

Abstract
Recent progress in genomics and molecular genetics has empowered novel approaches to closely study gene functions in disease-causing pathogens. In the human malaria parasite Plasmodium falciparum, the application of genome-based analyses, site-directed genome editing, and genetic systems that allow for temporal and quantitative regulation of gene and protein expression have been invaluable in defining the genetic basis of antimalarial resistance and elucidating candidate targets to accelerate drug discovery efforts. Using examples from recent studies, we review applications of some of these approaches in advancing our understanding of Plasmodium biology and illustrate their contributions and limitations in characterizing parasite genomic loci associated with antimalarial drug responses.
Keywords: Drug resistance, gene editing, genomics, genetic crosses, inducible expression, Plasmodium falciparum malaria
Plasmodium Biology and Investigations into Antimalarial Resistance and Modes of Action
Malaria, caused by Plasmodium parasites, remains a leading cause of morbidity and mortality especially in endemic resource-strained countries. According to World Health Organization estimates, there were 229 million cases and 409,000 malaria-related deaths worldwide in 2019, the vast majority of which occurred in sub-Saharan Africa [1]. Of the six human-infecting Plasmodium species, P. falciparum is the most virulent and lethal [2]. This species has a complex life cycle in human and female Anopheles mosquito hosts, with human infections comprising a haploid asexual phase divided into a pre-symptomatic liver stage that produces an estimated 10,000–30,000 merozoites per parasite over a one-week period, and an asexual blood stage (ABS) characterized by ~48-hour cycles of invasion, differentiation, mitotic replication, and egress. A proportion of ABS parasites develop into male and female gametocytes that, when ingested by a feeding mosquito, form gametes that develop into zygotes, ookinetes and subsequently oocysts on the midgut wall. Meiosis occurs during this period of sexual stage development in the mosquito. Inside these oocysts, haploid sporozoites form by sporogony and subsequently are released into the hemocoel, from where they migrate to the salivary glands. Sporozoites are then injected into a human host during a subsequent blood meal, where they enter the bloodstream and invade hepatocytes to initiate a new cycle of human infection. Strategies to control mosquito vector populations and to develop antimalarials active against ABS parasites and preferably additional life cycle stages are essential to curtail the burden of disease.
Chemotherapy has been the mainstay of malaria control and prophylaxis, with antimalarial drugs endowed with different modes of action developed in the past century (Box 1). However, the ability of P. falciparum in particular to develop resistance to these treatments has compromised their efficacy and raised the importance of using combinations as well as developing new drugs and identifying novel targets. The current World Health Organization-recommended first-line treatments of uncomplicated malaria, caused by ABS parasites, depend on artemisinin-based combination therapies (ACTs). These treatments comprise fast-acting semi-synthetic derivatives of the endoperoxide artemisinin (ART) the rapidly reduce the parasite biomass partnered with slower-acting but longer-lasting drugs to eliminate surviving parasites. The use of long-lasting insecticide-treated bed nets, indoor residual spraying, intermittent preventive treatment in infancy and pregnancy, and the global adoption of ACTs together have contributed to a significant reduction in malaria deaths between 2000 and 2015 [3]. However, this progress in malaria control has stalled, a situation aggravated by the spread of parasite and mosquito vector resistance to the front-line drugs and insecticides, respectively [4, 5]. In addition, the persistent threat of asymptomatic chronic infections that serve as parasite reservoirs that maintain the life cycle and allow for onward transmission [6], and the limited efficacy of the RTS,S/AS01 (see Glossary) candidate vaccine [7], necessitate sustained efforts towards achieving comprehensive malaria control. To expand the current antimalarial arsenal and overcome the shortcomings of ACTs, a detailed understanding of P. falciparum biology is crucial to identify druggable targets against which efficacious therapeutics can be developed.
Box 1. Antimalarials: Mode of Action and Mechanisms of Resistance
Antimalarial drugs in current clinical use are endowed with different modes of action and mechanisms of resistance [5, 8, 112]. QN, a former frontline antimalarial for P. falciparum used since the early 17th century, has been slow to select for resistance. This drug is currently used to treat pregnant women with uncomplicated P. falciparum malaria and provides an alternative to artesunate for severe malaria. QN has a complex mode of action that includes the inhibition of heme detoxification. The mechanism of QN resistance is multifactorial, and is partially associated with polymorphisms in pfcrt, pfmdr1 and potentially other genes. Similar to QN, CQ diffuses inside the parasite’s DV and prevents the biomineralization of toxic free heme into hemozoin. CQR is driven primarily by mutations in PfCRT (including K76T), which enable CQ efflux outside the DV, thus preventing drug accumulation at its primary site of action. ADQ, a 4-aminoquinoline structurally similar to CQ, also targets the heme detoxification pathway. Due to its ability to accumulate to high levels in the DV, ADQ and its metabolites are active against many CQ-resistant parasites. ADQ is currently an integral constituent of ACTs as a fixed dose combination with artesunate. Reduced parasite sensitivity to ADQ has been associated with polymorphisms in PfMDR1, including N86Y and D1246Y, which have been selected for by ADQ therapy in different malaria-endemic settings. PPQ, another 4-aminoquinoline and ACT partner drug (used in combination with DHA), also targets heme detoxification. PPQR is driven by point mutations in PfCRT (including F145I, T93S, I218F on the background of the Dd2 haplotype) and appears to be augmented via amplifications in PMII and PMIII.
Sulfadoxine plus pyrimethamine (SP) is a synergistic combination used for intermittent preventive treatment in pregnancy and infancy. SP interferes with folate biosynthesis, which is essential for DNA synthesis and parasite growth. Pyrimethamine inhibits PfDHFR while sulfadoxine inhibits PfDHPS. High-grade SP resistance occurs in mutant parasites harboring N51I, C59R and S108N point mutations in PfDHFR and A437G and K540E in PfDHPS.
Artemisinins are the current front-line antimalarials and are effective at killing ABS rings and trophozoites. ART and its derivatives are activated by Fe2+-heme, a byproduct of hemoglobin, and kill parasites by alkylating heme, proteins, and lipids. ARTR is driven by point mutations in PfK13, which reduce ring stage endocytosis of hemoglobin and enable parasite survival. The exact mechanism of action of the other ACT partner drugs lumefantrine and MFQ still remains unknown, although might in part involve inhibition of hemoglobin endocytosis. Recent efforts to identify new antimalarial compounds with low propensity to succumb to resistance have led to the discovery of “irresistible” compounds further discussed in Box 2.
Box 2. Irresistible Compounds as Attractive Antimalarial Candidates
Antimalarial drug development is currently focused on the need to identify compounds that can rapidly lower parasite biomass to stop disease progression, have multi-stage activity, and possess low propensity to develop resistance. Physicochemical and structural features that confer fast-killing and multi-target inhibition profiles could help make a compound less prone to succumbing to resistance. In a recent analysis of 48 diverse antimalarial compounds, acquisition of resistant P. falciparum parasites was repeatedly unsuccessful for 24 of these [113]. These compounds, fittingly dubbed ‘irresistibles’, present interesting candidates for drug development due to their lower likelihood of early failure from acquired drug resistance and strong correlation with fast-killing kinetics [113, 114]. Recent attempts to evaluate their resistance mechanisms and targets have involved selections with parasites that present with different chemosensitivity profiles [115] or a hypermutator line with an enhanced rate of mutation under drug pressure [116]. Proteomic and metabolomic approaches as well as selection experiments in a closely related organism like Toxoplasma gondii [117, 118] or a surrogate species like Saccharomyces cerevisiae [119] can provide additional methods to study these compounds.
Box 3. P. falciparum Crosses using Humanized Mice and Linkage Analyses
In the published P. falciparum genetic cross protocol utilizing human-liver chimeric immunodeficient (Rag2−/−/ IL2Rγ−/−) FRG huHep mice [14, 40], sporozoites resulting from meiotic recombination between distinct strains in infected mosquitoes are isolated from Anopheles salivary glands and injected into FRG huHep mice. These mice lack fumaryl acetoacetate hydrolase (FAH) that catabolizes toxic metabolites in blood and other tissues, resulting in liver damage. Cycling of NTBC (Nitisinone) results in depletion of mouse hepatocytes and repopulation of the mouse liver with human hepatocytes. These mice receive infusions of human RBCs, allowing parasites that emerge from human hepatocytes to initiate ABS development. These parasites are recovered and cloned for subsequent phenotyping and genotyping. QTL analysis is used to map loci associated with inheritance of a quantifiable phenotypic trait. A major single-gene trait will segregate phenotypic data into two groups, as shown for pfcrt for CQR, while a multi-gene, quantitative trait will exhibit a continuous phenotypic distribution, as seen for QN [46]. QTL is expressed as a log of the odds (LOD) score, which is the logarithm to base 10 of the ratio of the likelihood of a model with a QTL versus a model without a QTL. As exemplified in the cited studies, the QTL locus can be scanned for polymorphisms between parents in genes or gene expression levels to narrow down the candidate gene list. Candidate loci can then be validated by phenotyping genetically-edited parasites. An alternative approach is to directly phenotype and genotype progeny in bulk populations by applying selective pressure (alongside controls) and assessing allele frequency changes that associate with selection via bulk segregant analysis (also known as linkage group selection) [14, 26]. While the bulk segregant analysis is a high-throughput approach, only the clonal progeny QTL mapping can delineate how QTL loci interact. The expanded use of FRG huHep-based P. falciparum genetic crosses, complemented by emerging genomic tools, presents an exciting new frontier into research on antimalarial drug resistance and Plasmodium biology.
The complexity of the Plasmodium life cycle presents opportunities to develop drugs with prophylactic (liver stage), curative (ABS), and/or transmission-blocking (gametocyte stage) activity. Drug discovery efforts have focused predominantly on P. falciparum ABS parasites in part because of the ease of in vitro culture, and challenges in generating sufficient numbers of liver stages and gametocytes required to screen large compound libraries. Indeed, several physiological processes in ABS parasites, including host hemoglobin degradation and detoxification, folic acid biosynthesis, the mitochondrial electron transport chain, and pyrimidine biosynthesis, constitute important targets of antimalarials in clinical use or under development [8]. The development of reliable platforms for interrogating gametocytocidal and liver stage inhibitors has now broadened the scope of antimalarial drug discovery, and high-throughput phenotypic screening has led to the identification of new broad-range antimalarial chemical scaffolds with multi-stage activity [9–15]. Research into the modes of action of hits identified in these high-throughput phenotypic screens can then drive subsequent drug development and help identify mechanisms through which parasites can develop resistance.
The rapid acceleration in generating P. falciparum genome sequences and the implementation of other “omic” tools including chemoproteomics and metabolic fingerprinting have significantly enhanced approaches for studying critical physiological processes in P. falciparum parasites [16, 17]. As examples, chemoproteomic profiling identified phosphatidylinositol 4-kinase and cGMP-dependent protein kinase (PfPKG) as targets of potent antiplasmodial targets, as confirmed using resistance selection and conditional knockdown approaches [18, 19]. Genomic and genetic tools have also been invaluable in dissecting mechanisms of antimalarial drug resistance and other processes such as intracellular protein trafficking and red blood cell (RBC) egress and invasion [20–23]. This review recaps whole-genome approaches including in vitro resistance evolution, genome-wide association studies (GWAS), and experimental genetic crosses, along with landmark genetic findings, which have set the stage for current investigations into antimalarial drug resistance and Plasmodium biology.
Harnessing Plasmodium Genomes to Map Antimalarial Resistance Determinants
Genome-wide Association Studies of Antimalarial Resistance
Human-infecting Plasmodium species possess compact genomes of approximately 23–34 Mb, which in P. falciparum encode ~5,300 protein-coding genes, along with a varying number of sub-telomeric multigene families, spanning 14 chromosomes. Mining this genomics information has enabled the identification of genes contributing to various parasite traits, particularly those of antimalarial drug responses. GWAS investigations constitute a powerful genome-based approach to elucidating genetic determinants of antimalarial resistance (Figure 1A). The first P. falciparum GWAS report used geographically diverse isolates to characterize the parasite population structure, recombination rates, and loci under recent positive selection [24]. Another GWAS search for loci correlated with resistance to 13 antimalarials identified a new halofantrine resistance locus, PF3D7_1036300, whose overexpression was associated with decreased sensitivity to halofantrine, mefloquine (MFQ) and lumefantrine, and whose copy number amplification also mediated resistance [25]. Subsequent functional validation analyses using ΔPF3D7_1036300 parasites revealed increased sensitivity to the aryl-amino alcohols but not unrelated antimalarials, suggesting chemical class specificity. Analyses of field isolates revealed that the C591S allele of this locus strongly associated with halofantrine sensitivity among Senegalese samples and was linked to reduced sensitivity to lumefantrine in Kenya (reviewed in [26]). In another GWAS on culture-adapted isolates from Kenya, in vitro responses to 22 antimalarials mapped to loci including P. falciparum dihydrofolate reductase (pfdhfr, PF3D7_0417200), dihydropteroate synthetase (pfdhps, PF3D7_0810800), multidrug resistance protein 1 (pfmdr1, PF3D7_0523000), chloroquine resistance transporter (pfcrt, PF3D7_0709000) and the sodium/hydrogen exchanger (pfnhe-1, PF3D7_1303500), and reported a putative association between amodiaquine (ADQ) activity and single nucleotide polymorphisms (SNPs) in cysteine desulfurase (PF3D7_0716600) [27]. Another study on 10 common antimalarials using isolates from the China-Myanmar border region with a history of ART usage identified several loci associated with various drug responses including pfcrt and pfdhfr. This study also associated the autophagy-related protein 18 (PF3D7_1012900) with decreased parasite sensitivities to dihydroartemisinin (DHA), artemether and piperaquine (PPQ) [28]. Genome-wide associations between in vitro chloroquine (CQ)-sensitive and PPQ-resistant phenotypes and the C350R mutation in pfcrt have also been shown with isolates from French Guyana [29].
Figure 1. Genomic approaches for studying drug resistance.
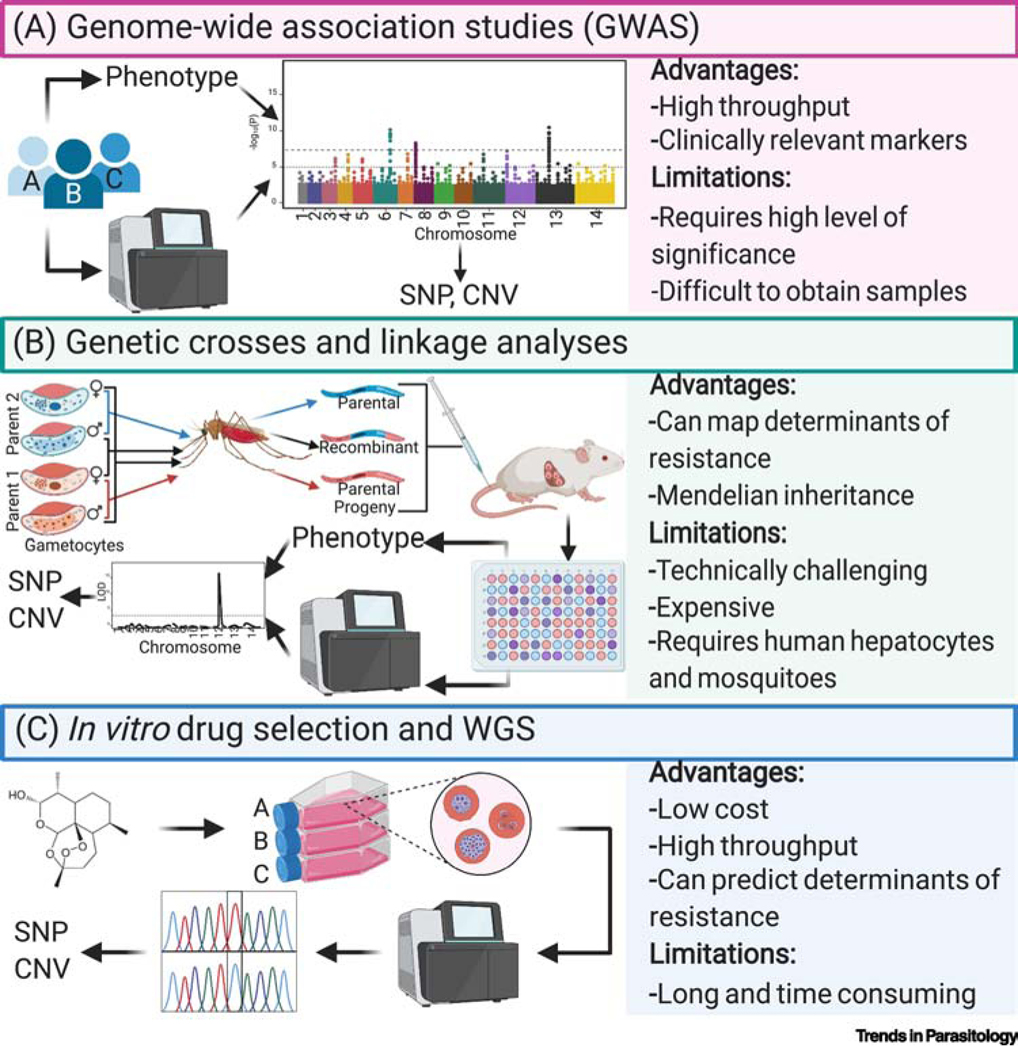
(A) Genome-wide association studies (GWAS): P. falciparum parasites are isolated from infected patients, profiled in vitro against antimalarial drugs of interest, and genotyped (using WGS, microsatellite markers or oligonucleotide arrays). These datasets are analyzed (as depicted visually in the Manhattan plot) to identify genetic variants associated with resistance. (B) Genetic crosses and linkage analyses: gametocytes from genetically distinct parental parasites are fed to Anopheles mosquitoes wherein they undergo sexual reproduction and genetic recombination, creating both parental and new recombinant genotypes. The resulting sporozoite haploid progeny are inoculated into FRG huHep mice by intravenous injection or mosquito bite, after which they develop inside the engrafted human hepatocytes and complete the liver to blood-stage transition after injection of human RBCs 6 to 7 days later. Progeny are cloned by limiting dilution and then genotyped and phenotyped. QTL analysis is conducted (as shown in the LOD plot) to localize the genetic determinant(s) of resistance and the polymorphisms that associate with the drug response variation. (C) in vitro drug selection and WGS (also known as in vitro evolution and whole-genome analysis [114]): antiplasmodial compounds are used to pressure P. falciparum parasites to select for recrudescent, resistant parasites. Genomic DNA (gDNA) of bulk cultures or their clones is sent for WGS, along with the parental strain, to identify genetic polymorphisms such as SNPs and CNVs that may underlie the resistance phenotype. WGS: whole-genome sequencing; SNP: single nucleotide polymorphism; CNV: copy number variation.
Epidemiological investigations into antimalarial resistance have also benefited from large-scale genetic field studies that have corroborated clinical evidence of increasing rates of ACT treatment failure. These include reports associating P. falciparum kelch13 (pfk13, PF3D7_1343700) mutations with slow parasite clearance in ART-treated patients [30, 31] and amplification of the hemoglobin protease plasmepsins (PM) II and III with PPQ resistance (PPQR) [32, 33]. Analyses of whole-genome variation in P. falciparum clinical samples from Asia and sub-Saharan Africa have also identified patterns of parasite population structure shaped by antimalarial selection pressure [34] and tracked the origins and dissemination of drug-resistant lineages and mutations [35].
Using Experimental Genetic Crosses and Linkage Analysis to Map Determinants
Experimental genetic crosses, which involve sexual stage recombination between two genetically distinct parasites to create recombinant progeny, are another powerful forward genetics tools to investigate the genetic basis of phenotypes including RBC invasion and growth, transmission, nutrient transport, and drug resistance, as well as studying the Plasmodium genome itself [14, 26] (Box 3). Marker-trait associations between phenotypic and genotypic data can be invoked from linkage analyses using quantitative trait locus (QTL) mapping, and the identified locus scanned to narrow down candidate genes (Figure 1B) [36–38]. Genetic determinants can then be validated by gene editing and phenotype profiling and examined in subsequent functional experiments. Although earlier studies of P. falciparum genetic crosses (Table 1) utilized splenectomized chimpanzees and monkeys to complete the human stages of the life cycle [26], recent crosses have used FRG-NOD human liver-chimeric (FRG huHep) mice that support P. falciparum liver and ABS infections [14, 39, 40].
Table 1.
Summary of Published Plasmodium falciparum Crosses
| Parent 1 | Parent 2 | Vertebrate Host | # of Unique Recombinant Progeny | Main Findings | Cross [Refs] | ||||
|---|---|---|---|---|---|---|---|---|---|
| Name | Origin (Year) | Phenotypes | Name | Origin (Year) | Phenotypes | ||||
| HB3 | Honduras (1980) | PYRR in vitro | 3D7 | Rwanda (1979)a | PYRS in vitro | Splenectomized chimpanzee | 15 | First to establish that P. falciparum genetic crosses can be conducted in non-human primates and mosquitoes; confirmed causal role of dhfr-ts SNPs in PYR resistance; associated pfmdr1 mutations with parasite response to MFQ. | [123] |
| Dd2 | Indochina (1980) | Verapamil-reversible CQR in vitro | HB3 | Honduras (1980) | Verapamil-irreversible CQS in vitro | Splenectomized chimpanzee | 35 | SNPs in pfcrt mediate CQ resistance; SNPs in pfmdr1 can modulate resistance to CQ; hemoglobin-derived peptide accumulation linked to CQR pfcrt alleles; loci associated with QN response and intracellular accumulation levels; SNPs in dhps mediate resistance to sulfadoxine; high expression of pfmdv-1 is important for gametocyte development; a region on chr. 12 is involved in regulating ABS cell cycle duration; cis and trans transcriptional regulation occur in P. falciparum. | [124] |
| 7G8 | Brazil (1980) | Aotus nancymaee non virulent in vivo | GB4 | Ghana (2000) | Aotus nancymaee - virulent in vivo | Splenectomized chimpanzee | 32 | PfRH5 is involved in ABS P. falciparum invasion of A. nanycmaee; an intergenic region between pfrh2a and pfrh2b is involved in P. falciparum alternative invasion pathways; SNPs in pfcrt and pfmdr1 contribute to resistance to ADQ. | [122] |
| GB4 | Ghana (2000) | CQR in vitro | NF54HT-GFP-luc | Rwanda (1979) | CQS, ARTS in vitro | FRG-NOD huHep mice | 4 | Proof-of-concept that FRG-NOD huHep mice can replace non-human primates for P. falciparum crosses. | [40] |
| 7G8 | Brazil (1980) | NF54HT-GFP-luc | Rwanda (1979) | CQS, ARTS in vitro | FRG-NOD huHep mice | 15 | Findings not yet reported. | [40] | |
| NHP*/ NHP4026 | Thailand-Myanmar border (2007) | CQR in vitro; Delayed clearance (but K13 WT) | NF54(HT-GFP-luc) | Rwanda (1979) | CQS, ARTS in vitro | FRG-NOD huHep mice | 84 | Can use a fresh clinical isolate as a cross parent in the huHep host system; geographical origin of parents may affect inbreeding and allele segregation; regions in chrs. 2, 13, and 14 are differentially selected in AlbuMAX vs serum media for ABS growth. | [40, 121] |
| 803 | Cambodia (2009–10) | Delayed clearance | GB4 | Ghana (2000) | ARTS in vitro | Splenectomized Aotus monkey | 27 | AS-treated K13 C580Y and C580 progeny showed similar in vivo clearance t1/2 in artemisinin-treated Aotus monkeys. | [44] |
| NHP1337 | Thailand-Myanmar border (2011) | Delayed clearance | MKK2835 | Thailand-Myanmar border (2003) | ARTS in vitro | FRG-NOD huHep mice | 60 | Geographical origin of parents may affect inbreeding and allele segregation; regions on chrs. 12 and 14 may compensate for ABS ARTR-associated fitness defects. | [39] |
The origin of 3D7, a clone of NF54, was recently mapped to Rwanda [36].
ABS, asexual blood stage; ADQ, amodiaquine; ART, artemisinin-resistant; ART, artemisinin-sensitive; chr., chromosome; CQ, chloroquine; GFP, green fluorescent protein; luc, luciferase; MFQ, mefloquine; PYR, pyrimethamine; QN, quinine; SNP, single nucleotide polymorphism
The first two genetic crosses, HB3×3D7 and HB3×Dd2, were used to map pyrimethamine and CQ resistance (CQR) to pfdhfr and pfcrt, respectively [26]. Analysis of the HB3×Dd2 cross progeny also indicated that pfmdr1 mutations can modulate the level of CQR [41] and linked hemoglobin-derived peptide accumulation to CQ-resistant pfcrt alleles [36]. The HB3×Dd2 cross also mapped segments putatively involved in quinine (QN) resistance, and pfdhps mutations that govern sulfadoxine resistance [14, 26, 37]. Investigations using the third cross, 7G8×GB4, mapped P. falciparum ABS invasion of Aotus nanycmaee RBCs to reticulocyte binding protein homologue 5 (pfrh5, PF3D7_0424100) and implicated an intergenic region between pfrh2a (PF3D7_1335400) and pfrh2b (PF3D7_1335300) in mediating an alternative invasion pathway [26, 38]. The 7G8×GB4 cross progeny also associated mutations in pfcrt and pfmdr1 with ADQ and CQ resistance [42]. A more recent cross between NF54(HT-GFP-luc) and NHP* (later called NHP4026) established that FRG huHep mice can replace non-human primates for P. falciparum crosses, and that fresh clinical isolates can be used as cross parents [40]. This cross was also used to map nutritional determinants, and identified regions in chromosomes 2, 13, and 14 that were differentially selected for ABS growth in lipid-enriched bovine serum albumin (Albumax) media versus media containing human serum [43]. In the fifth experimental cross between the 803 (PfK13 C580Y) and GB4 (PfK13 C580) strains, artesunate-treated PfK13 C580Y and C580 progeny displayed similar parasite clearances in Aotus monkeys despite their differential ART resistance (ARTR) phenotypes in vitro [44]. ABS progeny from the sixth reported cross between NHP1337 (PfK13 C580Y; ART-resistant) and MKK2835 (C580; ART-sensitive) revealed QTLs on chromosomes 12 and 14 favoring selection against the ART-resistant parent, which may indicate regions that compensate for fitness defects associated with in vitro mutant PfK13-mediated ARTR [39].
In characterizing the P. falciparum genome, genetic crosses have also been harnessed to assess recombination frequency, allele variability, de novo versus inherited copy number variations (CNVs), and transcriptional regulation using gene expression QTL [14, 26, 45]. Collective analyses of multiple crosses have indicated that indels are the most common form of polymorphism in the genome and crossover events are twice as frequent as non-crossover events. Furthermore, they showed that meiotic recombination can occur within CNVs encompassing drug resistance genes, revealing a mechanism for how parasites may compensate for fitness costs associated with resistance-conferring polymorphisms [45]. One advantage of genetic cross analysis in Plasmodium is that it is ‘simplified’ due to the Mendelian inheritance pattern with these haploid stages, as recombinant progeny will have a known allele of either parent at any locus. However, cost and demand for technical expertise with humanized mice and mosquitoes limit the routine use of P. falciparum genetic crosses, and inconsistencies in whether ABS parasites can maintain their ability to generate mature gametocytes is a complicating factor [14]. Genetic crosses with human Plasmodium spp. also require an in vitro culture system to obtain and phenotype progeny clones, excluding P. vivax from potential crosses, and restrict genotypes to two parents, which may cause genetic determinants to be missed and/or limit the influence of the genetic background. The genetic cross procedure itself introduces an unknown selective pressure that may also bias the resulting progeny pool [46].
Functional Genomic Analyses Using Mutant Parasite Screens
Phenotype-driven functional Plasmodium mutant screens have also revealed genes involved in all developmental stages [47–49], gametocytogenesis [50], apicoplast biogenesis [51], host cell remodeling [52], and mosquito transmission [53]. Three genome-wide Plasmodium mutant screens have been completed thus far, one in P. falciparum and two in the rodent malaria model species, P. berghei. A P. falciparum piggyBac transposon insertion saturation mutagenesis in the NF54 strain generated over 38,000 mutants created with mostly a single insertion per genome [49]. A mutagenesis index score and a mutagenesis fitness score were created to identify 2,680 of 5,399 genes as essential for ABS growth. Chemogenomic profiling of piggyBac mutants from a prior mutant library helped identify a group of seven DHA-sensitive mutants, including a pfk13 mutant that had a maximal expression in mid to late ring stages instead of the wild type early ring stage. This altered timing of expression was likely driven by the promoter of the drug selection cassette, as this cassette and its flanking piggyBac inverted terminal repeats were inserted into the endogenous putative pfk13 promoter [54]. This screen highlighted the power of the piggyBac genome-wide mutant library in identifying determinants of parasite susceptibility to selective agents. Genome-wide mutagenesis screens have also been completed in P. berghei with barcoded knockout mutants, and subsequent barcode counting on a next-generation sequencer [47, 48]. Analysis of the relative abundance of barcodes identified genes essential for ABS P. berghei growth and for the gametocyte to mosquito to liver to blood stage transitions. This system also enabled modeling of P. berghei liver stage metabolism to show major reprogramming at this stage, including 27 genes that represented seven metabolic systems - type II fatty acid synthesis and elongation, tricarboxylic acid, amino sugar, heme, lipoate, and shikimate metabolism - that were determined to be essential specifically in liver stages [48]. It is important to note, however, that these observed essentialities in the P. berghei model might not necessarily mirror those in P. falciparum. For example, while knockout mutants for genes encoding uridine-acetylglucosamine or N-acetylglucosamine-phosphate mutase show that UDP-N-acetyl-glucosamine synthesis is non-essential for P. berghei ABS parasites [47], unsuccessful attempts to knock out glucosamine-phosphate N-acetyltransferase [55] and the underrepresentation of transposon insertions into glucosamine-phosphate N-acetyltransferase, uridine-acetylglucosamine or N-acetylglucosamine-phosphate mutase genes [49] suggest that this biosynthetic pathway is essential for P. falciparum. Collectively, these functional genomic screens provide important insights into the essentiality and function of unannotated genes and help prioritize gene candidates identified in orthogonal screening experiments. Furthermore, barcoding of the mutant libraries allows for high-throughput profiling to investigate genetic determinants of phenotypes such as antimalarial drug resistance that may be elusive using in vitro selections.
In Vitro Drug Selection and Whole-Genome Sequence Analysis
Whole-genome sequencing (WGS) of parasite clones from in vitro evolution of resistance also exploits genome-wide information in identifying the genetic determinants that underlie antimalarial drug responses. [56]. During resistance selections, parasites are exposed to sublethal, continuous or dose-escalating drug pressure until the emergence of recrudescent resistant populations that show an increase in EC50 (the concentration required to inhibit parasite proliferation by 50%), characterized by a rightward shift in the concentration-response curves. Comparing genomes of the resistant clonal parasites to that of their untreated parent helps identify genome-encoded changes such as SNPs and CNVs that may underlie the resistance phenotype (Figure 1C). For instance, WGS analysis of resistant clones from in vitro selections using structurally diverse compounds identified CNVs and SNPs in 83 genes associated with resistance [57]. Drug pressuring with AN13762 [58], DSM265 [59], bortezomib or WLL [60–62], or DHA [63] has also traced in vitro resistance-conferring mutations to P. falciparum prodrug activation and resistance esterase (PF3D7_0709700), dihydroorotate dehydrogenase (PF3D7_0603300), the 20S proteasome β5 subunit (PF3D7_1011400), or coronin (PF3D7_1251200), respectively (Table 2). Selections with the imidazolopiperazines KAF156 and GNF179 also associated resistance with SNPs in the cyclic amine resistance locus (PF3D7_0321900), acetyl-CoA transporter (PF3D7_1036800), and UDP-galactose transporter (PF3D7_1113300) [21]. Other reports have demonstrated PfK13-independent resistance phenotypes arising from ART selections in vitro [64], increased copy number and mutations in P. falciparum purine nucleoside phosphorylase (PF3D7_0513300) that conferred resistance to transition-state analog inhibitors [65], and pfmdr1 mutations that confer resistance to hexahydroquinolines [66]. These examples, while not exhaustive of the large repertoire of in vitro evolution and WGS analysis, highlight the versatility of this approach.
Table 2.
Select Antiplasmodial Compounds Identified by Recent Drug Discovery Efforts
| Compound ID | Compound Class | Stage in Development | Putative Target | Target Location | Biological pathway | Pf Strain(s) for ABS | Resistance Mediator(s) | Refs |
|---|---|---|---|---|---|---|---|---|
| ACT-451840 | Piperazine | Phase I (NCT02223871, NCT02186002a) | Multidrug resistance 1 (PfMDR1)b | DV membrane | Unknown | Dd2, 7G8, NF54 | pfmdr1 (PF3D7_0523000) | [82] |
| AN13762 | Benzoxaborole | Pre-clinical | Cleavage and specificity factor homolog (PfCPSF3) | Nucleus | mRNA maturation | Dd2, 3D7 | Prodrug activation and resistance esterase (pfpare, PF3D7_0709700); SUMO-activating enzyme subunit 2 (pfuba2, Pf3D7_123700); E3 SUMO ligase (pfpias, Pf3D7_1360700), ring zinc-finger E3 ubiquitin ligase (Pf3D7_0529900); ubiquitin-activating enzyme 1 (Pf3D7_1350400); cleavage and polyadenylation specificity factor subunit 3 (pfcpsf3, PF3D7_1438500) | [57] |
| Bortezomib, WLL, WLW | Bortezomib: peptidyl boronic acid; WLL and WLW: peptide vinyl sulfones | Pre-clinical | P. falciparum 20S proteasome (β5 for bortezomib, β5 and β2 for WLL, β2 for WLW) | Proteasome | Ubiquitin-regulated protein degradation | Bortezomib: 3D7; WLL and WLW: Cam3.II K13WT, Cam3.II K13C580Y, V1/S K13WT, V1/S K13C580Y | Bortezomib: Pf 20S β5 (PF3D7_1011400); WLL: Pf 20S β6 (PF3D7_0518300) and β5; WLW: Pf 20S β2 (PF3D7_1328100), Pf 26S proteasome 19S regulatory particles (pfrpt4, PF3D7_1306400), (pfrpt5, PF3D7_1130400), (pfrpn6, PF3D7_1402300) | [59–61] |
| DSM265 | Triazolopyrimidine | Phase I/II (NCT02389348, NCT02573857, NCT02123290a) | Dihydroorotate dehydrogenase (PfDHODH) | Mitochondrion | Pyrimidine biosynthesis | Dd2, K1 | pfdhodh (PF3D7_0603300) | [58] |
| GNF-Pf-5640 | Hexahydroquinoline | Pre-clinical | Unknown | Unknown | Unknown | Dd2-B2 | pfmdr1 (PF3D7_0523000) | [65] |
| KAF156 and GNF179 | Imidazolopiperazines | KAF156: Phase II (NCT01753323, NCT04546633, NCT03167242a); GNF179: Pre-clinical | Unknown | Endoplasmic Reticulum/Golgi apparatus | Endoplasmic Reticulum-dependent protein processing | 3D7, Dd2 | Cyclic amine resistance locus (pfcarl, Pf3D7_0321900); UDP-galactose transporter (pfugt, PF3D7_1113300); acetyl-CoA transporter (pfact, PF3D7_1036800) | [20] |
| MB14 | 4-cyano-3-methylisoquinoline | Pre-clinical | Na+ efflux pump (PfATP4) | Parasite plasma membrane | ATP4-regulated Na+ homeostasis | 3D7 | non-SERCA-type Ca2+-transporting P-ATPase (pfatp4, PF3D7_1211900) | [66] |
| ML10 and MMV030084 | ML10: imidazopyridine; MMV030084: trisubstituted imidazole | Pre-clinical | cGMP-dependent protein kinase (PfPKG) | Cytosol and early secretory pathway | Parasite egress and host cell invasion | Dd2-B2 WT, Dd2-B2 TKL3KO | Tyrosine kinase-like protein 3 (tkl3, PF3D7_1349300) | [18, 68] |
ClinicalTrials.gov Identifier
Unknown if this is the primary target or resistance mediator
WGS of clones from in vitro selections can also inform target identification studies. For example, genome sequence data have revealed the Na+-dependent ATPase PfATP4 as the target of the multiple chemotypes including the recently described methylisoquinoline MB14 [67], cleavage and polyadenylation specificity factor subunit 3 as the target of benzoxaboroles [68], and PfPKG as the primary target of ML10 and MMV030084 [19, 69] (Table 2). As this approach is limited to compounds against which resistance can be easily generated, alternative methods are necessary for “irresistible” compounds (Box 2). Additionally, in vivo responses depend on the pharmacokinetics and pharmacodynamics of the drug thus restricting the translational value of this strategy. A recent proof-of-concept to compare the in vitro development of resistant P. falciparum parasites to that in an in vivo mouse model of P. falciparum infection [70] offers a good platform to interrogate the clinical relevance of identified genomic changes.
Genome Editing Approaches to Studying Antimalarial Resistance and Mode of Action
To demonstrate the power of genome-wide studies in elucidating functionally important loci in the P. falciparum genome, functional validation experiments using genetically-engineered parasites are critical. The advent of transfection-based genetic modification of P. falciparum enabled the first use of allelic exchange to confirm causal roles of drug resistance mediators, as shown for pfdhfr and pfdhps point mutations that mediate parasite resistance to pyrimethamine and sulfadoxine, respectively [71, 72]. Later, mutations in pfcrt were confirmed by allelic exchange to be the primary mediator of CQR [73]. Allelic exchange studies with polymorphic codons in pfmdr1 also implicated this ABC transporter in mediating resistance to MFQ and halofantrine [74]. Genetically-edited parasites can now be generated in a relatively efficient and scalable manner. One such method is site-specific zinc-finger nuclease (ZFN)-based editing in which parasites are transfected with a construct that encodes for pairs of customized deoxyribonucleic acid (DNA) sequence-specific zinc-finger proteins linked to a split endonuclease [75]. These DNA-binding proteins trigger double stranded breaks in the target sequence that trigger homology-directed DNA repair mechanisms using a plasmid-encoded donor sequence. This method of gene editing has helped elucidate the role of specific pfcrt and pfmdr1 mutations in multiple antimalarial resistance phenotypes [76–78], and also confirmed the essentiality of phosphatidylinositol 4-kinase and its role as a target of imidazopyrazines [79]. ZNFs also enabled the first and only documented evidence of gene editing in the human parasite P. vivax [80].
Gene disruptions in P. falciparum formerly involved extended culture periods of assessing integration via homologous recombination and single-site or double crossovers of an episomal plasmid, thus precluding large-scale genome manipulation efforts. CRISPR/Cas9 editing has circumvented this drawback by modifying a prokaryotic viral defense system to cleave a specific genomic sequence containing a unique 3’ protospacer adjacent motif (PAM) [81]. High-fidelity homologous recombination between the cleaved genomic locus and a donor sequence subsequently enables integration of the donor DNA [81, 82]. As examples, evaluation of PfPKG as a target of the imidazole compound MMV030084 [19], interaction of PfMDR1 with the piperazine-containing compound ACT-451840 [83], and the contribution of the PfK13 C580Y and R561H mutations to ARTR in vitro [82, 84], have all leveraged CRISPR/Cas9-based genome editing. CRISPR/Cas9 editing has also confirmed SNPs that confer resistance to the experimental antimalarials DSM265 [59], KAF156 and GNF179 [21], benzoxaboroles [68], and hexahydroquinolines [66]. In P. berghei, CRISPR/Cas9 editing was recently used to demonstrate that PfK13 mutations confer in vivo ARTR [85], and in a separate application was applied to ubiquitin hydrolase (PBANKA_0208800), which was found to modulate parasite susceptibility to ART and CQ [86].
CRISPR/Cas9 editing can also introduce epitope tags into genes of interest and help investigate their biological features. For example, CRISPR/Cas9 editing of a triple hemagglutinin (3xHA) epitope tag onto the adaptor protein 2 μ subunit (AP-2μ), followed by fluorescent and immune-electron microscopy, localized this trafficking protein to a compartment adjacent to the digestive vacuole (DV) and plasma membrane and enabled the identification of interacting partners including other AP-2 subunits, the K10 kelch-domain protein, and PfEHD, an effector of endocytosis and lipid mobilization [87]. Similarly, affinity purification of the Plasmodium translocon of exported (PTEX) protein with a CRISPR/Cas9-engineered 3×FLAG tag enabled its native structure to be solved by cryo-electron microscopy [88]. These direct observations of the PTEX structure provided evidence of its role as the gateway for the parasite’s exportome and yielded insights into the unique interactions between constituent proteins necessary for PTEX function. Nevertheless, transfection efficiency for P. falciparum remains low and complete editing of all transfected parasites is rare [81].
Selection-linked integration (SLI) has been developed as an alternative approach to generate parasites with targeted genomic integration events [89]. In this strategy, a promoter-less targeting region on the plasmid used for integration is linked with an additional selectable marker, separated by a 2A skip peptide that cleaves a single messenger RNA (mRNA) transcript into separate polypeptides. This additional selectable marker can be expressed only after single crossover recombination into the target locus, but because of the skip peptide, the marker is not attached to the target itself. Parasites carrying the integration can therefore be selected by using the additional integration-linked resistance marker (denoted as the SLI-resistance marker). The robustness of SLI has been validated via successful green fluorescent protein (GFP) fusion-based localization of native proteins, disruption of genes, knocking in an allele conferring ARTR, and functional analyses of known and newly identified essential genes at the protein level [89, 90].
Regulatable Systems in Antimalarial Resistance and Asexual Blood Stage Development
Typically, gene knockouts are only practical for dispensable genes or those whose products can be chemically and/or genetically compensated. Conditional expression systems that permit inducible regulation of RNA and protein levels offer promising alternatives to study essential genes. Target genes bearing a glucosamine-6-phosphate riboswitch (glmS) ribozyme in the 3’ untranslated region (UTR) can be efficiently knocked down in response to a glucosamine-6-phosphate inducer, leading to reduced protein levels [91] (Figure 2A). This system has enabled assessments of parasite sensitivity to the Plasmodium export element mimetic WEHI-916 using PMV knockdown lines [92], and hypersensitivity to pyrimethamine in parasites with attenuated expression of pfdhfr [91]. Target validation efforts through glmS ribozyme-mediated modification have also demonstrated deoxyhypusine synthase as a potential antiplasmodial target [93]. In addition, glmS-attenuated expression of eukaryotic translation initiation factor 2α kinase (PF3D7_0628200) has demonstrated its role in DHA-induced stress responses in the parasite endoplasmic reticulum [94]. This technique, however, can be confounded by glucosamine cytotoxicity due to its acidity and consequent ability to lower the pH of the medium [95].
Figure 2. Schematic representation of Plasmodium regulatable expression strategies.

(A) glmS riboswitch-based gene expression system: the targeting construct contains the glmS ribozyme at the 3’ end of the gene of interest. Addition of glucosamine activates the ribozyme, leading to cleavage of the mRNA and removal of the 3ʹ UTR, triggering rapid mRNA degradation by the proteasome, and a reduction in protein levels. (B) Dimerizable Cre (DiCre) system: the Cre recombinase is expressed as two inactive polypeptide moieties fused to FKBP12 and FRB, respectively. Rapamycin-induced dimerization leads to the association of the complementing Cre components and reconstitution of enzymatic activity. The Cre recombinase recognizes loxP sites (herein introduced via synthetic introns) to catalyze the excision (or insertion) of loxP-flanked DNA segments. This approach can be tailored to conditionally silence genes, introduce point mutations or generate fusions. (C) Destabilization domain-based regulation of protein expression, using Shield-1-protected protein-ddFKBP fusion proteins that degrade upon removal of Shield-1. (D) Knock-sideways system: FRB-mCherry fusion is localized in one subcellular compartment (in this example the nucleus) by a localization signal while the target protein, tagged with a FKBP-GFP fusion, is elsewhere in its native site of action (denoted herein as the cytoplasm). Addition of rapamycin stimulates heterodimerization of FRB and FKBP and results in mislocalization of the protein of interest, with possible phenotypic consequences. (E) aTc-inducible system: in this example a 10× tetracycline repressor protein (TetR) aptamer array is genetically encoded within the 3’ UTR of the target gene. A TetR-ATP-dependent RNA helicase DDX6 (DOZI) fusion protein is also expressed and binds the aptamer array mRNA, repressing translation of the target transcript. Addition of the tetracycline analog anhydrotetracycline (aTc) induces reversible allosteric interaction with the TetR-DOZI protein, disrupting the TetR-DOZI-aptamer interaction, and enabling mRNA expression of the target gene. Translation stops upon removal of aTc. Alternative designs include engineering aptamer arrays in the 5’ UTR or 5’ + 3’ UTRs [108]. aTc: anhydrotetracycline; FKBP: (FK506-binding protein); FRB: (binding domain of the FKBP-rapamycin-associated protein); term: terminus; loxPint: loxP site encoded in a synthetic intron; mC: mCherry; NLS: nuclear localization signal; Rapa: Rapamycin; S, Shield-1.
Genetic modification can also leverage sequence-specific recombination using the Cre recombinase that recognizes 34-nucleotide loxP sites and mediates excision or inversion of loxP-flanked sequences depending on the loxP orientation. The dimerizable Cre (DiCre) system (Figure 2B) involves expression of inactive Cre as two polypeptide moieties that fuse to FK506-binding protein (FKBP) 12 and FKBP rapamycin-binding (FRB) protein, respectively, with rapamycin-induced heterodimerization of the two Cre components restoring recombinase activity [96]. Following proof-of-principle experiments in P. falciparum ABS parasites, reports demonstrated the abrogation of merozoite invasion through DiCre-mediated conditional knockdown of cyclic AMP-dependent protein kinase A catalytic subunit (PF3D7_0934800) and actin-1 (PF3D7_1246200) expression [97, 98]. A marker-free NF54::DiCre parasite line has also been developed to conditionally delete genes across the life cycle [99]. More recently, a report on DiCre-mediated conditional disruption and allelic replacement of PfPKG demonstrated that the protein has no scaffolding or adaptor role in ABS development, and acts solely during merozoite egress [100]. Despite the relatively high efficiency of DiCre excision in P. falciparum, its removal of 3’ UTRs may not always reduce target protein levels if there is an alternative polyadenylation site that can stabilize the target mRNA [96]. Furthermore, given the low transfection efficiency in P. falciparum, it can take time to introduce two loxP sites that flank the desired target sequence. One recent innovation has been to introduce loxP sites as a part of silent synthetic introns (loxPint) [101]. The combined usage of loxPint-flanked kinase domains and the aforementioned NF54::DiCre parasite background has allowed the conditional knockdown of all 18 FIKK serine/threonine kinases, and the subsequent phosphoproteomic analyses of each kinase [102].
Protein levels can also be regulated by introducing unstable destabilizing domains, such as FKBP-based destabilization domain (DD) or an Escherichia coli DHFR destabilizing domain (DDD), into a protein of interest, thereby triggering proteasomal degradation (Figure 2C). This instability can be reversed by stabilizing compounds (Shield-1 and trimethoprim for DD and DDD fusion proteins, respectively), thus allowing tunable expression of protein levels. An earlier use of a FKBP protein destabilization domain (ddFKBP) in P. falciparum demonstrated that the swollen DV phenotype in falcipain-2 knockout lines could be rescued by expression of Shield-1-dependent falcipain-2-ddFKBP [103]. Transfected parasites expressing GFP fused to a DD have also been employed to investigate the anti-proteasome activity of novel peptidyl boronate hit compounds from a library of human proteasome inhibitors [61]. This strategy has also been used to assess the roles and essentiality of heat shock protein 70x and the receptor for activated c-kinase 1 [104, 105]. However, for some proteins, the degree of degradation may not be sufficient to produce a phenotype, in which case combining this system with another regulatable expression may tighten regulation. In addition, some proteins mistarget or do not function when tagged with DD, while others may not be degraded well as fusions [103].
Rapid mislocalization of proteins by the knock-sideways (KS) approach can also be used to study function [89]. KS is based on ligand-induced dimerization whereby the endogenous target is fused with FKBP, and FRB is fused to a signal that mediates trafficking to a different cellular location (Figure 2D). Upon addition of the ligand (often rapamycin or its analogs) that dimerizes FKBP and FRB, the target protein is directed away from its native site of action. This approach was validated through mislocalization and inducible inactivation of PfK13 [89]. More recently, the KS approach has been harnessed to investigate proteins within the PfK13 compartment that are essential for parasite survival. The combined use of KS and a quantitative BioID approach led these investigators to identify candidate proteins within the ARTR interactome [90]. Conditional inactivation through KS has also allowed for the evaluation of the role of vacuolar protein sorting-associated protein 45 (PF3D7_0216400) in host cell cytosol uptake [106] and phosphoinositide binding PH domain containing protein in merozoite attachment and invasion [107].
Blocking or permitting transcription of genes and then measuring the resulting cellular phenotype is also achievable using the tetracycline repressor protein (TetR), which binds to its associated DNA operator sequences, thereby blocking transcription [108]. This repression is reversible through allosteric interaction of TetR with tetracycline or its analog anhydrotetracycline (aTc). A recent adaptation of this approach is the TetR-DOZI aptamer system [108, 109], in which aTc-regulatable binding occurs between the TetR-DOZI fusion protein and 10×TetR RNA aptamer arrays encoded in the 3’ UTR of the gene of interest, leading to translation inhibition (Figure 2E). The TetR-DOZI system has been successfully leveraged in recent investigations of the druggability of PMV, PMIX and PMX as well as the nutrient-permeable channel EXP2 and the P. falciparum Niemann-Pick type C1-related protein [110, 111].
Concluding Remarks
Recent advances in genetic and genomic approaches to investigate Plasmodium biology have provided powerful tools to elucidate molecular determinants of antimalarial resistance and identify novel drug targets. The possibility of coupling site-specific editing techniques such as CRISPR/Cas9 with inducible systems heralds unprecedented robustness in validating genetic determinants of phenotypes including drug resistance, while the high-throughput scaling of genomic strategies enables increased analytical power (see Outstanding Questions). An established approach to studying antimalarials with an unknown mechanism of resistance is to first conduct in vitro selections with P. falciparum ABS parasites (the Dd2 B2 clone is often used), including a hypermutator line if needed. Uniquely barcoded panels of lines with SNPs or CNVs in known resistance determinants can also be drug pulsed as mixed cultures and the pool of surviving parasites barcode sequenced to assess whether known determinants can confer resistance. Libraries of knockout parasites (such as the piggyBac transposon-tagged library [49]) can also be screened, with the caveat that mutants of essential genes may not be available for profiling. A more resource-demanding, but high-yield alternative is to conduct genetic crosses between genotypically and phenotypically distinct clinical isolates, to investigate mechanisms of drug resistance that are elusive through in vitro drug selection, as well as other biological processes of interest. Candidate genes obtained by these methods can then be narrowed down by markers identified in GWAS, evidence of positive selection in field isolates, and targeted profiling of select barcoded mutants if available. Candidates can then be validated in a relatively high-throughput manner by conditional knockdown or editing of these genes (ideally in more than one genetic background). The continued refinement and use of these tools can be a powerful catalyst for ongoing research into discovering and developing new, safe, and efficacious antimalarials.
Outstanding Questions.
How can we increase experimental throughput in the identification of drug modes of action and mechanisms of resistance?
How can we close the gap between the lab and the field so that we can predict clinical resistance markers in vitro, and identify causal mechanisms as resistance emerges in real time? How can we best incorporate genetic crosses and functional genomic screens into this process, alongside the traditional GWAS and gene editing studies?
How does the genetic background influence parasite response to antimalarial treatment, and are there common genetic factors that make some parasites intrinsically less susceptible to drugs than others?
What methods can best maximize recombination and genetic diversity amongst progeny and minimize self-fertilization?
Highlights.
CRISPR/Cas9 and other site-specific gene editing techniques enable rapid and efficient generation of knockouts, targeted integrants, and allelic replacements of genes associated with malaria pathogenesis and drug resistance.
Regulatable genetic systems that quantitatively and temporally modulate expression levels facilitate mechanistic studies, especially those involving essential genes, and help distinguish on-target from off-target effects.
New or refined whole-genome based strategies such as functional genomic screens and genetic crosses in humanized mice now accompany traditional techniques including genome-wide association studies (GWAS) and gene editing to identify and characterize genetic determinants of resistance.
ACKNOWLEDGEMENTS
D.A.F. gratefully acknowledges support from the Medicines for Malaria Venture, the Bill & Melinda Gates Foundation (OPP1201387), the Department of Defense (W81WXH-15–2-0033 and W81XWH-19–1-0086) and the NIH (R01 AI109023, R01 AI124678, R01 AI147628, R33 AI127581). M.K. gratefully acknowledges support from the Japan Student Services Organization. The authors also thank members of the Malaria Drug Accelerator (MalDA) Consortium (led by Dr. Elizabeth Winzeler, OPP1054480) for their collaboration and stimulating discussions. Figures were created with BioRender.com.
GLOSSARY
- BioID:
A screening approach in which the protein of interest is fused to a biotin ligase that biotinylates proximal proteins, facilitating the identification of in situ protein interacting partners upon addition of biotin and subsequent biotin-affinity capture.
- FRG-NOD human-liver chimeric (FRG huHep) mouse:
Immunocompromised human liver-chimeric mice that can be inoculated with P. falciparum sporozoites and later injected with human RBCs to facilitate the mosquito to liver to blood stage transitions for the progeny of P. falciparum genetic crosses.
- Genome-wide Association Studies (GWAS):
A genetic approach involving an analysis of distinct field isolate genomes and their associated in vitro or in vivo phenotypes (including drug resistance) to determine genetic markers that strongly associate with those phenotypes and thus identify biomarkers for predictive purposes.
- Homologous recombination:
A mechanism for exchanging genetic information between two homologous double-stranded DNA sequences. In ABS parasites this mechanism can be leveraged to achieve allelic exchange following episomal plasmid-based single or double crossover events, or gene editing via the induction of ZFN- or CRISPR/Cas9-mediated double stranded breaks that trigger homology-directed repair using a donor template. Homologous recombination also creates recombinant progeny during meiosis between sexual stage parasites in mosquito midguts.
- Hypermutator line:
A P. falciparum Dd2 line with the D308A and E310A point mutations in the DNA polymerase - that ablate the polymerase’s proofreading (exonuclease) activity, which was earlier shown to increase the overall DNA mutation rate in P. berghei [120].
- Quantitative Trait Locus (QTL) mapping:
A statistical method of localizing chromosomal regions that affect the variation in quantitative traits by linking genotypic data (e.g SNPs) to phenotypic data (e.g. in vitro drug responses).
- RTS,S/AS01:
A recombinant malaria vaccine containing part of the P. falciparum circumsporozoite protein, co-expressed with hepatitis B surface antigen to induce anti-circumsporozoite antibodies and circumsporozoite-specific CD4-positive T cells associated with protection from P. falciparum infection and episodes of clinical malaria. RTS,S (co-administered with AS01 as an adjuvant) is the only malaria vaccine to reach Phase III testing and is currently in WHO pilot implementation in three African countries.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.World Health Organization; (2020) World Malaria Report 2020, WHO [Google Scholar]
- 2.Cowman AF, et al. (2016) Malaria: Biology and disease. Cell 167, 610–624 [DOI] [PubMed] [Google Scholar]
- 3.Gething PW, et al. (2016) Mapping Plasmodium falciparum mortality in Africa between 1990 and 2015. N Engl J Med 375, 2435–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.malERA: An updated research agenda for insecticide and drug resistance in malaria elimination and eradication. (2017) PLoS Med 14, e1002450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blasco B, et al. (2017) Antimalarial drug resistance: linking Plasmodium falciparum parasite biology to the clinic. Nat Med 23, 917–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen I, et al. (2016) “Asymptomatic” malaria: A chronic and debilitating infection that should be treated. PLoS Med 13, e1001942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duffy PE and Gorres JP (2020) Malaria vaccines since 2000: progress, priorities, products. NPJ Vaccines 5, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Phillips MA, et al. (2017) Malaria. Nat Rev Dis Primers 3, 17050. [DOI] [PubMed] [Google Scholar]
- 9.Alam MM, et al. (2019) Validation of the protein kinase PfCLK3 as a multistage cross-species malarial drug target. Science 365, eaau1682 [DOI] [PubMed] [Google Scholar]
- 10.Antonova-Koch Y, et al. (2018) Open-source discovery of chemical leads for next-generation chemoprotective antimalarials. Science 362, eaat9446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baragana B, et al. (2015) A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature 522, 315–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brancucci NM, et al. (2015) An assay to probe Plasmodium falciparum growth, transmission stage formation and early gametocyte development. Nat Protoc 10, 1131–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roth A, et al. (2018) A comprehensive model for assessment of liver stage therapies targeting Plasmodium vivax and Plasmodium falciparum. Nat Commun 9, 1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vendrely KM, et al. (2020) Humanized mice and the rebirth of malaria genetic crosses. Trends Parasitol 36, 850–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dorjsuren D, et al. (2021) Chemoprotective antimalarials identified through quantitative high-throughput screening of Plasmodium blood and liver stage parasites. Sci Rep 11, 2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cowell AN and Winzeler EA (2019) Advances in omics-based methods to identify novel targets for malaria and other parasitic protozoan infections. Genome Med 11, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kloehn J, et al. (2016) Using metabolomics to dissect host-parasite interactions. Curr Opin Microbiol 32, 59–65 [DOI] [PubMed] [Google Scholar]
- 18.Paquet T, et al. (2017) Antimalarial efficacy of MMV390048, an inhibitor of Plasmodium phosphatidylinositol 4-kinase. Sci Transl Med 9, eaad9735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vanaerschot M, et al. (2020) Inhibition of resistance-refractory P. falciparum kinase PKG delivers prophylactic, blood stage, and transmission-blocking antiplasmodial activity. Cell Chem Biol 27, 806–816 e808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flueck C, et al. (2019) Phosphodiesterase beta is the master regulator of cAMP signalling during malaria parasite invasion. PLoS Biol 17, e3000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lim MY, et al. (2016) UDP-galactose and acetyl-CoA transporters as Plasmodium multidrug resistance genes. Nat Microbiol 1, 16166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marapana DS, et al. (2018) Plasmepsin V cleaves malaria effector proteins in a distinct endoplasmic reticulum translocation interactome for export to the erythrocyte. Nat Microbiol 3, 1010–1022 [DOI] [PubMed] [Google Scholar]
- 23.Ross LS, et al. (2018) Emerging Southeast Asian PfCRT mutations confer Plasmodium falciparum resistance to the first-line antimalarial piperaquine. Nat Commun 9, 3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mu J, et al. (2010) Plasmodium falciparum genome-wide scans for positive selection, recombination hot spots and resistance to antimalarial drugs. Nat Genet 42, 268–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Tyne D, et al. (2011) Identification and functional validation of the novel antimalarial resistance locus PF10_0355 in Plasmodium falciparum. PLoS Genet 7, e1001383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Su XZ, et al. (2019) Plasmodium genomics and genetics: New insights into malaria pathogenesis, drug resistance, epidemiology, and evolution. Clin Microbiol Rev 32, e00019–00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wendler JP, et al. (2014) A genome wide association study of Plasmodium falciparum susceptibility to 22 antimalarial drugs in Kenya. PLoS One 9, e96486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z, et al. (2016) Genome-wide association analysis identifies genetic loci associated with resistance to multiple antimalarials in Plasmodium falciparum from China-Myanmar border. Sci Rep 6, 33891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pelleau S, et al. (2015) Adaptive evolution of malaria parasites in French Guiana: Reversal of chloroquine resistance by acquisition of a mutation in pfcrt. Proc Natl Acad Sci USA 112, 11672–11677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ariey F, et al. (2014) A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505, 50–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miotto O, et al. (2015) Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat Genet 47, 226–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amato R, et al. (2017) Genetic markers associated with dihydroartemisinin-piperaquine failure in Plasmodium falciparum malaria in Cambodia: A genotype-phenotype association study. Lancet Infect Dis 17, 164–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Witkowski B, et al. (2017) A surrogate marker of piperaquine-resistant Plasmodium falciparum malaria: A phenotype-genotype association study. Lancet Infect Dis 17, 174–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amambua-Ngwa A, et al. (2019) Major subpopulations of Plasmodium falciparum in sub-Saharan Africa. Science 365, 813–816 [DOI] [PubMed] [Google Scholar]
- 35.Cerqueira GC, et al. (2017) Longitudinal genomic surveillance of Plasmodium falciparum malaria parasites reveals complex genomic architecture of emerging artemisinin resistance. Genome Biol 18, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis IA, et al. (2014) Metabolic QTL analysis links chloroquine resistance in Plasmodium falciparum to impaired hemoglobin catabolism. PLoS Genet 10, e1004085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanchez CP, et al. (2014) A HECT ubiquitin-protein ligase as a novel candidate gene for altered quinine and quinidine responses in Plasmodium falciparum. PLoS Genet 10, e1004382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Campino S, et al. (2018) A forward genetic screen reveals a primary role for Plasmodium falciparum Reticulocyte Binding Protein Homologue 2a and 2b in determining alternative erythrocyte invasion pathways. PLoS Pathog 14, e1007436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X, et al. (2019) Genetic mapping of fitness determinants across the malaria parasite Plasmodium falciparum life cycle. PLoS Genet 15, e1008453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vaughan AM, et al. (2015) Plasmodium falciparum genetic crosses in a humanized mouse model. Nat Methods 12, 631–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patel JJ, et al. (2010) Chloroquine susceptibility and reversibility in a Plasmodium falciparum genetic cross. Mol Microbiol 78, 770–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sa JM, et al. (2009) Geographic patterns of Plasmodium falciparum drug resistance distinguished by differential responses to amodiaquine and chloroquine. Proc Natl Acad Sci USA 106, 18883–18889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumar S, et al. (2020) Bulk segregant approaches to nutritional genomics in Plasmodium falciparum. bioRxiv doi: 10.1101/2020.09.12.294736 [DOI] [Google Scholar]
- 44.Sa JM, et al. (2018) Artemisinin resistance phenotypes and K13 inheritance in a Plasmodium falciparum cross and Aotus model. Proc Natl Acad Sci USA 115, 12513–12518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miles A, et al. (2016) Indels, structural variation, and recombination drive genomic diversity in Plasmodium falciparum. Genome Res 26, 1288–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sen S and Ferdig M. (2004) QTL analysis for discovery of genes involved in drug responses. Curr Drug Targets Infect Disord 4, 53–63 [DOI] [PubMed] [Google Scholar]
- 47.Bushell E, et al. (2017) Functional profiling of a Plasmodium genome reveals an abundance of essential genes. Cell 170, 260–272 e268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stanway RR, et al. (2019) Genome-scale identification of essential metabolic processes for targeting the Plasmodium liver stage. Cell 179, 1112–1128 e1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang M, et al. (2018) Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 360, eaap7847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ikadai H, et al. (2013) Transposon mutagenesis identifies genes essential for Plasmodium falciparum gametocytogenesis. Proc Natl Acad Sci USA 110, E1676–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tang Y, et al. (2019) A mutagenesis screen for essential plastid biogenesis genes in human malaria parasites. PLoS Biol 17, e3000136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maier AG, et al. (2008) Exported proteins required for virulence and rigidity of Plasmodium falciparum-infected human erythrocytes. Cell 134, 48–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tewari R, et al. (2010) The systematic functional analysis of Plasmodium protein kinases identifies essential regulators of mosquito transmission. Cell Host Microbe 8, 377–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pradhan A, et al. (2015) Chemogenomic profiling of Plasmodium falciparum as a tool to aid antimalarial drug discovery. Sci Rep 5, 15930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cova M, et al. (2018) The Apicomplexa-specific glucosamine-6-phosphate N-acetyltransferase gene family encodes a key enzyme for glycoconjugate synthesis with potential as therapeutic target. Sci Rep 8, 4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rocamora F and Winzeler EA (2020) Genomic approaches to drug resistance in malaria. Annu Rev Microbiol 74, 761–786 [DOI] [PubMed] [Google Scholar]
- 57.Cowell AN, et al. (2018) Mapping the malaria parasite druggable genome by using in vitro evolution and chemogenomics. Science 359, 191–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sindhe KMV, et al. (2020) Plasmodium falciparum resistance to a lead benzoxaborole due to blocked compound activation and altered ubiquitination or sumoylation. mBio 11, e02640–02619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.White J, et al. (2019) Identification and mechanistic understanding of dihydroorotate dehydrogenase point mutations in Plasmodium falciparum that confer in vitro resistance to the clinical candidate DSM265. ACS Infect Dis 5, 90–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li H, et al. (2016) Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nature 530, 233–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xie SC, et al. (2018) Target validation and identification of novel boronate inhibitors of the Plasmodium falciparum proteasome. J Med Chem 61, 10053–10066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stokes BH, et al. (2019) Covalent Plasmodium falciparum-selective proteasome inhibitors exhibit a low propensity for generating resistance in vitro and synergize with multiple antimalarial agents. PLoS Pathog 15, e1007722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Demas AR, et al. (2018) Mutations in Plasmodium falciparum actin-binding protein coronin confer reduced artemisinin susceptibility. Proc Natl Acad Sci USA 115, 12799–12804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rocamora F, et al. (2018) Oxidative stress and protein damage responses mediate artemisinin resistance in malaria parasites. PLoS Pathog 14, e1006930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ducati RG, et al. (2018) Genetic resistance to purine nucleoside phosphorylase inhibition in Plasmodium falciparum. Proc Natl Acad Sci USA 115, 2114–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vanaerschot M, et al. (2017) Hexahydroquinolines are antimalarial candidates with potent blood-stage and transmission-blocking activity. Nat Microbiol 2, 1403–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gilson PR, et al. (2019) A 4-cyano-3-methylisoquinoline inhibitor of Plasmodium falciparum growth targets the sodium efflux pump PfATP4. Sci Rep 9, 10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sonoiki E, et al. (2017) A potent antimalarial benzoxaborole targets a Plasmodium falciparum cleavage and polyadenylation specificity factor homologue. Nat Commun 8, 14574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baker DA, et al. (2020) Targeting the malaria parasite cGMP-dependent protein kinase to develop new drugs. Front Microbiol 11, 602803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mandt REK, et al. (2019) In vitro selection predicts malaria parasite resistance to dihydroorotate dehydrogenase inhibitors in a mouse infection model. Sci Transl Med 11, eaav1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Triglia T, et al. (1998) Allelic exchange at the endogenous genomic locus in Plasmodium falciparum proves the role of dihydropteroate synthase in sulfadoxine-resistant malaria. EMBO J 17, 3807–3815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu Y, et al. (1996) Transformation of Plasmodium falciparum malaria parasites by homologous integration of plasmids that confer resistance to pyrimethamine. Proc Natl Acad Sci USA 93, 1130–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sidhu AB, et al. (2002) Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by pfcrt mutations. Science 298, 210–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reed MB, et al. (2000) Pgh1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum. Nature 403, 906–909 [DOI] [PubMed] [Google Scholar]
- 75.Straimer J, et al. (2012) Site-specific genome editing in Plasmodium falciparum using engineered zinc-finger nucleases. Nat Methods 9, 993–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dhingra SK, et al. (2019) Plasmodium falciparum resistance to piperaquine driven by PfCRT. Lancet Infect Dis 19, 1168–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim J, et al. (2019) Structure and drug resistance of the Plasmodium falciparum transporter PfCRT. Nature 576, 315–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Veiga MI, et al. (2016) Globally prevalent PfMDR1 mutations modulate Plasmodium falciparum susceptibility to artemisinin-based combination therapies. Nat Commun 7, 11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McNamara CW, et al. (2013) Targeting Plasmodium PI(4)K to eliminate malaria. Nature 504, 248–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moraes Barros RR, et al. (2015) Editing the Plasmodium vivax genome, using zinc-finger nucleases. J Infect Dis 211, 125–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee MCS, et al. (2019) Cutting back malaria: CRISPR/Cas9 genome editing of Plasmodium. Brief Funct Genomics 18, 281–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ghorbal M, et al. (2014) Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat Biotechnol 32, 819–821 [DOI] [PubMed] [Google Scholar]
- 83.Ng CL, et al. (2016) CRISPR-Cas9-modified pfmdr1 protects Plasmodium falciparum asexual blood stages and gametocytes against a class of piperazine-containing compounds but potentiates artemisinin-based combination therapy partner drugs. Mol Microbiol 101, 381–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Uwimana A, et al. (2020) Emergence and clonal expansion of in vitro artemisinin-resistant Plasmodium falciparum kelch13 R561H mutant parasites in Rwanda. Nat Med 26, 1602–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Simwela NV, et al. (2020) Plasmodium berghei K13 mutations mediate in vivo artemisinin resistance that is reversed by proteasome inhibition. mBio 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Simwela NV, et al. (2020) Experimentally engineered mutations in a ubiquitin hydrolase, UBP-1, modulate in vivo susceptibility to artemisinin and chloroquine in Plasmodium berghei. Antimicrob Agents Chemother 64, e02484–02419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Henrici RC, et al. (2020) The Plasmodium falciparum artemisinin susceptibility-associated AP-2 Adaptin μ subunit is clathrin independent and essential for schizont maturation. mBio 11, e02918–02919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ho CM, et al. (2018) Malaria parasite translocon structure and mechanism of effector export. Nature 561, 70–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Birnbaum J, et al. (2017) A genetic system to study Plasmodium falciparum protein function. Nat Methods 14, 450–456 [DOI] [PubMed] [Google Scholar]
- 90.Birnbaum J, et al. (2020) A Kelch13-defined endocytosis pathway mediates artemisinin resistance in malaria parasites. Science 367, 51–59 [DOI] [PubMed] [Google Scholar]
- 91.Prommana P, et al. (2013) Inducible knockdown of Plasmodium gene expression using the glmS ribozyme. PLoS One 8, e73783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sleebs BE, et al. (2014) Inhibition of Plasmepsin V activity demonstrates its essential role in protein export, PfEMP1 display, and survival of malaria parasites. PLoS Biol 12, e1001897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Aroonsri A, et al. (2019) Validation of Plasmodium falciparum deoxyhypusine synthase as an antimalarial target. PeerJ 7, e6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bridgford JL, et al. (2018) Artemisinin kills malaria parasites by damaging proteins and inhibiting the proteasome. Nat Commun 9, 3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Naik RS, et al. (2003) Glucosamine inhibits inositol acylation of the glycosylphosphatidylinositol anchors in intraerythrocytic Plasmodium falciparum. J Biol Chem 278, 2036–2042 [DOI] [PubMed] [Google Scholar]
- 96.Collins CR, et al. (2013) Robust inducible Cre recombinase activity in the human malaria parasite Plasmodium falciparum enables efficient gene deletion within a single asexual erythrocytic growth cycle. Mol Microbiol 88, 687–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Das S, et al. (2017) Multiple essential functions of Plasmodium falciparum actin-1 during malaria blood-stage development. BMC Biol 15, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wilde ML, et al. (2019) Protein kinase A Is essential for invasion of Plasmodium falciparum into human erythrocytes. mBio 10, e01972–01919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tiburcio M, et al. (2019) A novel tool for the generation of conditional knockouts to study gene function across the Plasmodium falciparum life cycle. mBio 10, e01170–01119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Koussis K, et al. (2020) Simultaneous multiple allelic replacement in the malaria parasite enables dissection of PKG function. Life Sci Alliance 3, e201900626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jones ML, et al. (2016) A versatile strategy for rapid conditional genome engineering using loxP sites in a small synthetic intron in Plasmodium falciparum. Sci Rep 6, 21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Davies H, et al. (2020) An exported kinase family mediates species-specific erythrocyte remodelling and virulence in human malaria. Nat Microbiol 5, 848–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Armstrong CM and Goldberg DE (2007) An FKBP destabilization domain modulates protein levels in Plasmodium falciparum. Nat Methods 4, 1007–1009 [DOI] [PubMed] [Google Scholar]
- 104.Blomqvist K, et al. (2017) Receptor for Activated C-kinase 1 (PfRACK1) is required for Plasmodium falciparum intra-erythrocytic proliferation. Mol Biochem Parasitol 211, 62–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cobb DW, et al. (2017) The exported chaperone PfHSP70x is dispensable for the Plasmodium falciparum intraerythrocytic life cycle. mSphere 2, e00363–00317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jonscher E, et al. (2019) PfVPS45 is required for host cell cytosol uptake by malaria blood stage parasites. Cell Host Microbe 25, 166–173 e165 [DOI] [PubMed] [Google Scholar]
- 107.Ebrahimzadeh Z, et al. (2019) A pan-apicomplexan phosphoinositide-binding protein acts in malarial microneme exocytosis. EMBO Rep 20, e47102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ganesan SM, et al. (2016) Synthetic RNA-protein modules integrated with native translation mechanisms to control gene expression in malaria parasites. Nat Commun 7, 10727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rajaram K, et al. (2020) Redesigned TetR-aptamer system to control gene expression in Plasmodium falciparum. mSphere 5, e00457–00420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Batista FA, et al. (2020) New directions in antimalarial target validation. Expert Opin Drug Discov 15, 189–202 [DOI] [PubMed] [Google Scholar]
- 111.Nasamu AS, et al. (2017) Plasmepsins IX and X are essential and druggable mediators of malaria parasite egress and invasion. Science 358, 518–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wicht KJ, et al. (2020) Molecular mechanisms of drug resistance in Plasmodium falciparum malaria. Annu Rev Microbiol 74, 431–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Corey VC, et al. (2016) A broad analysis of resistance development in the malaria parasite. Nat Commun 7, 11901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yang T, et al. (2021) MalDA, accelerating malaria drug discovery Trends Parasitol in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Goodman CD, et al. (2020) A single point mutation in the Plasmodium falciparum FtsH1 metalloprotease confers actinonin resistance. Elife 9, e58629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yoo E, et al. (2020) The antimalarial natural product salinipostin A identifies essential α/β serine hydrolases involved in lipid metabolism in P. falciparum parasites. Cell Chem Biol 27, 143–157 e145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Harding CR, et al. (2020) Genetic screens reveal a central role for heme metabolism in artemisinin susceptibility. Nat Commun 11, 4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rosenberg A, et al. (2019) Evolution of resistance in vitro reveals mechanisms of artemisinin activity in Toxoplasma gondii. Proc Natl Acad Sci USA 116, 26881–26891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.LaMonte GM, et al. (2020) Pan-active imidazolopiperazine antimalarials target the Plasmodium falciparum intracellular secretory pathway. Nat Commun 11, 1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Honma H, et al. (2016) Mutation tendency of mutator Plasmodium berghei with proofreading-deficient DNA polymerase δ. Sci Rep 6, 36971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Button-Simons KA, et al. (2019) Surprising variation in the outcome of two malaria genetic crosses using humanized mice: Implications for genetic mapping and malaria biology. bioRxiv doi: 10.1101/2019.12.13.871830 [DOI] [Google Scholar]
- 122.Hayton K, et al. (2008) Erythrocyte binding protein PfRH5 polymorphisms determine species-specific pathways of Plasmodium falciparum invasion. Cell Host Microbe 4, 40–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Walliker D, et al. (1987) Genetic analysis of the human malaria parasite Plasmodium falciparum. Science 236, 1661–1666 [DOI] [PubMed] [Google Scholar]
- 124.Wellems TE, et al. (1990) Chloroquine resistance not linked to mdr-like genes in a Plasmodium falciparum cross. Nature 345, 253–255 [DOI] [PubMed] [Google Scholar]