

INTRODUCTION:
Solitary juvenile polyps (JP) are characterized by a benign disease course with low recurrence rate but present with signs of intestinal inflammation. To better understand the underlying pathogenesis, we performed histological and molecular evaluation targeting distinct immune mechanisms.
METHODS:
Pediatric patients with JP (n = 12), with treatment-naïve inflammatory bowel disease (IBD; [n = 41]) as inflammatory control, and non-IBD controls (n = 14) were investigated. For a comparative analysis of infiltrating immune cells, a next-generation tissue microarray of biopsies was assembled, immunostained, and scored. Targeted transcriptional profiling was performed using a customized immunology panel.
RESULTS:
In JP, a predominant accumulation of neutrophils and eosinophils was observed. RNA expression profiles revealed increased levels of CXCL8, CXCL5, and CCL11 transcripts in JP, indicating an enhanced recruitment of neutrophils and eosinophils. Moreover, messenger RNA levels of the proinflammatory cytokine IL1b and the inflammation-amplifying receptor TREM1 were higher in JP, whereas we could not find signs of a functionally polarized Tcell response in JP when compared with IBD.
DISCUSSION:
Patients with JP and patients with treatment-naïve IBD have distinct cell infiltrates during active disease. The ample presence of eosinophils in JP supports neutrophil accumulation, which is responsible for the elevated release of calprotectin. Intriguingly, however, we were not able to identify a functionally polarized T-cell response in JP, which indicates that during the acute onset of inflammation in JP, a potent adaptive immune memory is not established. This may explain the low reoccurrence rate of JP.
INTRODUCTION
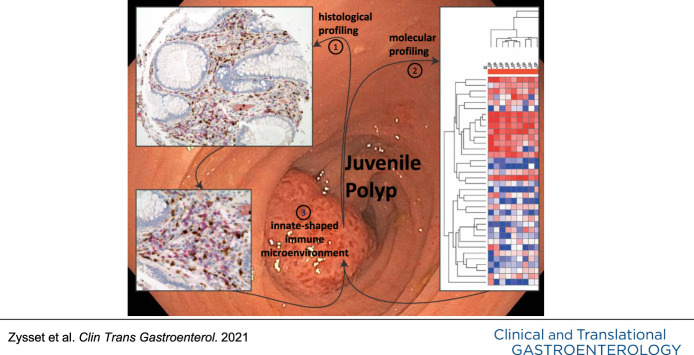
Juvenile polyps (JPs) are the most common colonic polyp in children and considered a benign entity (1,2). Epidemiological data are scarce; however, in up to 12% of pediatric patients who undergo colonoscopy because of hematochezia, a JP is identified as source of bleeding (3). JP seldom reoccurs after spontaneous dispatch or endoscopic removal (4). By contrast, inflammatory bowel disease (IBD) is a chronic relapsing inflammatory disease of the intestine. The etiology of IBD is multifactorial, and evidence suggests a disrupted interplay between host immune system and environmental factors at multiple levels (5). It is generally believed that IBD develops in genetically predisposed individuals because of aberrant immune responses of the adaptive and the innate immune system in reaction to luminal antigens.
Fecal calprotectin, a heterodimeric protein with the subunits S100A8 and A9 (also known as myeloid-related protein 8/14, MRP8/14), is an accepted stool marker for mucosal inflammation in IBD in adults and children (6) and is also elevated in JP (7–9). The main cellular source of calprotectin is neutrophils (10). In histological examinations, JP show high numbers of neutrophils (7). However, the underlying mechanisms leading to the neutrophil-driven immune response in JP are not well-described. JP and IBD both show local epithelial defects. The impaired barrier function with increased exposure to potentially inflammatory stimuli (11) may suggest a similar immune response in both entities.
Transcriptome analysis of adult IBD and control tissue revealed different expression levels of molecules involved in immune regulation and response between IBD and controls as well as between IBD subtypes (12,13). This observation allowed to deduce pathogenesis factors of the disease, such as a polarized T helper 2 (Th2) vs T helper 1 (Th1) immune response for ulcerative colitis (UC) and Crohn's disease (CD), respectively (13). We therefore aimed to uncover differences in the gene expression profiles in the affected colonic mucosa of patients with JP vs patients with pediatric IBD and noninflammatory (non-IBD) controls. Based on the histological findings, we decided to perform a targeted transcriptional approach to analyze the interactions between different immune cell subtypes.
We hypothesize that an inflammatory microenvironment, which supports innate immune responses is critical for the development and maintenance of JP. By contrast, the strong, functionally polarized activation of the adaptive immune response, notably of Tcell subsets (14), leads to the establishment of a chronic inflammation in IBD.
We analyze the inflammatory infiltrate of JP, treatment-naïve IBD patient samples without drug-related alterations in the inflammatory response and non-IBD controls (15). We were able to conclusively compare the immune microenvironment in JP in comparison with IBD and non-IBD controls. Furthermore, we provide novel insights into the involvement of the innate immune system in the pathology of JP and the exclusive role of eosinophils as supporters of neutrophil expansion. Collectively, the benign feature and high levels of calprotectin of JP may be explained by the lack of the establishment of a functional Tcell memory and the predominant neutrophil accumulation.
METHODS
Cohort and Samples
Sixty-seven biopsies from 12 JP, 14 non-IBD controls, and 41 patients with treatment-naïve pediatric IBD (18 CD and 23 UC) who underwent diagnostic colonoscopy at the Children's Hospital, Lucerne, or at the Children's Hospital, Bern, between 2007 and 2013 were retrieved. Patients with more than 1 polyp or polyposis syndromes were excluded. IBD diagnosis was based on the revised Porto criteria (16). For patients with IBD, biopsies from the most inflamed area were used. For CD, only patients with histological activity in colonic biopsies were considered because of the known site-specific differences in the composition of immune cells in the terminal ileum and colon (17). As controls, biopsies were used from pediatric patients, where IBD was ruled out based on findings of macroscopically and microscopically normal mucosa (further referred as “non-IBD”). Main indications for colonoscopy in patients with non-IBD were rectal bleeding, recurrent abdominal pain, or diarrhea. The definite diagnosis in these patients was functional gastrointestinal disorders, including obstipation, irritable bowel syndrome, and functional abdominal pain.
Clinical data taken as part of clinical routine were collected from patients' charts, including sex, age at diagnosis, rectal bleeding at first presentation, disease location, and activity scores (Pediatric Crohn's Disease Activity Index (18); Pediatric Ulcerative Colitis Activity Index (19)) for patients with IBD at diagnosis and laboratory workup at diagnosis.
The Ethics Committee of Bern granted ethical approval (KEK-BE: 200/2014).
Next-generation tissue microarray
Hematoxylin and eosin (H&E) slides were retrieved from the diagnostic archive and scanned using slide scanner (3DHistech, Budapest, Hungary). Then, each whole slide image corresponding to polyps and colonic mucosa of patients with non-IBD and IBD was digitally annotated using a tissue microarray annotation tool of 0.6 mm in diameter in 5 representative tissue areas. One area was annotated to represent a tissue punch for embedding into the tissue microarray and the remaining areas for subsequent molecular analysis. Annotated scans were aligned to the corresponding tissue blocks in an automated tissue microarrayer (Grand Master; 3DHistech, Budapest, Hungary), and corresponding regions of interest were punched out. One punch was transferred into recipient next-generation Tissue Microarray (ngTMA) block and the remaining punches into Eppendorf tubes for subsequent RNA extraction. Generating ngTMA allowed serial staining of all tissue samples for H&E and immunohistochemistry (20). The ngTMA block was sectioned at 4 μm, and after deparaffinization and rehydration, standard H&E and immunohistochemistry staining was performed (see Supplementary Table 1, http://links.lww.com /CTG/A624). A pathologist (M.M.), blinded to the diagnosis and clinical data, reviewed and scored all histology slides.
H&E stainings were scored for inflammatory activity on a 3-point-scale (1 = none/mild, 2 = moderate, and 3 = severe) according to the activity definition used in the PAID (pattern, activity, interpretation, dysplasia) scheme (21). The inflammatory infiltrate as a whole was used as a nominator and the abundance of the single-cell types as a numerator to characterize the composition of the inflammatory infiltrate. The proportion of eosinophils of the inflammatory infiltrate was determined on H&E slides. For neutrophils, T cells, B cells, and plasma cells, the appropriate cell markers myeloperoxidase (MPO), CD3, CD20, and CD138, respectively, were analyzed on serial ngTMA sections. Cell counting per punch was performed for T-regulatory cells (FoxP3) and activated T cells (CD30) because of their low abundance. Macrophage (CD68) density of the punch was scored as 1 = low, 2 = moderate, and 3 = high. Costainings of the described markers with the subunit S100A8 of calprotectin allowed us to determine the cellular source of calprotectin.
Nanostring analysis
Total RNA from 3 punches from biopsies of each patient was purified with a column-based approach as described by Oberli et al. (22) RNA concentration was determined by measuring the 260 nm absorbance on a NanoDrop One (Thermo Scientific, Waltham, MA), and 100 ng of each RNA sample was used for transcriptional profiling using a custom designed nCounter PlexSet (nanostring, Seattle, WA) (see Supplementary Table 2, http://links.lww.com/CTG/A624).
Statistical analysis
For all data, the Student t test, Wilcoxon rank sum test, or χ2 test were used as indicated. NanoString Raw counts were normalized using the R package NanoStringNorm. Normalized counts were log-transferred, and the differential gene expression analysis was performed using the edgeR package. Each patient group was compared with each other individually. P values were adjusted according to the Benjamini-Hochberg correction test. P values < 0.05 were considered to be statistically significant. PC analysis was performed in R (stats package), and data were visualized using the ggplot2 package. Heat map of the gene expression data was generated using the Morpheus app (https://software.broadinstitute.org/morpheus), using 1 minus Pearson correlation for both, columns and rows. Data were analyzed using R v3.5.0.
RESULTS
Initial clinical and laboratory presentation in patients with JP are nonspecific
We found that most JP presented with signs of rectal bleeding as the leading symptom (Table 1), which was also true for non-IBD controls and patients with UC. Systemic inflammatory markers such as hemoglobin, thrombocytes, erythrocyte sedimentation rate, C-reactive protein, and albumin were variable and mostly in normal range in our patients with JP and UC. By contrast, patients with CD had an increase in systemic inflammatory markers and decreased hemoglobin and albumin values.
Table 1.
Baseline characteristics of study cohort
| Non-IBD control | JP | CD | UC | |
| N (male) | 14 (7) | 12 (7) | 18 (13) | 23 (11) |
| Age, yr, mean (SD) | 7.72 (4.93) | 5.83 (3.72) | 9.6 (3.47) | 10.16 (3.88) |
| Rectal bleeding at first presentation, n (%) | 9 (64) | 10 (83) | 7 (39) | 17 (74) |
| PCDAI score at diagnosis, n, 10 to 30 mild: >30 moderate/severe | 6: 12 | |||
| PUCAI score at diagnosis, n, 10 to <30 mild: 30 to <65 moderate: >65 severe | 3: 18: 2 | |||
| Paris classification at diagnosis, n (%) | ||||
| L1: distal ileal +/− limited cecal disease | 4 (20) | |||
| L2: colonic | 4 (20) | |||
| L3: ileocolonic | 9 (45) | |||
| L4a: upper disease proximal to the ligament of Treitz | 3 (15) | |||
| E1: proctitis | 1 (4) | |||
| E2: left-sided | 3 (13) | |||
| E3: extensive | 5 (22) | |||
| E4: pancolitis | 14 (61) | |||
| Laboratory values at diagnosis, mean (SD) | ||||
| Hemoglobin, g/L | 128.2 (9.0) | 123.5 (13.3) | 107.1 (14.8) | 109.7 (20.3) |
| Thrombocytes, g/L | 376.6 (83.8) | 327.4 (84.0) | 481.2 (107.7) | 400.8 (101.2) |
| C-reactive protein, mg/dL | 1 (0) | 10.6 (27.3) | 32.6 (14.7) | 14.7 (25.4) |
| Erythrocyte sedimentation rate, mm/hr | 10 (2.8) | 10.1 (6.4) | 47.5 (35.9) | 21.1 (14.2) |
| Albumin, g/L | 40.3 (2.5) | 39.3 (2.1) | 30.3 (5.0) | 36.1 (6.6) |
CD, Crohn's disease; JP, juvenile polyps; IBD, inflammatory bowel disease; PCDAI, Pediatric Crohn's Disease Activity Index; PCUAI, Pediatric Ulcerative Colitis Activity Index; UC, ulcerative colitis.
Fecal calprotectin levels were available from 33 patients at diagnosis; however, because of different (lateral flow and enzyme-linked immunosorbent assay-based) laboratory assays used in our cohort, the indicated levels were not directly comparable. The levels ranged from 25 to 277 mg/kg in 4 non-IBD controls, 110–934 mg/kg in 7 patients with JP, 101–5,432 mg/kg in 15 patients with UC, and 473–3,000 mg/kg in 8 patients with CD. As the cutoff was 50 mg/kg in all assays, all patients with JP and IBD had per definition raised levels. None of non-IBD controls, 3 patients with JP, 13 patients with UC, and 8 patients with CD had calprotectin values > 10× ULN.
ngTMA analysis reveals a pronounced accumulation of neutrophils and eosinophils in JP
Because we were interested in factors supporting local infiltration and survival of inflammatory cells, we identified the most inflamed mucosal tissue sections in patients with IBD for further analysis (Figure 1a). The cellular composition of the inflammatory infiltrates is substantially different between patients with JP and patients with IBD at the site of maximum histopathological inflammatory activity. In patients with JP, we detected a prominent infiltration by neutrophils and eosinophils, whereas in patients with IBD, the cellular infiltrates were dominated by T cells, plasma cells, and B cells (Figure 1a,b; see Supplementary Figure 1, http://links.lww.com/CTG/A620). There was no difference in the presence of eosinophils between CD and UC. Furthermore, we found CD68+ macrophages at comparable frequencies in all tissue samples (see Supplementary Figures 1, http://links.lww.com/CTG/A620 and 2, http://links.lww.com/CTG/A621).
Figure 1.

(a) Representative ngTMA H&E staining's of non-IBD controls (non-IBD ctrl), solitary JP, CD, and UC tissue samples. (b) Calprotectin (red) and MPO (brown) costaining. (a–b) Representative micrographs at 20× and 40× magnification. Bars indicate 100 and 50 μm, respectively. Arrowheads show examples of calprotectin and MPO double-positive cells. (c) Cellular composition of inflammatory infiltrates. Relative abundance of B cells, eosinophils, neutrophils, plasma cells, and T cells in the indicated groups. (d) Relative frequency of either calprotectin positive neutrophils or macrophages within the indicated groups. CD, Crohn's disease; JP, juvenile polyps; IBD, inflammatory bowel disease; MPO, myeloperoxidase; UC, ulcerative colitis
Next, we defined the cellular source of calprotectin by performing costaining of calprotectin and inflammatory cell markers. Calprotectin reactivity was most often associated with the presence of neutrophils (MPO+); however, there was no difference in the frequency of calprotectin+ neutrophils between the JP and IBD groups (Figure 1c,d). In addition, macrophages expressed calprotectin (23), however, to a much lesser extent than neutrophils (Figure 1d; see Supplementary Figure 1, http://links.lww.com/CTG/A620). No coexpression was seen for T cells, B cells, eosinophils, epithelial, and endothelial cells.
Different Tcell subsets are known to trigger and perpetuate intestinal damage, including t-bet (TBX21)-expressing Th1 cells, which support proinflammatory cell-mediated immune reactions and CD30+ and GATA3-expressing Th2 cells, whereby the latter produce eosinophil-activating cytokines, such as IL-5 and IL-13 (24). In non-IBD controls, no CD30+ cells were spotted and only a few FoxP3+ regulatory T cells (Tregs), whereas all 3 disease groups (JP, UC, and CD) showed similar numbers of these cells (see Supplementary Figure 3, http://links.lww.com/CTG/A622).
Collectively, histological analysis showed that JP has a distinct inflammatory infiltrate, which consists mainly of neutrophils and eosinophils, whereas in pediatric IBD, cells of the adaptive immune cells dominate (T cells, B cells, and plasma cells). Neutrophils and, to a much lesser extent, macrophages were the sole source of calprotectin.
Transcriptional profiling reveals disease-specific alteration in JP compared with CD and UC
In addition, we aimed to determine whether distinct cellular inflammatory infiltrates in JP and IBD can be attributed to a differential expression of chemokines and cytokines. To this end, we isolated total RNA from tissue punches of all patients and non-IBD controls for gene expression analysis. To decipher the molecular pathways involved in the maintenance of inflammation, we selected the most inflammed areas of tissue, which resulted in similar inflammation scores based on histopathological evaluation (see Supplementary Figure 4, http://links.lww.com/CTG/A623).
The transcriptional profiling was performed using a custom-designed NanoString PlexSet consisting of a panel of 40 genes involved in immune pathways (see Supplementary Table 2, http://links.lww.com/CTG/A624). The selection of the genes was based on the histopathological assessment in a targeted approach to identify underlying pathogenetic mechanisms. We excluded 4 genes (MPO, IL17F, IL23A, and TREM2) from the analysis because their expression was below the detection limit. In contrast to the histological evaluation, we could not detect any substantial messenger RNA (mRNA) levels of MPO. This might be due to the fact that MPO is mainly produced during maturation of neutrophils and stored in azurophilic granules in mature neutrophils (25). Principal component (PC) analysis revealed a clear difference in the transcriptional profile between JP and non-IBD control group. The IBD subtype groups, UC and CD, clustered in between the JP and the non-IBD control group (Figure 2a). Hierarchical clustering analysis of the complete dataset clustered the JP group in line with the PC analysis separately compared with the non-IBD control and the IBD group (Figure 2b). Significant gene expression differences were found between JP vs non-IBD control group, JP and CD, and JP and UC in 55.6% (20 genes), 58.3% (21 genes), and 52.8% (19 genes), respectively. There was no significant change between patients with treatment-naïve pediatric CD and patients with UC besides a trend toward an increased mRNA expression of IL17A in UC patient samples. Mainly, genes encoding cytokines and chemokines which are involved in the regulation of recruitment, activation, and survival of neutrophils, such as CXCL8 (26), colony-stimulating factor (CSF) 2, CSF3 (27), CXCL5 (28) (Figure 3a), and eosinophils (CCL11 and CCR3) (Figure 3b) were upregulated in JP. Because CCR3 is also expressed on Th2 cells (29), a preferential shift to local Th2 cell differentiation in JP compared with IBD cannot be excluded. Indeed, the IBD group tends to have higher ratio (TBX21/GATA3) of transcripts levels toward the Th1 cell-specific transcription factor TBX21 compared with JP and non-IBD control (Figure 3c).
Figure 2.

(a) PC analysis of non-IBD controls vs solitary JP vs IBD, including CD and UC of the log2-transformed counts per million (CPM) of the NanoString gene expression analysis and (b) heat map of the log2-transformed CPM, including k means clustering of all genes analyzed. CD, Crohn's disease; JP, juvenile polyps; IBD, inflammatory bowel disease; UC, ulcerative colitis.
Figure 3.

(a–f) NanoString Gene expression analysis of non-IBD controls, solitary JP, CD, and UC tissue samples. Box and whiskers are representative of the log2-transformed CPM gene expression values of the indicated groups. #Indicates the adjusted P-value of the indicated groups in comparison with the non-IBD control group. *Indicates the adjusted P-value among JP, CD, and UC groups with */#P < 0.05, **/##P < 0.01, ***/###P < 0.001, and ****/####P < 0.0001. No indication means P > 0.05 and is considered not significant. CD, Crohn's disease; JP, juvenile polyps; IBD, inflammatory bowel disease; UC, ulcerative colitis.
Next, we analyzed the transcriptional profile of proinflammatory markers. In the selected regions, we found high levels of calprotectin transcripts (S100A8/A9) with significantly increased expression in JP samples (Figure 3d). As expected, the inflammatory profiles of JP tend to exhibit a mostly innate-driven proinflammatory gene expression profile, notably, increased transcript levels of the IL1ß and TREM1 genes. By contrast, UC and CD preferably display a preferential upregulation of mRNA's generally associated with a preferential Th1 CD4 Tcell differentiation, including IFNγ and the IFNγ inducible gene CXCL10/IP10, which encodes the ligand for CXCR3 (Figure 3e).
The mononuclear phagocyte system represents a major regulator of the local tissue homeostasis in the intestinal lamina propria. Hence, we also examined the transcriptional profile targeting the mononuclear phagocyte system, particularly, monocytes and macrophages. In line with the histology data, the total presence of macrophages does not seem to be altered in JP vs pediatric IBD based on CD68 transcription level between groups (Figure 3f). Interestingly, the JP group seems to have a reduced influx of CCR2+ monocytes but increased CSF2/GM-CSF expression, which critically supports survival, proliferation, and differentiation of myeloid cells during injury (30).
Together these results reveal a distinct gene expression profile between JP and IBD biopsies but also between JP and non-IBD control biopsies. The aforementioned gene expression profiles are in line with the findings by immunohistochemistry and support the notion that in JP, the enhanced recruitment, activation, and survival of neutrophils, and eosinophils critically contribute to the pathogenesis, whereas in pediatric IBD components of the adaptive Tcell response, notably, IFNγ and CCXL10/IP10 are prominently expressed.
DISCUSSION
In pediatric practice, JP symptoms are nonspecific and can resemble IBD, especially UC, at initial presentation with raised calprotectin and rectal bleeding. JP is an inflammatory condition that does not usually reoccur, whereas IBD is a chronic relapsing inflammatory disease of the intestine.
To shed light on the pathogenesis of JP, we first determined the composition of the inflammatory infiltrate and the source of calprotectin in intestinal tissue samples. JP tissue shows a predominant accumulation of innate immune cells, dominated by neutrophils and eosinophils. Immunohistological costaining identified neutrophils and macrophages as main sources of calprotectin. Previously, it was documented that colonic biopsies from areas adjacent to JP have normal mucosa (31). Thus, other relevant regional sources of calprotectin in JP are unlikely, and a solitary JP alone explains the high calprotectin found in stool samples of JP. Fecal calprotectin in JP can be even as high as in IBD. In IBD, however, affected mucosal area is often substantially larger, which can explain a cumulative higher level of fecal calprotectin despite a lower local density of neutrophils. In line with this assumption, the mRNA expression of S100A8/A9, the subunits of calprotectin, were significantly higher in JP than IBD tissue samples in our cohort. The excessive expression of S100A8/A9 in JP in comparison with IBD may be one reason for the neutrophil rich infiltrate in JP because S100A8/A9 exhibits chemotactic function and enhances leukocyte recruitment (32) as well as exhibits an antiapoptotic effect on neutrophils (33).
To obtain further insight into the mechanisms that lead to the preferential accumulation of neutrophils and eosinophils in JP, we selectively looked for factors known to promote the recruitment, activation, and survival of these 2 cell types. CXCL8/IL-8, a chemokine supporting neutrophil migration, was previously reported to be elevated in inflamed tissue of patients with IBD (26,34). In addition, in our cohort, the pediatric IBD samples showed significantly increased CXCL8 mRNA levels in comparison with non-IBD control samples. More interestingly, CXCL8 mRNA levels in JP significantly exceeded the levels in IBD. Hence, we can speculate that JP reflects an acute and transient event in an otherwise healthy environment and that the high neutrophil influx may even prevent chronicity by avoiding or suppressing the recruitment and activation of adaptive immune cells. The cascade leading to the establishment of a local adaptive immune cell memory —and to an enhanced risk of relapsing disease —is interrupted.
Furthermore, we found significantly increased levels of TREM-1 and IL-1ß mRNAs in JP. TREM-1 is mainly expressed by neutrophils and inflammatory macrophages and amplifies the production of proinflammatory mediators, including CXCL8 (35). Hence, the activation of TREM-1 may help to sustain a neutrophil-driven inflammation in JP. IL1ß is known to be expressed not only by macrophages and neutrophils but also by gastrointestinal eosinophils (36). Thus, in JP, the accumulation of eosinophils may contribute to the observed high level of IL1ß and thereby also support neutrophil influx and survival (37).
Eosinophils were detected in high frequencies in JP based on histochemical evaluation and indirectly by the detection of increased CCR3 transcript levels. In line with this observation, we also detected high levels of CCL11 transcripts, which indicates enhanced recruitment of eosinophils in JP compared with IBD. The exact role of eosinophils in the pathogenesis of JP is so far unknown and is subject of further investigations; however, we can assume that eosinophils might support an enhanced recruitment of neutrophils through the production of CXCL5/ENA78 (38).
We also saw a significant increased CSF2/GM-CSF mRNA expression in JP compared with the other groups. CSF2 supports macrophages differentiation but also eosinophil and neutrophil survival (30). More importantly, the high levels of CSF2 transcripts in JP may also help restore immune hemostasis by limiting the activation of the adaptive immune response through the regulation of antigen presenting cells and their interactions with T cells (30). The low mRNA expression of IFNγ and CXCL10 indicates only moderate T-cell recruitment and Th1 priming (39) in JP compared with IBD. Hence, our data indicate that JP can be characterized as an isolated event with a predominant recruitment and activation of neutrophils and eosinophils in the absence of an adaptive local Th1 response.
Based on the immunohistochemical evaluation and the NanoString analysis, no difference in the frequency of CD68+ macrophages was observed. On a transcriptional level, however, we detected reduced levels of CCR2 transcripts in JP biopsies. This observation argues that in JP only limited proinflammatory CCR2+ monocytes are recruited and that the constant presence of mainly resident intestinal macrophages may result in a highly attenuated priming and expansion of CD4 T cells (40). Intriguingly, tissue-resident CD4 T cells were characteristically observed in IBD and identified as critical mediators of relapsing inflammatory disease (41). Our findings may explain the transient nature of the local inflammation in JP without progression toward a chronic relapsing disease. However, to gain a more complete understanding of the exact underlying differences in the immunological mechanisms operative in the pathogenesis of JP vs IBD, future studies will be required to directly confirm the absence of a functionally polarized, tissue-resident adaptive Tcell memory in JP, as indicated by our results.
In conclusion, we elucidate the inflammatory infiltrate in JP tissue with its unique expression of genes involved in the recruitment (S100A8/A9, CXCL8), activation (TREM1, CXCL5/ENA78), and survival (CSF2) of the innate immune cell subsets. For the clinician, it is reassuring that the limited expansion of the strong but spatially highly restricted site of inflammation in JP may be sufficient to cause a raise in calprotectin and rectal bleeding. This occurs without the formation of a local adaptive immunological memory, which may be a prerequisite for a chronic relapsing inflammatory disease, such as IBD. Furthermore, the benign etiology of JP can be explained by the composition of the specific immune infiltrate, and hence, emerging stool markers reflecting the inflammatory response may distinguish between JP and IBD before proceeding to colonoscopy in the near future.
CONFLICT OF INTEREST
Guarantor of the article: Christiane Sokollik, MD.
Specific author contributions: D.Z., M.M., C.M., and C.S.: conceptualized and designed the study. J.S., S.S., and C.S.: provided patient material. D.Z., I.Z., and C.S.: performed statistical analysis. D.Z., M.M., C.M., and C.S.: were involved in the interpretation of the results and discussion. D.Z., M.M., and C.S.: drafted the article. All authors critically revised the manuscript and approved the final version of the article.
Financial support: The project was supported by grants to C.S. (Fondazione Ettore e Valeria Rossi) and C.M. (SNSF 310030_170084).
Potential competing interests: None to declare.
Study Highlights.
WHAT IS KNOWN
✓ Solitary juvenile polyps (JPs) are benign but present with obvious signs of intestinal inflammation.
WHAT IS NEW HERE
✓ The immunological microenvironment in JP is characterized by an innate immune cell signature.
✓ No evidence for a functionally polarized T-cell response is seen in JP as is characteristic for inflammatory bowel disease.
TRANSLATIONAL IMPACT
✓ The observed immune response may explain the benign feature and low-recurrence rate of JP.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Silvia Rihs for the RNA extraction and the technical support, Fabian Mairinger for the help with the Nanostring analysis, and José Galván for the immunohistochemical stainings.
Footnotes
SUPPLEMENTARY MATERIAL accompanies this paper at http://links.lww.com/CTG/A620, http://links.lww.com/CTG/A621, http://links.lww.com/CTG/A622, http://links.lww.com/CTG/A623, http://links.lww.com/CTG/A624
Contributor Information
Daniel Zysset, Email: daniel.zysset@pathology.unibe.ch.
Matteo Montani, Email: matteo.montani@pathology.unibe.ch.
Johannes Spalinger, Email: johannes.spalinger@luks.ch.
Susanne Schibli, Email: susanne.schibli@insel.ch.
Inti Zlobec, Email: inti.zlobec@pathology.unibe.ch.
Christoph Mueller, Email: christoph.mueller@pathology.unibe.ch.
REFERENCES
- 1.Kapetanakis AM, Vini D, Plitsis G. Solitary juvenile polyps in children and colon cancer. Hepatogastroenterology 1996;43:1530–1. [PubMed] [Google Scholar]
- 2.Nugent KP, Talbot IC, Hodgson SV, et al. Solitary juvenile polyps: Not a marker for subsequent malignancy. Gastroenterology 1993;105:698–700. [DOI] [PubMed] [Google Scholar]
- 3.Thakkar K, Alsarraj A, Fong E, et al. Prevalence of colorectal polyps in pediatric colonoscopy. Dig Dis Sci 2012;57:1050–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fox VL, Perros S, Jiang H, et al. Juvenile polyps: Recurrence in patients with multiple and solitary polyps. Clin Gastroenterol Hepatol 2010;8:795–9. [DOI] [PubMed] [Google Scholar]
- 5.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012;491:119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henderson P, Casey A, Lawrence SJ, et al. The diagnostic accuracy of fecal calprotectin during the investigation of suspected pediatric inflammatory bowel disease. Am J Gastroenterol 2012;107:941–9. [DOI] [PubMed] [Google Scholar]
- 7.Olafsdottir I, Nemeth A, Lörinc E, et al. Value of fecal calprotectin as a biomarker for juvenile polyps in children investigated with colonoscopy. J Pediatr Gastroenterol Nutr 2016;62:43–6. [DOI] [PubMed] [Google Scholar]
- 8.Pauley-Hunter RJ, Kunnath S, Wolff K, et al. Fecal calprotectin and pediatric juvenile polyps. J Pediatr Gastroenterol Nutr 2015;60:e30–1. [DOI] [PubMed] [Google Scholar]
- 9.Zijlstra M, Pluimakers V, Nikkels P, et al. Elevated faecal calprotectin does not differentiate between inflammatory bowel disease and a juvenile polyp. J Pediatr Gastroenterol Nutr 2016;62:e22–3. [DOI] [PubMed] [Google Scholar]
- 10.Hessian PA, Edgeworth J, Hogg N. MRP-8 and MRP-14, two abundant ca(2+)-binding proteins of neutrophils and monocytes. J Leukoc Biol 1993;53:197–204. [PubMed] [Google Scholar]
- 11.Peterson LW, Artis D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat Rev Immunol 2014;14:141–53. [DOI] [PubMed] [Google Scholar]
- 12.Costello CM, Mah N, Häsler R, et al. Dissection of the inflammatory bowel disease transcriptome using genome-wide cdna microarrays. PLoS Med 2005;2:e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christophi GP, Rong R, Holtzapple PG, et al. Immune markers and differential signaling networks in ulcerative colitis and Crohn's disease. Inflamm Bowel Dis 2012;18:2342–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iboshi Y, Nakamura K, Ihara E, et al. Multigene analysis unveils distinctive expression profiles of helper t-cell-related genes in the intestinal mucosa that discriminate between ulcerative colitis and Crohn's disease. Inflamm Bowel Dis 2014;20:967–77. [DOI] [PubMed] [Google Scholar]
- 15.Arijs I, De Hertogh G, Machiels K, et al. Mucosal gene expression of cell adhesion molecules, chemokines, and chemokine receptors in patients with inflammatory bowel disease before and after infliximab treatment. Am J Gastroenterol 2011;106:748–61. [DOI] [PubMed] [Google Scholar]
- 16.Levine A, Koletzko S, Turner D, et al. Espghan revised porto criteria for the diagnosis of inflammatory bowel disease in children and adolescents. J Pediatr Gastroenterol Nutr 2014;58:795–806. [DOI] [PubMed] [Google Scholar]
- 17.Howell KJ, Kraiczy J, Nayak KM, et al. DNA methylation and transcription patterns in intestinal epithelial cells from pediatric patients with inflammatory bowel diseases differentiate disease subtypes and associate with outcome. Gastroenterology 2018;154:585–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hyams J, Markowitz J, Otley A, et al. Evaluation of the pediatric Crohn disease activity index: A prospective multicenter experience. J Pediatr Gastroenterol Nutr 2005;41:416–21. [DOI] [PubMed] [Google Scholar]
- 19.Turner D, Hyams J, Markowitz J, et al. Appraisal of the pediatric ulcerative colitis activity index (PUCAI). Inflamm Bowel Dis 2009;15:1218–23. [DOI] [PubMed] [Google Scholar]
- 20.Zlobec I, Suter G, Perren A, et al. A next-generation tissue microarray (ngtma) protocol for biomarker studies. J Vis Exp 2014:51893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feakins RM. Inflammatory bowel disease biopsies: Updated British society of gastroenterology reporting guidelines. J Clin Pathol 2013;66:1005–26. [DOI] [PubMed] [Google Scholar]
- 22.Oberli A, Popovici V, Delorenzi M, et al. Expression profiling with RNA from formalin-fixed, paraffin-embedded material. BMC Med Genomics 2008;1:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukunaga S, Kuwaki K, Mitsuyama K, et al. Detection of calprotectin in inflammatory bowel disease: Fecal and serum levels and immunohistochemical localization. Int J Mol Med 2018;41:107–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tarkowski M. Expression and a role of CD30 in regulation of t-cell activity. Curr Opin Hematol 2003;10:267–71. [DOI] [PubMed] [Google Scholar]
- 25.Vanhamme L, Zouaoui Boudjeltia K, Van Antwerpen P, et al. The other myeloperoxidase: Emerging functions. Arch Biochem Biophys 2018;649:1–14. [DOI] [PubMed] [Google Scholar]
- 26.Mazzucchelli L, Hauser C, Zgraggen K, et al. Expression of interleukin-8 gene in inflammatory bowel disease is related to the histological grade of active inflammation. Am J Pathol 1994;144:997–1007. [PMC free article] [PubMed] [Google Scholar]
- 27.Ina K, Kusugami K, Hosokawa T, et al. Increased mucosal production of granulocyte colony-stimulating factor is related to a delay in neutrophil apoptosis in inflammatory bowel disease. J Gastroenterol Hepatol 1999;14:46–53. [DOI] [PubMed] [Google Scholar]
- 28.Z'Graggen K, Walz A, Mazzucchelli L, et al. The c-x-c chemokine ena-78 is preferentially expressed in intestinal epithelium in inflammatory bowel disease. Gastroenterology 1997;113:808–16. [DOI] [PubMed] [Google Scholar]
- 29.Sallusto F, Mackay CR, Lanzavecchia A. Selective expression of the eotaxin receptor ccr3 by human t helper 2 cells. Science 1997;277:2005–7. [DOI] [PubMed] [Google Scholar]
- 30.Däbritz J. Granulocyte macrophage colony-stimulating factor and the intestinal innate immune cell homeostasis in Crohn's disease. Am J Physiol Gastrointest Liver Physiol 2014;306:G455–65. [DOI] [PubMed] [Google Scholar]
- 31.Malandra M, Kaur S, Chogle A. Utility of routine colonic biopsies in pediatric colonoscopic polypectomy for benign juvenile hamartomatous polyps. J Pediatr Gastroenterol Nutr 2017;64:555–8. [DOI] [PubMed] [Google Scholar]
- 32.Pruenster M, Kurz AR, Chung KJ, et al. Extracellular MRP8/14 is a regulator of beta2 integrin-dependent neutrophil slow rolling and adhesion. Nat Commun 2015;6:6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Atallah M, Krispin A, Trahtemberg U, et al. Constitutive neutrophil apoptosis: Regulation by cell concentration via s100 a8/9 and the mek-erk pathway. PLoS One 2012;7:e29333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bruno ME, Rogier EW, Arsenescu RI, et al. Correlation of biomarker expression in colonic mucosa with disease phenotype in Crohn's disease and ulcerative colitis. Dig Dis Sci 2015;60:2976–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bouchon A, Dietrich J, Colonna M. Cutting edge: Inflammatory responses can be triggered by trem-1, a novel receptor expressed on neutrophils and monocytes. J Immunol 2000;164:4991–5. [DOI] [PubMed] [Google Scholar]
- 36.Jung Y, Wen T, Mingler MK, et al. Il-1beta in eosinophil-mediated small intestinal homeostasis and iga production. Mucosal Immunol 2015;8:930–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov 2012;11:633–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Persson T, Monsef N, Andersson P, et al. Expression of the neutrophil-activating CXC chemokine ena-78/cxcl5 by human eosinophils. Clin Exp Allergy 2003;33:531–7. [DOI] [PubMed] [Google Scholar]
- 39.Bonecchi R, Bianchi G, Bordignon PP, et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 t helper cells (th1s) and th2s. J Exp Med 1998;187:129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol 2011;11:762–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zundler S, Becker E, Spocinska M, et al. Hobit- and blimp-1-driven CD4(+) tissue-resident memory t cells control chronic intestinal inflammation. Nat Immunol 2019;20:288–300. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.