

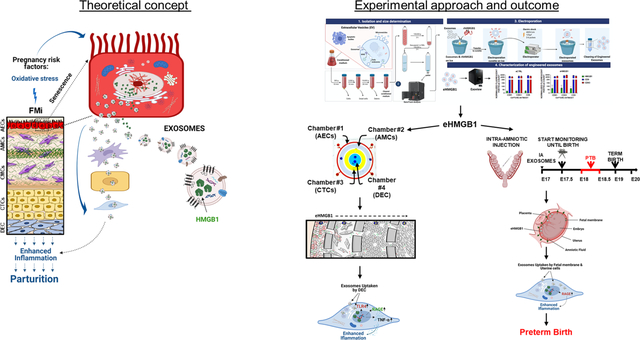
Abstract
Preterm birth (PTB; < 37 weeks of gestation) impacts ~ 11% of all pregnancies and contributes to 1 million neonatal deaths worldwide annually. An understanding of the feto-maternal (F-M) signals that initiate birthing (parturition) at term is critical to design strategies to prevent their premature activation, resulting in PTB. Although endocrine and immune cell signaling are well-reported, fetal-derived paracrine signals capable of transitioning quiescent uterus to an active state of labor are poorly studied. Recent reports have suggested that senescence of the fetal amnion membrane coinciding with fetal growth and maturation generates inflammatory signals capable of triggering parturition. This is by increasing the inflammatory load at the feto-maternal interface (FMi) tissues (i.e., amniochorion-decidua). High mobility group box 1 protein (HMGB1), an alarmin, is one of the inflammatory signals released by senescent amnion cells via extracellular vesicles (exosomes; 40–160 nm). Increased levels of HMGB1 in the amniotic fluid, cord and maternal blood are associated with term and PTB. This study tested the hypothesis that senescent amnion cells release HMGB1, which is fetal signaling capable of increasing FMi inflammation, predisposing them to parturition. To test this hypothesis, exosomes from amnion epithelial cells (AECs) grown under normal conditions were engineered to contain HMGB1 by electroporation (eHMGB1). eHMGB1 was characterized (quantity, size, shape, markers and loading efficiency), and its propagation through FMi was tested using a four-chamber microfluidic organ-on-a-chip device (FMi-OOC) that contained four distinct cell types (amnion and chorion mesenchymal, chorion trophoblast and decidual cells) connected through microchannels. eHMGB1 propagated through the fetal cells and matrix to the maternal decidua and increased inflammation (receptor expression [RAGE and TLR4] and cytokines). Furthermore, intra-amniotic injection of eHMGB1 (containing 10 ng) into pregnant CD-1 mice on embryonic day 17 led to PTB. Injecting carboxyfluorescein succinimidyl ester (CFSE)-labeled eHMGB1, we determined in vivo kinetics and report that eHMGB1 trafficking resulting in PTB was associated with increased FMi inflammation. This study determined that fetal exosome mediated paracrine signaling can generate inflammation and induce parturition. Besides, in vivo functional validation of FMi-OOC experiments strengthens the reliability of such devices to test physiologic and pathologic systems.
Keywords: DAMPs, pregnancy, parturition, organ-on-chip, fetal membrane, feto-maternal interface
Graphical Abstract
INTRODUCTION
Spontaneous preterm birth (PTB), birth before the 37th completed week of pregnancy, is a major pregnancy complication that affects ~11% of all pregnancies worldwide1. PTB is a complex syndrome with multiple etiologies and pathologic pathways. When mothers are exposed to risk factors such as infection during pregnancy, the feto-maternal (F-M) inflammatory response overrides the immune tolerance that maintains pregnancy, leading to PTB2, 3. The fetal inflammatory response is a major contributor to this process4. Inflammation results from a complex group of tissue-specific molecular and cellular interactions5; however these interactions are not well delineated in the process or parturition. To better understand the mechanisms of PTB and reduce its risk, an understanding of the inflammatory signals that induce labor is needed6–8. Immune cell activation and migration have been reported in feto-maternal interface (FMi) tissues (maternal: myometrium, decidua, and cervix; fetal: fetal membrane [amniochorion] and placenta) that override immune tolerance at the FMi in response to both endocrine and fetal signals of organ maturation9, 10. However, significant knowledge gaps exist in our understanding of how signals from the fetus, specifically the fetal inflammatory response, affects the uterus to transition from a relaxed to laboring state to facilitate birth.
Inflammatory mediators produced by FMi are different in term and preterm tissues, suggesting distinct tissue-level mechanistic events causing immune activation11–13. Using amnion membranes (the innermost lining of the uterine cavity) as a model, we have previously reported two distinct mechanisms, namely, senescence and epithelial-to-mesenchymal transition (EMT) of amnion epithelial cells (AECs), generating inflammation, observed in both humans and mouse models14, 15. Senescence and EMT are physiological responses to intrauterine oxidative stress (OS) experienced by the amnion at term to promote parturition. However, the premature activation of senescence in response to pregnancy risk factors such as infection is pathologic. Similarly, both placental and decidual senescence reported during pregnancy are also capable of producing sterile inflammation16–18. Inflammatory mediators within the amnion membrane include damage-associated molecular pattern markers (DAMPs)19–26. DAMPs released from OS-induced amnion membranes and placenta include high-mobility group box 1 protein (HMGB1)27–29, heat-shock protein 7030, cell-free fetal (cff) DNA telomere fragments (TFs)21, and uric acid23. Amnion senescence coincides with fetal growth, maturation, and inflammatory signal increase that likely indicate fetal readiness for parturition.
HMGB1 is a well-reported DAMP that is associated with both normal and adverse pregnancy outcomes13, 29, 31–34. Increased HMGB1 in various biological compartments has been observed in pathologic pregnancies28, 31, 32, 34–37. OS-induced nuclear injury to AECs causes translocation of HMGB1 to the cytoplasm, where it is often modified and secreted either directly or after being packaged by extracellular vesicles (exosomes; typically 40–160 nm in diameter)38. As a paracrine signaler, HMGB1 is pro-inflammatory and causes damage to other FMi cells by increasing inflammation27, 29, 38, 39. Based on its function as a pro-inflammatory mediator, we hypothesize that senescent fetal-derived HMGB1 is a fetal signaling mechanism that can amplify the inflammatory immune responses within FMi tissues to promote parturition40–45. FMi tissues in this study is defined as the interface between human fetal membranes (amniochorion) and maternal decidua. Exosome-based HMGB1 delivery can overcome the short half-life of this molecule in the systemic circulation, making exosomes an ideal vehicle for paracrine signaling in the F-M environment46. However, the functional impact of senescent fetal cell-derived exosomal HMGB1 on pregnancy and parturition is unclear.
To test the mechanistic function of this exosomal HMGB1 and to overcome technical challenges of testing the functions of exosomes in vivo, we engineered AEC-derived exosomes to contain HMGB1 by electroporation (eHMGB1). Then, using an organ-on-chip device that mimics the human in vivo FMi, we determined the kinetics of eHMGB1 propagation between four different FMi cell layers and the changes in the production of inflammatory molecules in these cell layers. In addition, we tested in vivo trafficking of eHMGB1 and its ability to induce parturition by injecting them intra-amniotically in a pregnant mouse model. In both the organ-chip model and mouse model, we compared the effect of recombinant HMGB1 (rHMGB1) and eHMGB1. We report that eHMGB1 can traffic far more efficiently between the FMi cell layers, cause inflammation, and induce PTB.
In this study, we tested the hypothesis that, at term, OS-induced senescent AECs package HMGB1 as inflammatory cargo within exosomes, which propagates to the maternal uterine cells, increases inflammation, and functions as one of the fetal signals to initiate parturition.
METHODS
IRB Approval –
No subjects were recruited or consented for this study. Placental specimens used for this study were deidentified and considered as discarded human specimens that do not require institutional review board (IRB) approval. Placental specimens were collected from John Sealy Hospital at the University of Texas Medical Branch at Galveston, Texas, USA in accordance with the relevant guidelines and regulations of approved protocols for various studies (UTMB 11–251; University of Texas Medical Branch at Galveston).
Utilizing the Fetal-Maternal interface Organ-on-chip (FMi-OOC) model to mimic Fetal-Maternal interface (FMi) to study exosomes trafficking during pregnancy
Device design
The FMi-OOC devices were designed and manufactured at Texas A&M University Electrical and Computer Engineering laboratory as previously described47–49.The microfluidic FMi-OOC is composed of four poly(dimethylsiloxane) (PDMS) concentric circles that each form a cell culture chamber and connected through arrays of microfluidic channels (Fig. 1c and e–f). Each cell culture chamber is 250 μm in height and has widths of 600 μm for chamber#1, 2000 μm for chamber#2 and #3, and 3000 μm for chamber#4. These chambers are interconnected through an array of 24 microchannels (5 μm in height, 30 μm in width, and 300–600 μm in length (Fig. 1e). An on-chip reservoir block placed on top of the cell culture chamber layer is comprised of multiple 4 mm diameter and 2 mm deep reservoirs, where each reservoir is aligned in such a way that they are placed on top of the inlets and outlets of each cell culture chamber in the main cell culture layer. The center culture chamber has one reservoir on top of it and the outermost culture chamber has four reservoirs on top of it, while the two middle culture chambers have two reservoirs each on top of them, to provide sufficient cell culture medium to all parts of the cell culture chambers evenly. Before using the FMi-OOC, the devices were sterilized with 70% ethanol for 15 min, washed 3 times with PBS, filled with type IV basement membrane collagen matrigel (Corning matrigel basement membrane matrix, DEV-free; 1:25 in media), and incubated at 37°C with 5% CO2 overnight. Diluted type IV collagen basement membrane matrigel was used to fill the microchannels to mimic the amnion and chorion basement membranes in utero.
Fig. 1. Fetal and maternal interface (FMi) recreation using OOC device.

(a) An illustration of fetal maternal interface (FMi) tissue in utero anatomy (black box). FMi consists of several different cell types (zoom out box), fetal derived cells: amnion epithelial cells (AECs), the most closest cells to fetus; amnion mesenchymal cells (AMCs); chorion mesenchymal cells (CMCs); chorion trophoblast cells (CTCs); maternal derived cells: decidua cells (DEC). (b) FMi in Histology. Close-up images for each cell type displayed. Scale bar, 100 μm. (c) Schematic illustration of the FMi-OOC highlighting the four concentric cell culture chambers and block reservoir filled with color dye in each of the corresponding cell culture layers. Close up schematic illustration of the cells within the FMi-OOC separated by arrays of 24 microchannels. (d) Representative histological image comparing the structure of FMi tissue to FMi-OOC device. Scale bar, 100 μm. (e) Cross-sectional view of the cell culture chambers showing the chamber height (250 μm), individual chamber diameters (mimicking in utero thickness), and the interconnecting microchannel lengths. (f) Image of the microfabricated FMi-OOC where each cell culture chamber was filled with different color dye for easy visualization. AECs (chamber 1), AMCs (chamber 2), CTCs (chamber 3) and DEC (chamber 4) were seeded for exosome trafficking study.
After this process, the cell chambers were rinsed with PBS to remove extra matrigel, and the devices were loaded with cells in the different culture chambers depending on the experiment (Fig. 1f and Sup. Fig. 5a).
Culturing human fetal membrane (amniochorion) and maternal uterine cells
Placental fetal membranes were collected from women with term, not-in-labor, delivery (>38 weeks), and without any pregnancy-related complications. Fetal membrane-derived cells were cultured as described previously, including amnion epithelial cells (AECs)50,51, amnion mesenchymal cells (AMCs)15, chorion mesenchymal cells (CMCs)52, chorion trophoblast cells (CTCs)52, maternal decidua cells (DEC),51 and myometrial cells (MYO)48. The characteristics and the viabilities of each cell type in the FMi-OOC device is previously described49.
Generating a tetraspanin CD9-red fluorescence protein (CD9-RFP) expressing stable AEC cell line
AECs were seeded (100,000 cells) in a 12-well plate cultured in keratinocyte serum-free medium (KSFM, Gibco™, Dublin, Ireland) and incubated at 37°C with 5% CO2. The next day, the cells were infected with 10 multiplicity of infection (MOI) of pCT-CD9-RFP lentivirus (SBI, CA, USA) and incubated for 72 hours (h) at 37°C with 5% CO2. Culture media were replaced with fresh KSFM and the infected cells were allowed to recover for an additional 48 h. Cells were then transferred to a 6-well plate and infected cells were selected with 1μg/mL of puromycin for a week while changing the puromycin-containing KSFM every 2–3 days. CD9-RFP-AECs cell lines were passaged up to 10 times before the experiments.
HMGB1 co-localization with CD9-RFP-AECs exosomes
HMGB1 green fluorescence protein (HMGB1-GFP) plasmid DNA (Vector Builder Inc., Chicago, IL, USA) was transiently transfected into CD9-RFP-expressing AEC cells with lipofectamine (Invitrogen, Waltham, MA, USA) according to manufacturer’s instruction by co-incubating them for 16 h at 37°C in a 5% CO2 incubator. These cells were then treated with cigarette smoke extract (CSE) at 1:50 dilution to induce oxidative stress, as described previously53–55. HMGB1-GFP and CD9-RFP expression was monitored for co-localization via time-lapse video recording and Z-stack capturing with a Keyence microscope (Keyence Corp., Osaka, Japan) (Sup. Fig. 2).
AEC-derived exosome isolation and purification
In this study, we define exosomes as extracellular vesicles with a particle size between 30 – 160 nm in diameter. Exosomes were isolated and purified by a defined centrifugation process as described previously38, 56. Briefly, frozen media was thawed overnight at 4°C, and sequentially centrifuged at 300 g for 10 min, 2000 g for 20 min at 4°C. Supernatants were concentrated in the Amicon® ultra-15 centrifugal tube of 100,000 nominal molecular weight limit (NMWL) for 30 min at 4000 g. Concentrated media was collected then centrifuged at 10,000 g for 30 min at 4°C. Supernatants were filtered through 0.2 μm NalgeneTM syringe filter (Thermo Scientific, Waltham, MA, USA), and ultracentrifuged at 100,000 g in a type 70.1 Ti rotor (Beckman Coulter, Brea, CA, USA) for 2 hr at 4°C. Supernatants were discarded and pellets were resuspended in ice-cold PBS, and centrifuged at same speed for another 1 hr to clean the exosomes. Then, pellets were resuspended in the PBS, aliquoted and stored at −80°C for further usage.
Electroporation for loading HMGB1 into the exosomes
A total of 109 AEC-derived exosomes and 1 μg of human recombinant HMGB1 (rhHMGB1, R&D Systems, Inc., Minneapolis, MN, USA) were mixed in 400 μl of electroporation buffer (1.15 mM potassium phosphate, pH 7.2, 25 mM potassium chloride, 21% Optiprep), and were electroporated using a Gene Pulser Xcell Electroporation System (Bio-Rad, Hercules, CA, USA) as previously described57, 58. Briefly, the mixtures were transferred in a single 4-mm cuvette and electroporated at 400 V, 125 μF, and ∞ ohms with 2 pulses, and immediately transferred to ice. After electroporation, the exosomes were kept on ice and washed twice with ice-cold PBS to remove excess rhHMGB1 and electroporation buffer using Amicon® ultra-15 centrifugal tube (NMWL 100,000) at 1000 g for 10 min. After washing, collected exosomes were pooled, aliquoted and stored at –80°C for further analysis. As control exosomes, 109 exosomes from the same batch were electroporated using the same conditions with PBS (Endotoxin-Free Dulbecco’s PBS (1X), w/o Ca++ & Mg++, MilliporeSigma, Burlington, MA, USA) instead of rhHMGB1.
Exosome characterization
Exosome size distribution and concentration measurements
Nanoparticle tracking analysis (NTA) was performed with NanoSight NS300 (Malvern Panalytical Ltd., Malvern, UK) to measure the sizes and concentrations of the exosomes as described previously38, 56 (Sup. Fig. 3a). The analysis settings were optimized at the day of the experiment and remained constant between the samples. Exosomes were diluted in filtered ultrapure water (ELGA,Bucks Marlow, UK) before running through the instrument.
Western blot analysis
Exosomes were lysed and analyzed as described previously with some modifications56. Briefly, the lysates were mixed with 4 × loading buffer (Bio-Rad) in non-reducing conditions and without heating. 50 μl lysates were loaded into each well of 4–15% gradient polyacrylamide gels (Bio-Rad) and subjected to electrophoresis. Subsequently, the proteins were transferred to polyvinylidene fluoride (PVDF) membranes (Bio-Rad) by semi-dry electrophoretic transfer (Bio-Rad), and the membranes were blocked in 5% nonfat dry milk/TBST (Tris-Buffered Saline, 0.1% Tween20) for 2 h at room temperature (RT). The membranes were then incubated with primary antibodies of CD63 (Novus Biologicals, Centennial, CO, USA, Clone: MX-49.129.5, [NBP2–32830]) and CD81 (Cell Signaling Technology, Cat. No: MAB6435, Lot. No: 531413) diluted at 1:400 in 5% nonfat dry milk /TBST overnight at 4°C, with secondary antibodies (SouthernBiotech, Birmingham, AL, USA, Cat. No: 1030–05, Lot: K3515-T566, DF: 1:15000) for 1 h at RT. Protein bands were visualized using an Enhanced Chemiluminescent Western Blotting solution (Bio-Rad) with ChemiDoc™ Imaging System (Bio-Rad).
Transmission electron microscopy (TEM) analyses
To visualize the size and the integrity of the electroporated exosomes, TEM negative staining was performed as described previously56 with modifications. Briefly, 20 μl of exosomes were dropped onto 200 mesh formvar/carbon-coated grids (Electron Microscopy Sciences, Inc., Hatfield, PA) and allowed to absorb to the formvar for 10 min. Exosomes were then treated with 2% aqueous uranyl acetate (Electron Microscopy Sciences) for 1 min and allowed to dry up for 10 min at RT under a heating light. The samples were viewed and imaged with a Phillips (FEI, Hillsboro, OR, USA) CM-100 transmission electron microscope.
HMGB1 concentration measurement in the exosomes
HMGB1 concentration in electroporated exosomes were measured by ELISA (IBL International Corp., Hamburg, Germany). Electroporated control (eCTRL) and HMGB1 exosomes (eHMGB1) were lysed with a 10 × radio-immunoprecipitation assay (RIPA) lysis buffer (0.5 M Tris, pH 8.0; 1.50 M NaCl: 10% (v/v) Triton X-100; 10 mM EDTA, pH 8.0; and 10% (w/v) Sodium dodecyl sulfate-SDS) supplemented with protease (MilliporeSigma), phosphatase (Thermo Scientific) inhibitor cocktails, and phenylmethylsulfonyl fluoride (PMSF, Honeywell Fluka, Charlotte, USA) for 5 min at RT while vortexing intermittently. HMGB1 ELISA was performed according to the manufacturer’s instruction and color development was measured with a Synergy™ H4 plate reader (BioTek™, Winooski, VT, USA). The results of the ELISA were used to determine the amount of HMGB1 (ng) per exosome.
ExoView detection of HMGB1
To validate and determine the loading efficiency of the electroporation methods, exosomes were analyzed using the ExoView platform (NanoView, Boston, MA, USA) following the manufacture’s procedure with modifications. ExoView allows the detection of specific cargo protein at a single-vesicle level. Briefly, 35 μl of exosomes (1×109/ml) from eCTRL and eHMGB1 were diluted in solution A (NanoView Biosciences) and incubated on tetraspanin microarray chips placed in a 24-well plate overnight at RT. Each chip was pre-coated with CD9, CD63, CD81 antibodies and MIgG control antibodies. Solutions and buffers provided by the manufacturer for ExoView experiments are proprietary of the company and their exact composition is not known to these investigators. The following day, unbound exosomes were washed 3 times for 3 min at 500 rpm shaker in solution A. Exosomes bound to the capture spots were then fixed and permeabilized with the ExoView Cargo Kit according to the manufacturers protocol. Breifly the bound exosomes were fixed with solution C (NanoView Biosciences) for 10 min, washed as previously described, and lysed in solution D (NanoView Biosciences) for 10 min and washed again as described.
HMGB1 primary antibody (Cell Signaling Technology, Danvers, MA, USA) was labeled with Alexa Fluor™ 555 labeling kit (Invitrogen) according to manufacturer’s instruction. Briefly, 1 μg HMGB1 antibody was labeled with 5 μl of Alexa Fluor™555 for 5 min at RT, followed by quenching with 5 μl of blocking reagent (Invitrogen) and used immediately after the labeling. Then exosomes were co-stained with AF555 conjugated HMGB1 antibody (Cell Signaling Technology) (0.3 μg per chip), CD9 and CD63 antibodies diluted in Blocking solution (NanoView Biosciences) for 1 h at RT in the dark. The tetraspanin microarray chips were then sequentially washed 3 times for 5 min in solution A, and in solution B (NanoView Biosciences), and 5 times for 5 min in Milli-Q water (ELGA) at 500 rpm shaker. Then, the chips containing exosomes were carefully dried from the final water wash and placed on absorbent paper and then imaged on the ExoView R100 instrument (NanoView Biosciences) using the nScan 2.9.3 acquisition software. The size distribution, concentration, and HMGB1 loading efficiency were calculated using nanoViewer 2.9.3 provided by NanoView Bioscience, and output was displayed and stored on an Excel spreadsheet.
EV-Track
We have submitted all relevant data of our experiments to the EV-TRACK knowledgebase (EV-TRACK ID: EV200186)59.
Uptake of fluorescently labeled AEC-derived exosomes by maternal decidual & myometrial cells
The green fluorescent lipophilic tracer DiO (Invitrogen) was used for labeling exosome membranes as described previously60. Briefly, DiO was added to the exosomes at a final concentration of 10 μM in PBS and incubated for 30 min at 37°C. To remove free DiO, exosomes were washed with ice-cold PBS twice using an Amicon® ultra-15 centrifugal tube (NMWL 100,000) by centrifugation at 1000 g for 10 min for each wash. Human DEC and MYO cells were seeded in exosome-depleted complete media in a 8-well slide glass at a density of 20,000 cells and cultured overnight. The following day, 6 × 108 DiO-labeled AEC-exosomes were added to the cells and incubated for 4 h and 8 h at 37°C with 5% CO2. Cells were fixed with 4% paraformaldehyde (PFA) and counterstained with vimentin (Abcam, Cambridge, MA, USA, Cat. No: ab92547, Lot No: GR3186827–13) diluted at 1:300 in 3% bovine serum albumin (BSA Gemini, Bio-Products, West Sacramento, CA, USA)/ TBST and DAPI (Invitrogen). Z-stack images were taken with a Keyence microscope (Keyence Corp.) and co-localization of the vimentin and DiO were analyzed with the RGB Profiler from ImageJ (open source).
FMi-OOC model to monitor fetal exosome trafficking and inflammation propagation
FMi-OOC setup for monitoring exosome trafficking
The FMi-OOC was used to monitor exosomes trafficking from the fetal amnion to maternal decidua since the PDMS material does not absorb exosomes (Sup. Fig. 1). For this purpose, CD9-RFP-AECs (300,000 cells) producing red fluorescent exosomes were seeded in chamber #1 (outermost) for testing endogenous exosome trafficking, AMCs (40,000 cells) were seeded in chamber #2, CTCs (300,000 cells) were seeded in chamber #3, and maternal DECs (10,000 cells) were seeded in chamber #4 (innermost) (Fig. 1f). Epithelial and the mesenchymal cells ratio is mimicking the fetal membrane cells correlation in vivo. Cells were seeded a day before exosome treatment to ensure proper attachment of the device surface. The next day, DiO-labeled exosomes (6×108 exosomes, exogenous) were added to chamber #1. Endogenous and exogenous exosome trafficking were monitored via time-lapse imaging with a Keyence microscope (Keyence corp.) (Sup. Fig. 4a–b).
FMi-OOC setup for monitoring exosome-induced inflammation
In order to monitor exosomes trafficking-induced functional changes in the FMi-OOC model, fetal-derived AMCs (20,000 cells) were seeded in chamber #1, CMCs (35,000 cells) in chamber #2, CTCs (300,000 cells) in chamber #3, and maternal DECs (10,000 cells) in chamber #4, all in exosome-depleted media (Sup. Fig. 5a). Physiological concentration of HMGB1 (30.6 ng/ml as seen in amniotic fluid during labor)35 containing eHMGB1 or eCTRL (1.85 × 108 exosomes) were added to the AMC chamber (chamber #1) and incubated for 24 h, 48 h, and 72 h. After the incubation period, the culture media was manually collected from each chamber for cytokine analysis using immunoassay. Cells were fixed with 4% PFA and immunostained for receptor for advanced glycation endproducts (RAGE) and Toll-like receptor 4 (TLR-4) (see next section for more details).
Immunocytochemical staining
After fixing, the cells in the FMi-OOC devices were blocked with 3% BSA/TBST for 30 min at RT, and then incubated with primary antibodies for RAGE (ab3611, Abcam, Cat. No: ab3611, Lot: GR3327202–1) and TLR4 (ab22048, Abcam, Cat. No: ab22048, Lot: GR55869–1), diluted at 1:200 in 3% BSA/TBST for overnight at 4°C. The next day, the cells were washed 3 times for 10 min with 1 × TBST and incubated with Alexa Fluor secondary antibodies (Alexa Fluor® 488, Abcam, Cat. No: ab150073, Lot: GR269274–4 and Alexa Fluor® 594, Invitrogen, Cat. No: ab150080, Lot: GR3323881–1, respectively) diluted in BSA/TBST for 1 h at RT. Then, cells were stained with DAPI (Invitrogen), washed 3 times for 10 min, and the FMi-OOC devices filled with cold 1xPBS. Fluorescence images were taken with a Keyence microscope (Keyence corp.) and the mean signal intensity were measured with ImageJ.
Immunoassay
To detect human inflammatory cytokine in the culture media from the FMi-OOC model, an immunoassay was performed using MILLIPLEX human cytokine panel (Millipore, Burlington, MA, USA) for tumor necrosis factor-alpha (TNF-α) according to manufacturer’s instructions as described previously50, 61.
Intra-amniotic injection of eHMGB1 to pregnant mice to determine trafficking and function
Mice model was chosen to further investigate the fetal cell derived eHMGB1 during pregnancy in vivo study, because the placentas of humans and mice have several similarities such as both considered hemochorial despite having some structural differences48. Moreover, our group has previously reported exosomes can traffic between fetal to maternal and maternal to fetal side using tdTomato mice model62. All mouse experiments were conducted in agreement with a protocol approved by the Institutional Animal Care and Use Committee (IACUC) at UTMB. CD-1 timed pregnant mice were purchased from Charles River Laboratories (Wilmington, MA, USA). Animals arrived on gestation day (E) 14 and allowed to rest in the vivarium of UTMB until E17 under a circadian cycle (12 h and 12 h, light and dark, respectively). On E17, animals were subjected to a mini-laparotomy as described previously62 and each gestational sac was injected with 25 μl of the following treatments: PBS (control) (Endotoxin-Free Dulbecco’s PBS (1X), w/o Ca++ & Mg++, MilliporeSigma), 10 ng of rhHMGB1 (R&D Systems), eCTRL (equal number of exosomes with eHMGB1), and 10 ng of eHMGB1 containing HMGB1. 10 ng of HMGB1 dose per amniotic sac was chosen because Gomez-Lopez N et al. has previously shown that similar dose of rhHMGB1 per amniotic sac leads to PTB25. Carboxyfluorescein succinimidyl ester (CFSE) alone was injected as a control for non-specific background caused by the exosomes labeling. Animals were monitored hourly until recovery under the heating lamps. Animals were monitored using Wansview cameras (Shenzhen Wansview Technology Co., Ltd, Shenzhen, China) to determine the timing of delivery.
For in vivo exosome trafficking, eCTRL and eHMGB1 were labeled with CFSE as described previously63. Briefly, exosomes were incubated in 7.5 μM CFSE at 37°C for 30 min, and then diluted in PBS containing 5% BSA. To remove excess CFSE, exosomes were washed with ice-cold PBS twice using an Amicon® ultra-15 centrifugal tube (NMWL 100,000) by centrifugation at 1000 g for 10 min for each wash. Fluorescently labeled exosomes were then injected into E17 pregnant mice intra-amniotically. After 4 h and 24 h of exosome injection, fetal and maternal tissues were collected for immunohistochemical staining.
Immunohistochemical staining
Tissues collected were fixed in 4% PFA overnight at 4°C then cryoprotected in 30% sucrose and stored at 4°C for 24 h. The next day, tissues were embedded in Tissue-Tek (optimal cutting temperature) OCT compound (Sakura Finetek, Tokyo, Japan)62, 64. Frozen sections of 10 μm were air-dried at RT to allow tissues to adhere to slide glasses (Matsunami Glass, Osaka, Japan), and blocked with 3% BSA/TBST for 1 h at RT. Then, tissues were stained with RAGE (Abcam) at a 1:200 dilution in 3% BSA/TBST overnight at 4°C. The next day, the tissues were incubated with secondary antibody (Alexa Fluor® Plus 594, Abcam) for 3 h at RT followed by DAPI staining. CFSE-labeled exosomes and RAGE staining were visualized with a Keyence microscope (Keyence corp.).
Statistical analysis
Statistical analysis was performed using the GraphPad Prism 8.0 software (GraphPad, San Diego, CA). Statistical parameters associated with each figure are reported in the figure legends. All data are reported as the mean ± SEM. Statistical significance in differences between experimental groups was assessed as following: unpaired t-test for ELISA and ExoView, Fisher’s exact test for the rates of preterm birth and pup mortality, and Mann–Whitney U-test for gestational age. All other statistical comparisons were carried out using two-way ANOVA. Throughout all figures, the following notations were used: *P < 0.05, **P < 0.01, and ***P < 0.001. Significance was considered at P < 0.05.
RESULTS
Fetal-Maternal interface Organ-on-chip (FMi-OOC) model development
FMi-OOC device was used to study fetal cell derived exosomes tracking into the maternal cells. Figure 1 shows the development of FMi-OOC device mimicking in utero FMi structure. Figure 1 a–b shows fetal and maternal derived cell types and their spatial location in the tissue during pregnancy. Figure 1c shows a schematic of the FMi organ-on-a-chip (FMi-OOC) device used in this study, which is composed of four ring-shaped cell culture chambers (#1–4, visualized by different colors) connected through arrays of microfluidic channels (Fig. 1c and e–f). In this design, the outside chamber #1 contains CD9-RFP-AECs producing red fluorescent exosomes, followed by amnion mesenchymal cells (AMCs; chamber #2), chorion trophoblast cells (CTCs; chamber #3), and lastly maternal decidua cells (DEC; chamber #4) (Fig. 1c and f). Figure 1c (not drawn to scale) shows a more detailed view of the design, where the microfluidic channel array interconnecting each ring-shaped cell culture compartments is illustrated. This FMi-OOC structure is similar to that of the human fetal membrane-decidual interface (e.g., the FMi) (Fig. 1d). We first tested the device for any undesired exosome adsorption to the polydimethyl siloxane (PDMS) surface of the FMi-OOC device. Supplemental figure 1a shows that when the device was loaded with carboxyfluorescein succinimidyl ester (CFSE)-labeled exosomes and sampled after 24 h and 72 h, no noticeable difference in fluorescence was observed. This suggests that no or a minimum level of exosomes are lost to the surface of the device, and hence the PDMS-based FMi-OOC can be utilized to study exosomal trafficking across the FMi-OOC.
Transiently expressed HMGB1 is packaged into AEC-derived exosomes under OS conditions
Before conducting fetal cell-derived exosome trafficking into the maternal side and the functional studies using the FMi-OOC device, fetal-derived exosomal cargo protein HMGB1 was explored in the fetal cell-derived exosomes. HMGB1 has been previously shown to translocate from the nucleus to the cytoplasm in primary AECs due to OS an in in vitro experiment38. To further validate these findings, we transiently transfected GFP-tagged HMGB1 (HMGB1-GFP) into RFP-tagged CD9-expressing stable AECs (CD9-RFP-AECs). In Figure 2a, we show HMGB1 (green) in the nucleus and the exosome-enriched marker CD9-RFP (red) in the cytoplasm (upper left panel, Control). Cigarette smoke extract (CSE)-induced OS forced transiently expressed HMGB1-GFP to translocate from the nucleus to the cytoplasm in AECs (Fig. 2a; Sup. Fig. 2a) and promoted the co-localization of HMGB1-GFP and CD9-RFP at 6 h and 24 h (Fig. 2a; Sup. Fig. 2b). We further confirmed the co-localization of HMGB1-GFP with CD9-RFP via orthogonal views (Fig. 2b) and line graphs (Fig. 2c) from Z-stack images. 3D visualization of HMGB1-GFP and CD9-RFP were shown in ImarisViewer (Fig. 2d). These data supported the hypothesis that OS induces translocation of HMGB1, its packaging into exosomes within the cytoplasm, and subsequent release that validates further testing of exosomal delivery of HMGB1 and its potential functional impact on recipient maternal uterine cells.
Fig.2. Co-localization of HMGB1-GFP with red fluorescent exosomes (CD9-RFP) in fetal AECs under oxidative stress condition.
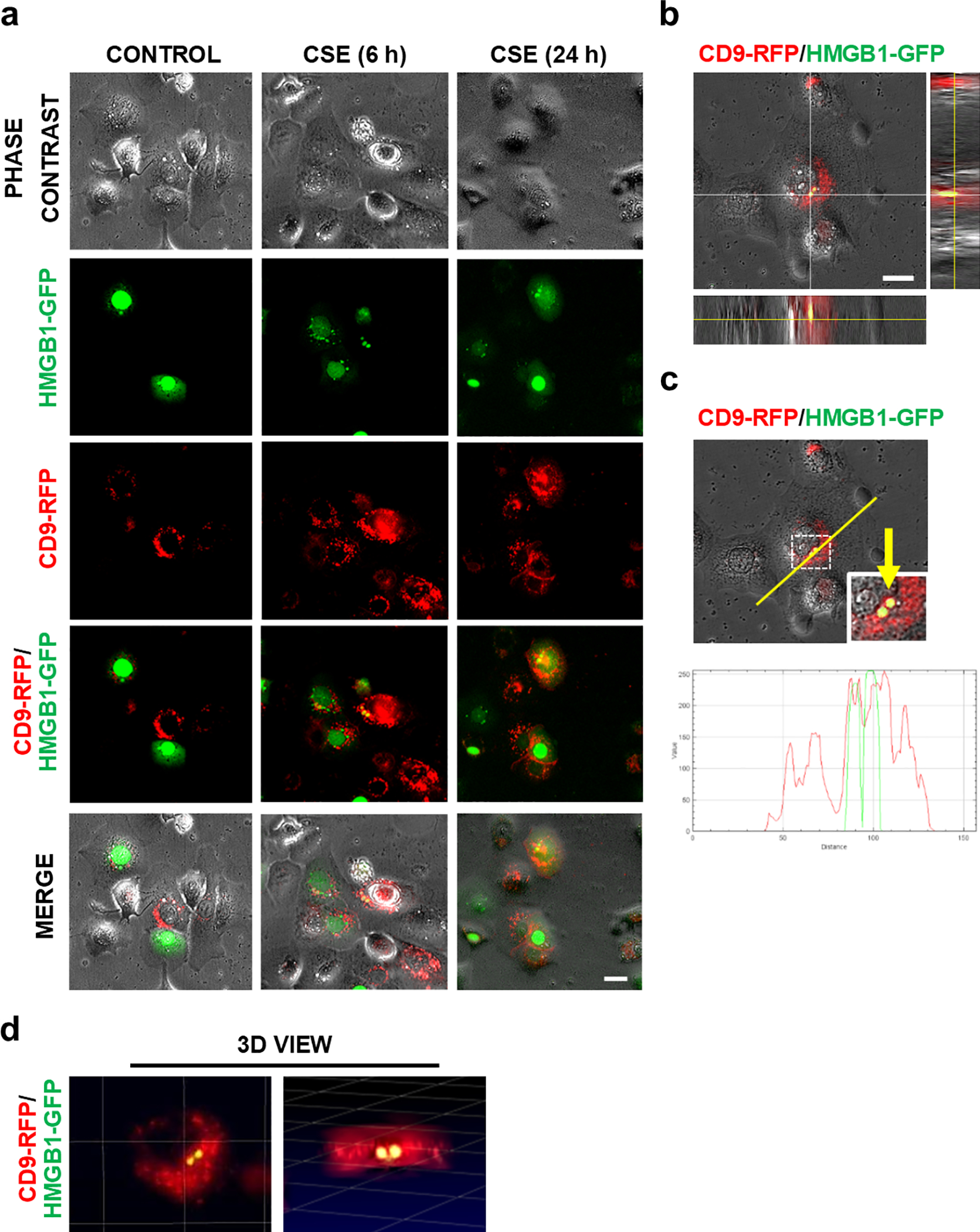
(a) Red fluorescent exosome-producing CD9-RFP-AECs were transiently transfected with HMGB1-GFP. OS was induced in these cells with CSE (1:50 diluted) to determine HMGB1 loading into the exosomes. HMGB1-GFP expression and their translocation was monitored via time-lapse microscopy. Representative images at indicated time periods are shown (n=3). (b) Co-localization of HMGB1-GFP with CD9-RFP displayed in orthogonal view and (c) in line graphs, which show the topographical profile of the pixel intensity levels of each fluorescent signal (RFP and GFP) along with freely positioned yellow lines using representative Z-stack images taken by Keyence microscope. (d) 3D view of the co-localization of HMGB1-GFP with CD9-RFP using ImarisViewer on Z-stack images. Scale Bars, 20μm.
Uptake of AEC-derived exosomes by maternal decidual and myometrial cells
Prior to conducting exosome trafficking and functional studies using the FMi organ-chip model, we tested the ability of maternal decidual and myometrial cells to uptake AEC-derived exosomes in a 2D culture system. The co-localization of the lipophilic tracer DiO-labeled AEC-exosomes (green color) and intermediate filament vimentin (red color) showed the internalization of the AEC-derived exosomes by maternal cells (both decidual and myometrial cells) after 4 h and 8 h (Fig. 3). Line graphs (shown on the right of images) of selected regions further confirmed DiO-labeled exosomes (green) being co-localized within the cytoplasm (vimentin; red), which is indicated by the overlap between the red and green lines, suggesting that the fetal AEC-derived exosomes indeed can be internalized by the maternal side of cells.
Fig. 3. Fetal membrane AECs-derived exosome uptake into maternal decidua and myometrial cells.
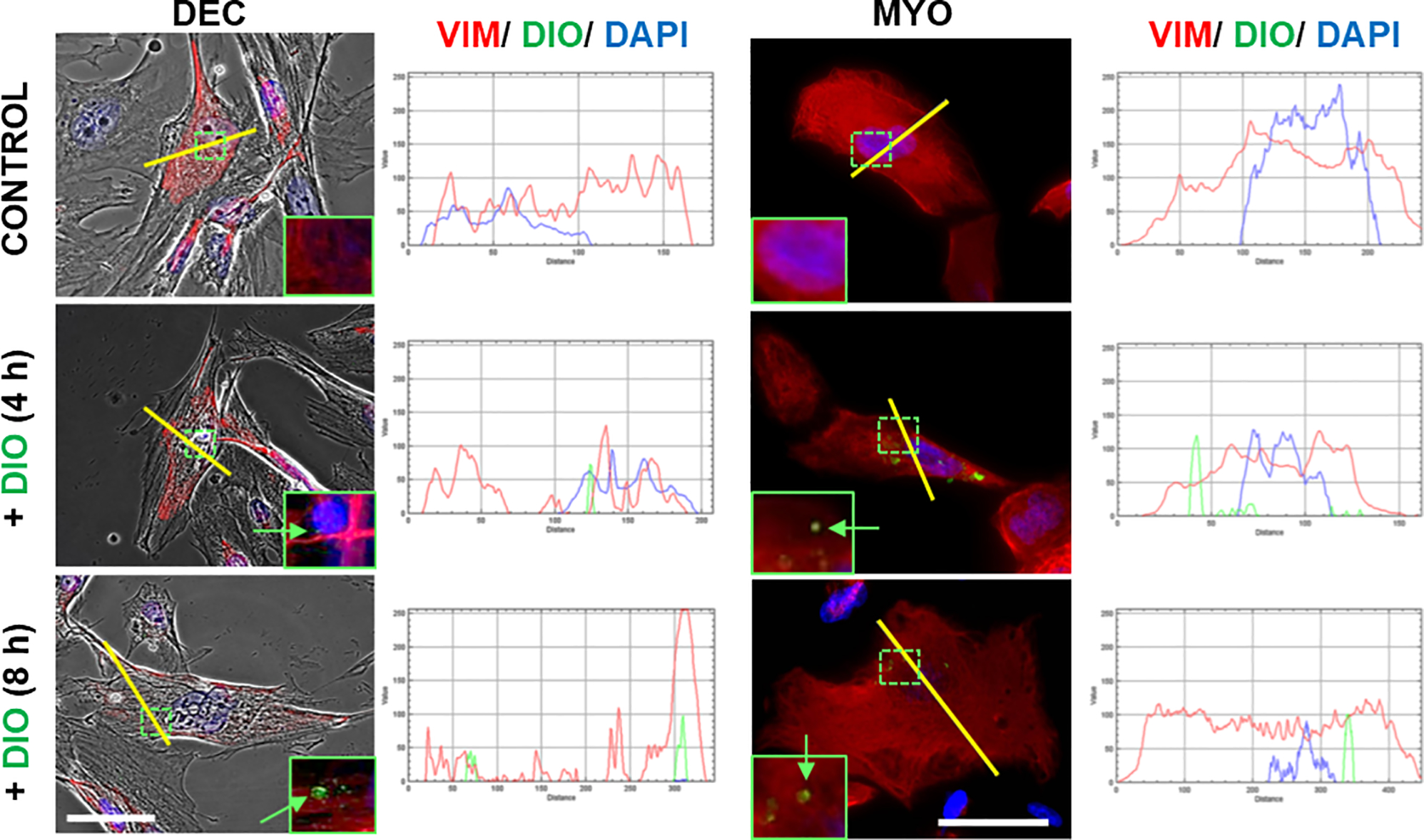
Representative images showing the maternal decidual and myometrial cells taking up DiO-labeled AEC-derived exosomes (green), and the co-localization of DiO and vimentin (red) labels at indicated time periods (n=3). Yellow line indicates the area where co-localization of DiO with vimentin is shown in the line graphs. Scale Bars, 50μm. DEC-decidua, MYO-myormetrial. Close up images in the insets.
Preparation and characterization of HMGB1-containing engineered exosomes
AEC-derived exosomes grown under normal culture conditions were used to engineer them to contain HMGB1 (eHMGB1) as cargo. Prior to their engineering, the size (127 nm ± 4 nm), concentration (2.6 × 1011 ± 3.3 × 107 particles/ml) and morphology of AEC-derived exosomes were measured with nanoparticle tracking analysis (NTA) and transmission electron microscope (TEM), respectively (Sup. Fig. 3a–b). Next, rHMGB1 was loaded into the AEC-derived exosomes via electroporation (Fig. 4a). The characteristics of the electroporated control (eCTRL) and eHMGB1 were first compared to non-electroporated parent exosomes (naïve). The size and morphology of the exosomes were unaffected by the electroporation, as shown by NTA, TEM analyses, and ExoView, respectively (Fig. 4b–c; Sup. Fig. 3c). As shown in Figure 4d, regardless of electroporation, preparation methods or the presence of HMGB1, all exosomes (eCTRL, eHMGB1, and naïve) expressed tetraspanin markers CD81 and CD63, confirming that no changes in surface markers occurred to the exosomes. HMGB1 concentration in the eHMGB1 was measured using ELISA, and it was determined that the eHMGB1 contained approximately 20 times more HMGB1 after electroporation (203.68 ± 16.84 ng/ml) compared to their non-electroporated naïve exosomes (7.71 ± 3.37 ng/ml) (P < 0.001) (Fig. 4e). The presence of HMGB1 (green) inside the exosome was further validated with ExoView analysis by double-staining exosome tetraspanin markers CD9 (blue) and CD63 (red) in the same samples (Fig. 4f). The fact that AF555 signal (green signals with ExoView) from the anti-HMGB1 is detected significantly higher in the eHMGB1 than in the eCTRL while the exosome marker proteins (anti-CD9-AF488 and anti-CD63-AF647; blue and red signals with ExoView, respectively), being expressed in similar amounts in both samples, shows the successful loading of the HMGB1 into the exosomes. The localization of the HMGB1 within a tetraspanin positive exosome is also implied by the positive selection method, wherein exosomes which display the common tetraspanins are captured from both samples and then those captured exosomes are probed for the cargo protein (Fig. 4f).
Fig. 4. Engineering and characterization of eHMGB1.
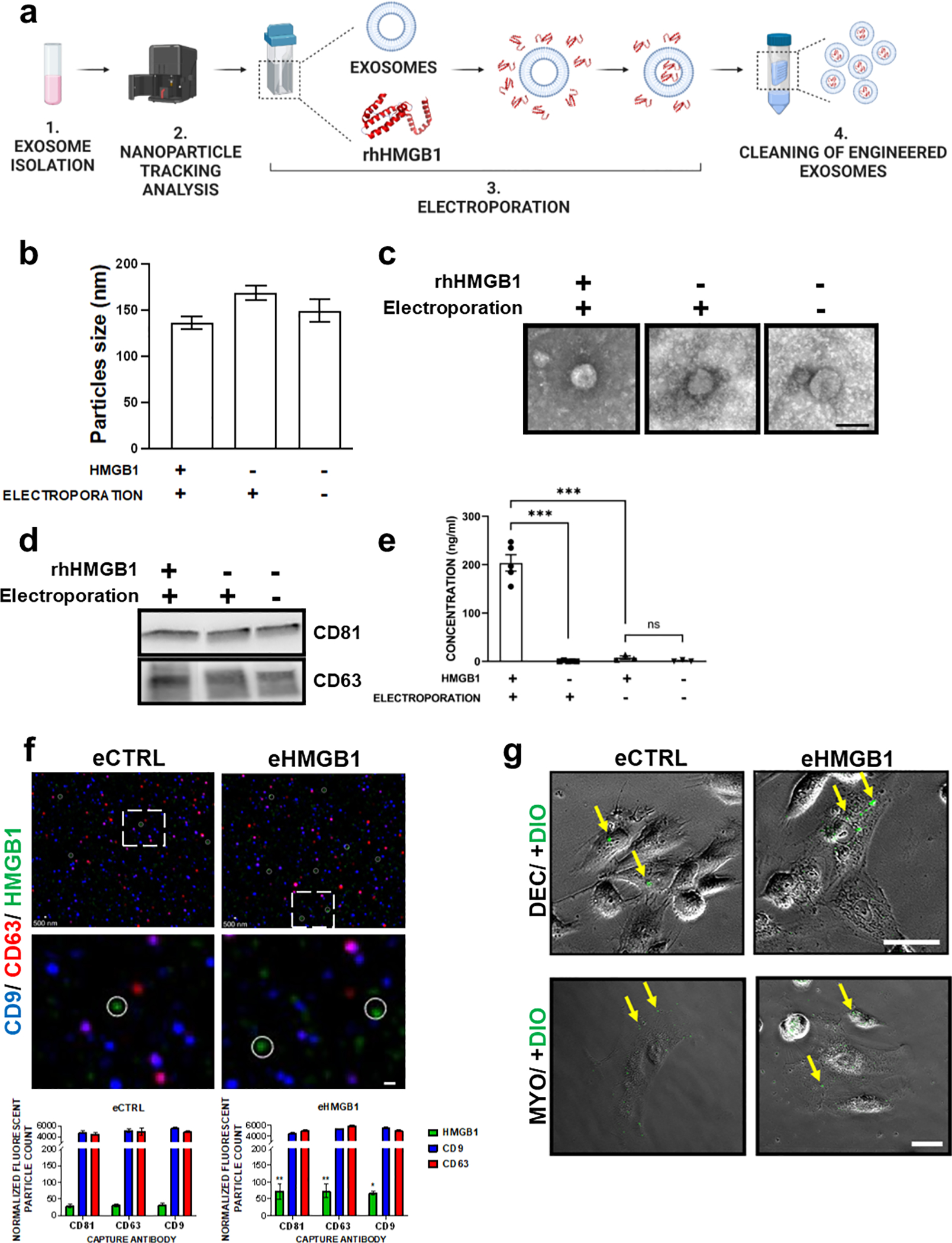
(a) Schematic illustration of engineering exosomes to contain HMGB1 via electroporation. (b) Nanoparticle tracking analysis (NTA) using ZetaView for analyzing the size distribution of AEC-derived engineered vs. naïve exosomes (n=3). (c) Representative transmission electron microscopy (TEM) images of the engineered vs. non-electroporated naïve exosomes. Scale bar, 60 nm. (d) Western blotting analysis of exosome markers (CD63 and CD81). (e) ELISA analysis of HMGB1 in engineered vs. naïve exosomes (n=5). The data are presented as the means ± SEM. ***P < 0.001, ns: not significant, unpaired T-test. (f) ExoView analysis of engineered eCTRL vs. eHMGB1. Representative composite images, HMGB1 (green, circled), exosome markers CD9 (blue) and CD63 (red). Close up images in the bottom. Scale bars, 500 nm (scale bars are not utilized for sizing in this image). Bottom graph shows ExoView counts of immunofluorescent stained positive exosomes for all markers detected. The data are presented as the means ± SEM. **P < 0.01, *P < 0.05. Multiple comparison two-way analysis of variance (ANOVA). (g) Representative phase contrast images showing the uptake of DiO-labeled engineered exosomes, eCTRL and eHMGB1 (6×108 exosomes) by maternal decidual and myometrial cells after 4 h of incubation. Yellow arrows pointing to the internalized exosomes. Scale bars, 50μm
Next, we tested whether electroporation diminishes the capacity of exosomes to be internalized by the maternal cells. As shown in Figure 4g, DiO-labeled exosomes co-cultured with both decidual and myometrial cells showed exosome uptake by the maternal cells within 4 h, as can be seen by phase-contrast/fluorescent microscopy images of exosomes (green color) being within the maternal cells for both naïve exosomes (eCTRL) and electroporated exosomes (eHMGB1). This shows that electroporating exosomes with specific cargo within them does not alter the properties of exosomes or impact their uptake capability by maternal cells.
AEC-derived exosomes traffic across the FMi-OOC
Prior to testing eHMGB1-induced inflammatory changes in the FMi cellular layers, we first determined the ability of both endogenously generated and exogenously introduced exosomes to traffic through the device (i.e., move from one chamber to the neighboring chamber) (Fig. 5a). Endogenous exosomes are tested as they are the actual particles trafficking in utero, and exogenous eHMGB1 was tested as the experimental counterpart. CD9-RFP-AECs were seeded and grown in the outermost chamber (chamber #1), producing and releasing red fluorescent exosomes (endogenous). DiO-labeled (green fluorescence) exogenous exosomes (eHMGB1) were also loaded into the outermost chamber (chamber #1) so that trafficking of both exosomes could be tested simultaneously. Time-lapse fluorescence imaging (both red and green channels) of all chambers were conducted (Fig. 5b–c). Both types of exosomes propagated between chambers through the microchannels, either together with migrating cells or as free-floating vesicles. Both the endogenous and exogenous exosomes propagated from the AEC compartment (chamber #1) to the AMC compartment (chamber #2) within 24 h, then to the CTC compartment (chamber #3) by 48 h, and finally to the DEC compartment (chamber #4) by 72 h (Fig. 5b–c; Sup. Fig. 4a–b). These data confirm that fetal AECs-derived exosomes can traffic through the FMi-OOC in a stepwise fashion over time to reach the maternal side within 72 h.
Fig. 5. Feto-maternal interface (FMi) OOC device used in the study and how exosomes traffic through the FMi cell layers.
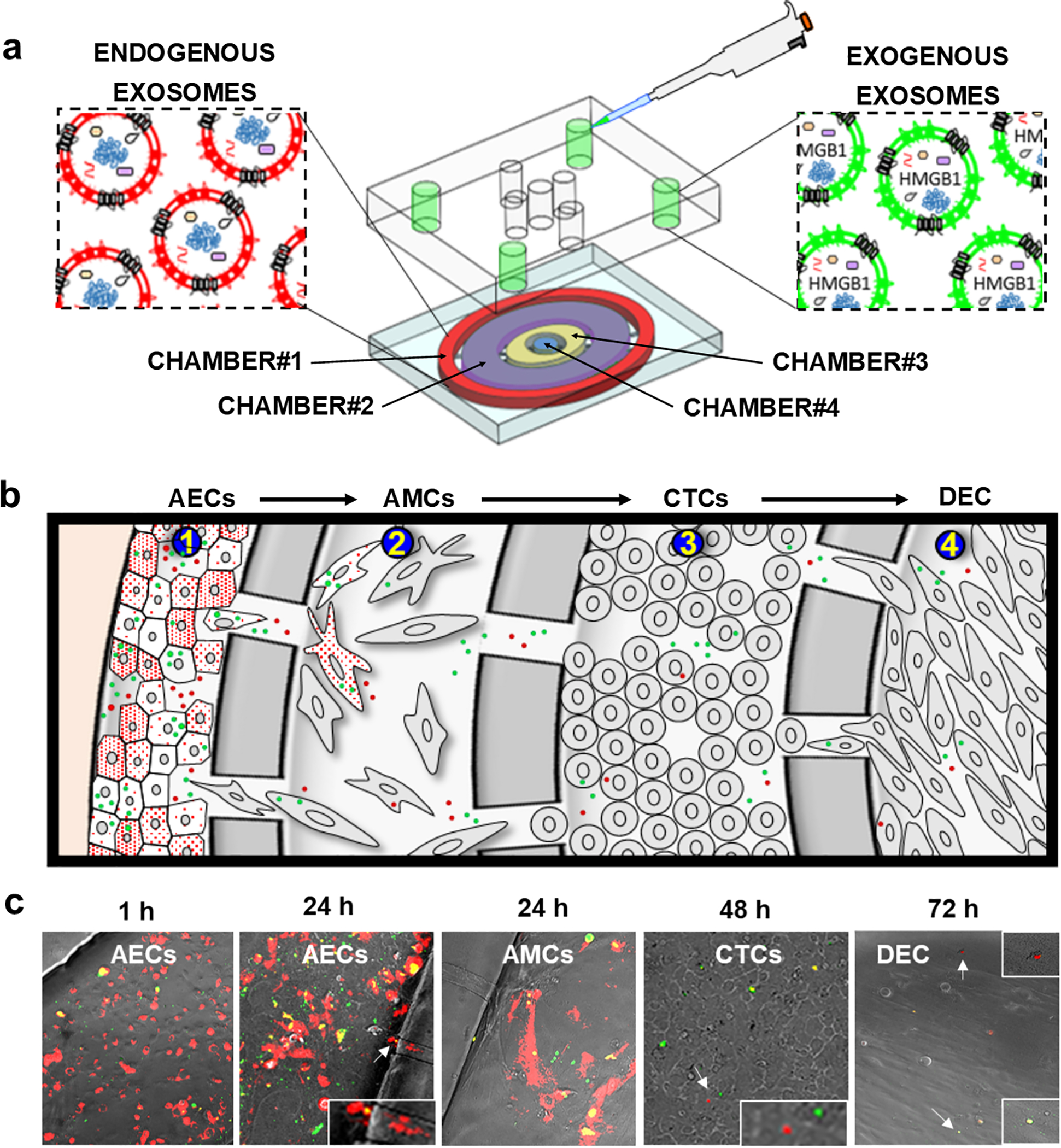
(a) Schematic illustration of the FMi-OOC device, along with how DiO-labeled (green) exogenous exosomes are loaded into the outermost AEC compartment (chamber#1), where red fluorescent exosomes producing CD9-RFP-AECs cells are seeded so that both endogenous exosomes and engineered exosomes can be monitored for their trafficking. (b) Cartoon illustration of exosomes (red and green) trafficking throughout the cell layers. (c) Representative images of endogenous and exogenous exosomes trafficking through the fetal membrane cell layers (AMCs and CTCs, chamber#2 and chamber#3, respectively), and finally reaching the maternal DEC layer (chamber#4). Close up images are shown in the insets.
eHMGB1 trafficking activates RAGE, TLR4, and inflammatory cytokines production throughout the FMi-OOC
After confirming the trafficking of both endogenous and exogenous exosomes in the FMi-OOC, functional studies were conducted with eHMGB1. Patient-derived primary fetal membrane cells (AMCs, CMCs, and CTCs) and maternal DEC cells were seeded in the FMi-OOC device for this experiment (Sup. Fig. 5a). AMCs (chamber #1) were treated with either eCTRL (1.85 × 108 exosomes) or eHMGB1 (containing 30.6 ng/ml, 1.85 × 108 exosomes), and the expressions of the HMGB1 receptors TLR4 (red) and RAGE (green), along with cytokine production, were measured within the FMi-OOC (Fig. 6a). Signals from AMCs treated with eCTRL or eHMGB1 trafficked to the next compartment and subsequent internalization induced a significant increase in TLR4 (P < 0.001) and RAGE expression (P < 0.001) in the CTCs by 24 h (Fig. 6a–b) and in the DEC by 72 h (RAGE; P = 0.028) (Fig. 6c) compared to eCTRL. eHMGB1 trafficking also induced significant production of the pro-inflammatory cytokine TNF-α in the CTCs (P = 0.01), but in the other chambers did not reach statistical significance during a 24 h period (Sup. Fig. 5b).
Fig. 6. eHMGB1 trafficking increases RAGE and TLR4 receptors throughout fetal membrane cells.
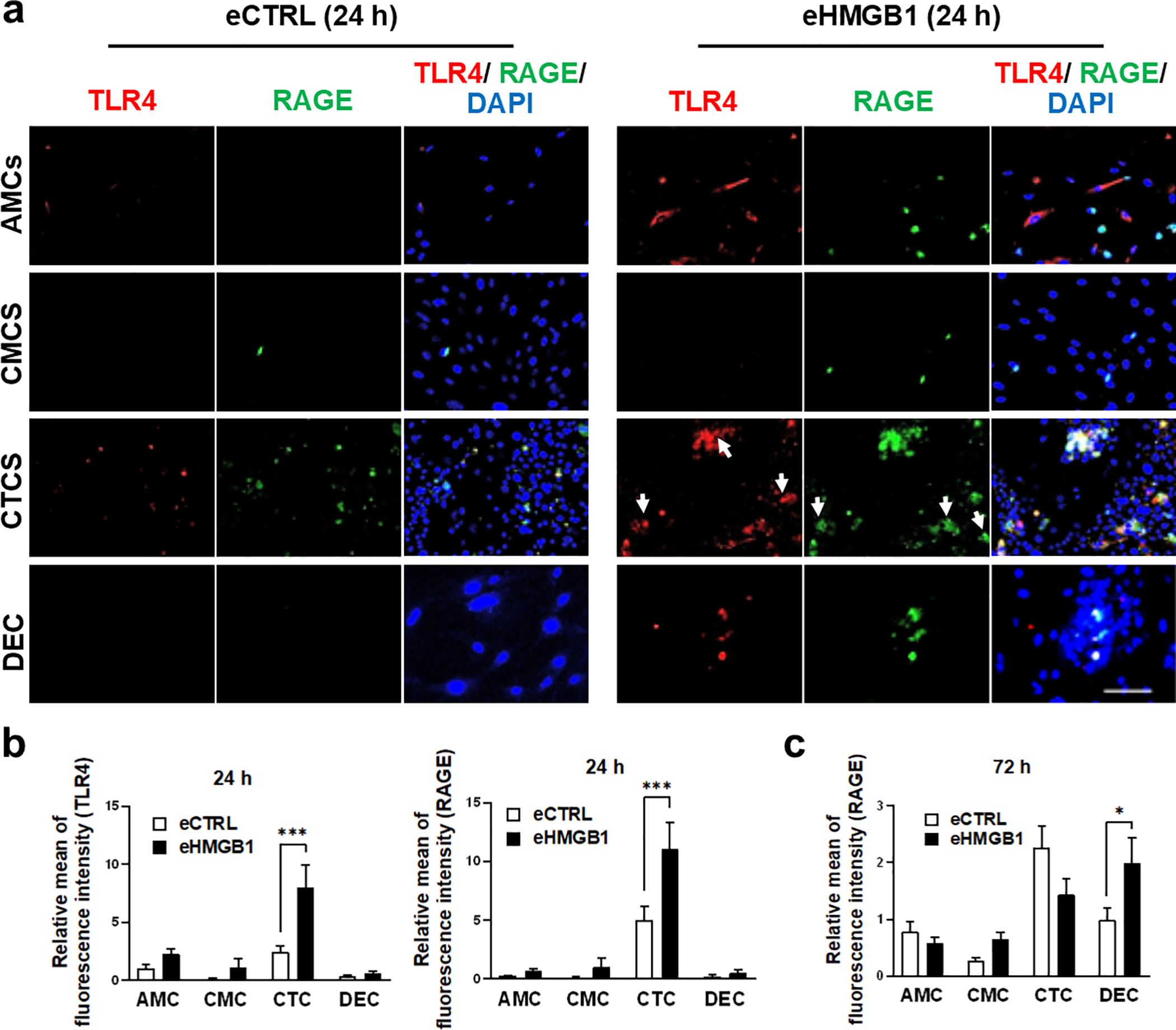
(a) Representative fluorescent images of TLR4 and RAGE receptor activation in each cell layers. The AMCs (chamber#1) were treated with either eCTRL or eHMGB1 for 24 h, and then cells were double stained for TLR4 and RAGE as a marker for inflammatory signal propagation and activation of its receptors throughout each cell layer. Scale bar, 100 μm. (b) TLR4 and RAGE fluorescence signal intensities were measured at 24 h post treatment by ImageJ (n=3). The data are presented as means ± SEM. ***P < 0.001, two-way analysis of variance (ANOVA). (c) RAGE fluorescence signal intensity was measured at 72 h post treatment by ImageJ (n=3). The data is presented as means ± SEM. P=0.028, two-way analysis of variance (ANOVA).
Additionally, FMi-OOCs designed without microchannels, thus no interconnection between the four cell culture chambers, showed eHMGB1-induced changes in AMCs only. However, no changes were seen in the remaining chambers as lack of interconnecting microchannels prevented eHMGB1 migration to the neighboring chambers (Sup. Fig.6), confirming that the observed pro-inflammatory changes in the other chambers were likely due to migrating eHMGB1. To note, this approach cannot confirm any paracrine factors such as cytokines released by AECs. Furthermore, addition of rHMGB1 to chamber #1 and subsequent propagation of either rHMGB1 and/or inflammatory mediators induced by rHMGB1 in AMCs (chamber #1) induced similar changes in the expressions of TLR4 and RAGE; however, 48 h were required to produce these changes (Sup. Fig. 7). These data suggest that exosomal HMGB1 propagates faster across the FMi and induce differential labor-associated pro-inflammatory changes in each cell type.
eHMGB1-induced PTB in mice
After determining that eHMGB1 can cross through the fetal cell layers and reach the maternal decidua layer to produce proinflammatory changes using the FMi-OOC device as an in vivo-like in vitro model system, the functional validity of eHMGB1 trafficking in an in vivo model was tested. E17 mice were chosen for eHMGB1 injection for the following reasons: 1) the gestational period in this model is 19.5–20.5 days and delivery before this time point is considered as preterm and 2) we have shown that senescence and EMT peaks around E18 when senescence-associated inflammation and DAMPs start to propagate from fetal membrane cells to maternal tissues16. Senescence indicates completion of fetal maturation and a fetal-specific biological timing of pregnancy. Therefore, providing an inflammatory thrust using eHMGB1 (mimicking premature senescence-associated inflammation) a day prior to the expected day of their increase (E18) is expected to induce PTB. Survival surgeries were performed and eHMGB1 were injected intra-amniotically on E17 mice into each embryo, and animals were monitored until delivery (Fig. 7a). Video recordings were collected and analyzed for timing of birth. In this study, delivery occurring before or on E18.5 was considered as a PTB and those occurring on E19.5 ± 0.5 as a term birth. We report that 80% of animals (4/5) injected with eHMGB1 delivered preterm compared to none in the PBS injected animals (control) group (Fig. 7b). PTB rates were 20% (1/5) in eCTRL and 50% (2/4) in rHMGB1 injected animals (Fig. 7b). PTB rates were highest when injected with eHMGB1 (P < 0.001) (Fig. 7b), supporting our hypothesis that the packaging of HMGB1 in exosomes may provide safe and secure delivery of signals, rather than simple diffusion, to generate a functional outcome. Moreover, the average gestation days of eHMGB1-injected animals were significantly decreased compared to the PBS controls (E18.5 vs. E19.5, respectively, P = 0.039) (Fig. 7c). After delivery, the pup mortality rate at birth was observed to be higher when treated with rHMGB1 (47%) compared to when treated with eHMGB1 (37%) and lower in eCTRL (21%) and PBS control (9%) cases (Fig. 7d).
Fig. 7. eHMGB1-induced PTB in mice.
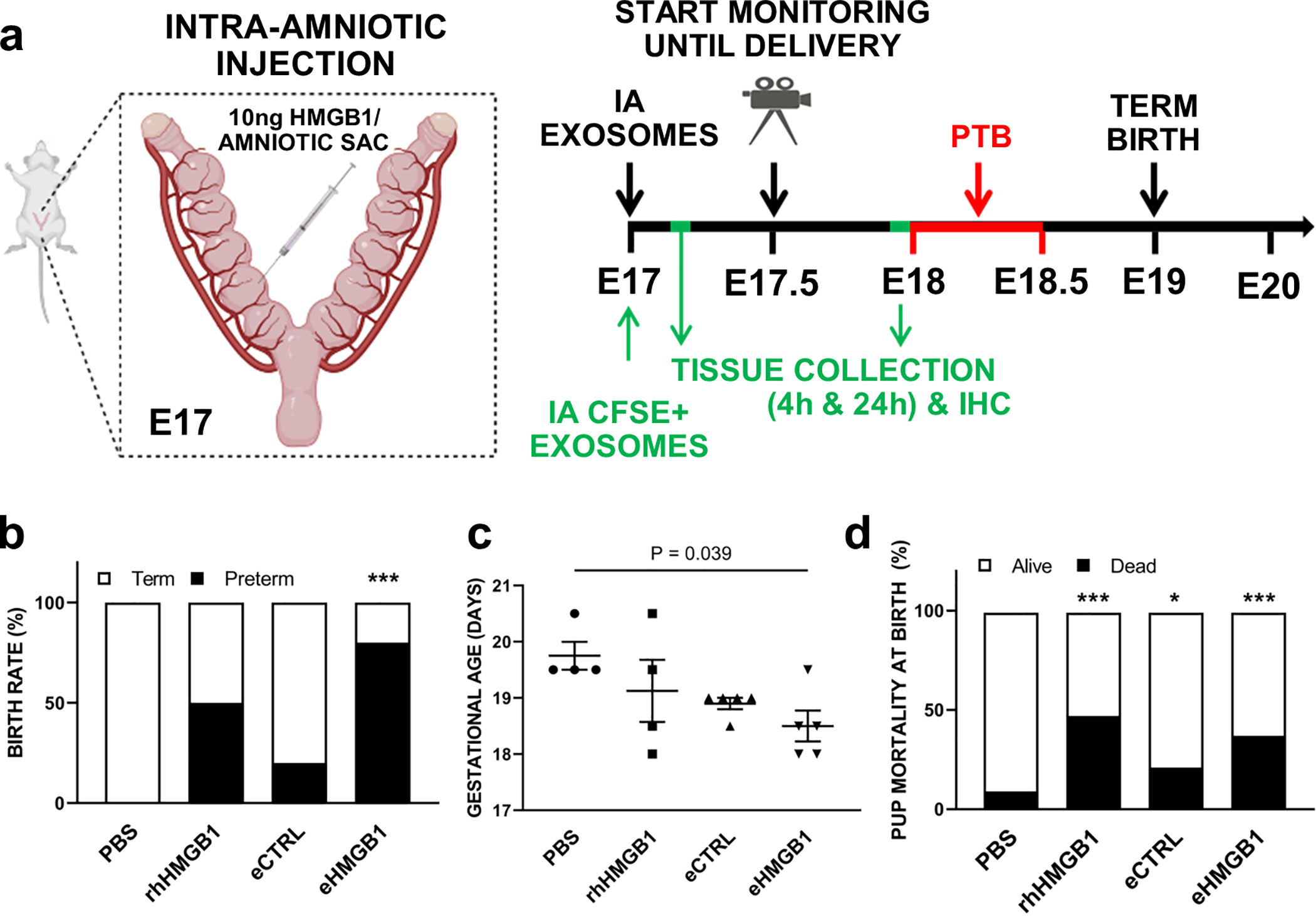
(a) Schematic diagram of the in vivo experimental design. E17 pregnant mice were injected with 10 ng of HMGB1 per amniotic sac and monitored for delivery with video recordings. Some group of the mice were euthanized at indicated time periods for in vivo exosomes trafficking analyses. (b) E17 pregnant mice were injected intra-amniotically (IA) with either 25 μl of PBS (n=4), rhHMGB1 (10 ng per amniotic sac) (n=4), eCTRL (n=5), or eHMGB1 (10 ng per amniotic sac) (n=5), and video monitored until the delivery. Fisher’s exact test was used for the statistical significance assessments (***P < 0.001 for PBS vs. eHMGB1). (c) The gestational days are presented as means ± SEM. Mann-Whitney test. (d) The rate of pup mortality at birth. Pup mortality rate observed higher in rhHMGB1 and eHMGB1, slight increase in eCTRL compared to PBS controls (***P < 0.001, P = 0.028, respectively). IA, Intra-amniotic.
In vivo trafficking of eHMGB1 from amniotic fluid to fetal membranes and uterus increases RAGE expression
Since eHMGB1 injection induced PTB in our murine model, we hypothesized that eHMGB1 can traffic from the fetal side to the maternal side and increase inflammation along its way. To further test this possibility, either CFSE-labeled engineered exosomes (eCTRL and eHMGB1) or controls (PBS and CFSE only) were injected into the amniotic cavity of CD-1 mice (Fig. 8a, labeled green). After the injection, the animals were sacrificed at 4 h and 24 h post-injection and subjected to tissue collection for immunohistochemistry (IHC) analysis. At 4 h and 24 h, fluorescent CFSE signals were detected in fetal membranes from both eCTRL and eHMGB1 treatments, but not in fetal membranes from PBS and CFSE only treated animals, indicating exosome trafficking (Fig. 8a). CFSE-labeled exosomes were detected in the uterine tissues of eCTRL and eHMGB1 treatments at 4 h and 24 h but not in the controls (PBS and CFSE) (Fig. 8c). Collected fetal membrane and uterine tissues were further analyzed for RAGE expression to check the functional changes caused by fluorescently labeled eHMGB1 trafficking. There were no apparent differences observed between the groups in the fetal membrane tissues (Fig. 8b). However, at both 4 h and 24 h, higher expression of RAGE was detected in uterine tissues treated with eHMGB1 compared to those of the eCTRL, PBS and CFSE treated tissues (Fig. 8d), suggesting eHMGB1 trafficking caused labor-associated inflammatory signals in maternal uterine tissues along the way.
Fig. 8. In vivo trafficking of eHMGB1 from amniotic fluid to fetal membranes and uterus in mice, where increase in RAGE expression associated with eHMGB1 was observed.
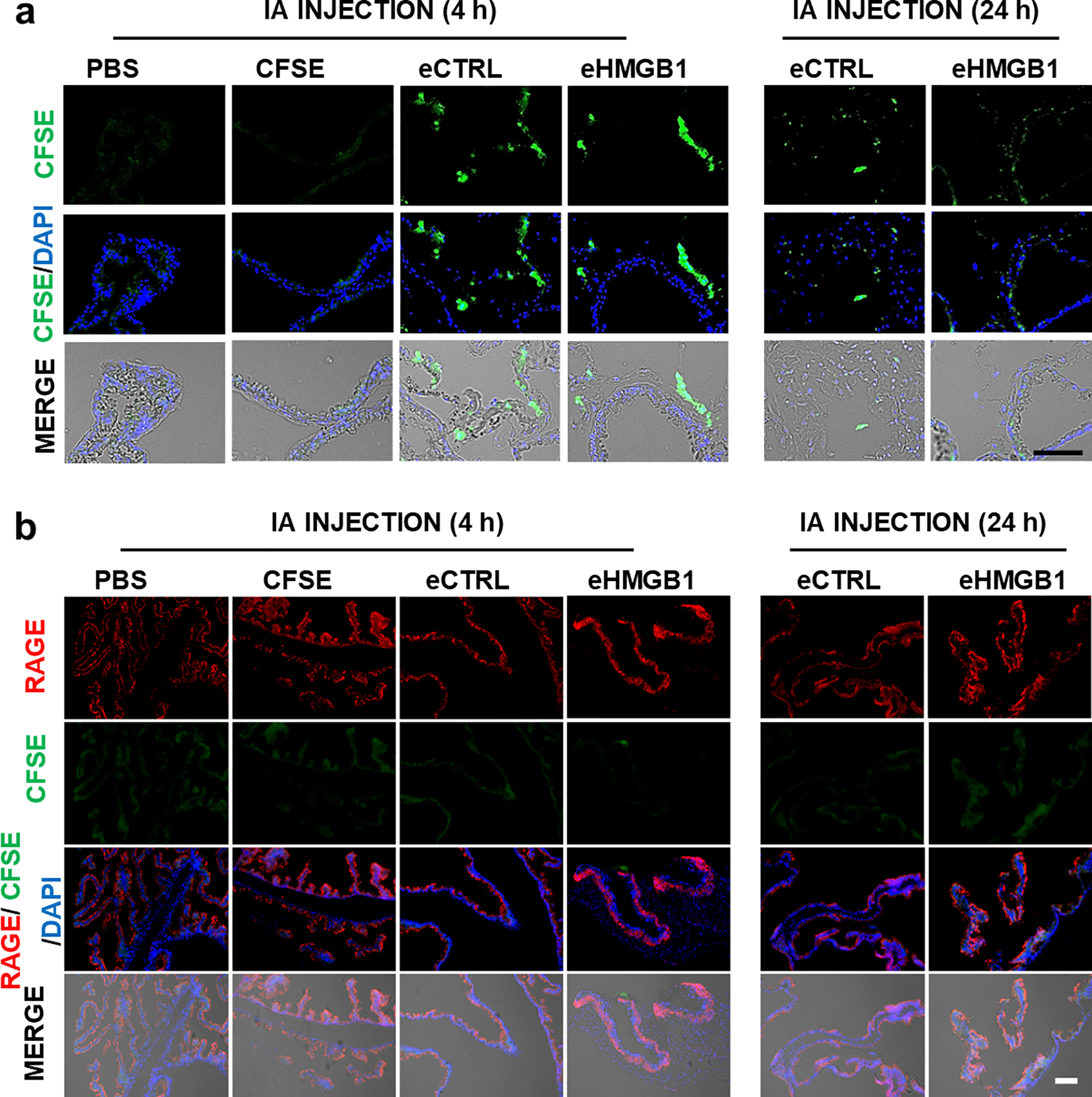

(a) Representative images showing intra-amniotically injected (at E17) CFSE-labeled engineered exosomes (eCTRL vs. eHMGB1 containing 10ng HMGB1) localization in the fetal membrane at indicated time periods. PBS and CFSE results are shown as controls. (b) Representative images of immunohistochemical analysis for RAGE expression in the fetal membrane tissue sections. (c) Representative images of CFSE-labeled engineered exosomes (eCTRL vs. eHMGB1 containing 10ng HMGB1) trafficking in the uterus tissues at indicated time periods. (d) Representative images of immunohistochemical analysis of RAGE expression in the uterine tissue sections. IA, Intra-amniotic. Scale bars, 100 μm.
DISCUSSION
Communication between the fetus and the mother is paramount during pregnancy. FMi (between the placenta and decidua basalis and fetal membranes and decidua parietalis) are critical junctions where this communication is translated functionally to maintain pregnancy or determine the timing of birth. Multiple factors, including endocrine, paracrine immune, and mechanical, are involved in biological communications between the two systems. Due to the cellular complexity of the FMi, the lack of animal models that can precisely mimic human FMi, and the difficulty in obtaining samples without any confounding physiological (labor and delivery) and pathological (PTB) factors has been a major hindrance in filling the knowledge gaps in the field of obstetrics. The placental/decidual interface is better studied than the fetal membrane/decidual interface and contributing to the lack of understanding in regard to fetal signaling inducing PTB. Using a four-chamber FMi-OOC device, together with the technology to engineer HMGB1-encapsulating exosomes, we tested the hypothesis that senescent fetal cell-derived exosomes carrying DAMPs (i.e., HMGB1) can function as a fetal signal and cause inflammatory changes across the FMi, immune cell activation, and trigger parturition. The principal findings from this study are as follows: 1. senescent amnion cell-derived exosomes package HMGB1 with the exosomes, 2. electroporated HMGB1 containing exosomes (eHMGB1) maintain their integrity and contain functional HMGB1 as an inflammatory cytokine, 3. the kinetics of eHMGB1 propagation in vitro using the FMi-OOC model was determined, 4. propagation of eHMGB1-induced RAGE and TLR4 expression, as well as inflammation of the FMi cells were observed, and 5. intra-amniotic injection of eHMGB1 into mice caused RAGE activation in the F-M uterine tissues and induced PTB. Taken together, these data confirm that paracrine signaling by amnion exosomes can cause immune activation at the FMi, predisposing to parturition.
HMGB1 is a non-histone chromatin-associated protein that binds double-stranded DNA, stabilizing nucleosomes during DNA repair and recombination65. Nuclear injury or other stressors experienced by the cell due to cellular senescence or cell death increase the passive release of extracellular HMGB1 or via active secretion to produce inflammation. Although HMGB1-mediated inflammation is primarily established by an array of receptors, RAGE and TLR4 are well-documented receptors shown to promote the effects of HMGB166. HMGB1 through TLR4 and its adaptor, myeloid differentiation factor 2 (MD-2), or through RAGE can activate nuclear factor-κB (NF-kB) and cause cytokine production66, 67. In our FMi-OOC model, as eHMGB1 propagates through the FMi cell layers RAGE and TLR4 expression and cytokine production increased; similar changes were seen in uterine tissues in vivo after eHMGB1 injection, leading to PTB. Importantly, rHMGB1 produced similar functions with a lesser effect in both in vitro and in vivo models, and this diminished effect is likely due to its much shorter half-life in the extracellular environment compared to exosomes. An NMR-based study has shown that the half-life of disulfide and thiol HMGB1 ranged from ~17 min (in human serum and saliva) to 3 h (in cell culture medium)68. Therefore, stimulation of cells with a single dose is not expected to produce sustained effects compared to their transport within the exosomal lumen. During pregnancy, exosomal paracrine signaling events are likely more efficient than the systemic spread of various signalers, where multiple factors such as half-life, presence of proteolytic factors, inhibitors, and other regulatory molecules can impede their transport and function.
In our FMi-OOC model, an increase in receptor activation and cytokine production can also result from the trafficking of biochemicals between chambers. Fetal membrane AMCs are vulnerable to inflammatory insults69, and their activation by eHMGB1 can cause an influx of cytokines that can propagate along with eHMGB1. Cellular movement is also expected between chambers. The observed increase in receptor expressions and inflammatory activation in subsequent chambers can result from both trafficking eHMGB1 and inflammatory mediators and cells. Thus, FMi-OOC is advantageous in determining intercellular interactions as observed in utero that is otherwise not possible in a 2D or even 2-cell transwell cultures. We have previously shown that inhibiting HMGB1 function using glycyrrhizin can reduce cytokine production in AECs27, and similar blocking experiments may reveal the differential roles of eHMGB1 vs. trafficking biochemicals and/or cell generated in response to eHMGB1 activation.
In our study, chorion trophoblasts were most vulnerable to eHMGB1 treatment, whereas decidual cells did not show a statistical change with markers tested within 24 h. The following multiple factors could have contributed to the lack of response in 24 h: 1) time required for enough eHMGB1 to reach the decidua was not achieved or 2) continuous perfusion of eHMGB1 in the first chamber may still be needed to sustain function at a distance. The significant inflammatory response observed in decidual cells after 72 h confirms the kinetics of eHMGB1 propagation. However, the latter is likely the scenario during in utero signaling. Constant shedding of exosomes containing HMGB1 and other DAMPs by senescent cells are likely to induce F-M signaling to ensure sustained uterine inflammatory activation. This is required to ensure the transition of quiescent tissues to an active state of labor. In this experimental model, we tested eHMGB1 as the sole cargo and showed its potential functional impact. In a senescent cell-derived exosome, other DAMPs and senescence-associated secretory phenotype (SASP) markers or other mediators of inflammation, such as MAPKs, are also expected to be cargo38. Collectively, they can achieve the inflammatory threshold required for labor initiation. In support of this, Gomez-Lopez’s group has shown that intra-amniotic injection of HMGB1 induces inflammasome activation to enhance local inflammation associated with PTB19.
In addition to the biological impact of exosomal HMGB1 signaling and functional effects, we introduced the potential of electroporation or electro-permeabilization of exosomes to carry a protein cargo. Electroporation has been previously used for cellular uptake of specific proteins to determine their functional studies70–72. Engineering exosomes using electroporation has been widely used for loading them with siRNA and miRNA and drugs, such as doxorubicin73–75. Temporary hydrophilic pores created by the electric field increase exosomal membrane permeability for content packaging, and the pores are closed immediately after electroporation to restore exosome membrane integrity76. Some major concerns using this approach are integrity of exosomes and changes electroporation can cause to cargo protein structure and function. TEM data and functional analysis of eHMGB1 show that the exosomes used in our model carry HMGB1 capable of eliciting the desired function in recipient cells. In vivo, compared to either naïve or rHMGB1, eHMGB1 injection effectively induced PTB while increasing its receptor expression along the path of its trafficking, supporting that HMGB1 is functional within the electroporated exosomes. However, the amount of functional cargo and the efficiency of loading is not compared with other approaches of cargo loading. Other technologies such as EXPLOR have been known to improve protein cargo loading efficiency77, and this technology is being tested in reproductive biology62. In addition to exosome engineering, we have overcome limitations of our prior OOC devices for fetal membranes that were initially designed to test only two cell types and technically challenging for cellular imaging47, 78. The presented FMi-OOC can be used to determine cell and exosome trafficking, analyte kinetics, and can be modified for dynamic studies where sustained flow of DAMPs as expected in utero can be studied.
Recent advances in exosome-mediated communication has helped to advance knowledge on paracrine signaling at the FMi and to decipher mechanisms of fetal-derived communication signals, their propagation, and function. In this study, we modeled HMGB1 as a signaler based on its reported physiologic and pathological roles in normal birth and pregnancy complications, such as PTB. Other DAMPs and pro-parturition markers can also be signaled via exosomes. Although we were able to show function at a target site of our interest, other destinations and specificity of cargo delivery by exosomes and function at various sites are unclear. This study provides some interesting points. First, in vivo functional validation of FMi-OOC experiments strengthens the utility and need of such OOC devices. Its usefulness to test physiologic and pathologic systems can potentially lead to avoidance of animal models in certain experimental setups. Second, we demonstrate the usefulness of engineered exosomes to carry specific functionally active protein cargo. This approach can be used for delivering specific drugs during pregnancy and/or the use of exosomes as a biomarker indicative of high-risk pregnancy. In summary, we report that fetal cell-derived exosomes can distribute HMGB1, produce inflammation in recipient cells, and finally activate maternal uterine tissues, predisposing them to parturition.
Supplementary Material
Acknowledgments
Funding: This study was supported by funding from the NIH / NIAID grant 5R21AI140249–02 to R. Menon. L. Richardson was supported by a postdoctoral fellowship through the Regulatory Science in Environmental Health and Toxicology Training Grant (T32 ES026568) from the National Institute of Environmental Health Sciences (NIEHS) of the National Institutes of Health (NIH).
REFERENCES
- 1.Beck S , Wojdyla D , Say L, et al. The worldwide incidence of preterm birth: a systematic review of maternal mortality and morbidity. Bull World Health Organ 2010;88:31–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.PrabhuDas M , Bonney E , Caron K, et al. Immune mechanisms at the maternal-fetal interface: perspectives and challenges. Nat Immunol 2015;16:328–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Racicot K , Kwon JY , Aldo P , Silasi M , Mor G. Understanding the complexity of the immune system during pregnancy. Am J Reprod Immunol 2014;72:107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gotsch F , Romero R , Kusanovic JP, et al. The fetal inflammatory response syndrome. Clin Obstet Gynecol 2007;50:652–83. [DOI] [PubMed] [Google Scholar]
- 5.MEDZHITOV R Inflammation 2010: new adventures of an old flame. Cell 2010;140:771–6. [DOI] [PubMed] [Google Scholar]
- 6.Challis JR , Bloomfield FH , Bocking AD, et al. Fetal signals and parturition 28. J Obstet Gynaecol Res 2005;31:492–99. [DOI] [PubMed] [Google Scholar]
- 7.Mendelson CR. Minireview: fetal-maternal hormonal signaling in pregnancy and labor 28. Mol Endocrinol 2009;23:947–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Menon R Oxidative stress damage as a detrimental factor in preterm birth pathology 14. Front Immunol 2014;5:567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Erlebacher A Immunology of the maternal-fetal interface. Annu Rev Immunol 2013;31:387–411. [DOI] [PubMed] [Google Scholar]
- 10.Lash GE. Molecular Cross-Talk at the Feto-Maternal Interface. Cold Spring Harb Perspect Med 2015;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hadley EE , Richardson LS , Torloni MR , Menon R. Gestational tissue inflammatory biomarkers at term labor: A systematic review of literature. Am J Reprod Immunol 2018;79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Behnia F , Sheller S , Menon R. Mechanistic Differences Leading to Infectious and Sterile Inflammation 3. Am J Reprod Immunol 2016. [DOI] [PubMed] [Google Scholar]
- 13.Brien ME , Boufaied I , Bernard N , Forest JC , Giguere Y , Girard S. Specific inflammatory profile in each pregnancy complication: a comparative study. Am J Reprod Immunol 2020:e13316. [DOI] [PubMed] [Google Scholar]
- 14.Menon R , Behnia F , Polettini J , Richardson LS. Novel pathways of inflammation in human fetal membranes associated with preterm birth and preterm pre-labor rupture of the membranes. Semin Immunopathol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richardson LS , Taylor RN , Menon R. Reversible EMT and MET mediate amnion remodeling during pregnancy and labor. Sci Signal 2020;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonney EA , Krebs K , Saade G, et al. Differential senescence in feto-maternal tissues during mouse pregnancy. Placenta 2016;43:26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cox LS , Redman C. The role of cellular senescence in ageing of the placenta. Placenta 2017;52:139–45. [DOI] [PubMed] [Google Scholar]
- 18.Hirota Y , Daikoku T , Tranguch S , Xie H , Bradshaw HB , Dey SK. Uterine-specific p53 deficiency confers premature uterine senescence and promotes preterm birth in mice. J Clin Invest 2010;120:803–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plazyo O , Romero R , Unkel R, et al. HMGB1 Induces an Inflammatory Response in the Chorioamniotic Membranes That Is Partially Mediated by the Inflammasome. Biol Reprod 2016;95:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romero R , Miranda J , Chaiworapongsa T, et al. Prevalence and clinical significance of sterile intra-amniotic inflammation in patients with preterm labor and intact membranes. Am J Reprod Immunol 2014;72:458–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Polettini J , Behnia F , Taylor BD , Saade GR , Taylor RN , Menon R. Telomere Fragment Induced Amnion Cell Senescence: A Contributor to Parturition? PLoS One 2015;10:e0137188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phillippe M Cell-Free Fetal DNA, Telomeres, and the Spontaneous Onset of Parturition. Reprod Sci 2015;22:1186–201. [DOI] [PubMed] [Google Scholar]
- 23.Mulla MJ , Myrtolli K , Potter J, et al. Uric acid induces trophoblast IL-1beta production via the inflammasome: implications for the pathogenesis of preeclampsia. Am J Reprod Immunol 2011;65:542–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nadeau-Vallee M , Obari D , Palacios J, et al. Sterile inflammation and pregnancy complications: a review. Reproduction 2016;152:R277–R92. [DOI] [PubMed] [Google Scholar]
- 25.Gomez-Lopez N , Romero R , Plazyo O, et al. Intra-Amniotic Administration of HMGB1 Induces Spontaneous Preterm Labor and Birth. Am J Reprod Immunol 2016;75:3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buhimschi CS , Baumbusch MA , Dulay AT, et al. Characterization of RAGE, HMGB1, and S100beta in inflammation-induced preterm birth and fetal tissue injury. Am J Pathol 2009;175:958–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bredeson S , Papaconstantinou J , Deford JH, et al. HMGB1 promotes a p38MAPK associated non-infectious inflammatory response pathway in human fetal membranes. PLoS One 2014;9:e113799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baumbusch MA , Buhimschi CS , Oliver EA, et al. High Mobility Group-Box 1 (HMGB1) levels are increased in amniotic fluid of women with intra-amniotic inflammation-determined preterm birth, and the source may be the damaged fetal membranes. Cytokine 2016;81:82–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Menon R , Behnia F , Polettini J , Saade GR , Campisi J , Velarde M. Placental membrane aging and HMGB1 signaling associated with human parturition. Aging (Albany NY) 2016;8:216–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andocs G , Meggyeshazi N , Balogh L, et al. Upregulation of heat shock proteins and the promotion of damage-associated molecular pattern signals in a colorectal cancer model by modulated electrohyperthermia. Cell Stress Chaperones 2015;20:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stephen GL , Lui S , Hamilton SA, et al. Transcriptomic profiling of human choriodecidua during term labor: inflammation as a key driver of labor. Am J Reprod Immunol 2015;73:36–55. [DOI] [PubMed] [Google Scholar]
- 32.Girard S , Heazell AE , Derricott H, et al. Circulating cytokines and alarmins associated with placental inflammation in high-risk pregnancies. Am J Reprod Immunol 2014;72:422–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dubicke A , Andersson P , Fransson E, et al. High-mobility group box protein 1 and its signalling receptors in human preterm and term cervix. J Reprod Immunol 2010;84:86–94. [DOI] [PubMed] [Google Scholar]
- 34.Romero R , Chaiworapongsa T , Alpay SZ, et al. Damage-associated molecular patterns (DAMPs) in preterm labor with intact membranes and preterm PROM: a study of the alarmin HMGB1. J Matern Fetal Neonatal Med 2011;24:1444–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Menon R , Taylor BD. Exploring Inflammatory Mediators in Fetal and Maternal Compartments During Human Parturition. Obstet Gynecol 2019;134:765–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Romero R , Chaiworapongsa T , Savasan ZA, et al. Clinical chorioamnionitis is characterized by changes in the expression of the alarmin HMGB1 and one of its receptors, sRAGE. J Matern Fetal Neonatal Med 2012;25:558–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang B , Koga K , Osuga Y, et al. High mobility group box 1 (HMGB1) levels in the placenta and in serum in preeclampsia. Am J Reprod Immunol 2011;66:143–48. [DOI] [PubMed] [Google Scholar]
- 38.Sheller-Miller S , Urrabaz-Garza R , Saade G , Menon R. Damage-Associated molecular pattern markers HMGB1 and cell-Free fetal telomere fragments in oxidative-Stressed amnion epithelial cell-Derived exosomes. J Reprod Immunol 2017;123:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Menon R Human fetal membranes at term: Dead tissue or signalers of parturition? Placenta 2016;44:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang W , Tang Y , Li L. HMGB1, a potent proinflammatory cytokine in sepsis. Cytokine 2010;51:119–26. [DOI] [PubMed] [Google Scholar]
- 41.Lange SS , Vasquez KM. HMGB1: The Jack-of-All-Trades Protein Is a Master DNA Repair Mechanic. Mol Carcinog 2009;48:571–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lotze MT , Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 2005;5:331–42. [DOI] [PubMed] [Google Scholar]
- 43.Pisetsky DS , Gauley J , Ullal AJ. HMGB1 and Microparticles as Mediators of the Immune Response to Cell Death. Antioxid Redox Sign 2011;15:2209–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamada S , Maruyama I. HMGB1, a novel inflammatory cytokine. Clin Chim Acta 2007;375:36–42. [DOI] [PubMed] [Google Scholar]
- 45.Menon R , Mesiano S , Taylor RN. Programmed Fetal Membrane Senescence and Exosome-Mediated Signaling: A Mechanism Associated With Timing of Human Parturition. Front Endocrinol (Lausanne) 2017;8:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yim N , Choi C. Extracellular vesicles as novel carriers for therapeutic molecules. BMB Rep 2016;49:585–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Richardson L , Jeong S , Kim S , Han A , Menon R. Amnion membrane organ-on-chip: an innovative approach to study cellular interactions. FASEB J 2019:fj201900020RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richardson L , Kim S , Menon R , Han A. Organ-On-Chip Technology: The Future of Feto-Maternal Interface Research? Front Physiol 2020;11:715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Richardson LS, Kim S, Han A, Menon R. Modeling ascending infection with a feto-maternal interface organ-on-chip. Lab Chip. 2020. November 24;20(23):4486–4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lavu N , Richardson L , Radnaa E, et al. Oxidative stress-induced downregulation of glycogen synthase kinase 3 beta in fetal membranes promotes cellular senescencedagger. Biol Reprod 2019;101:1018–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hadley EE , Sheller-Miller S , Saade G, et al. Amnion Epithelial Cell Derived Exosomes Induce Inflammatory Changes in Uterine Cells. Am J Obstet Gynecol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jin J , Richardson L , Sheller-Miller S , Zhong N , Menon R. Oxidative stress induces p38MAPK-dependent senescence in the feto-maternal interface cells. Placenta 2018;67:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ayad MT , Taylor BD , Menon R. Regulation of p38 mitogen-activated kinase-mediated fetal membrane senescence by statins. Am J Reprod Immunol 2018:e12999. [DOI] [PubMed] [Google Scholar]
- 54.Menon R , Fortunato SJ , Yu J, et al. Cigarette smoke induces oxidative stress and apoptosis in normal term fetal membranes. Placenta 2011;32:317–22. [DOI] [PubMed] [Google Scholar]
- 55.Menon R , Fortunato SJ. Distinct pathophysiologic pathways induced by in vitro infection and cigarette smoke in normal human fetal membranes. Am J Obstet Gynecol 2009;200:334–38. [DOI] [PubMed] [Google Scholar]
- 56.Sheller S , Papaconstantinou J , Urrabaz-Garza R, et al. Amnion-Epithelial-Cell-Derived Exosomes Demonstrate Physiologic State of Cell under Oxidative Stress. PLoS One 2016;11:e0157614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lamichhane TN , Raiker RS , Jay SM. Exogenous DNA Loading into Extracellular Vesicles via Electroporation is Size-Dependent and Enables Limited Gene Delivery. Mol Pharm 2015;12:3650–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mendt M , Kamerkar S , Sugimoto H, et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Consortium E-T , Van Deun J , Mestdagh P, et al. EV-TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat Methods 2017;14:228–32. [DOI] [PubMed] [Google Scholar]
- 60.Zheng Y , Tu C , Zhang J , Wang J. Inhibition of multiple myelomaderived exosomes uptake suppresses the functional response in bone marrow stromal cell. Int J Oncol 2019;54:1061–70. [DOI] [PubMed] [Google Scholar]
- 61.Sheller-Miller S , Radnaa E , Arita Y, et al. Environmental pollutant induced cellular injury is reflected in exosomes from placental explants. Placenta 2020;89:42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sheller-Miller S , Choi K , Choi C , Menon R. Cre-Reporter Mouse Model to Determine Exosome Communication and Function during Pregnancy. Am J Obstet Gynecol 2019. [DOI] [PubMed] [Google Scholar]
- 63.Sheller-Miller S , Trivedi J , Yellon SM , Menon R . Exosomes Cause Preterm Birth in Mice: Evidence for Paracrine Signaling in Pregnancy. Scientific reports 2019;9:608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Enkhtuya R , Sato T , Wakasugi M, et al. The scaffold protein JLP plays a key role in regulating ultraviolet B-induced apoptosis in mice. Genes Cells 2014;19:350–8. [DOI] [PubMed] [Google Scholar]
- 65.Kang R , Zhang Q , Zeh HJ III, LOTZE MT, TANG D. HMGB1 in cancer: good, bad, or both? Clin Cancer Res 2013;19:4046–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang H , Wang H , Ju Z, et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J Exp Med 2015;212:5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Deng M , Tang Y , Li W, et al. The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 2018;49:740–53 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zandarashvili L , Sahu D , Lee K, et al. Real-time kinetics of high-mobility group box 1 (HMGB1) oxidation in extracellular fluids studied by in situ protein NMR spectroscopy. J Biol Chem 2013;288:11621–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sato BL , Collier ES , Vermudez SA , Junker AD , Kendal-Wright CE. Human amnion mesenchymal cells are pro-inflammatory when activated by the Toll-like receptor 2/6 ligand, macrophage-activating lipoprotein-2. Placenta 2016;44:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lambert H , Pankov R , Gauthier J , Hancock R. Electroporation-mediated uptake of proteins into mammalian cells. Biochem Cell Biol 1990;68:729–34. [DOI] [PubMed] [Google Scholar]
- 71.Graziadei L , Burfeind P , Bar-Sagi D. Introduction of unlabeled proteins into living cells by electroporation and isolation of viable protein-loaded cells using dextran-fluorescein isothiocyanate as a marker for protein uptake. Anal Biochem 1991;194:198–203. [DOI] [PubMed] [Google Scholar]
- 72.Knudsen JR , Henriquez-Olguin C , Li Z , Jensen TE. Electroporated GLUT4–7myc-GFP detects in vivo glucose transporter 4 translocation in skeletal muscle without discernible changes in GFP patterns. Exp Physiol 2019;104:704–14. [DOI] [PubMed] [Google Scholar]
- 73.Gilligan KE , Dwyer RM. Engineering Exosomes for Cancer Therapy. Int J Mol Sci 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kamerkar S , LeBleu VS , Sugimoto H, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017;546:498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tian Y , Li S , Song J, et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014;35:2383–90. [DOI] [PubMed] [Google Scholar]
- 76.Familtseva A , Jeremic N , Tyagi SC. Exosomes: cell-created drug delivery systems. Mol Cell Biochem 2019;459:1–6. [DOI] [PubMed] [Google Scholar]
- 77.Yim N , Ryu SW , Choi K, et al. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module. Nat Commun 2016;7:12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Richardson L , Gnecco J , Ding T, et al. Fetal Membrane Organ-On-Chip: An Innovative Approach to Study Cellular Interactions. Reprod Sci 2020;27:1562–69. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.