

Abstract
Glioblastoma, the deadliest form of primary brain tumor, remains a disease without cure. Treatment resistance is in large part attributed to limitations in the delivery and distribution of therapeutic agents. Over the last 20 years, numerous preclinical studies have demonstrated the feasibility and efficacy of stem cells as antiglioma agents, leading to the development of trials to test these therapies in the clinic. In this review we present and analyze these studies, discuss mechanisms underlying their beneficial effect and highlight experimental progress, limitations and the emergence of promising new therapeutic avenues. We hope to increase awareness of the advantages brought by stem cells for the treatment of glioblastoma and inspire further studies that will lead to accelerated implementation of effective therapies.
Keywords: : enzyme/prodrug, exosomes, glioblastoma, immunologic cell death, mesenchymal stem cells, nanoparticles, neural stem cells, oncolytic virotherapy, stem cells, therapeutic stem cells
Lay abstract
Glioblastoma is the deadliest and most common form of brain tumor, for which there is no cure. It is very difficult to deliver medicine to the tumor cells, because they spread out widely into the normal brain, and local blood vessels represent a barrier that most medicines cannot cross. It was shown, in many studies over the last 20 years, that stem cells are attracted toward the tumor and that they can deliver many kinds of therapeutic agents directly to brain cancer cells and shrink the tumor. In this review we analyze these studies and present new discoveries that can be used to make stem cell therapies for glioblastoma more effective to prolong the life of patients with brain tumors.
Practice points.
Stem cells (SCs) have a natural tumor-tropic migratory behavior and can be modified to deliver a variety of therapeutic agents and efficiently distribute their cargo into brain tumors, pursuing invading streams of tumor cells deep into the brain parenchyma.
Over the last 20 years, neural stem cells (NSCs), mesenchymal stem cells (MSCs) and induced NSCs have been developed as therapeutic agents for the treatment of glioblastoma.
There is a need for more mechanistic studies to determine the molecular determinants for optimal efficiency of therapeutic NSCs. This will allow for the development of more NSCs as clinical-grade agents. So far, only one NSC line (HB1.F3.CD) has been approved for use in clinical trials for glioblastoma.
There is a great need for a more rigorous characterization of therapeutic MSCs and for adopting standardized protocols across the scientific community, to ensure the safety of therapeutic MSCs and the reproducibility of studies across laboratories.
SCs have been developed for use in several therapeutic strategies, the most successful so far being represented by enzyme/prodrug and oncolytic virus delivery. Phase I clinical trials using these strategies are currently underway (NCT02015819, NCT04657315, NCT03072134, NCT03896568).
The efficiency of SC therapies depends on the number of therapeutic cells that are distributed throughout the tumor. Repeated administrations are needed to increase efficiency. Noninvasive or minimally invasive techniques for delivery of SCs are preferred, but need perfecting.
Exciting progress comes from using SCs conjugated with nanoparticles loaded with chemotherapeutics or nucleic acids, bringing the advantages of decreased systemic drug toxicity and the potential to molecularly target currently ‘undruggable’ glioma-specific factors.
SCs are prolific exosome-producing cells. Engineering SCs to produce glioma-targeted exosomes loaded with nucleic acids, proteins or chemotherapeutics represents a very promising emerging strategy.
SCs represent a viable and promising solution that can greatly improve upon current treatment strategies for glioblastoma. Concerted efforts to perfect therapeutic approaches with SCs from the scientific community, clinicians and industry will accelerate discovery and bring hope to patients burdened by this devastating disease.
The beginnings of therapeutic stem cells
The existence of stem cells as proliferating, specialized cells present in the periventricular spaces of the embryonic brain, postulated to give rise to terminally differentiated neurons and glia, was first recognized by the Swiss neurologist Wilhelm His (1874) [1,2]. The presence, properties and potential of these cells have fascinated the neuroscience community ever since. Studies in the mid-20th century established them as the source of both neurons and macroglia in the brain. Toward the end of the 20th century, when numerous elegant lineage-tracing studies demonstrated the ability of certain stem cells to become and potentially replenish any cell type in the CNS, neuroscientists agreed to name these cells ‘neural stem cells’ (NSCs) [3–6]. Recognition of the therapeutic potential of NSCs for various neurodegenerative diseases and traumatic injuries of the CNS was practically instantaneous. Early preclinical studies have demonstrated the potential use of NSCs in lysosomal storage diseases [7], Huntington’s disease [8], Parkinson’s disease [9,10], multiple sclerosis [11], amyotrophic lateral sclerosis [12] and spinal cord injury [13].
Today, more than 40 clinical trials are testing the use of NSCs for the treatment of a variety of neurological disorders, including glioblastoma (clinicaltrials.gov). Translation of preclinical findings into approved therapies has, however, been hindered by several barriers. Among these are ethical concerns raised against the use of fetal-derived cells and adverse consequences encountered when using unapproved stem cell interventions [14]. Clinical testing of stem cell therapies has also been slowed down by the enormous cost and effort required to develop approved ‘off the shelf’ stem cells as new investigational drugs, effort that entails creating efficient collaborations between scientists, clinicians, industry, regulatory bodies and funding agencies [15].
Malignant gliomas are the most common primary brain tumors, with an incidence of ∼6/100,000 in the years 2010–2014 [16]. The majority of gliomas (61.5%) are glioblastomas (GBM), the most aggressive form, for which no cure exists. The standard of care (SOC), which includes maximal safe tumor resection followed by radiation and chemotherapy, has remained relatively unchanged since 2005 [17]. Recently, it was demonstrated that the addition to SOC of tumor treating fields, low-intensity alternating electric fields delivered through transducer arrays placed on the patient’s scalp, increased overall survival in primary GBM from 16 months to 20.9 months [18]. While tumor treating fields have been approved by the US FDA as adjuvant therapy for newly diagnosed GBM, their use in clinical practice has so far remained limited [19,20]. The median life expectancy for patients diagnosed with GBM remains dismal at 15–17 months [16].
Following SOC, tumors invariably recur and progress to an increasingly aggressive form. Recurrence of GBM is attributed to many factors: the heterogeneous makeup of the tumors, making them difficult to target with single agents; the blood–brain barrier (BBB) and abnormal intratumoral vascularization that prevent drug distribution; the highly invasive and infiltrative nature of the disease, with glioma cells penetrating the brain parenchyma at great distance from the bulk of the tumor; the presence of glioma stem cells (GSCs), also referred to as brain tumor-initiating cells or recurrence-initiating stem cells [21], which are resistant to chemotherapy and radiotherapy and can initiate tumor formation; and an immunosuppressive tumor microenvironment which permits tumor growth. GSCs share many characteristics with NSCs, including high proliferative potential, association with blood vessels, telomerase activity, diverse progeny and similar gene expression profiles including NES, CD133, OLIG2, SOX2, SOX4, ITGA6, CD15, L1CAM, BMI1, A2B5, CD44 and others [22–26].
The idea that at the origin of GBM lies an NSC gone awry has long been suggested, but never entirely proven [27]. There exists, nonetheless, evidence that this may be the case, at least in a subset of tumors. For example, it was demonstrated that GBMs that are in contact with the subventricular zone (SVZ) and reach the cortex are more likely to be multifocal at initial diagnosis and/or to progress to multifocal disease at recurrence [28], suggesting a mechanism by which transformed cells in the SVZ (either NSCs or their progeny) migrate out toward the cortex, similar to the inside-out migration of neural progenitor cells (NPCs) guided by radial glia during cortical development [29].
Telomerase activity, required for the maintenance of telomere length, is a characteristic of both stem cell compartments and cancerous cells, serving important functions that promote cell survival and proliferation [30]. Germline and somatic mutations in the promoter of the TERT gene result in increased TERT promoter activity and are frequently observed in a number of solid cancers, including primary brain tumors, primarily in adults [31,32]. A compelling study in support of the notion that NSCs may represent the cell of origin in GBM analyzed the number of mutations present in the tumor and the tumor-free SVZ as well as in normal brain in 30 patients with GBM and other primary or metastatic tumors [33]. The authors reported that 42.3% of patients had at least one somatic mutation in the coding or promoter region of TERT that was shared between the tumor and tumor-free SVZ. The tumor-free SVZ of some GBM patients (5/16) also harbored driver mutations commonly found in GBM (in TP53, PTEN, EGFR and RB1), at a rate of about 1% of that found in the matched tumor. Mutations in the SVZ were localized to GFAP+ astrocyte-like stem cells, suggesting a lineage relationship between the SVZ NSCs and cells in the bulk of the tumor, in support of a scenario in which NSCs are at the origin of GBM, at least in some patients. TERT promoter mutations, the most common mutations identified in this study, were also found in normal brain tissue, albeit at lower frequency than in the tumor-free SVZ [33].
Our understanding of what constitutes a NSC – namely, a cell that can self-renew and generate both differentiated neurons and glia – has evolved over the years. During development, radial glia of the ventricular zone and SVZ are at the origin of all neurons and macroglia in the growing brain [34]. In the adult brain, neurogenic zones persist in the SVZ and the subgranular zone of the hippocampus, where NSCs are represented by specialized astrocytes: B cells in the SVZ and radial astrocytes in the hippocampus. NSCs give rise to intermediate progenitor cells, which further divide to generate NPCs and oligodendrocyte precursor cells [34,35]. NPCs are also multipotent progenitors that proliferate but have a more limited capacity of self-renewal and can differentiate into at least two different cell lineages [36]. Importantly, when cultured in vitro using the Neurosphere Assay, in serum free media supplemented with EGF and FGF, neurospheres derived from one NSC are a heterogeneous population of NSCs and NPCs that can give rise to neurons, oligodendrocytes and astrocytes [37–39].
Interestingly, a recent study using single-cell RNA sequencing (scRNA-Seq) of GBM samples collected from 11 patients identified a subpopulation of radial glia with high invasive potential, which also harbored a GSC genetic signature and displayed the mitotic somatic translocation phenotype characteristic of developmental radial glia [40]. Whether this reflects a potential origin of GBMs from arrested embryonic tissue that was reactivated during carcinogenesis is difficult to say; nonetheless, these findings support the idea that malignancy makes use of developmental mechanisms to promote growth and invasion. This study, as well as other recent studies using the powerful scRNA-Seq technology, highlights the enormously heterogeneous nature of GBMs and the widespread expression of GSC markers [41,42,43,44,45]. Comparing scRNA-Seq signatures between primary GSC-enriched GBM cells and human fetal brain NPCs, Couturier et al. described a conserved neural trilineage GBM hierarchy that is centered around glial progenitor-like cells, the most chemoresistant and tumorigenic cells, and mirrors the hierarchy of progenitor cells in normal brain development [45].
A defining feature of NPCs is their ability to migrate long distances. The CXCL12 chemokine and its cognate signaling receptor CXCR4, originally identified as a homeostatic chemokine/receptor complex that regulates hematopoietic stem cell trafficking, activates one of the main signaling pathways that drives NPC migration in the developing CNS and also guides migration of GSCs [46,47]. CXCL12 is highly expressed in the SVZ [47,48] and in the bone marrow hematopoietic stem cell niche, where it promotes migration of primitive hematopoietic cells [49]. Production of CXCL12 is increased inside tumors in response to hypoxia and irradiation [50]. High levels of CXCL12 are also found in pseudopallisades (a hallmark histological feature of WHO grade IV GBM), regions that also harbor GSCs [51,52]. GSCs and NSCs thus have shared migratory behavior, due at least in part to their responsiveness to CXCL12 chemotaxis.
Rodent NSCs transplanted into the brain exhibit an intrinsic migratory behavior, following developmental pathways [53,54]. Human NSCs (hNSCs) behave similarly. When hNSCs, generated from the periventricular telencephalic region of a 15-week-old human fetus and immortalized by retroviral transduction with v-myc, were injected into the lateral ventricles of newborn mice, they joined migrating endogenous mouse neuroblasts in the rostral migratory stream and differentiated into olfactory neurons [55]. Implanted hNSCs were also found to differentiate into oligodendrocytes, astrocytes or cerebellar granule cells depending on the site of transplantation, demonstrating their multipotency. Interestingly, it was observed that while the immortalized hNSCs proliferated well in vitro, once transplanted into mouse brains their expression of v-myc was downregulated and the cells stopped dividing [55]. The underlying cause of this behavior is not known, but is assumed to be a consequence of normal developmental mechanisms that induce mitotic arrest during differentiation [55]. Similar observations were made with mouse immortalized NSCs [56,57].
Considering the pressing need to find better strategies for intratumoral distribution of therapeutic agents, scientists tested immortalized NSCs in a mouse model of GBM [58]. It was elegantly demonstrated that when murine NSCs (C17.2 cells derived from the cerebellum of 4-day-old mice and immortalized with v-myc [59]) were injected into the tumor or at a distance from the tumor (in the contralateral hemisphere, intraventricularly or intravenously), they became distributed widely throughout the tumor, albeit with lower efficacy, following systemic administration [58]. NSCs also migrated toward invasive streams of glioma cells deep into the brain parenchyma. This tumor-tropic migratory behavior was not altered when cells were transduced to express a therapeutic enzyme, providing proof of principle that NSCs have the potential to deliver therapeutic agents in GBM.
Origin of stem cells used for the treatment of brain tumors
In addition to NSCs, mesenchymal stem cells (MSCs) and induced NSCs (iNSCs), derived from pluripotent stem cells or transdifferentiated from somatic cells, have been shown to display tumor-tropic behavior and distribute extensively in intracranial gliomas. Many preclinical trials have tested these cells for their ability to distribute throughout the tumor and deliver a variety of therapeutic agents: bioactive proteins, viruses, cytokines, antibodies, toxins or nanoparticles. The majority of these studies tested the use of human stem cells, though some used rodent stem cells. Immortalized human NSCs were most commonly used, followed by MSCs derived either from bone marrow, adipose tissue, umbilical cord or amniotic fluid, and one study used iNSCs transdifferentiated from fibroblasts. Rodent stem cells were employed similarly. Experimental details of the different studies are summarized in Supplementary Table 1. Interestingly, all but one study with human NSCs used cells obtained from fetal human brain and immortalized with the v-myc oncogene. Transduction of v-myc allowed for easy, long-term in vitro propagation and prevented terminal differentiation of these cells in the presence of serum [60].
The HB1.F3 cell line was derived from the telencephalon of a human female fetus at 15 weeks’ gestation. Cells were initially propagated on polylysine-coated tissue culture plates in Dulbecco’s modified Eagle medium supplemented with 5% horse serum, and transduced with a replication-incompetent retroviral vector encoding v-myc. Successfully transduced cells were plated at clonal density and several clones were selected, including HB1.F3 [61]. HB1.F3 cells were transduced to express cytosine deaminase (CD) [58], generating the HB1.F3.CD cell line that was subsequently extensively characterized and FDA-approved for use in clinical trials [62]. HB1.F3.CD cells are easily propagated in culture media with 10% fetal bovine serum. Early experiments showed that HB1.F3 cells expressed NES and the ABCG2 transporter, and upon exposure to 5% bovine serum differentiated into neurons expressing Neurofilament (NF-L, NF-M and NF-H), astrocytes (expressing GFAP) and oligodendrocytes (expressing MBP) [60]. Other hNSC lines used are the ReNCell lines derived from 10-week gestation fetal midbrain (ReNCell VM) or 14-week fetal cortex (ReNCell CX), expanded on laminin-coated plates in serum-free defined NSC media supplemented with EGF and FGF [63]. ReNCells were also immortalized with a retrovirus encoding v-myc (ReNCell VM) or c-myc (ReNCell CX) [63] and are currently commercially available. ReNCells differentiate upon growth factor withdrawal into early neurons expressing TUBB3, dopaminergic neurons expressing TH, GFAP+ astrocytes and oligodendrocytes expressing GALC [63]. Despite expression of the myc oncogene, these cells have not been shown to produce tumors in the brains of experimental animals or in clinical trials, and most of them do not persist in the brain. The differentiation potential of the HB1.F3.CD line has not recently been tested; nonetheless, it was demonstrated to be safe and effective in trials for GBM [78].
The main function of NSCs used for the treatment of GBM is to migrate deep into the tumor and deliver therapeutic agents. It was demonstrated that NSCs derived from different parts of the mouse and human CNS have different proliferation and differentiation potential and express transcription factors specific for their region of origin [39,64,65]. However, it was also reported that upon culture, the expression of many transcription factors that indicate positional identity of the NSCs was downregulated or lost, and these changes altered the differentiation potential of these cells [66,67]. It is likely that such changes affect functional aspects of NSCs used for GBM as well; rigorous mechanistic studies in this direction may lead to findings that will improve the efficiency of therapeutic NSCs.
MSCs are cells capable of differentiating into tissues derived from the embryonic mesoderm, such as adipose tissue, cartilage, bone and muscle [68]. MSCs can be found in a great variety of tissues. Therapeutic MSCs have been derived from bone marrow, adipose tissue, umbilical cord blood and even placenta, and have the advantage of being abundant, easy to isolate and propagate; they also have high tumor-tropic migratory potential and can be donor matched. Some studies report that unmodified MSCs can promote tumor growth in other cancers [69]. However, so far, preclinical studies using MSCs for the treatment of GBM have not reported tumor formation (Supplementary Table 1).
NSCs appeared to be better suited in their ability to distribute throughout brain tumors and deliver oncolytic viruses (OVs) when compared with MSCs [70]. HB1.F3.CD cells were more efficient in this process than ReNCells [71]. The underlying mechanisms for this therapeutic advantage have so far not been explored. Regrettably, the authors did not specify whether the ReNCells used were midbrain- or cortex-derived (ReNCell-VM or ReNCell-CX) [71].
iNSCs, generated by transdifferentiation of somatic cells by transient expression of specific transcription factors, can be derived from the patient’s own cells, evading a potential immune response, as may be encountered when using allogeneic NSCs. Both mouse and human iNSCs have been shown to be effective in preclinical studies for GBM [72,73]. The primary safety concern for therapeutic NSCs is their potential to induce tumor formation, especially when implanted into the brain of patients with established tumors that generate a microenvironment permissive for tumor growth. It was demonstrated in a syngeneic mouse model that iNSCs were safe in this respect and did not induce tumor formation, unlike induced pluripotent stem cells and embryonic stem cells, which generated aggressive, deadly tumors [74]. iNSCs have not yet been tested in the clinic for GBM. Considering the rapid progression of GBM, concerns arise whether such an approach would allow for a timely and in-depth quality and safety analysis to validate and expand autologous iNSCs to the necessary quantities needed for therapeutic use, notwithstanding the costs associated with such an endeavor. Nonetheless, transdifferentiation of somatic cells may be a powerful tool for drug delivery. If factors that promote optimal NSC migration and intratumoral distribution are uncovered and engineered in iNSCs, with thorough characterization and testing, the therapeutic NSC toolbox could be considerably expanded. Detailed strategies for generation of iNSCs have been described in a recently published review [75].
For clinical use and to achieve FDA approval, each newly established stem cell line must be prepared following good manufacturing practice protocols and requires extensive testing of safety and therapeutic properties according to standards of good clinical practice. When rigorous procedural practices are not followed – as was the case in some for-profit clinics carrying out unproven stem cell interventions for a variety of conditions – and in the absence of regulatory oversight, as encountered in some countries, administration of stem cells can lead to major complications, including tumor formation [76,77], embolism, vision loss, infectious events, autoimmune reactions, stroke, brain hemorrhage and even death [14].
To ensure the safety of stem cell-based therapies, it is critical to determine what happens to the stem cells once they have been delivered. As mentioned above, early studies have tracked the fate of NSCs following administration into the brain, demonstrating their ability to differentiate into neural, astrocyte and oligodendrocyte lineages [55]. NSCs immortalized with v-myc, derived from either human or mice, proliferated well in vitro but stopped dividing once transplanted into the brain, possibly due to developmental cues that induced differentiation [55–57]. Analysis of brain tissue from patients treated with HB1.F3.CD cells also illustrated that NSCs found in the brain were not proliferating [78]. Unfortunately, more recent studies have not systematically analyzed the phenotype of stem cells distributed into the brains of tumor-bearing animals. For most strategies employed, therapeutic stem cells are engaged in a suicide mission and die after the delivery of their cargo.
The fate of injected stem cells is more relevant when using cells engineered to secrete cytokines, antibodies or toxins that do not affect their viability. One of the few studies that described the fate of bone marrow iNSCs, modified to secrete IL-23, a cytokine that activates the antitumor immune defense, illustrated that these BM-NSCs-IL-23 cells, when injected into the brains of tumor-bearing mice, can express neuronal-, astrocyte- or oligodendrocyte-specific markers (TUBB3, GFAP and MYELIN, respectively), suggesting that they differentiated into the respective lineages [79]. It is critical that future studies address the phenotype and fate of therapeutic stem cells with more rigor and consistency, especially when using MSCs, a rather loosely defined cell population derived from different tissues in a variety of ways. The lack of standardized protocols and methods to characterize therapeutic stem cells can lead to therapeutic failures and decreased confidence in this promising strategy. Indeed, MSCs are referred to as a ‘double-edged sword’ as a result of their ability to both promote and inhibit tumor growth [80]. An increasing number of scientists point to the need to thoroughly characterize this heterogeneous population of cells and establish gold standards for the derivation and use of MSCs – standards shared between researchers, clinicians and industry – to allow for proper comparison between different studies and for scientific progress [81].
Routes of stem cell administration
An important parameter in stem cell-based therapies for GBM is the route of administration, which must be designed to ensure that sufficient cells can reach the tumor and provide therapeutic benefit. Local administration has been the preferred method employed, most commonly through injection into the tumor or tumor resection cavity in both preclinical (Supplementary Table 1) and clinical studies [78,82]. Intraventricular administration has also been proven to result in good intratumoral distribution of stem cells [58]. Quantitative analysis and 3D reconstructions of the distribution of NSCs following intraventricular administration in mice demonstrated that these cells migrated efficiently toward tumors, even in the case of multifocal tumors located in different hemispheres [83]. NSCs tended to locate around the tumor margin but were also found within the tumor core. Some cells aggregated in the ventricles, and this was thought to be due either to the speed of the injection or to a cell suspension that was too concentrated [83]. Intraventricular administration of stem cells brings several advantages to this treatment modality, allowing for dose escalation by overcoming the space limitations of densely cellular environments, repeated administrations through the placement of a minimally invasive intraventricular catheter (a common neurosurgical procedure) and possible increased viability of administered NSCs within the cerebrospinal fluid.
The distribution of NSCs into the tumor following intravenous or intracranial administration of increasing numbers of cells has been systematically quantified in a mouse model of GBM [84]. This analysis revealed, as observed before, that higher doses of NSCs yielded better tumor coverage, but only up to a point, after which the percentage of actual NSCs present in the tumors declined with both the intracranial and the intravenous administration route. The authors theorized that the rate-limiting factors could be tumor dependent or related to the administration technique; high density of cells may lead to aggregation or decreased survival that would limit intratumoral migration and distribution. It was also found that larger tumors attracted more cells, due likely to chemotactic tumor-derived cytokines. Systemic intravenous administration required about ten-times more NSCs than intracranial injections to reach the same degree of tumor coverage [84], a requirement that needs to be carefully considered when the number of therapeutic cells is limited or when they are very costly to produce.
Local administration of stem cells into the brain is invasive, and repeated administration may lead to complications. An attractive alternative route of noninvasive administration of therapeutic stem cells is represented by intranasal delivery, a method that takes advantage of the anatomical and physiological properties of the nasal mucosa, allowing for transport alongside the olfactory or trigeminal nerves and through the perivascular pathway within the CNS, avoiding the transport restrictions imposed by the BBB and the clearance of therapeutic cells following systemic administration [85]. This method has been established and optimized over the years in several studies, showing promising results in preclinical brain tumor models, albeit with less efficacy than other administration routes [86–90].
Another emerging strategy to deliver stem cells for the treatment of brain tumors is represented by the intra-arterial route. A limitation of intravenous stem cell administration is the entrapment in the lungs of the majority of cells, decreasing the efficacy of the treatment. Intra-arterial administration can overcome this limitation, especially when using endovascular selective intra-arterial techniques, closely targeted to the tumor site [91]. MSCs loaded with OVs and administered into the carotid artery were successfully used in preclinical models of GBM [92] and are currently being tested in a clinical trial using the conditionally replicating oncolytic adenovirus Delta24-RGD (NCT03896568, Supplementary Table 2). This strategy takes advantage of the tumor-tropism of the MSCs, which limits spread of the virus to other organs, and also of the ability of MSCs to cross the BBB and distribute widely in the tumor.
Therapeutic strategies & their mechanisms of action
The first preclinical studies using stem cells for the treatment of malignant glioma emerged 20 years ago, testing four different therapeutic strategies: enzyme/prodrug [58], OV [93], cytokine therapy [94] and delivery of proapoptotic molecules [95]. Today, these mechanisms are still being tested, optimized, fine-tuned and combined with other treatment modalities to overcome limitations and improve efficacy. Pilot/feasibility and Phase I clinical trials using NSCs loaded with OVs or delivering enzymes for intratumoral prodrug conversion are underway (Supplementary Table 2). Exciting new therapeutic avenues have been opened by the tremendous progress in bioengineering technologies, generating nanoparticles and nanorods that can be conjugated to glioma-tropic stem cells for drug delivery, and targeted photothermal ablation therapy. We describe these studies in the following sections, highlighting experimental progress, limitations and efforts to translate these strategies into the clinic.
Stem cells in enzyme/prodrug strategies
Enzyme/prodrug strategies for cancer treatment have long been pursued in an effort to artificially create selective and local cytotoxicity for tumor cells, while leaving normal cells unharmed [96]. The most widely used combinations are the herpes simplex virus thymidine kinase (HSV-TK) with gancyclovir (GCV) and the bacterial CD with 5-fluorocytosine (5-FC). The HSV-TK enzyme converts GCV into GCV monophosphate, which is further phosphorylated to GCV triphosphate, a toxic antimetabolite that undergoes erroneous incorporation into DNA, leading to the death of dividing cells (Figure 1A). Cells lacking HSV-TK can still be targeted for apoptosis through a phenomenon called the ‘bystander effect’, which entails transport of the active drug through gap junctions from neighboring cells, enhancing the cytotoxic response. Similarly, the bacterial enzyme CD is able to convert the nontoxic prodrug 5-FC into the powerful cytotoxic compound 5-fluorouracil (5-FU), which primarily inhibits the production of the thymidine required for DNA replication, thus killing dividing cells exposed to it (Figure 1B). Enzyme/prodrug gene therapy strategies using viral vectors have been extensively explored for the treatment of GBM for over 40 years but still face many challenges, mainly related to the distribution of the viruses into the tumor [97]. The high migratory capacity of NSCs and their ability to distribute throughout the tumor and tumor satellites may serve to improve this therapeutic strategy.
Figure 1. . Mechanisms of action employed in enzyme/prodrug strategies for the treatment of glioblastoma.
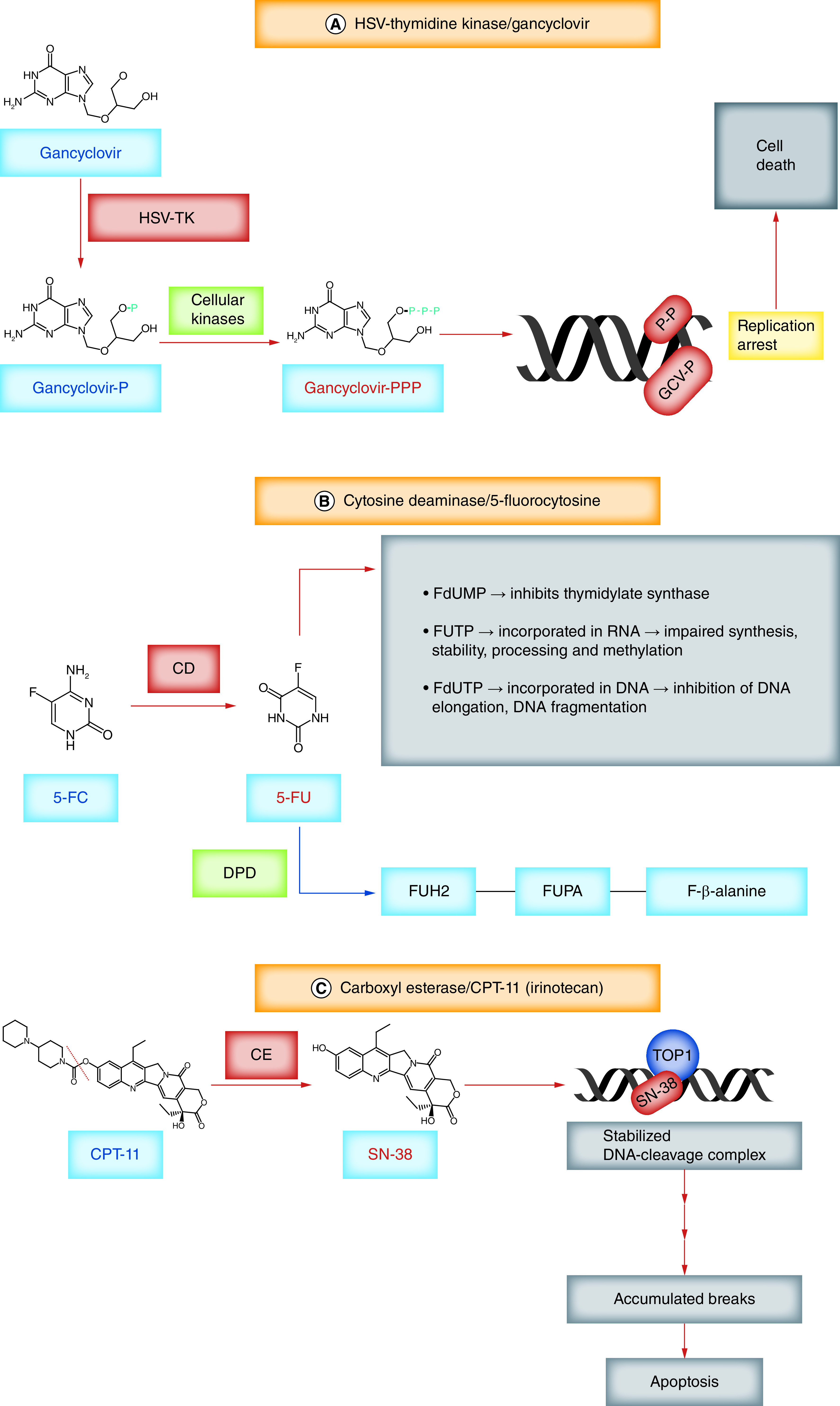
(A) HSV-TK can phosphorylate not only thymidine but also gancyclovir, generating gancyclovir monophosphate. Cellular kinases convert it into gancyclovir triphosphate, which can be integrated into newly synthesized DNA, arresting further DNA synthesis and inducing cell death. (B) CD converts 5-FC into 5-FU. To exert its cytotoxic function 5-FU is anabolized into fluoropyrimidine nucleotides (FdUMP, FUTP and FdUTP). FdUMP is a suicide inhibitor of TS, binding and inactivating it. TS converts dUMP to dTMP; inhibiting TS results in nucleotide imbalance, excess dUTP and lack of dTMP, leading to DNA damage. FUTP is extensively incorporated into nuclear and cytoplasmic RNA, leading to impaired RNA synthesis, stability, processing and methylation. FdUTP, when incorporated into DNA, inhibits DNA elongation and induces DNA fragmentation. 5-FU is deactivated and converted to FUH2 through the catalytic action of DPD, the initial and rate-limiting step in the catabolism of 5-FU. FUH2 can be further degraded to FUPA and subsequently to the nontoxic amino acid FBAL. (C) CE converts the water-soluble compound CPT-11 into the more potent, lipophilic metabolite SN38. During the DNA synthesis phase of the cell cycle, TOP1 attaches to the 3′ end of the cleaved DNA and forms a reversible DNA-TOP1 cleavage complex (TOP1cc). SN-38 binds to TOP1 and stabilizes this complex, halting DNA synthesis and leading to the accumulation of single-strand DNA breaks which trigger apoptosis.
5-FC: 5-Fluorocytosine; 5-FU: 5-Fluorouracyl; CD: Cytosine deaminase; CE: Carboxyl esterase; dTMP: Deoxythymidine monophosphate; dUMP: Deoxyuridine monophosphate; DPD: Dihydropyrimidine dehydrogenase; FBAL: Fluoro-β-alanine; FdUMP: 5-Fluoro-2′-deoxyuridine 5′-monophosphate; FdUTP: 5-fluoro-2′-deoxyuridine 5′-triphosphate; FUH2: Fluoro-5,6-dihydrouracil; FUPA: Fluoro-β-ureidopropionate; FUTP: 5-Fluorouridine 5′-triphosphate; GCV-P: Gancyclovir monophosphate; HSV-TK: Herpes simplex virus thymidine kinase; TOP1: Topoisomerase 1; TOP1cc: TOP1 cleavage complex; TS: Thymidylate synthase.
A proof-of-principle study using an in vitro coculture model demonstrated that NSCs expressing HSV-TK can induce the death of glioma cells when combined with GCV, and that this cytotoxic effect was dependent on cell-to-cell contact and on the presence of the gap junction protein connexin 43 [98]. The bystander effect of HSV-TK delivered by NSCs was also demonstrated in a mouse model in which C6 glioma cells were implanted with NSCs transduced with HSV-TK (NSC-TK) [99]. Long-term survival was observed in all animals treated with NSC-TK, whereas all control animals succumbed before 30 days. It was also demonstrated that human bone marrow-derived MSCs transduced to express HSV-TK and then injected into mouse brains at the time of glioma cell implantation induced long-term survival in all animals [100]. In a rat glioma model, the same MSC-TK cells improved the survival of tumor-bearing animals, with 40% of animals becoming tumor-free [100]. These studies demonstrate that delivery of HSV-TK by either NSCs or MSCs provides a significant survival benefit in animal models of GBM. One caveat associated with such studies comes from the simultaneous administration of tumor cells and therapeutic stem cells, a scenario that cannot be applied in the clinic. Other animal studies have administered stem cells at later times following tumor cell implantation and demonstrated a therapeutic effect, albeit with lower efficacy (Supplementary Table 1).
A pioneering preclinical study that illustrated the migration of mouse and human NSCs toward intracranial gliomas tested the ability of immortalized mouse NSCs (C17.2) that were transduced with CD to induce glioma cell death in the presence of 5-FC [58,59]. Results demonstrated the feasibility of this approach and tumor reduction upon intratumoral administration of the transduced NSCs; proliferating NSCs were also killed by the treatment [58]. Human NSC cells (HB1.F3) were also transduced to express CD, generating HB1.F3-CD cells that were tested for their migration potential and tumor killing efficacy in mice injected with Daoy medulloblastoma cells [101]. Survival was not analyzed in this study; however, HB1.F3-CD cells injected into established tumors, when combined with 5-FC, were able to significantly reduce the tumor volume (by 74%) after 3 weeks of treatment, demonstrating a strong bystander effect from the CD-producing NSCs [101]. Adipose tissue-derived MSCs (AT-MSCs) were transduced with yeast-derived CD (CDy) and UPRT, generating CDy-AT-MSCs, which migrated toward gliomas when implanted at a distance [102]. UPRT catalyzes the conversion of 5-FU to 5-fluorouridine monophosphate, which is more cytotoxic than 5-FU. It was demonstrated that CDy-AT-MSCs improved the survival of tumor-bearing rats in a dose-dependent manner, showing an almost linear relationship between survival and the number of MSCs administered. It was further illustrated that repeated administration of increased numbers of CDy-AT-MSCs, combined with continuous intraventricular delivery of 5-FC via osmotic pump, further improved survival, with 88% of animals becoming tumor free [102].
Inactivation of 5-FU is dependent on the activity of the rate-limiting enzyme, DPD, that converts 5-FU to fluoro-5,6-dihydrouracil, which is further metabolized to fluoro-β-alanine (Figure 1B). Several studies have identified an inverse correlation between the sensitivity to 5-FU treatment and levels of DPD, as well as increased toxicity of 5-FU in patients with decreased levels of DPD, oftentimes due to inactivating mutations of DPD [103]. It was also demonstrated that high levels of DPD expression in glioma cells can render therapies with hCD-MSC/5-FC inefficient, warranting assessment of DPD levels prior to the start of treatment [104]. Pharmacological inhibitors of DPD may also improve the efficacy of 5-FU-based therapies, especially in local administrations with low systemic load.
The transition from preclinical findings to clinical testing first requires rigorous, diligent, in-depth safety analyses. The 2013 study by Aboody et al. thoroughly characterizes the properties of the human NSC line HB1.F3.CD, which was derived from HB1.F3 cells and modified to produce cytosine deaminase [62,105]. HB1.F3.CD cells were approved by the FDA as an Investigational New Drug for therapeutic use in human clinical trials, and to this day represent the only human NSC line used in clinical trials for GBM [62]. It was demonstrated that over 32 passages, HB1.F3.CD cells had a stable normal karyotype, included just one copy each of the v-myc and CD genes, retained tropism toward glioma cell lines, even in the presence of dexamethasone, and expressed the CD protein and HLA-I antigens but not (or very low levels of) HLA-II. When injected into the brains of naive immunocompromised or immunocompetent mice (up to 1 × 106 cells), no tumor formation was observed; no human cells were detected in the brain or other organs at 4 and 12 weeks after injection, indicating that the HB1.F3.CD cells either did not survive or were cleared by immune cells (or both). When the cells were administered to mice with intracranial tumors and combined with 5-FC, tumor volume was significantly decreased in animals receiving the highest dose of NSCs (1 × 105) [62]. This study highlights the fact that therapeutic efficacy is intimately dependent on the number of NSCs available in the tumor. The pilot/feasibility clinical study designed to use these cells in patients with recurrent high-grade glioma (rHGG) (NCT01172964, Supplementary Table 2), planned a classic 3+3 dose escalation regimen (1 × 107 to 5 × 107 cells and 5-FC from 75 to 150 mg/kg/day, administered for 7 days and starting 4 days after surgery) with injection of NSCs into the walls of the resection cavity. Results from 15 patients found steady-state levels of 5-FU in the brain, much higher than in the blood, indicating local production of 5-FU by the NSCs [106]. Overall, administration of HB1.F3.CD was safe, resulted in no detection of NSCs outside the brain and elicited no humoral immune responses. The median overall survival (OS) at the highest dose level was 15.4 months, compared with patients on doses 1–2 with OS of 2.9 months [78]. Distribution of NSCs was analyzed in postmortem brains by v-myc PCR. Several selected sites from brain sections from a male patient, who had died at some time after surgery, tested positive for v-myc by PCR, indicating the presence of HB1.F3.CD cells. In situ hybridization with XX and XY probes (the NSCs are female) in combination with PCNA staining confirmed the presence of the injected NSCs and showed they were not proliferating. It was estimated that the NSCs traveled about 11 cm from the site of injection [78]. These results are indeed exciting proof of safety and of principle, with added optimism for the efficacy and identification of live, possibly still therapeutic, cells more than 2 months after administration. The follow-up Phase I study (NCT02015819), started in 2014 and designed to include 18 patients with rHGG, has been modified to include an intraventricular Rickham catheter, placed at the time of surgery, to be used for the administration of subsequent doses of NSCs every 2 weeks, followed each time by a 7-day course of oral 5-FC. Leucovorin was also added to the protocol to enhance the cytotoxic effects of 5-FU.
Another enzyme/prodrug combination used for cancer treatment is carboxyl esterase (CE) and CPT-11 (irinotecan), a water-soluble, semisynthetic derivative of camptothecin. CPT-11 has antitumor activity on its own; however, its lipophilic metabolite SN-38 is a more potent cytotoxic agent. Camptothecin and its derivatives induce DNA damage during the S-phase of the cell cycle by inhibiting TOP-1, forming a stable ternary DNA–TOP1 cleavage complex (TOP1cc) and inducing apoptosis. CPT-11 is metabolized to SN-38 by CE, an enzyme naturally present in the liver, albeit at low concentrations (Figure 1C). High expression of CE in the tumor results in efficient conversion of CPT-11 into SN-38 and high cytotoxic effect, mitigating systemic toxicity. Human adipose tissue-derived MSCs transduced to express the rabbit CE (hAT-MSC.rCE) were tested in a rat brainstem GBM model, in a protocol that entailed two injections of hAT-MSC.rCE at 2-week intervals with systemic administration of CPT-11 for 3 weeks [107]. Survival analysis showed a modest improvement (24 vs 19 days) in animals treated with hAT-MSC.rCE, highlighting the difficulty of treatment for brainstem gliomas. Administration of CE/CPT-11 was also analyzed in a combinatorial strategy using amniotic fluid-derived MSCs (AF-MSCs), transduced to express the antiangiogenic molecule endostatin and/or a secreted form of CE. AF-MSCs were injected either intracranially together with glioma cells, or subcutaneously after surgical removal of 90% of established tumors. AF-MSCs transduced with both CE and endostatin inhibited progression of brain and flank tumors more strongly than AF-MSC expressing only endostatin or CE, and induced decreased vessel density in the tumor, a lower proliferation index and increased apoptosis; unfortunately, no survival data were presented [108]. A mouse model of medulloblastoma, in which tumors were generated in neonatal mice with intracerebellar administration of viruses encoding Shh and Mycn, genes known to induce medulloblastoma, was used to test the antitumor effect of NSC-CE/CPT-11 therapy. Human HB1.F3.CD cells were transduced to express the rabbit CE, which is more effective in metabolizing CPT-11 to SN-38 than the human CE, generating HB1.F3.CD.rCE cells. Animals with established tumors, documented by MRI, were injected into the cerebellum with HB1.F3.CD.rCE, then treated systemically with CPT-11. A second dose of NSCs was administered 1 week later. This regimen resulted in a decreased rate of tumor growth, as calculated from MRI data, and a decrease in tumor size when compared with controls at the end of the experiment (postnatal day 71) [109]. Although survival analysis would be difficult in such a model due to the variability in viral transduction and tumor formation, the study illustrates that NSC-mediated CE/CPT-11 enzyme/prodrug therapy for medulloblastoma is feasible and warrants further optimization.
Administration of the rabbit CE in humans may result in an inflammatory response, reducing the efficacy of the stem cell treatment. To generate a potent human isoform of CE, Wierdl et al. analyzed the structural properties of the rabbit CE enzyme responsible for its increased enzymatic activity and constructed a genetically enhanced version of the human liver CE1. They generated hCE1m6, an enzyme 70-fold more efficient in converting CPT-11 to SN18 than the original CE1 [110]. The HB1.F3.CD human NSC line was transduced with replication-deficient adenovirus to transiently produce hCE1m6, generating HB1.F3.CD.hCE1m6 cells. These were compared with HB1.F3.CD.rCE cells in vitro as well as in vivo, using Es1e/SCID mice with plasma CE levels comparable to those in humans. It was demonstrated that HB1.F3.CD.hCE1m6 cells were as efficient as HB1.F3.CD.rCE in converting CPT-11 and had similar migratory behavior and cytotoxic activity against glioma cells. In vitro degranulation assays with the two enzymes using human peripheral blood mononuclear cells indicated that hCE1m6 had lower immunogenic potential than the rabbit counterpart [111]. The HB1.F3.CD.rCE cells have been approved for use in clinical trials [112].
Stem cells carrying proapoptotic molecules
Dysregulated apoptotic pathways are a hallmark of malignancy. The TP53 tumor suppressor gene, which initiates intrinsic apoptotic pathways in response to cellular stressors including genomic aberrations and DNA damage, is often inactivated in cancer and very frequently in GBM [113,114]. Malignant cells evade apoptotic signals and therapeutically induced DNA damage and continue to proliferate. Activation of the extrinsic apoptotic pathway via ligands like TRAIL or FasL, which bind to cell-surface death receptors, can induce activation of the caspase enzymatic cascade, leading to apoptosis independent of TP53. TRAIL has therefore been extensively explored as a promising anticancer agent, as it specifically targets tumor cells, leaving normal cells unharmed [115,116,117]. Soluble TRAIL is very rapidly cleared from systemic circulation, and strategies for extending its stability and delivery by linking it to nanoparticles or stem cell carriers have been intensely studied [118].
If cells express the TRAIL ligand on their surface, they can induce apoptosis in neighboring cells. It was demonstrated that antigen-specific CD4+ T cells expressing TRAIL can kill glioma cells in vitro [119]. Primary mouse NSCs derived from the forebrain of embryonic mice modified to express human TRAIL were able to specifically induce apoptosis in human GBM cells in mouse orthotopic xenografts, while sparing the NSC population [95]. Also, mouse NPCs (C17.2), transduced to express a secreted form of TRAIL (sTRAIL) were able to decrease viability of human glioma cell lines through caspase-mediated apoptosis [120]. This effect was synergistically increased if NPC-sTRAIL was combined with miR-21 knockdown. It had been previously reported that miR-21 is highly expressed in human GBM and that its knockdown led to apoptosis of GBM cell lines [121]. A synergistic effect was found when combining NPCs (C17.2) with temozolomide treatment. Temozolomide sensitized cells to TRAIL-mediated apoptosis by increasing death receptor expression, activating the checkpoint kinase Chk1 and arresting cells in G2/M [122].
While many glioma cells are sensitive to TRAIL-mediated apoptosis, some (especially GSCs) are not. Resistance to TRAIL has been linked to several mechanisms, including upregulation of antiapoptotic proteins of the BCL-2 family, inactivating mutations and epigenetic silencing of caspase 8, or upregulation of FLIP, a molecule that blocks the formation of the death-inducing signaling complex (DISC) [115]. In some cells, resistance to TRAIL-induced apoptosis can be overcome by simultaneous administration of the proteasome inhibitor bortezomib, which results in enhanced caspase 8 activation that depends on accumulation of the cyclin-dependent kinase inhibitor p21CIP1 and inhibition of cyclin-dependent kinase activity (CDK1/2) [123]. In vitro analysis of multiple primary astrocytoma cell lines treated with TRAIL and bortezomib demonstrated that bortezomib sensitized these cells to TRAIL-induced apoptosis by increasing the expression of the signaling death receptors TRAIL-R1 or TRAIL-R2, enhancing formation of the DISC complex, increasing recruitment of caspase 8 to DISC and decreasing recruitment of the inhibitory protein FLIP (Figure 2) [124]. Balyasnikova et al. tested the combinatorial use of TRAIL, delivered by NSCs, with bortezomib in a preclinical orthotopic glioma model [125]. This study demonstrated that NSC-TRAIL, when injected at a 1:1 ratio with tumor cells at the time of tumor initiation, was able to induce long-term survival in 80% of mice. This was increased to 100% long-term survival when bortezomib was added. The result was dose and time dependent; lowering the number of NSCs or administering NSCs into established tumors decreased the number of long-term survivors. Administration of bortezomib increased expression of the TRAIL-R2 receptor in vitro, which could account for the increased sensitivity to TRAIL. However, this effect was not reproduced in vivo, possibly due to poor penetration of the drug into the tumor, following systemic administration [125].
Figure 2. . Stem cells carrying proapoptotic molecules.
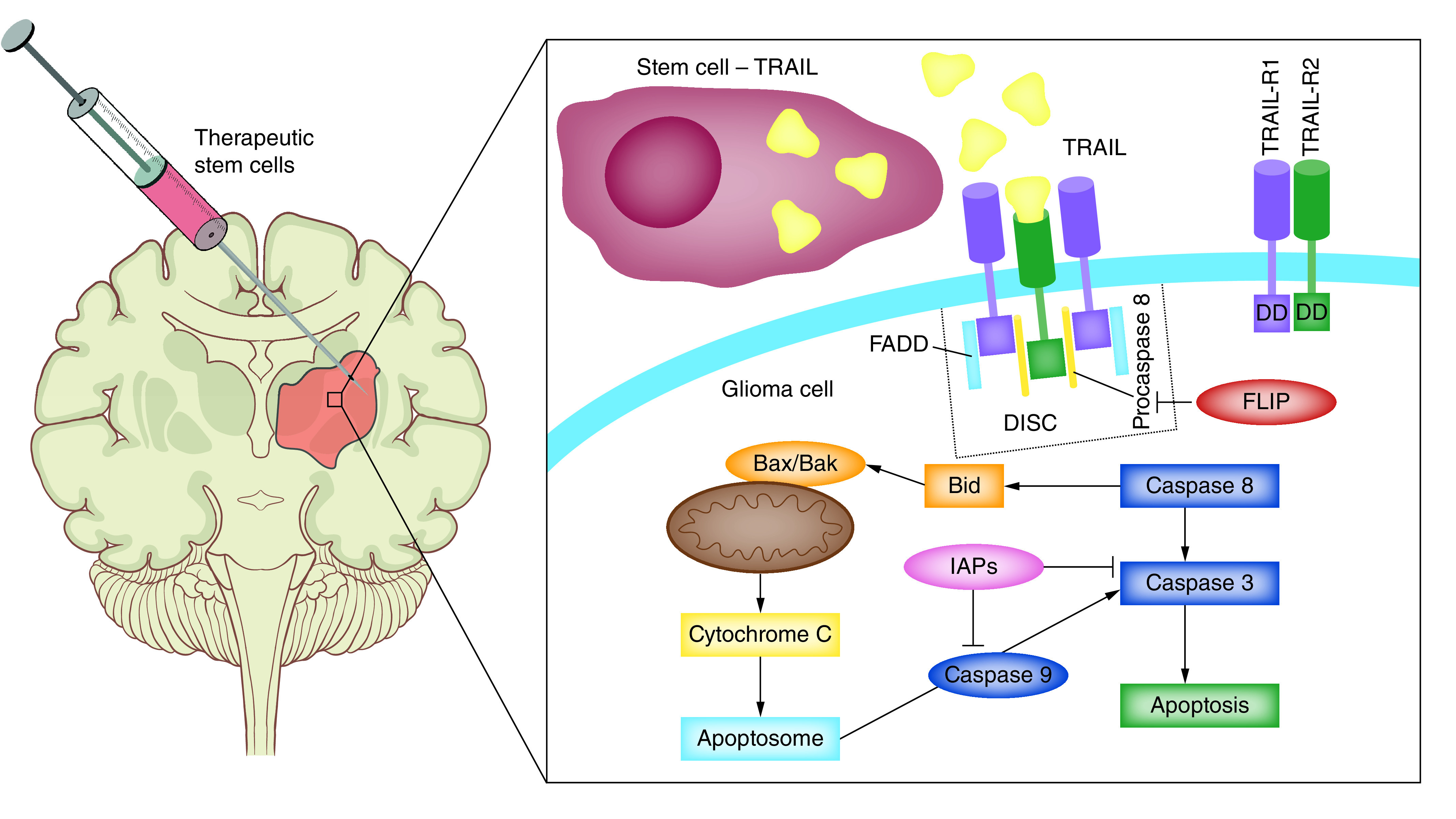
Intratumoral delivery of TRAIL activates the extrinsic apoptotic pathway. Binding of TRAIL to the death receptors DR4 (TRAIL-R1) and DR5 (TRAIL-R2) results in formation of DISC and proteolytic cleavage of procaspase 8. This triggers activation of the effector caspase 3, leading to apoptosis. Activation of the initiator caspase 8 results also in activation of Bax and Bak inducing release of cytochrome C from the mitochondria and further activation of the effector caspases. Resistance to TRAIL-induced apoptosis may be caused by excess FLIP. FLIP binds to FADD and caspase-8 and inhibits formation of DISC and activation of caspase 8. The proteasome inhibitor bortezomib increases sensitivity of tumor cells to TRAIL-induced apoptosis by inhibiting recruitment of FLIP to DISC and increasing expression of the two signaling TRAIL receptors (TRAIL-R1 and TRAIL-R2).
DISC: Death-inducing signaling complex; FADD: FAS-associated death domain protein; TRAIL: TNF-related apoptosis-inducing ligand.
TRAIL was also delivered into the brains of tumor-bearing rats via iron oxide magnetic nanoparticles (NP-TRAILs), and it was shown that these were effective in inducing apoptosis of glioma cells and significantly extended the median survival (MS) of tumor-bearing rats when compared with rats treated with free soluble TRAIL [126]. NP-TRAILs were also able to induce apoptosis and increase radiation sensitivity and efficacy of bortezomib in patient-derived GSCs (HF2414, HF2584), cells refractory to TRAIL-induced apoptosis [126].
In addition to established allogeneic NPC lines that may be in limited supply and may induce immunological rejection, other stem cells have been explored as carriers for TRAIL, providing an avenue for autologous cell therapies for GBM. Human adipose-derived MSCs transduced with TRAIL using biodegradable nanoparticles were able to migrate toward intracranial tumors, and selectively induced glioma cell death via caspase-mediated apoptosis, while sparing normal astrocytes. Treatment with these cells significantly extended the MS of glioma bearing animals, especially after repeated intratumoral administrations [127].
TRAIL-mediated apoptosis of glioma cells has also been accomplished using iNSCs. Mouse embryonic fibroblasts were transdifferentiated with lentiviral expression constructs of Brn2, Sox2 and FoxG1 to generate iNSCs. Glioma tropism of these cells was demonstrated in vitro and in vivo and when transduced to express sTRAIL [72]. Administration of iNSC-sTRAIL 3 days after implantation of patient-derived cells (GBM8) extended the survival of tumor-bearing animals from 37 to 59 days [72]. A subsequent study from the same group reported the generation of human iNSCs (h-iNSCTE) by transdifferentiating and immortalizing human fibroblasts with lentiviral vectors encoding SOX2 and human telomerase (h-TERT) [73]. Transcriptionally, these cells resembled brain-derived NSCs and migrated selectively toward GBM cells and spheroids in vitro in a CXCR4-dependent manner. When injected into the brains of immunodeficient mice, the iNSCs persisted for up to 3 weeks but were eventually cleared. These cells were then transduced to express either a secreted form of TRAIL (h-iNSCTE-sTRAIL) or TK (h-iNSCTE-TK) and tested as therapeutic agents. The h-iNSCTE-TRAIL reduced viability of glioma cells in vitro and extended the MS of mice bearing intracranial xenografts when equal numbers of tumor cells and h-iNSCTE-TRAIL were implanted at the same time. Intratumoral administration of h-iNSCTE-TK, when combined with GCV, also increased the MS of glioma-bearing mice. The group went on to demonstrate that postsurgical treatment with h-iNSCTE-TK cells, encapsulated in extracellular matrix hydrogel that was placed into the tumor cavity, followed by GCV administration (a paradigm that more closely resembles the situation in the clinic) also resulted in increased MS of the animals [73].
An interesting development of cell-mediated therapy for GBM is represented by the use of modified tumor cells to deliver therapeutic agents into the tumor. This is based from observations of the ‘self-seeding’ behavior of cancer cells, where metastatic cells migrate back toward the primary tumor or cells from the primary tumor migrate toward established metastases [128]. Glioma cells, especially GSCs, share many characteristics with NSCs, including migration and invasion toward CXCL12 and TGFb [129,130]. Genetically modified glioma cells have therefore been tested for their ability to deliver proapoptotic molecules for the treatment of GBM and other metastatic cancers [131]. Two strategies for this approach were designed: allogeneic ‘off the shelf’ GBM cells, intrinsically resistant to TRAIL-mediated apoptosis and expressing sTRAIL), or autologous glioma cells with CRISPR/Cas9-mediated genetic deletion of the two TRAIL signaling receptors TRAIL-R1(DR4) and TRAIL-R2(DR5) and lentiviral expression of sTRAIL. The cells were also modified to express HSV-TK to render them sensitive to GCV-mediated elimination, yet insensitive to the actions of TRAIL. Mice with established intracranial tumors injected with these cells showed increased MS, efficiency being further enhanced with GCV administration [131].
Overall, these studies demonstrate that stem cells represent efficient vehicles for delivery of TRAIL into tumors and that the therapeutic effect is dependent on the tumor’s sensitivity to TRAIL-mediated apoptosis. This strategy may be useful in combinatorial approaches with agents that mitigate resistance to TRAIL, and other synergistic cytotoxic drugs.
Stem cells & oncolytic virotherapy
Oncolytic viral therapy either uses viral vectors that are able to selectively replicate in tumor cells and induce tumor cell lysis that is propagated outwards from the site of virus administration, while sparing normal brain cells, or employs viruses that are replication-deficient and are used for the delivery of therapeutic genes [132,133]. In addition to the direct tumor lytic effect, OVs strongly stimulate the innate immune response through viral antigens (pathogen-associated molecular patterns) and the antitumor immune response through the release of damage-associated molecular patterns, tumor-associated antigens and neoantigens, inducing immunogenic cell death (ICD) (Figure 3) [134]. The idea to use viruses to treat malignancies emerged from observations that concomitant viral infections (e.g., influenza) had beneficial effects on the progression of blood malignancies. For almost a century, numerous viruses have been modified and optimized for use in the treatment of cancer [135]. Today, exquisitely tailored adenoviruses, herpes simplex virus, vaccinia, measles, retroviruses and reoviruses are being tested in clinical trials as emerging antiglioma therapies that bring hope for a cure [136]. The most compelling of these, PVS-RIPO, TOCA511 and DNX-2401, have been granted an expedited drug review process by the FDA [136]. PVS-RIPO, a recombinant nonpathogenic poliovirus that is recognized by the poliovirus receptor CD155, present on many tumor cells, has been proven safe in a Phase I trial with WHO grade IV malignant glioma (NCT01491893), patients showing an increased survival rate compared with historical controls [137]. Repeated administration of the virus in patients in whom the disease recurred was also concluded to be safe, with encouraging efficacy results [138]. The TOCA511 virus is a replicating γ-retrovirus, based on a modified murine leukemia virus, that also encodes CD. In the TOCA511 ascending Phase I trial (NCT01470794), 56 patients with rHGG were treated with intracavity administration of TOCA511 and orally administered 5-FC; durable complete responses were observed, with the trial entering Phase III [139]. DNX-2401 (Delta-24-RGD), a replication-competent oncolytic adenovirus, has been tested in a Phase I trial in 37 patients with rHGG; evidence of long-term survival, direct oncolytic effects and immunological antitumor effects was observed [140]. In Phase III, the TOCA511 trial (NCT02414165) studied 403 patients with rHGG or anaplastic astrocytoma and compared TOCA511 with SOC. The results indicated compelling improvement of OS in patients with secondary recurrence, and favorable trends in the IDH1-mutant and anaplastic astrocytoma population. Overall, however, there were no significant differences in OS between the two arms [141]. Exciting results have also been reported from Japan with the third-generation oncolytic herpes virus G47Δ, which has been engineered for enhanced viral growth and MHC-I presentation in infected tumor cells [142]. A Phase II trial with this virus in 13 patients with recurrent or residual GBM revealed an increased survival rate in virus-treated patients compared with historical controls and also increased antitumor immune responses, with elevated infiltration of T cells at the injection sites [143].
Figure 3. . Stem cells and oncolytic virotherapy.
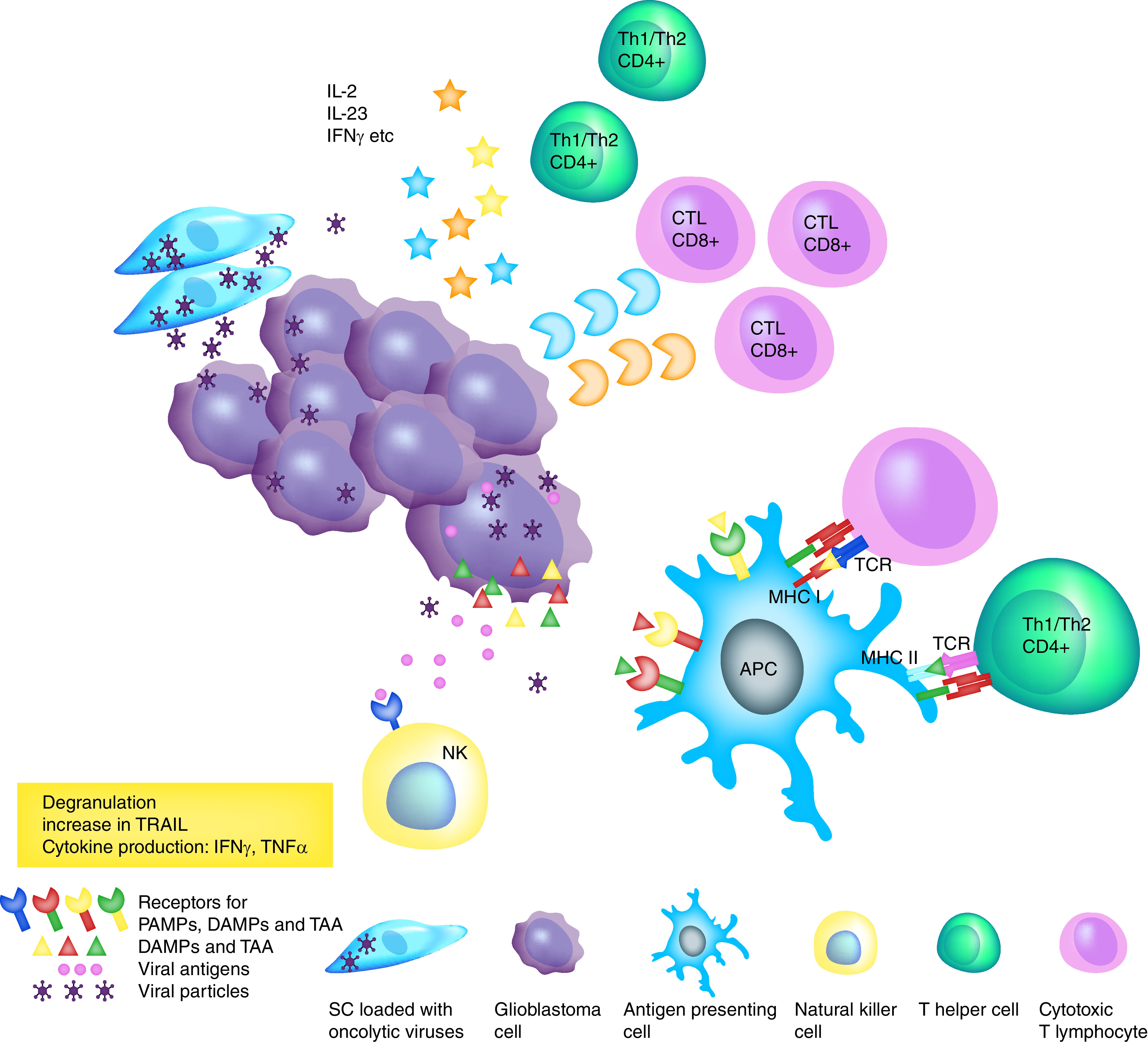
Intratumoral delivery of oncolytic viruses by SCs results in virus-induced glioma tumor cell death and also in immunogenic cell death through activation of the innate and adaptive antitumor immune responses. Dying cells release viral antigens (PAMPs), DAMPs and TAAs. These antigens activate NK cells that induce direct tumor cell killing through production of granzyme and perforin, and also induce apoptosis through the release of TRAIL, TNF-α and IFN-γ. DAMPs and TAA activate antigen-presenting cells that travel to the draining lymph nodes, where they cross-present antigens to naive T lymphocytes and activate them. Activated T helper and cytotoxic T lymphocytes migrate into the tumor and release cytokines, perforin and granzyme that amplify the cytotoxic effect.
APC: Antigen presenting cell; DAMP: Damage-associated molecular pattern; NK: Natural killer; PAMP: Pathogen-associated molecular pattern; SC: Stem cell; TAA: Tumor-associated antigen; TRAIL: TNF-related apoptosis-inducing ligand.
Taken together, these clinical studies are indeed exciting, and provide hope that oncolytic virotherapy may provide therapeutic benefit. Nonetheless, limitations exist, particularly in relation to the limited spatial distribution of the virus following injection, especially in large tumors and those that invade at a distance and necessitate multiple rounds of injections at different co-ordinates. As a result, many OVs designed to replicate specifically in glioma cells that were shown to be safe in early clinical trials did not advance to Phase III studies [144]. Tumor-tropic stem cells may be able to improve viral distribution and enhance therapeutic efficacy, especially in tumors that are difficult to reach surgically.
Numerous preclinical studies have demonstrated an advantage of using stem cells as vehicles for the delivery of OVs in intracranial gliomas. Several strategies have been employed to allow specific replication of conditionally replicating adenoviruses (CRAds) in glioma cells: deleting a viral region responsible for inactivating p53, as in the ONYX-015 virus [145], or deleting the E1A viral region that binds to RB, allowing for replication in cells with defects in the retinoblastoma pathway, frequently altered in GBM, as in the Delta-24 virus [146]. Later it was established that ONYX-015 was also active in cells with intact p53, and the selectivity for tumor cells was dependent on a tumor-specific viral RNA export mechanism [147]. The Delta-24 virus was further modified to express the RGD sequence (arginine–glycine–aspartic acid), improving infectivity of tumor cells, and evolved into the very promising Δ24-RGD.
The ability of human MSCs transduced with Δ24-RGD to deliver the virus was tested in an elegant preclinical trial of GBM, in which hMSCs-Δ24-RGD were administered intra-arterially [92]. The authors showed extensive distribution of hMSC-Δ24-RGD throughout the tumor. They observed increased expression of viral proteins over time and the cytopathic death of hMSCs after 11 days, suggesting movement of the virus from hMSCs to glioma cells. Administration of two rounds of 1.5 million hMSCs infected with Δ24-RGD increased the MS of tumor-bearing animals from 42 to 75 days, with 37.5% of animals becoming long-term survivors and showing complete remission of the tumors [92].
Results of a Phase I clinical trial with Δ24-RGD for recurrent GBM (NCT00805376) have recently been reported with promising results: 20% of patients who received one intratumoral injection of Δ24-RGD viral particles (1 × 107 to 3 × 1010) survived more than 3 years after treatment, with three patients (12%) showing a >95% reduction in the enhancing tumor [140]. One group of patients received intratumoral administration of Δ24-RGD viral particles through a catheter placed in the tumor at the time of biopsy and the resection was performed 2 weeks later. This allowed for analysis of local treatment effect; the results showed that the viral particles replicated and spread in the resected tumor, which was infiltrated with immune cells, including CD8+ and T-bet+, indicative of antitumor immune activation and possible decreased T cell exhaustion [140]. The study did not record dose-limiting toxicities, and the treatment-related adverse effects were rare and mild.
Considering these promising results with Δ24-RGD and the improved efficiency demonstrated with hMSC delivery of Δ24-RGD in preclinical trials [92,148] a new Phase I clinical trial was designed to administer Δ24-RGD with allogeneic MSCs in patients with rHGG (NCT03896568, Supplementary Table 2). The main goals of the study are to determine the maximal tolerated dose of bone marrow hMSCs carrying Δ24-RGD, using a transfemoral superselective endovascular intracranial administration route, and also to determine the local toxicity of this treatment and the distribution of hMSCs-DNX2401 and viral particles within the tumor. The trial is scheduled to end in 2022. If targeted intra-arterial administration, a method used for other cancers [91], results in efficient distribution of therapeutic stem cells within the tumor, it will open up new possibilities for minimally invasive, repetitive administrations of therapeutic cells for the treatment of GBM.
Further modifications of DNX2401 resulted in third-generation CRAd vectors like ICOVIR15, which encodes an enhanced E1A promoter for optimal viral function while maintaining a small genome size [149]. ICOVIR17 was generated from ICOVIR15, modified to express hyaluronidase for enhanced intratumoral distribution of the virus [150]. ICOVIR17 was further enhanced with an RGD insertion on the fiber shaft, rather than the fiber knob, for increased tumor cell infectivity, generating VCN-01 [151,152]. Administration of MSCs loaded with ICOVIR17 significantly enhanced MS in tumor-bearing animals when compared with virus administration alone; the presence of the hyaluronidase in ICOVIR17 also conferred an increased survival advantage over ICOVIR15. Encapsulating ICOVIR17-infected MSCs in a synthetic extracellular matrix that was implanted in the post-resection cavities of mice with intracranial tumors also showed higher efficacy compared with administration of virus alone [153]. Similarly, encapsulated MSCs carrying the oncolytic herpes virus G47Δ killed tumor cells more efficiently than administration of virus alone, and arming G47Δ to express TRAIL further enhanced the survival of tumor-bearing mice [154].
Myxomavirus is a virus pathogenic to European rabbits, but not other vertebrates, making it a safe therapeutic OV. This virus was used to infect adipose-derived MSCs, generating hADMSC-vMyxo cells. Treatment of glioma-bearing animals with hADMSC-vMyxo induced their long-term survival, and repeated administrations of MSCs enhanced the therapeutic benefit [155].
Specific replication of CRAds in glioma cells was also accomplished by employing the human CXCR4 promoter to drive expression of the viral E1 gene (CRAd-CXCR4-RGD and CRAd-CXCR4–5/3); hMSCs infected with these viruses were able to migrate into the tumor and deliver a higher dose of viral particles, especially at distant pockets of migrating glioma cells, than free viral particles [156]. The CXCR4 promoter allowed replication of this virus in both hMSCs and tumor cells, but not in normal astrocytes. Toxicity of the virus was significantly lower in hMSCs than in tumor cells [156].
Survivin, a member of the inhibitor of apoptosis family, is specifically expressed not only in cancer cells and cancer stem cells but also in normal stem cells, where it promotes survival and proliferation [157]. Generating CRAds that drive expression of the E1A viral replication gene under the control of the survivin promoter allows for great specificity of the oncolytic effect. CRAd-S-pk7 was engineered with survivin promoter-dependent replication and a polylysine modification (pk7) that binds to heparin sulfate proteoglycans expressed on the surface of tumor cells, facilitating viral entry. This virus was able to infect both NSCs and human glioma cells with high efficiency, allowed NSCs to migrate into orthotopic implanted tumors in mice and reduced the size of glioma flank tumors by about 50% compared with administration of loose viral particles [158]. The same CRAd-S-pk7 vector was used to optimize parameters of viral loading of NSCs to achieve relevant therapeutic efficacy that can be explored clinically [159]. The study demonstrated that loading the adenovirus onto NSC carriers at a low dose (10–50 viral particles per cell) allowed virus replication, with reduced lysis of NSCs but efficient killing of glioma cells. This strategy mitigated virus-induced neuroinflammation, enhanced tumor-tropic migration of NSCs and prolonged the MS of tumor-bearing mice by about 30% compared with administration of loose viral particles, in an orthotopic glioma model [159]. It was then demonstrated that the human NSC immortalized cell line ReNCell was more effective in delivering viral particles to glioma cells compared with hMSCs [70]. The ReNCells released more viral particles than MSCs following viral infection and displayed enhanced migration toward tumor cells. When implanted in mice with U87MG intracranial tumors, ReNCells laden with CRAd-S-pk7 were more effective than hMSCs in killing glioma cells, improving the MS of tumor-bearing mice [70]. A further study found that the HB1.F3.CD human NSC line displayed increased efficacy compared with ReNCells in delivering the CRAd-S-pk7 to U87MG and improved the MS of tumor-bearing animals [71]. It remains unknown why NSCs have higher capacity for OV delivery than other cells. Until the underlying mechanisms are identified, therapeutic stem cells will need to be individually and thoroughly characterized, keeping in mind that even in the most strictly controlled and optimized preclinical studies there is variability from one study to the next.
Knowing that tropism of stem cells toward gliomas is promoted by the CXCL12/CXCR4 signaling axis and that expression of CXCL12 in gliomas is induced by hypoxia and irradiation [50,51,160], Dey et al. tested whether overexpression of CXCR4 on the human NSC line HB1.F3.CD would improve intratumoral OV delivery, following repeated intranasal administration of NSCs infected with CRAd-S-pk7 [87]. The authors demonstrated that hypoxic preconditioning and transduction of NSCs with CXCR4 promoted migration of NSCs toward glioma cells and increased survival of mice bearing intracranial xenografts treated with radiation therapy (XRT) prior to intranasal delivery of HB1.F3.CD infected with CRAd-S-pk7 [87]. Interestingly, treatment with XRT and OV-loaded NSCs in the absence of hypoxia preconditioning or expression of CXCR4 did not improve survival over XRT alone. Previously, it was shown that HB1.F3.CD cells infected with CRAd-S-pk7 were effective in improving the survival of mice with GBM43 xenografts when radiation was administered after intracranial delivery of NSCs, but less so when XRT was administered prior to NSCs [161]. These studies illustrate that the timing of NSC administration relative to radiation therapy and the route of administration are critical factors to consider when combining OV-NSCs with XRT.
To improve the delivery of stem cells using the intranasal route, Spencer et al. tested two methods of reducing the clearance of NSC from the nasal cavity and improving transmigration of NSCs through the nasal epithelium into the brain: administration of a biodegradable fibrin- and thrombin-based glue into the nasal cavity immediately after the administration of NSCs loaded with OV; or administration of methimazole, a drug used to treat hyperthyroidism which also causes a thinning of the nasal epithelium [90]. Using the same HB1.F3.CD NSC line, which overexpressed CXCR4 and was loaded with CRAd-S-pk7, the authors demonstrated that both the fibrin glue and methimazole decreased the clearance of intranasally administered NSCs and that methimazole treatment increased migration into the tumor and extended the survival of tumor-bearing mice treated with OV-loaded NSCs. In the absence of methimazole, intranasal delivery of OV-NSCs induced no survival benefit [90].
In preparation for clinical use, the HB1.F3.CD NSC cell line, loaded with CRAd-S-pk7 (now named NSC-CRAd-S-pK7) has been extensively tested in laboratory animals for tumor coverage and distribution, safety, viral delivery, persistence in immunocompetent semipermissive hosts and efficacy when combined with SOC [161-164]. It was demonstrated that administration of NSC-CRAd-S-pK7 is safe, results in extensive coverage of the tumor area by migrating NSCs and is more effective when administered prior to radiation and temozolomide therapy. Evidence was presented in support of a mechanism by which pretreatment with CRAd-S-pk7 impaired the DNA damage response and increased radiosensitivity of glioma cells [161].
The human HB1.F3.CD loaded with the CRAd-survivin-pK7 OV has been approved by the FDA as a clinical-grade agent. Preliminary safety data analysis of the first-in-human Phase I clinical trial for primary grade III and IV gliomas using NSCs as delivery agents for OVs (NCT03072134, Supplementary Table 2), has recently been reported [82]. The study enrolled 13 patients with resectable or unresectable tumors, with the primary goal of establishing safety and toxicity of the NSC-CRAd-S-pK7 therapy when combined with SOC, including a dose escalation regimen with three doses of NSCs: 50, 100 and 150 million cells, corresponding to 6.25 × 1010, 1.25 × 1011 and 1.875 × 1011 viral particles, respectively, which were injected at the site of tumor biopsy or resection, followed by standard radiotherapy and temozolomide treatment. The study also aims to follow tumor progression by MRI and measure changes in cytokines and T cell subgroups [82]. Results from this study, scheduled to complete at the end of 2021, are eagerly anticipated.
Therapeutic cytokines, antibodies & toxins delivered by stem cells
Developing effective immunotherapy strategies for GBM has been the goal of numerous preclinical and clinical studies over many decades, with limited success so far. New enthusiasm followed successes in the treatment of other solid tumors with checkpoint inhibitors, and sustained efforts to replicate such results in GBM are ongoing [165]. The promise of long-term efficacy by eliciting durable antitumor immunological memory is a highly desired outcome, especially in a cancer that inevitably recurs. Immunotherapy for gliomas needs to address the severe tumor-induced immune suppression as well as the minimal tolerance for inflammation in the brain [166]. In an effort to boost the antitumor immune response and aid antiglioma vaccine therapies, cytokine therapy has been pursued in preclinical and clinical trials for GBM. The most promising results were shown with delivery of IL-2, IL-4, IL-12 and GMCSF [167,168]. In addition to antigen-presenting cells that bring the added advantage of expressing costimulatory proteins which are able to directly activate immune cells, glioma-tropic stem cells represent good vehicles for cytokine delivery.
IL-4 strongly activates the antitumor immune system in a variety of cancers [169,170]. Expression of the IL-4 receptor has been found extensively on GBM cells, and not on normal neurons and astrocytes, making this receptor a good target for specific therapeutic intervention [171]. The therapeutic potential of mouse and rat NPCs, genetically modified to produce IL-4, has been tested in immunocompetent preclinical models of glioma [94]. Mouse NPCs were generated from the cortex of a postnatal day 1 mouse and transduced with a retrovirus to express IL-4 [94]. It was demonstrated that mouse NPC-IL4 induced long-term survival in the majority of mice, both when injected into the brain simultaneously with tumor cells and when injected 5 days later into established tumors. Similarly, NPCs isolated from the striatum of a rat and immortalized with a temperature-sensitive variant of the SV-40 large T antigen, transduced to express IL-4, induced increased survival of animals with new or established tumors when compared with control animals and even when compared with retrovirus-mediated in vivo transfer of IL-4 vectors [94].
IL-12, a cytokine with important roles in modulating cross-talk between the native and adaptive immune systems, induces proliferation of T lymphocytes and natural killer cells and stimulates production of other cytokines, especially IFN-γ. These activities result in a strong antitumor response in numerous cancers [172,173]. It was shown that intracranial administration of mouse NSCs transduced to express IL-12 improved survival in immunocompetent C57BL/6 mice [174]. Importantly, animals that survived the first administration of tumors were able to reject tumors implanted into the contralateral hemisphere 3 months later, demonstrating the development of antitumor immunological memory. Increased infiltration of CD4+ and CD8+ T cells into the tumor was also observed [174]. Similarly, in a rat glioma model, intratumoral administration of human NSCs that were isolated from the hippocampi of 3- to 5-month-old human embryos and transduced to express IL-12 significantly improved the MS of tumor-bearing rats: from 17 to 87 days when NPCs were administered at the same time as tumor cells, or to 73 days when NPCs were administered 5 days later [175]. Increased infiltration with T cells was also observed. In this study the large survival benefit was likely due to the high number of administered hNSCs, 10- and 30-fold higher than the number of tumor cells injected [175]. MSCs derived from either mouse bone marrow or human umbilical cord, modified to express IL-12, were also found to provide therapeutic benefit in preclinical glioma models [176,177].
Evidence that cytokine delivery by stem cells induces ICD of glioma cells in preclinical models is provided by an elegant study with mouse bone marrow-derived NSC-like cells transduced to express IL-23 (BM-NSC-IL23), a cytokine that induces IL-12 and IFN-γ production [79]. Over half (60%) of mice treated with BM-NSC-IL23 showed long-term survival (>60 days) in immune-competent B6 mice, while the MS of control animals was about 30–35 days. Antibody depletion of CD8+ cytotoxic T cells, CD4+ T cells or NK cells reduced the survival benefit of tumor-bearing mice, demonstrating that cytotoxic T cells were the primary cells responsible for the observed effect with minor contributions from CD4+ and NK cells [79]. When surviving animals were rechallenged with glioma cell implantation, they rejected the tumors and became tumor free, indicating the establishment of long-term antitumor immunity; this process was associated with increased levels of IFN-γ. Studies using MSCs that delivered IL-18, IFN-β or both cytokines also showed a therapeutic effect and establishment of long-term antitumor immunity in glioma-bearing rats, an effect associated with increased intratumoral production of IL-2 and IFN-γ [178,179]. Combined delivery of IL-7 by MSCs with peripheral immunization with IFN-γ-transduced irradiated tumor cells also resulted in decreased tumor size in a rat model of glioma [180]. MSCs transduced to express IFN-β had a direct cytotoxic effect on mouse glioma cells and human primary GBM cells and induced survival benefit in tumor-bearing mice, an effect that was more pronounced when MSC-IFN-β were placed in the postresection cavity and encapsulated in a synthetic extracellular matrix [181]. The melanoma differentiation-associated gene-7 (mda-7/IL-24) cytokine, expressed in terminally differentiated cells and less so in malignant cells, also induces a direct, specific cytotoxic effect in a variety of tumor cells and increases sensitivity to radiotherapy [182]. It has been modified for increased stability and expression of luciferase for in vivo luminescence tracking, generating the multifunctional protein SML7 [183]. Primary mouse NSCs transduced to deliver SML7 induced a reduction in intracranial glioma growth; coadministration of sTRAIL by NSCs further improved this beneficial effect [183].
BMP4, a critical developmental growth factor in embryogenesis, was reported to inhibit the tumorigenic potential of GSCs and increase survival of glioma-bearing animals when administered into the tumor as BMP4-coated acrylic beads [184]. Treatment with human adipose-derived MSCs (hAMSCs), transduced with a retroviral vector to express BMP4 and administered systemically into the left cardiac ventricle of mice with intracranial gliomas (U87 cells), also elicited great survival benefit, with over 75% of tumor-bearing animals becoming tumor free [185]. To increase safety of the therapeutic MSCs, avoiding possible insertional mutagenesis or other viral antigen-induced inflammatory responses, hAMSCs were transfected to express BMP4 using polymer nanoparticles (NP-BMP4.hAMSCs) [186]. Conditioned medium from these cells decreased the viability of patient-derived human GSCs in vitro. Intranasal administration of NP-BMP4.hAMSCs in rats bearing intracranial tumors with GSCs significantly improved MS: from ~14 to ~17 days. It was also demonstrated using fluorescence imaging that both intravenous and intranasal administration of these MSCs resulted in extensive distribution into the tumor [186]. The difference in the extent to which the two BMP4.hAMSC preparations increased survival in the mouse versus the rat model was large: 75% of mice were tumor free versus a ∼3-day increase in MS in rats. However, the studies cannot be properly compared due to other variations in the two experimental paradigms. The difference could be explained by the different distribution of MSCs following intracardiac versus intranasal delivery, but is most likely due to the different tumor cells used, highlighting the fact that impressive results when using the U87 GBM cell line may not reflect efficacy on primary patient tumor GSCs.
Another strategy for cancer immunotherapy emerged in the late 1970s, kindled by advances in toxinology and immunology, when hybrid molecules were constructed from potent toxins conjugated to antitumor antibodies to specifically target tumor cells, leaving normal cells unharmed [187]. Debinski et al. demonstrated that the receptor for IL-13 is amply expressed on numerous GBM cell lines, and that a chimeric protein made of the human IL-13 and a modified form of Pseudomonas exotoxin (PE), PE38QQR, had a powerful cytotoxic effect on GBM cells that was neutralized by IL-13 [188]. It was later discovered that IL-13 binds with high affinity to IL13Rα2, which is present almost exclusively on tumor cells, acts as a decoy receptor and has a higher affinity for IL-13 than IL13Rα1, a receptor found on most normal cells [189]. IL13-PE38QQR was embraced with great enthusiasm and developed as an investigational drug. A Phase III, multicenter, randomized controlled study in first-recurrence GBM patients, comparing bis-chloroethylnitrosourea (BCNU) wafers with IL13-PE38QQR administered by convection-enhanced delivery through a catheter placed into the resection cavity, found no significant difference in OS between the two groups [190]. The authors had concerns about the distribution of the drug (especially to tumor cells that invaded away from the resection cavity), the short half-life of the chimeric protein and the heterogeneous intratumoral expression of IL13Ra2. To overcome these limitations, a stem cell-based distribution strategy was designed. Because PE acts by inactivating the elongation factor EF-2, which leads to inhibition of protein synthesis, EF-2 can be modified to prevent the toxin from exerting its effect. Human NSCs, genetically altered to express a toxin-resistant form of EF-2 and transduced to express IL-13-PE, were tested in a mouse model of GBM for their ability to inhibit tumor growth [191]. Administration of IL-13-PE via toxin-resistant hNSCs (hNSC.IL13-PE) embedded in synthetic extracellular matrix and placed in the postresection cavity of glioma-bearing mice increased MS of the mice by about 60% [191].
Gain of function of EGF signaling – through amplification of EGFR or expression of the mutant, constitutively active form of EGFRvIII – represents one of the most common and best-characterized tumor-specific features of GBM [192], extensively pursued in a variety of targeted antiglioma therapies. Antibody targeting of EGFR and EGFRvIII leads to internalization and silencing of receptor-mediated signaling, inhibiting tumor growth. Preclinical studies with intratumoral delivery of MSCs modified to express a single-chain antibody against EGFRvIII on their surface demonstrated increased survival of glioma-bearing animals treated with these cells [193]. However, the enthusiasm for using EGFRvIII as a therapeutic target in GBM was unfortunately curbed by the results of advanced clinical trials with a peptide vaccine (Rindopepimut/CDX110) developed to target EGFRvIII – results that demonstrated extensive immune editing following treatment with CDX110, with 82% of treated patients losing EGFRvIII expression at recurrence [194]. The large, randomized, multicenter ACT IV study with Rindopepimut for the treatment of patients with newly diagnosed EGFRvIII-positive GBM was discontinued as it did not produce significant OS improvements [195]. The failure of this trial illustrates the difficulty in designing specific therapies for GBM, even when unique tumor-associated antigens are identified and carefully targeted; disease progression leads to the development of a continuously moving target that is increasingly difficult to manage.
Stem cell trafficking of therapeutic nanoparticles
Chemotherapeutic drugs are highly toxic to normal tissues. Intratumoral administration of therapeutic agents has significant advantages over systemic administration, allowing for increased therapeutic index and concentration of the active drug in the tumor tissue, while reducing off-target effects. However, effective distribution and intratumoral retention of the drug following injection (or local placement of hydrogels or other drug-laden scaffolds) is hindered by limited diffusion capacity and adverse pressure gradients that rapidly clear the drug from the tumor. Combining NSC delivery of chemotherapeutics with nanotechnology allows for more efficient distribution of drugs throughout the tumor, including to invading margins and tumor satellites, as well as decreased clearance and release of targeted agents where they are most effective.
Nanoparticles are synthetically generated vesicles with sizes as small as 10 nm and as large as 1000 nm. They are prepared from organic (lipids, polymers, dendrimers) or inorganic (metallic, ceramic) materials, can be solid or colloidal and have applications in numerous biomedical fields, including theranostic applications of drug delivery, gene therapy, immunomodulation, imaging of treatment and disease progression, targeted radiotherapy and photodynamic therapy, modulating release of bioactive molecules, minimizing toxicity and enhancing biocompatibility. Many advances have been made over the last 15 years in the field of nanotechnology, and increased translation into cancer treatment is justifiably anticipated [196]. An excellent review of the use of nanoparticles in brain cancer has recently been published [197]. In this section we will highlight preclinical studies that have employed nanoparticles conjugated to stem cells to improve treatment of GBM.
Systemic administration of nanoparticles for cancer therapies still faces many challenges related to efficient delivery, distribution and retention in brain tumors, especially due to nanoparticle sequestration by the reticuloendothelial system, renal clearance, transport through the BBB and overcoming the elevated intratumoral interstitial fluid pressure, as well as phagocytosis by resident microglia and tumor-associated macrophages. Stem cell-mediated delivery of therapeutic nanoparticles is able to overcome these roadblocks. A proof-of-concept study by Roger et al. demonstrated that poly-lactic acid nanoparticles and lipid nanocapsules can be uploaded onto human bone marrow-derived MSCs and intracranially delivered into mice with established U87 orthotopic brain tumors [198].
Nanoparticles conjugated to NSCs have been used in an intracranial mouse model of GBM, demonstrating that distribution and retention of nanoparticles within the tumor was enhanced when nanoparticles were surface bound to NSCs and administered either adjacent to the tumor, in the contralateral hemisphere or intravenously [199]. These nanoparticles were large (∼800 nm), with high drug-loading capacity, manufactured from polystyrene and conjugated to biotinylated NSCs (HB1.F3 cells) via streptavidin, resulting in substantial nanoparticle cargo onto the NSCs –approximately 175 nanoparticles per NSC [199]. In a preclinical model of ovarian carcinoma, using the same human NSC line (HB1.F3), treatment of tumor-bearing animals with NSCs loaded with silica–platinum nanoparticles was able to induce a higher level of platimun in tumors compared with treatment with nanoparticles or drug alone [200]. Silica nanoparticles loaded with doxorubicin (DOX), named a silica nanorattle, were loaded onto human bone marrow-derived MSCs using antibody-mediated conjugation (CD73, CD90), as a delayed-release ‘time bomb’ for antiglioma therapy, and tested for use in a mouse flank tumor model with U251 glioma cells [201]. MSC administration of DOX showed high-level, broad distribution and prolonged retention compared with injection of free DOX or NP-DOX, and induced increased apoptosis in U251 glioma cells.
Biotin/avidin conjugation of NSCs (HB1.F3) to docetaxel-loaded nanoparticles was employed in a preclinical mouse model of triple-negative breast cancer [202]. In this study the nanoparticles, manufactured using a pH-responsive biotinylated polymer (PEG-PDPAEMA) were conjugated to biotinylated NSCs via an avidin linker. Acidic pH (6.3–6.9) is found in the extracellular space of many solid tumors, including GBM [203–206], making it an ideal signal for the unloading of nanoparticles’ cargo. This study demonstrated that docetaxel-loaded nanoparticles conjugated to NSCs induced a significant decrease in tumor proliferation and vascularization after 7 days when compared with free docetaxel nanoparticles, which were rapidly cleared from the tumor [202].
DOX-loaded pH-sensitive mesoporous silica nanoparticles (DOX-NPs) have been tested in a preclinical intracranial glioma model on their own or when loaded onto human NSCs (HB1.F3.CD) and injected either into the tumor or in the contralateral hemisphere (5 mm away) [207]. It was demonstrated that these nanoparticles, when loaded onto NSCs, delayed release of the drug by about 20 h, presumably until the DOX-NPs were exposed to the acidic lysosomal compartment inside the NSCs. During this time, the tumor-tropic capacity of HB1.F3.CD was not compromised and they were able to reach the tumor to widely distribute and release DOX, inducing the death of both tumor cells and the NSCs. When compared with the intratumoral administration of loose DOX-NPs, NSCs loaded with DOX-NPs were able to significantly increase the MS of tumor-bearing mice [207].
In vitro experiments have also demonstrated that NSCs can be loaded with magnetic spinning disks, 2 μm in diameter, made of several layers of cobalt, iron and boron separated by platinum and covered in gold [208]. In the absence of a magnetic field, these disks are innocuous to the cells. Application of a rotating magnetic field, however, causes the disks to spin and inflict mechanical damage inside the cells. In coculture experiments of NSCs loaded with spinning disks and glioma cells (U87), sequential application of the magnetic field allowed for release of the disks by NSCs and subsequent uptake by glioma cells, which were killed when the rotating magnetic field was applied again [208].
An innovative use of NSCs as vehicles for glioma therapeutic agents was illustrated by Mooney et al. with the use of gold nanorods for plasmonic photothermal ablation of flank tumors in mice [209]. It was demonstrated that human NSCs (HB1.F3) induced a wider, more homogeneous distribution of these nanorods in flank tumors compared with administration of free gold nanorods and reduced tumor recurrence following photoablative treatment. We anticipate seeing this strategy tested in intracranial GBM preclinical models.
Future perspective
Looking back at the last 20 years, the field of stem cell-mediated delivery of antiglioma therapies has advanced considerably. Numerous preclinical studies demonstrated that stem cell delivery of several therapeutic agents results in improved tumor targeting, distribution and efficacy when compared with other methods. Early clinical trials are testing the distribution and safe dose for stem cells that deliver OVs or are used in enzyme/prodrug strategies (NCT02015819, NCT04657315, NCT03072134, NCT03896568).
Inspired by successes in the treatment of other solid tumors with immune checkpoint inhibitors, extensive and sustained efforts to develop effective immunotherapy strategies for GBM are underway [165]. OVs have the proven capacity to induce ICD [134] and can also be regarded as a form of immunotherapy. Following successful early clinical trials, many OVs received an expedited drug review process by the FDA to be advanced to large-scale randomized trials [136]. Study after study demonstrated that delivery of OVs by NSCs results in enhanced tumor coverage, distribution and efficacy when compared with delivery of free virus, advocating for efforts to fast track many more trials using this strategy into the clinic [92,153,154,159]. The first-in-human clinical trial using NSCs as delivery agents for OVs will soon be completed and the results are eagerly awaited. The prospects of success with stem cell-based oncolytic virotherapy bring added hope for the induction of long-term immunological memory that would halt recurrence of the disease early on. It is anticipated that more trials with combination therapies using stem cell delivery of OVs and/or immunostimulatory agents will soon be conducted. Combinatorial strategies using multimodal therapeutic stem cells are likely to be more successful than current therapies. Successful targeting of GBM will require combination therapy with multiple agents amenable to adaptation as the tumor progresses.
Developing an expanded repository of clinically approved stem cells, able to deliver a variety of antiglioma agents with different mechanisms of action, would bring significant improvements to currently available therapies. There is a great need to conduct more rigorous and mechanistic studies to identify factors that determine the stem cell properties leading to their increased efficiency as therapeutic vehicles. These findings will allow for targeted and reproducible generation of cells, permitting real comparisons between different studies and leading to accelerated progress. Due to the rapid expansion of the use of MSCs in translational studies, there is an imminent need to rigorously define this heterogeneous population of cells and establish gold standards for the procedures used to derive and use MSC-based therapies to ensure their safety and efficacy [81].
Exciting progress comes from the field of nanotechnology, with enhanced biomaterials being developed for drug and nucleic acid delivery. Conjugation of NSCs to nanoparticles has proven to be feasible and more effective in delivering chemotherapeutics in preclinical models of glioma than direct administration of nanoparticles. The acidic intratumoral environment brings an added bonus for pH-responsive nanoparticles, facilitating local unloading of their cargo. Great hope is provided by the ability of nanoparticles to deliver nucleic acids into gliomas. Oligonucleotides targeting mRNA, regulatory RNAs and disease-specific mutations are emerging as a new class of targeted therapeutics with great potential to improve the outcome for GBM patients [210]. Studies are focused on improving the stability of modified oligonucleotides, evading endosomal entrapment and improving delivery and distribution of these agents into brain tumors [210]. This strategy, if optimized for use with stem cell-mediated delivery, would overcome many current therapeutic limitations in gliomas. Targeting glioma-specific genes with siRNAs or CRISPR/Cas9 vectors represents a powerful therapeutic approach, as many of the drivers of malignant glioma and of the GSC phenotype are developmental transcription factors generally considered ‘undruggable’. Stem cells conjugated via a pH-sensitive bond to nanoparticles loaded with nucleic acids may provide a viable avenue to overcome current limitations.
An emerging field with exciting potential for the treatment of GBM is based on the ability of stem cells to deliver proteins and nucleic acids via exosomes [211,212]. MSCs and NSCs are cell types known to be some of the most prolific producers of exosomes [213,214]. Exosomes from genetically engineered MSCs have been used to deliver prodrugs, miRNAs or tumor suppressors to glioma cells in vivo and in vitro, demonstrating therapeutic effects [215–219]. Treatment with modified stem cells that produce exosomes in situ may provide increased efficacy by evading hepatic and renal clearance and allowing for improved homing and distribution of the exosomes throughout the tumor.
Finally, in vitro brain tumor organoid models [220] provide improved 3D systems to test the efficacy of stem cell therapies for GBM [221]. These models will likely be instrumental for the much-needed in-depth mechanistic analyses and for identification of molecular players and pathways that orchestrate optimal and reproducible parameters for stem cell therapies.
In this review we aimed to present a comprehensive view of the progress in stem cell therapies for GBM in the 20 years since their initiation. We presented and analyzed mechanisms underlying the biological effects of therapeutic stem cells, highlighting experimental progress, limitations, the importance of critical experimental parameters and the emergence of promising new therapeutic avenues. The hope is to increase awareness of the advantages brought by stem cells for the treatment of GBM and inspire further studies that will lead to accelerated implementation of effective therapies. Treatment failure in GBM is in large part due to limitations in the delivery of therapeutic agents into invasive intracranial tumors. Stem cells represent a viable and promising solution for this problem that is worthwhile perfecting further.
Acknowledgments
We thank the Undergraduate Research Opportunity Program at the University of Michigan for supporting the work of MK and ZS.
Footnotes
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.futuremedicine.com/doi/suppl/10.2217/cns-2020-0026
Financial & competing interests disclosure
Grant support was provided by NIH/NINDS R21NS107879 (AC). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Breunig JJ, Haydar TF, Rakic P. Neural stem cells: historical perspective and future prospects. Neuron 70(4), 614–625 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.His W. Die Entwickelung des menschlichen Gehirns: während der ersten Monate. S. Hirzel; (1904). [Google Scholar]
- 3.Gage FH, Ray J, Fisher LJ. Isolation, characterization, and use of stem cells from the CNS. Annu. Rev. Neurosci. 18(1), 159–192 (1995). [DOI] [PubMed] [Google Scholar]
- 4.Weiss S, Reynolds BA, Vescovi AL, Morshead C, Craig CG, Van Der Kooy D. Is there a neural stem cell in the mammalian forebrain? Trends Neurosci. 19(9), 387–393 (1996). [DOI] [PubMed] [Google Scholar]
- 5.Temple S. Division and differentiation of isolated CNS blast cells in microculture. Nature 340(6233), 471–473 (1989). [DOI] [PubMed] [Google Scholar]
- 6.Temple S. The development of neural stem cells. Nature 414(6859), 112–117 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Snyder EY, Taylor RM, Wolfe JH. Neural progenitor cell engraftment corrects lysosomal storage throughout the MRS VII mouse brain. Nature 374(6520), 367–370 (1995). [DOI] [PubMed] [Google Scholar]
- 8.Armstrong RJ, Watts C, Svendsen CN, Dunnett SB, Rosser AE. Survival, neuronal differentiation, and fiber outgrowth of propagated human neural precursor grafts in an animal model of Huntington’s disease. Cell Transplant. 9(1), 55–64 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Svendsen CN, Caldwell MA, Shen J et al. Long-term survival of human central nervous system progenitor cells transplanted into a rat model of Parkinson’s disease. Exp. Neurol. 148(1), 135–146 (1997). [DOI] [PubMed] [Google Scholar]
- 10.Olanow C, Fahn S. Fetal nigral transplantation as a therapy for Parkinson’s disease. : Restorative Therapies in Parkinson’s Disease, SpringerScience+Business Media, LLC, NY, USA, 93–118 (2006). [Google Scholar]
- 11.Yandava BD, Billinghurst LL, Snyder EY. ‘Global’ cell replacement is feasible via neural stem cell transplantation: evidence from the dysmyelinated shiverer mouse brain. Proc. Natl Acad. Sci. USA 96(12), 7029–7034 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silani V, Cova L, Corbo M, Ciammola A, Polli E. Stem-cell therapy for amyotrophic lateral sclerosis. Lancet 364(9429), 200–202 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Teng YD, Lavik EB, Qu X et al. Functional recovery following traumatic spinal cord injury mediated by a unique polymer scaffold seeded with neural stem cells. Proc. Natl Acad. Sci. USA 99(5), 3024–3029 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bauer G, Elsallab M, Abou-El-Enein M. Concise review: a comprehensive analysis of reported adverse events in patients receiving unproven stem cell-based interventions. Stem Cells Transl. Med. 7(9), 676–685 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aboody K, Capela A, Niazi N, Stern JH, Temple S. Translating stem cell studies to the clinic for CNS repair: current state of the art and the need for a Rosetta stone. Neuron 70(4), 597–613 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Ostrom QT, Cote DJ, Ascha M, Kruchko C, Barnholtz-Sloan JS. Adult glioma incidence and survival by race or ethnicity in the United States from 2000 to 2014. JAMA Oncol. 4(9), 1254–1262 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stupp R, Mason WP, Van Den Bent MJ et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352(10), 987–996 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Stupp R, Taillibert S, Kanner A et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA 318(23), 2306–2316 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lassman AB, Joanta-Gomez AE, Pan PC, Wick W. Current usage of tumor treating fields for glioblastoma. Neuro Oncol. Adv. 2(1), vdaa069 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fabian D, Guillermo Prieto Eibl MDP, Alnahhas I et al. Treatment of glioblastoma (GBM) with the addition of tumor-treating fields (TTF): a review. Cancers 11(2), 174 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Osuka S, Van Meir EG. Overcoming therapeutic resistance in glioblastoma: the way forward. J. Clin. Invest. 127(2), 415–426 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh SK, Clarke ID, Terasaki M et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 63(18), 5821–5828 (2003). [PubMed] [Google Scholar]
- 23.Ligon KL, Huillard E, Mehta S et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron 53(4), 503–517 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bao S, Wu Q, Li Z et al. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 68(15), 6043–6048 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beier D, Schulz JB, Beier CP. Chemoresistance of glioblastoma cancer stem cells-much more complex than expected. Mol. Cancer 10(1), 128 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calinescu AA, Yadav VN, Carballo E et al. Survival and proliferation of neural progenitor derived glioblastomas under hypoxic stress is controlled by a CXCL12/CXCR4 autocrine positive feedback mechanism. Clin. Cancer Res. 23(5), 1250–1262 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanai N, Alvarez-Buylla A, Berger MS. Neural stem cells and the origin of gliomas. N. Engl. J. Med. 353(8), 811–822 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Lim DA, Cha S, Mayo MC et al. Relationship of glioblastoma multiforme to neural stem cell regions predicts invasive and multifocal tumor phenotype. Neuro Oncol. 9(4), 424–429 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rakic P, Lombroso PJ. Development of the cerebral cortex: i. Forming the cortical structure. J. Am. Acad. Child Adolesc. Psychiatry 37(1), 116–117 (1998). [DOI] [PubMed] [Google Scholar]
- 30.Roake CM, Artandi SE. Regulation of human telomerase in homeostasis and disease. Nat. Rev. Mol. Cell Biol. 21(7), 384–397 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vinagre J, Almeida A, Pópulo H et al. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 4(1), 1–6 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Killela PJ, Reitman ZJ, Jiao Y et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl Acad. Sci. USA 110(15), 6021–6026 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JH, Lee JE, Kahng JY et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 560(7717), 243–247 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. Neurons derived from radial glial cells establish radial units in neocortex. Nature 409(6821), 714–720 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 32, 149–184 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley JG, Mitchell BD, Kempermann G, Macklis JD. Adult neurogenesis and repair of the adult CNS with neural progenitors, precursors, and stem cells. Prog. Neurobiol. 75(5), 321–341 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Reynolds BA, Rietze RL. Neural stem cells and neurospheres – re-evaluating the relationship. Nat. Methods 2(5), 333–336 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Wakeman DR, Hofmann MR, Teng YD, Snyder EY. Neural progenitors. : Human Adult Stem Cells. SpringerScience+Business Media, LLC, NY, USA, 1–44 (2009). [Google Scholar]
- 39.Horiguchi S, Takahashi J, Kishi Y et al. Neural precursor cells derived from human embryonic brain retain regional specificity. J. Neurosci. Res. 75(6), 817–824 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Bhaduri A, Di Lullo E, Jung D et al. Outer radial glia-like cancer stem cells contribute to heterogeneity of glioblastoma. Cell Stem Cell 26(1), 48–63. e46 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Darmanis S, Sloan SA, Croote D et al. Single-cell RNA-seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep. 21(5), 1399–1410 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Venteicher AS, Tirosh I, Hebert C et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355(6332), eaai8478 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Filbin MG, Tirosh I, Hovestadt V et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360(6386), 331–335 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neftel C, Laffy J, Filbin MG et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178(4), 835–849. e821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Couturier CP, Ayyadhury S, Le PU et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Comm. 11(1), 1–19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tiveron M-C, Cremer H. CXCL12/CXCR4 signalling in neuronal cell migration. Curr. Opin. Neurobiol. 18(3), 237–244 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Goffart N, Kroonen J, Di Valentin E et al. Adult mouse subventricular zones stimulate glioblastoma stem cells specific invasion through CXCL12/CXCR4 signaling. Neuro Oncol. 17(1), 81–94 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hira VV, Molenaar RJ, Breznik B, Lah T, Aronica E, Van Noorden CJ. Immunohistochemical detection of neural stem cells and glioblastoma stem cells in the subventricular zone of glioblastoma patients. J. Histochem. Cytochem. 69(5), 349–364 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature 505(7483), 327–334 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tabatabai G, Frank B, Möhle R, Weller M, Wick W. Irradiation and hypoxia promote homing of haematopoietic progenitor cells towards gliomas by TGF-β-dependent HIF-1α-mediated induction of CXCL12. Brain 129(9), 2426–2435 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Komatani H, Sugita Y, Arakawa F, Ohshima K, Shigemori M. Expression of CXCL12 on pseudopalisading cells and proliferating microvessels in glioblastomas: an accelerated growth factor in glioblastomas. Int. J. Oncol. 34(3), 665–672 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Inukai M, Hara A, Yasui Y, Kumabe T, Matsumoto T, Saegusa M. Hypoxia-mediated cancer stem cells in pseudopalisades with activation of hypoxia-inducible factor-1α/Akt axis in glioblastoma. Hum. Pathol. 46(10), 1496–1505 (2015). [DOI] [PubMed] [Google Scholar]
- 53.Mckay R. Stem cells in the central nervous system. Science 276(5309), 66–71 (1997). [DOI] [PubMed] [Google Scholar]
- 54.Gage FH. Mammalian neural stem cells. Science 287(5457), 1433–1438 (2000). [DOI] [PubMed] [Google Scholar]
- 55.Flax JD, Aurora S, Yang C et al. Engraftable human neural stem cells respond to development cues, replace neurons, and express foreign genes. Nat. Biotechnol. 16(11), 1033–1039 (1998). [DOI] [PubMed] [Google Scholar]
- 56.Martinez-Serrano A, Lundberg C, Björklund A. Use of conditionally immortalized neural progenitors for transplantation and gene transfer to the CNS. : Isolation, Characterization and Utilization of CNS Stem Cells. Springer-Verlag Berlin Heidelberg, Germany, 151–168 (1997). [Google Scholar]
- 57.Snyder EY. Neural stem-like cells: developmental lessons with therapeutic potential. Neuroscientist 4(6), 408–425 (1998). [Google Scholar]
- 58.Aboody KS, Brown A, Rainov NG et al. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc. Natl Acad. Sci. USA 97(23), 12846–12851 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• First evidence in a mouse model of glioblastoma (GBM) that murine and human neural stem cells (NSCs) transduced to express therapeutic molecules and administered either in the contralateral hemisphere, into the lateral ventricle or intravenously, migrate from a distance towards established tumors and even towards invading streams of GBM cells.
- 59.Ryder EF, Snyder EY, Cepko CL. Establishment and characterization of multipotent neural cell lines using retrovirus vector-mediated oncogene transfer. J. Neurobiol. 21(2), 356–375 (1990). [DOI] [PubMed] [Google Scholar]
- 60.Kim SU. Human neural stem cells genetically modified for brain repair in neurological disorders. Neuropathology 24(3), 159–171 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Ryu JK, Choi HB, Hatori K et al. Adenosine triphosphate induces proliferation of human neural stem cells: role of calcium and p70 ribosomal protein S6 kinase. J. Neurosci. Res. 72(3), 352–362 (2003). [DOI] [PubMed] [Google Scholar]
- 62.Aboody KS, Najbauer J, Metz MZ et al. Neural stem cell-mediated enzyme/prodrug therapy for glioma: preclinical studies. Sci. Transl. Med. 5(184), 184ra159–184ra159 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Donato R, Miljan EA, Hines SJ et al. Differential development of neuronal physiological responsiveness in two human neural stem cell lines. BMC Neurosci. 8(1), 36 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ostenfeld T, Joly E, Tai Y-T et al. Regional specification of rodent and human neurospheres. Brain Res. Dev. Brain Res. 134(1–2), 43–55 (2002). [DOI] [PubMed] [Google Scholar]
- 65.Kelly TK, Karsten SL, Geschwind DH, Kornblum HI. Cell lineage and regional identity of cultured spinal cord neural stem cells and comparison to brain-derived neural stem cells. PLoS ONE 4(1), e4213 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hack M, Sugimori M, Lundberg C, Nakafuku M, Götz M. Regionalization and fate specification in neurospheres: the role of Olig2 and Pax6. Mol. Cell. Neurosci. 25(4), 664–678 (2004). [DOI] [PubMed] [Google Scholar]
- 67.Santa-Olalla J, Baizabal JM, Fregoso M, Del Carmen Cardenas M, Covarrubias L. The in vivo positional identity gene expression code is not preserved in neural stem cells grown in culture. Eur. J. Neurosci. 18(5), 1073–1084 (2003). [DOI] [PubMed] [Google Scholar]
- 68.Chamberlain G, Fox J, Ashton B, Middleton J. Concise review: mesenchymal stem cells: their phenotype, differentiation capacity, immunological features, and potential for homing. Stem Cells 25(11), 2739–2749 (2007). [DOI] [PubMed] [Google Scholar]
- 69.Klopp AH, Gupta A, Spaeth E, Andreeff M, Marini Iii F. Concise review: dissecting a discrepancy in the literature: do mesenchymal stem cells support or suppress tumor growth? Stem Cells 29(1), 11–19 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ahmed AU, Tyler MA, Thaci B et al. A comparative study of neural and mesenchymal stem cell-based carriers for oncolytic adenovirus in a model of malignant glioma. Mol. Pharm. 8(5), 1559–1572 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ahmed AU, Thaci B, Tobias AL et al. A preclinical evaluation of neural stem cell–based cell carrier for targeted antiglioma oncolytic virotherapy. J. Natl Cancer Inst. 105(13), 968–977 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bagó JR, Alfonso-Pecchio A, Okolie O et al. Therapeutically engineered induced neural stem cells are tumour-homing and inhibit progression of glioblastoma. Nat.Commun. 7, 10593 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bagó JR, Okolie O, Dumitru R et al. Tumor-homing cytotoxic human induced neural stem cells for cancer therapy. Sci. Transl. Med. 9(375), eaah6510 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gao M, Yao H, Dong Q et al. Tumourigenicity and immunogenicity of induced neural stem cell grafts versus induced pluripotent stem cell grafts in syngeneic mouse brain. Sci. Rep. 6(1), 1–13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lettry V, Hagler SB, Khagi S, Hingtgen SD. Tumor-homing stem cell therapy for brain cancer. Curr. Surg. Rep. 5, 28 (2017). [Google Scholar]
- 76.Amariglio N, Hirshberg A, Scheithauer BW et al. Donor-derived brain tumor following neural stem cell transplantation in an ataxia telangiectasia patient. PLoS Med. 6(2), e1000029 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Berkowitz AL, Miller MB, Mir SA et al. Glioproliferative lesion of the spinal cord as a complication of ‘stem-cell tourism’. N. Engl. J. Med. 375(2), 196–198 (2016). [DOI] [PubMed] [Google Scholar]
- 78.Portnow J, Synold TW, Badie B et al. Neural stem cell-based anticancer gene therapy: a first-in-human study in recurrent high-grade glioma patients. Clin. Cancer Res. 23(12), 2951–2960 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• First report of a pilot clinical trial (NCT01172964) using therapeutic human NSCs (HB1.F3.CD) for the treatment of GBM. Brain levels of 5-fluorouracil (5-FU) were much higher than in the blood, indicating local production of 5-FU by the NSCs.
- 79.Yuan X, Hu J, Belladonna ML, Black KL, John SY. Interleukin-23-expressing bone marrow-derived neural stem-like cells exhibit antitumor activity against intracranial glioma. Cancer Res. 66(5), 2630–2638 (2006). [DOI] [PubMed] [Google Scholar]; • Demonstrated that mesenchymal stem cells (MSCs) expressing IL-23 delivered into brain tumors of mice induced long-term survival. When cytotoxic CD8+ T cells were depleted (and, to a lesser degree, CD4+ T cells), the survival benefit decreased, indicating tumor immune rejection.
- 80.Liang W, Chen X, Zhang S et al. Mesenchymal stem cells as a double-edged sword in tumor growth: focusing on MSC-derived cytokines. Cell. Mol. Biol. Lett. 26(1), 1–25 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bajetto A, Thellung S, Dellacasagrande I, Pagano A, Barbieri F, Florio T. Cross talk between mesenchymal and glioblastoma stem cells: communication beyond controversies. Stem Cells Transl. Med. 9(11), 1310–1330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lesniak MS, Stupp R, Sachdev S et al. ACTR-36. A Phase I study of neural stem cells loaded with an oncolytic adenovirus for patients with newly diagnosed malignant glioma: preliminary safety and data analysis. Neuro Oncol. 19(Suppl. 6), vi8 (2017). [Google Scholar]
- 83.Gutova M, Flores L, Adhikarla V et al. Quantitative evaluation of intraventricular delivery of therapeutic neural stem cells to orthotopic glioma. Front. Oncol. 9, 68 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Barish ME, Herrmann K, Tang Y et al. Human neural stem cell biodistribution and predicted tumor coverage by a diffusible therapeutic in a mouse glioma model. Stem Cells Transl. Med. 6(6), 1522–1532 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li G, Bonamici N, Dey M, Lesniak MS, Balyasnikova IV. Intranasal delivery of stem cell-based therapies for the treatment of brain malignancies. Expert Opin. Drug Deliv. 15(2), 163–172 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Balyasnikova IV, Prasol MS, Ferguson SD et al. Intranasal delivery of mesenchymal stem cells significantly extends survival of irradiated mice with experimental brain tumors. Mol. Ther. 22(1), 140–148 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dey M, Yu D, Kanojia D et al. Intranasal oncolytic virotherapy with CXCR4-enhanced stem cells extends survival in mouse model of glioma. Stem Cell Rep. 7(3), 471–482 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Showed that intranasal delivery of NSCs overexpressing CXCR4 loaded with oncolytic virus, combined with radiotherapy, increased survival of tumor bearing animals versus radiation alone. Intranasal delivery of therapeutic stem cells (SCs) will allow for repeated noninvasive administrations of SCs.
- 88.Gutova M, Shahmanyan D, Oganesyan D et al. Intranasal delivery of therapeutic neural stem cells to target intracerebral glioma. Enliven: J. Stem Cells Regen. Med. 1(1), 1–7 (2015). [Google Scholar]
- 89.Chastkofsky MI, Pituch KC, Katagi H et al. Mesenchymal stem cells successfully deliver oncolytic virotherapy to diffuse intrinsic pontine glioma. Clin. Cancer Res. 27(6), 1766–1777 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Spencer D, Yu D, Morshed RA et al. Pharmacologic modulation of nasal epithelium augments neural stem cell targeting of glioblastoma. Theranostics 9(7), 2071 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Srinivasan VM, Lang FF, Kan P. Intraarterial delivery of virotherapy for glioblastoma. Neurosurg. Focus 50(2), E7 (2021). [DOI] [PubMed] [Google Scholar]
- 92.Yong RL, Shinojima N, Fueyo J et al. Human bone marrow-derived mesenchymal stem cells for intravascular delivery of oncolytic adenovirus Δ24-RGD to human gliomas. Cancer Res. 69(23), 8932–8940 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Herrlinger U, Woiciechowski C, Sena-Esteves M et al. Neural precursor cells for delivery of replication-conditional HSV-1 vectors to intracerebral gliomas. Mol. Ther. 1(4), 347–357 (2000). [DOI] [PubMed] [Google Scholar]
- 94.Benedetti S, Pirola B, Pollo B et al. Gene therapy of experimental brain tumors using neural progenitor cells. Nat. Med. 6(4), 447–450 (2000). [DOI] [PubMed] [Google Scholar]; •• NSCs modified to express IL-4 and injected into brain tumors in mice and rats induced long-term survival in both models. Upon tumor rechallenge in the contralateral hemisphere, long-term surviving rats rejected the tumor, indicating establishment of immunological memory.
- 95.Ehtesham M, Kabos P, Gutierrez MA et al. Induction of glioblastoma apoptosis using neural stem cell-mediated delivery of tumor necrosis factor-related apoptosis-inducing ligand. Cancer Res. 62(24), 7170–7174 (2002). [PubMed] [Google Scholar]
- 96.Moolten FL. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res. 46(10), 5276–5281 (1986). [PubMed] [Google Scholar]
- 97.Hossain JA, Marchini A, Fehse B, Bjerkvig R, Miletic H. Suicide gene therapy for the treatment of high-grade glioma: past lessons, present trends, and future prospects. Neuro Oncol. Adv. 2(1), vdaa013 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Uhl M, Weiler M, Wick W, Jacobs AH, Weller M, Herrlinger U. Migratory neural stem cells for improved thymidine kinase-based gene therapy of malignant gliomas. Biochem. Biophys. Res. Commun. 328(1), 125–129 (2005). [DOI] [PubMed] [Google Scholar]
- 99.Li S, Tokuyama T, Yamamoto J, Koide M, Yokota N, Namba H. Bystander effect-mediated gene therapy of gliomas using genetically engineered neural stem cells. Cancer Gene Ther. 12(7), 600–607 (2005). [DOI] [PubMed] [Google Scholar]
- 100.Li S, Gu C, Gao Y et al. Bystander effect in glioma suicide gene therapy using bone marrow stromal cells. Stem Cell Res. 9(3), 270–276 (2012). [DOI] [PubMed] [Google Scholar]
- 101.Kim S-K, Kim SU, Park IH et al. Human neural stem cells target experimental intracranial medulloblastoma and deliver a therapeutic gene leading to tumor regression. Clin. Cancer Res. 12(18), 5550–5556 (2006). [DOI] [PubMed] [Google Scholar]
- 102.Altanerova V, Cihova M, Babic M et al. Human adipose tissue-derived mesenchymal stem cells expressing yeast cytosinedeaminase::uracil phosphoribosyltransferase inhibit intracerebral rat glioblastoma. Int. J. Cancer 130(10), 2455–2463 (2012). [DOI] [PubMed] [Google Scholar]
- 103.Van Kuilenburg AB. Dihydropyrimidine dehydrogenase and the efficacy and toxicity of 5-fluorouracil. Eur. J. Cancer 40(7), 939–950 (2004). [DOI] [PubMed] [Google Scholar]
- 104.Chung T, Na J, Kim Y-I et al. Dihydropyrimidine dehydrogenase is a prognostic marker for mesenchymal stem cell-mediated cytosine deaminase gene and 5-fluorocytosine prodrug therapy for the treatment of recurrent gliomas. Theranostics 6(10), 1477 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kim SU, Nagai A, Nakagawa E et al. Production and characterization of immortal human neural stem cell line with multipotent differentiation property. Methods Mol. Biol. 438, 103–121 (2008). [DOI] [PubMed] [Google Scholar]
- 106.Portnow J, Badie B, Synold TW et al. A first-in-human study of neural stem cells (NSCs) expressing cytosine deaminase (CD) in combination with 5-fluorocytosine (5-FC) in patients with recurrent high-grade glioma. J. Clin. Oncol. 31(Suppl. 15), 2018 2013). [Google Scholar]
- 107.Choi SA, Lee JY, Wang K-C et al. Human adipose tissue-derived mesenchymal stem cells: characteristics and therapeutic potential as cellular vehicles for prodrug gene therapy against brainstem gliomas. Eur. J. Cancer 48(1), 129–137 (2012). [DOI] [PubMed] [Google Scholar]
- 108.Yin J, Kim J-K, Moon J-H et al. hMSC-mediated concurrent delivery of endostatin and carboxylesterase to mouse xenografts suppresses glioma initiation and recurrence. Mol. Ther. 19(6), 1161–1169 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gutova M, Shackleford GM, Khankaldyyan V et al. Neural stem cell-mediated CE/CPT-11 enzyme/prodrug therapy in transgenic mouse model of intracerebellar medulloblastoma. Gene Ther. 20(2), 143–150 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wierdl M, Tsurkan L, Hyatt J et al. An improved human carboxylesterase for enzyme/prodrug therapy with CPT-11. Cancer Gene Ther. 15(3), 183–192 (2008). [DOI] [PubMed] [Google Scholar]
- 111.Metz MZ, Gutova M, Lacey SF et al. Neural stem cell-mediated delivery of irinotecan-activating carboxylesterases to glioma: implications for clinical use. Stem Cells Transl. Med. 2(12), 983–992 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mooney R, Majid AA, Batalla J, Annala AJ, Aboody KS. Cell-mediated enzyme prodrug cancer therapies. Adv. Drug Deliv. Rev. 118, 35–51 (2017). [DOI] [PubMed] [Google Scholar]
- 113.Levine AJ. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 20(8), 471–480 (2020). [DOI] [PubMed] [Google Scholar]
- 114.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455(7216), 1061–1068 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Von Karstedt S, Montinaro A, Walczak H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer 17(6), 352–366 (2017). [DOI] [PubMed] [Google Scholar]
- 116.Micheau O, Shirley S, Dufour F. Death receptors as targets in cancer. Br. J. Pharmacol. 169(8), 1723–1744 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ashkenazi A, Pai RC, Fong S et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Invest. 104(2), 155–162 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Belkahla H, Herlem G, Picaud F et al. TRAIL–NP hybrids for cancer therapy: a review. Nanoscale 9(18), 5755–5768 (2017). [DOI] [PubMed] [Google Scholar]
- 119.Dörr J, Waiczies S, Wendling U, Seeger B, Zipp F. Induction of TRAIL-mediated glioma cell death by human T cells. J. Neuroimmunol. 122(1–2), 117–124 (2002). [DOI] [PubMed] [Google Scholar]
- 120.Corsten MF, Miranda R, Kasmieh R, Krichevsky AM, Weissleder R, Shah K. MicroRNA-21 knockdown disrupts glioma growth in vivo and displays synergistic cytotoxicity with neural precursor cell–delivered S-TRAIL in human gliomas. Cancer Res. 67(19), 8994–9000 (2007). [DOI] [PubMed] [Google Scholar]
- 121.Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 65(14), 6029–6033 (2005). [DOI] [PubMed] [Google Scholar]
- 122.Hingtgen S, Ren X, Terwilliger E, Classon M, Weissleder R, Shah K. Targeting multiple pathways in gliomas with stem cell and viral delivered S-TRAIL and temozolomide. Mol. Cancer Ther. 7(11), 3575–3585 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lashinger LM, Zhu K, Williams SA, Shrader M, Dinney CP, Mcconkey DJ. Bortezomib abolishes tumor necrosis factor–related apoptosis-inducing ligand resistance via a p21-dependent mechanism in human bladder and prostate cancer cells. Cancer Res. 65(11), 4902–4908 (2005). [DOI] [PubMed] [Google Scholar]
- 124.Koschny R, Holland H, Sykora J et al. Bortezomib sensitizes primary human astrocytoma cells of WHO grades I to IV for tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Clin. Cancer Res. 13(11), 3403–3412 (2007). [DOI] [PubMed] [Google Scholar]
- 125.Balyasnikova IV, Ferguson SD, Han Y, Liu F, Lesniak MS. Therapeutic effect of neural stem cells expressing TRAIL and bortezomib in mice with glioma xenografts. Cancer Lett. 310(2), 148–159 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Perlstein B, Finniss SA, Miller C et al. TRAIL conjugated to nanoparticles exhibits increased anti-tumor activities in glioma cells and glioma stem cells in vitro and in vivo. Neuro Oncol. 15(1), 29–40 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jiang X, Fitch S, Wang C et al. Nanoparticle engineered TRAIL-overexpressing adipose-derived stem cells target and eradicate glioblastoma via intracranial delivery. Proc. Natl Acad. Sci. USA 113(48), 13857–13862 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Norton L, Massagué J. Is cancer a disease of self-seeding? Nat. Med. 12(8), 875–878 (2006). [DOI] [PubMed] [Google Scholar]
- 129.Ehtesham M, Winston J, Kabos P, Thompson R. CXCR4 expression mediates glioma cell invasiveness. Oncogene 25(19), 2801–2806 (2006). [DOI] [PubMed] [Google Scholar]
- 130.Liu Z, Kuang W, Zhou Q, Zhang Y. TGF-β1 secreted by M2 phenotype macrophages enhances the stemness and migration of glioma cells via the SMAD2/3 signalling pathway. Int. J. Mol. Med. 42(6), 3395–3403 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Reinshagen C, Bhere D, Choi SH et al. CRISPR-enhanced engineering of therapy-sensitive cancer cells for self-targeting of primary and metastatic tumors. Sci. Transl. Med. 10(449), eaao3240 (2018). [DOI] [PubMed] [Google Scholar]
- 132.Kaufmann JK, Chiocca EA. Glioma virus therapies between bench and bedside. Neuro Oncol. 16(3), 334–351 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wollmann G, Ozduman K, Van Den Pol AN. Oncolytic virus therapy of glioblastoma multiforme–concepts and candidates. Cancer J. 18(1), 69 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol. Res. 2(4), 295–300 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol. Ther. 15(4), 651–659 (2007). [DOI] [PubMed] [Google Scholar]
- 136.Martikainen M, Essand M. Virus-based immunotherapy of glioblastoma. Cancers 11(2), 186 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Desjardins A, Gromeier M, Herndon JE et al. Recurrent glioblastoma treated with recombinant poliovirus. N. Engl. J. Med. 379(2), 150–161 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Desjardins A, Gromeier M, Herndon JE et al. Oncolytic polio/rhinovirus recombinant (PVSRIPO) against WHO grade IV malignant glioma (MG): experience with retreatment of survivors from the Phase I trial. J. Clin. Oncol. 37(Suppl. 15), 2060–2060 (2019). [Google Scholar]
- 139.Cloughesy TF, Landolfi J, Vogelbaum MA et al. Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511+ Toca FC. Neuro Oncol. 20(10), 1383–1392 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lang FF, Conrad C, Gomez-Manzano C et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent malignant glioma. J. Clin. Oncol. 36(14), 1419 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cloughesy T, Petrecca K, Walbert T et al. LTBK-08. TOCA 511 & TOCA FC versus standard of care in patients with recurrent high grade glioma. Neuro Oncol. 21(Suppl. 6), vi284–vi284 (2019). [Google Scholar]
- 142.Todo T, Martuza RL, Rabkin SD, Johnson PA. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl Acad. Sci. USA 98(11), 6396–6401 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Todo T. ATIM-14. Results of Phase II clinical trial of oncolytic herpes virus G47Δ in patients with glioblastoma. Neuro Oncol. 21(Suppl. 6), vi4–vi4 (2019). [Google Scholar]
- 144.Foreman PM, Friedman GK, Cassady KA, Markert JM. Oncolytic virotherapy for the treatment of malignant glioma. Neurotherapeutics 14(2), 333–344 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chiocca EA, Abbed KM, Tatter S et al. A Phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol. Ther. 10(5), 958–966 (2004). [DOI] [PubMed] [Google Scholar]
- 146.Fueyo J, Gomez-Manzano C, Alemany R et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 19(1), 2–12 (2000). [DOI] [PubMed] [Google Scholar]
- 147.O’Shea CC, Johnson L, Bagus B et al. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell 6(6), 611–623 (2004). [DOI] [PubMed] [Google Scholar]
- 148.Gopakumar S, Gumin J, Daou M, Ledbetter D, Kerrigan BP, Lang FF. Stem cell delivery of oncolytic adenovirus DNX-2401 following surgical resection for the treatment of glioblastoma in a murine model. Neurosurgery 66(Suppl. 1), nyz310_312 (2019). [Google Scholar]
- 149.Rojas JJ, Guedan S, Searle PF et al. Minimal RB-responsive E1A promoter modification to attain potency, selectivity, and transgene-arming capacity in oncolytic adenoviruses. Mol. Ther. 18(11), 1960–1971 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Guedán Carrió S, Gros Vidal A, Mercadé Gil ME, Cascalló Piqueras M, Alemany Bonastre R. Hyaluronidase expression by an oncolytic adenovirus enhances its intratumoral spread and supresses tumor growth. Mol. Ther. 18(7), 1275–1283 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rodríguez-García A, Giménez-Alejandre M, Rojas JJ et al. Safety and efficacy of VCN-01, an oncolytic adenovirus combining fiber HSG-binding domain replacement with RGD and hyaluronidase expression. Clin. Cancer Res. 21(6), 1406–1418 (2015). [DOI] [PubMed] [Google Scholar]
- 152.Vera B, Martínez-Vélez N, Xipell E et al. Characterization of the antiglioma effect of the oncolytic adenovirus VCN-01. PLoS ONE 11(1), e0147211 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Martinez-Quintanilla J, He D, Wakimoto H, Alemany R, Shah K. Encapsulated stem cells loaded with hyaluronidase-expressing oncolytic virus for brain tumor therapy. Mol. Ther. 23(1), 108–118 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Duebgen M, Martinez-Quintanilla J, Tamura K et al. Stem cells loaded with multimechanistic oncolytic herpes simplex virus variants for brain tumor therapy. J. NatlCancer Inst. 106(6), dju090 (2014). [DOI] [PubMed] [Google Scholar]
- 155.Josiah DT, Zhu D, Dreher F, Olson J, Mcfadden G, Caldas H. Adipose-derived stem cells as therapeutic delivery vehicles of an oncolytic virus for glioblastoma. Mol. Ther. 18(2), 377–385 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sonabend AM, Ulasov IV, Tyler MA, Rivera AA, Mathis JM, Lesniak MS. Mesenchymal stem cells effectively deliver an oncolytic adenovirus to intracranial glioma. Stem Cells 26(3), 831–841 (2008). [DOI] [PubMed] [Google Scholar]
- 157.Warrier NM, Agarwal P, Kumar P. Emerging importance of survivin in stem cells and cancer: the development of new cancer therapeutics. Stem Cell Rev. Rep. 16(5), 828–852 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tyler MA, Ulasov I, Sonabend A et al. Neural stem cells target intracranial glioma to deliver an oncolytic adenovirus in vivo. Gene Ther. 16(2), 262–278 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ahmed AU, Thaci B, Alexiades NG et al. Neural stem cell-based cell carriers enhance therapeutic efficacy of an oncolytic adenovirus in an orthotopic mouse model of human glioblastoma. Mol. Ther. 19(9), 1714–1726 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Demonstrated that human NSCs loaded with oncolytic virus (CRAd-S-pk7) and administered into the brain tumors of mice can increase survival by about 50% more than administration of naked viral particles alone.
- 160.Zhao D, Najbauer J, Garcia E et al. Neural stem cell tropism to glioma: critical role of tumor hypoxia. Mol. Cancer Res. 6(12), 1819–1829 (2008). [DOI] [PubMed] [Google Scholar]
- 161.Tobias AL, Thaci B, Auffinger B et al. The timing of neural stem cell-based virotherapy is critical for optimal therapeutic efficacy when applied with radiation and chemotherapy for the treatment of glioblastoma. Stem Cells Transl. Med. 2(9), 655–666 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lin D, Najbauer J, Salvaterra PM et al. Novel method for visualizing and modeling the spatial distribution of neural stem cells within intracranial glioma. Neuroimage 37, S18–S26 (2007). [DOI] [PubMed] [Google Scholar]
- 163.Morshed RA, Gutova M, Juliano J et al. Analysis of glioblastoma tumor coverage by oncolytic virus-loaded neural stem cells using MRI-based tracking and histological reconstruction. Cancer Gene Ther. 22(1), 55–61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Thaci B, Ahmed AU, Ulasov IV et al. Pharmacokinetic study of neural stem cell-based cell carrier for oncolytic virotherapy: targeted delivery of the therapeutic payload in an orthotopic brain tumor model. Cancer Gene Ther. 19(6), 431–442 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Fecci PE, Sampson JH. The current state of immunotherapy for gliomas: an eye toward the future: JNSPG 75th anniversary invited review article. J. Neurosurg. 131(3), 657–666 (2019). [DOI] [PubMed] [Google Scholar]
- 166.Calinescu A-A, Kamran N, Baker G, Mineharu Y, Lowenstein PR, Castro MG. Overview of current immunotherapeutic strategies for glioma. Immunotherapy 7(10), 1073–1104 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Okada H, Pollack IF. Cytokine gene therapy for malignant glioma. Expert Opin. Biol. Ther. 4(10), 1609–1620 (2004). [DOI] [PubMed] [Google Scholar]
- 168.Sikorski CW, Lesniak MS. Immunotherapy for malignant glioma: current approaches and future directions. Neurol. Res. 27(7), 703–716 (2005). [DOI] [PubMed] [Google Scholar]
- 169.Tepper RI, Pattengale PK, Leder P. Murine interleukin-4 displays potent anti-tumor activity in vivo. Cell 57(3), 503–512 (1989). [DOI] [PubMed] [Google Scholar]
- 170.Okada H, Banchereau J, Lotze MT. Interleukin-4. In: The Cytokine Handbook (4th Edition). Thomson AW, Lotze MT (Eds). Academic Press, Cambridge, MA, USA, 227–262 (2003). [Google Scholar]
- 171.Joshi BH, Leland P, Asher A, Prayson RA, Varricchio F, Puri RK. In situ expression of interleukin-4 (IL-4) receptors in human brain tumors and cytotoxicity of a recombinant IL-4 cytotoxin in primary glioblastoma cell cultures. Cancer Res. 61(22), 8058–8061 (2001). [PubMed] [Google Scholar]
- 172.Brunda MJ, Luistro L, Warrier RR et al. Antitumor and antimetastatic activity of interleukin 12 against murine tumors. J. Exp. Med. 178(4), 1223–1230 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Colombo MP, Trinchieri G. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 13(2), 155–168 (2002). [DOI] [PubMed] [Google Scholar]
- 174.Ehtesham M, Kabos P, Kabosova A, Neuman T, Black KL, John SY. The use of interleukin 12-secreting neural stem cells for the treatment of intracranial glioma. Cancer Res. 62(20), 5657–5663 (2002). [PubMed] [Google Scholar]
- 175.Yang S-Y, Liu H, Zhang J-N. Gene therapy of rat malignant gliomas using neural stem cells expressing IL-12. DNA Cell Biol. 23(6), 381–389 (2004). [DOI] [PubMed] [Google Scholar]
- 176.Hong X, Miller C, Savant-Bhonsale S, Kalkanis SN. Antitumor treatment using interleukin-12-secreting marrow stromal cells in an invasive glioma model. Neurosurgery 64(6), 1139–1147 (2009). [DOI] [PubMed] [Google Scholar]
- 177.Ryu CH, Park S-H, Park SA et al. Gene therapy of intracranial glioma using interleukin 12–secreting human umbilical cord blood-derived mesenchymal stem cells. Hum. Gene Ther. 22(6), 733–743 (2011). [DOI] [PubMed] [Google Scholar]
- 178.Xu G, Jiang X-D, Xu Y et al. Adenoviral-mediated interleukin-18 expression in mesenchymal stem cells effectively suppresses the growth of glioma in rats. Cell Biol. Int. 33(4), 466–474 (2009). [DOI] [PubMed] [Google Scholar]
- 179.Xu G, Guo Y, Seng Z, Cui G, Qu J. Bone marrow-derived mesenchymal stem cells co-expressing interleukin-18 and interferon-β exhibit potent antitumor effect against intracranial glioma in rats. Oncol. Rep. 34(4), 1915–1922 (2015). [DOI] [PubMed] [Google Scholar]
- 180.Gunnarsson S, Bexell D, Svensson A, Siesjö P, Darabi A, Bengzon J. Intratumoral IL-7 delivery by mesenchymal stromal cells potentiates IFNγ-transduced tumor cell immunotherapy of experimental glioma. J. Neuroimmunol. 218(1–2), 140–144 (2010). [DOI] [PubMed] [Google Scholar]
- 181.Choi SH, Stuckey DW, Pignatta S et al. Tumor resection recruits effector T cells and boosts therapeutic efficacy of encapsulated stem cells expressing IFNβ in glioblastomas. Clin. Cancer Res. 23(22), 7047–7058 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Dent P, Yacoub A, Hamed HA et al. MDA-7/IL-24 as a cancer therapeutic: from bench to bedside. Anticancer Drugs 21(8), 725 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hingtgen S, Kasmieh R, Elbayly E et al. A first-generation multi-functional cytokine for simultaneous optical tracking and tumor therapy. PLoS ONE 7(7), e40234 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Piccirillo S, Reynolds BA, Zanetti N et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 444(7120), 761–765 (2006). [DOI] [PubMed] [Google Scholar]
- 185.Li Q, Wijesekera O, Salas SJ et al. Mesenchymal stem cells from human fat engineered to secrete BMP4 are nononcogenic, suppress brain cancer, and prolong survival. Clin. Cancer Res. 20(9), 2375–2387 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Mangraviti A, Tzeng SY, Gullotti D et al. Non-virally engineered human adipose mesenchymal stem cells produce BMP4, target brain tumors, and extend survival. Biomaterials 100, 53–66 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Cavaillon J-M. Historical links between toxinology and immunology. Pathog. Dis. 76(3), fty019 (2018). [DOI] [PubMed] [Google Scholar]
- 188.Debinski W, Obiri NI, Powers SK, Pastan I, Puri RK. Human glioma cells overexpress receptors for interleukin 13 and are extremely sensitive to a novel chimeric protein composed of interleukin 13 and pseudomonas exotoxin. Clin. Cancer Res. 1(11), 1253–1258 (1995). [PubMed] [Google Scholar]
- 189.Thaci B, Brown CE, Binello E, Werbaneth K, Sampath P, Sengupta S. Significance of interleukin-13 receptor alpha 2–targeted glioblastoma therapy. Neuro Oncol. 16(10), 1304–1312 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kunwar S, Chang S, Westphal M et al. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro Oncol. 12(8), 871–881 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Stuckey DW, Hingtgen SD, Karakas N, Rich BE, Shah K. Engineering toxin-resistant therapeutic stem cells to treat brain tumors. Stem Cells 33(2), 589–600 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Heimberger AB, Suki D, Yang D, Shi W, Aldape K. The natural history of EGFR and EGFRvIII in glioblastoma patients. J. Transl. Med. 3(1), 1–6 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Balyasnikova IV, Ferguson SD, Sengupta S, Han Y, Lesniak MS. Mesenchymal stem cells modified with a single-chain antibody against EGFRvIII successfully inhibit the growth of human xenograft malignant glioma. PLoS ONE 5(3), e9750 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Babu R, Adamson DC. Rindopepimut: an evidence-based review of its therapeutic potential in the treatment of EGFRvIII-positive glioblastoma. Core Evid. 7, 93 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Gerstner ER. ACT IV: the final act for rindopepimut? Lancet Oncol. 18(10), 1294–1296 (2017). [DOI] [PubMed] [Google Scholar]
- 196.Martins JP, Das Neves J, De La Fuente M et al. The solid progress of nanomedicine. Drug Deliv. Transl. Res. 10(3), 726–729 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Bhargav AG, Mondal SK, Garcia CA, Green JJ, Quiñones-Hinojosa A. Nanomedicine revisited: next generation therapies for brain cancer. Adv. Ther. (2020). [Google Scholar]
- 198.Roger M, Clavreul A, Venier-Julienne M-C et al. Mesenchymal stem cells as cellular vehicles for delivery of nanoparticles to brain tumors. Biomaterials 31(32), 8393–8401 (2010). [DOI] [PubMed] [Google Scholar]
- 199.Mooney R, Weng Y, Tirughana-Sambandan R et al. Neural stem cells improve intracranial nanoparticle retention and tumor-selective distribution. Future Oncol. 10(3), 401–415 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Cao P, Mooney R, Tirughana R et al. Intraperitoneal administration of neural stem cell–nanoparticle conjugates targets chemotherapy to ovarian tumors. Bioconjug. Chem. 28(6), 1767–1776 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Li L, Guan Y, Liu H et al. Silica nanorattle–doxorubicin-anchored mesenchymal stem cells for tumor-tropic therapy. ACS Nano 5(9), 7462–7470 (2011). [DOI] [PubMed] [Google Scholar]
- 202.Mooney R, Weng Y, Garcia E et al. Conjugation of pH-responsive nanoparticles to neural stem cells improves intratumoral therapy. J. Control. Rel. 191, 82–89 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Filatova A, Seidel S, Böğürcü N, Gräf S, Garvalov BK, Acker T. Acidosis acts through HSP90 in a PHD/VHL-independent manner to promote HIF function and stem cell maintenance in glioma. Cancer Res. 76(19), 5845–5856 (2016). [DOI] [PubMed] [Google Scholar]
- 204.Gillies RJ, Raghunand N, Garcia-Martin ML, Gatenby RA. pH imaging. IEEE Eng. Med. Biol. Mag. 23(5), 57–64 (2004). [DOI] [PubMed] [Google Scholar]
- 205.Corbet C, Feron O. Tumour acidosis: from the passenger to the driver's seat. Nat. Rev. Cancer 17(10), 577 (2017). [DOI] [PubMed] [Google Scholar]
- 206.Hjelmeland AB, Wu Q, Heddleston J et al. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 18(5), 829–840 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Cheng Y, Morshed R, Cheng SH et al. Nanoparticle-programmed self-destructive neural stem cells for glioblastoma targeting and therapy. Small 9(24), 4123–4129 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Human NSCs carrying pH-sensitive nanoparticles loaded with doxorubicin and injected into the tumor or in the contralateral hemisphere distributed widely into the tumor, induced apoptosis of tumor cells and improved survival of GBM-bearing animals more than injection of doxorubicin nanoparticles alone.
- 208.Muroski ME, Morshed RA, Cheng Y et al. Controlled payload release by magnetic field triggered neural stem cell destruction for malignant glioma treatment. PLoS ONE 11(1), e0145129 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Mooney R, Roma L, Zhao D et al. Neural stem cell-mediated intratumoral delivery of gold nanorods improves photothermal therapy. ACS Nano 8(12), 12450–12460 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Krichevsky AM, Uhlmann EJ. Oligonucleotide therapeutics as a new class of drugs for malignant brain tumors: targeting mRNAs, regulatory RNAs, mutations, combinations, and beyond. Neurotherapeutics 16(2), 319–347 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Han C, Sun X, Liu L et al. Exosomes and their therapeutic potentials of stem cells. Stem Cells Int. 2016, 7653489 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Pullan JE, Confeld MI, Osborn JK, Kim J, Sarkar K, Mallik S. Exosomes as drug carriers for cancer therapy. Mol. Pharm. 16(5), 1789–1798 (2019). [DOI] [PubMed] [Google Scholar]
- 213.Chivet M, Hemming F, Fraboulet S, Sadoul R. Emerging role of neuronal exosomes in the central nervous system. Front. Physiol. 3, 145 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Yeo RWY, Lai RC, Zhang B et al. Mesenchymal stem cell: an efficient mass producer of exosomes for drug delivery. Adv. Drug Deliv. Rev. 65(3), 336–341 (2013). [DOI] [PubMed] [Google Scholar]
- 215.Altanerova U, Jakubechova J, Benejova K et al. Prodrug suicide gene therapy for cancer targeted intracellular by mesenchymal stem cell exosomes. Int. J. Cancer 144(4), 897–908 (2019). [DOI] [PubMed] [Google Scholar]
- 216.Katakowski M, Buller B, Zheng X et al. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 335(1), 201–204 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Lang FM, Hossain A, Gumin J et al. Mesenchymal stem cells as natural biofactories for exosomes carrying miR-124a in the treatment of gliomas. Neuro Oncol. 20(3), 380–390 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Demonstrated that repeated administration of MSC-derived exosomes carrying miR-124a increased median survival in mice bearing intracranial xenografts.
- 218.Yang Z, Shi J, Xie J et al. Large-scale generation of functional mRNA-encapsulating exosomes via cellular nanoporation. Nat. Biomed. Eng. 4(1), 69–83 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Munoz JL, Bliss SA, Greco SJ, Ramkissoon SH, Ligon KL, Rameshwar P. Delivery of functional anti-miR-9 by mesenchymal stem cell–derived exosomes to glioblastoma multiforme cells conferred chemosensitivity. Mol. Ther. Nucleic Acids 2, e126 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ogawa J, Pao GM, Shokhirev MN, Verma IM. Glioblastoma model using human cerebral organoids. Cell Rep. 23(4), 1220–1229 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Carey-Ewend AG, Hagler SB, Bomba HN, Goetz MJ, Bago JR, Hingtgen SD. Developing bioinspired three-dimensional models of brain cancer to evaluate tumor-homing neural stem cell therapy. Tissue Engineering Part A (2020) (Epub ahead of print). [DOI] [PubMed] [Google Scholar]