebrospinal fluid (ACSF) at 3.5°C composed of the following (in mM): 126 NaCl, 3 KCl, 0.5 CaCl2, 5 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, 15 glucose, 0.3 kynurenic acid, with pH adjusted to 7.35–7.40, with 95% O2-5% CO2, 305–315 mOsm/kg. Hippocampal slices were maintained in oxygenated ACSF at 30°C for 60 min, and experiments performed at 25°C. Patch pipettes were pulled from borosilicate glass capillaries (KG-33; King Precision Glass) using a Flaming/Brown micropipette puller P97 (Sutter Instruments) and had resistances between 5 and 7 MΩ. The internal pipette solution consisted of (in mM): 110 KCl, 2 EGTA, 2 MgCl2, 10 HEPES, 4 Na2-ATP, 2 Na2-GTP, 10 Tris2 phosphocreatine, and 10 tetrapotassium pyrophosphate adjusted to 7.3 pH using KOH at 280 mOsm. Electrophysiological recordings in the slice were performed in the whole-cell patch clamp configuration. Neurons were visually identified with a Nikon FN-1 microscope equipped with a ×40 water-immersion objective with infrared differential interference contrast optics and camera. Contrast dye loaded into the pipette was used to routinely confirm intact dendrites and axons of the neurons. Neurons without visible axons or with shortly-cut axons were not examined. Current and voltage recordings were acquired using a HEKA EPC-9 amplifier and Pulse software (HEKA/Instrutech, Port Washington, NY). Membrane capacitance, series resistance, and input resistance were monitored by applying 5 mV depolarizing voltage steps. Input resistance was determined as the slope of the linear section of the current-voltage plot of recordings that did not contain action potential firing. Membrane time-constants were measured from an exponential fit to subthreshold voltage responses from −60 pA to +60 pA current steps in 20 pA increments. The resulting membrane capacitance calculated by the current-clamp step protocol (Golowasch et al., 2009) was consistent with that acquired by the Pulse software. Drugs were delivered to the bath chamber using a multi-channel perfusion system. All solutions were continuously bubbled with 95% O2/5% CO2.
For voltage-clamp recordings, DGGCs were held at −75 mV and CA1 pyramidal cells were held at −65 mV, reflecting their different typical resting potentials. Whole-cell current signals were digitized at 5-10 kHz and filtered at 500 Hz–2 kHz. The bath recording solution consisted of (in mM): 124 NaCl, 3 KCl, 1.5 MgCl2, 2.4 CaCl2, 1.25 Na2H2PO4, 26 NaHCO3, and 15 glucose, adjusted to 7.4 pH and 295-305 mOsm. M current (IM) was studied in hippocampal neurons with pipette access resistances <20 MΩ under wholecell mode.
Mature DGGCs selected for analysis exhibited input resistances of 350-450 MΩ, and displayed baseline membrane time constants (τmembrane) of 25 ± 2 ms. For M-current recordings, the potential was held at −25 mV, followed by 600 ms hyperpolarizing steps decrementing from −30 to −80 mV. IM amplitudes were measured at −55 mV (the maximum amplitude of the deactivating current) as the relaxation (τ ~100 ms) of the deactivating current sensitive to the M-channel specific blocker XE991 (20 μM). As previously noted (Carver & Shapiro, 2019), the product of the series resistance (10-20 MΩ) multiplied by the pre-pulse current clamped at −25 mV (200-300 pA) yielded an estimate of the whole-cell voltage error, which we estimated to average 2-6 mV. For pipette and bath solutions for which the major cations are K+ and Na+, respectively, the junction potential error (which was not corrected) was typically between −2 and − 4 mV (Bernheim, Beech, & Hille, 1991). Therefore, the true voltage at which the deactivating relaxation was quantified was estimated to be between −54 and − 60 mV for each cell.
For current-clamp recordings, to derive active discharge properties of neurons, a holding current was injected to maintain a membrane potential of −75 mV and square waves of current added in +20 pA steps for 500 ms with 10 s between each step, up to a maximum of 300 pA. The individual properties of action potential waveforms were analyzed using AxoGraph and in-house software. Action potentials were detected with a derivative threshold of 20 mV/ms. Then, 10-90% peak rise times were calculated and action potential width measured at 10% of the peak. At each value of injected current, initial and final spike frequency were determined as the measurement of frequency between the first two action potentials or last two action potentials, respectively.
2.8 |. Experimental design and statistical analysis
Group data are expressed as the mean ± SEM. Statistical comparisons of parametric measures, including electrophysiology data, were performed using an independent two-tailed Student’s t-test followed by Tukey’s HSD test post hoc. For data in which normal distribution could not be assumed, the Mann-Whitney U-test was performed. We estimated numbers of mice for behavioral tests so as to detect significance (p < .05) at a power of 0.8. The effect size was based on previously tested differences in pilocarpine or pentylenetetrazol seizure susceptibility between control and retigabine-treated (1 mg/kg, s.c.) mice. In all statistical tests, the criterion for statistical significance was p < .05.
3 |. RESULTS
3.1 |. Characterization of non-SE hyperexcitability in the hippocampus
Previous work in our lab found that after a chemoconvulsant-induced seizure, KCNQ2 and KCNQ3 mRNA were significantly upregulated in the hippocampus, as measured by whole hippocampus lysates (Zhang & Shapiro, 2012). However, the types of hippocampal neurons that responded by upregulation of KCNQ2 or KCNQ3 could not be previously specified. Therefore, we decided to determine the region-specific outcomes of hyperexcitability to the hippocampal network. For this study, we focused on expression of the KCNQ2 subunit, given that KCNQ2 has a more significant role than KCNQ3 in control over discharge properties, such as after hyperpolarization, spike-frequency adaptation, resonance and epileptogenesis in the cortex and hippocampus (Peters et al., 2005; Soh et al., 2014; Tzignounis and Nicoll, 2008). We used the chemoconvulsant pilocarpine as a model of limbic hyperexcitability. Pilocarpine induces convulsant activity in vivo by nonspecifically stimulating muscarinic acetylcholine receptors, which among other targets, strongly suppresses M current (IM). In order to block peripheral muscarinic acetylcholine receptors, 1 mg/kg scopolamine methyl nitrate was administered 30 min prior to pilocarpine or saline vehicle. Adult, 2-month old Swiss-Webster mice were used for pilocarpine experiments (Borges et al., 2003), and a dose-response curve compiled using the Racine scale of motor seizures (EC50 245 ± 3 mg/kg; Figure 1a). The mice used were derived from a transgenic line of KCNQ2 mRNA-EGFP reporter mouse, in which the EGFP gene was inserted upstream of the KCNQ2 promoter with a bacterial artificial chromosome (Gong et al., 2003). Therefore, we were able to differentiate the dynamic transcription of KCNQ2 mRNA in hippocampus subregions after hyperexcitability. In order to investigate the pre-epileptogenic hyperexcitability of the hippocampus in further experiments, pilocarpine was administered to naïve mice at its EC30 (230 mg/kg), a dose that induces modest brain hyperexcitation without SE seizures (Gröticke, Hoffman, & Löscher, 2007, see Methods). The pilocarpine-induced motor activity terminated after 1 hr without requiring anticonvulsant intervention. Our rationale for using a lower dose of pilocarpine was to investigate the early-stage response of the hippocampus to a challenge of limited limbic hyperexcitability rather than convulsive SE, which drastically alters non-temporal seizure circuits and causes excessive seizures without a clearly identifiable origin, as well as affecting the transcription of a host of genes (Curia, Longo, Biagini, Jones, & Avoli, 2008; Grabenstatter, Russek, & Brooks-Kayal, 2012; Sloviter, Zappone, Bumanglag, Norwood, & Kudrimoti, 2007).
FIGURE 1.
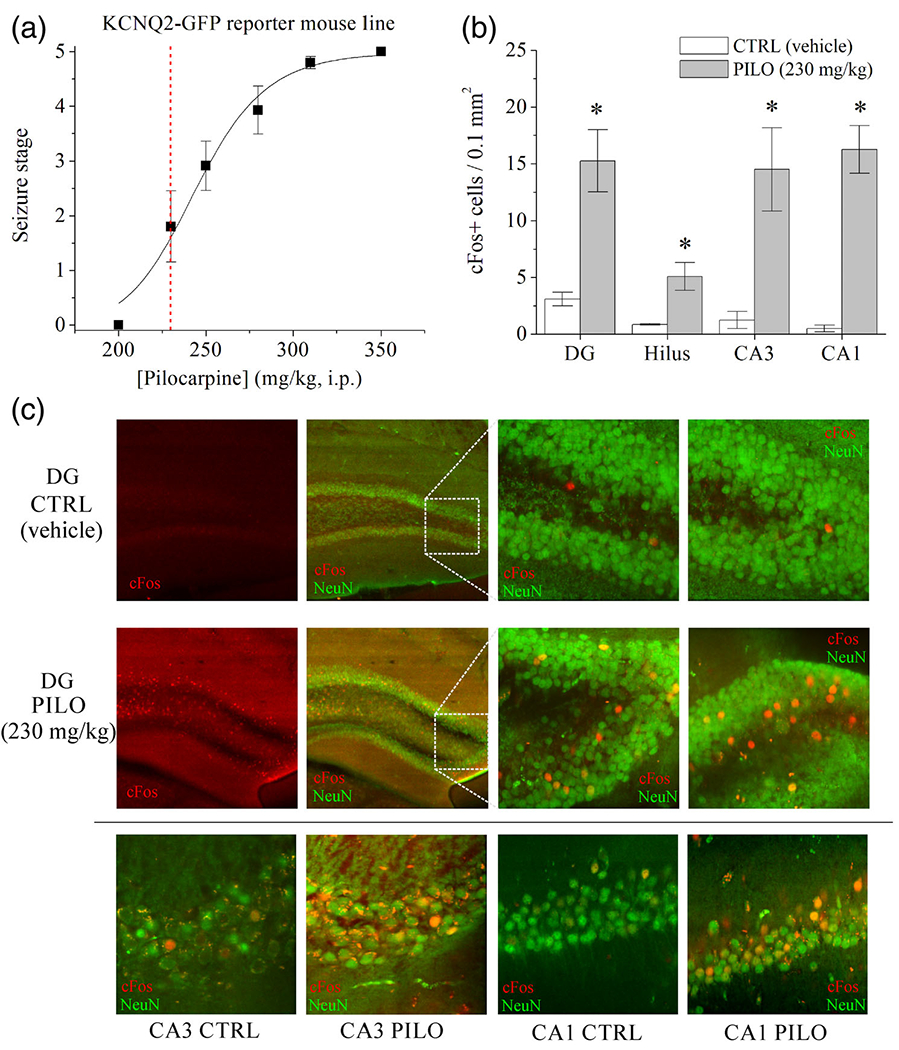
Characterization of KCNQ2 mRNA-EGFP reporter mouse line and increased cFos activity in the hippocampus after hyperexcitability. (a) Dose-response curve for maximum behavioral seizure severity (Racine scale) of the KCNQ2 mRNA-EGFP reporter mouse line after injection of pilocarpine (mg/kg, i.p.). Mice were administered 1 mg/kg scopolamine 30 min. before pilocarpine. Mean group data were pooled from male and female mice (n = 8-12 animals per concentration), of which there were no significant sex differences. The red dashed line denotes the 230 mg/kg subthreshold dose to be used in subsequent experiments. (b) Bars summarize cFos + immunoreactive cell counts from each hippocampal subregion of vehicle- and pilocarpine-challenged (230 mg/kg) mice. *p < .05. n = 3-4 mice per subregion. (c) Shown are confocal microscopy images of cFos (red) immunohistochemistry in (upper panels) DG and (lower panels), under ×40 magnification, of CA1, CA3, and DG principal neurons in vehicle- and pilocarpine-treated mice, 2 hr after injection. Hippocampal slices were co-stained for Neu-N (green). Data represent mean ± SEM [Color figure can be viewed at wileyonlinelibrary.com]
Administration of 230 mg/kg pilocarpine did not produce spontaneous, recurrent seizures or chronic epilepsy (Gröticke et al., 2007; Mazzuferi, Kumar, Rospo, & Kaminski, 2012). However, to confirm that hyperexcitability of principal neurons occurred, we examined cFos activity in the hippocampus 2 hr after administration of vehicle (control) or pilocarpine (Figure 1b,c). Control mice displayed very sparse cFos immunoreactivity across the hippocampus. In contrast, 230 mg/kg pilocarpine administration increased the number of cFos-positive neurons in the DG (p = .012, n = 4-5), CA3 (p = .011, n = 4), and CA1 (p < .001, n = 4) hippocampal regions, comparatively. Interestingly, we also observed an increase in the number of cFos+ hilar neurons after pilocarpine (p = .026, n = 4-5) (Figure 1b). Despite the lack of overt tonic-clonic seizures from animals in this model, these findings demonstrate significant neuronal activation in response to sub-seizure chemoconvulsant stimulation (Greene et al., 2018; Harvey & Sloviter, 2005). Previous work has demonstrated robust cFos activity in neurons of DG and hilus 15 min-2 hr after seizures in mice (Barton, Klein, Wolf, & Whit, 2001; Peng & Houser, 2005). Parvalbumin+ (PV) interneurons have demonstrated delayed Fos activation 2 hr after seizure (Peng & Houser, 2005). Likewise, we chose to examine PV+ interneuron activation due to their previously documented regulation by KCNQ2-containing channels (Soh et al., 2018). Hippocampus was colabeled for PV and cFos from control and pilocarpine-challenged mice (Figure S1). Two hours after 230 mg/kg pilocarpine, PV interneurons did not exhibit significant activation of cFos in any of the three regions of CA1, CA3, or DG. Therefore, we demonstrate that pre-epileptic hyperexcitability, even without SE, significantly enhances cFos activity in the principal neurons, but not PV interneurons, of the hippocampus. We also examined mossy fiber sprouting using immunohistochemistry with an antibody for zinc transporter-3 (ZnT3), which labels the axon terminals of DGGCs as a measure of pathophysiological rewiring of the hippocampus (Sutula, Xiao-Xian, Cavazos, & Scott, 1988; Hester and Danzer, 2013). As expected, we did not observe any significant changes in ZnT3 labeling within the hilus (p = .077), granule cell layer (p = .520), or molecular layer (p = .103) of the DG, between control and pilocarpine-treated mice after 48 hr (n = 3 mice per group, Figure S2).
3.2 |. KCNQ2 transcription is upregulated in CA1 and CA3, but not DG after chemoconvulsant-induced hyperexcitability
To investigate the issue of KCNQ2-containing channel plasticity at the protein level, we performed immunoblot analysis of KCNQ2 protein that compared naïve mice with mice challenged with chemo-convulsants. Hippocampus tissues were collected from male mice 48 hr after administration of either vehicle, 230 mg/kg pilocarpine, or the GABAA receptor antagonist, pentylenetetrazol (PTZ), which induces brain hyperexcitability by a disparate mechanism of action from pilocarpine (Huang et al., 2001; Squires, Saederup, Crawley, Skolnick, & Paul, 1984). In naïve mice, PTZ (60 mg/kg, s.c.) resulted in myoclonic jerks and clonic forelimb seizure activity within 10 min of administration, but tonic-clonic seizures and tonic hindlimb extensions were not observed at this dose. To maintain parity with the other two groups, scopolamine methyl nitrate was injected 30 min prior to delivery of PTZ to mice. Immunoblot analysis of whole hippocampus lysate demonstrated significant increases in KCNQ2 protein levels 48 hr after administration of either chemoconvulsant, compared to vehicle-injected control mice (Figure 2a). Compared to control mice, pilocarpine challenge exhibited a 4.22 ± 0.60 fold increase in KCNQ2 protein (p < .001), and PTZ challenge produced a 4.05 ± 0.23 fold increase in KCNQ2 protein (p < .0001; n = 5 mice per group). Therefore, the previously increased transcription of KCNQ2 mRNA observed after hyperexcitation of the brain in mice (Zhang & Shapiro, 2012), is here demonstrated to result in approximately fourfold greater KCNQ2 protein expression in two distinct models of hyperexcitability.
FIGURE 2.
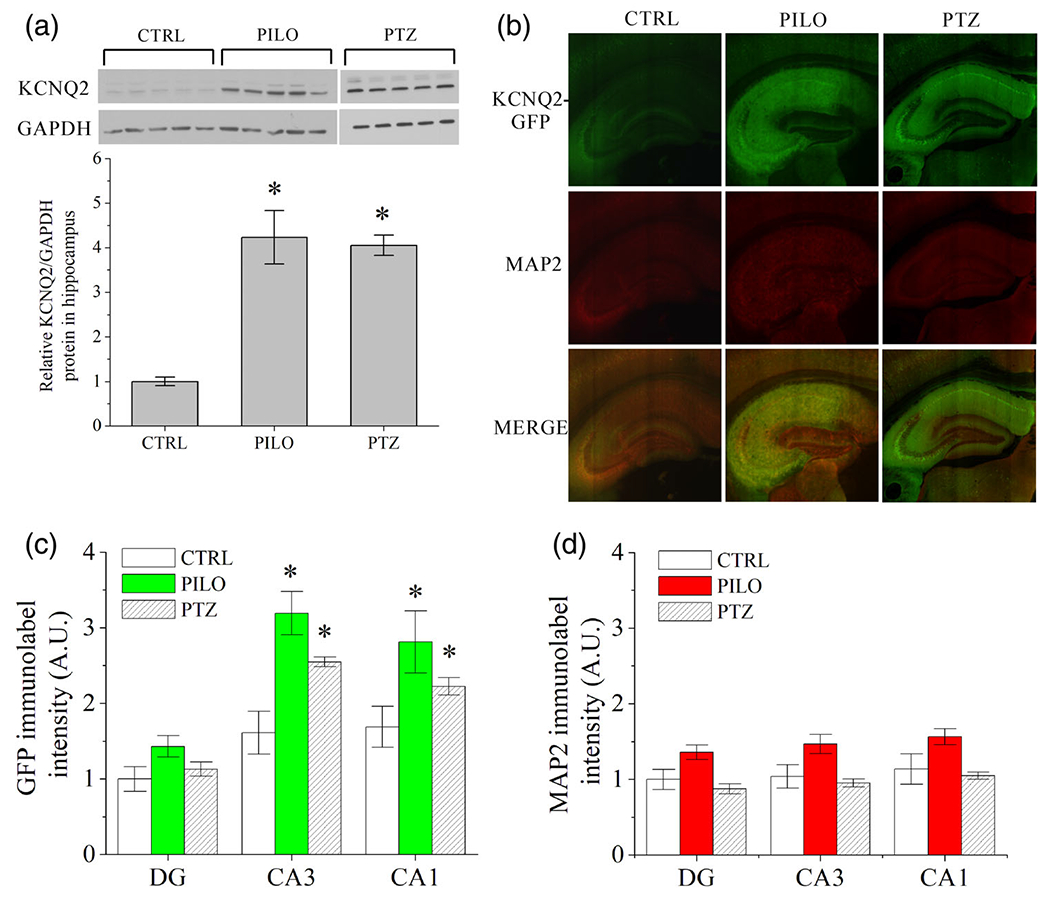
Hippocampal sub-region specific upregulation of KCNQ2 protein and mRNA after chemoconvulsant-induced hyperexcitability. (a) Immunoblot analysis of whole hippocampus lysates from KCNQ2 mRNA-EGFP reporter mice revealed significant increase in KCNQ2 protein in pilocarpine (230 mg/kg) and PTZ (60 mg/kg) treated male mice compared with vehicle injected controls (n = 5 mice per group). Each band in the western blot image depicts whole hippocampus protein lysate from distinct mice. (b) Representative ×4 confocal images of KCNQ2-EGFP reporter dorsal hippocampus 48 hr after vehicle (CTRL), pilocarpine (PILO, 230 mg/kg), or pentylenetetrazol (PTZ, 60 mg/kg) administration. EGFP and MAP2 were detected with immunofluorescence. (c) Quantification of DG, CA3, and CA1 subregion fluorescence of α-GFP label or (d) α-MAP2 label as depicted in b (n = 5-7 mice per group). Chemoconvulsant-treated CA3 and CA1 subregions exhibited significantly greater EGFP immunolabel intensity compared with control, whereas MAP2 was only marginally, but not significantly, altered in all three subregions. Data represent mean ± SEM *p < .05 [Color figure can be viewed at wileyonlinelibrary.com]
We then sought to determine if limbic hyperexcitability increases the expression and function of KCNQ2-containing channels in a cell-type specific manner. To explore this issue, we used the KCNQ-EGFP reporter mice. Whereas this reporter transgene is an indirect measure of KCNQ2 transcription, this method enables comparison of neuron-specific changes within intact sections of the hippocampus without digesting the tissue, as would be necessary for quantitative real-time PCR measures of mRNA. Moreover, we opted to quantify mapped expression levels using immunofluorescent antibody labeling of EGFP rather than direct fluorescence (Whitmire et al., 2017, Figure 2). This was due to concerns of degraded signal quality and quenching of the intrinsic protein fluorescence after the PFA fixation of tissue, which was necessary for processing (Schnell, Dijk, Sjollem, & Giepmans, 2012). Brain sections were co-stained with anti-MAP2 as an index of neuronal density (Seki, 2002; Whitmire et al., 2017). After 48 hr, both CA3 and CA1 regions of pilocarpine-challenged mice exhibited significantly greater regional EGFP-labeled intensity than controls (CA3: p = .003; CA1: p = .028), indicative of increased transcription of KCNQ2 mRNA (Figure 2b,c, n = 5-7 mice per group). However, there were no significant changes between control and pilocarpine-treated animals in the DG (p = .069). MAP2-labeled intensity was not significantly different between control and chemoconvulsant-challenged mice (Figure 2d, DG: p = .084; CA3: p = .334; CA1: p = .593). In addition, we did not observe sex-specific differences in the EGFP signal among control (p = .578, n = 4 mice per group) or among pilocarpine-challenged animals (p = .221, n = 3-4 mice per group).
As a separate measure of mRNA transcription, we used chromogenic in situ hybridization of hippocampal sections comparing control and pilocarpine-challenged animals. KCNQ3 forms heteromeric tetra-meric channels with KCNQ2, of varying combinations of KCNQ2 and KCNQ3 subunits, as well as homomeric KCNQ3 channels (Carver & Shapiro, 2019). Since there was not a KCNQ3 mRNA-EGFP reporter mouse available, we probed both KCNQ2 and KCNQ3 mRNA simultaneously (Figure S3). Similar to the observations in the EGFP-reporter mice, KCNQ2 transcription was increased in CA1 (control: 0.53 ± 0.10, pilocarpine: 0.86 ± 0.03, p = .031, n = 3) and CA3 (control: 0.55 ± 0.07, pilocarpine: 0.85 ± 0.09, p = .045, n = 3) pyramidal neurons, as determined by probe particle density. Intriguingly however, KCNQ3 transcription was not significantly changed in DG (p = .752), CA3 (p = .415), or CA1 (p = .148) after pilocarpine challenge (n = 3 per group). This is consistent with previous studies suggesting that KCNQ2, rather than KCNQ3, is the main regulatory M channel subunit for modulation of excitability in hippocampus principal neurons (Soh et al., 2014).
To more precisely examine neuron-specific alterations in KCNQ2 transcription, we compared the cell-by-cell KCNQ2-EGFP reporter signal at higher magnification with confocal microscopy (Figure 3). This analysis involved measuring the immunofluorescence intensity in individual principal neurons, totaling between 50 and 200 cells per region in each animal, which were quantified as a group average per animal (see Methods). We normalized the immunolabeled EGFP signal by the MAP2 signal (Figure 3c,f,i). Cellular level quantification revealed increases in KCNQ2 expression within CA3 and CA1 pyramidal neurons after chemoconvulsant-induced hyperexcitability. Control CA3 pyramidal neurons displayed a mean EGFP/MAP2 intensity of 1.26 ± 0.08, whereas CA3 pyramidal neurons from pilocarpine-challenged mice displayed a mean intensity of 1.87 ± 0.07 (p < .0001, n = 7 mice per group). Control CA1 pyramidal neurons displayed a mean EGFP/MAP2 intensity of 1.04 ± 0.09, whereas CA1 pyramidal neurons from pilocarpine-challenged mice displayed a mean intensity of 1.41 ± 0.17 (p = .039, n = 7 mice per group). Interestingly, not every DGGC demonstrated apparent labeling by our α-GFP antibodies. We quantified KCNQ2 expression from DGGCs that exhibited greater than twofold of the mean signal intensity compared to the background fluorescence as a threshold. DGGCs did not exhibit significant differences in normalized EGFP intensity after 48 hr between the control group (EGFP/MAP2: 0.78 ± 0.06) and pilocarpine-challenged group (pilocarpine EGFP/MAP2: 0.88 ± 0.02, p = .161, n = 7 mice per group) (Figures 3g-i).
FIGURE 3.
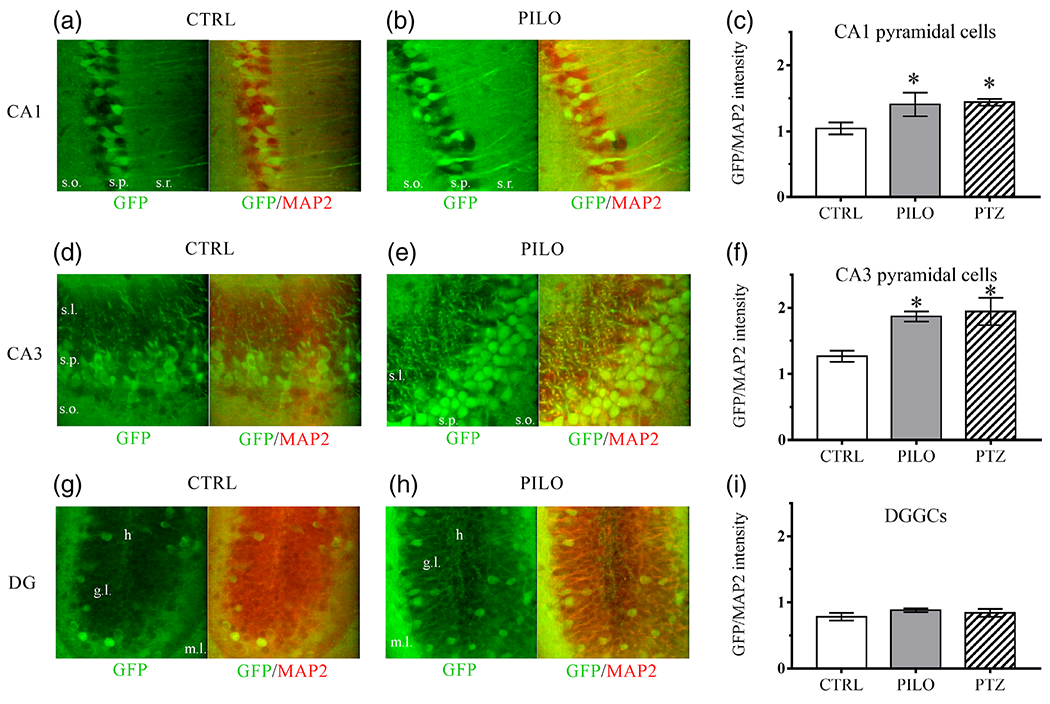
Hippocampal cell-specific differences in KCNQ2 expression after chemoconvulsant hyperexcitability, as detected by the KCNQ2 mRNA-EGFP reporter mouse line. Representative ×40 magnification images of α-GFP or α-GFP and α-MAP2 merged immunofluorescence of hippocampal principal neurons from the three major subregions. Brain slices are from adult male mice 48 hr after vehicle-control or 230 mg/kg pilocarpine injection in adult male mice for CA1 pyramidal neurons (a-b; s.o., stratum oriens; s.p., stratum pyramidale; s.r., stratum radiatum), CA3 pyramidal neurons (d, e; s.l., stratum lucidum; s.p., stratum pyramidale; s.o., stratum oriens), and DGGCs (g, h; h, hilus; g.l, granule cell layer; m.l., molecular layer). Right-most bar graphs show quantification of normalized EGFP/MAP2 immunolabel intensity 48 hr after vehicle (CTRL), 230 mg/kg pilocarpine (PILO), or 60 mg/kg PTZ induced hyperexcitability in CA1 pyramidal neurons (c), CA3 pyramidal neurons (f), and DGGCs (i). Bar graphs are the averaged group values derived from individual cell-by-cell quantification of fluorescence intensity at ×40. n = 5-7 mice per group. Data represent mean ± SEM *p < .05 [Color figure can be viewed at wileyonlinelibrary.com]
To determine if the observed upregulation of KCNQ2 transcription was artificially specific for the mechanism of action of pilocarpine, a separate group of age-matched mice were challenged with PTZ (60 mg/kg) to induce acute seizures of distinct origin (Figures 2 and 3). Forty-eight hours after PTZ induction of clonic seizures in five mice, we again observed significant upregulation of KCNQ2 expression, as assayed by the KCNQ2-EGFP reporter mice, in CA3 (EGFP/MAP2: 1.94 ± 0.2, p = .005, n = 5) and CA1 (EGFP/MAP2: 1.44 ± 0.05, p = .006, n = 5) principal neurons, similar to that seen in mice challenged with pilocarpine (Figure 3c, f). Furthermore, as in the pilocarpine-challenged mice, EGFP immunolabel intensity was not significantly different between DGGCs from controls and the PTZ-treated group (EGFP/MAP2: 0.84 ± 0.06, p = .508, n = 5) (Figure 3i). Therefore, we conclude that an initial hyperexcitability challenge to the brain is sufficient to induce selective upregulation of KCNQ2 transcription in the hippocampus, independent of the mechanism of action of the chemoconvulsant.
In addition to the glutamatergic principal neurons, we investigated KCNQ2 immunoreactivity within hilar neurons after pilocarpine challenge. Forty-eight hours after 230 mg/kg pilocarpine, the number of EGFP+ hilar neurons significantly increased (CTRL: 0.8 ± 0.2 cells/0.1 mm2; PILO: 5.7 ± 0.4 cells/0.1 mm2, p = .012; n = 5) (Figure S1). We hypothesize this to be a binary expression of KCNQ2, whereas most of the hilar interneurons demonstrate little to no KCNQ2 immunoreactivity at basal conditions in control mice, similar to our observations in DGGCs, which we address below. In the case of interneurons, increased KCNQ2 expression may be in response to increased neuronal firing during hyperactivity, as we observed changes to cFos immunoreactivity as well (see Discussion).
3.3 |. KCNQ2 upregulation is not present 7 days after hyperexcitability
Other groups have documented an increase in seizure susceptibility in the hippocampus after seizures, both from morphological contributions (Scharfman & Pierce, 2012) and via alterations in HCN channel expression and function (Brennan, Baram, & Poolos, 2016; Jung, Warner, Pitsch, Becker, & Poolos, 2011). We hypothesized that the increase in KCNQ2 expression after hyperexcitability was a transient event, since chemoconvulsant challenge promotes downstream seizure susceptibility. To investigate the time-dependent window of increased KCNQ2 expression in the hippocampus, we examined hippocampi from KCNQ2 mRNA-EGFP reporter mice 7 days after pilocarpine challenge (Figure S4). Immunolabeled EGFP levels were not significantly different between the principal neurons of the CA1, CA3, or DG subregions between control and pilocarpine-challenged animals after 7 days. Therefore, we conclude that the KCNQ2 mRNA upregulation observed at 48 hr after hyperexcitability subsides over time, returning to near basal levels after 1 week. This observation sheds light on the ion channel-based mechanisms of altered seizure susceptibility after epileptiform network activity. This issue is further explored in Discussion.
3.4 |. Dentate gyrus exhibits no alteration in KCNQ2 expression during mild hyperexcitability, but KCNQ2 is significantly upregulated after severe seizure activity
We found it intriguing that the expression of KCNQ2 was not significantly altered in DGGCs, whereas CA3 and CA1 pyramidal neurons consistently displayed upregulation of KCNQ2 expression after sub-threshold hyperexcitability. Strikingly, we found obvious heterogeneous labeling of KCNQ2 mRNA using the KCNQ2 mRNA-EGFP reporter mice among DGGCs. The neurons located proximal to the molecular layer (typically more mature DGGCs) displayed significantly greater intensity than a majority of the neurons in the granule cell layer. Whereas a majority of CA1 and CA3 pyramidal neurons demonstrated expression of KCNQ2 mRNA, as assayed with the KCNQ2 mRNA-EGFP reporter mice, a relatively small proportion of DGGCs appeared immunoreactive for EGFP. The heterogenous expression of KCNQ2 mRNA in DGGCs could be related to the random insertion of the transgene into the genome (Schmidt, Kus, Gong, & Heintz, 2013), and therefore be an artificial outcome of the expression mapping. Alternatively, such differences in KCNQ2-reporter expression among cell-types could rather be related to differential signaling-processing demands amongst the neurons. We previously reported that DGGCs and CA1 pyramidal neurons have starkly different mechanisms by which muscarinic acetylcholine receptors regulate IM and neuronal excitability (Carver & Shapiro, 2019). Therefore, further investigation of the gradation in pilocarpine-induced hyperexcitability in the DG was warranted. We hypothesized that the generation of synchronous hyperexcitability resulting in SE seizures would secondarily induce greater KCNQ2 transcription in DGGCs by overriding the intrinsic inhibitory mechanisms of the DG. In mice challenged with 300 mg/kg pilocarpine to induce SE, KCNQ2 upregulation of expression was significantly weakened in CA1 (2.7 ± 0.3 fold reduction, p = .002, n = 5) and CA3 (3.3 ± 0.4 fold reduction, p = .001, n = 5) regions at 48 hr, resulting in a reduction in overall fluorescence intensity compared to controls (Figure 4a), largely due to substantial excitotoxic cell death (Borges et al., 2003; Gröticke et al., 2007; Turski et al., 1984). Furthermore, MAP2 immunoreactivity decreased in CA1 (1.8 ± 0.3 fold reduction, p = .014, n = 5) and CA3 (1.8 ± 0.1 fold reduction, p = .021, n = 5), indicative of cell loss after epileptogenic events (Schartz et al., 2016; Yan et al., 2012). Interestingly after SE, we also observed that the CA2 region remained strongly labeled for KCNQ2 mRNA, distinct from the surrounding pyramidal neurons in which the EGFP signal was ablated (Figure 4a). However, DGGCs from SE mice demonstrated both significantly increased overall KCNQ2-labeled intensity (vs. control: p < .001, vs. 230 mg/kg PILO: p < .001, n = 5-12 mice per group; Figure 4b,c). Furthermore, there were a substantially greater number of EGFP+ DGGCs from mice challenged with 300 mg/kg pilocarpine to induce SE (49.2 ± 2.1% EGFP+ cells), significantly above control mice (9.6 ± 0.6% EGFP+, vs. SE p < .001, n = 5) and even above that of mice challenged with 230 mg/kg pilocarpine (12.3 ± 1.1% EGFP+ cells, vs. SE: p < .001, n = 5) (Figure 4d). Comparatively, there were no significant differences in the number of EGFP+ DGGCs between control mice and those which were challenged with 230 mg/kg pilocarpine to induce moderate hyperexcitability (p = .120, n = 5). Therefore, cell-selective upregulation of KCNQ2 expression in DGGCs may depend on the severity of the hyperexcitability to the hippocampus, and to whether or not the network is compromised due to SE network exacerbations. Specifically, upregulation of KCNQ2 expression can only occur in neurons that survive a deleterious insult that induces excitotoxicity, represented here by the stark contrast of signal between the types of principal neurons. We recently report a similar result concerning cortical neuron plasticity in responding to and surviving an adverse insult (Vigil et al., 2019).
FIGURE 4.
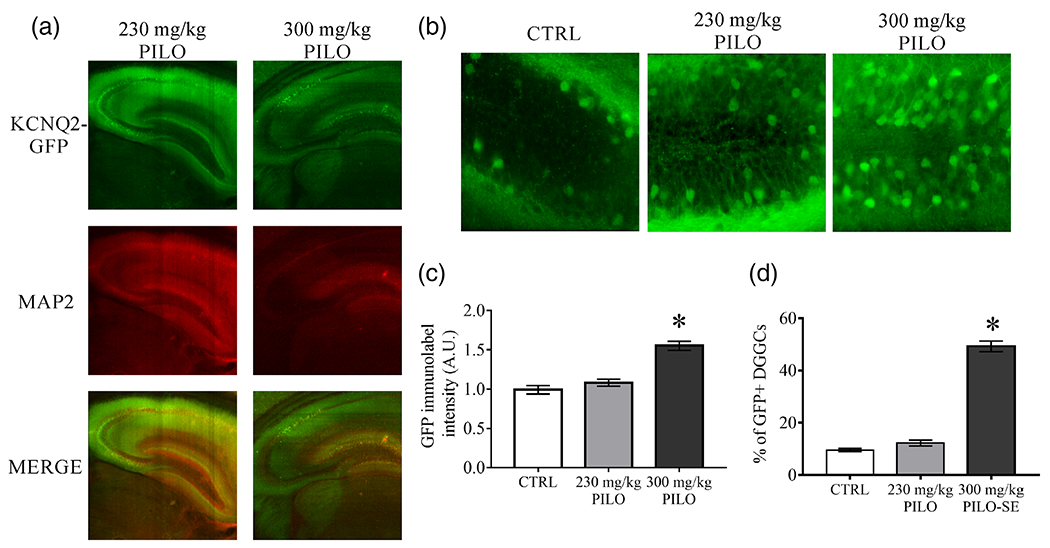
Changes in KCNQ2 transcription for DGGCs differ according to the severity of hyperexcitability in the hippocampus. (a) Representative images of immunostaining for KCNQ2 mRNA-linked EGFP and MAP2 in whole hippocampus 48 hr after 230 mg/kg (subconvulsive) or 300 mg/kg (SE) pilocarpine. (b) Representative images of the DGGC layer exhibiting substantially greater numbers of KCNQ2 mRNA-linked, EGFP+ DGGCs after 300 mg/kg PILO, as in (a). (c) DGGC individual cell quantification for KCNQ2 mRNA-EGFP reporter mean fluorescent intensity values of mice injected with vehicle (CTRL) or 48 hr after pilocarpine-induced hyperexcitability. (d) Individual cell counts under ×40 magnification images for KCNQ2 mRNA-linked, EGFP+ DGGCs, with selection criteria determined as cells that exhibited signal intensity twofold greater than the background fluorescent intensity. There were no significant differences between control and 48 hr PILO GFP+ cell quantity, however, the number of DGGCs was significantly increased in PILO (300 mg/kg) mice, compared to controls.*p < .05 [Color figure can be viewed at wileyonlinelibrary.com]
3.5 |. Non-seizure hyperexcitability induces higher action potential frequency in DGGCs
Despite lack of significant change in KCNQ2 expression of DGGCs after non-seizure hyperexcitability, we observed increased cFos activity in those neurons (Figure 1b,c). In order to determine if hippocampal neurons are truly more excitable after a subconvulsant dose of pilocarpine, we examined action potential firing properties of DGGCs using current-clamp electrophysiology in the brain slice. As above, hippocampi were isolated 48 hr after mice were injected with vehicle or pilocarpine (230 mg/kg), and brain slices prepared as previously described (Carver & Shapiro, 2019). DGGCs from pilocarpine-challenged mice demonstrated significantly increased action potential firing over a range of injected current (60-220 pA) (Figure 5a,b). Furthermore, DGGCs from pilocarpine-treated mice exhibited significantly reduced rheobase (41 ± 2 pA) compared to control mice (66 ± 3 pA, p < .001, n = 12-15 cells), indicative of a lowered depolarization threshold (Figure 5c). Passive membrane properties did not significantly change for input resistance (control Rin = 404 ± 28 MΩ; pilocarpine Rin = 371 ± 41 MΩ; p = .491, n = 10-17 cells) and membrane time-constant (control τmembrane = 27.7 ± 1.9 ms; pilocarpine τmembrane = 25.6 ± 3.0 ms; p = .540, n = 10-17 cells) between control DGGCs and pilocarpine-challenged DGGCs. It is known that KCNQ2-deficiency reduces the spike-frequency adaptation of many types of principal neurons and PV+ interneurons (Carver & Shapiro, 2019; Niday et al., 2017; Soh et al., 2014; Soh et al., 2018). In order to determine if spike-frequency adaptation is altered by pilocarpine (via muscarinic receptor-induced hyperexcitability), we investigated the interspike intervals in DGGCs within slices 48 hr after pilocarpine challenge (Figure 5d). Across a wide range of the current-injection, the initial instantaneous spike frequency was significantly increased in pilocarpine-challenged mice compared to control mice. However, the final spike frequency was similar between the groups, suggesting that whereas DGGCs exhibit initial increase to firing upon excitation, the spike-frequency adaptation remains intact. Overall, these data show that DGGC function is significantly altered after a single, non-SE challenge with pilocarpine.
FIGURE 5.
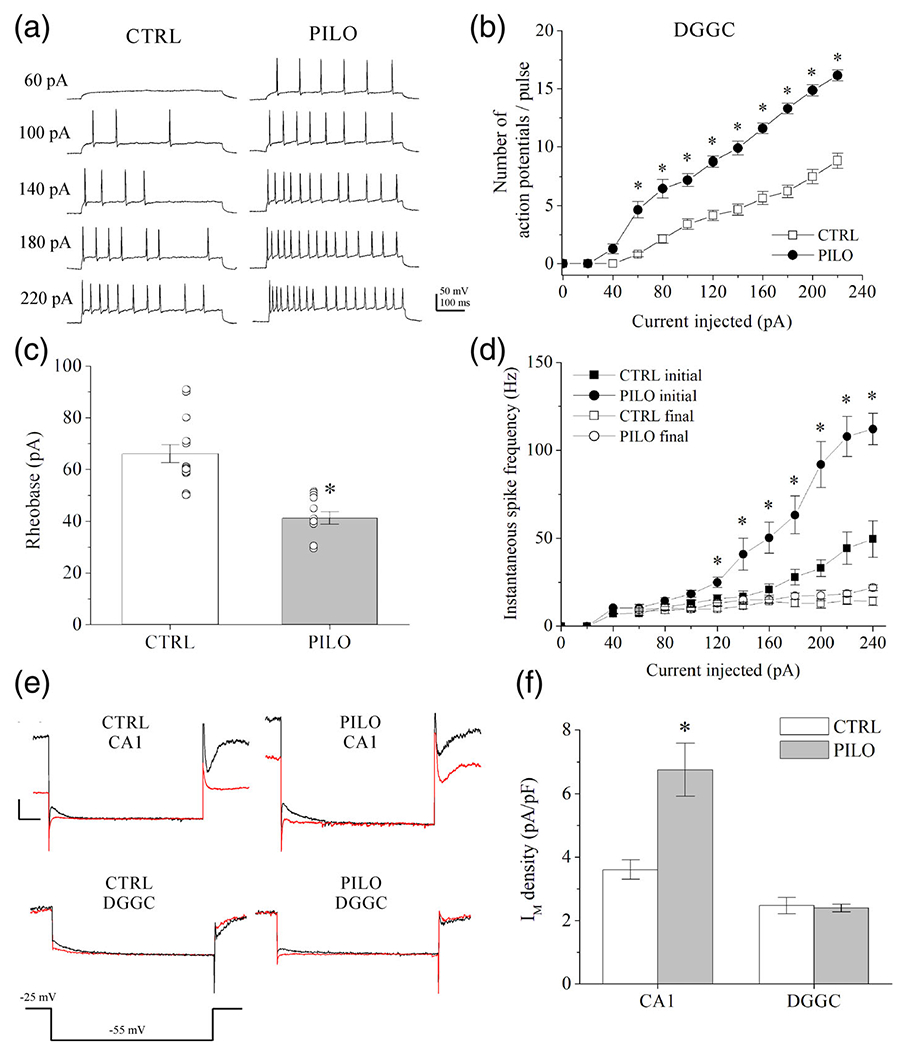
Action potential properties and IM in DGGCs and CA1 pyramidal neurons after hyperexcitability observed using brain slice electrophysiology. (a) Shown are representative current-clamp recordings of evoked action potential firing in mouse DGGCs from brain slice, 48 hr after vehicle (CTRL) or pilocarpine (PILO, 230 mg/kg) administration. (b) Summarized action potential frequencies in DGGCs recorded in current-clamp mode as depicted in A. (c) Summarized current amplitudes required to evoke a single action potential (rheobase) in DGGCs. (d) Summarized are the initial and final instantaneous spike frequency of action potentials from DGGCs in response to increasing current injection (see Methods). (e) Shown are representative current traces of deactivating IM at −55 mV in control solution (black) or during bath-application of XE-991 (20 μM) (red) in voltage-clamp recordings of CA1 pyramidal neurons or DGGCs. (f) Bars summarize IM density (pA/pF) of CA1 pyramidal neurons or DGGCs in comparison of control and pilocarpine-injected mice, as depicted in (e). IM amplitude was measured as the current sensitive to the M-channel blocker 20 μM XE-991. Currents are normalized to cell capacitance as an index of cell membrane size. CA1 pyramidal neurons displayed significant increase to IM amplitude 48 hr after pilocarpine, whereas DGGCs demonstrated no significant changes between control and pilocarpine mice. Data represent mean ± SEM. n = 8-15 cells per group. *p < .05 versus CTRL [Color figure can be viewed at wileyonlinelibrary.com]
3.6 |. Hyperexcitability results in increased IM amplitude in CA1 pyramidal neurons, but not in DGGCs
To characterize the functional effect of hyperexcitability on IM, we performed further patch-clamp electrophysiology on hippocampal neurons in brain slice from control and pilocarpine-challenged mice, under whole-cell voltage clamp. The voltage-clamp protocol was used to measure deactivation currents in either CA1 pyramidal neurons or DGGCs (Figure 5e,f), as previously described (Carver & Shapiro, 2019). Cells were maintained at a pre-pulse holding potential of −25 mV prior to a step to −55 mV to enable assay of the deactivating IM amplitudes (see Methods for voltage-error correction). The M-channel blocker XE-991 (20 μM) was applied at the end of each recording, and IM was measured as the deactivating current that was sensitive to XE-991. In CA1 neurons from mice challenged with pilocarpine (230 mg/kg), IM amplitude was significantly greater than in mice administered vehicle as control (control: 3.61 ± 0.3 pA/pF, pilocarpine: 6.75 ±0.83 pA/pF, p = .003, n = 15-16 cells per group) (Figure 5e). However, despite the changes in spike activity reported above, DGGCs did not exhibit significant differences in IM between control and pilocarpine-challenged groups (control: 2.47 ± 0.26 pA/pF, pilocarpine: 2.40 ± 0.13 pA/pF, p = .86, n = 7-16 cells per group) (Figure 5f). Therefore, the changes in IM current amplitudes parallel the effects on KCNQ2 mRNA and protein upregulation observed after pilocarpine-induced hyperexcitability.
3.7 |. Traumatic insult to the brain increases KCNQ2 transcription in the hippocampus
Since we observed KCNQ2 upregulation with different classes of chemoconvulsant, we postulated that brain hyperexcitability after traumatic brain injury (TBI) could also result in increased KCNQ2 transcription in the hippocampus. After TBI, it is thought that an initial cascade of excitotoxicity-mediated cell death and damage exacerbates or even initiates the process of epileptogenesis (Algattas & Huang, 2013; Hunt, Boychuk, & Smith, 2013), and moreover, the severity of injury is strongly associated with the risk of developing epilepsy (Christensen et al., 2009; Lowenstein, 2009). Previous studies report incidence of epilepsy between 7 and 39% from closed-head TBI injuries and greater than 50% of people after penetrating TBI (Annegers, Hauser, Coan, & Rocca, 1998; Englander et al., 2003). In experimental models of TBI, mice develop post-traumatic seizures 8-10 weeks or more after TBI, due to the secondary cascade from injury (Hunt et al., 2009), but seizures can also manifest in the acute phase after the initial TBI (Nilsson et al., 1994). Furthermore, selective neuronal death occurs in the hippocampus 1-7 days after TBI that promotes further hyperexcitability (Golarai, Greenwood, Feeney, & Connor, 2001). We used the controlled cortical impact (CCI) model, as recently described (Vigil et al., 2019). Forty-eight hours after CCI-TBI, we investigated changes in relative KCNQ2 transcription in hippocampus using the KCNQ2 mRNA-EGFP reporter mice. Ispilateral and contralateral hippocampi were immunostained with α-GFP and α-MAP2 antibodies and compared KCNQ2-linked EGFP expression in each neuronal subregion, as before (Figure 6). The principal neurons from each three subregions of DG (p = .035), CA3 (p = .010), and CA1 (p = .031) exhibited significantly increased KCNQ2 expression on the ipsilateral hemisphere of the TBI, compared with the contralateral hippocampus. This is intriguing, given that the CCI injury was delivered without breaking the skull, and notable damage and swelling were only observed in the cortex. Based on these findings, initial injury localized to the cortex confers KCNQ2 upregulation in the hippocampus within 48 hr of the trauma.
FIGURE 6.
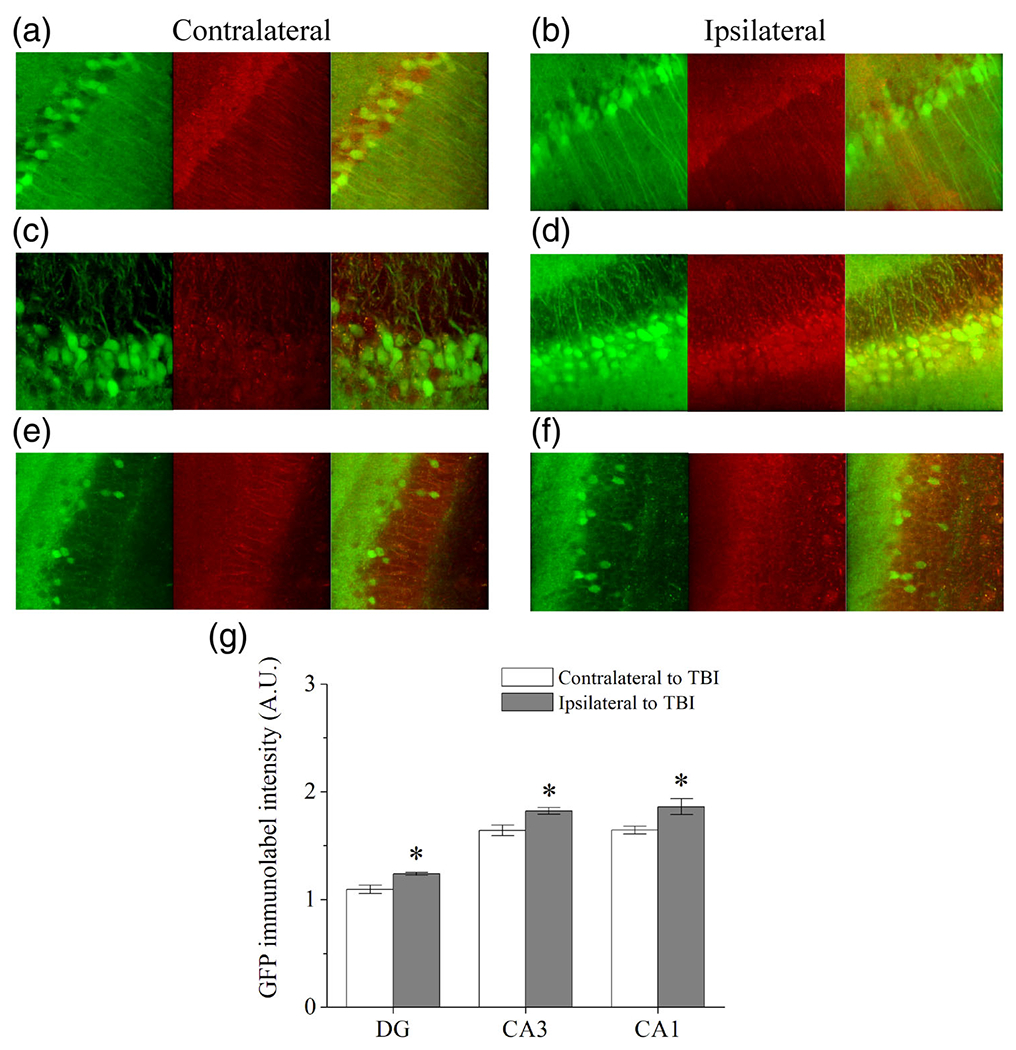
Traumatic brain injury induces KCNQ2 upregulation of expression in the ipsilateral hippocampal hemisphere. Panels depict immunolabeling of EGFP (green), MAP2 (red) and merged immunofluorescence of contralateral and ipsilateral hippocampal hemispheres in (a,b) CA1, (c,d) CA3, and (e,f) DG regions from KCNQ2 mRNA-EGFP mice 48 hr after receiving CCI-TBI. (g) Bars summarize the KCNQ2 mRNA-linked EGFP intensities from contralateral and ipsilateral hippocampi after TBI. *p < .05 ipsilateral versus contralateral. Group data represent mean ± SEM [Color figure can be viewed at wileyonlinelibrary.com]
3.8 |. M-channel activators reduce seizure susceptibility subsequent to pilocarpine-induced hyperexcitability
M-channel activators like retigabine (RTG) are anti-convulsants that augment channel opening and increase the neuronal inhibition in control of seizures (Gunthorpe et al., 2012; Kumar et al., 2016; Wickenden, Yu, Zou, Jegla, & Wagoner, 2000). We hypothesized that enhancement of IM with RTG via increased opening of KCNQ2-containing M channels would provide greater protection from seizures after an initial hyperexcitable event. Using the low-dose pilocarpine hyperexcitability model used above, we examined secondary seizure susceptibility in conjunction with the anticonvulsant efficacy of RTG to prevent seizure behavior in vivo (Figure 7). After 48 hr, naïve (1 mg/kg scopolamine methyl nitrate only) or scopolamine + pilocarpine-challenged (230 mg/kg) mice were subsequently challenged with 60 mg/kg PTZ (Figure 7b). In this assay, we used PTZ for the secondary induction of acute seizures due to more rapid onset of excitatory activity and greater delineation of the progression of motor seizure activity, as well as testing of anticonvulsant drug efficacy (Blanco et al., 2009; Ferraro et al., 1999). Latencies to myoclonic jerking, generalized forelimb clonus, and maximal tonic-clonic behavioral seizures were recorded. For naïve control animals (without prior pilocarpine-induced hyperexcitability), these seizure latencies were 6 min 22 s ± 24 s for jerking, 15 min 23 s ± 1 min 56 s for clonus, and 26 min 49 s ± 1 min 37 s for tonic-clonic seizure, respectively (Figure 7b). In addition, the naïve animals experienced low incidence of seizures in response to this dose of PTZ, and minimal tonic-clonic activity was observed (Figure 7c-e). In comparison, pilocarpine-challenged animals exhibited significantly reduced latency at all three stages of PTZ-induced seizure, indicative of greater susceptibility (Figure 7b). Latencies for pilocarpine animals were 2 min 34 s ± 39 s for jerking (vs. control: p < .001), 4 min 7 s ± 51 s for clonus (vs. control: p = .006), and 6 min 8 s ± 1 min 55 s for tonic-clonic activity (vs. control: p < .001, n = 7-15 mice per group). Furthermore, there were a significantly greater number of mice from the pilocarpine-challenged group in which severe tonic seizures progressed to mortality after tonic hindlimb extension (Figure 7d,e). In another cohort, RTG (1 mg/kg, s.c.) was administered 15 min prior to PTZ challenge in comparison of control and pilocarpine-treated mice. RTG administration did not significantly alter PTZ seizure susceptibility in naïve animals, and the percentage of mice experiencing clonic and tonic episodes was similar with or without RTG treatment. However, for pilocarpine-challenged animals, RTG significantly prolonged latency to myoclonic jerking (11 min 7 s ± 49 s, vs. pilocarpine: p = .003, n = 7 mice per group) and clonic episodes (24 min 10 s ± 3 min 44 s, vs. pilocarpine: p = .001, n = 7 mice per group) (Figure 7b), including lower incidence of forelimb clonus, no incidence of tonic-clonic activity, and all mice were protected from tonic seizures and survived (Figure 7c,d). Therefore, initial pilocarpine-induced hyperexcitability conferred subsequently greater efficacy of RTG in anticonvulsant control of seizures.
FIGURE 7.
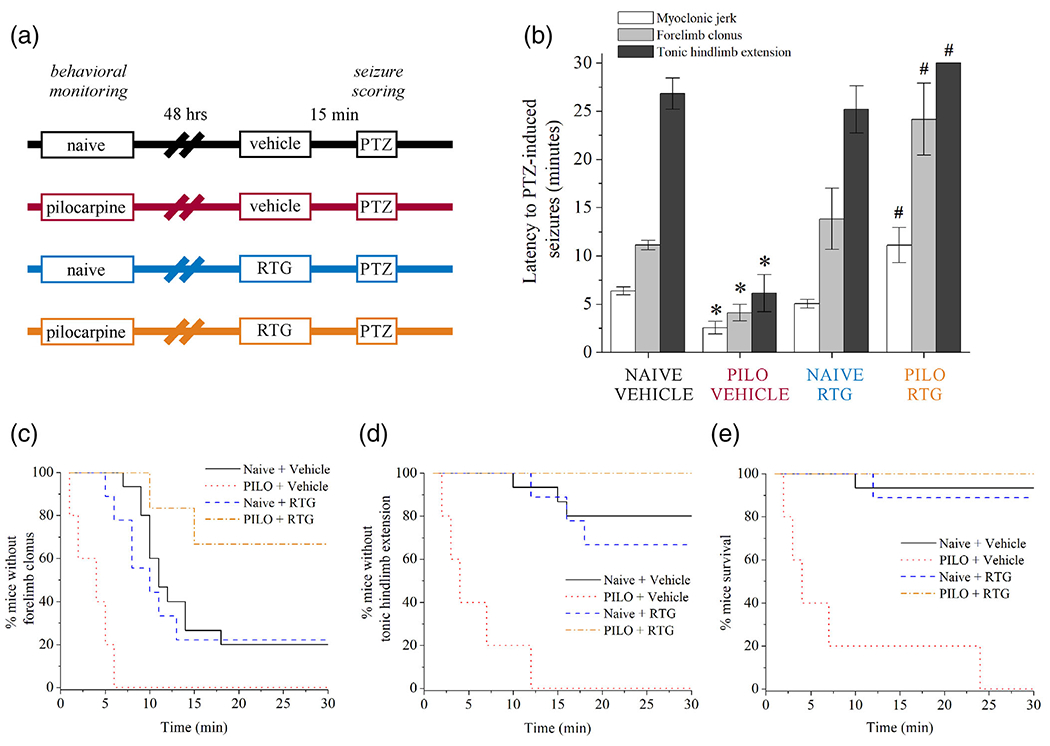
M-channel openers produce greater protection from seizures after induction of hyperexcitability. (a) Experimental design schematic for GABAA receptor antagonist PTZ (60 mg/kg, s.c.) challenge of seizure susceptibility in mice 48 hr after control (naïve, scopolamine only) or pilocarpine (230 mg/kg) administration. (b) Bars summarize the latency to PTZ-induced seizures from the groups of mice depicted in (a). PILO-induced hyperexcitability mice (red) demonstrated greater seizure susceptibility compared with CTRL (black). CTRL mice treated with RTG (1 mg/kg) 15 min before PTZ (blue) did not exhibit altered sensitivity profiles. PILO mice treated with RTG before PTZ (orange) displayed significantly improved seizure latency and were protected from clonic and tonic-clonic seizures. (c) Shown is the Kaplan-Meier fractional curve of percentage of mice without forelimb clonus over time. (d) Shown is the Kaplan-Meier fractional curve of percentage of mice without tonic hindlimb extension (criterion for tonic-clonic seizure) over time. (e) Shown is the Kaplan-Meier fractional curve of percentage of mice surviving PTZ-induced seizures over time. Data in panel b represent mean ± SEM. n = 7-15 mice per group. *p < .05 versus CTRL. #p < .05 versus PILO [Color figure can be viewed at wileyonlinelibrary.com]
3.9 |. KCNQ2 deficiency in the DG results in greater seizure susceptibility
The DG serves as the putative “gate-keeper” for the hippocampal circuit (Scharfman and Myers, 2016; Krook-Magnuson et al., 2015), and maladaptive changes in the DG contribute to the overall epileptogenic transformation of the temporal lobe (Hester and Danzer, 2013). Our previous observation was that a subthreshold hyperexcitability challenge to mice significantly altered DGGC neuronal firing properties. We also found that persistent hyperexcitability (SE) was required to induce KCNQ2 transcriptional upregulation in DGGCs. We also wondered if a deficit in KCNQ2-containing channels within DGGCs would critically alter the inhibitory control of network excitability. Deletion of KCNQ2 from mouse CA1 pyramidal neurons is sufficient to induce hyperexcitability that progresses into cortical epileptogenic activity (Soh et al., 2014). We previously reported that KCNQ2-deficient DGGCs have increased action potential firing and reduced spike-frequency adaptation (Carver & Shapiro, 2019); however, in vivo network activity has not yet been examined. Therefore, we explored if hypofunction of DG-specific KCNQ2-containing channels contributes to seizure susceptibility using the PTZ model. To effectively accomplish this, we turned to mouse lines in which conditional and regional expression of KCNQ2 could be controlled within the brain. This would allow us the opportunity to distinguish the significance of the DGGCs during propagation of in vivo circuit hyperexcitability. To achieve DG-targeted KCNQ2 deficiency, we crossed Cre-POMC mice (McHugh et al., 2007) with mice floxed for KCNQ2 (Soh et al., 2014). Cre+ mice either possessed a single loxP allele (KCNQ2+/flox) or homozygous for loxP flank of KCNQ2 (KCNQ2flox/flox) resulting in a knockdown of KCNQ2 protein, as described previously (Carver & Shapiro, 2019). At 2 months of age, mice were injected with PTZ (60 mg/kg, s.c.) and monitored for behavioral motor seizures for 1 hr (Figure 8). We did not observe sex-specific differences in seizure susceptibility between male and female of the same genotype (Figure 8a). KCNQ2−/− mice demonstrated significantly reduced to PTZ-induced seizures compared to KCNQ2++/+ mice, indicative of greater hyperexcitability (Figure 8a). For myoclonic jerks, KCNQ2+/+ mice exhibited an average latency of 6 min 1 s ± 29 s, whereas KCNQ2−/− mice had a latency of 4 min 28 s ± 30 s (p = .037, n = 9 per group). For forelimb clonus, KCNQ2+/+ mice had an average latency of 14 min 40 s ± 2 min 15 s, compared to a latency of 7 min 28 s ± 42 s for KCNQ2−/− mice (p = .017, n = 9 per group). For tonic-clonic seizure activity, KCNQ2+/+ mice displayed an average latency of 24 min 53 s ± 2 min 20 s, whereas KCNQ2−/− mice displayed a latency of 11 min 6 s ± 43 s (p < .001, n = 9 per group). In group data pooled from both sexes, the haplo-insufficient (KCNQ2+/−) and KCNQ2-deficient (KCNQ2−/−) mice exhibited significantly faster progression to myoclonic jerking and forelimb clonic seizure behavior compared to KCNQ+/+ controls (Figure 8c). Furthermore, KCNQ2−/− mice demonstrated high incidence of tonic hindlimb extension and mortality after PTZ, whereas KCNQ2+/+ animals produced little or no tonic seizure activity and no mortalities (Figure 8c-e). Therefore, KCNQ2 deficiency within DGGCs may exert a profound impact on seizure susceptibility and progression of hyperexcitability to epilepsy throughout the brain.
FIGURE 8.
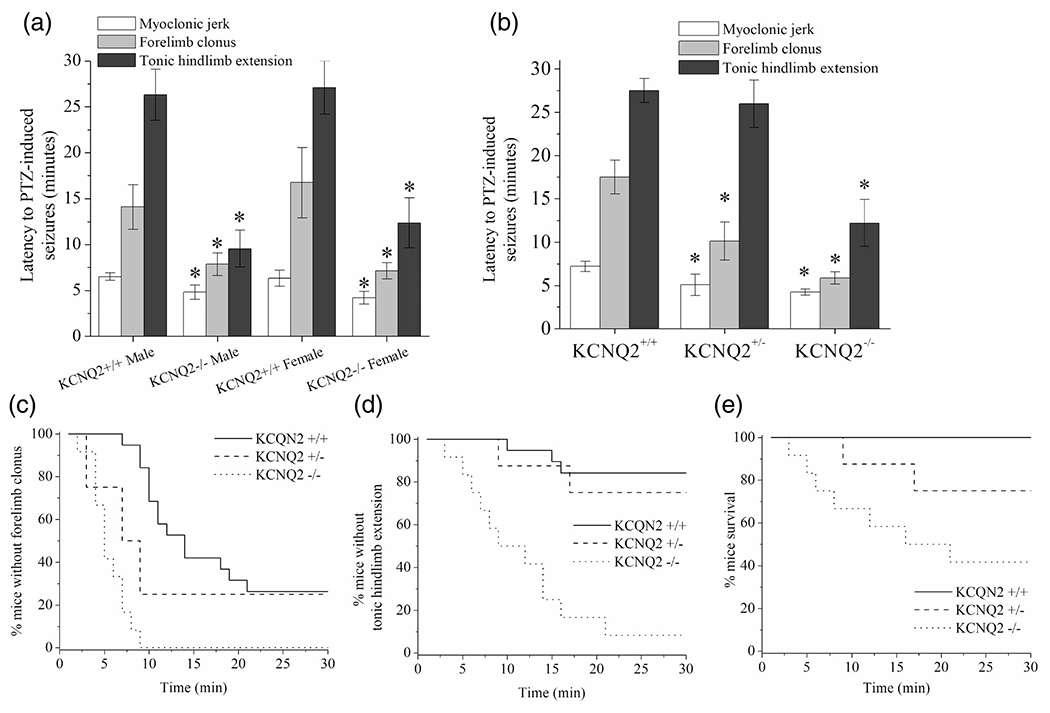
KCNQ2-deficiency exclusive to DGGCs results in greater seizure susceptibility. (a) Bars summarize the latency to seizures in Cre-POMC-/Q2flox/flox (KCNQ2+/+) or Cre-POMC+/Q2+/flox (KCNQ2+/−) or Cre-POMC+/Q2flox/flox (KCNQ2−/−) mice after injection of PTZ (60 mg/kg, s.c.). Seizures were categorized in three stages involving myoclonic jerks, followed by forelimb clonus, and finally tonic hindlimb extension. Mice not exhibiting a category of seizing behavior were scored as 30 min. For that value. Mice deficient for KCNQ2 DGGCs exhibited significantly greater seizure susceptibility. (b) Bars summarize the PTZ-induced seizure latency for Cre-POMC+ male mice of either WT (KCNQ2+/+) and either one allele (KCNQ2+/−), or both alleles deleted (KCNQ−/−) in the DG. KCNQ2-deficient mice exhibited significantly reduced latency to clonic and tonic-clonic seizure progression. (c) Shown is the Kaplan-Meier fractional curve of percentage of mice without forelimb clonus over time. (d) Shown is the Kaplan-Meier fractional curve of percentage of mice without tonic hindlimb extension (criterion for tonic-clonic seizure) over time. (e) Shown is the Kaplan-Meier fractional curve of percentage of mice surviving PTZ-induced seizures over time. Data in panels a and b represent mean ± SEM. n = 9-12 mice per group. *p < .05 versus KCNQ2+/+
4 |. DISCUSSION
The therapeutic prevention of epileptogenesis has been extensively investigated in the attempt to diminish or eliminate the deleterious, pathophysiological changes that contribute to excitotoxicity cascades and seizures (Löscher & Brandt, 2010). Anticonvulsant drugs that are effective in controlling epileptic seizures do not necessarily demonstrate efficacy as antiepileptogenic agents. In epileptogenesis, the precursor “latent period” that occurs prior to synchronous and recurrent seizure is no longer viewed as a time of “silence,” but of significance to the progression of epilepsy disease (Scharfman, 2019). During this time, the hippocampus undergoes significant plasticity in expression and/or function of ion channels, growth factors, receptors and other signaling molecules, increased metabolic demand, as well as changes in synaptic circuitry and behavior (Bernard, 2012; Dudek & Staley, 2012; Löscher, 2002). Activity-dependent upregulation of inhibitory receptors in the hippocampus has been shown to facilitate greater anticonvulsant control of seizure susceptibility in pre-clinical models (Carver et al., 2014; Vezzani et al., 2000). Similarly, we hypothesize that compensatory M channel upregulation occurs after G protein-coupled receptor-induced stimulation in the pre-epileptic hippocampus. Surprisingly, we discovered that such M-channel plasticity occurs under multiple types of hippocampal hyperexcitability involving broad network excitation.
Genetic loss-of-function KCNQ mutations have been well-described in relationship to epilepsy (Cooper et al., 2000; Peters et al., 2005; Singh et al., 1998; Niday et al., 2017); however, the contribution of M channels to nongenetic epileptogenesis has remained unidentified until now. We have elucidated novel M-channel plasticity in the hippocampus due to hyperexcitability, which precedes epileptogenesis in adult mammalian brain. Across multiple models, including chemoconvulsant and TBI-induced hyperexcitability, we found a conserved regulatory response that results in increased regional KCNQ2 transcriptional expression, protein expression, and IM amplitudes (Table 1). The functional significance of the observed increase in KCNQ2 expression is increased neuronal inhibition, which can be further augmented with drugs that potentiate IM. Using in situ hybridization, we confirmed that KCNQ2 transcription was increased after hyperexcitability challenge to the brain; however, KCNQ3 transcription was not altered. This is consistent with our previous finding of lack of KCNQ3 transcription in the cortex after TBI, despite increases to KCNQ2 (Vigil et al., 2019). As KCNQ3 is abundantly co-assembled with KCNQ2 in neurons, any specified role of KCNQ3 plasticity in neuromodulation of hippocampus has yet to be revealed (Soh et al., 2014). KCNQ2-deficient DGGCs, with a higher fraction of KCNQ3 homomers, demonstrate hyperexcitability (Carver & Shapiro, 2019), which confers greater seizure susceptibility in vivo (Figure 8). We have yet to explore expressional changes of KCNQ3 after severe epileptogenic hyperexcitability; for example, by the pilocarpine-induced SE model. There may be differential KCNQ3 expression profiles according to varying degrees of network hyperexcitability, as we observed with KCNQ2 in DGGCs (Figure 4). Despite this, our results suggest that transcription of KCNQ2 is significantly more sensitive to neuromodulation occurring in, and through, the hippocampus. The unilateral increase in KCNQ2 but not KCNQ3 suggests that KCNQ2 homomers may be more prevalently expressed in neurons after hyperexcitability. If KCNQ2 homomers were displacing the KCNQ2/3 heteromers after stimulation, one would expect IM to decrease due to the differential apparent affinity of PIP2 (Li, Gamper, Hilgemann, & Shapiro, 2005; Zhang et al., 2003). However, we measured a significant increase in IM amplitude from CA1 pyramidal neurons in which KCNQ2 transcription had increased after hyperexcitability. Therefore, it is possible that an additive number of channels in the cell is contributing to greater IM, and excitability changes in the network are being regulated by the increase in KCNQ2-containing channels, which may also involve reconfiguration of the axon initial segment itself (Lezmy etal., 2017).
TABLE 1.
Summary of hyperexcitability-induced changes in KCNQ2 transcription in the mouse hippocampus, as detected by KCNQ2 mRNA-EGFP reporter mice
| Hippocampal subregion | PILO (230 mg/kg) | PILO (300 mg/kg) | PTZ (60 mg/kg) | CCI-TBI | Composite response to hyperexcitability |
|---|---|---|---|---|---|
| CA1 pyramidal neurons | Increase | Decrease (cytotoxicity) | Increase | Increase (ipsilateral) | Increase |
| CA3 pyramidal neurons | Increase | Decrease (cytotoxicity) | Increase | Increase (ipsilateral) | Increase |
| DGGCs | No significant change | Increase | No significant change | Increase (ipsilateral) | Context-dependent |
The KCNQ2 mRNA-EGFP transgenic mouse enabled semi-quantitative determination of regional and cell-specific changes in KCNQ2 mRNA expression. Our findings demonstrate that in mice, KCNQ2 mRNA and protein are increased in the hippocampus in a neuron-specific manner. Furthermore, KCNQ2 expression is differentially altered based on the severity and extent of muscarinic receptor-induced hyperexcitability. CA1 and CA3 pyramidal cells are highly susceptible to cell death after temporal lobe epileptic seizures, whereas DGGCs and CA2 neurons are comparatively protected by factors yet to be uncovered (de Lanerolle et al., 2003; Gröticke et al., 2007; Henshall & Meldrum, 2012; Steve et al., 2014). In this study, DGGCs retained near-basal expression of KCNQ2 during moderate pilocarpine-induced excitability; however, KCNQ2 expression was significantly increased in response to severe SE activity (Figure 4). The differential effect of seizure severity on KCNQ2 expression in the DGGCs may reflect the degree to which the filtering function of the DG is compromised by synchronous hyperexcitability of the neurons (Hester and Danzer, 2013). Despite no detectable change in KCNQ2 transcription in DGGCs as a result of moderate hyperexcitability, we recorded significant changes to DGGC action potential discharge properties. This was in conjunction with observed elevation of cFos activity in the DGGCs. After pilocarpine-induced hyperexcitability, the overall number of evoked action potentials increased, as well as demonstrating an increased initial spike frequency. However, these neurons still exhibited spike-frequency adaptation, denoting that inhibitory neuromodulation may be conserved or further augmented. Enhancement of IM resulting in increase in the afterhypolarization area thus promotes greater excitability from relief of accumulated inactivation of Na+ channels, previously explored by us and others (Carver & Shapiro, 2019; Vervaeke, Gun, Agdestein, Hu, & Storm, 2006). Indeed, gain-of-function mutations in KCNQ2 or KCNQ3 subunits that result in epileptic encephalopathy have been linked to neuronal network hyperexcitability through distinct mechanisms of channel dysfunction (Miceli et al., 2015).
We have shown that the transcriptional regulation of the KCNQ2 gene is correlated with increases in IM amplitude in CA1 pyramidal neurons. This is in agreement with a previous investigation linking expressional changes in mRNA and M-channel function (Mucha et al., 2010). In DGGCs, however, the lack of mRNA upregulation and lack of inhibitory M-current compensation after initial insult could partially explain why DGGCs exhibit increase in excitation, as measured by electrophysiology properties in brain slice. This could be compounded with activity-dependent alterations in GABAA receptor plasticity and chloride homeostasis, also contributing to the discharge properties of DGGCs (Dengler et al., 2017; Kapur & Macdonald, 1997; Maguire & Mody, 2009; Sørensen et al., 2018). Therefore, the M channel-dependent inhibition of network excitation may be significant to the pathophysiology and progression of temporal lobe epilepsy.
In addition to the impactful pre-ictal contributions by CA1 and DG principal cells (Fujita, Toyoda, Thamattoor, & Buckmaster, 2014), mossy cells and GABAergic interneurons also significantly affect hippocampal excitability (Bui et al., 2018; Liu, Yu, Liu, He, & Peng, 2014). The tri-synaptic principal neurons demonstrated significant KCNQ2 expressional changes, however, their response to our model of hyperexcitability is complex due to their heterogeneity of expression and function. We did not observe significant cFos activation of PV+ interneurons after moderate pilocarpine challenge (Figure S1), but cFos was activated in the hilus. Deletion of KCNQ2/3 from PV+ interneurons increases hyperexcitability and seizure susceptibility, however deletion of KCNQ2/3 from somatostatin+ interneurons does not significantly alter their firing properties in younger mice (Soh et al., 2018). Others have shown that loss of KCNQ2 function induces excitatory activity of PV+ interneurons of the CA1 during an early stage of development in which GABA depolarizes neurons (Uchida et al., 2017). Therefore, the role of IM in control of excitability may change significantly across development as synaptic GABAergic signaling switches from excitatory to inhibitory. In the adult brain, a hyperexcitability challenge and direct stimulation of IM-containing interneurons may transiently increase local GABA release to glutamatergic cells, but may deplete or impair long-term signaling and fine-tuning of inhibition. In fully adult mice, there is a well-described role of IM control of interspike interval in O-LM somatostatin+ interneurons, and block of KCNQ2/3 increases interneuron excitability (Lawrence et al., 2006). In investigation of activity-dependent changes to KCNQ2 within hilar interneurons, we observed a significantly increased number of KCNQ2 immunoreactive cells during the window in which principal cell expression was altered (Figure S1). However, due to the wide heterogeneity of classes of interneurons across the hippocampus (Pelkey et al., 2017), more investigation is required to fully clarify this picture. Future studies will be useful to directly classify interneuron subtypes in conjunction with KCNQ2 expressional changes in response to moderate and severe hyperexcitability to the hippocampus.
Retigabine/ezogabine is a prototypic M-channel opener previously approved for the treatment of seizures (Large et al., 2012). Although effective as an anti-convulsant, the poor specificity of RTG among KCNQ2-5 channel subtypes has contributed to increased risks of urinary retention and central nervous system complications, since KCNQ4 and KCNQ5 are expressed widely in smooth muscle (Bruggemann, Mackie, Martin, Cribbs, & Byron, 2011; Evseev et al., 2013; Mackie et al., 2008; Soldovieri, Miceli, & Taglialatela, 2011), and thus was primarily restricted as adjunctive treatment of partial onset seizures (Daniluk, Cooper, Stender, & Kowalczyk, 2016). Evidence suggests that increase of IM by RTG or flupirtine may exert neuroprotective effects in reducing neuronal cell death in states of metabolic stress and depletion (Boscia, Annunziato, & Taglialatela, 2006; Gamper et al., 2006). However, this has yet to be confirmed with in vivo models in which cell death is prompted due to epileptiform activity spurring excitotoxicity and other metabolically-demanding cascades. Greater KCNQ2/3 channel selectivity has been achieved with experimental derivatives of RTG that may address some associated risks (Kumar et al., 2016), but require further investigation. Following an initial hyperexcitable event, we found that KCNQ2 upregulation increases the efficacy of M channels as a therapeutic target for protection from further seizure insults (Figure 7). In our rodent model of secondary hyperexcitability challenge with PTZ, RTG was highly efficacious in preventing seizures after an initial insult had already taken place (Figure 7b). In comparison, mice without anticonvulsant protection were more greatly affected by PTZ after the initial hyperexcitability, demonstrating tonic-clonic seizures and high incidence of mortality. Despite increase in KCNQ2 expression, the hyperexcitability provoked by 230 mg/kg pilocarpine facilitated greater seizure susceptibility to PTZ challenge 48 hours later. Any inhibitory protection resulting from the concomitant increase in KCNQ2 expression is overwhelmed by far stronger pro-epileptic mechanisms downstream of the pilocarpine-induced excitation. However, retigabine-mediated reduction in seizure susceptibility in this model can only be explained by an increase in the number of M channels. Therefore, greater anticonvulsant control over seizures with retigabine was afforded by the opening of additional M channels expressed in the brain, compared to control animals treated with retigabine, which did not achieve anyimprovement in anti-seizure efficacy. Therefore, hyperexcitability-dependent plasticity of M-channel expression could be leveraged to promote the inhibitory control of progressive seizure activity (Mazarati, Wu, Shin, Kwon, & Sankar, 2008). Based on our findings of time-dependent increase in hippocampal KCNQ2 function after hyperexcitability, the utility of M-channel openers in preventing seizures during periods of heightened susceptibility remains viable.
With our mouse model, we have determined that hyperexcitability-induced increase in KCNQ2 expression in the hippocampus is a transient event. Seven days after hyperexcitability, KCNQ2 mRNA expression subsides to baseline levels. This could either be a result of cessation of the cascades induced by the initial hyper-excitability event or, alternatively, an opposing and delayed signaling mechanism that inhibits further sustained expression of KCNQ2. This could explain the pro-excitatory changes in HCN, which might be more long-lasting (Arnold, McMurray, Gray, & Johnston, 2019; Powell et al., 2008; Shah, Anderson, Leung, Lin, & Johnston, 2004), and these alterations eventually “win-out” over upregulation of M-channel expression (George, Abbott, & Siegelbaum, 2009). Our previous discovery of the involvement of calcineurin/NFAT signaling in KCNQ2/3 transcriptional expression in neurons suggests the possibility that complementary negative regulation of NFAT or its phosphorylation state could result in suppression of KCNQ2 mRNA (Zhang & Shapiro, 2012). Other regulatory mechanisms of KCNQ2 and KCNQ3 activity-dependent transcriptional regulation have been described in sensory neurons including both the transcriptional activator Sp1, and the transcriptional repressor, REST (Mucha et al., 2010), but it is unclear if these mechanisms are conserved in central nervous system neurons. These pathways have yet to be thoroughly tested in an epileptogenesis mouse model, but may yield further insight to the regulatory role of M channels in hyperexcitability. Overall, intrinsic M-channel plasticity may confer compensatory control over pathophysiological excitation and potentially reveal successful strategies for preventing epilepsy.
Supplementary Material
Funding information
Center for Scientific Review, Grant/Award Number: T32 HL007446; Morrison Trust, Grant/Award Number: Morrison Trust Grant; National Institute of Neurological Disorders and Stroke, Grant/Award Numbers: R01 NS043394, R01 NS094461
ACKNOWLEDGMENTS
This research was supported by National Institute of Neurological Disorders and Stroke R01 NS094461 and R01 NS043394 (M.S.S.), Morrison Trust Grant (M.S.S.), and an institutional training fellowship T32 HL007446 (C.M.C.). The authors would like to thank Dr. Anastasios Tzingounis (University of Connecticut) for the kind donation of KCNQ2 flox mice.
Footnotes
CONFLICT OF INTEREST
The authors declare no competing financial interests.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
REFERENCES
- Algattas H, & Huang JH (2013). Traumatic brain injury pathophysiology and treatments: Early, intermediate, and late phases post-injury. International Journal of Molecular Sciences, 15, 309–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkadhi KA (2019). Cellular and molecular differences between area CA1 and the dentate gyrus of the hippocampus. Molecular Neurobiology, 56, 6566–6580. 10.1007/s12035-019-1541-2, epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Annegers JF, Hauser WA, Coan SP, & Rocca WA (1998). A population-based study of seizures after traumatic brain injuries. The New England Journal of Medicine, 338, 20–24. [DOI] [PubMed] [Google Scholar]
- Arnold EC, McMurray C, Gray R, & Johnston D (2019). Epilepsy-induced reduction in HCN channel expression contributes to an increased excitability in dorsal, but not ventral, hippocampal CA1 neurons. eNeuro, 6(2), e0036–e0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton ME, Klein BD, Wolf HH, & Whit HS (2001). Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Research, 47, 217–227. [DOI] [PubMed] [Google Scholar]
- Bernard C (2012, 2012). Alterations in synaptic function in epilepsy. In Noebels JL, Avoli M, Rogawski MA, Olsen RW, & Delgado-Escueta AV (Eds.), Jasper’s basic mechanisms of the epilepsies (4th ed.). Bethesda (MD): National Center for Biotechnology Information. [PubMed] [Google Scholar]
- Bernheim L, Beech DJ, & Hille B (1991). A diffusible second messenger mediates one of the pathways coupling receptors to calcium channels in rat sympathetic neurons. Neuron, 6, 859–867. [DOI] [PubMed] [Google Scholar]
- Bierbower SM, Choveau FS, Lechleiter JD, & Shapiro MS (2015). Augmentation of M-type (KCNQ) potassium channels as a novel strategy to reduce stroke-induced brain injury. The Journal of Neuroscience, 35(5), 2101–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, & Steinlein OK (1998). A potassium channel mutation in neonatal human epilepsy. Science, 279, 403–406. [DOI] [PubMed] [Google Scholar]
- Blanco MM, Dos SJ Jr., Perez-Mendes P, Kohek SR, Cavarsan CF, Hummel M, … Mello LE (2009). Assessment of seizure susceptibility in pilocarpine epileptic and nonepileptic Wistar rats and of seizure reinduction with pentylenetetrazole and electroshock models. Epilepsia, 50(4), 824–831. [DOI] [PubMed] [Google Scholar]
- Borges K, Gearing M, McDermott DL, Smith AB, Almonte AG, Wainer BH, & Dingledine R (2003). Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Experimental Neurology, 182, 21–34. [DOI] [PubMed] [Google Scholar]
- Boscia F, Annunziato L, & Taglialatela M (2006). Retigabine and flupirtine exert neuroprotective actions in organotypic hippocampal cultures. Neuropharmacology, 51, 283–294. [DOI] [PubMed] [Google Scholar]
- Brennan GP, Baram TZ, & Poolos NP (2016). Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels in epilepsy. Cold Spring Harbor Perspectives in Medicine, 6(3), a022384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, & Adams PR (1980). Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature, 283, 673–676. [DOI] [PubMed] [Google Scholar]
- Brown DA, & Passmore GM (2009). Neural KCNQ (Kv7) channels. British Journal of Pharmacology, 156, 1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruggemann LI, Mackie AR, Martin JL, Cribbs LL, & Byron KL (2011). Diclofenac distinguishes among homomeric and heteromeric potassium channels composed of KCNQ4 and KCNQ5 subunits. Molecular Pharmacology, 79(1), 10–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui AD, Nguyen TM, Limouse C, Kim HK, Szabo GG, Felong S, … Soltesz I (2018). Dentate gyrus mossy cells control spontaneous convulsive seizures and spatial memory. Science, 359, 787–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver CM, & Shapiro MS (2019). Gq-coupled muscarinic receptor enhancement of KCNQ2/3 channels and activation of TRPC channels in multimodal control of excitability in dentate gyrus granule cells. The Journal of Neuroscience, 39, 1566–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver CM, Wu X, Gangisetty O, & Reddy DS (2014). Perimenstrual-like hormonal regulation of extrasynaptic δ-containing GABAA receptors mediating tonic inhibition and neurosteroid sensitivity. The Journal of Neuroscience, 34, 14181–14197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen J, Pedersen MG, Pedersen CB, Sidenius P, Olsen J, & Vestergaard M (2009). Long-term risk of epilepsy after traumatic brain injury in children and young adults: A population-based cohort study. Lancet, 373, 1105–1110. [DOI] [PubMed] [Google Scholar]
- Cooper EC, Aldape KD, Abosch A, Barbaro NM, Berger MS, Peacock WS, … Jan LY (2000). Colocalization and coassembly of two human brain M-type potassium channel subunits that are mutated in epilepsy. Proceedings of the National Academy of Sciences of the United States of America, 97, 4914–4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Nixon KM, Alvestad RM, Valenzuela CF, & Browning MD (2005). Differential expression of NMDA receptor subunits and splice variants among the CA1, CA3 and dentate gyrus of the adult rat. Molecular Brain Research, 135, 104–111. [DOI] [PubMed] [Google Scholar]
- Curia G, Longo D, Biagini G, Jones RSF, & Avoli M (2008). The pilocarpine model of temporal lobe epilepsy. Journal of Neuroscience Methods, 172, 143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniluk J, Cooper JA, Stender M, & Kowalczyk A (2016). Survey of physicians’ understanding of specific risks associated with retigabine. Drugs Real World Outcomes, 3, 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao AT, Zagaar MA, Levine AT, & Alkadhi KA (2016). Comparison of the effect of exercise on late-phase LTP of the dentate gyrus and CA1 of Alzheimer’s disease model. Molecular Neurobiology, 53, 6859–6868. [DOI] [PubMed] [Google Scholar]
- de Lanerolle NC, Kim JH, Williamson A, Spencer SS, Zaveri HP, Eid T, & Spencer DD (2003). A retrospective analysis of hippocampal pathology in human temporal lobe epilepsy: Evidence for distinctive patient subcategories. Epilepsia, 44, 677–687. [DOI] [PubMed] [Google Scholar]
- Dengler CG, Yue C, Takano H, & Coulter DA (2017). Massively augmented hippocampal dentate granule cell activation accompanies epilepsy development. Scientific Reports, 7, 42090. 10.1038/srep42090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek FE, & Staley KJ (2012, 2012.). The time course and circuit mechanisms of acquired epileptogenesis. In Noebels JL, Avoli M, Rogawski MA, Olsen RW, & Delgado-Escueta AV (Eds.), Jasper’s basic mechanisms of the epilepsies (4th ed.). Bethesda (MD): National Center for Biotechnology Information (US). [PubMed] [Google Scholar]
- Englander J, Bushnik T, Duong TT, Cifu DX, Zafonte R, Wright J, … Bergman W (2003). Analyzing risk factors for late posttraumatic seizures: A prospective, multicenter investigation. Archives of Physical Medicine and Rehabilitation, 84, 365–373. [DOI] [PubMed] [Google Scholar]
- Evseev AI, Semenov I, Archer CR, Medina JL, Dube PH, Shapiro MS, & Brenner R (2013). Functional effects of KCNQ K+ channels in airway smooth muscle. Frontiers in Physiology, 4, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro TN, Golden GT, Smith GG, St Jean P, Schork NJ, Mulholland N, … Berrettini WH (1999). Mapping loci for pentylenetetrazol-induced seizure susceptibility in mice. The Journal of Neuroscience, 19, 6733–6739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita S, Toyoda I, Thamattoor AK, & Buckmaster PS (2014). Preictal activity of subicular, CA1, and dentate gyrus principal neurons in the dorsal hippocampus before spontaneous seizures in a rat model of temporal lobe epilepsy. The Journal of Neuroscience, 34(50), 16671–16687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, & Shapiro MS (2015). KCNQ channels. In Zheng J, & Trudeau MC (Eds.), Handbook of ion channels (1st ed.). Boca Raton, FL: CRC, 275–306. [Google Scholar]
- Gamper N, Zaika O, Li Y, Martin P, Hernandez CC, Perez MR, … Shapiro MS (2006). Oxidative modification of M-type (K+) channels as a mechanism of cytoprotective neuronal silencing. The EMBO Journal, 25(20), 4996–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George MS, Abbott LF, & Siegelbaum SA (2009). HCN hyperpolarization-activated cation channels inhibit EPSPs by interactions with M-type K+ channels. Nature Neuroscience, 12(5), 577–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golarai G, Greenwood AC, Feeney DM, & Connor JA (2001). Physiological and structural evidence for hippocampal involvement in persistent seizure susceptibility after traumatic brain injury. The Journal of Neuroscience, 21, 8523–8537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golowasch J, Thomas G, Taylor AL, Patel A, Pineda A, Khalil C, Nadim F (2009). Membrane capacitance measurements revisited: dependence of capacitance value on measurement method in nonisopotential neurons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Zhen C, Doughty ML, Losos K, Didkovsky N, Schambra UB, … Heintz N (2003). A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature, 425, 917–925. [DOI] [PubMed] [Google Scholar]
- Grabenstatter HL, Russek SJ, & Brooks-Kayal AR (2012). Molecular pathways controlling inhibitory receptor expression. Epilepsia, 53 Suppl 9, 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene DL, & Hoshi N (2017). Modulation of Kv7 channels and excitability inthe brain. Cellular and Molecular Life Sciences, 74, 495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene DL, Kosenko A, & Hoshi N (2018). Attenuating M-current suppression in vivo by a mutant Kcnq2 gene knock-in reduces seizure burden and prevents status-epilepticus-induced neuronal death and epileptogenesis. Epilepsia, 59, 1908–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gröticke I, Hoffman K, & Löscher W (2007). Behavioral alterations in the pilocarpine model of temporal lobe epilepsy in mice. Experimental Neurology, 207, 329–349. [DOI] [PubMed] [Google Scholar]
- Gunthorpe MJ, Large CH, & Sankar R (2012). The mechanism of action of retigabine (ezogabine), a first-in-class K+ channel opener for the treatment of epilepsy. Epilepsia, 53(3), 412–424. [DOI] [PubMed] [Google Scholar]
- Hadley JK, Noda M, Selyanko AA, Wood IC, Abogadie FC, & Brown DA (2000). Differential tetraethylammonium sensitivity of KCNQ1-4 potassium channels. British Journal of Pharmacology, 129, 413–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey BD, & Sloviter RS (2005). Hippocampal granule cell activity and c-Fos expression during spontaneous seizures in awake, chronically epileptic, pilocarpine-treated rats: Implications for hippocampal epileptogenesis. The Journal of Comparative Neurology, 488, 442–463. [DOI] [PubMed] [Google Scholar]
- Henshall DC, & Meldrum BS (2012). Cell death and survival mechanisms after single and repeated brief seizures. In Noebels JL, Avoli M, Rogawski MA, Olsen RW, & Delgado-Escueta AV (Eds.), Jasper’s basic mechanisms of the epilepsies (4th ed.). Bethesda (MD): National Center for Biotechnology Information (US). [PubMed] [Google Scholar]
- Hester MS, & Danzer SC (2013). Accumulation of abnormal adult-generated hippocampal granule cells predicts seizure frequency and severity. Journal of Neuroscience, 33, 8926–8936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JC, Zhang Y, Takagi N, Gurd JW, Wallace MC, Zhang L, & Eubanks JH (1998). Decreased expression and functionality of NMDA receptor complexes persist in the CA1, but not in the dentate gyrus after transient cerebral ischemia. Journal of Cerebral Blood Flow and Metabolism, 18, 768–775. [DOI] [PubMed] [Google Scholar]
- Huang RQ, Bell-Horner CL, Dibas MI, Covey DF, Drewe JA, & Dillon GH (2001). Pentylenetetrazole-induced inhibition of recombinant gamma-aminobutyric acid type a (GABA(A)) receptors: Mechanism and site of action. The Journal of Pharmacology and Experimental Therapeutics, 298(3), 986–995. [PubMed] [Google Scholar]
- Hunt RF, Boychuk JA, & Smith BN (2013). Neural circuit mechanisms of post-traumatic epilepsy. Frontiers in Cellular Neuroscience, 7, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt RF, Scheff SW, & Smith BN (2009). Posttraumatic epilepsy after controlled cortical impact injury in mice. Experimental Neurology, 215, 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Warner LN, Pitsch J, Becker AJ, & Poolos NP (2011). Rapid loss of dendritic HCN channel expression in hippocampal pyramidal neurons following status epilepticus. The Journal of Neuroscience, 31 (40), 14291–14295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur J, & Macdonald RL (1997). Rapid seizure-induced reduction of benzodiazepine and Zn2+ sensitivity of hippocampal dentate granule cell GABAA receptors. The Journal of Neuroscience, 17, 7532–7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinger F, Gould G, Boehm S, & Shapiro MS (2011). Distribution of M-channel subunits KCNQ2 and KCNQ3 K+ channels in rat hippocampus. NeuroImage, 58, 761–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook-Magnuson E, Armstrong C, Bui A, Lew S, Oijala M, & Soltesz I (2015). In vivo evaluation of the dentate gate theory in epilepsy. The Journal of Physiology, 593, 2379–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M, Reed N, Liu R, Aizenman E, Wipf P, & Tzounopoulos T (2016). Synthesis and evaluation of potent KCNQ2/3-specific channel activators. Molecular Pharmacology, 89, 667–677. [DOI] [PubMed] [Google Scholar]
- Large CH, Sokal DM, Nehlig A, Gunthorpe MJ, Sankar R, Crean CS, … White HS (2012). The spectrum of anticonvulsant efficacy of retigabine (ezogabine) in animal models: Implications for clinical use. Epilepsia, 53, 425–436. [DOI] [PubMed] [Google Scholar]
- Lavenex P, & Banta Lavenex P (2013). Building hippocampal circuits to learn and remember: Insights into the development of human memory. Behavioural Brain Research, 254, 8–21. [DOI] [PubMed] [Google Scholar]
- Lawrence JJ, Saraga F, Churchill JF, Statland JM, Travis KE, Skinner FK, & McBain CJ (2006). Somatodendritic Kv7/KCNQ/M channels control interspike interval in hippocampal interneurons. The Journal of Neuroscience, 26,12325–12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lezmy J, Lipinsky M, Khrapunsky Y, Patrich E, Shalom L, Peretz A, … Attali B (2017). M-current inhibition rapidly induces a unique CK2-dependent plasticity of the axon initial segment. Proceedings of the National Academy of Sciences of the United States of America, 114 (47), E10234–E10243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gamper N, Hilgemann DW, & Shapiro MS (2005). Regulation of Kv7 (KCNQ) K+ channel open probability by phosphatidylinositol 4,5-bisphosphate. The Journal of Neuroscience, 25, 9825–9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidster K, Jefferys JG, Blumcke I, Crunelli V, Flecknell P, Frenguelli BG, … Prescott MJ (2016). Opportunities for improving animal welfare in rodent models of epilepsy and seizures. Journal of Neuroscience Methods, 260, 2–25. [DOI] [PubMed] [Google Scholar]
- Liu YQ, Yu F, Liu WH, He XH, & Peng BW (2014). Dysfunction of hippocampal interneurons in epilepsy. Neuroscience Bulletin, 30(6), 985–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W (2002). Animal models of epilepsy for the development of anti-epileptogenic and disease-modifying drugs. A comparison of the pharmacology of kindling and post-status epilepticus models of temporal lobe epilepsy. Epilepsy Research, 50, 105–123. [DOI] [PubMed] [Google Scholar]
- Löscher W, & Brandt C (2010). Prevention or modification of epileptogenesis after brain insults: Experimental approaches and translational research. Pharmacological Reviews, 62(4), 668–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein DH (2009). Epilepsy after head injury: An overview. Epilepsia, 50(Suppl2), 4–9. [DOI] [PubMed] [Google Scholar]
- Mackie AR, Brueggemann LI, Henderson KK, Shiels AJ, Cribbs LL, Scrogin KE, & Byron KL (2008). Vascular KCNQ potassium channels as novel targets for the control of mesenteric artery constriction by vasopressin, based on studies in single cells, pressurized arteries, and in vivo measurement of mesenteric vascular resistance. The Journal of Pharmacology and Experimental Therapeutics, 325(2), 475–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire J, & Mody I (2009). Steroid hormone fluctuations and GABA(A) R plasticity. Psychoneuroendocrinology, 34 Suppl 1, S84–S90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazarati A, Wu J, Shin D, Kwon YS, & Sankar R (2008). Antiepileptogenic and antiictogenic effects of retigabine under conditions of rapid kindling: An ontogenic study. Epilepsia, 49, 1777–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzuferi M, Kumar G, Rospo C, & Kaminski RM (2012). Rapid epileptogenesis in the mouse pilocarpine model: Video-EEG, pharmacokinetic and histopathological characterization. Experimental Neurology, 238, 156–167. [DOI] [PubMed] [Google Scholar]
- McHugh TJ, Jones MW, Quinn JJ, Balthasar N, Coppari R, Elmquist JK, … Tonegawa S (2007). Dentate gyrus NMDA receptors mediate rapid pattern separation in the hippocampal network. Science, 317, 94–99. [DOI] [PubMed] [Google Scholar]
- Miceli F, Soldovieri MV, Ambrosino P, De Maria M, Migliore M, Migliore R, & Taglialatela M (2015). Early-onset epileptic encephalopathy caused by gain-of-function mutations in the voltage sensor of Kv7.3 and Kv7.3 potassium channel subunits. The Journal of Neuroscience, 35(9), 3782–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucha M, Ooi L, Linley JE, Mordaka P, Dalle C, Robertson B, … Wood IC (2010). Transcriptional control of KCNQ channel genes and regulation of neuronal excitability. The Journal of Neuroscience, 30, 13235–13245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niday Z, Hawkins VE, Soh H, Mulkey DK, & Tzingounis AV (2017). Epilepsy associated KCNQ2 channels regulate multiple intrinsic properties of layer 2/3 pyramidal neurons. Journal of Neuroscience, 37, 576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto-Gonzalez JL, & Jensen K (2013). BDNF depresses excitability of parvalbumin-positive interneurons through an M-like current in rat dentate gyrus. PLoS ONE, 8(6), e67318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson P, Ronne-Engström E, Flink R, Ungerstedt U, Carlson H, & Hillered L (1994). Epileptic seizure activity in the acute phase following cortical impact trauma in rat. Brain Research, 637, 227–232. [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Chittajallu R, Craig MT, Tricoire L, Wester JC, & McBain CJ (2017). Hippocampal GABAergic inhibitory interneurons. Physiological Reviews, 97(4), 1619–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z, & Houser CR (2005). Temporal patterns of fos expression in the dentate gyrus after spontaneous seizures in a mouse model of temporal lobe epilepsy. The Journal of Neuroscience, 25, 7210–7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters HC, Hu H, Pongs O, Storm JF, & Isbrandt D (2005). Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nature Neuroscience, 8, 51–60. [DOI] [PubMed] [Google Scholar]
- Poolos NP, & Johnston D (2012). Dendritic ion channelopathy in acquired epilepsy. Epilepsia, 9, 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell KL, Ng C, O’Brien TJ, Xu SH, Williams DA, Foote SJ, & Reid CA (2008). Decreases in HCN mRNA expression in the hippocampus after kindling and status epilepticus in adult rats. Epilepsia, 49(10), 1686–1695. [DOI] [PubMed] [Google Scholar]
- Racine RJ (1972). Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalography and Clinical Neurophysiology, 32, 281–294. [DOI] [PubMed] [Google Scholar]
- Reddy DS, & Kuruba R (2013). Experimental models of status epilepticus and neuronal injury for evaluation of therapeutic interventions. International Journal of Molecular Sciences, 14, 18284–18318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruifrok AC, & Johnston DA (2001). Quantification of histochemical staining by color deconvolution. Analytical and Quantitative Cytology and Histology, 23(4), 291–299. [PubMed] [Google Scholar]
- Sampath D, Valdez R, White AM, & Raol YH (2017). Anticonvulsant effect of flupirtine in an animal model of neonatal hypoxic-ischemic encephalopathy. Neuropharmacology, 123, 126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE (2019). The dentate gyrus and temporal lobe epilepsy: An “exciting” era. Epilepsy Currents, 19(4), 249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, & Myers CE (2016). Corruption of the dentate gyrus by “dominant” granule cells: Implications for dentate gyrus function in health and disease. Neurobiology of Learning and Memory, 129, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, & Pierce JP (2012). New insights into the role of hilar ectopic granule cells in the dentate gyrus based on quantitative anatomic analysis and three-dimensional reconstruction. Epilepsia, 53, 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schartz ND, Herr SA, Madsen L, Butts SJ, Torres C, Mendez LB, & Brewster AL (2016). Spatiotemporal profile of Map2 and microglial changes in the hippocampal CA1 region following pilocarpine-induced status epilepticus. Scientific Reports, 6, 24988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EF, Kus L, Gong S, & Heintz N (2013). BAC transgenic mice and the GENSAT database of engineered mouse strains. Cold Spring Harbor Perspectives in Medicine, 2013(3), 2013. 10.1101/pdb.top073692 [DOI] [PubMed] [Google Scholar]
- Schnell U, Dijk F, Sjollem KA, & Giepmans BNG (2012). Immunolabeling artifacts and the need for live-cell imaging. Nature Methods, 9, 152–158. [DOI] [PubMed] [Google Scholar]
- Schroeder BC, Hechenberger M, Weinreich F, Kubisch C, & Jentsch TJ (2000). KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. Journal of Biological Chemistry, 275, 24089–24095. [DOI] [PubMed] [Google Scholar]
- Seki T (2002). Expression patterns of immature neuronal markers PSA-NCAM, CRMP-4 and NeuroD in the hippocampus of young adult and aged rodents. Journal of Neuroscience Research, 70, 327–334. [DOI] [PubMed] [Google Scholar]
- Shah MM, Anderson AE, Leung V, Lin X, & Johnston D (2004). Seizure-induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron, 44, 495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MS, Roche JP, Kaftan EJ, Cruzblanca H, Mackie K, & Hille B (2000). Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K+channels that underlie the neuronal M current. Journal of Neuroscience, 20, 1710–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloviter RS, Zappone CA, Bumanglag AV, Norwood BA, & Kudrimoti H (2007). On the relevance of prolonged convulsive status epilepticus in animals to the etiology and neurobiology of human temporal lobe epilepsy. Epilepsia, 48(Suppl 8), 6–10. [DOI] [PubMed] [Google Scholar]
- Singh NA, Charlier C, Stauffer D, DuPont BR, Leach RJ, Melis R, … Leppert M (1998). A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nature Genetics, 18, 25–29. [DOI] [PubMed] [Google Scholar]
- Singh NA, Otto JF, Dahle EJ, Pappas C, Leslie JD, Vilaythong A, … Leppert MF (2008). Mouse models of human KCNQ2 and KCNQ3 mutations for benign familial neonatal convulsions shows seizures and neuronal plasticity without synaptic reorganization. The Journal of Physiology, 586, 3405–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh H, Pant R, LoTurco JJ, & Tzingounis AV (2014). Conditional deletions of epilepsy-associated KCNQ2 and KCNQ3 channels from cerebral cortex cause differential effects on neuronal excitability. The Journal of Neuroscience, 34, 5311–5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh H, Park S, Ryan K, Springer K, Maheshwari A, & Tzingounis AV (2018). Deletion of KCNQ2/3 potassium channels from PV+ interneurons leads to homeostatic potentiation of excitatory transmission. eLife, 7, e38617. 10.7554/eLife.38617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldovieri MV, Miceli F, & Taglialatela M (2011). Driving with no brakes: Molecular pathophysiology of potassium channels. Physiology, 26, 365–376. [DOI] [PubMed] [Google Scholar]
- Sørensen AT, Ledri M, Melis M, NikitdouLedri L, Andersson M, & Kokaia M (2018). Altered chloride homeostasis decreases the action potential threshold and increases hyperexcitability in hippocampal neurons. eNeuro, 4(6). 10.1523/ENEURO.0172-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squires RF, Saederup E, Crawley JN, Skolnick P, & Paul SM (1984). Convulsant potencies of tetrazoles are highly correlated with actions on GABA/benzodiazepine/picrotoxin receptor complexes in brain. Life Sciences, 35(14), 1439–1444. [DOI] [PubMed] [Google Scholar]
- Steve TA, Jirsch JD, & Gross DW (2014). Quantification of subfield pathology in hippocampal sclerosis: A systematic review and metaanalysis. Epilepsy Research, 108(8), 1279–1285. [DOI] [PubMed] [Google Scholar]
- Sutula T, Xiao-Xian H, Cavazos J, & Scott G (1988). Synaptic reorganization in the hippocampus induced by abnormal functional activity. Science, 239, 1147–1150. [DOI] [PubMed] [Google Scholar]
- Takano H, & Coulter DA (2012). Imaging of hippocampal circuits in epilepsy. In Noebels JL, Avoli M, Rogawski MA, Olsen RW, & Delgado-Escueta AV (Eds.), Jasper’s basic mechanisms of the epilepsies (4th ed.). Bethesda: National Center for Biotechnology Information. [PubMed] [Google Scholar]
- Trieu BH, Kramar EA, Cox CD, Jia Y, Wang W, Call CM, & Lynch G (2015). Pronounced differences in signal processing and synaptic plasticity between piriform-hippocampal network stages: A prominent role for adenosine. The Journal of Physiology, 593, 2889–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turski WA, Cavalheiro EA, Bortolotto ZA, Mello LM, Schwarz M, & Turski L (1984). Seizures produced by pilocarpine in mice: A behavioral, electroencephalographic and morphological analysis. Brain Research, 321, 237–253. [DOI] [PubMed] [Google Scholar]
- Tzingounis AV, Heidenreich M, Kharkovets T, Spitzmaul G, Jensen HS, Nicoll RA, & Jentsch TJ (2010). The KCNQ5 potassium channel mediates a component of the afterhyperpolarization current in mouse hippocampus. Proceedings of the National Academy of Sciences of the United States of America, 107, 10232–10237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzingounis AV, & Nicoll RA (2008). Contribution of KCNQ2 and KCNQ3 to the medium and slow afterhyperpolarization currents. Proceedings of the National Academy of Sciences of the United States of America, 105, 19974–19979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida T, Lossin C, Ihara Y, Deshimaru M, Yanagawa Y, Koyama S, & Hirose S (2017). Abnormal γ-aminobutyric acid neurotransmission in a Kcnq2 model of early onset epilepsy. Epilepsia, 58(8), 1430–1439. [DOI] [PubMed] [Google Scholar]
- Vervaeke K, Gun N, Agdestein C, Hu H, & Storm JF (2006). Kv7/KCNQ/M-channels in rat glutamatergic hippocampal axons and their role in regulation of excitability and transmitter release. The Journal of Physiology, 576, 235–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Moneta D, Mulé F, Ravizza T, Gobbi M, & French-Mullen J (2000). Plastic changes in neuropeptide Y receptor subtypes in experimental models of limbic seizures. Epilepsia, 41(Suppl 6), S115–S121. [DOI] [PubMed] [Google Scholar]
- Vigil FA, Bozdemir E, Bugay V, Espinoza L, Veraza RJ, Chun SH, … Shapiro MS (2019). Prevention of brain damage after traumatic brain injury by pharmacological enhancement of KCNQ (Kv7) K+ currents. Journal of Cerebral Blood Flow and Metabolism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, … Luo Y (2012). RNAscope®: A novel in situ RNA analysis platform for formalin-fixed paraffin-embedded tissues. The Journal of Molecular Diagnostics, 14, 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, … McKinnon D (1998). KCNQ2 and KCNQ3 potassium channel subunits: Molecular correlates of the M-channel. Science, 282, 1890–1893. [DOI] [PubMed] [Google Scholar]
- Wang Y, & Mattson MP (2014). L-type Ca2+ currents at CA1 synapses, but not CA3 or dentate granule neuron synapses, are increased in 3xTgAD mice in an age-dependent manner. Neurobiology of Aging, 35, 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmire LE, Ling L, Bugay V, Carver CM, Timilsina S, Chuang HH, … Brenner R (2017). Downregulation of KCNMB4 expression and changes in BK channel subtype in hippocampal granule neurons following seizure activity. PLoS ONE, 12(11), e0188064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickenden AD, Yu W, Zou A, Jegla T, & Wagoner PK (2000). Retigabine, a novel anti-convulsant, enhances activation of KCNQ2/Q3 potassium channels. Molecular Pharmaceutics, 58, 591–600. [DOI] [PubMed] [Google Scholar]
- Xiong ZQ, & Stringer JL (2000). Extracellular pH responses in CA1 and the dentate gyrus during electrical stimulation, seizure discharges, and spreading depression. Journal of Neurophysiology, 83, 3519–3524. [DOI] [PubMed] [Google Scholar]
- Yan XX, Cai Y, Zhang XM, Luo XG, Cai H, Rose GM, & Patrylo RR (2012). BACE1 elevation is associated with aberrant limbic axonal sprouting in epileptic CD1 mice. Experimental Neurology, 235(1), 228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Craciun LC, Mirshahi T, Rohacs T, Lopes CM, Jin T, & Logothetis DE (2003). PIP2 activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron, 37, 963–975. [DOI] [PubMed] [Google Scholar]
- Zhang H, Liu Y, Xu J, Zhang F, Liang H, Du X, & Zhang H (2013). Membrane microdomain determines the specificity of receptor-mediated modulation of Kv7/M potassium currents. Neuroscience, 254, 70–79. [DOI] [PubMed] [Google Scholar]
- Zhang J, & Shapiro MS (2012). Activity-dependent transcriptional regulation of M-type (Kv7) K+ channels by AKAP79/150-mediated NFAT actions. Neuron, 76, 1133–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.