

Abstract
Dysregulated translation is a common feature of cancer. Uncovering its governing factors and underlying mechanism are important for cancer therapy. Herein, we report that enhancer of zeste homolog 2 (EZH2), previously known as a transcription repressor and lysine methyltransferase, could directly interact with fibrillarin (FBL) to exert its role in translational regulation. We demonstrate that EZH2 enhances rRNA 2′-O methylation (2′-O-Me) via its direct interaction with FBL. Mechanistically, EZH2 strengthens the FBL-NOP56 interaction and facilitates the assembly of box C/D small nucleolar ribonucleoprotein (box C/D snoRNP). Strikingly, EZH2 deficiency impairs the translation process globally and reduces internal ribosome entry site (IRES)-dependent translation initiation in cancer cells. Our findings reveal a previously unrecognized role of EZH2 in cancer-related translational regulation.
Keywords: EZH2, FBL, box C/D snoRNP, 2′-O methylation, translation
Introduction
Ribosome biogenesis is a precisely coordinated process that involves multiple transcriptional and post-transcriptional steps to produce functional ribosomes1. In particular, post-transcriptional modifications of rRNA stabilize its structure and determine the functional fidelity of mature ribosome2, 3. As the most prevalent modification in rRNA, 2′-O methylation (2′-O-Me) occurs at over 100 residues in human rRNA and contributes to the translational capacity of ribosomes4. Despite this fact, the impact of 2′-O-Me on translational control of carcinogenesis needs to be further studied.
As an important complex mediating 2′-O-Me, the box C/D small nucleolar ribonucleoprotein (box C/D snoRNP) consists of a guiding box C/D small nucleolar RNA (snoRNA) and four core proteins, fibrillarin (FBL), NOP56, NOP58 and SNU135, 6. FBL is the only known methyltransferase that catalyzes 2′-O-Me of rRNA in a site-specific manner7–9. Additionally, FBL also participates in 18S rRNA maturation10 and acts as a protein methyltransferase to methylate histone H2A at glutamine 104 (H2AQ104me)11. Through different interacting partners or modifications, targeting FBL leads to different biological consequences in various cellular processes12–15. More importantly, compelling evidences demonstrate the dysregulation of FBL in multiple cancer types, which contributes to the overactivated translation in cancer16–18.
Enhancer of zeste homolog 2 (EZH2) is a lysine methyltransferase directing histone H3 lysine 27 tri-methylation (H3K27me3) and the catalytic subunit of Polycomb Repressive Complex 2 (PRC2)19, 20. Besides its roles in development and cell fate specification21–23, EZH2 is also well known as an oncogene in solid tumors like prostate cancer (PCa) and breast cancer24, 25. Although the oncogenic role of EZH2 mainly relies on its lysine methyltransferase activity and is PRC2-dependent, emerging findings support the notion that EZH2 could also regulate cancer development in a way independent of its methyltransferase activity26–28.
Results
EZH2 directly interacts with FBL in the nucleus
Our co-immunoprecipitation/mass spectrometry (co-IP/MS) data29 suggested that FBL is a potential interacting partner of EZH2. To validate this interaction, we first performed co-IP assay followed by immunoblot analysis in C4–2 PCa cell line. Immunoprecipitation of EZH2 pulled down FBL, as well as two known EZH2 binding partners EED and SUZ12 (Fig. 1a). However, reciprocal immunoprecipitation of FBL only pulled down EZH2 (Fig. 1b). Using a PCa patient-derived xenograft (PDX) model LuCaP 35CR, we further confirmed the EZH2-FBL interaction in tissues (Fig. 1c and 1d). To investigate whether this interaction is direct, purified proteins of GST-tagged EZH2/EED and Flag-tagged FBL were used for GST pull down assay. The GST-EZH2, but not the negative control GST-EED, could pull down Flag-FBL in vitro, suggesting that EZH2 and FBL directly interact with each other, independently of DNA or RNA (Fig. 1e and Extended Data Fig. 1a). The AlphaLISA assay using purified EZH2 and FBL proteins revealed the binding affinity of EZH2-FBL interaction as 841.5 nM (Fig. 1f and Extended Data Fig. 1b), comparable to the previously reported EZH2-EED binding affinity of 380 nM30. As a nucleolar marker, FBL proteins are enriched in the nucleoli. Intriguingly, immunostaining revealed an accumulation of endogenous EZH2 inside the nucleoli of C4–2 cells, co-localized with endogenous FBL (Extended Data Fig. 1c). This observation was supported by Proximity Ligation Assay (PLA) in PDX, in which the in situ EZH2-FBL interaction was visualized by PLA dots within the nuclei (Fig. 1g). To explore the importance of this interaction during PCa progression, PLA was next conducted in PCa tissue microarray (TMA) slides. As shown in Fig. 1h and Extended Data Fig. 1d, the PLA dots were rare in primary PCa types, but enriched in advanced ones like CRPC (Castration-Resistant PCa) and NEPC (Neuroendocrine PCa), indicating a positive correlation between EZH2-FBL interaction and PCa stages. Above all, these data indicated a direct EZH2-FBL interaction.
Figure 1. EZH2 directly interacts with FBL in PCa cells and tissues.
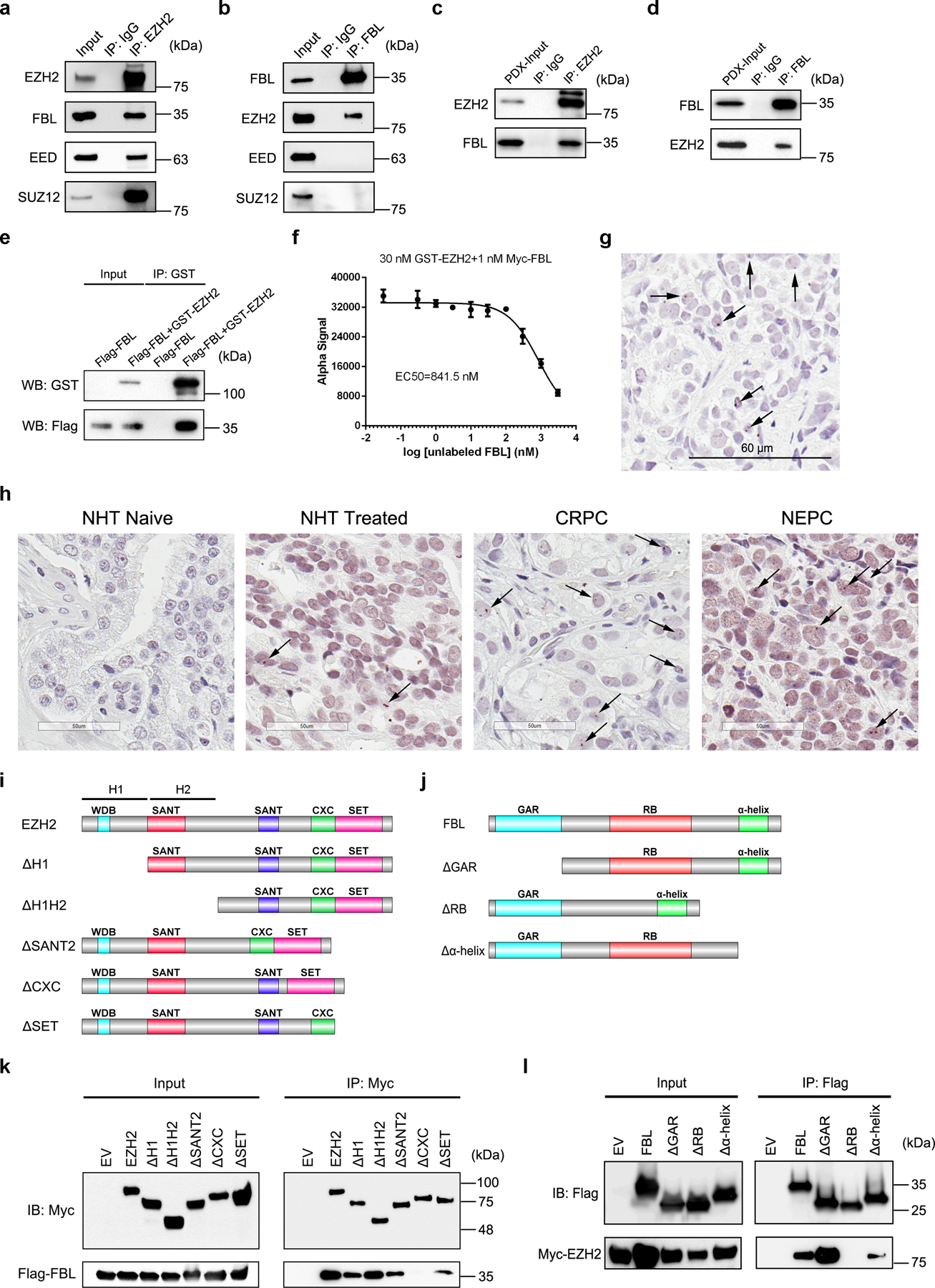
(a, b) C4–2 cells were lysed and subjected to co-IP assay using anti-EZH2 (a) or anti-FBL antibody (b), followed by western blot analysis with indicated antibodies. Rabbit IgG was set as a negative control.
(c, d) PCa PDX LuCaP 35CR tissues were lysed and subjected to co-IP assay using anti-EZH2 (c) or anti-FBL antibody (d), followed by western blot analysis with indicated antibodies. Rabbit IgG was set as a negative control.
(e) Purified proteins of GST-tagged EZH2 and Flag-tagged FBL were subjected to GST pull down assay, followed by western blot analysis with indicated antibodies.
(f) Inhibition of GST-tagged EZH2 and Myc-tagged FBL binding by unlabeled FBL in AlphaLISA displacement assay. Data represent Mean ± SD for n=3 biologically independent experiments.
(g) Image of Proximity Ligation Assay showing an interaction between EZH2 and FBL in PDX tumor. Positive staining corresponds to immunoperoxidase staining (brown dots marked by arrows). Scale bar=60 μm.
(h) Representative images of Proximity Ligation Assay showing an interaction between EZH2 and FBL in different types of PCa tissues. Positive staining corresponds to immunoperoxidase staining (brown dots marked by arrows). Scale bar=50 μm.
(i) Domain organization of EZH2 protein and its truncation mutants. The homology domain 1 (H1) contains WDB domain, while the homology domain 2 (H2) contains the first SANT domain.
(j) Domain organization of FBL protein and its truncation mutants.
(k) Co-IP of Flag-tagged FBL with full-length or truncation mutants of Myc-tagged EZH2, followed by western blot analysis with indicated antibodies.
(l) Co-IP of Myc-tagged EZH2 with full-length or truncation mutants of Flag-tagged FBL, followed by western blot analysis with indicated antibodies.
The assays in a-e, g-h and k-l have been performed three times with similar results. Statistical source data and unprocessed blots are provided in Source data Fig. 1.
EZH2 mainly contains two SANT protein-protein interaction domains, one cysteine-rich CXC domain and an enzymatic SET domain (Fig. 1i). FBL consists of a glycine and arginine-rich (GAR) domain, an RNA binding (RB) domain and a α-helix domain (Fig. 1j). To determine the domains being involved in EZH2-FBL interaction, serially truncated mutants of Myc-tagged EZH2 and Flag-tagged FBL constructs were co-transfected with their full-length partner plasmids into HEK293T cells for co-IP analyses. As revealed in Fig. 1k, except EZH2ΔCXC, all other truncated mutants and full-length of EZH2 pulled down full-length FBL. Meanwhile, only FBLΔRB was not able to pull down full-length EZH2 (Fig. 1l). These findings suggest that CXC domain of EZH2 and RB domain of FBL are essential for the EZH2-FBL interaction.
FBL-mediated H2AQ104me modification and 18S rRNA processing were unaffected upon EZH2 inhibition
We next wondered what role EZH2 may play by interacting with FBL. Since both EZH2 and FBL have histone methyltransferase activities, we first sought to determine whether the H2AQ104me marks were affected by EZH2. Immunofluorescence in C4–2 cells revealed that the nuclear staining of H2AQ104me was significantly attenuated by FBL siRNAs, not by EZH2 depletion (Extended Data Fig. 1e). Furthermore, we monitored the interaction of FBL with histone H2A by co-IP assay. Equal amount of histone H2A was pulled down by FBL in either control or EZH2-knockdown cells (Extended Data Fig. 1f), suggesting that EZH2 has no impact on FBL-H2A interaction.
As a mediator in rRNA processing, FBL participates in the release of a 414 nt fragment from the 5′-external transcribed spacer (5′-ETS) of pre-rRNA, which is a crucial step in 18S rRNA maturation10. Northern blot analysis revealed an accumulation of 30S rRNA intermediate in FBL-deficient cells which resulted in a markedly reduced production of 18SE and 18S rRNAs. In comparison, no evident defect was observed in EZH2-deficient cells (Extended Data Fig. 1g). Thus, EZH2 affects neither H2AQ104me modification nor 18S rRNA processing.
EZH2 alters rRNA methylation status through interaction with FBL
Given that FBL is the only known methyltransferase regulating rRNA 2′-O-Me, we next wondered whether EZH2-FBL interaction would affect 2′-O-Me status of rRNA. To test this hypothesis, total RNA was digested into nucleosides and subjected to Mass Spectrometry analysis to quantify the ratios of 2′-O methylated adenosine (Am) and guanine (Gm)31. As depicted in Fig. 2a, a global reduction of 2′-O-Me was observed in EZH2-deficient cells as compared to control. Moreover, overexpression of RNAi-resistant full-length EZH2 or EZH2ΔSET, but not EZH2ΔCXC, rescued the reduced 2′-O-Me induced by EZH2 repression (Fig. 2b), suggesting that EZH2 regulates 2′-O-Me independently of its lysine methyltransferase activities while its interaction with FBL is essential.
Figure 2. EZH2 modulates rRNA 2′-O-Me by interacting with FBL.
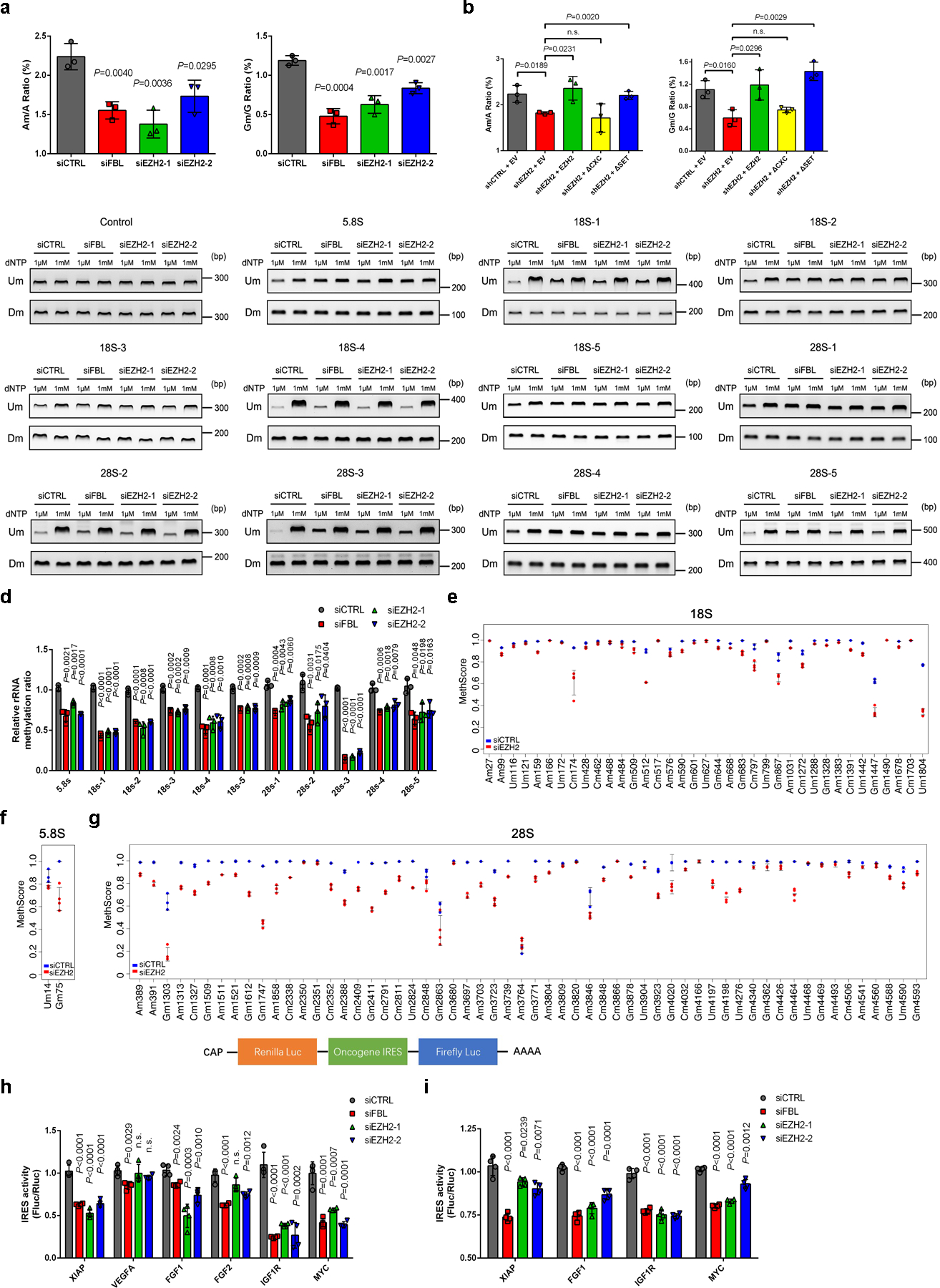
(a) The 2′-O-Me levels of adenosine (A) and guanosine (G) in total RNA were quantified by LC-MS/MS. Data represent Mean ± SD from n=3 biologically independent experiments. Am: 2′-O methylated adenosine; Gm: 2′-O methylated guanosine.
(b) Rescue assay showing that ectopic expression of full-length EZH2 or EZH2ΔSET, but not EZH2ΔCXC, restored the 2′-O-Me levels in EZH2-deficient cells, as measured by LC-MS/MS. Data represent Mean ± SD from n=3 biologically independent experiments.
(c) RTL-P assay to detect the 2′-O-Me level in rRNA. Total RNAs were subjected to reverse transcription (RT) with RT primer at low (1 μM) or high (1 mM) concentration of dNTP, respectively. The obtained cDNA was then amplified with primer pairs corresponding to upstream (Um) or downstream (Dm) regions of specific methylation site(s). This assay has been performed three times with similar results.
(d) Densitometric analysis of data from c were shown as signal intensity ratio of PCR products at low dNTP (1 μM) over high dNTP (1 mM) level. Methylation levels in control cells were set close to 1. Data represent Mean ± SD from n=3 biologically independent experiments.
(e-g) MethScore values for each 2′-O methylated nucleotide in 18S (e), 5.8S (f) and 28S (g) rRNAs from control and EZH2-deficient C4–2 cells. MethScore is equal to the ratio of 2′-O-Me at each modified nucleotide. Data represent Mean ± SD from n=3 biological replicates for control cells and n=4 for EZH2-deficient cells. The detailed results were shown in Supplementary Table 1.
(h) Each of the indicated reporter plasmids was transfected into C4–2 cells and IRES-dependent translation (Fluc/Rluc) from IRES elements of six cancer-relevant genes was measured through dual-luciferase assay. Data represent Mean ± SD from n=4 biologically independent experiments.
(i) RNA was transcribed in vitro from each of the indicated plasmids and transfected into C4–2 cells, followed by dual-luciferase assay. Data represent Mean ± SD from n=4 biologically independent experiments.
For all relevant panels, unless otherwise stated, statistical significance was determined by two-tailed Student’s t-test.
Statistical source data and unprocessed blots are provided in Source data Fig. 2.
Next, the RTL-P [Reverse Transcription at Low deoxyribonucleoside triphosphate (dNTP) concentrations followed by PCR] method was conducted to determine the effect of EZH2 on rRNA methylation32. A total of twelve primer pairs distributed throughout three rRNA types were designed, with eleven of which contain one or several sites reported to be methylated before, and one negative control contains no known methylation site9. Depletion of either FBL or EZH2 resulted in significantly reduced 2′-O-Me levels in all rRNA fragments detected except for the negative control (Fig. 2c and 2d). In addition, overexpression of FBL rescued the down-regulation of rRNA methylation induced by EZH2 depletion (Extended Data Fig. 2a and 2b), indicating that EZH2 modulates 2′-O-Me through FBL.
To systematically study the dynamics of 2′-O-Me at all known methylated nucleotides following EZH2 knockdown, a quantitative RiboMeth-seq method33 was further applied. As expected, depletion of EZH2 decreased the frequency of 2′-O-Me at most of the known sites (Fig. 2e–2g), with a pattern comparable to the previously reported result upon FBL knockdown9. To be specific, the 2′-O-Me level was significantly decreased in 87 sites (P < 0.05; 31 in 18S, 2 in 5.8S and 54 in 28S rRNAs) (Supplementary Table 1). Similar to FBL suppression, inhibition of EZH2 also led to down-regulation of 2′-O-Me in a site-specific manner (Extended Data Fig. 2c–2e).
One of the key consequences of rRNA 2′-O-Me is to modify translation initiation modalities from CAP-dependent towards IRES-dependent translation9, 16, 34. To investigate whether EZH2 could control initiation mode of translation, a dual-luciferase reporter with a CAP-driven renilla luciferase (Rluc) and a Poliovirus (PV) IRES-driven firefly luciferase (Fluc) was introduced to evaluate efficiency of CAP- and IRES-dependent translation. After EZH2 knockdown, the luciferase activity/mRNA ratios showed a reduction in both CAP- and IRES-dependent translations (Extended Data Fig. 2f and 2g), indicating an impaired protein synthesis. The Fluc/Rluc ratio that reflects the IRES activity was also significantly decreased, suggesting a stronger inhibition of IRES-dependent translation compared to CAP-dependent translation (Extended Data Fig. 2h). Importantly, overexpression of FBL in EZH2-deficient cells showed rescued translational efficiency and re-activation of IRES-dependent translation (Extended Data Fig. 3f–3h), confirming that FBL is critical for EZH2-mediated translational control. Numerous cancer-relevant genes contain IRES elements in mRNA, which are often utilized by tumor cells to promote survival35. We then cloned six IRES sequences from oncogenes in PCa and inserted into a bi-cistronic luciferase vector for analyses. As expected, four of the six oncogene IRESs showed significantly reduced activities in EZH2-deficient cells (Fig. 2h). Since the conventional plasmid DNA-based assessment of IRES activity may lead to misinterpretations when RNA splicing or a cryptic promoter is present in the proposed sequence36, we further transfected the in vitro transcribed RNA from each vector directly into C4–2 cells for assaying IRES activity. Consistently, all four EZH2-mediated IRESs identified in Fig. 2h also underwent decreased activities after EZH2 depletion through the RNA-based assessment (Fig. 2i). Above all, these data reveal the positive role of EZH2 in rRNA methylation, which subsequently enhances IRES-dependent translation.
EZH2 facilitates box C/D snoRNP assembly by strengthening FBL-NOP56 interaction
We next investigated how EZH2 regulates the function of FBL. Immunoblot analysis showed that EZH2 depletion did not affect FBL expression (Extended Data Fig. 3a). Similarly, FBL suppression did not alter EZH2 and H3K27me3 levels (Extended Data Fig. 3b). Since EZH2 can methylate non-histone proteins37, we wondered whether FBL is a substrate of EZH2. Immunoprecipitated FBL proteins from control or EZH2-deficient C4–2 cells were subjected to immunoblot analysis to visualize their lysine methylation levels. Although a portion of FBL lysines were tri-methylated, the methylation level was barely changed after EZH2 depletion (Extended Data Fig. 3c), excluding the possibility that EZH2 can methylate FBL.
We then focused on the assembly of box C/D snoRNP complex, whose formation is indispensable for FBL to exert 2′-O methyltransferase activity. To investigate whether EZH2 affects the binding of FBL with three other protein components of box C/D snoRNP, endogenous FBL and its interacting partners were coimmunoprecipitated from either control or EZH2-deficient C4–2 cells, and their abundance was compared by immunoblot analysis. As shown in Fig. 3a, EZH2 depletion did not affect the expression of any snoRNP component. Interestingly, when equal amounts of FBL proteins were pulled down from both control and EZH2-deficient cell lysates, much less NOP56 proteins were coimmunoprecipitated in EZH2-deficient cells, while the amounts of NOP58 and SNU13 binding to FBL were unaltered. These results were also observed in Ezh1/Ezh2 double-knockout mouse extraembryonic endoderm stem (XEN) cells (Extended Data Fig. 3d). Previous reports revealed that FBL interacts with NOP56 before being co-assembled into pre-mature snoRNP complex and this step is crucial for the box C/D snoRNP biogenesis5, 38. We then hypothesized that EZH2 may enhance FBL-NOP56 interaction. To test this hypothesis, the AlphaLISA displacement assay was conducted using purified FBL, NOP56 and EZH2 proteins. With the increased concentration of EZH2 protein, the alpha signal representing the affinity of FBL-NOP56 interaction was significantly enhanced till reached the hook point at peak, suggesting that EZH2 could facilitate the binding of FBL to NOP56 (Fig. 3b and Extended Data Fig. 3e). As a negative control, increased EED concentration did not affect FBL-NOP56 interaction affinity (Extended Data Fig. 3f), further validating that EZH2 regulates box C/D snoRNP functions independent of PRC2.
Figure 3. EZH2 bridges FBL-NOP56 interaction by binding to both proteins.
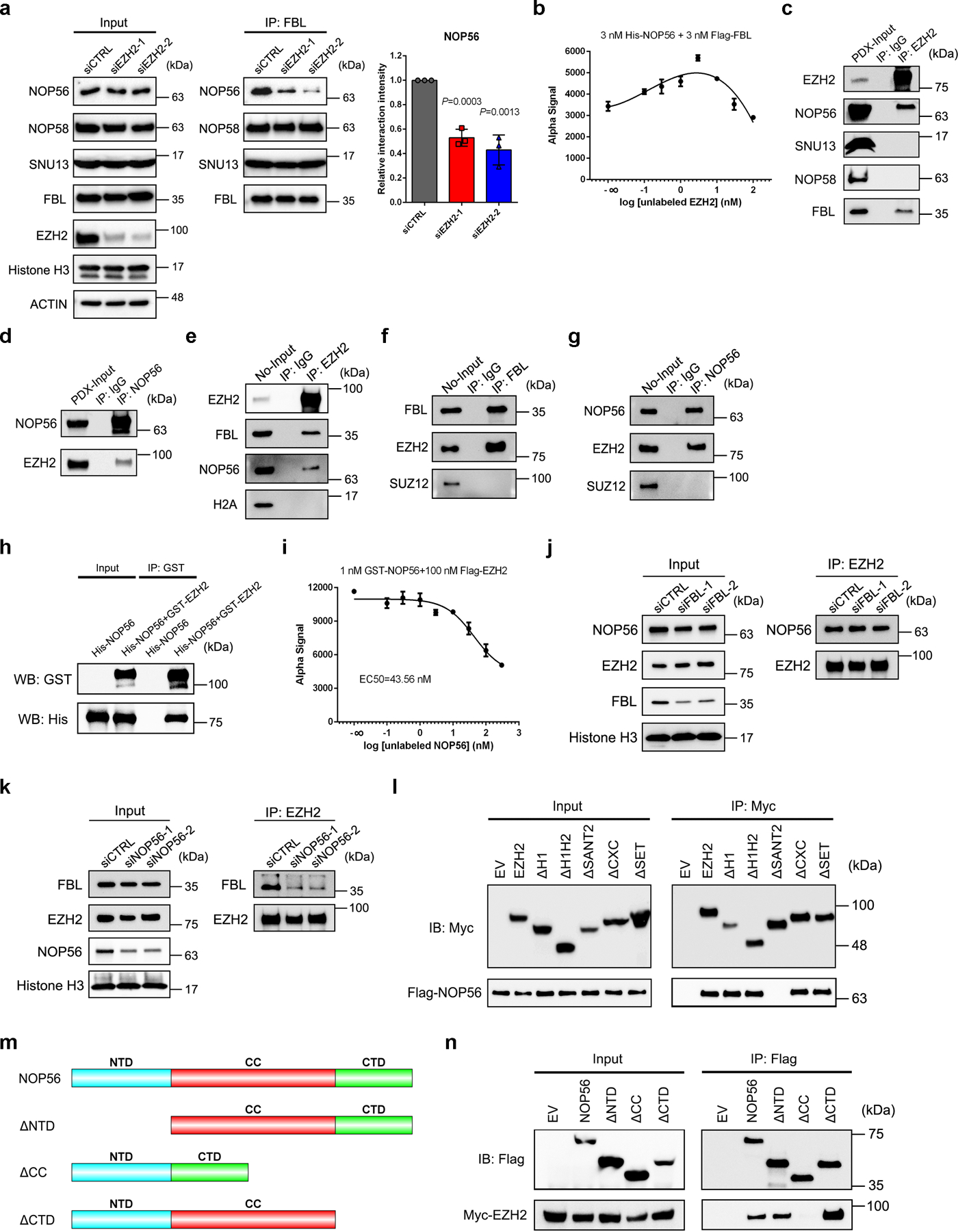
(a) Co-IP of box C/D snoRNP components NOP56, NOP58 and SNU13 with FBL in control and EZH2-deficient C4–2 cells, followed by western blot analysis with indicated antibodies. Graph represents the relative NOP56 protein level coimmunoprecipitated with FBL in each group. Data represent Mean ± SD (n=3 biologically independent measurements). Interaction intensity at control group was set as 1. Statistical significance was determined by two-tailed Student’s t-test.
(b) AlphaLISA displacement assay showing that FBL-NOP56 interaction is strengthened by EZH2. Data represent Mean ± SD for n=3 biologically independent experiments.
(c, d) PDX tissues were lysed and subjected to co-IP assay using anti-EZH2 (c) or anti-NOP56 (d) antibody, followed by western blot analysis with indicated antibodies. Rabbit IgG was set as a negative control.
(e-g) Purified nucleoli (No) from C4–2 cells were lysed and subjected to co-IP assay using anti-EZH2 (e), anti-FBL (f) or anti-NOP56 antibody (g), followed by western blot analysis with indicated antibodies. Rabbit IgG was set as a negative control.
(h) Purified proteins of GST-tagged EZH2 and His-tagged NOP56 were subjected to GST pull down assay, followed by western blot analysis with indicated antibodies.
(i) Inhibition of GST-tagged NOP56 and Flag-tagged EZH2 binding by unlabeled NOP56 in AlphaLISA displacement assay. Data represent Mean ± SD for n=3 biologically independent experiments.
(j) Co-IP of NOP56 with EZH2 in control and FBL-deficient C4–2 cells, followed by western blot analysis with indicated antibodies.
(k) Co-IP of FBL with EZH2 in control and NOP56-deficient C4–2 cells, followed by western blot analysis with indicated antibodies.
(l) Co-IP of Flag-tagged NOP56 with full-length or truncation mutants of Myc-tagged EZH2, followed by western blot analysis with indicated antibodies.
(m) Domain organization of NOP56 protein and its truncation mutants.
(n) Co-IP of Myc-tagged EZH2 with full-length or truncation mutants of Flag-tagged NOP56, followed by western blot analysis with indicated antibodies.
The assays in c-h, j-l and n have been performed three times with similar results. Statistical source data and unprocessed blots are provided in Source data Fig. 3.
Since potential EZH2-NOP56 interaction has been previously indicated in HEK293T cells28, we next hypothesized that EZH2 may reinforce the FBL-NOP56 interaction by binding to both proteins. In line with this hypothesis, co-IP assay using PDX tumors validated the interactions of EZH2 with NOP56 and FBL, but not NOP58 and SNU13 (Fig. 3c and 3d). In addition, co-IP assay using C4–2 nucleolar fractions further revealed these interactions at specific subcellular fractions (Extended Data Fig. 3g and Fig. 3e–3g). GST pull down assay with purified EZH2 and NOP56 proteins confirmed that EZH2-NOP56 interaction is also direct (Fig. 3h). As assessed by AlphaLISA assay, the binding affinity of EZH2-NOP56 interaction equals to 43.56 nM (Fig. 3i and Extended Data Fig. 3h). To expand this observation, co-IP assays were conducted in C4–2 cells upon FBL/NOP56 depletion. Interestingly, FBL depletion had no significant impact on EZH2-NOP56 interaction (Fig. 3j), while depleting NOP56 substantially decreased the amounts of FBL coimmunoprecipitated with EZH2 (Fig. 3k). These data suggest that EZH2 has a higher affinity for NOP56 than for FBL. Using our previously generated EZH2 truncation constructs (Fig. 1i), we observed that SANT2 domain of EZH2 is required for EZH2-NOP56 interaction (Fig. 3l). NOP56 protein possesses a N-terminal (NTD) domain, a coiled-coil (CC) domain and a C-terminal (CTD) domain (Fig. 3m). We confirmed that CC domain of NOP56 is responsible for NOP56-EZH2 interaction while its NTD domain is involved in NOP56-FBL interaction (Fig. 3n and Extended Data Fig. 3i).
We next sought to determine the existence of EZH2-FBL-NOP56 trimer by bis(sulfosuccinimidyl) (BS3) crosslinking assay. BS3 crosslinks interacting proteins together if there are nearby lysine residues from individual proteins39. Hence, the protein-protein interactions could be determined by band shift in SDS-PAGE. Without crosslinking, all three proteins in the mixed sample were separated as monomers by SDS-PAGE (Extended Data Fig. 4a, left lane of each panel). Interestingly, they shifted to an analogous location after BS3 treatment, indicating the formation of protein complexes (Extended Data Fig. 4a, right lane of each panel). Mass Spectrometry analysis further confirmed the presence of all three proteins in the shifted species (Extended Data Fig. 4b). To explore the effect of EZH2 on box C/D snoRNP assembly in vivo, the size-exclusion chromatography analyses were carried out using nuclear extracts from control and EZH2-deficient C4–2 cells. As predicted, all four snoRNP components were co-eluted with EZH2 at fractions 6–22 in control cells (Fig. 4a). Remarkably, EZH2 inhibition led to an altered pattern of FBL/NOP56/NOP58/SNU13 elution profile towards smaller size fractions (Fig. 4a and Extended Data Fig. 4c–4f), reflecting the impairment of snoRNP assembly. In parallel, different fractions from control group were subjected to co-IP assay. As shown in Extended Data Fig. 4g, EZH2 could only pull-down FBL and NOP56 in fractions containing all three proteins, confirming the existence of these interactions at the co-eluted fractions.
Figure 4. Formation of EZH2-FBL-NOP56 trimer facilitates box C/D snoRNP assembly.
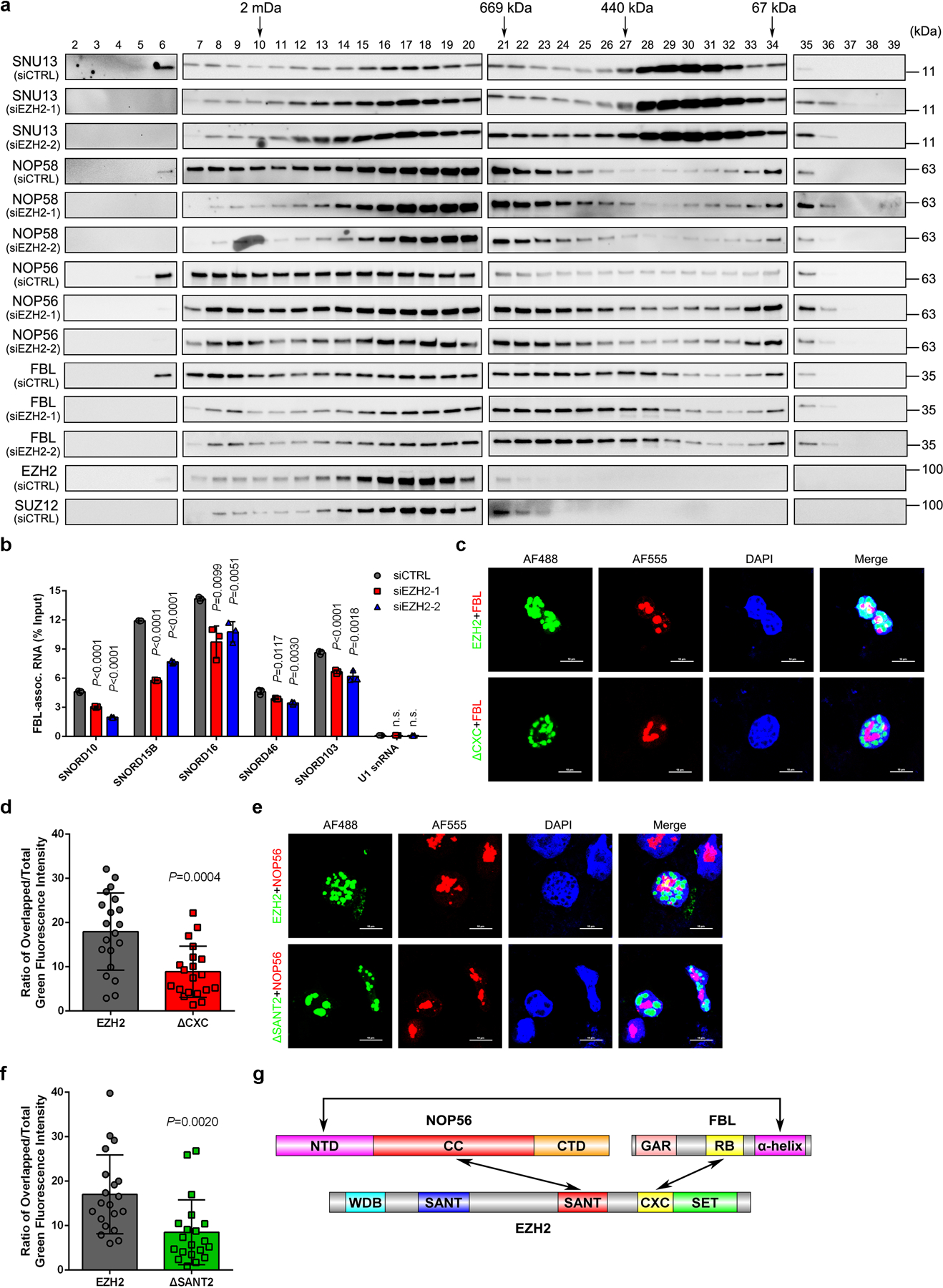
(a) Nuclear extracts from control and EZH2-deficient C4–2 cells were subjected to size-exclusion and the protein levels of FBL, NOP56, NOP58 and SNU13 were determined by western blot analysis in all samples. Protein distributions of EZH2 and SUZ12 in control cells were also detected as references. This assay has been performed three times with similar results.
(b) RIP-qPCR assay to monitor the binding of snoRNAs to FBL in control and EZH2-deficient C4–2 cells. Data represent Mean ± SD from n=3 biologically independent experiments. U1 snRNA, which is not a binding target of FBL, was used as a control.
(c, d) Representative fluorescence images of C4–2 cells expressing GFP-EZH2 or GFP-EZH2ΔCXC. The nucleoli were co-stained using anti-FBL antibody followed by an Alexa Fluor 555-conjugated secondary antibody and the nuclei were visualized by DAPI (Scale bar: 10 μm). Graph represents the nucleolar proportion of EZH2 estimated by the ratio of nucleolar GFP intensity to nuclear GFP intensity of individual cells (Mean ± SD, n=20 cells analyzed over 3 independent experiments).
(e, f) Representative fluorescence images of C4–2 cells expressing GFP-EZH2 or GFP-EZH2ΔSANT2. The nucleoli were co-stained using anti-NOP56 antibody followed by an Alexa Fluor 555-conjugated secondary antibody and the nuclei was visualized by DAPI (Scale bar: 10 μm). Graph represents the nucleolar proportion of EZH2 estimated by the ratio of nucleolar GFP intensity to nuclear GFP intensity of individual cells (Mean ± SD, n =20 cells analyzed over 3 independent experiments).
(g) Schematic diagram of EZH2-FBL-NOP56 trimer.
For all relevant panels, unless otherwise stated, statistical significance was determined by two-tailed Student’s t-test.
Statistical source data and unprocessed blots are provided in Source data Fig. 4.
We next investigated the impact of EZH2 on box C/D snoRNP assembly by detecting the change of FBL-snoRNA association upon EZH2 inhibition. To assess this, FBL RNA immunoprecipitation-qPCR (RIP-qPCR) assay was performed and the enrichment of five co-precipitating snoRNAs that have sequences sufficient in length to design qPCR primers was detected. As revealed in Fig. 4b, EZH2 knockdown led to a significant reduction in FBL’s ability to interact with all snoRNAs tested, suggesting that loss of EZH2 affects the box C/D snoRNP assembly and subsequently impedes the interaction of FBL with snoRNAs.
To determine whether interactions with two snoRNP members affect subnuclear localization of EZH2, C4–2 cells expressing GFP-EZH2 or GFP-EZH2ΔCXC were visualized by immunofluorescence. Staining of endogenous FBL was used to delineate the boundary of nucleolus. As shown in Fig. 4c, loss of binding capacity to FBL significantly reduced the nucleolar distribution of EZH2ΔCXC. The ratio of overlapped/total GFP fluorescence intensity, which reflects the nucleolar/nuclear percentage, was much lower in EZH2ΔCXC group compared to group of full-length EZH2 (Fig. 4d). Similarly, loss of NOP56 binding also impaired the nucleolar distribution of EZH2ΔSANT2 (Fig. 4e and 4f), in which case endogenous NOP56 was immunostained to visualize the nucleoli.
Taken together, these findings indicate the involvement of EZH2 during box C/D snoRNP assembly. Combined with the established evidence that α-helix domain of FBL is responsible for FBL-NOP56 interaction38, the domain interaction diagram of EZH2-FBL-NOP56 trimer was illustrated (Fig. 4g).
EZH2 modulates gene expression at both transcription and translation levels
In addition to the well-established role of EZH2 as a transcription regulator, our findings strongly indicate the relevance of EZH2 with translational control. We first investigated the relationship between EZH2 expression and nucleolar activity in cancer since EZH2 can enter nucleoli. Immunohistochemistry (IHC) assay was conducted in serially sectioned PCa TMA slides using anti-EZH2 antibodies to detect EZH2 expression and anti-FBL/anti-NOP56 antibodies to stain nucleoli. With elevated EZH2 expression, the nucleoli were significantly enlarged in PCa cells (Fig. 5a and 5b, Extended Data Fig. 5a and 5b). In addition, the nucleolar number per cell was also positively correlated with EZH2 expression in multiple PCa types (Fig. 5c and Extended Data Fig. 5c). Transmission electron microscope (TEM) was then conducted to visualize the nucleolar architecture. Consistently, markedly smaller nucleoli were observed in EZH2-deficient C4–2 cells as compared with control cells (Fig. 5d). To evaluate whether EZH2 expression affects the global protein synthesis, Puromycylation assay was performed to detect the puromycin incorporation into ribosome-bound nascent chain40. Similar to that of FBL9, EZH2 depletion reduced the de novo protein synthesis broadly (Fig. 5e), while EZH2 overexpression promoted global protein synthesis (Extended Data Fig. 5d).
Figure 5. EZH2 regulates the translation process in a positive manner.
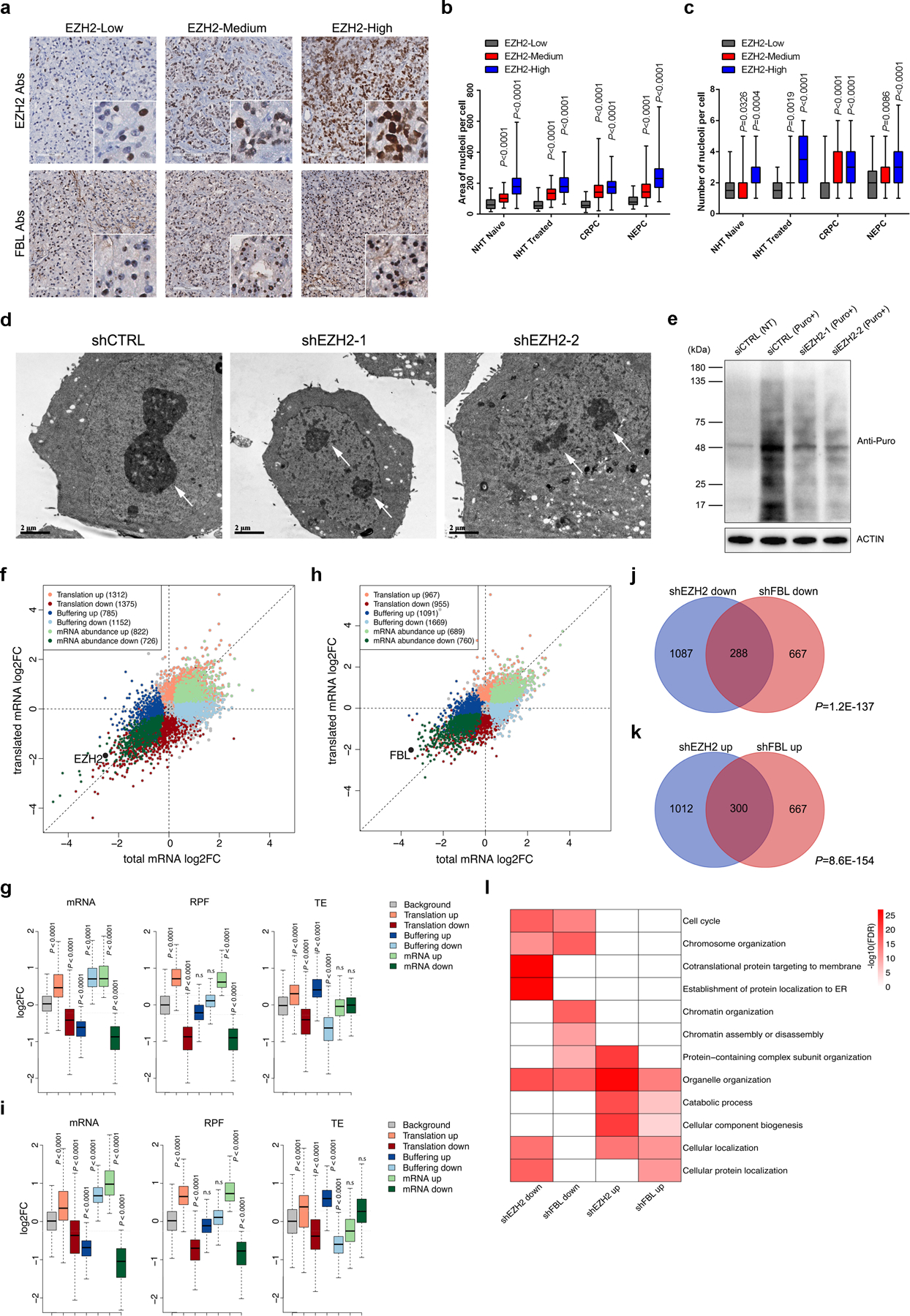
(a) Representative Immunohistochemistry staining images of PCa TMA slides using indicated antibodies (Abs).
(b, c) Graph showing the relative nucleolar area (b) and nucleolar number (c) per cell by counting 60 cells from three TMA cores representing each group. The ends of box are upper and lower quartiles and box spans the interquartile range. Median is marked by a vertical line inside the box and whiskers represent for the highest and lowest observations. NHT, neoadjuvant hormonal therapy; CRPC, castration-resistant PCa; NEPC, neuroendocrine PCa.
(d) Representative electron micrographs showing nucleolus in control or EZH2-deficient C4–2 cells (Scale bar: 2 μm). Arrows indicate nucleoli.
(e) Global protein synthesis in control and EZH2-deficient C4–2 cells were detected by Puromycylation assay followed by western blot. Expression of β-actin was used as reference.
(f-i) Scatter plot to show expression changes of mRNA levels and RPFs between control and EZH2-deficient C4–2 cells (f) or FBL-deficient C4–2 cells (h). Genes were colored according to their regulation mode defined by anota2seq. A threshold of absolute fold change (FC) > 1.2, P < 0.05 was used. Box plots of g and i showed the log2 fold change (Log2FC) of the mRNA, RPF and TE in f and h, respectively. The ends of the box are the upper and lower quartiles and the box spans the interquartile range. The median is marked by a vertical line inside the box and the whiskers represent for 1.5x inter-quartile range. P values were calculated by two-tailed Wilcoxon rank-sum test. A threshold of the absolute Log2FC >0.3, P <0.05 was used.
(j, k) Venn diagram to show overlap between genes from “translation” mode upon EZH2 or FBL deficiency. P values were calculated by one-tailed Fisher’s exact test.
(l) GO enrichment analysis of genes from “translation” mode upon EZH2 or FBL deficiency.
For all relevant panels, unless otherwise stated, statistical significance was determined by two-tailed Student’s t-test.
The assays in a and e have been performed three times with similar results. Statistical source data and unprocessed blots are provided in Source data Fig. 5.
To deeply understand the contribution of EZH2 in the translation process, we applied ribosome profiling (Ribo-seq) to sequence the ribosome-protected fragments (RPFs) on C4–2 cells undergoing EZH2 depletion. Meanwhile, the mRNA level in each group was measured by RNA-seq. The anota2seq algorithm was introduced to calculate the differences in translational efficiency (TE) by conjoint analyses of Ribo-seq and RNA-seq data41, 42. Anota2seq classifies genes into three groups: 1) “Translation”, referring to genes with dramatic change in RPFs but with little change in mRNA; 2) “Buffering”, referring to genes with changes in mRNA without corresponding changes in RPFs; 3) “mRNA abundance”, referring to genes with congruent changes in mRNA and RPFs. Both “translation” and “buffering” groups represent for genes with TE alteration while the “mRNA abundance” group contains genes that are solely regulated at transcriptional level.
After depleting EZH2, 2,687 genes were identified in “translation” group, 1,937 in “buffering” group, and 1,548 in “mRNA abundance” group (Fig. 5f and Supplementary Table 2). The expression changes of each group matched the desired setting, confirming the effectiveness of classification algorithm (Fig. 5g). To compare the role of EZH2 and FBL in translational regulation, Ribo-seq in FBL-deficient C4–2 cells was performed in parallel (Fig. 5h, 5i and Supplementary Table 2). In line with the predictions, genes in “translation” group of EZH2- and FBL-deficient cells were significantly overlapped (Fig. 5j and 5k), and co-enriched in multiple cellular pathways (Fig. 5l). With respect to “buffering” genes which also underwent TE alteration but were deeply affected at mRNA level, slightly significant overlap could still be observed despite about 3.5-fold smaller number of overlapping genes (Extended Data Fig. 5e and 5f), while these genes were hardly co-enriched into any pathway (Extended Data Fig. 5g). These data suggest that EZH2 and FBL share the function in translational control, but may vary in terms of transcriptional regulation.
Although the above evidences support the notion that EZH2 mainly regulates the translation process through an FBL-related mechanism, it is possible that EZH2 may bind to mRNA directly and affect its translation28. To evaluate the global impact of EZH2-mRNA interaction on translation, we used a publicly available EZH2 RIP-seq data from C4–2 cells28 to define EZH2-bound mRNAs for the gene set enrichment analysis (GSEA) and tested whether these genes were enriched with TE changes upon EZH2 depletion. As shown in Extended Data Fig. 5h, no significant enrichment could be observed, suggesting that mRNAs showing TE changes differ from EZH2-bound mRNAs. Only a minor portion of EZH2-bound mRNAs overlapped with the TE-altered groups, with the numbers smaller than expected by random chance (Extended Data Fig. 5i). In addition, the majority of TE-altered transcripts (e.g. IFT81) showed weaker EZH2 RIP-seq signals as compared with Input, while only a few EZH2-bound transcripts (e.g. TP53) showed alteration in translation (Extended Data Fig. 5j and 5k). Collectively, we demonstrated the dominant role of EZH2 in translational regulation, which is largely independent of its mRNA-binding capacities.
EZH2 promotes the IRES-dependent translation of XIAP
We have shown above that EZH2 could modulate the translation initiation modality towards IRES-dependent translation. To extend this observation to a global scale, we performed GSEA of TE alteration upon EZH2 knockdown using a recently published IRES gene set43. In line with our previous findings, a significant enrichment of IRES genes was observed, indicating a weakened IRES activity in EZH2-deficient cells (Fig. 6a and Extended Data Fig. 6a). Polysome profiling assay was further conducted to monitor cytoplasmic translation. The polysomal fractions correspond to mRNA bound to more than one ribosome and therefore contain mRNA engaging in active translation. As depicted in Fig. 6b and 6c, EZH2-deficient C4–2 cells exhibited a significantly decreased proportion of polysomes. Next, the mRNA distribution of four EZH2-mediated IRES-containing genes identified above was examined. Despite that FGF1 mRNA was undetectable in C4–2 cells, a significantly reduced enrichment of XIAP/MYC/IGF1R mRNA in polysomal fractions was found in EZH2-deficient cells (Fig. 6d–6f). We then focused on XIAP because it plays an oncogenic role in PCa44 and contains a well-characterized IRES element in mRNA45. XIAP was one of the top 30 IRES-containing genes with suppressed TE after EZH2 depletion (Extended Data Fig. 6a), and its mRNA did not bind to EZH2 (Fig. 6g). According to our Ribo-seq data, EZH2 or FBL depletion resulted in a down-regulation in TE of XIAP that counteracts its change in mRNA (Fig. 6g). Consistently, suppression of either EZH2 or FBL led to dramatic decrease of XIAP in various PCa cell lines at protein level (Fig. 6h and Extended Data Fig. 6b), but not at mRNA level (Fig. 6i and Extended Data Fig. 6c). To further confirm this observation, we treated C4–2 cells with different EZH2 inhibitors. Similar to EZH2 RNAi, EZH2 expression inhibitor DZNep effectively decreased XIAP at protein level, while EZH2 enzymatic inhibitor GSK126 or EPZ6438 had no impact on XIAP expression, suggesting that EZH2 regulates XIAP expression independently of its lysine methyltransferase activity (Fig. 6j and Extended Data Fig. 6d). We next treated control and EZH2-deficient cells with cycloheximide (CHX), but did not observe apparent difference in XIAP protein half-lives (Extended Data Fig. 6e), indicating that XIAP protein stability was not affected upon EZH2 suppression.
Figure 6. EZH2 promotes XIAP IRES-dependent translation.
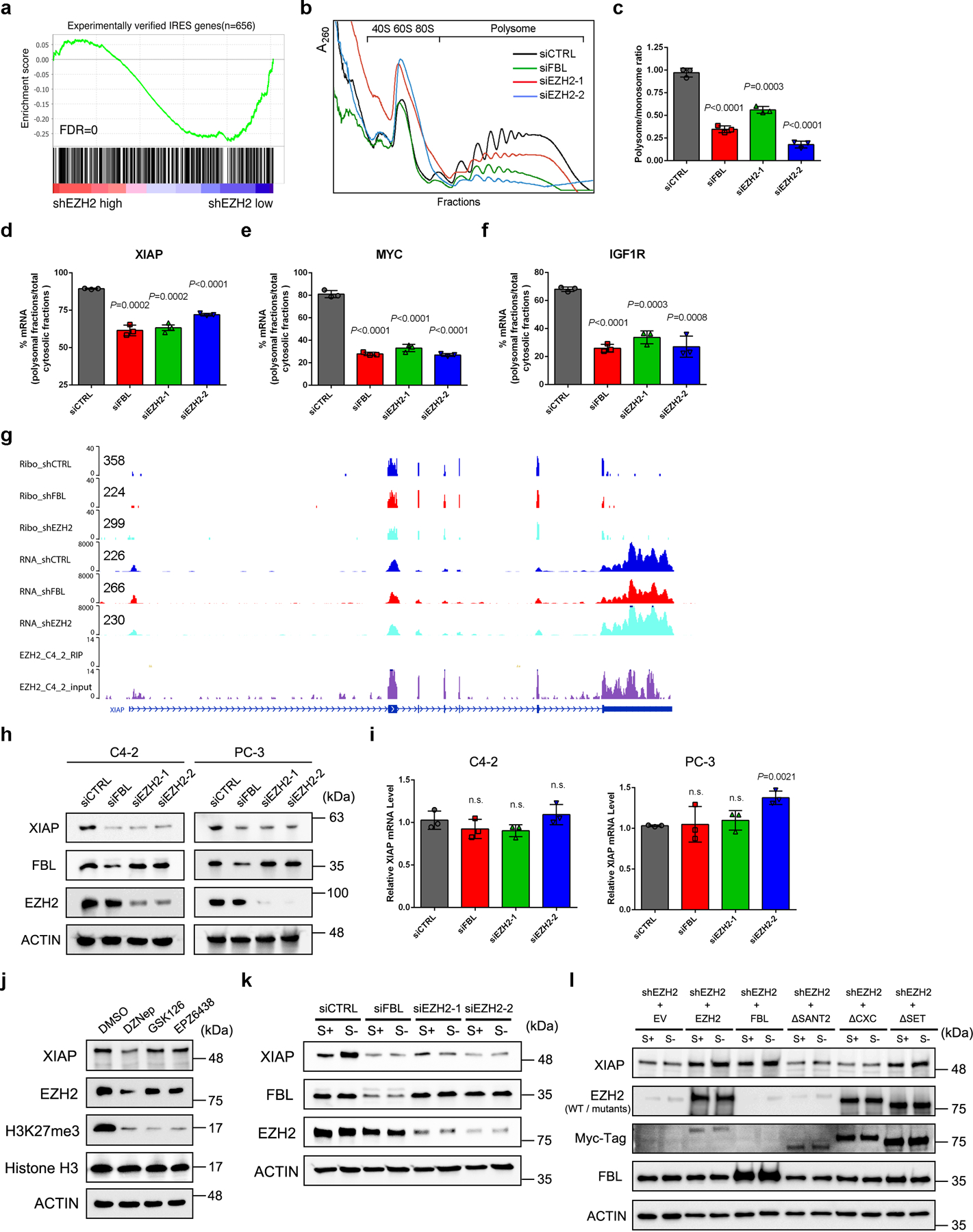
(a) GSEA analysis using a list of 656 IRES genes showed a negative enrichment of putative IRES genes associated with the TE changes after EZH2 knockdown.
(b) Cytoplasmic polysome patterns of control, FBL-deficient and EZH2-deficient C4–2 cells were denoted.
(c) Quantification of the ratio of polysomes to the monosomes (80S). Data represent Mean ± SD from n=3 biologically independent experiments.
(d-f) Quantification of the ratio of polysomal-bound XIAP (d), MYC (e) and IGF1R (f) mRNA to the total cytoplasmic mRNA of their own. Data represent Mean ± SD from n=3 biologically independent experiments.
(g) Representative genome browser tracks to show Ribo-seq, RNA-seq and public EZH2 RIP-seq data around the XIAP locus. The normalized Ribo-seq and RNA-seq signals at 5′ upstream of XIAP gene were labeled on the left of each gene track.
(h) Western blot analysis of XIAP protein level upon FBL or EZH2 depletion in C4–2 and PC-3 cells.
(i) RT-qPCR analysis of XIAP mRNA level upon FBL or EZH2 depletion in C4–2 and PC-3 cells. Data represent Mean ± SD from n=3 biologically independent experiments.
(j) Western blot analysis of XIAP protein level in C4–2 cells after treatment of various EZH2 inhibitors as indicated.
(k) Serum starvation assay followed by western blot to detect the induction of XIAP expression in control, FBL-deficient and EZH2-deficient C4–2 cells.
(l) Rescue assay showing that ectopic expression of EZH2ΔSET, but not EZH2ΔSANT2 or EZH2ΔCXC, restored induction of XIAP expression in EZH2-deficient cells, as measured by western blot. It is noted that the EZH2ΔSANT2 mutant could not be detected by anti-EZH2 antibody used here since this antibody targets to residues in SANT2 domain specifically.
For all relevant panels, unless otherwise stated, statistical significance was determined by two-tailed Student’s t-test.
The assays in h and j-l have been performed three times with similar results. Statistical source data and unprocessed blots are provided in Source data Fig. 6.
IRES-dependent translation of XIAP was found to be activated after serum starvation, whereas the canonical CAP-dependent translation was block46. To determine whether EZH2 regulates XIAP expression through mediating IRES activity, C4–2 cells were cultured in serum-free medium for 24 h followed by immunoblot analyses. In accordance with previous finding, serum deprivation substantially up-regulated XIAP at protein level (Fig. 6k and Extended Data Fig. 6f). However, much weaker induction was observed in EZH2 or FBL-deficient cells, indicating an impaired IRES activity. Moreover, a rescued up-regulation of XIAP protein was observed in EZH2-deficient cells overexpressing EZH2ΔSET, but not EZH2ΔCXC or EZH2ΔSANT2, suggesting that interactions with FBL and NOP56 are essential for EZH2 to mediate IRES-dependent translation of XIAP (Fig. 6l).
FBL and NOP56 are critical in EZH2-driven tumorigenesis of PCa
FBL has been reported to be overexpressed in PCa and is critical for tumorigenesis17, 18, while the role of NOP56 during PCa progression is unexplored. Here, we found that PCa tumors at more aggressive grades frequently expressed higher levels of FBL and NOP56 (Fig. 7a and 7b). Survival analysis using JHMI PCa dataset47, 48 revealed that the median survival time of high FBL or NOP56 expression group was significantly shorter than that of low expression group (Fig. 7c and 7d). In addition, FBL- or NOP56-deficient PCa cells were less potent in migration and invasion (Extended Data Fig. 7a–7d), and produced much smaller tumors compared to control cells (Extended Data Fig. 7e and 7f). EZH2 is well-established as a diagnostic and prognostic biomarker of PCa49. Remarkably, the association of EZH2 with unfavorable outcome is more significant in FBL-High PCa patients than in FBL-Low PCa patients (Fig. 7e and 7f). Meanwhile, phenotypically similar plots could be depicted when PCa patients were divided into NOP56-High and -Low groups (Fig. 7g and 7h). To examine the effort of the two box C/D snoRNP members on oncogenic role of EZH2, the IC50 shift assay was performed in control and FBL/NOP56-deficient cells using either DZNep or GSK126. FBL or NOP56 suppression significantly increased the IC50 of DZNep, but had no effect on IC50 of GSK126 (Fig. 7i–7l), suggesting that FBL and NOP56 are key factors involved in non-methyltransferase function of EZH2. Intriguingly, ectopic expression of EZH2ΔSET, but not the other mutants, rescued the EZH2-deficient cell growth with an effect comparable with that of full-length EZH2 (Fig. 7m), suggesting that the non-methyltransferase function of EZH2 is enough to promote PCa cell growth.
Figure 7. Contributions of FBL and NOP56 in EZH2-driven PCa tumorigenesis.
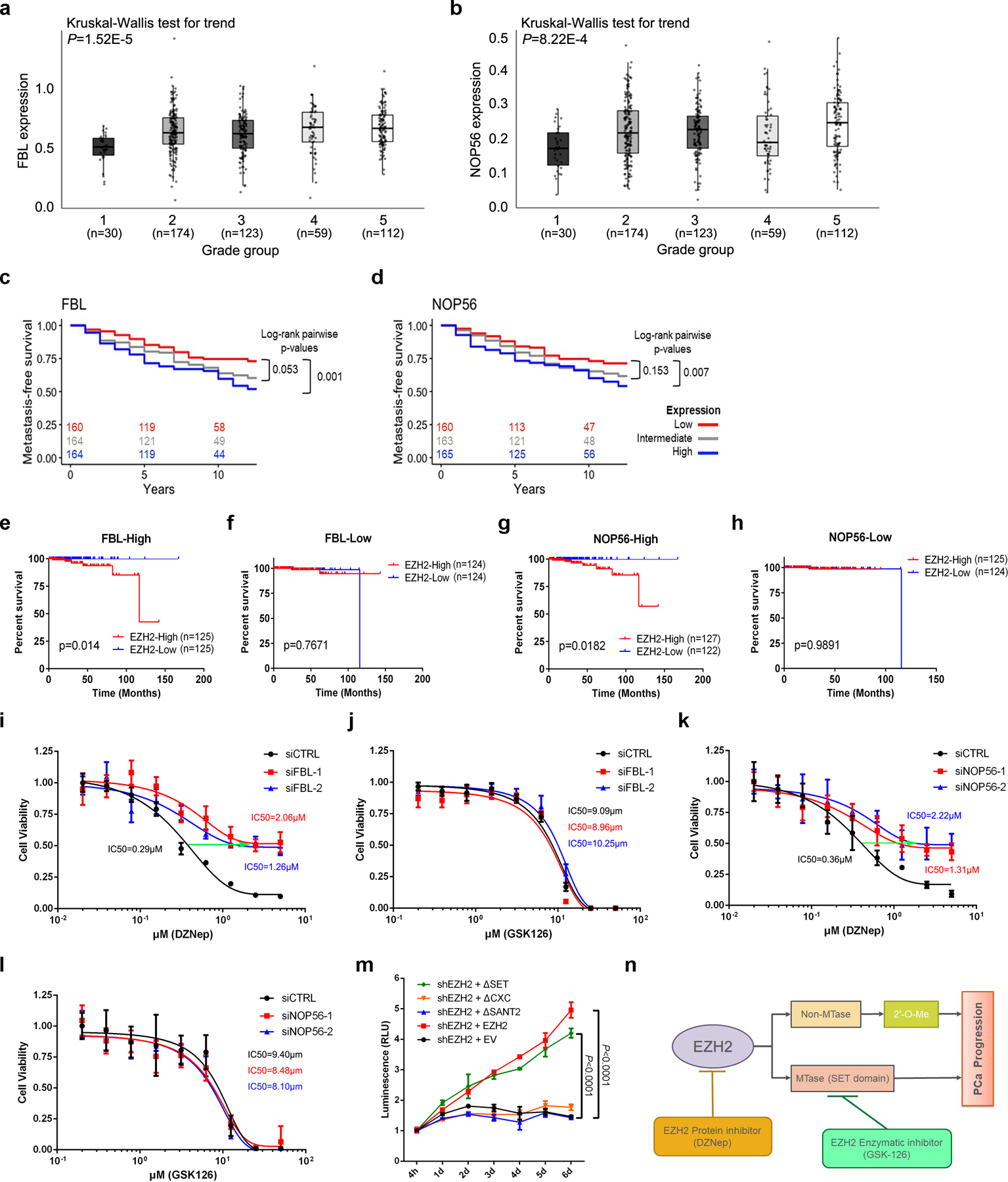
(a, b) Comparison of FBL (a) or NOP56 (b) mRNA levels in PCa patient samples with different Gleason grades using JHMI cohort. The ends of the box are the upper and lower quartiles and the box spans the interquartile range. The median is marked by a vertical line inside the box and the whiskers represent for 1.5x interquartile range. The significance of trend was calculated by two-sided Kruskal-Wallis test. ‘n’ represents the number of patients included in the analyses.
(c, d) The association between FBL (c) or NOP56 (d) expression and metastasis-free survival time of PCa patients was analyzed by Kaplan–Meier analysis using JHMI cohort. For each group, the number of patients remained at each time-point was indicated.
(e, f) The association between EZH2 expression and survival time of PCa patients with high FBL expression (e) or low FBL expression (f) was analyzed by Kaplan-Meier analysis using TCGA dataset. ‘n’ represents the number of patients included in the analyses.
(g, h) The association between EZH2 expression and survival time of PCa patients with high NOP56 expression (g) or low NOP56 expression (h) was analyzed by Kaplan-Meier analysis using TCGA dataset. ‘n’ represents the number of patients included in the analyses.
(i, j) IC50 shift assay showing the IC50 curve of EZH2 inhibitors DZNep (i) and GSK126 (j) in control or FBL-deficient C4–2 cells. Data represent Mean ± SD from n=6 biologically independent experiments.
(k, l) IC50 shift assay showing the IC50 curve of EZH2 inhibitors DZNep (k) and GSK126 (l) in control or NOP56-deficient C4–2 cells. Data represent Mean ± SD from n=6 biologically independent experiments.
(m) Cell viability assay was used to assess the proliferative capacity of EZH2-deficient C4–2 cells overexpressing full-length or truncated EZH2. Data represent Mean ± SD from n=3 biologically independent experiments. Statistical significance was determined by two-tailed Student’s t-test.
(n) Schematic diagram depicting two distinctive functions of EZH2 during cancer development. PCa, prostate cancer; MTase, methyltransferase.
Statistical source data are provided in Source data Fig. 7.
All these findings could be refined in Fig. 7n. Both lysine methyltransferase-dependent and -independent functions of EZH2 contribute to the progression of PCa. As critical interacting partners, FBL and NOP56 are cofactors to exert EZH2 function independently of EZH2 lysine methyltransferase activity. Thus, as shown by survival analyses, elevated FBL/NOP56 expression increases the risky outcome predicted by EZH2 overexpression in PCa patients. In addition, selective impact of FBL/NOP56 inhibition on drug efficacy of DZNep, but not GSK126, reflects the specific role of both proteins in lysine methyltransferase-independent regulation mediated by EZH2.
Discussion
In this study, we disclosed a previously unknown role of EZH2 in rRNA 2′-O-Me by modulating the functionality of FBL. Our data suggested that EZH2 could facilitate box C/D snoRNP assembly by strengthening FBL-NOP56 interaction. This finding could also explain why EZH2 does not affect the role of FBL in methylating histone H2A, which does not rely on the formation of snoRNP complex11. Similar to that of 2′-O-Me, a specific U3 snoRNP complex is needed for FBL to participate in 18S rRNA processing10. However, other than the box C/D snoRNPs dedicated to 2′-O-Me, additional assembly factors are required during the U3 snoRNP assembly and a specific protein component Rrp9/U3–55K is existed in the mature U3 snoRNP50. All these factors may affect the involvement of EZH2 in 18S rRNA processing.
A highlight of this study is to extend the function of EZH2 to the translation level. In contrast to its canonical role as a transcriptional silencer, the regulatory role of EZH2 in translation process is positive, as revealed by Puromycylation assay and polysome profiling data. With the help of Ribo-seq analyses, the EZH2-regulated transcripts are capable of further classification based on the modes of regulation. Notably, 75% of the translation-affected genes were classified into two TE-altered groups, suggesting that most of the EZH2-regulated transcripts do not change their protein levels concordantly with the changes in mRNA. In contrast to the high similarity between “translation” groups in EZH2- and FBL-deficient cells, a lot more differences were observed in “buffering” groups of two cell types. This is not unexpected, given that the buffering genes are deeply modulated in transcription, at which level EZH2 exerts its transcriptional regulator function independent of FBL.
The current EZH2-targeting strategy in cancer treatment mainly focuses on blocking of lysine methyltransferase activity of EZH2, which could not completely inhibit the progression of solid tumors51, 52. Our results revealed the significance of non-lysine methyltransferase role of EZH2 in oncogenesis, which could provide distinctive perspectives for the development of EZH2-targeting strategies specialized in elimination of its non-methyltransferase roles. In conclusion, we demonstrated that EZH2 plays a dual role in regulating transcription and translation processes in EZH2-high cells, i.e., cancer cells, and then promotes cancer progression by accelerating ribosome functions and contributing to the translational control (Extended Data Fig. 8).
Methods
Cell culture and Antibodies
Human PCa cell lines PC-3, DU145, 22RV1, LNCaP and VCaP were purchased from ATCC, while C4–2 cell line is a generous gift from Dr. Leland Chung. The C4–2, PC-3, DU145, 22RV1 and LNCaP cells were grown in RPMI 1640 medium supplemented with 10% FBS, while VCaP cells were grown in DMEM medium supplemented with 10% FBS. HEK293T cells were purchased from ATCC and cultured in DMEM medium supplemented with 10% FBS. Control and Ezh1/Ezh2 double-knockout XEN cells were provided by Dr. Sundeep Kalantry and grown in MEM-alpha medium with 20% ES qualified FBS, 1 mM Sodium Pyruvate (Invitrogen), 2 mM L-Glutamine (Invitrogen), 1×NEAA (Invitrogen) and 100 μM β-Mercaptoethanol (Invitrogen). All cells were maintained at 37 °C and 5% CO2 in a humidified atmosphere. The primary antibodies used in this paper were listed in Supplementary Table 3.
Transfection
All the Silencer siRNAs used in this study were purchased from Thermo Fisher (siEZH2–1: s4916, siEZH2–2: s4917; siFBL-1: s4820, siFBL-2: s4821; siNOP56–1: s20641, NOP56–2: s20642). Lipofectamine RNAiMAX (Invitrogen) were utilized for siRNA transfection according to the procedure of manufacturer. Cells were collected at 3–5 days post-transfection.
All the shRNA vectors were purchased from Sigma (shEZH2–1: TRCN0000286227, targeting 3′-UTR region for rescue assay; shEZH2–2: TRCN0000040077; shFBL-1: TRCN0000292354, shFBL-2: TRCN0000292351; shNOP56–1: TRCN0000148851, shNOP56–2: TRCN0000276500). For cell transfection, shRNA vectors and helper plasmids were transfected into target cells using Lipofectamine 3000 (Invitrogen) following its protocol.
Lentivirus construction
Lentivirus was packaged by co-transfection of shRNA constructs with helper plasmids pVSVG and psPAX2 into HEK293T cells. The medium was renewed at 24 h post-transfection and the supernatants containing viruses were collected at 48 h post-transfection. The lentivirus was stored in −80 °C until further use.
EZH2 inhibitors treatment
GSK126 was purchased from BioVision, DZNep and EPZ6438 were purchased from Selleck Chemicals. C4–2 cells were treated with the EZH2 inhibitors at a concentration of 5 μM for 72 h until further use.
Western blot (WB)
Protein samples were separated electrophoretically by SDS-PAGE, and semi-dry transferred to PVDF membranes (Roche). The membranes were blocked for 30 min in Tris-buffered saline-Tween 20 (TBST) with 5% nonfat milk. Thereafter, immunoblottings were performed with primary antibodies for 2 h at room temperature. After washing for three times, the membranes were incubated with Clean-Blot IP Detection Reagent (Thermo Fisher, 1:500 dilution, for co-IP WB) or goat anti-mouse/rabbit IgG (H+L)-HRP secondary antibody (GenDEPOT, 1:5000 dilution, for normal WB) for 1 h. The signals were developed using western ECL Substrate (Bio-Rad) and captured by a Bio-Rad imaging system. The relative protein level was evaluated using ImageJ software.
Real-time (RT)-qPCR analysis
The total RNA isolation was prepared using RNeasy Plus Mini Kit (Qiagen). The obtained RNA was then reverse transcribed into cDNA using Maxima H Minus First Strand cDNA Synthesis Kit (Thermo). Each cDNA sample was amplified using Universal SYBR Green Supermix (Bio-Rad) in the QuantStudio 6 Flex Real-time PCR System (GE Healthcare) following manufacturer’s instructions. The primers used for RT-qPCR analysis were summarized in Supplementary Table 4. The relative RNA level was calculated using the 2−ΔΔCt method with the Ct values normalized using GAPDH as an internal control unless noted otherwise.
GST pull down
To perform GST pull down experiment, 1 μg recombinant GST-tagged EZH2/EED protein was mixed with 1 μg purified protein of interest in 1 ml NP-40 lysis buffer (GenDEPOT) with a protease and phosphatase inhibitor (Thermo). The mixture was incubated with Glutathione Sepharose 4B (GE Healthcare) for 1 h at room temperature. Then, the protein-attached beads were collected by centrifugation followed by repeated washing. To denature proteins, beads were added to 2× reducing SDS-sample buffer (Bio-Rad) and heated at 95 °C for 10 min. Protein samples were subjected to western blot assay for further analysis. The recombinant proteins used were listed in Supplementary Table 5.
Co-immunoprecipitation (co-IP) assay
Whole-cell lysate was obtained by lysing cells in the NP-40 lysis buffer (GenDEPOT) with a protease and phosphatase inhibitor (Thermo). The lysate was kept on ice for 15 min followed by sonication at 5 s on and 5 s off for 60 s. Insoluble material was removed by centrifugation. Lysates were pre-incubated with Dynabeads protein A/G (Invitrogen) to eliminate nonspecific binding. Then, antibodies were mixed into lysates with new-added Dynabeads and incubated at 4 °C overnight. The immune complexes were collected using magnetic separator and washed three times with lysis buffer. To denature proteins, beads were added to 2× reducing SDS-sample buffer (Bio-Rad) and heated at 95 °C for 10 min. Protein samples were subjected to western blot assay for further analysis.
AlphaLISA assay
The AlphaLISA assay was performed according to the User’s Guide (PerkinElmer). Assay development and optimization were carried out in a white 96-well half area plate (Greiner) and all incubation steps were performed in dark at room temperature. In brief, protein cross-titration experiment was firstly performed by testing multiple concentration combinations of two proteins in a matrix to detect the optimal analyte association. Then, a third protein or an untagged version of one or the other protein was introduced to the displacement assay to affect the existing association between beads. The alpha signals reflecting the binding affinity were measured in a Tecan plate reader in AlphaLISA detection mode. All the tagged or untagged recombinant proteins used in this assay were ordered from OriGene and the corresponding Donor and Acceptor beads were selected based on the tags of protein pairs. The recombinant proteins used were listed in Supplementary Table 5.
Tissue microarrays (TMAs) and Immunohistochemistry (IHC) staining
Prostate tumor biopsies were retrieved from Vancouver Prostate Centre tissue bank and used to construct TMAs as published previously53. This protocol was approved by the office of research ethics in the University of British Columbia. IHC was performed by Ventana Discovery XT autostainer (Ventana) with anti-EZH2, anti-FBL and anti-NOP56 antibodies as reported54. All stained slides were scanned by a Leica SCN400 scanner and digital images were evaluated and scored by a pathologist, Dr. Ladan Fazli. EZH2 histology score (H-score) was calculated by the Aperio ImageScope software based on both intensity (no staining, low, moderate and high intensity staining) and percentage (0–16%, 17–33%, 34–66% and 67–100%) of the IHC signals, according to the manufacturer’s instructions (Leica Biosystems). The primary antibodies used for IHC were listed in Supplementary Table 3.
Proximity ligation assay (PLA)
To determine if EZH2-FBL protein interaction exists in situ, proximity ligation assay (PLA) was conducted manually with the Duolink PLA Brightfield Kit (Sigma) according to manufacturer’s protocol. Briefly, baked tissue sections were deparaffinized in CitriSolv, permeabilized to enhance detection of nuclear proteins, and treated in citrate buffer to retrieve antigen. After peroxidase quenching and blocking, anti-FBL and anti-EZH2 antibodies were applied to tissue overnight at room temperature in a humidity chamber. To detect EZH2-FBL protein complexes, tissue was then incubated with the following according to manufacturer’s specifications: PLA probes (37 °C for 1 h), ligation solution (37 °C for 30 min), amplification solution (37 °C for 2 h), and developed with substrate solution (room temperature for 30 min). Tissues were then counterstained with hematoxylin, dehydrated, and mounted. For analysis, slides were digitally scanned, and positive staining was quantified with Aperio ImageScope. This protocol was approved by the office of research ethics in the University of British Columbia.
BS3 crosslinking assay
Purified recombinant human EZH2, FBL and NOP56 proteins (Supplementary Table 5) were purchased from Origene and exchanged into K150 buffer (20 mM KH2PO4, pH 7.4, 10% glycerol, 150 mM KCl, 0.01% NP40 and 1 mM BME) to avoid non-protein sourced primary amine groups in the crosslinking mixture, which can react with the crosslinking reagent, BS3 (bis[sulfosuccinimidyl] suberate, Thermo).
To explore the physical interaction between three proteins, EZH2, FBL and NOP56, all present in solution at 2 μM, were incubated at room temperature for 30 min in the absence or the presence of 800 μM BS3 crosslinker in 25 µL K150 buffer. Following incubation, the crosslinking experiments were quenched by addition of 2 μL 1 M Tris-HCl, pH 8.0, and the samples were analyzed on SGS-PAGE. Subsequent western blot analysis was carried out with antibodies separately recognizing EZH2, FBL and NOP56 to indicate the location of the cross-linked species that includes all three proteins on the SDS-PAGE. Gel band containing cross-linked proteins was further subjected to Mass Spectrometry analyses to validate the existence of these three proteins.
Size-Exclusion Chromatography
Nuclear extracts from control and EZH2-deficient C4–2 cells were obtained using NE-PER nuclear extraction kit (Thermo), and 5 mg of nuclear protein was concentrated to 200 μL using Ultra-0.5 Centrifugal Filter (Amicon). The concentrated solution was then loaded into Pharmacia SMART System for size-exclusion. A total of 48 fractions with a volume of 50 μL for each fraction were finally collected, and eluted fractions were subjected to western blot and co-IP assays.
Isolation of Nucleoli
Nucleoli were isolated from PCa cells by using a newly developed method55. In brief, freshly harvested C4–2 cells were collected into a 15 mL tube and resuspended with 3 mL pre-cooled solution I (0.5 M sucrose, 3 mM MgCl2 with one tablet of Thermo protease inhibitors/50 mL). After sonication on ice, the cell suspension was underlaid with 3 mL of pre-cooled solution II (1 M sucrose, 3 mM MgCl2 with one tablet of Thermo protease inhibitors/50 mL). The obtained sucrose gradient was centrifuged at 1800 g for 10 min and the resulting pellet contained the isolated nucleoli. To assess the purity of the isolated nucleoli, samples of whole cell lysate, the cytoplasm + nucleoplasm pellet, and the isolated nucleoli were collected, followed by protein gel staining and western blot analysis. After validation, lysates from the purified nucleoli were subjected to co-IP assay for further investigation.
Northern blot (NB)
The Northern blot assay was conducted using NorthernMax-Gly kit (Thermo) following manufacturer’s procedure. In brief, cellular RNA (10–20 μg) was separated on agarose gel and transferred to positively charged nylon membrane (Thermo). After UV crosslinking, membranes were hybridized with biotin-labeled oligonucleotides (Supplementary Table 4) at 37 °C for 16 h in a hybridizer oven. Non-radioactive signals were amplified and visualized by Biotin Chromogenic Detection kit (Thermo).
Quantification of rRNA methylation level using LC-MS/MS
Quantitative analysis of rRNA methylation level was carried out using a previously described method with revision56. Briefly, a total of 1 μg total RNA was digested by nuclease P1 (NEB) in 40 μL of buffer containing 50 mM sodium acetate at 37 °C for 1 h, followed by the addition of NH4HCO3 (1 M, 3 μl, Sigma) and alkaline phosphatase (0.5 U, Promega). After an additional incubation at 37 °C for 1 h, the solution was diluted three times and subjected into LC-MS/MS at Metabolomics Core Facility of Northwestern University. The purified adenosine, guanosine and their 2′-O methylated forms were synthesized in IDT and used as references in MS analysis.
Determination of rRNA methylation level by Reverse Transcription at Low dNTP concentrations followed by PCR (RTL-P) method
The RTL-P approach32 is based on a principle that the 2′-O-methylated nucleotide could impede reverse transcription (RT) reaction at a low dNTP concentration. Therefore, the RT reaction with a low dNTP concentration should produce shorter cDNA products than that with a high dNTP concentration. Subsequently, PCR amplification was performed with different PCR primer pairs targeting upstream (Um) or downstream (Dm) of specific methylation site(s). For RNAs with high level of 2′-O-Me, the quantity of the RT primer/Um primer PCR products is less than those RNAs with low level of methylation in the presence of low dNTP concentration. Meanwhile, this quantity is similar in the presence of high concentration of dNTP.
To detect the change of 2′-O-Me levels after EZH2 knockdown, the total RNA was extracted using RNeasy Plus Mini Kit (Qiagen). RT was performed using specific reverse primers targeting a rRNA sequence upstream to one or several methylation site (s), in presence of either a low level (1 μM) or a high level (1 mM) of dNTP. The RT reaction mixture contains 200 ng RNA, 200 U M-MLV reverse transcriptase (Promega), 10 mM RT primers, 0.5 U RNase Inhibitor (Promega) and dNTP (low/high, Invitrogen). The obtained cDNA was added to two separate amplification reactions using two pairs of specific primers targeting upstream and downstream of methylation site (s) respectively. The PCR mixture contains 10 μL amfisure PCR Master Mix (GenDEPOT), 2 μL cDNA and 10 mM PCR primers. The PCR products were then separated on 1% agarose gels and visualized by a Bio-Rad imaging system. DNA band signal intensities were analyzed using ImageJ software. The methylation ratio of each group was determined from densities of PCR bands obtained using high and low dNTP concentrations. The RT and PCR primers designed for RTL-P assay were listed in Supplementary Table 4.
Detection of rRNA methylation by RiboMeth-seq
RiboMeth-seq analyses were performed in control and EZH2-deficient C4–2 cells based on a previously described protocol33. Biological triplicates were used for control cells while a total of 4 replicates (2 for siEZH2–1 and 2 for siEZH2–2) were used for siEZH2 cells. In brief, the total RNA was extracted using RNeasy Plus Mini Kit (Qiagen), followed by alkaline hydrolysis into short fragments. After fragmentation, size of RNA fragments (30–200 nt) generated was assessed by capillary electrophoresis using a PicoRNA chip on Bioanalyzer 2100 (Agilent). Fragmented RNA was 3′-end dephosphorylated directly using 5 U of Antarctic Phosphatase (NEB). After inactivation of the phosphatase, RNA fragments were phosphorylated at the 5′-end using T4 PNK (NEB) and 1 mM ATP. End-repaired RNA fragments were then purified using RNeasy MinElute Cleanup kit (Qiagen) according to the manufacturer’s recommendations. The eluted RNA was subjected to BGI for library preparation and sequencing using the DNBseq platform with a 50 bp single-end read mode.
Bioinformatic pipeline of the RiboMeth-seq is similar to the previous published methods57, 58. In brief, the adapter of each read was trimmed by Cutadapt. The reads were then mapped to the rRNA with bowtie2 in End-to-End module. Only uniquely mapped reads were used to do the further analysis. The bam files were converted into bed files using the function bamtobed in the package bedtools. Bedtools was also used to calculate the number of the 5′ and 3′ reads in the bed files. The coverage of reads number were used to compute the methylation score (MethScore) at nucleotide resolution in the 2’-O-Me sites. To be specific, the MethScore which presents the fraction of 2’-O-methylated molecules were calculated by R scripts for each site of 18S and 28S rRNAs from the coverage information of 5′-end reads. Twelve neighboring nucleotides (+/−6 nt window) were taken into account to calculate the 2′-O-Me scores. For 5.8S rRNA, to avoid the negative value for MethScore, an optimized method with 4 neighboring nucleotides (+/−2 nt window) from the 5′- end reads and 3′- end reads were used to calculate the MethScore. The Unix and R scripts were downloaded from https://github.com/FlorianPichot/RiboMethSeqPipeline/58.
Dual-luciferase assay
The pcDNA3-RLuc-PolIRES-FLuc plasmid was a gift from Nahum Sonenberg59. The bi-cistronic luciferase vectors containing IRES elements of cancer-relevant genes were generated by subcloning the coding sequence for firefly luciferase (Fluc) from pGEM-LUC (Promega), the coding sequence for renilla luciferase (Rluc) from pRL-CMV (Promega) and IRES sequence of oncogenes into pcDNA3.1 vector. The IRES sequences of XIAP60, VEGFA61, FGF162, FGF263, IGF1R64 and MYC65 were determined based on the references cited. Dual-luciferase assays were performed at 24 h post-transfection using the Dual-Glo luciferase reagent (Promega) according to the manufacturer’s instructions, and a Tecan plate reader.
In vitro transcription
In vitro synthesis of capped RNA was achieved by mMESSAGE mMACHINE Kit (Thermo). Plasmid DNA (XIAP, FGF1, IGF1R and MYC) was linearized prior to transcription using XhoI (NEB). After linearization, the plasmid DNA was recycled at a concentration of 0.5–1 μg/μL. The capped, runoff RNA transcripts were then synthesized by using recombinant T7 RNA polymerase. To enhance the stability of the synthesized RNA, polyadenylation of RNA was conducted using Poly(A) Tailing Kit (Thermo) according to the specifications of the manufacturer. For RNA transfection and measurement, C4–2 cells were seeded in 96-well plate the day prior to transfection. Cells were transfected with RNA (400 ng/well), cultured for ∼8 h at 37 °C, followed by measurement using the Dual-Glo luciferase reagent (Promega).
Polysome profiling
C4–2 cells were incubated with 100 μg/ml cycloheximide (Sigma) for 15 min at 37 °C, and harvested in Polysome Extraction Buffer (20 mM Tris-HCl, pH 7.5, 100 mM KCl, 5 mM MgCl2, 0.3 % Igepal CA-630). Nuclei and debris were pelleted by centrifugation at 10,000 rpm for 10 min at 4 °C. The supernatant (ribosomal particles) was then separated on 10–50% sucrose gradient by ultracentrifugation at 39,000 rpm for 1.5 h at 4 °C. Ribosome distribution was analyzed by measuring the absorbance of each fraction at OD 260 nm (A260) by UV photometry. RNA from each fraction was isolated with TRIzol (Invitrogen) and subjected to RT-qPCR assay for further analysis.
RNA immunoprecipitation (RIP)-qPCR analysis
The RIP experiment was performed in triplicates using the EZ-Magna RIP kit (Millipore) with the procedure provided by the manufacturer. Control or EZH2-deficient C4–2 cells were lysed in RIP lysis buffer and incubated with anti-FBL antibody-magnetic beads mixture at 4 °C overnight to enrich RNA binding protein. The RNA bound to FBL protein were collected by Proteinase K digestion, followed by purification and reverse transcription into cDNA. Then, qPCR assay was performed to measure the %Input of snoRNAs in each group using the primers presented in Supplementary Table 4.
Immunofluorescence (IF) assay
To detect the co-localization of endogenous EZH2 and FBL, C4–2 cells were cultured on coverslips for 24 h. Cells were fixed in pre-cooled methanol for 15 min and permeabilized in phosphate-buffered saline (PBS) containing 0.5% Triton X-100 for 10 min at room temperature. Slides were rinsed thrice with PBS and blocked with 5% BSA for 30 min. Then, the slides were incubated with rabbit anti-EZH2 antibody and mouse anti-FBL antibody at 4 °C overnight. Slides were rinsed thrice with PBS, followed by exposure to Alexa Fluor 488 conjugated goat anti-rabbit antibody and Alexa Fluor 555 conjugated goat anti-mouse antibody (all from Invitrogen) for additional 1 h at 37 °C. Cells were co-stained in the dark with DAPI (Invitrogen) and mounted using ProLong Diamond Antifade Mountant (Invitrogen). Immunostained cells were viewed using Nikon A1R confocal microscope.
For observation of localization of EZH2 truncation mutants, C4–2 cells were transfected with either GFP-full length EZH2 or GFP-truncated EZH2 plasmids. At 48 h post-transfection, the IF assay was performed as described above except for the use of mouse anti-GFP antibody and rabbit anti-FBL/anti-NOP56 antibody as primary antibodies, and Alexa Fluor 488 conjugated goat anti-mouse antibody and Alexa Fluor 555 conjugated goat anti-rabbit antibody (all from Invitrogen) as secondary antibodies.
Transmission Electron Microscope (TEM)
Pellet samples from control and EZH2-deficient C4–2 cells were fixed in 2.5% glutaraldehyde for 12 h and post-fixed in osmium tetroxide for another 2 h. Samples were then embedded in resin mixture of Embed 812 kit. After routine processing, samples were sectioned on a Leica Ultracut UC6 ultramicrotome. The 70 nm sections were collected on 200 mesh copper grids and stained with uranyl acetate and Reynolds lead citrate. These sections were further examined and imaged on a Hitachi7600 transmission electron microscope (TEM).
Puromycylation assay
Global protein synthesis was analyzed by puromycylation assay as described before40. Briefly, cells were pre-treated with elongation inhibitor cycloheximide (CHX, Sigma) at a concentration of 355 μM for 15 min to freeze polysomes. Then, puromycin was added into the culture medium to a final concentration of 91 μM. After treatment for 5 min at 37 °C, the whole-cell protein extracts were immediately prepared and puromycin incorporation into the nascent chain was detected by western blot using a specific anti-puromycin antibody (Supplementary Table 3).
Ribosome profiling (Ribo-seq) and RNA sequencing (RNA-seq)
Ribosome profiling (Ribo-seq) experiment was performed in triplicates following the method described previously66. Briefly, 4 million cells were rapidly harvested and lysed under conditions that maintain in vivo ribosome positions on mRNAs. Before proceeding with nuclease footprinting, a portion of the undigested cell lysate was kept for the preparation of RNA-seq library. The rest cell lysates were subjected to nuclease footprinting with RNase I (Thermo), and ribosomes were recovered by ultracentrifugation. Ribosome footprints were purified and ligated to a single-stranded linker. The RNA-DNA hybrid molecules were used as templates for reverse transcription, followed by gel-based purification of full-length products. The first-strand reverse transcription products were then circularized. Finally, a double-stranded DNA library for Illumina sequencing was constructed from the single-stranded cDNA circles by means of a PCR amplification. The ribosome profiling libraries were subjected to Novogene for sequencing using the Illumina TruSeq platform with a 150 bp pair-end reads mode.
For data analysis, the software PEAR67 was used to merge and integrate the reads and quality information of pair-end reads, followed by the removal of adapters. The barcodes were removed using Cutadapt and the reads were split into different samples based on their barcode sequences. Then the reads mapped to the human rRNA were removed by the bowtie methods. The UMI information combined with the SeqKit rmdup method68 was utilized to remove PCR duplication. After that, clean reads from three biological RNA-seq and Ribo-seq replicates were mapped to the human hg19 genome and assigned to the exons of genes using TopHat69. Only the uniquely mapped reads were used for the further analysis. Read counts for each gene were calculated by the htseq-count function of the tool HTSeq70. Subsequently, Fragments Per Kilobase of transcript per Million mapped reads (FPKM) were calculated and normalized using quantile normalization procedures in R (https://www.r-project.org/). Since the log-ratio based approach to capture changes in translational efficiency may lead to spurious correlation, we used the anota2seq algorithm to determine the changes in mRNA, polysome-associated RNA, and translational efficiency41, 42. The p-value < 0.05 and the other default thresholds were set for analysis. The anota2seq separates the protein coding genes into three groups including translation, buffering and mRNA abundance, while the remaining genes were regarded as background genes. For genes belonging to more than one group, a priority order was established as follows: changes in translational efficiency leading to altered protein levels will belong to the translation group; transcripts that change their mRNA abundance but are not in the translation group will be in the mRNA abundance group; buffering (offset) mRNAs are not in the former two groups.
The functional enrichment analysis was conducted using the R package clusterProfiler71. The wiq function of DANPOS72 was used to perform quantile normalization of wig file between different samples. The normalized wig files were converted into bigwig files using wigToBigWig. Bigwig files from the Ribo-seq and RNA-seq were visualized using the washU genome browser (http://epigenomegateway.wustl.edu/)73.
Analysis of public EZH2 RIP-seq data
High-throughput EZH2 RIP-seq data (GSE63230) in C4–2 cell lines was downloaded for conjoint analyses with our Ribo-seq data. Using 1.5-fold change as cutoff, a total of 2,760 genes were identified as EZH2 mRNA-binding targets in C4–2 cells. These EZH2 RIP target genes were used as a gene set for Gene Set Enrichment Analysis (GSEA) and Venn diagram analysis with our Ribo-seq results. The bigwig format files transformed from the mapped bam files were visualized in the washU genome browser.
Analysis of XIAP protein stability by CHX treatment assay
C4–2 cells were cultured in 6-well plate and exposed to CHX at a concentration of 25 μg/mL. Cell lysates were harvested at specific times after CHX addition, followed by western blot assay to detect the protein level of XIAP at each time-point. The percentage of XIAP protein remaining at the indicate points was quantified by densitometry after normalization with β-actin. The amount of the XIAP protein at the zero time in each group was considered as 1.
Murine prostate tumor xenograft model and PDX tumors
Five-week-old male Balb/c athymic nude mice were purchased from Model Animal Research Center of Nanjing University, and housed in individually ventilated microisolator cages. Nude mice were divided into five groups of 8 mice each. Animal experiments were approved by the Animal Care and Use Committee of Sun Yat-sen University.
For subcutaneous tumor model, each mouse was injected subcutaneously in the right flank with 4 × 106 of PC-3 cells (control, FBL-deficient or NOP56-deficient) suspended in 100 μL of PBS. Tumor volumes were measured by length (a), width (b) and calculated as tumor volume = MIN(a)2 × MAX(b) × 0.52. Fifty days after the tumor cell injection, the mice were sacrificed and tumor xenografts were removed, photographed, and stored at −80 °C.
The LuCaP 35CR patient-derived xenograft (PDX) was kindly provided by Eva Corey (University of Washington).
Wound healing assay
PC-3 Cells were cultured in 35 mm dish with 3-well Culture-Insert (Ibidi) till grown to 90% confluency. After removing the inserts gently, dishes were re-filled with serum-free RPMI 1640 medium and maintained in a humidified atmosphere at 37 °C. Pictures were taken under microscope at 0 h/24 h and the distance of cell migration was measured by ImageJ software.
Boyden chamber invasion assay
The invasiveness of PCa cells was assessed by their ability to pass through Matrigel-coated Transwell inserts (Millipore). To perform this assay, the upper surface of the Transwell chambers was coated with Matrigel matrix (Corning) diluted in RPMI 1640 medium at a dilution of 1: 20. PC-3 Cells (1×104) diluted in 300 μL of serum-free 1640 medium were seeded into the upper compartments of the chambers. Meanwhile, the lower compartments of the chambers were filled with 800 μL of 1640 medium with 10% FBS. After 24 h, invasive cells that had migrated from Matrigel to the lower surface of the chamber were fixed in methanol, stained with 0.1% crystal violet (Sigma) and subjected to microscopic inspection. The number of invasive cells was calculated as the average number of cells counted in 5 random fields per filter.
IC50 shift assay
Control or siRNA-treated C4–2 cells were seeded in 96-well plates and treated at drug concentration gradients for 72 h. Bioluminescence was measured to quantify cell viability using CellTiter-Glo Luminescent Cell Viability Assay Kit (Promega) and was read on a Tecan plate reader. The cell proliferation curve was drawn and fit by the bioluminescence to drug concentration. Half-maximum inhibitory concentration (IC50) was calculated with non-linear fitting.
Cell growth rescue assay
EZH2-deficient C4–2 cells were seeded in 96-well plates and transfected with plasmids overexpressing full-length or truncated mutants of EZH2. The CellTiter-Glo Luminescent Cell Viability Assay (Promega) was applied to each plate from 48 h post-transfection. The bioluminescence was read on a Tecan plate reader.
Visualization of protein domain structures
The domain structure of protein was illustrated using Domain Graph (DOG) 2.0 software74.
Survival analyses
The Johns Hopkins Medical Institute (JHMI) cohort which contains Gene expression profiling data for a total of 531 PCa patients was used for analyses of FBL/NOP56 expression in different grades of PCa by R package47, 48, while both JHMI and TCGA cohorts were used for survival analyses. The Kaplan-Meier survival analysis was carried out using GraphPad Prism (version 6.0). Statistical significance of the difference between survival curves for different groups of patients was assessed using log-rank test.
Statistics and reproducibility
Statistical analysis was performed using GraphPad Prism (version 6.0) or R and presented as means ± SD. Unless otherwise specified, the P values were obtained using two-tailed Student’s t-tests for comparison of two datasets or by analysis and variance (ANOVA) where appropriate. Statistical data were considered significant if P < 0.05. The results were reproducible and conducted with established internal controls. When feasible, experiments were repeated three or more times and yielded similar results. We have indicated the n values used for each analysis in the figure captions. All cell lines used were authenticated and routinely screened for Mycoplasma.
Extended Data
Extended Data Fig. 1. EZH2 interacts with FBL, but does not affect H2AQ104me modification and 18S rRNA processing.
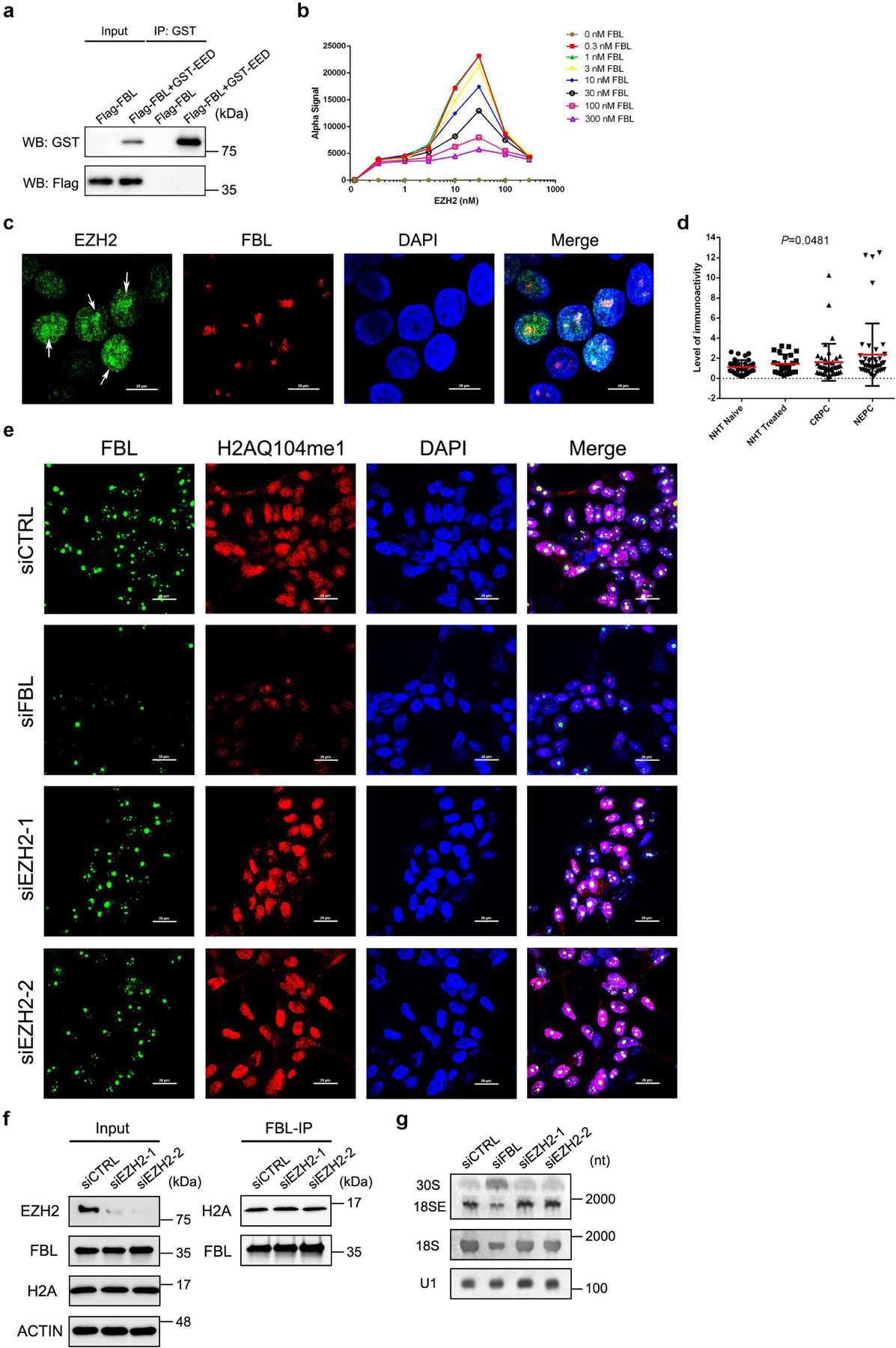
(a) Purified proteins of GST-tagged EED and Flag-tagged FBL were subjected to GST pull down assay, followed by western blot analysis.
(b) AlphaLISA cross-titration assay to determine the optimal protein concentration combination of EZH2 and FBL. A hook point is reached at 30 nM GST-tagged EZH2 and 1 nM Myc-tagged FBL.
(c) Representative fluorescence images of C4–2 cells with antibodies against endogenous EZH2 (Green) and FBL (Red). The nuclei were visualized by DAPI (Scale bar: 20 μm). Arrows indicate the enrichment of EZH2 signals in the nucleolus.
(d) Graph showing the immunoactivity values based on the PLA results from four PCa types. Data represent Mean ± SD from 38 samples in NHT Naïve group, 24 sample in NHT treated group, 42 samples in CRPC group and 43 samples in NEPC group. Statistical significance was determined by One-way ANOVA.
(e) Representative confocal images of immunofluorescence staining of C4–2 cells with antibodies against FBL (Green) and H2AQ104me1 (Red). Nuclei were stained with DAPI. Scale bars: 20 μm.
(f) Co-IP assay using anti-FBL antibody showing binding of endogenous FBL with histone H2A.
(g) Northern blot of RNA from C4–2 cells upon FBL or EZH2 suppression. The 30S and 18SE processing intermediates were detected using the ITS-1 probe while the 18S rRNA and U1 snRNA were hybridized by their corresponding probes.
The assays in a-c and e-g have been performed three times with similar results. Statistical source data and unprocessed blots are provided in Source data Extended data Fig. 1.
Extended Data Fig. 2. EZH2 modulates rRNA 2′-O-Me by interacting with FBL.
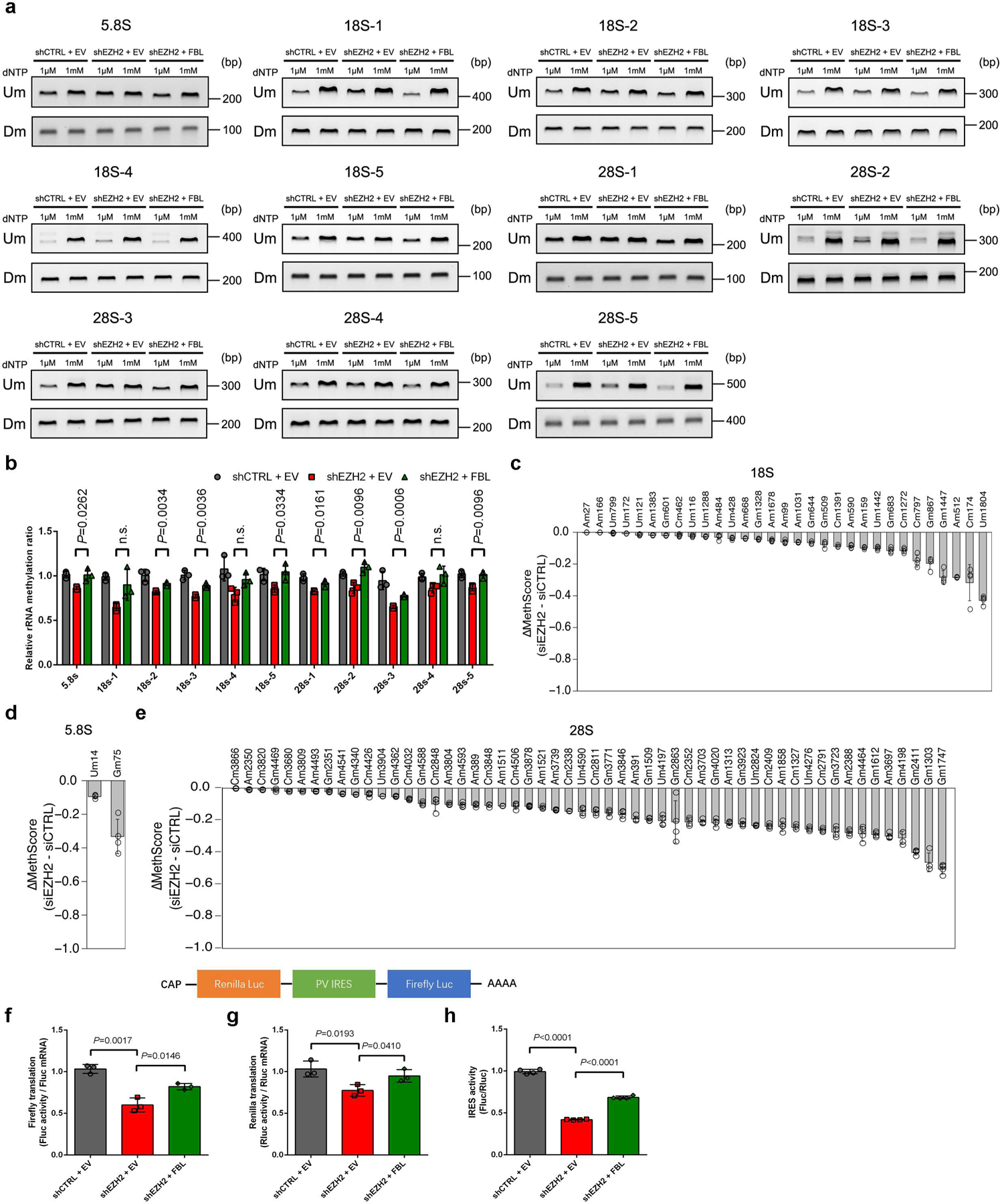
(a) RTL-P assay to detect the 2′-O-Me level in 12 areas in rRNA. Total RNAs were extracted and subjected to reverse transcription (RT) with RT primer at low (1 μM) or high (1 mM) concentration of dNTP, respectively. The obtained cDNA was then amplified with primer pairs corresponding to upstream (Um) or downstream (Dm) regions of specific methylation site(s). This assay has been performed three times with similar results.
(b) Densitometric analysis of data from a were shown as signal intensity ratio of PCR products at low dNTP (1 μM) over high dNTP (1 mM) level. Methylation levels in control cells were set close to 1. Data represent Mean ± SD from n=3 biologically independent experiments.
(c-e) For the 87 sites in which the 2′-O-Me level was significantly decreased upon EZH2 inhibition (detailed information provided in Supplementary Table 1), the MethScore obtained in control C4–2 cells was subtracted from the one in EZH2-deficient C4–2 cells. Sites are shown in order of increasing difference in MethScore for the 18S (c), 5.8S (d), and 28S (e) rRNAs. Data represent Mean ± SD from n=4 biologically independent experiments.
(f, g) Translation efficiency of firefly (f) and renilla luciferase (g) reporters was evaluated as the ratio of luciferase activity over mRNA levels. Luciferase activities were detected by dual-luciferase assay using a bi-cistronic luciferase reporter construct as shown above, while the luciferase mRNA levels were measured by RT-qPCR assay. Data represent Mean ± SD from n=3 biologically independent experiments.
(h) The Poliovirus (PV) IRES activity was calculated as the ratio of firefly luciferase activity over renilla luciferase activity. Data represent Mean ± SD from n=4 biologically independent experiments.
For all relevant panels, unless otherwise stated, statistical significance was determined by two-tailed Student’s t-test.
Statistical source data and unprocessed blots are provided in Source data Extended data Fig. 2.
Extended Data Fig. 3. EZH2 bridges FBL-NOP56 interaction by binding to both proteins.
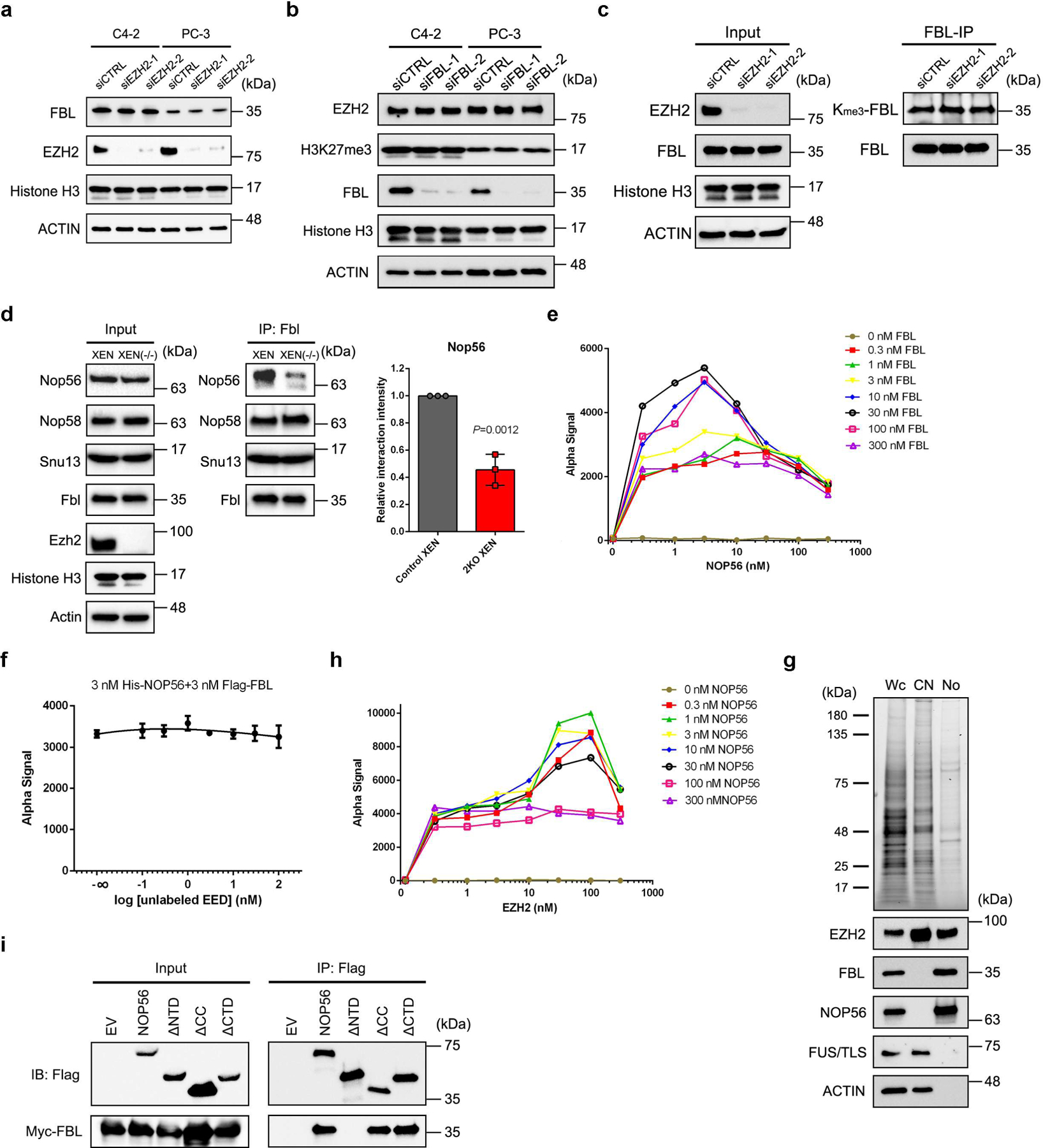
(a) Western blot analysis of FBL protein level in PCa cell lines upon EZH2 knockdown.
(b) Western blot analysis of EZH2 and H3K27me3 levels in PCa cell lines upon FBL knockdown.
(c) Equal amount of FBL protein in control and EZH2-deficient C4–2 cells were pulled down using anti-FBL antibody followed by western blot to detect its trimethyl-lysine (Kme3) level.
(d) Co-IP of Nop56, Nop58 and Snu13 with Fbl in control and Ezh1/Ezh2 double-knockout XEN cells, followed by western blot analysis with indicated antibodies. Graph represents the relative Nop56 protein level coimmunoprecipitated with Fbl in each group. Data represent Mean ± SD (n=3 biologically independent measurements). Interaction intensity at control XEN group was set as 1. Statistical significance was determined by two-tailed Student’s t-test.
(e) AlphaLISA cross-titration assay to determine the optimal protein concentration combination of FBL and NOP56. A hook point is reached at 3 nM His-tagged NOP56 and 30 nM Flag-tagged FBL. To achieve best results, a combination of 3 nM His-tagged NOP56 and 3 nM Flag-tagged FBL was used for the subsequent experiments.
(f) AlphaLISA displacement assay showing that FBL-NOP56 interaction is unaffected by EED. Data represent Mean ± SD for n=3 biologically independent experiments.
(g) After nucleolar isolation, proteins in each fraction were separated by SDS-PAGE and visualized by UV (upper panel). Distributions of EZH2, FBL (nucleolar marker), NOP56, FUS/TLS (nucleoplasmic marker) and β-actin (cytoplasmic marker) in each fraction were detected by western blot (lower panel). Wc: whole cells; CN: cytoplasm + nucleoplasm; No: nucleoli.
(h) AlphaLISA cross-titration assay to determine the optimal protein concentration combination of Flag-tagged-EZH2 and GST-tagged-NOP56. A hook point is reached at 100 nM Flag-tagged-EZH2 and 1 nM GST-tagged-NOP56.
(i) Co-IP of Myc-tagged FBL with full-length or truncation mutants of Flag-tagged NOP56.
The assays in a-c, e and g-i have been performed three times with similar results. Statistical source data and unprocessed blots are provided in Source data Extended data Fig. 3.
Extended Data Fig. 4. EZH2 alters box C/D snoRNP assembly.
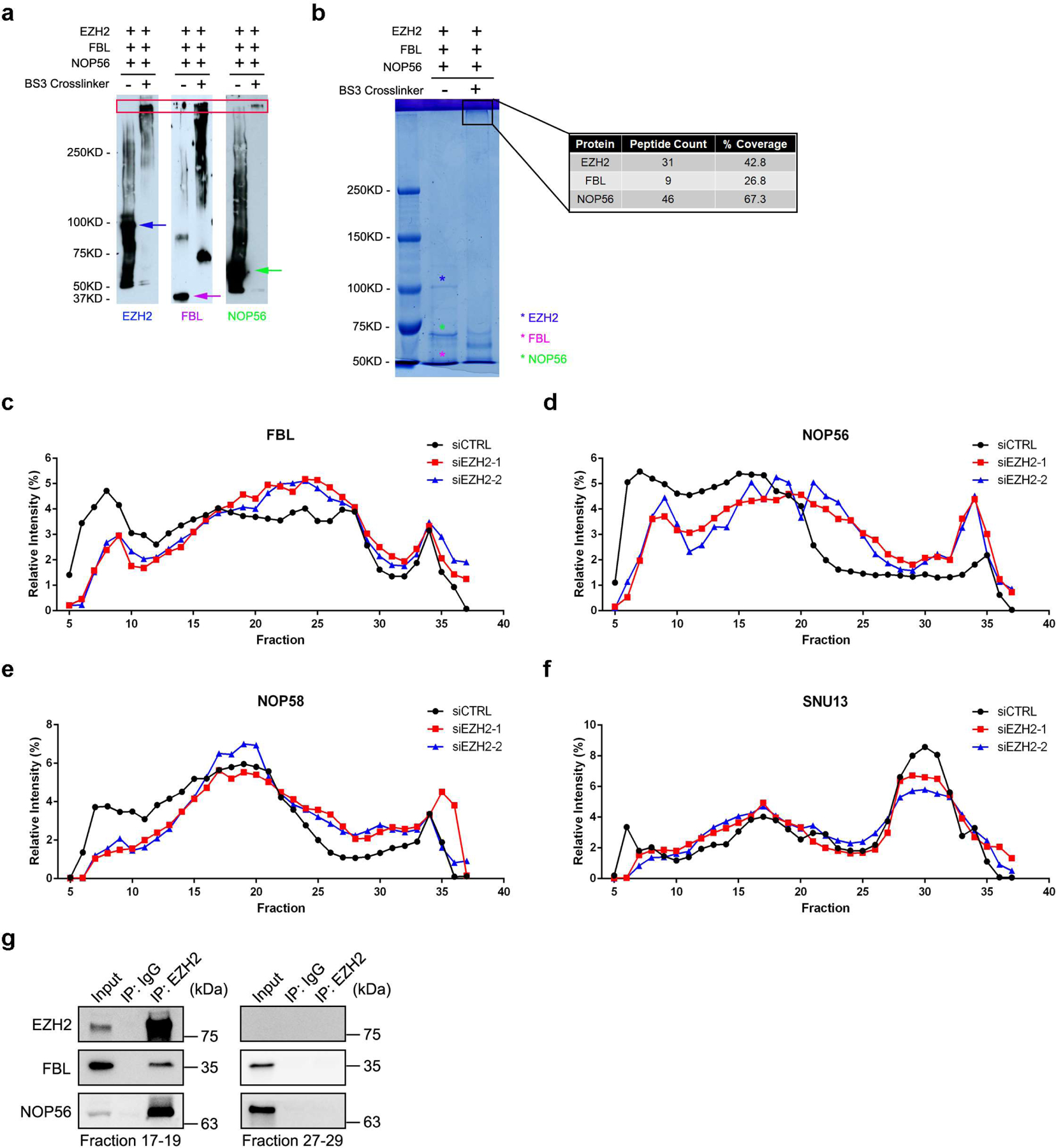
(a) Mixtures of recombinant EZH2, FBL and NOP56 proteins with or without BS3 crosslinking were subjected to SDS-PAGE, followed by western blot analysis using their own antibodies to visualize the location of cross-linked species.
(b) Gel band containing cross-linked proteins was subjected to Mass Spectrometry analyses to detect the presence of all three proteins.
(c-f) Densitometric analysis of data from Fig. 4a were shown as distribution proportion of FBL (c)/NOP56 (d)/NOP58 (e)/SNU13 (f) protein in each fraction.
(g) Fractions 17–19 and 27–29 from control C4–2 cell nuclear extracts were subjected to co-IP assay using anti-EZH2 antibody.
The assays in a-b and g have been performed three times with similar results. Statistical source data and unprocessed blots are provided in Source data Extended data Fig. 4.
Extended Data Fig. 5. EZH2 regulates the translation process independently of its mRNA-binding capacities.
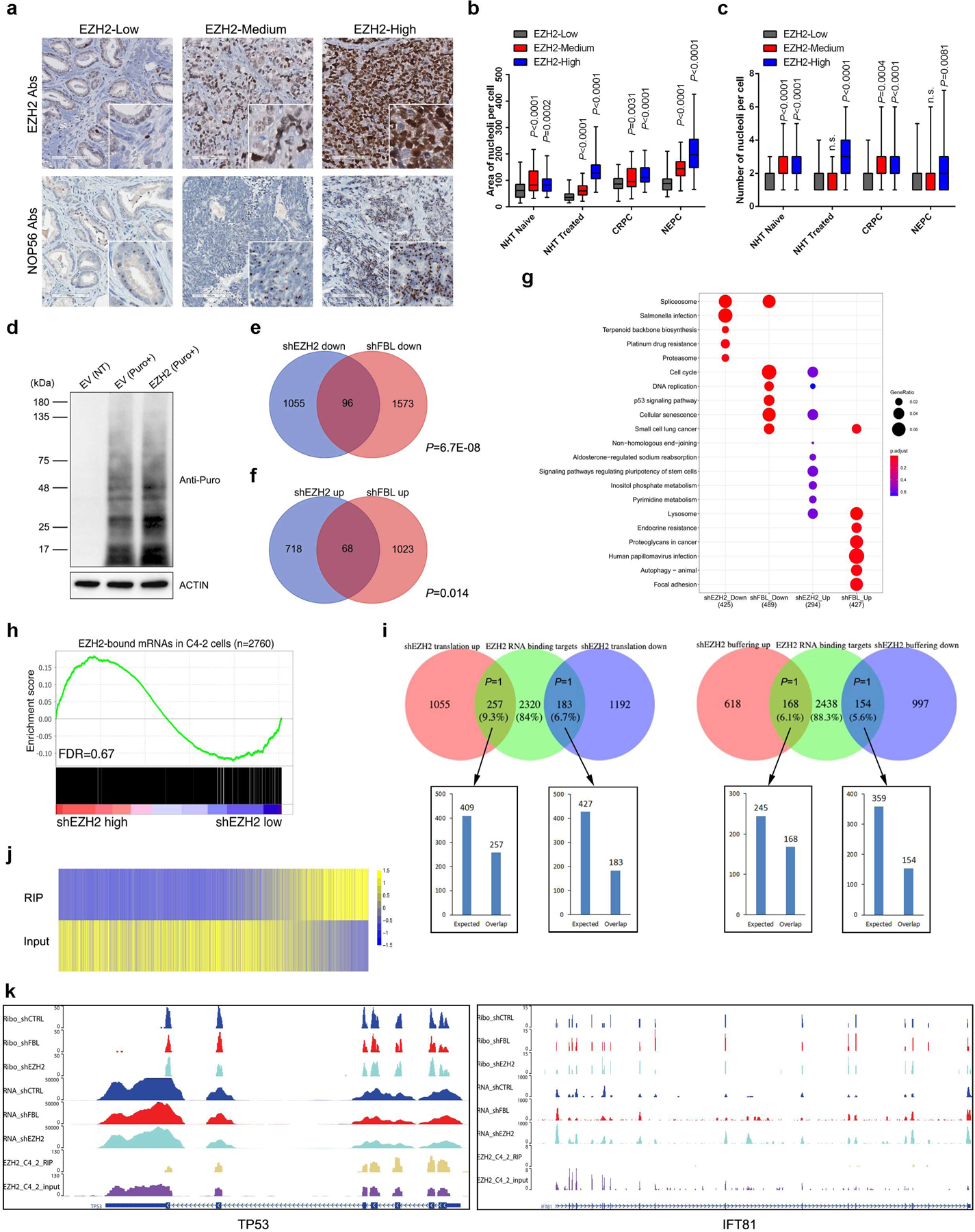
(a) Representative Immunohistochemistry staining images of PCa TMA slides using the indicated antibodies (Abs).
(b, c) Graph showing the relative nucleolar area (b) and nucleolar number (c) per cell by counting 60 cells from three TMA cores representing each group. The ends of box are the upper and lower quartiles and box spans the interquartile range. Median is marked by a vertical line inside the box and whiskers represent for the highest and lowest observations. NHT, neoadjuvant hormonal therapy; CRPC, castration-resistant PCa; NEPC, neuroendocrine PCa.
(d) Global protein synthesis in control and EZH2-overexpressing C4–2 cells were detected by Puromycylation assay followed by western blot. Expression of β-actin was used as reference.
(e, f) Venn diagram to show overlap between genes from “buffering” mode upon EZH2 or FBL deficiency. P values were calculated by one-tailed Fisher’s exact test.
(g) KEGG pathway analysis of genes from “buffering” mode upon EZH2 or FBL deficiency.
(h) GSEA analysis of genes bound by EZH2 to test their enrichment with TE changes after EZH2 knockdown.
(i) Venn diagram to show overlap between genes from two TE-altered groups after EZH2 knockdown and EZH2 RNA binding targets in C4–2 cells. P values were calculated by one-tailed Fisher’s exact test.
(j) Heatmap to show EZH2 RIP-seq and Input signals for TE-altered genes in EZH2-deficient C4–2 cells.
(k) Representative genome browser tracks to show Ribo-seq, RNA-seq and public EZH2 RIP-seq data at the loci of TP53 (a known mRNA binding target of EZH2) and IFT81 (an identified TE-altered gene after EZH2 inhibition). For all relevant panels, unless otherwise stated, statistical significance was determined by two-tailed Student’s t-test.
The assays in a and d have been performed three times with similar results. Statistical source data and unprocessed blots are provided in Source data Extended data Fig. 5.
Extended Data Fig. 6. EZH2 promotes XIAP IRES-dependent translation.
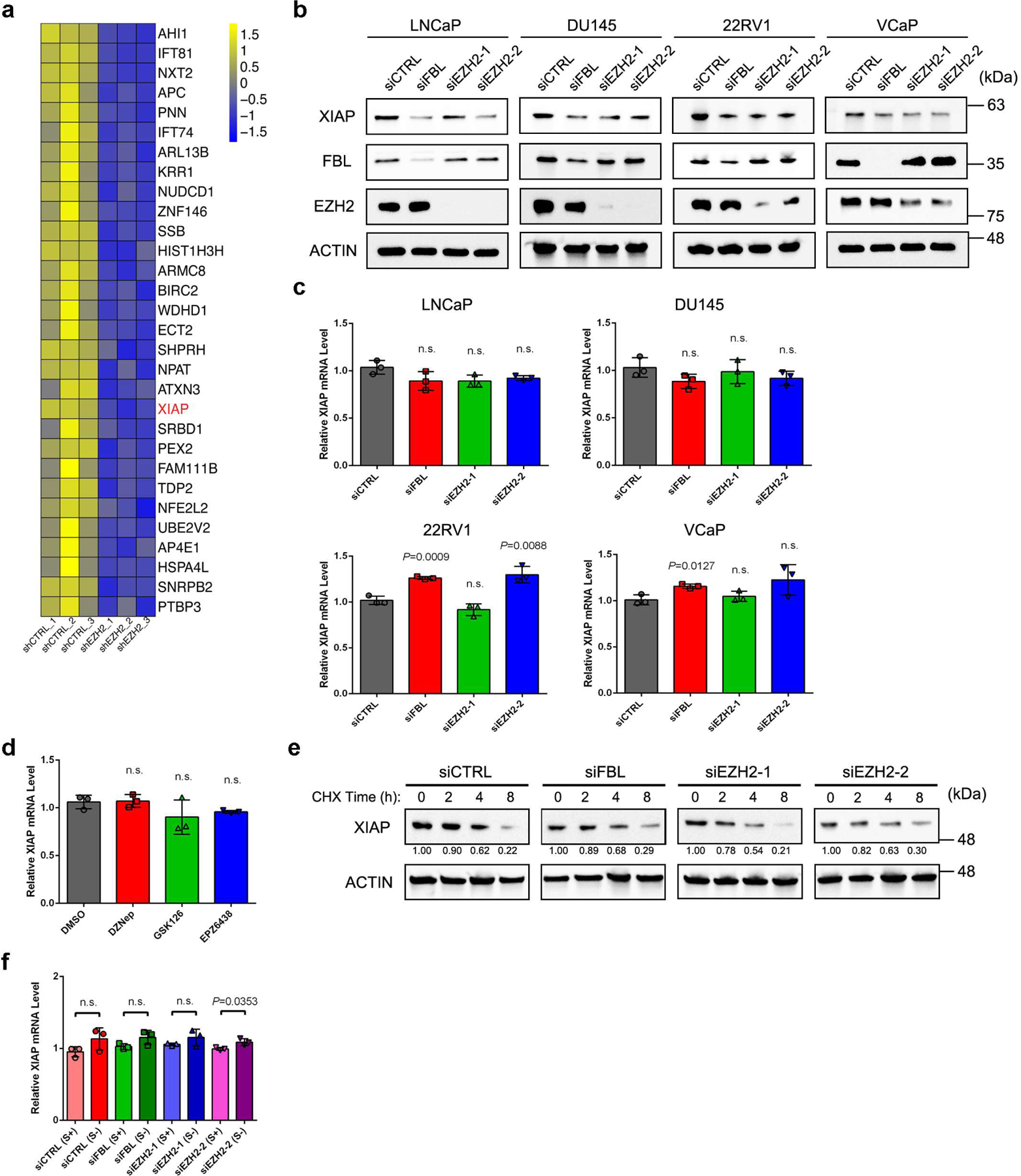
(a) Heatmap to show TE changes for the top 30 IRES genes which underwent TE down-regulation after EZH2 inhibition, as revealed from gene set enrichment analysis (GSEA).
(b) Western blot analysis of XIAP protein level upon FBL or EZH2 depletion in LNCaP, DU145, 22RV1 and VCaP cells.
(c) RT-qPCR analysis of XIAP mRNA level upon FBL or EZH2 depletion in LNCaP, DU145, 22RV1 and VCaP cells. Data represent Mean ± SD from n=3 biologically independent experiments.
(d) RT-qPCR analysis of XIAP mRNA level in C4–2 cells after treatment of various EZH2 inhibitors as indicated. Data represent Mean ± SD from n=3 biologically independent experiments.
(e) CHX treatment assay was performed to monitor the degradation of XIAP protein in control, FBL-deficient and EZH2-deficient cells. The relative protein level is shown under the bands.
(f) RT-qPCR analysis of XIAP mRNA level upon serum starvation in control, FBL-deficient and EZH2-deficient C4–2 cells. Data represent Mean ± SD from n=3 biologically independent experiments.
For all relevant panels, unless otherwise stated, statistical significance was determined by two-tailed Student’s t-test.
The assays in b and e have been performed three times with similar results. Statistical source data and unprocessed blots are provided in Source data Extended data Fig. 6.
Extended Data Fig. 7. Contributions of FBL and NOP56 in PCa tumorigenesis.
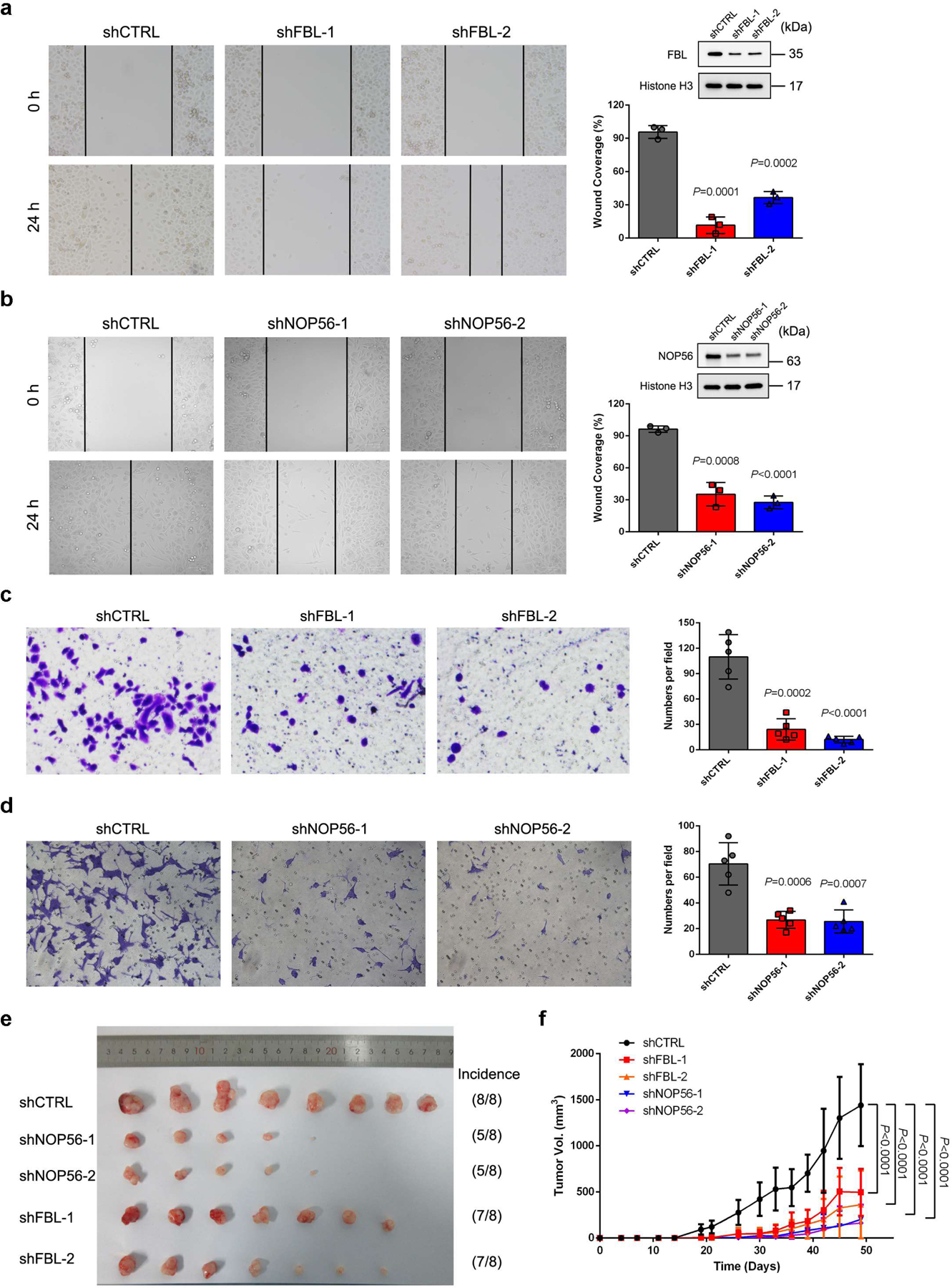
(a) Wound healing assay was conducted to evaluate the migration potential of PC-3 cells after FBL depletion. The healing of wounded cell layer was monitored under a microscope every 24 h. Graph showing the rate of filling of the scratched area by cells. Data represent Mean ± SD from n=3 biologically independent experiments. The knockdown efficiency of FBL was validated by western blot.
(b) Wound healing assay was conducted to evaluate the migration potential of PC-3 cells after NOP56 depletion. The healing of wounded cell layer was monitored under a microscope every 24 h. Graph showing the rate of filling of the scratched area by cells. Data represent Mean ± SD from n=3 biologically independent experiments. The knockdown efficiency of NOP56 was validated by western blot.
(c, d) Boyden chamber invasion assay was performed to determine the invasive capability of PC-3 cells after FBL depletion (c) or NOP56 depletion (d). Graph showing the number of migrated cells passing through Matrigel at 24 h. Data represent Mean ± SD from n=5 random fields per filter.
(e, f) Tumor formation in nude mice injected with control, FBL-deficient or NOP56-deficient PC-3 cells. The images of xenograft tumors at the end point of measurement were shown in e. Tumor volume was measured by caliper twice a week and plotted in f. Data represent Mean ± SD from n=8 tumors in each group. Statistical significance was determined by two-way ANOVA.
For all relevant panels, unless otherwise stated, statistical significance was determined by two-tailed Student’s t-test.
Statistical source data and unprocessed blots are provided in Source data Extended data Fig. 7.
Extended Data Fig. 8. Model of the implication of EZH2 in control of gene expression.

EZH2 plays a dual-role to regulate gene expression. On one hand, EZH2 inhibits DNA transcription by catalyzing H3K27me3 marks in a PRC2-dependent manner; On the other hand, EZH2 activates mRNA translation by enhancing the functionality of FBL through a non-lysine methyltransferase role. Hence, EZH2 could exert its oncogenic functions by coordination of transcriptional inhibition (i.e., tumor suppressors) and promotion of translation (i.e., pro-oncogenic, anti-apoptotic, and survival proteins) during cancer progression.
Supplementary Material
Supplementary Table 1 Detailed information of RiboMETH-seq data.
(a) MethScore information for each 2′-O-Me site in 18S rRNA;
(b) MethScore information for each 2′-O-Me site in 5.8S rRNA;
(c) MethScore information for each 2′-O-Me site in 28S rRNA.
Supplementary Table 2 List of translation-affected genes upon 13 EZH2 or FBL inhibition.
(a) Quantification of translational efficiency of the coding genes from the Ribo-seq combined with RNA-seq upon EZH2 inhibition;
(b) Quantification of translational efficiency of the coding genes from the Ribo-seq combined with RNA-seq upon FBL inhibition.
Supplementary Table 3 Antibodies used in this study.
Supplementary Table 4 Oligonucleotides used for this study.
Supplementary Table 5 Recombinant proteins used for this study.
Acknowledgements
We thank Center for Advanced Microscopy/Nikon Imaging Center of Northwestern University for assistance with confocal microscopy. We thank Vancouver Prostate Centre for assistance with IHC and PLA assays. We appreciate Metabolomics Core Facility of Northwestern University for assistance with LC-MS/MS to detect 2′-O-Me status of RNA. We thank Dr. Jonathan C. Trinidad and the Laboratory for Biological Mass Spectrometry facility at IUB for conducting the Mass Spectrometry analysis of crosslink samples.
This work was supported by a startup funding provided by the Northwestern University. Part of effort for Q.C. was supported by Northwestern University, U.S. Department of Defense (W81XWH-17-1-0357, W81XWH-19-1-0563 and W81XWH-20-1-0504), American Cancer Society (RSG-15-192-01), NIH/NCI (R01CA208257 and Prostate SPORE P50CA180995 DRP) and Northwestern Univ. Polsky Urologic Cancer Institute. K.C. was supported by startup fundings provided by The Houston Methodist Research Institute and Boston Children’s Hospital. W.Z. was supported by National Natural Science Foundation of China (81972651, 31771630, 81802974, and 81702784), Guangdong Innovative and Entrepreneurial Research Team Program (2016ZT06S029), Natural Science Foundation of Guangdong Province (2017A030310228, 2017A030312009 and 2016A030313238), Medical Scientific Research Foundation of Guangdong Province of China (A2017110), and the Special funds for Dapeng New District Industry Development (KY20160309). H.N. was supported by National Institutes of Health grant R35GM124765. Z.J. was supported by R00 CA 207865 from National Cancer Institute. E.M.S. was supported in part by the National Institutes of Health grant 5U01CA196390, the Prostate Cancer Foundation, Department of Defense grant W81XWH-15-1-0661 (E.M.S. and T.L.L.). A.B.W. was supported by the 2019 Urology Care Foundation Residency Research Award Program and the Russell Scott, Jr., MD Urology Research Fund.
Footnotes
Competing interests
No potential conflict of interest to disclose.
Data availability
Next generation sequencing data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under accession codes GSE159004 (RiboMETH-seq) and GSE143975 (Ribo-seq/RNA-seq). Previously published EZH2 RIP-seq data that were re-analyzed here are available under accession code GSE63230. Source data are provided with this paper. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
References
- 1.Pelletier J, Thomas G & Volarevic S Ribosome biogenesis in cancer: new players and therapeutic avenues. Nature reviews. Cancer 18, 51–63 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Polikanov YS, Melnikov SV, Soll D & Steitz TA Structural insights into the role of rRNA modifications in protein synthesis and ribosome assembly. Nature structural & molecular biology 22, 342–344 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma S & Lafontaine DLJ ‘View From A Bridge’: A New Perspective on Eukaryotic rRNA Base Modification. Trends in biochemical sciences 40, 560–575 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Monaco PL, Marcel V, Diaz JJ & Catez F 2’-O-Methylation of Ribosomal RNA: Towards an Epitranscriptomic Control of Translation? Biomolecules 8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Massenet S, Bertrand E & Verheggen C Assembly and trafficking of box C/D and H/ACA snoRNPs. RNA biology 14, 680–692 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Falaleeva M, Welden JR, Duncan MJ & Stamm S C/D-box snoRNAs form methylating and non-methylating ribonucleoprotein complexes: Old dogs show new tricks. BioEssays : news and reviews in molecular, cellular and developmental biology 39 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shubina MY, Musinova YR & Sheval EV Nucleolar Methyltransferase Fibrillarin: Evolution of Structure and Functions. Biochemistry. Biokhimiia 81, 941–950 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Rodriguez-Corona U, Sobol M, Rodriguez-Zapata LC, Hozak P & Castano E Fibrillarin from Archaea to human. Biology of the cell 107, 159–174 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Erales J et al. Evidence for rRNA 2’-O-methylation plasticity: Control of intrinsic translational capabilities of human ribosomes. Proceedings of the National Academy of Sciences of the United States of America 114, 12934–12939 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kass S, Tyc K, Steitz JA & Sollner-Webb B The U3 small nucleolar ribonucleoprotein functions in the first step of preribosomal RNA processing. Cell 60, 897–908 (1990). [DOI] [PubMed] [Google Scholar]
- 11.Tessarz P et al. Glutamine methylation in histone H2A is an RNA-polymerase-I-dedicated modification. Nature 505, 564–568 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li D et al. Activity dependent LoNA regulates translation by coordinating rRNA transcription and methylation. Nature communications 9, 1726 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iyer-Bierhoff A et al. SIRT7-Dependent Deacetylation of Fibrillarin Controls Histone H2A Methylation and rRNA Synthesis during the Cell Cycle. Cell reports 25, 2946–2954 e2945 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Ren X et al. Maintenance of Nucleolar Homeostasis by CBX4 Alleviates Senescence and Osteoarthritis. Cell reports 26, 3643–3656 e3647 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Nachmani D et al. Germline NPM1 mutations lead to altered rRNA 2’-O-methylation and cause dyskeratosis congenita. Nature genetics 51, 1518–1529 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marcel V et al. p53 acts as a safeguard of translational control by regulating fibrillarin and rRNA methylation in cancer. Cancer cell 24, 318–330 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koh CM et al. Alterations in nucleolar structure and gene expression programs in prostatic neoplasia are driven by the MYC oncogene. The American journal of pathology 178, 1824–1834 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Su H et al. Elevated snoRNA biogenesis is essential in breast cancer. Oncogene 33, 1348–1358 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Cao R et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298, 1039–1043 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Margueron R & Reinberg D The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Plath K et al. Role of histone H3 lysine 27 methylation in X inactivation. Science 300, 131–135 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Boyer LA et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122, 947–956 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ezhkova E et al. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 136, 1122–1135 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Varambally S et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419, 624–629 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Kleer CG et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proceedings of the National Academy of Sciences of the United States of America 100, 11606–11611 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee ST et al. Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Molecular cell 43, 798–810 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Xu K et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 338, 1465–1469 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Y et al. EZH2 cooperates with gain-of-function p53 mutants to promote cancer growth and metastasis. The EMBO journal 38 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao Q et al. The central role of EED in the orchestration of polycomb group complexes. Nature communications 5, 3127 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han Z et al. Structural basis of EZH2 recognition by EED. Structure 15, 1306–1315 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Qin W et al. Quantitative time-resolved chemoproteomics reveals that stable O-GlcNAc regulates box C/D snoRNP biogenesis. Proceedings of the National Academy of Sciences of the United States of America 114, E6749–E6758 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong ZW et al. RTL-P: a sensitive approach for detecting sites of 2’-O-methylation in RNA molecules. Nucleic acids research 40, e157 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marchand V, Blanloeil-Oillo F, Helm M & Motorin Y Illumina-based RiboMethSeq approach for mapping of 2’-O-Me residues in RNA. Nucleic acids research 44, e135 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruggero D Translational control in cancer etiology. Cold Spring Harbor perspectives in biology 5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walters B & Thompson SR Cap-Independent Translational Control of Carcinogenesis. Frontiers in oncology 6, 128 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Eden ME, Byrd MP, Sherrill KW & Lloyd RE Demonstrating internal ribosome entry sites in eukaryotic mRNAs using stringent RNA test procedures. RNA 10, 720–730 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gan L et al. Epigenetic regulation of cancer progression by EZH2: from biological insights to therapeutic potential. Biomarker research 6, 10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lechertier T, Grob A, Hernandez-Verdun D & Roussel P Fibrillarin and Nop56 interact before being co-assembled in box C/D snoRNPs. Experimental cell research 315, 928–942 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Mattson G et al. A practical approach to crosslinking. Molecular biology reports 17, 167–183 (1993). [DOI] [PubMed] [Google Scholar]
- 40.David A et al. Nuclear translation visualized by ribosome-bound nascent chain puromycylation. The Journal of cell biology 197, 45–57 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oertlin C et al. Generally applicable transcriptome-wide analysis of translation using anota2seq. Nucleic acids research 47, e70 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Larsson O, Sonenberg N & Nadon R Identification of differential translation in genome wide studies. Proceedings of the National Academy of Sciences of the United States of America 107, 21487–21492 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao J et al. IRESbase: A Comprehensive Database of Experimentally Validated Internal Ribosome Entry Sites. Genomics, proteomics & bioinformatics (2020). [DOI] [PMC free article] [PubMed]
- 44.Krajewska M et al. Elevated expression of inhibitor of apoptosis proteins in prostate cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 9, 4914–4925 (2003). [PubMed] [Google Scholar]
- 45.Lewis SM & Holcik M IRES in distress: translational regulation of the inhibitor of apoptosis proteins XIAP and HIAP2 during cell stress. Cell death and differentiation 12, 547–553 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Holcik M, Lefebvre C, Yeh C, Chow T & Korneluk RG A new internal-ribosome-entry-site motif potentiates XIAP-mediated cytoprotection. Nature cell biology 1, 190–192 (1999). [DOI] [PubMed] [Google Scholar]
- 47.Ross AE et al. Tissue-based Genomics Augments Post-prostatectomy Risk Stratification in a Natural History Cohort of Intermediate-and High-Risk Men. European urology 69, 157–165 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Kaur HB et al. Association of tumor-infiltrating T-cell density with molecular subtype, racial ancestry and clinical outcomes in prostate cancer. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc 31, 1539–1552 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang YA & Yu J EZH2, an epigenetic driver of prostate cancer. Protein & cell 4, 331–341 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rothe B et al. Implication of the box C/D snoRNP assembly factor Rsa1p in U3 snoRNP assembly. Nucleic acids research 45, 7455–7473 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Q et al. Antihistamine Drug Ebastine Inhibits Cancer Growth by Targeting Polycomb Group Protein EZH2. Molecular cancer therapeutics (2020). [DOI] [PMC free article] [PubMed]
- 52.Kim J et al. Polycomb- and Methylation-Independent Roles of EZH2 as a Transcription Activator. Cell reports 25, 2808–2820 e2804 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- 53.Yu Y et al. Progesterone receptor expression during prostate cancer progression suggests a role of this receptor in stromal cell differentiation. The Prostate 75, 1043–1050 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Xie N et al. The expression of glucocorticoid receptor is negatively regulated by active androgen receptor signaling in prostate tumors. International journal of cancer 136, E27–38 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Li ZF & Lam YW A new rapid method for isolating nucleoli. Methods Mol Biol 1228, 35–42 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Jia G et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nature chemical biology 7, 885–887 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marchand V et al. Next-Generation Sequencing-Based RiboMethSeq Protocol for Analysis of tRNA 2’-O-Methylation. Biomolecules 7 (2017). [Google Scholar]
- 58.Pichot F et al. Holistic Optimization of Bioinformatic Analysis Pipeline for Detection and Quantification of 2’-O-Methylations in RNA by RiboMethSeq. Frontiers in genetics 11, 38 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Poulin F, Gingras AC, Olsen H, Chevalier S & Sonenberg N 4E-BP3, a new member of the eukaryotic initiation factor 4E-binding protein family. The Journal of biological chemistry 273, 14002–14007 (1998). [DOI] [PubMed] [Google Scholar]
- 60.Holcik M & Korneluk RG Functional characterization of the X-linked inhibitor of apoptosis (XIAP) internal ribosome entry site element: role of La autoantigen in XIAP translation. Molecular and cellular biology 20, 4648–4657 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huez I et al. Two independent internal ribosome entry sites are involved in translation initiation of vascular endothelial growth factor mRNA. Molecular and cellular biology 18, 6178–6190 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martineau Y et al. Internal ribosome entry site structural motifs conserved among mammalian fibroblast growth factor 1 alternatively spliced mRNAs. Molecular and cellular biology 24, 7622–7635 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vagner S et al. Alternative translation of human fibroblast growth factor 2 mRNA occurs by internal entry of ribosomes. Molecular and cellular biology 15, 35–44 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meng Z, Jackson NL, Shcherbakov OD, Choi H & Blume SW The human IGF1R IRES likely operates through a Shine-Dalgarno-like interaction with the G961 loop (E-site) of the 18S rRNA and is kinetically modulated by a naturally polymorphic polyU loop. Journal of cellular biochemistry 110, 531–544 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nanbru C et al. Alternative translation of the proto-oncogene c-myc by an internal ribosome entry site. The Journal of biological chemistry 272, 32061–32066 (1997). [DOI] [PubMed] [Google Scholar]
- 66.McGlincy NJ & Ingolia NT Transcriptome-wide measurement of translation by ribosome profiling. Methods 126, 112–129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang J, Kobert K, Flouri T & Stamatakis A PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shen W, Le S, Li Y & Hu F SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PloS one 11, e0163962 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Trapnell C, Pachter L & Salzberg SL TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anders S, Pyl PT & Huber W HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yu G, Wang LG, Han Y & He QY clusterProfiler: an R package for comparing biological themes among gene clusters. Omics : a journal of integrative biology 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen K et al. DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome research 23, 341–351 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li D, Hsu S, Purushotham D, Sears RL & Wang T WashU Epigenome Browser update 2019. Nucleic acids research 47, W158–W165 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ren J et al. DOG 1.0: illustrator of protein domain structures. Cell research 19, 271–273 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 Detailed information of RiboMETH-seq data.
(a) MethScore information for each 2′-O-Me site in 18S rRNA;
(b) MethScore information for each 2′-O-Me site in 5.8S rRNA;
(c) MethScore information for each 2′-O-Me site in 28S rRNA.
Supplementary Table 2 List of translation-affected genes upon 13 EZH2 or FBL inhibition.
(a) Quantification of translational efficiency of the coding genes from the Ribo-seq combined with RNA-seq upon EZH2 inhibition;
(b) Quantification of translational efficiency of the coding genes from the Ribo-seq combined with RNA-seq upon FBL inhibition.
Supplementary Table 3 Antibodies used in this study.
Supplementary Table 4 Oligonucleotides used for this study.
Supplementary Table 5 Recombinant proteins used for this study.
Data Availability Statement
Next generation sequencing data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under accession codes GSE159004 (RiboMETH-seq) and GSE143975 (Ribo-seq/RNA-seq). Previously published EZH2 RIP-seq data that were re-analyzed here are available under accession code GSE63230. Source data are provided with this paper. All other data supporting the findings of this study are available from the corresponding author on reasonable request.