

Supplemental Digital Content is available in the text.
Keywords: Air Pollution, causal, disparities, life expectancy, PM2.5, NO2, O3, propensity score
Background:
Many studies have reported associations of air pollutants and death, but fewer examined multiple pollutants, or used causal methods. We present a method for directly estimating changes in the distribution of age at death using propensity scores.
Methods:
We included all participants in Medicare from 2000 to 2016 (637,207,589 person-years of follow-up). We fit separate logistic regressions modeling the probability of death at each year of age from 65 to 98 or older as a function of exposure to particulate matter less tha 2.5 µM in diameter (PM2.5), NO2, and O3, using separate propensity scores for each age. We estimated the propensity score using gradient boosting. We estimated the distribution of life expectancy at three counterfactual exposures for each pollutant.
Results:
The estimated increase in mean life expectancy had the population been exposed to 7 versus 12 µg/m3 PM2.5 was 0.29 years (95% CI = 0.28, 0.30). The change in life expectancy had the population been exposed to 10 versus 20 ppb of NO2 was −0.01 years (95% CI = −0.015, −0.006). The increase in mean life expectancy had the population been exposed to 35 versus 45 ppb of O3 was 0.15 years (95% CI = 0.14, 0.16). Each of these effects was independent and additive.
Conclusions:
We estimated that reducing PM2.5 and O3 concentrations to levels below current standards would increase life expectancy by substantial amounts compared with the recent increase of life expectancy at age 65 of 0.7 years in a decade. Our results are not consistent with the hypothesis that exposure to NO2 decreases life expectancy.
Survival data are commonly analyzed using proportionate hazard models, which estimate effects of variables on the instantaneous rate of events, not the mean time to event. Accelerated failure time models can estimate mean age at death but are rarely used in health studies because they require assuming a parametric distribution of life expectancies, which may not accurately represent the true distribution in humans. Moreover, they only estimate exposure association with the mean age at death, rather than with the distribution of life expectancies. We recently introduced a method to overcome these limitations.1 Our method allows one to estimate the effect of an exposure on the entire distribution of life expectancy without making any assumption about that distribution. It allows the effects of exposure and confounders to differ at each age and hence affect the shape as well as the mean of the distribution. Since the change in life expectancy is a more intuitive concept to policy makers than change in the instantaneous mortality rate, this approach has advantages. In addition, understanding how pollution affects the shape of the distribution of age at death provides insights that estimates of mean effects lack.
A large body of literature has reported that long-term exposure to air pollution is associated with higher mortality rates.2–10,11,12 For particulate matter less tha 2.5 µM in diameter (PM2.5), this is supported by a substantial toxicologic literature, showing particle exposure produces endothelial dysfunction, atherosclerosis, systemic inflammation, decreased plaque stability, and electrocardiogram abnormalities.13–21 Ozone effects are also supported by toxicology.22–24 Fewer toxicologic studies are available that study the role of NO2 near current concentrations. The 2015 Global Burden of Disease study included ambient PM2.5 and tropospheric ozone (O3) exposure among the largest worldwide contributors to avoidable early deaths.25 Recent studies have reported associations between PM2.5 and mortality at concentrations below the 2012 US EPA National Ambient Air Quality Standard or WHO guideline.26–28 Fewer studies have reported associations of long-term exposure to O3 and NO2, but the number has recently grown substantially.12,29–31
These studies have been criticized for not using causal modeling and assuming that the effects of confounders remain proportionate as the cohort ages. Recent exceptions using propensity score methods include a study by Abu Awad (2019), who focused on over 10 million movers among Medicare enrollees, and a doubly robust additive hazard model applied to Medicare enrollees in the Southeast United States by Wang (2017).32,33 Another exception is the study by Wu (2020)34 who studied 570 million person-years of observation among Medicare enrollees. They provide strong evidence of the causal link between long-term PM2.5 exposure and mortality. However, these papers only examined exposure to PM2.5, leaving the effects of gaseous pollutants, including their potential to confound the PM2.5 effects, unknown. None of them examined the effects of pollution on life expectancy or the shape of the distribution of life expectancy (i.e., how many early deaths occur at what ages), and none of them let the propensity score vary for each year of age, allowing confounders to have different effects at different ages.
The recent meetings of EPA’s Clean Air Scientific Advisory Committee have highlighted the importance of these issues.35 The Chair of the committee rejected the findings of multiple papers reporting associations with mortality, indicating that traditional epidemiologic methods of controlling for confounding do not inform causality, and only studies using causal methods can be admitted.36 Consequently, EPA recently proposed to maintain the current PM2.5 and O3 standards despite multiple studies showing deaths and hospital admissions are associated with those pollutants at levels below the current standards. Besides the assumption of the same hazard rate for each year of age, to extrapolate Cox models to changes in life expectancy requires additional assumptions and the use of life tables derived from populations different from the cohorts. Since proportionate hazard models are not collapsible, this is problematic.
Causal modeling methods represent a valuable approach to advance the argument for causality. They try to make an observational study closely mimic a randomized trial. Specifically, standard observational studies compare high exposed people with one level of confounders to low exposed people with different levels of confounders and seek to control for the difference in confounders statistically. Causal methods seek, as in a randomized trial, to first make exposure independent of confounders so that the subsequent analysis can compare what would have happened had people had high versus low exposure in a weighted or matched pseudo-population with no differences in confounders by exposure level. This seems closer to most people’s conception of what a causal contrast is. In addition, causal methods provide marginal estimates of the effects of exposure, that is, ones that do not depend on the distribution of the covariates in the study population.37 As such, their use in quantitative risk assessments, such as the Global Burden of Disease estimates, is more straightforward. Specifically, the coefficients of a standard Cox regression analysis, when applied to an individual, produces the marginal effect of an increment in exposure, holding all covariates constant in that individual. However, because of the lack of collapsibility of the proportionate hazard model, the mean of the individual marginal effects is not the population marginal effect.38 In contrast, causal approaches do produce population marginal effect estimates.39 Hence the traditional estimate of attributable risk as (RR-1)/RR is only valid if the RR is estimated using the marginal effect of exposure in the population, which inverse probability of exposure weighting (IPW) analyses provide.
Here, we present a causal model to estimate the marginal effect of air pollution exposure on the distribution of life expectancy in the United States. This model makes no assumption about the distributional form of life expectancy and, under appropriate conditions, is a causal estimate. To our knowledge, it is the first to apply causal methods to the study of long-term effects of three major air pollutants. We have applied this approach to estimate the effect of annual air pollution (PM2.5, NO2, O3) on life expectancy in the Medicare cohort in the contiguous United States between 2000 and 2016.
DATA AND METHODS
Medicare Cohort
We obtained data on all Medicare participants in the United States during 2000–2016 from the Center for Medicare and Medicaid Services.40 Medicare covers over 95% of the population ≥65 years of age in the United States. Participants aged 65 years or higher and alive on January 1 of the year following their enrollment in Medicare were entered into the open cohort for survival, and follow-up periods were calendar years. Use of this data was approved by the Harvard School of Public Health Human Subjects Committee.
Covariates
From the Medicare file for each calendar year, we extracted the age, sex, race, ZIP code of residence for that year, whether they were covered by Medicaid that year, and date of death (or censoring) of each participant. Age, Medicaid status, and ZIP code were updated annually. Race and sex were self-reported at enrollment. This file is publicly available from the Centers for Medicare and Medicaid Services.40
We obtained small area–level social, economic, and housing characteristic variables from the U.S. Census Bureau 2000 and 2010 Census Summary File 341 at the ZIP code tabulation–area level (ZCTA). Variables were updated each year by linearly extrapolating between the census years. In addition, the county-level percentage of people who ever smoked and their mean body mass index scores were obtained from the CDC Behavioral Risk Factor Surveillance Survey,42 which were then assigned to each ZCTA within the county and updated each year. From the Dartmouth Health Atlas, we obtained percentage of Medicare participants who had a hemoglobin A1c test, a low-density lipoprotein cholesterol (LDL-C) test, a mammogram, an eye exam, and a visit to a primary care physician for each year in each hospital catchment area in the United States and assigned it to all ZCTAs in that area.43 We also computed the distance from each ZIP code centroid to the nearest hospital. To capture long-term smoking history of Medicare participants in each ZIP code, we used the Medicare data to compute their hospitalization rate for lung cancer by ZIP code for each year. This risks overcontrol because air pollution has been associated with increased risk of lung cancer.
Finally, to account for the potential for omitted confounders that vary over time, we included year as a covariate as well.
Exposure Assessment
We assigned annual exposure to PM2.5, NO2, and O3 to each participant based on their residential ZIP Code for each year. The exposures came from models we fit that provided daily estimates for a 1 km grid of the contiguous United States.44,45 The models used data from predictions of chemical transport models (GEOS-Chem, CMAQ, CAMS, and MERRA-2), meteorologic data from NOAA, land-use terms from the National Land Cover Dataset, road density data from the Census, traffic data from ESRI, and satellite-based measures of aerosol optical depth, NO2, O3, NDVI (a measure of greenness), surface reflectance, and absorbing aerosol index. Using these variables, we trained three models: a neural network, a random forest, and a gradient boosting machine to United States Environmental Protection Agency (EPA) Air Quality System monitoring data from the contiguous United States to generate daily predictions of each of these three pollutants (PM2.5, O3, and NO2) on a 1 × 1 km grid. We combined the predictions from the three models for each pollutant in a nonlinear geographically weighted regression to generate a single prediction per day per grid cell for each pollutant. The models performed well, with 10-fold cross-validation on held out monitoring sites yielding an out of sample R2’s of 0.89 for PM2.5, 0.86 for O3, and 0.84 for NO2 for annual average predictions of each. The daily O3 predictions were for the 8-hour daily maximum, the NO2 predictions were for the 1-hour maximum, and PM2.5 predictions were for the daily mean. Predictions for all grid cells whose centroids were inside the Zipcode boundary were averaged for each year and assigned to participants in that Zipcode in that year.
Statistical Methods
We fit a separate logistic regression for death at each year of age, conditional on the participant having survived until that age, as a function of long-term exposure to each of the three pollutants. If we control for covariates in each of these age-specific models, then the effect of pollutants and covariates can also vary by age, freeing us from the proportionate hazard assumption.
The function of randomization in a trial is to ensure that the exposure of interest is independent of the covariates. Propensity score methods seek to recover that property in observational studies by making the distribution of the exposure independent of the covariates.39,46 For a continuous exposure, the generalized propensity score first models the dependence of exposure on covariates.37 The probability density of the residual for each observation is the probability density of the subject receiving their observed exposure level at that age given their covariates at that age. We used this to create inverse probability weights for analyses. In IPW, this density is the denominator of the IPW, and the numerator is the marginal probability density of exposure.47 If the logistic regression for surviving each year of age is weighted by the IPW for that age, and the analysis used to derive the weights was correctly specified (i.e., it included the necessary interactions and accounted for nonlinearity), the exposure should be independent of the covariates in the weighted sample. Then, if all confounders were included, the analysis should provide a causal estimate just as randomized treatment would. This has several advantages over traditional methods. First, it acts in the same manner as randomization, albeit with more uncertainty since we must estimate the probability of exposure instead of assigning it. Second, it provides marginal estimates of the effect of exposure, whereas when we control covariates in the model for the outcome, the estimated effect of exposure is conditional. The marginal estimate allows us to consider a causal contrast: what would happen underexposure a, compared with under exposure a′. Finally, using a propensity score-based approach allowed us more flexibility to fit the effect of confounders. By using gradient boosting, we fit a model that allowed highly nonlinear effects of confounders and high-order interactions among confounders if the data indicated that was necessary. We fit a separate gradient boosting model for each year of age and each pollutant to ensure the weights properly captured such features. For each year of age, the gradient boosting machine included 100 trees with a depth of 5 for each tree. The covariates included in the propensity score model included, for each pollutant, the other two pollutants. The other covariates are those described above. These covariates were updated each year, so time trends due to, for example, trends in income, population density, or racial composition in each ZIP code were captured by these variables, with the calendar year term capturing trends due to other factors.
These covariates were chosen to control for different factors that may be confounders of the association between air pollution and mortality. Specifically, the percent of the population that is Black or Hispanic controls for area level racial composition that may be predictive of the presence, or presence upwind, of pollution sources; individual race controls for any confounding by race on an individual level. We used percent with less than a high school education, percent below poverty, median household income, and individual Medicaid eligibility control for income-related socio-economic status that may predict air pollution concentrations; median housing value and percent of owner-occupied housing controls for neighborhood wealth effects, which may also predict air pollution; percent with annual checkup, with annual LDL measurement, with annual eye exam, with annual mammogram, with annual hemoglobin A1c measurement, and distance to nearest hospital are used to capture confounding due to access to medical care; percent of smokers and lung cancer rate (which should be proportional to population pack-years) to capture any confounding by current smoking or by pack-years of smoking; we used body mass index to capture any confounding by obesity. This is indicated in a Directed Acyclic Graph (eFigure 1; http://links.lww.com/EDE/B800).
Using these weights, we fit the logistic regression for death at age t with only exposure as the predictor. We used a sandwich estimate of the standard errors, which can account for spatial correlation in the residuals or the weights. We performed separate analyses for Whites and Blacks, using separate propensity score models for each race.
The above approach estimates the population marginal probability of dying in age t and exposure E in the population surviving to that age. Hence, the probability of dying at age 65 at exposure E would be P65.E. The probability of dying at age 66 given one had survived to 66 would be P66,E, and hence the unconditional probability of dying at age 66 and exposure E would be (1-P65.E) P66,E, the product of the probability of surviving to age 66 by the probability of dying at that age having survived until it. In general, the marginal probability of dying at age t is 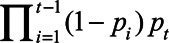 (1). We used this to estimate the percent of the population expected to die at each year of age, at three counterfactual exposure levels for each pollutant (i.e., what would have occurred had everyone been exposed to each level), as well as the mean loss of life expectancy between the lowest and highest exposure for each pollutant, and the percent of the population that died at age ≤75, or at age >85. The levels were chosen for each pollutant to approximate the range from the 25th percentile to the 75th percentile of observed exposures.
(1). We used this to estimate the percent of the population expected to die at each year of age, at three counterfactual exposure levels for each pollutant (i.e., what would have occurred had everyone been exposed to each level), as well as the mean loss of life expectancy between the lowest and highest exposure for each pollutant, and the percent of the population that died at age ≤75, or at age >85. The levels were chosen for each pollutant to approximate the range from the 25th percentile to the 75th percentile of observed exposures.
Clearly the percent of the population expected to die at each age depends on multiple coefficients. To estimate the uncertainty in that number, we sampled the coefficients predicting each pi in equation (1) from a normal distribution using the estimated coefficients for each age i, and the sandwich estimate of variance. This produces an estimate of pt, the percent of the population that would die at age t at exposure E. We repeated this process 10,000 times and used the empirical 95% confidence interval of those estimates as our uncertainty bounds.
RESULTS
Table 1 shows descriptive statistics for the data. This was an open cohort with persons entering each year, and the statistics shown include all years. The mean age was 75 years, and Medicaid covered 12.9% of participant-years. There were 637,207,589 person-years of follow-up. The mean concentration of PM2.5 was 10 μg/m3, O3 38.7 ppb, and NO2 17.8 ppb during the period 2000–2016. The IPW model was able to capture the nonlinear time trends in PM2.5 (eFigure 2; http://links.lww.com/EDE/B800). The increase in mean life expectancy had the population been exposed to 7 versus 12 µg/m3 of PM2.5 was 0.29 years (95% CI = 0.28, 0.30). Table 2 shows the estimated effects of the pollutants. The increase in mean life expectancy had the population been exposed to 35 versus 45 ppb of O3 was 0.15 years (95% CI = 0.14, 0.16). The change in life expectancy had the population been exposed to 10 versus 20 ppb of NO2 was −0.01 years (95% CI = −0.015, −0.006). Each of these effects was independent.
TABLE 1.
Descriptive Statistics of Person-year Data in the Medicare Cohort, 2000–2016
| Variable | 25% | 50% | 75% | Mean |
|---|---|---|---|---|
| Individual covariates | ||||
| Black (person yrs) | 53,518,588 (8.4%) | |||
| White (person yrs) | 544,082,779 (85.4%) | |||
| Other (person yrs) | 39,606,222 (6.2%) | |||
| Male | 42.8% | |||
| Age | 69 | 74 | 80 | 75 |
| Medicaid eligible | 12.9% | |||
| Sociodemographic variables | ||||
| Median income ($) | 38,000 | 48,000 | 63,000 | 53,000 |
| Percent living in poverty | 5.3 | 7.9 | 11.8 | 9.5 |
| Percent owner-occupied housing | 60 | 70 | 79 | 68 |
| Median house value ($) | 98,600 | 150,400 | 240,300 | 200,139 |
| Percent < high school | 14.2 | 22.6 | 33.7 | 25.3 |
| Percent Black | 1.1 | 3.7 | 12.0 | 11.0 |
| Percent Hispanic | 2.1 | 5.3 | 14.6 | 12.6 |
| Population density (people-km−2) | 167 | 967 | 3,353 | 3,396 |
| Behavioral risk factors | ||||
| Ever smoker (%) | 41.8 | 46.2 | 50.4 | 46.2 |
| BMI (kgm−2) | 26.8 | 27.3 | 28.0 | 27.5 |
| Medicare lung cancer hospitalization rate (×10−5) | 19 | 33 | 49 | 39 |
| Access to care | ||||
| Percent annual checkup | 74.4 | 79.0 | 82.1 | 77.8 |
| HbA1c (% screened) | 80.5 | 83.7 | 86.3 | 83.1 |
| LDL-C (% screened | 76.1 | 80.1 | 83.5 | 79.5 |
| Mammogram (%) | 59.2 | 63.9 | 68.2 | 63.7 |
| Eye exam (%) | 63.9 | 67.1 | 70.0 | 67.4 |
| Distance to nearest hospital (km) | 2 | 3.9 | 8.1 | 6.5 |
| Air pollution | ||||
| PM2.5 (µg/m3) | 7.9 | 9.8 | 12.0 | 10.0 |
| O3 (ppb) | 36.4 | 38.7 | 40.9 | 38.7 |
| NO2 (ppb) | 11.8 | 17.8 | 26.1 | 19.8 |
BMI indicates body mass index; LDL-C, low-density lipoprotein cholesterol.
TABLE 2.
Life Expectancy at Three Counterfactual Exposures for Each Pollutant (i.e., Had the Entire Population Been Exposed at That Concentration) Controlling for the Other Pollutants and Covariatesa Using Inverse Probability Weighting Separately for Each Year of Age
| Counterfactual Exposure Level | Mean Life Expectancy (Years) | % Dying Age 75 or Younger (%) | % Living Past Age 85 (%) |
|---|---|---|---|
| PM2.5 (µg/m3) | |||
| 7 | 82.8 | 22.5 | 41.9 |
| 10 | 82.6 | 23.1 | 41.2 |
| 12 | 82.5 | 23.6 | 40.7 |
| O3v (ppb) | |||
| 35 | 82.75 | 22.7 | 41.7 |
| 40 | 82.67 | 22.9 | 41.4 |
| 45 | 82.59 | 23.1 | 41.0 |
| NO2 (ppb) | |||
| 10 | 82.7 | 22.8 | 41.5 |
| 15 | 82.7 | 22.8 | 41.5 |
| 20 | 82.7 | 22.8 | 41.5 |
| Blacks | |||
| PM2.5 (µg/m3) | |||
| 7 | 81.39 | 28.3 | 35.1 |
| 10 | 81.07 | 29.7 | 33.7 |
| 12 | 80.85 | 30.6 | 32.8 |
| O3 (ppb) | |||
| 35 | 81.05 | 29.7 | 33.7 |
| 40 | 81.02 | 29.8 | 33.6 |
| 45 | 80.98 | 30.0 | 33.4 |
| NO2 (ppb) | |||
| 10 | 81.00 | 29.9 | 33.4 |
| 15 | 81.01 | 29.8 | 33.5 |
| 20 | 81.02 | 29.8 | 33.5 |
| Whites | |||
| PM2.5 (µg/m3) | |||
| 7 | 82.88 | 22.10 | 42.40 |
| 10 | 82.69 | 22.82 | 41.55 |
| 12 | 82.56 | 23.32 | 40.98 |
| O3 (ppb) | |||
| 35 | 82.83 | 22.3 | 42.2 |
| 40 | 82.74 | 22.6 | 41.8 |
| 45 | 82.65 | 22.8 | 41.3 |
| NO2 (ppb) | |||
| 10 | 82.77 | 22.5 | 41.9 |
| 15 | 82.77 | 22.5 | 41.9 |
| 20 | 82.76 | 22.5 | 41.8 |
Age specific models control for race, sex, Medicaid eligibility, calendar year, median household income, median home value, percent of owner-occupied housing, percent persons age ≥65 living in poverty, population density, percent of persons with <high school education, lung cancer rate, body mass index, percent of smokers in population, percent of population who are Hispanic, percent of population who are Black, percent of persons age ≥65 who had an annual checkup, who had a mammogram, who had an eye exam, who had a hemoglobin A1c test, and who had a LDL cholesterol test, and distance to nearest hospital. For each pollutant, the model includes the other two pollutants.
LDL indicates low-density lipoprotein.
The estimated effects of air pollution were modified by race (eFigure 3; http://links.lww.com/EDE/B800). Among Black participants, the increase in life expectancy comparing 7 to 12 µg/m3 was 0.54 years (95% CI = 0.50, 0.58) versus 0.33 years (95% CI = 0.32, 0.34) in Whites. In contrast, for O3, the increase in life expectancy had Blacks been exposed to 35 ppb versus 45 ppb was 0.07 years (95% CI = 0.04, 0.11) versus 0.18 years (95% CI = 0.17, 0.19) in Whites. In Blacks, the difference in life expectancy contrasting 10 to 20 ppb of NO2 was −0.02 years (95% CI = −0.03, −0.00) versus 0.01 years (95% CI = 0.00, 0.02) in Whites.
eFigure 4A (http://links.lww.com/EDE/B800) shows the distribution of age at death at the three different counterfactual PM2.5 concentrations and eFigure 4B (http://links.lww.com/EDE/B800) shows the difference in the probability of death at each age contrasting exposure at 10 versus 7 µg/m3. The main effect of exposure is not to shift the mode of the distribution; rather, the increased deaths at higher exposures occur predominantly between ages 65 and 75. These figures present not the rate of death at each age, but the fraction of the population that survived to, and died at, that age. Since people only die once, a higher probability of dying between ages 65 and 75 necessarily means a lower probability of dying at older ages. Hence, the reduced mortality probability at higher ages is simply a reflection of fewer people living long enough to die at, for example, 90. In contrast (eFigure 5A,B; http://links.lww.com/EDE/B800), the increased death rates from higher O3 are predominantly between ages 75 and 85. Hence the early deaths at higher O3 concentrations occur at later ages, and the reduction in mean life expectancy is less. eFigure 6 (http://links.lww.com/EDE/B800) shows a similar plot for NO2, with little change in death rates at any age between NO2z counterfactual levels.
We also computed the probability of dying by age 75 across the counterfactuals for each exposure by summing the area under the distribution curve between age 65 and age 75. For PM2.5, 23.6% of the population would have died by age 75 had they all been exposed at the current standard, versus 22.5% at 7 µg/m3. For O3, 23.1% would have died by age 75 at 45 ppb versus 22.7% at 35 ppb, although for NO2 it was 22.8% at both 10 and 20 ppb.
DISCUSSION
We believe this article adds to the existing literature on air pollution and mortality in multiple ways. Methodologically, it adds to the modest literature using causal modeling, and is, to our knowledge, the first to do so coadjusting for three pollutants. It estimates the probability of dying at each year of age, a more intuitive concept than a hazard rate. It allows the effects of exposure and covariates to vary with each year of age, eliminating the assumption of proportional hazards, and estimates how exposure affects the distribution of life expectancy without assuming a particular form for that distribution, or that the effect of pollution is the same at all ages. It more flexibly controls for potential confounding by using machine learning to incorporate potential nonlinearities and interactions in the dependence of air pollutants on the potential confounders, and it allows the effects of those covariates to vary by year of age and calendar year.
Substantively, it reports associations of O3 and PM2.5 with early death, and importantly that those pollutants change the shape of the distribution of life expectancy rather than simply shifting its mean. In particular, we find that PM2.5 is particularly associated with more people dying between 65 and 75, although for O3 the early deaths occur between 75 and 85. For both pollutants, fewer people survive to die in their late 80s or 90s because they died earlier. In contrast, we found a minimal change in life expectancy with higher exposure to NO2. eFigure 6 (http://links.lww.com/EDE/B800) illustrates the essentially null effect.
This study confirms previous reports that the effect of PM2.5 on Black Americans is greater than on White Americans.26,48 In contrast, we estimated that O3’s effects are larger for Whites. Black Americans, who live disproportionately in urban areas, are exposed to more traffic related PM2.5 and less O3, on average, than Whites are. If traffic particles are more toxic, that could account for a higher effect of PM2.5 in Blacks. Alternatively, the other stressors that Blacks are subject to may impair compensatory responses to particles. Similarly, a reason Blacks have lower O3 exposure than Whites is that they are more likely to live in high traffic neighborhoods and, hence, more NOx quenching of O3. Hence, there can be negative confounding of O3 by traffic pollution not captured by the NO2 and PM2.5 terms in the model. Since PM2.5 measures all particle mass, of which traffic particles are a modest fraction, control for PM2.5 may not completely capture their effect. Of course, chance cannot be ruled out as an explanation for these racial differences in response, which seems particularly likely for NO2.
How important are these effects? Between 2008 and 2017, life expectancy at age 65 increased from 17.4 to 18.1 years. Compared with a 0.7 years increase in life expectancy over a decade, a 0.29 years increase in life expectancy from lowering PM2.5 concentrations from 12 to 7 µg/m3 is substantial. The estimated O3 effects on life expectancy, at 20% of the decadal increase, are also substantial, and independent of the PM2.5 effects. For PM2.5, if everyone in Medicare were exposed at the currently permissible concentration, an additional 1.1% of them would die before the age of 75 compared with if they were all exposed at 7 µg/m3. There are 48 million Americans over the age of 65, so this is a substantial number of avoidable early deaths. For O3, the effects are smaller but still substantial. And clearly complying with standards on maximum daily O3 concentrations do not ensure long-term average concentrations are low enough to avoid excess deaths.
Moreover, we know how to accomplish the exposure reductions to reduce these early deaths. Indeed, during the study period 16% of the population was already exposed to 7 µg/m3 or less PM2.5. Installing scrubbers on coal-fired electric generating units or replacing them with gas or renewable generators will reduce sulfate particles. Requiring NOx controls to be well maintained and run all year will reduce secondary organic aerosols and ozone, updating the heavy-duty truck particle and NOx standards, which have not been changed in 19 years, will reduce primary and secondary particles and ozone. Retrofitting particle filters on Diesel engines is an off the shelf technology, already implemented for some buses and trucks. Tighter controls on wood burning furnaces and stoves are also possible with existing technology.
Studies using Cox proportionate hazard models have reported positive associations of mortality with PM2.5, NO2, and O3, and particularly with PM2.5 and O3. For particles, a recent meta-analysis of 52 cohort studies reported an association with mortality.11 The ESCAPE study examined PM2.5 and NO2 and reported an association with PM2.5 but not NO2.49 In contrast, the CANCHEC study reported associations with all three pollutants,3 although a longer term follow-up only reported associations with PM2.5 and O3.50 Hence, this study’s general findings do not diverge from those.
Observational studies, including ones using propensity scores, are subject to the risk of omitted confounders. We used a rich list of covariates but this does not exclude the possibility of confounding by an unmeasured one. One key advantage to using area level exposure, rather than personal exposure, is that personal exposure is subject to confounding by personal covariates, whereas neighborhood exposure is confounded by neighborhood covariates. For example, persons who spend more time in traffic have greater personal pollution exposure than their neighbors, but also more stress, a potential confounder. That is why Weisskopf and Webster refer to neighborhood exposures as instrumental variables for the more confounded personal exposure.51 In contrast, suppose neighborhoods with higher obesity rates had more air pollution. If a person with low body weight moved to such a neighborhood, she would receive the higher exposure, because it is neighborhood-level obesity, not personal obesity, that is associated with pollution, and therefore the confounder. Hence our greater use of neighborhood-level confounders is an advantage for the exposure we used. More individual-level covariates could be useful to allow better assessment of effect modification, however, and this is a limitation of our study.
Because we fit the propensity score model separately for each age, control for neighborhood-level confounders is strengthened. By using gradient boosting, we incorporate interactions, and allow for the effects of confounders to vary by calendar year and year of age, and control for time trends in omitted predictors. Nevertheless, inference is still dependent on the assumption that we have captured all confounders. This is why causal inference still requires judgment, and examination of biologic plausibility based on mechanistic controlled exposure studies in humans and animals.
That evidence is strongest for PM2.5, where random assignment of people to receiving a true or sham home air-filtering device demonstrated that sham filtration resulted in higher concentrations of CRP and 8-OHdG, and higher blood pressure.52 In animal studies PM2.5 increased atherosclerosis and decreased the stability of atherosclerotic plaque,20,53–55 increased oxidative stress,56–58 produced proarrhythmic changes in ECGs,59 worsened the response to ischemia,60 and impaired lung clearance.61
For O3, a cohort study reported an association between long-term O3 exposure and factor VII coagulant activity,62 a chamber study reported that O3 affected fibrinolytic activity,63 and a toxicology study reported that following O3 exposure, isolated coronary vessels exhibited greater basal tone, enhanced susceptibility to serotonin stimulation, and impaired response to acetylcholine.23 A review of toxicologic studies found decreased heart rate, metabolism, blood pressure, and cardiac output when rats were exposed to typical concentrations of O3.13 A panel study found that O3 was associated with increased levels of C reactive protein, fibrinogen, 8-hydroxy-2'-deoxyguanosine, plasminogen activator inhibitor 1, and decreased heart rate variability.64
Toxicologic evidence for NO2 effects at concentrations near those observed in this study is largely absent, which supports our finding of no noticeable estimated effect on life expectancy. However, some studies have found associations of NO2, and more needs to be done to understand its true effects. Regarding PM2.5 and O3, the robust propensity score modeling using machine learning and substantial mechanistic data provide complementary evidence that the association we have observed is likely causal, and the public health impacts are substantial.
Supplementary Material
Footnotes
This research was supported by EPA grant RD-83587201, and NIH grant P30 ES000002.
J.D.S. declares that he is an expert witness for the U.S. Department of Justice in cases relating to Clean Air Act violations. The other authors report no conflicts of interest.
Supplemental digital content is available through direct URL citations in the HTML and PDF versions of this article (www.epidem.com).
REFERENCES
- 1.Schwartz JD, Wang Y, Kloog I, Yitshak-Sade M, Dominici F, Zanobetti A. Estimating the effects of PM2.5 on life expectancy using causal modeling methods. Environ Health Perspect. 2018;126:127002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beelen R, Raaschou-Nielsen O, Stafoggia M, et al. Effects of long-term exposure to air pollution on natural-cause mortality: an analysis of 22 European cohorts within the multicentre ESCAPE project. Lancet. 2014;383:785–795. [DOI] [PubMed] [Google Scholar]
- 3.Crouse DL, Peters PA, Hystad P, et al. Ambient PM2.5, O3, and NO2 exposures and associations with mortality over 16 years of follow-up in the canadian census health and environment cohort (CanCHEC). Environ Health Perspect. 2015;123:1180–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kioumourtzoglou MA, Schwartz J, James P, Dominici F, Zanobetti A. PM2.5 and mortality in 207 US cities: modification by temperature and city characteristics. Epidemiology. 2016;27:221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krewski D, Jerrett M, Burnett RT, et al. Extended follow-up and spatial analysis of the American Cancer Society study linking particulate air pollution and mortality. Res Rep Health Eff Inst. 2009:5–114. [PubMed] [Google Scholar]
- 6.Lepeule J, Laden F, Dockery D, Schwartz J. Chronic exposure to fine particles and mortality: an extended follow-up of the Harvard Six Cities study from 1974 to 2009. Environ Health Perspect. 2012;120:965–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385:117–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Puett RC, Hart JE, Yanosky JD, et al. Chronic fine and coarse particulate exposure, mortality, and coronary heart disease in the Nurses’ Health Study. Environ Health Perspect. 2009;117:1697–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi L, Zanobetti A, Kloog I, et al. Low-concentration PM2.5 and mortality: estimating acute and chronic effects in a population-based study. Environ Health Perspect. 2016;124:46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Lee M, Liu P, et al. Doubly robust additive hazards models to estimate effects of a continuous exposure on survival. Epidemiology. 2017;28:771–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vodonos A, Awad YA, Schwartz J. The concentration-response between long-term PM2.5 exposure and mortality; a meta-regression approach. Environ Res. 2018;166:677–689. [DOI] [PubMed] [Google Scholar]
- 12.Crouse DL, Peters PA, Hystad P, et al. Ambient PM2.5, O3, and NO2 exposures and associations with mortality over 16 years of follow-up in the Canadian Census Health and Environment Cohort (CanCHEC). Environ Health Perspect. 2015;123:1180–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watkinson WP, Campen MJ, Nolan JP, Costa DL. Cardiovascular and systemic responses to inhaled pollutants in rodents: effects of ozone and particulate matter. Environ Health Perspect. 2001;109suppl 4539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adar SD, Klein R, Klein BE, et al. Air Pollution and the microvasculature: a cross-sectional assessment of in vivo retinal images in the population-based multi-ethnic study of atherosclerosis (MESA). PLoS Med. 2010;7:e1000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bräuner EV, Forchhammer L, Møller P, et al. Indoor particles affect vascular function in the aged: an air filtration-based intervention study. Am J Respir Crit Care Med. 2008;177:419–425. [DOI] [PubMed] [Google Scholar]
- 16.Brook RD. Cardiovascular effects of air pollution. Clin Sci (Lond). 2008;115:175–187. [DOI] [PubMed] [Google Scholar]
- 17.Brook RD, Urch B, Dvonch JT, et al. Insights into the mechanisms and mediators of the effects of air pollution exposure on blood pressure and vascular function in healthy humans. Hypertension. 2009;54:659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gareus R, Kotsaki E, Xanthoulea S, et al. Endothelial cell-specific NF-kappaB inhibition protects mice from atherosclerosis. Cell Metab. 2008;8:372–383. [DOI] [PubMed] [Google Scholar]
- 19.Hansen CS, Sheykhzade M, Møller P, et al. Diesel exhaust particles induce endothelial dysfunction in apoE-/- mice. Toxicol Appl Pharmacol. 2007;219:24–32. [DOI] [PubMed] [Google Scholar]
- 20.Soares SR, Carvalho-Oliveira R, Ramos-Sanchez E, et al. Air pollution and antibodies against modified lipoproteins are associated with atherosclerosis and vascular remodeling in hyperlipemic mice. Atherosclerosis. 2009;207:368–373. [DOI] [PubMed] [Google Scholar]
- 21.Sun Q, Yue P, Kirk RI, et al. Ambient air particulate matter exposure and tissue factor expression in atherosclerosis. Inhal Toxicol. 2008;20:127–137. [DOI] [PubMed] [Google Scholar]
- 22.Miller DB, Ghio AJ, Karoly ED, et al. Ozone exposure increases circulating stress hormones and lipid metabolites in humans. Am J Respir Crit Care Med. 2016;193:1382–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paffett ML, Zychowski KE, Sheppard L, et al. Ozone inhalation impairs coronary artery dilation via intracellular oxidative stress: evidence for serum-borne factors as drivers of systemic toxicity. Toxicol Sci. 2015;146:244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu W, Wages PA, Devlin RB, Diaz-Sanchez D, Peden DB, Samet JM. SRC-mediated EGF receptor activation regulates ozone-induced interleukin 8 expression in human bronchial epithelial cells. Environ Health Perspect. 2015;123:231–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forouzanfar MH, Alexander L, Anderson HR, et al. GBD 2013 Risk Factors Collaborators; Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;386:2287–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Q, Dominici F, Schwartz JD. Air pollution and mortality in the medicare population. N Engl J Med. 2017;377:1498–1499. [DOI] [PubMed] [Google Scholar]
- 27.Yang C, Yang H, Guo S, et al. Alternative ozone metrics and daily mortality in Suzhou: the China Air Pollution and Health Effects Study (CAPES). Sci Total Environ. 2012;426:83–89. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Kloog I, Coull BA, Kosheleva A, Zanobetti A, Schwartz JD. Estimating causal effects of long-term PM2.5 exposure on mortality in New Jersey. Environ Health Perspect. 2016;124:1182–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Danesh Yazdi M, Wang Y, Di Q, Zanobetti A, Schwartz J. Long-term exposure to PM2.5 and ozone and hospital admissions of Medicare participants in the Southeast USA. Environ Int. 2019;130:104879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Q, Wang Y, Zanobetti A, et al. Air pollution and mortality in the Medicare population. N Engl J Med. 2017;376:2513–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turner MC, Jerrett M, Pope CA, III, et al. Long-term ozone exposure and mortality in a large prospective study. Am J Respir Crit Care Med. 2015;193:1134–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Awad YA, Di Q, Wang Y, et al. Change in PM2.5 exposure and mortality among Medicare recipients: combining a semi-randomized approach and inverse probability weights in a low exposure population. Environ Epidemiol. 2019;3:e054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y, Lee M, Liu P, et al. Doubly robust additive hazards models to estimate effects of a continuous exposure on survival. Epidemiology. 2017;28:771–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu X, Braun D, Schwartz J, et al. Evaluating the impact of long-term exposure to fine particulate matter on mortality among the elderly. Sci Adv. 2020;6:eaba5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.CASAC. CASAC Review of the EPA’s Integrated Science Assessment for Particulate Matter. 2019.
- 36.CASAC. Public teleconference on particulate matter (PM). In: EPA Chartered Clean Air Scientific Advisory Committee (CASAC). 2019. [Google Scholar]
- 37.Imai K, van Dyke DA. Causal inference with general treatment regimes: generalizing the propensity score. J Am Stat Assoc. 2004;99:854–866. [Google Scholar]
- 38.Greenland S, Pearl J. Adjustments and their consequences – collapsibility analysis using graphical models. Int Stat Rev. 2011;79:401–426. [Google Scholar]
- 39.Robins JM, Hernán MA, Brumback B. Marginal structural models and causal inference in epidemiology. Epidemiology. 2000;11:550–560. [DOI] [PubMed] [Google Scholar]
- 40.RESDAC. Denominator File. 2018.
- 41.Bureau USC. US. Census 2000. Summary File 3 (SF 3). 2010.
- 42.CDC. Behavioral Risk Factor Surveillance System. BRFSS 2013 Survey Data and Documentation. 2013.
- 43.Wennberg J, Cooper M. The Dartmouth Atlas of Health Care. 1996.Chicago, IL: American Hospital Publishing; [PubMed] [Google Scholar]
- 44.Di Q, Amini H, Shi L, et al. An ensemble-based model of PM2.5 concentration across the contiguous United States with high spatiotemporal resolution. Environ Int. 2019;130:104909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Di Q, Amini H, Shi L, et al. Assessing NO2 concentration and model uncertainty with high spatiotemporal resolution across the contiguous United States using ensemble model averaging. Environ Sci Technol. 2020;54:1372–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hernán MA, Alonso A, Logan R, et al. Observational studies analyzed like randomized experiments: an application to postmenopausal hormone therapy and coronary heart disease. Epidemiology. 2008;19:766–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lunceford JK, Davidian M. Stratification and weighting via the propensity score in estimation of causal treatment effects: a comparative study. Stat Med. 2004;23:2937–2960. [DOI] [PubMed] [Google Scholar]
- 48.Weaver AM, McGuinn L, Neas L, et al. Neighborhood sociodemographic effects on the associations between long-term PM2.5 exposure and cardiovascular outcomes and diabetes mellitus. Environ Epidemiol. 2019;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beelen R, Raaschou-Nielsen O, Stafoggia M, et al. Effects of long-term exposure to air pollution on natural-cause mortality: an analysis of 22 European cohorts within the multicentre ESCAPE project. Lancet. 2014;383:785–795. [DOI] [PubMed] [Google Scholar]
- 50.Cakmak S, Hebbern C, Pinault L, et al. Associations between long-term PM2.5 and ozone exposure and mortality in the Canadian Census Health and Environment Cohort (CANCHEC), by spatial synoptic classification zone. Environ Int. 2018;111:200–211. [DOI] [PubMed] [Google Scholar]
- 51.Weisskopf MG, Webster TF. Trade-offs of personal versus more proxy exposure measures in environmental epidemiology. Epidemiology. 2017;28:635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chuang HC, Ho KF, Lin LY, et al. Long-term indoor air conditioner filtration and cardiovascular health: a randomized crossover intervention study. Environ Int. 2017;106:91–96. [DOI] [PubMed] [Google Scholar]
- 53.Sun Q, Wang A, Jin X, et al. Long-term air pollution exposure and acceleration of atherosclerosis and vascular inflammation in an animal model. JAMA. 2005;294:3003–3010. [DOI] [PubMed] [Google Scholar]
- 54.Tzeng HP, Yang RS, Ueng TH, Liu SH. Upregulation of cyclooxygenase-2 by motorcycle exhaust particulate-induced reactive oxygen species enhances rat vascular smooth muscle cell proliferation. Chem Res Toxicol. 2007;20:1170–1176. [DOI] [PubMed] [Google Scholar]
- 55.Araujo JA, Barajas B, Kleinman M, et al. Ambient particulate pollutants in the ultrafine range promote early atherosclerosis and systemic oxidative stress. Circ Res. 2008;102:589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chuang HC, Ho KF, Lin LY, et al. Long-term indoor air conditioner filtration and cardiovascular health: a randomized crossover intervention study. Environ Int. 2017;106:91–96. [DOI] [PubMed] [Google Scholar]
- 57.Lundbäck M, Mills NL, Lucking A, et al. Experimental exposure to diesel exhaust increases arterial stiffness in man. Part Fibre Toxicol. 2009;6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun Q, Yue P, Ying Z, et al. Air pollution exposure potentiates hypertension through reactive oxygen species-mediated activation of Rho/ROCK. Arterioscler Thromb Vasc Biol. 2008;28:1760–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hemmingsen JG, Rissler J, Lykkesfeldt J, et al. Controlled exposure to particulate matter from urban street air is associated with decreased vasodilation and heart rate variability in overweight and older adults. Part Fibre Toxicol. 2015;12:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wellenius GA, Coull BA, Godleski JJ, et al. Inhalation of concentrated ambient air particles exacerbates myocardial ischemia in conscious dogs. Environ Health Perspect. 2003;111:402–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sigaud S, Goldsmith CA, Zhou H, et al. Air pollution particles diminish bacterial clearance in the primed lungs of mice. Toxicol Appl Pharmacol. 2007;223:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Green R, Broadwin R, Malig B, et al. Long- and short-term exposure to air pollution and inflammatory/hemostatic markers in midlife women. Epidemiology. 2016;27:211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kahle JJ, Neas LM, Devlin RB, et al. Interaction effects of temperature and ozone on lung function and markers of systemic inflammation, coagulation, and fibrinolysis: a crossover study of healthy young volunteers. Environ Health Perspect. 2015;123:310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chuang KJ, Chan CC, Su TC, Lee CT, Tang CS. The effect of urban air pollution on inflammation, oxidative stress, coagulation, and autonomic dysfunction in young adults. Am J Respir Crit Care Med. 2007;176:370–376. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.