

Abstract
Fragile X Syndrome (FXS) is one of the most common monogenic forms of autism and intellectual disability. Preclinical studies in animal models have highlighted the potential of pharmaceutical intervention strategies for alleviating the symptoms of FXS. However, whether treatment strategies can be tailored to developmental time windows that define the emergence of particular phenotypes is unknown. Similarly, whether a brief, early intervention can have long-lasting beneficial effects, even after treatment cessation, is also unknown. To address these questions, we first examined the developmental profile for the acquisition of associative learning in a rat model of FXS. Associative memory was tested using a range of behavioral paradigms that rely on an animal’s innate tendency to explore novelty. Fmr1 knockout (KO) rats showed a developmental delay in their acquisition of object-place recognition and did not demonstrate object-place-context recognition paradigm at any age tested (up to 23 weeks of age). Treatment of Fmr1 KO rats with lovastatin between 5 and 9 weeks of age, during the normal developmental period that this associative memory capability is established, prevents the emergence of deficits but has no effect in wild-type animals. Moreover, we observe no regression of cognitive performance in the FXS rats over several months after treatment. This restoration of the normal developmental trajectory of cognitive function is associated with the sustained rescue of both synaptic plasticity and altered protein synthesis. The findings provide proof of concept that the impaired emergence of the cognitive repertoire in neurodevelopmental disorders may be prevented by brief, early pharmacological intervention.
INTRODUCTION
Fragile X Syndrome (FXS) is a major heritable cause of intellectual disability and one of the most common single-gene causes of autism spectrum disorder (ASD), with 30 to 50% of boys clinically diagnosed with ASD (1). It affects about 1:4000 boys and 1:6000 to 8000 girls. FXS has numerous co-occurring conditions including anxiety disorders, sensory hypersensitivity, and seizures (1). FXS is usually diagnosed around 3 years of age as a result of a delay in language development (2). However, early diagnosis through genetic screening suggests early symptom development in agreement with data from cellular phenotypes in rodent models (2–4). FXS is typically caused by an expansion of a trinucleotide repeat (CGG) in the promoter region of the FMR1 gene that leads to silencing of the gene and no protein expression (5), although de novo mutations that are predicted to alter protein function also cause FXS (6, 7).
There is abundant preclinical evidence that an array of functional impairments in FXS arise from a disruption of cellular biochemistry and physiology that is correctable with pharmacological interventions [for reviews see (2, 8–10)]. Furthermore, on the basis of knowledge of critical periods in sensory system development and language acquisition (11, 12), it has been suggested that therapeutic success would be improved by starting treatments early, before major symptoms develop (13). However, it is unknown whether effective treatments would need to be maintained throughout life.
Numerous clinical trials have taken place for FXS using newly developed compound or repurposed drugs (14). There are numerous advantages of drug repurposing in the treatment of disease, including speed of translation and cost implications associated with new drug development (15). When assessing the feasibility of initiating chronic treatments in infancy, an obvious concern is safety. For these reasons, there has been interest in the possibility of repurposing drugs with a known safety profile in children. Two examples of such candidate drugs are arbaclofen, a γ-aminobutyric acid type B (GABAB) receptor agonist used for the treatment of spasticity in cerebral palsy, and lovastatin, a 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor used for the treatment of hypercholesterolemia. These compounds have been shown to correct the alterations in synaptic plasticity, neuronal morphology, neuronal excitability and behavior in a mouse model of FXS (16, 17). The precise mechanisms underlying this rescue in Fmr1 knockout (KO) mice are not known; however, increasing GABAB activation with arbaclofen alters the excitability of neurons and, via the inhibition of glutamate release, reduces the activation of the metabotropic glutamate receptor (mGluR) 5 (18)—two key cellular processes thought to underlie FXS pathophysiology (9). Lovastatin regulates the membrane association of Ras and subsequently mildly reduces the downstream activation of the extracellular signal–regulated kinases (ERKs) 1 and 2 (17). A recent human study evaluating the effects of arbaclofen in children and adolescents with FXS suggested that age may indeed be a critical variable in determining the effect of the treatment; arbaclofen had therapeutic effects in children that were lost in adolescent/adults with FXS (19). Open-label phase 2 clinical trials with lovastatin in FXS were promising (20); follow-up studies have not been reported yet (NCT02680379, NCT02998151, and NCT02642653). The combined age range in these trials is 8 to 55 years of age.
As noted above, a key question for the potential treatment of FXS is the age at which treatment should begin to maximize effectiveness. In this study, we tested the hypothesis that lovastatin treatment restricted to early development could permanently prevent impairments in cognitive function in an animal model of FXS. We chose lovastatin because it can be orally administered in food, has been shown to ameliorate circuit deficits in Fmr1 KO mice, and has a known safety profile in children (17).
Key to addressing this question is knowledge of the development of cognitive abilities in animal models. Robust behavioral paradigms independent from reward-based learning that can be used repeatedly in the same animals are therefore required. Therefore, to assess cognition across juvenile development in rats, we used a battery of spontaneous object exploration “novelty preference” tasks. These tasks are based on rodents’ innate preference to explore novel over familiar stimuli in their environment (21) and can be used to infer memory for the familiar stimulus (22). The four task variants used in the current study test the ability to discriminate between novel and familiar objects [object recognition (OR)] (22); novel and familiar configurations of objects and local contextual cues within the testing box [object-context recognition (OCR)] (23, 24); novel and familiar configurations of objects and their spatial locations within the testing box [object-place recognition (OPR)] (25); or novel and familiar configurations of objects, their spatial locations, and the local context [object-place-context recognition (OPCR)] (25). These tasks were chosen for several reasons. First, we have previously reported that, whereas adult Fmr1 KO rats show intact short-term recognition (2 to 5 min) in the OR, OCR, and OPR tasks, unlike their wild-type (WT) littermates, they do not show intact short-term memory in the OPCR task (26). Therefore, we could assess whether treatment during juvenile development would ameliorate the deficits in OPC memory in Fmr1 KO rats. Second, the tasks are based on spontaneous behavior and do not depend on the acquisition of task rules and stimulus-reward contingencies. This means that the same animal can be tested on multiple occasions (with different objects) to assess recognition memory at different ages. Third, the spontaneous exploration protocol has been used successfully in juvenile rats to characterize the ontogeny of recognition memory abilities in rats. These studies have shown that OR memory emerges before postnatal day 17 (P17) in the Long Evans Hooded (LEH) rat (27), whereas OCR emerges at around P17 or P26, depending on the types of cues used to define context (28). In contrast, OPR (29) and OPCR (30) emerge later during juvenile development. Last, short-term (2 to 5 min) recognition memory in the four different task variants described above is thought to depend on different overlapping brain circuits (31). Whereas the complete circuitry supporting recognition memory in each task is not fully resolved, it is generally agreed that the perirhinal cortex is required for OR, whereas the entorhinal cortex, hippocampus, and medial prefrontal cortex (mPFC) are not (although there is debate concerning the role of the hippocampus at longer retention intervals and its normal role in the intact brain) (23–25, 31–34). For the version of the OCR task described here (in which context is signaled by local nonpolarizing cues), the postrhinal cortex and lateral entorhinal cortex (LEC) are necessary (24, 34, 35). The LEC, together with its connections with the mPFC, is required for memory in the OPR task (33, 34), whereas for the OPCR task, the hippocampus is required, together with the LEC and its connections with mPFC (25, 32, 33).
In this study, we examine the developmental emergence of associative memory using these object exploration tasks in a rat model of FXS. We then examine whether deficits in associative memory can be prevented by early treatment with lovastatin and whether any benefits of treatment persist after treatment termination. Last, we examine whether lovastatin treatment corrects alterations at cellular levels in hippocampus and mPFC, two regions known to be involved in the tasks used in the study and known to have altered function in FXS (36, 37).
RESULTS
WT LEH rats show distinct developmental trajectories in different spontaneous object exploration tasks
As the first step to determining the emergence of cognitive abilities in rats, we conducted a longitudinal study to examine the developmental time course over which WT LEH rats exhibit nonassociative and associative recognition memory capabilities by examining their performance on a battery of four spontaneous exploration-based tasks from 4 weeks old to adulthood. In all tasks, memory is inferred on the basis of the amount of time that an animal spends, preferentially exploring the novel object or novelty based on an object’s location and/or context (Fig. 1A). At 4 to 6 weeks old, rats exhibit novelty preference in both the nonassociative OR task and the associative OCR task (Fig. 1, B and C), indicating memory of objects and object-context associations. Memory was observed at all subsequent ages tested for these tasks. In contrast, novelty preference was not observed in the associative OPR and OPCR tasks at 4 to 6 weeks of age but was present at 7 to 9 weeks and at later ages (Fig. 1, D and E). This indicates that the circuits necessary for these associative memory processes are not fully mature until 7 weeks of age. Total exploration time did not differ between time points for any task (Fig. 1, B to E).
Fig. 1. WT LEH rats show distinct developmental trajectories in different spontaneous object exploration tasks.
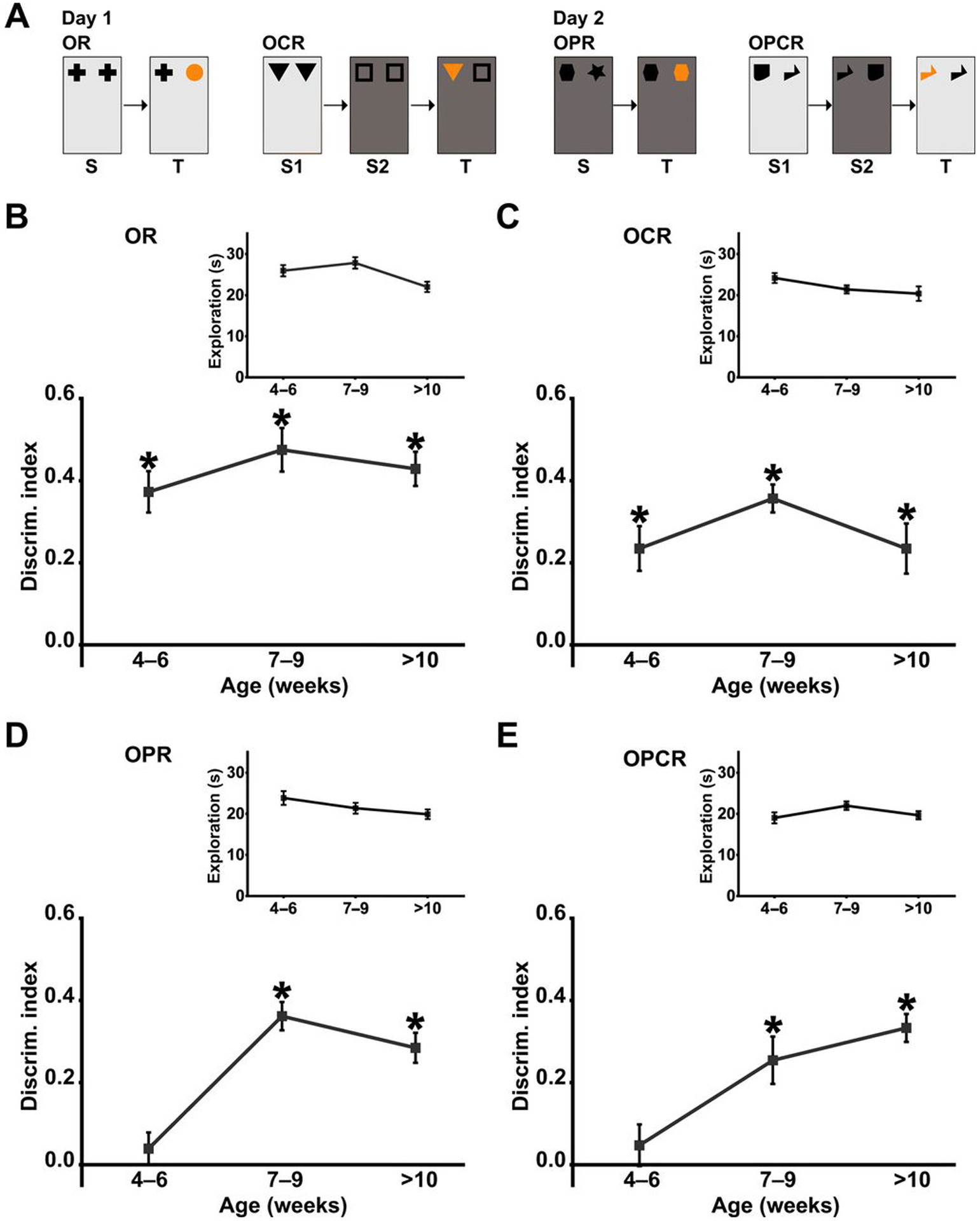
(A) Schematic of the spontaneous object exploration tasks used. S, sample phase; T, test phase. Light and dark gray backgrounds denote distinct contexts, and orange symbols denote novel object/object association. Discrimination index (DI) over time for WT LEH rats in (B) OR, (C) OCR, (D) OPR, and (E) OPCR tasks. Insets (B to E) Total object exploration time during the test phase for each task. For all tasks, n(4–6) = 13, n(7–9) = 13, n(>10) = 11; *P < 0.05 difference from chance (DI = 0) based on one-sample t tests. P values from one-sample t tests have been corrected for false discovery rate using the Benjamini-Hochberg procedure. For details on t, df, and P values for one-sample t tests, see table S1.
Loss of FMRP leads to selective deficits in OPR and OPCR memory in Fmr1 KO rats
We generated an Fmr1 KO rat line on the outbred LEH strain (Fig. 2A) to determine whether the developmental trajectory of associative memory was altered as a result of chronic deletion of the Fmr1 gene. In Fmr1 KO LEH rats, no Fmr1 transcripts were detected by reverse transcription polymerase chain reaction (RT-PCR) (Fig. 2B). This was also confirmed by RNA sequencing. Similarly, no protein was detected using either Western blotting or immunohistochemistry (Fig. 2, C and D), confirming the complete absence of Fragile X protein (FMRP). In agreement with previous findings in the mouse (33) and Sprague-Dawley (SD) rats (26), FMRP is expressed throughout postnatal development in WT LEH rats (Fig. 2E).
Fig. 2. Loss of FMRP leads to selective deficits in OPR and OPCR memory in Fmr1 KO rats.
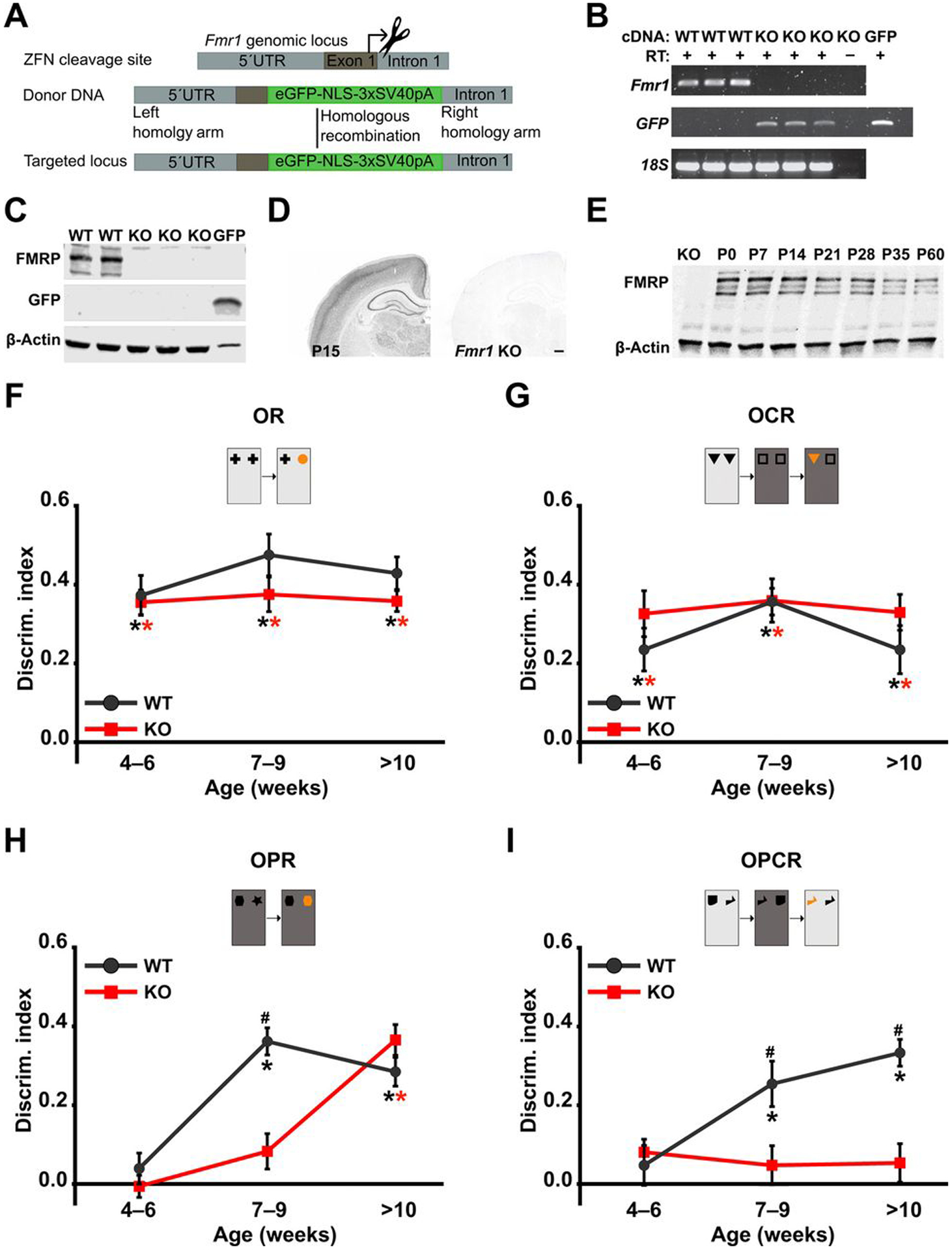
(A) Zinc finger nuclease (ZFN)–mediated disruption of Fmr1. Diagrams illustrate the target site for ZFN cleavage, donor DNA sequence including enhanced green fluorescent protein (eGFP) and a nuclear localization signal flanked by 5′ and 3′ homology recombination arms for homology-directed repair, and the resulting targeted locus (top). 5′UTR, 5′ untranslated region. (B) RT-PCR for Fmr1 and eGFP mRNA in WT and Fmr1 KO rats. Lanes 1 to 3, samples from three WT rats; lanes 4 to 6, samples from three Fmr1 KO rats; lane 7, RT control from one Fmr1 KO rat; lane 8, GFP-positive control. cDNA, complementary DNA. (C) Western blot analyses of FMRP and GFP expression in WT and Fmr1 KO rats. Lanes 1 to 2, samples from two WT rats; lanes 3 to 5, samples from three Fmr1 KO rats; lane 6, positive control for GFP. (D) Immunohistochemical localization of FMRP in P15 WT and Fmr1 KO rats. Scale bar, 500 μm. (E) Western blot analysis of FMRP expression in hippocampal homogenates from WT littermates over postnatal development compared with P15 Fmr1 KO rat. DI at different ages for WT and Fmr1 KO rats in (F) OR, (G) OCR, (H) OPR, and (I) OPCR tasks. For all tasks, nWT(4–6) = 13, nWT(7–9) = 13, nWT(>10) = 11, nKO(4–6) = 12, nKO(7–9) = 12, nKO(>10) = 11; *P< 0.05 difference from chance (DI = 0), black for WT and red for KO; #P< 0.05 difference between groups. Linear mixed effect (LME) models were fitted to the data (for details, see tables S7 and S8). P values from one-sample t tests and post hoc two-sample t tests have been controlled for the false discovery rate using the Benjamini-Hochberg procedure. For details on t, df, and P values for one-sample t tests and post hoc two-sample t tests, see table S1.
At a behavioral level, our results showed that adult (3 to 4 months old) Fmr1 KO rats on the LEH background had selective deficits in the OPCR task but intact novelty preference in the OR, OCR, and OPR tasks (fig. S1). This agrees with our previous findings in Fmr1 KO SD rats (26), indicating that this pattern of cognitive deficits is shared across outbred strains of adult rats.
Repeated testing during juvenile development (4 to 23 weeks) revealed an emergence of OR and OCR ability in Fmr1 KO LEH rats indistinguishable from that seen in WT animals (Fig. 2, F and G). In contrast, the ability of Fmr1 KO rats to show significant memory in OPR was delayed by 2 to 4 weeks compared to WT rats (Fig. 2H). As predicted from our findings in adult rats, as shown in fig. S1, (26), the Fmr1 KO rats did not exhibit OPCR memory at any age (Fig. 2I). For each of the four tasks, the total object exploration time in the sample and test phases did not differ between genotypes across different time points (fig. S2), indicating that the deficits were not secondary to changes in motivation or ability to explore objects.
Transient treatment with lovastatin restores WT-like developmental trajectory of OPR and OPCR memory in Fmr1 KO rats and has sustained effects on memory
Previous studies in the mouse model of FXS have shown that continuous, chronic treatments can correct behavioral phenotypes such as hyperactivity in an open field (17, 38), susceptibility to audiogenic seizures (16, 17), and impairments in startle response and inhibitory avoidance (38). However, the effects of a transient treatment, initiated early in postnatal life, on cognitive impairments are still mostly unexplored. We next tested whether a transient administration of the drug lovastatin to juvenile rats would correct the cognitive deficits described above. Lovastatin was chosen for three reasons. First, by inhibiting the ERK signaling pathway (fig. S3), lovastatin has been shown to correct the excessive protein synthesis, which is a core pathophysiology of FXS, and numerous cellular and circuit-based phenotypes associated with the loss of FMRP in mice. Second, lovastatin is a U.S. Food and Drug Administration–approved drug for the treatment of a familial form of hypercholesterolaemia (39) and has a known safety profile in children. Third, lovastatin can easily be administered through the diet (17).
We tested the effect of administering a 5-week oral free-feeding laboratory chow protocol, with and without lovastatin (100 mg/kg), initiated at 4 weeks, before the emergence of associative recognition in the OPR and OPCR tasks in WT rats (Fig. 3A). Treatment condition (lovastatin versus control diet) had no effect on food intake, and weight gain did not differ between genotypes or treatment conditions (fig. S4). Moreover, the total object exploration time did not differ between experimental groups (fig. S5). However, the lovastatin treatment restored the normal developmental emergence of OPR and OPCR abilities in Fmr1 KO rats without altering that of WT rats (Fig. 3, B and C). At 7 to 9 weeks of age, both WT and Fmr1 KO animals on lovastatin showed novelty preference similar to WT animals on control chow in the OPR and OPCR tasks, whereas Fmr1 KO animals on control chow showed no evidence of memory on these tasks at that age (Fig. 3D). OR and OCR abilities, which are not impaired in the Fmr1 KO rats, were not affected by the lovastatin treatment in either genotype (fig. S6). Next, to determine whether the ability of Fmr1 KO rats to perform OPCR requires continuous exposure to lovastatin, we terminated the treatment at 9 weeks of age and tested the rats again at 14 and 23 weeks of age. Treated Fmr1 KO rats maintained their ability to perform the OPCR task even at 14 and 23 weeks of age, 5 and 14 weeks after termination of lovastatin treatment, respectively (Fig. 3E). Moreover, the animals’ ability to perform the OPR remained unaffected after removal of lovastatin (see Fig. 3B). These results suggest that the restoration of the developmental trajectory of associative recognition performance by lovastatin treatment persists long after treatment termination.
Fig. 3. Transient treatment with lovastatin restores WT-like developmental trajectory of OPR and OPCR memory in Fmr1 KO rats and has sustained effects on both memory and cellular pathophysiology.
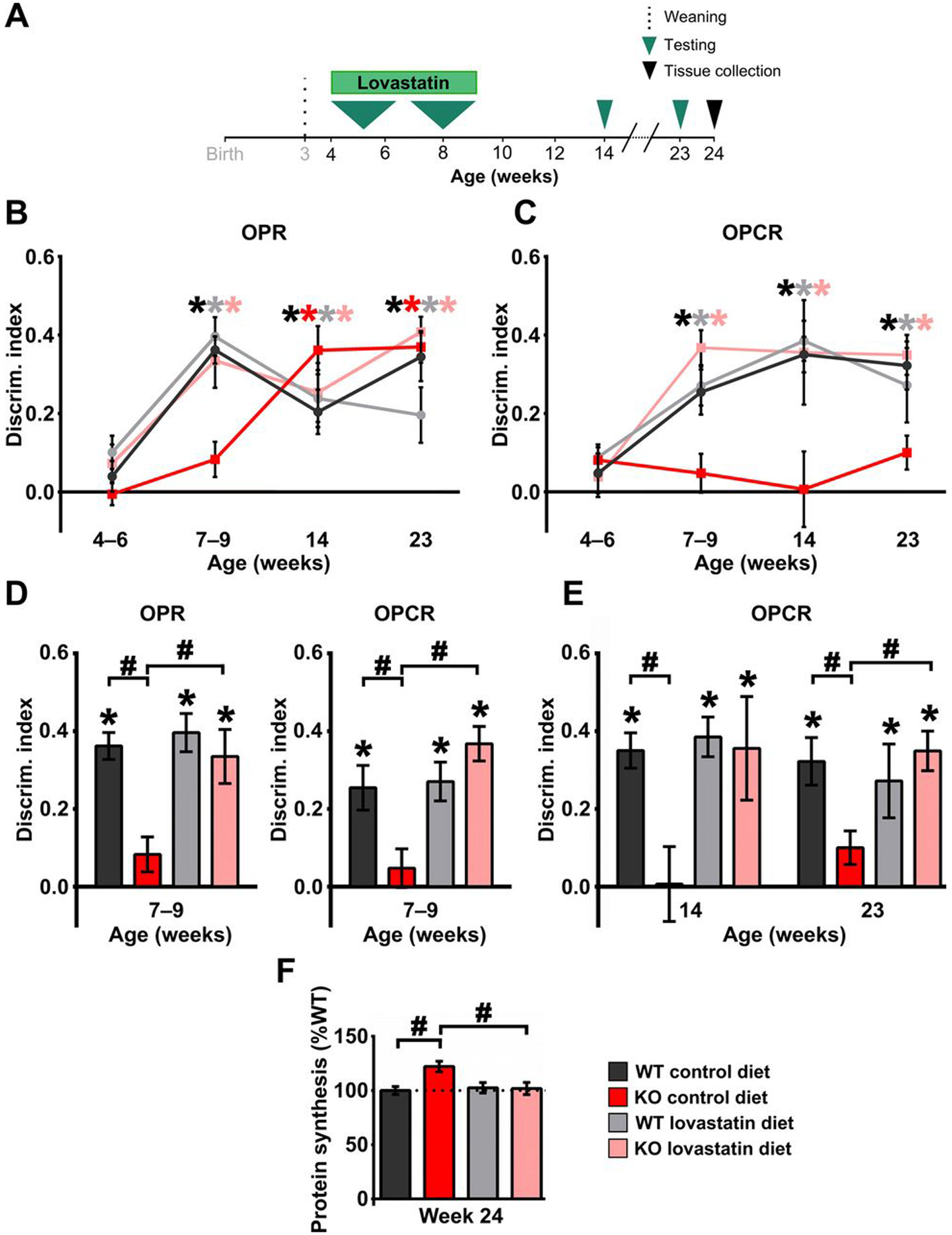
(A) Experimental time course for longitudinal assessment of cognitive development in WT and Fmr1 KO rats treated with lovastatin between 4 and 9 weeks of age. DI at different ages for WT and Fmr1 KO rats treated with control or lovastatin diet in (B) OPR and (C) OPCR tasks. (D) Effect of lovastatin treatment on OPR (left) and OPCR (right) memory tested at 7 to 9 weeks of age in WT and Fmr1 KO rats. (E) Effect of lovastatin treatment on OPCR memory in WT and Fmr1 KO rats tested at 14 and 23 weeks of age. (F) Effect of lovastatin treatment on hippocampal basal protein synthesis levels in WT and Fmr1 KO rats measured at 24 weeks of age, after behavioral testing was complete. Sample sizes: OPR 4 to 6 weeks: nWTcontrol = 13, nWTlova = 12, nKOcontrol = 12, nKOlova = 12; OPR 7 to 9 weeks: nWTcontrol = 13, nWTlova = 12, nKOcontrol = 12, nKOlova = 12; OPR 14 weeks: nWTcontrol = 10, nWTlova = 11, nKOcontrol = 11, nKOlova = 8; OPR 23 weeks: nWTcontrol = 11, nWTlova = 10, nKOcontrol = 11, nKOlova = 7; OPCR 4 to 6 weeks: nWTcontrol = 13, nWTlova = 12, nKOcontrol = 12, nKOlova = 12; OPCR 7 to 9 weeks: nWTcontrol = 13, nWTlova = 12, nKOcontrol = 12, nKOlova = 12; OPCR 14 weeks: nWTcontrol = 11, nWTlova = 11, nKOcontrol = 11, nKOlova = 8; OPCR 23 weeks: nWTcontrol = 10, nWTlova = 10, nKOcontrol = 11, nKOlova = 8; for protein synthesis, n = 6 for all groups. *P< 0.05 difference from chance (DI = 0), black for WT and red for KO; #P< 0.05 difference between groups. LMEs were fitted to the behavioral data (for details, see tables S7 and S9), and two-way analysis of variance (ANOVA) with post hoc two-sample t tests was used to analyze the effect of lovastatin on hippocampal protein synthesis levels (for details, see table S6F). P values from one-sample t tests and post hoc two-sample t tests have been controlled for the false discovery rate using the Benjamini-Hochberg procedure. For details on behavioral data t, df, and P values for one-sample t tests, see table S3, and for post hoc two-sample t tests, see table S4.
Early, brief lovastatin treatment corrects increased basal protein synthesis levels in Fmr1 KO hippocampus, 15 weeks after drug removal
We next tested whether the same brief lovastatin exposure produced sustained correction of the elevated protein synthesis that is known to result from the loss of FMRP (40–42). Because OPCR is a hippocampus-dependent task (32), we examined basal protein synthesis in hippocampal slices from the rats used in our behavioral study, harvested at 24 weeks of age. Fmr1 KO rats on control diet showed an increase in basal protein synthesis relative to WT rats even at this old age. The temporary lovastatin treatment between 4 and 9 weeks of age corrected basal protein synthesis in Fmr1 KO rats without affecting WT rats (Fig. 3F).
Lovastatin prevents the emergence of prefrontal cortex plasticity deficits associated with the loss of FMRP
We next tested whether the same regimen of brief, early lovastatin treatment could prevent the emergence of an age-dependent phenotype in synaptic plasticity associated with the loss of FMRP (43). As mPFC connections are required for OPCR [referred to by Chao et al. (33) as OCP task], and Fmr1 KO mice exhibit an age-dependent deficit in long-term potentiation (LTP) of synaptic responses in this region (43), we assessed LTP in the mPFC of Fmr1 KO rats. Our results show that, whereas LTP at layers 2 to 5 synapses of mPFC was not affected by the loss of FMRP at 4 to 6 weeks of age (Fig. 4A), LTP in Fmr1 KO rats was impaired at 10 to 12 weeks of age (Fig. 4B). Lovastatin treatment from 5 weeks of age prevented the emergence of this LTP deficit as measured in 10- to 12-week-old animals (Fig. 4C).
Fig. 4. Lovastatin prevents the emergence of plasticity deficits associated with the loss of FMRP.
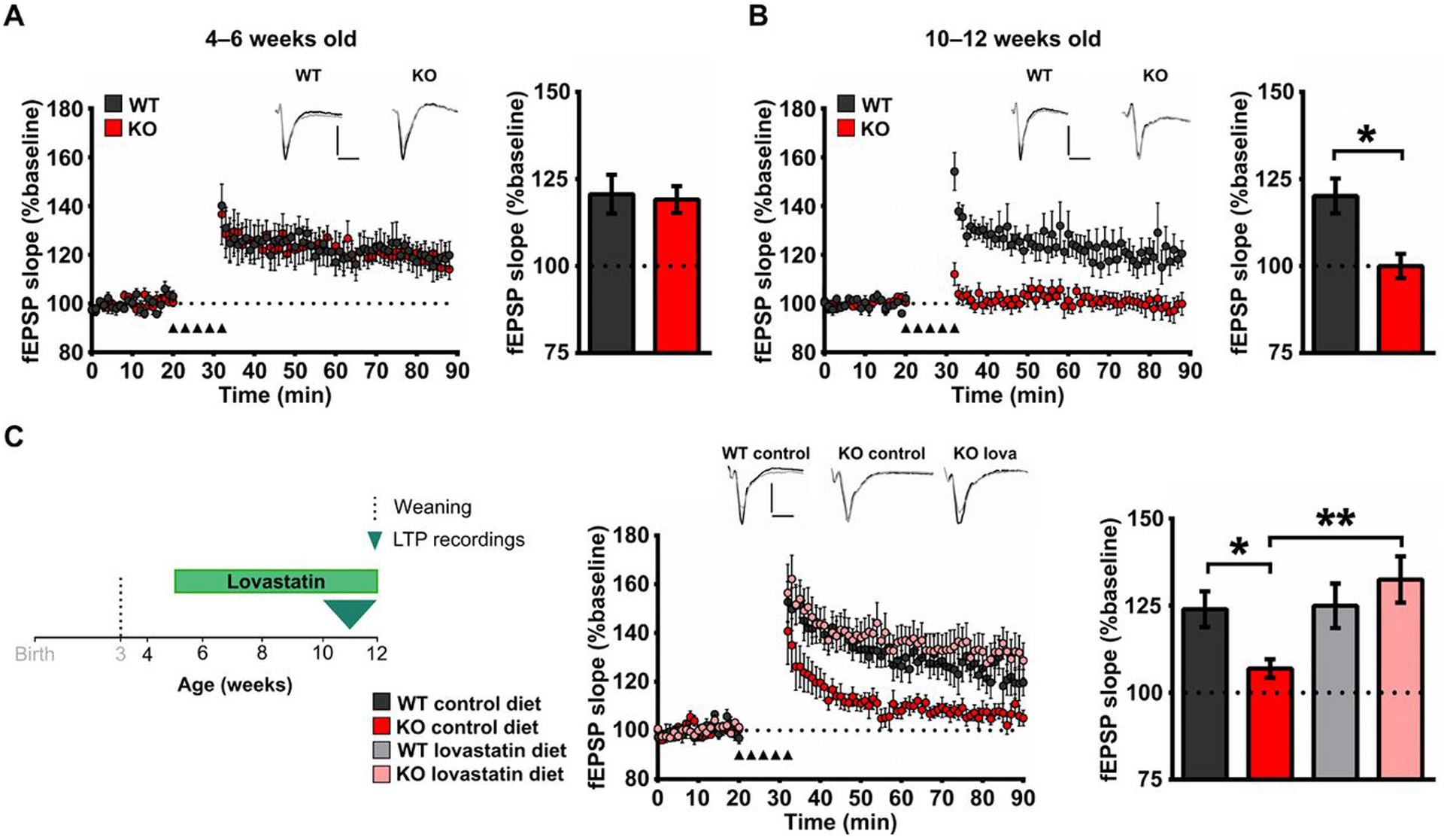
(A) Left: Time course plotting field excitatory postsynaptic potential (fEPSP) slopes normalized to baseline after LTP induction in layers 2 to 5 synapses in the prelimbic mPFC taken from 4- to 6-week-old WT and Fmr1 KO rats. Right: Averages of fEPSP slopes normalized to baseline during the last 20 min of the recording (70 to 90 min). (B) Left: Time course plotting fEPSP slopes normalized to baseline after LTP induction at synapses from layer 2/3 inputs onto layer 5 neurons in the prelimbic mPFC taken from 10- to 12-week-old WT and Fmr1 KO rats. Right: Averages of fEPSP slopes normalized to baseline during the last 20 min of the recording (70 to 90 min). (C) Left: Experimental time course for assessment of the effect of lovastatin (lova) treatment beginning at 5 weeks of age on WT and Fmr1 KO plasticity in the prelimbic mPFC. Middle: Time course plotting averages of fEPSP slopes normalized to baseline after LTP induction in layers 2 to 5 synapses in prelimbic mPFC slices taken from 10- to 12-week-old WT and Fmr1 KO treated with either control or lovastatin diet. Right: Averages of fEPSP slopes normalized to baseline during the last 20 min of recordings (70 to 90 min). Insets: Example traces showing synaptic responses during baseline (black trace) and at 80 to 90 min (gray trace). Scale bar, 0.5 mV; 5 ms. Sample sizes: LTP 4 to 6 weeks, n = 6 for WT and KO; LTP 10 to 12 weeks, n = 7 for WT and KO; for lovastatin effects on LTP, nWTcontrol = 9, nWTlova = 7, nKOcontrol = 9, nKOlova = 8; *P < 0.05; **P < 0.01 difference between groups; two-way ANOVA with post hoc two-sample t tests was used for data analyses (for details, see table S6, G and H). P values for post hoc two-sample t tests have been controlled for the false discovery rate using the Benjamini-Hochberg procedure.
Lovastatin has previously been shown to rescue the exaggerated mGluR-dependent long-term depression (LTD) found in mouse hippocampus at 1 month of age (17). To determine whether a similar effect on hippocampal LTD was present in our animals, we first examined whether LTD was exaggerated at 4 to 5 weeks and at 9 to 10 weeks (the age at which lovastatin is terminated in our experiments). Although we confirmed that 3,5-dihydroxyphenylglycine (DHPG)–induced LTD is exaggerated in Fmr1 KO LEH rats at 4 to 5 weeks of age, no difference was found at 9 to 10 weeks. Therefore, the effects of lovastatin treatment in our paradigm could not be examined. These data suggest that the downstream mechanisms supporting mGluR-dependent LTD change during postnatal development (fig. S7).
DISCUSSION
This study sought to test the hypothesis that early and transient therapeutic intervention can produce long-lasting benefits on cognition in a rat model of FXS. Our results demonstrate that early lovastatin treatment for 5 weeks, initiated before OPR and OPCR capabilities emerge, restored the normal developmental trajectory of these cognitive abilities in Fmr1 KO rats. Furthermore, the ability to perform associative recognition in these tasks persisted for at least 14 weeks after the end of treatment. These findings indicate that brief, early treatment not only prevents the emergence of cognitive deficits but also these beneficial effects on cognition are sustained long after the end of treatment. They also suggest that lovastatin treatment during an early developmental time window might rescue the normal development of the neural circuits underlying these behaviors and that FMRP, lacking in these animals, might not be needed for their maintenance.
Natural history studies of the symptomatology of individuals with neurodevelopmental disorders suggest deficits change over the lifespan. A challenge for animal studies modeling these disorders is to effectively capture developmental trajectories, especially for cognitive abilities (44, 45). Ideally, this requires longitudinal behavioral testing to assess cognitive ability across development in the same animal. In this study, we demonstrate distinct developmental trajectories for different types of recognition memory in WT LEH rats using a battery of four spontaneous exploration-based tasks. The ability to exhibit nonassociative memory for objects and associative OCR memory is apparent by 4 to 6 weeks of age [consistent with previous findings (28)]. In contrast, we find that the ability to exhibit associative OPR and OPCR memory does not emerge until 7 weeks of age [but see (30)]. Whereas the Fmr1 KO rats showed intact memory at each time point in the early-developing OR and OCR tasks, the loss of FMRP specifically affected the later-developing cognitive abilities, with a delay in the OPR task, and inability to show memory in the OPCR task at any age. OPR and OPCR require the coordination of a number of intact brain circuits, including the prefrontal cortex (32–34). The observed abnormalities in OPR and OPCR are paralleled by an age-dependent deficit in LTP in the prefrontal cortex. Our finding that LTP is intact at 4 to 6 weeks, but impaired at 10 to 12 weeks of age in Fmr1 KO rats, suggests that the role of FMRP in mediating this form of plasticity changes over postnatal development. A similar conclusion can be drawn from our finding that mGluR-dependent LTD in CA1 is increased in Fmr1 KO rats at 1 month but not 2 months of age. Future experiments to determine whether the targets of FMRP change over this period or whether the mRNAs associated with polyribosomes differ (see below) (46) should be considered.
The characterization of developmental trajectories of cognitive function in rodent models of neurodevelopmental disorders is important because it provides a temporal framework for designing experiments to determine whether potential therapeutic interventions either prevent the emergence of deficits or reverse established deficits. On the basis of this framework, we treated animals with lovastatin starting at 4 weeks of age and showed that this intervention prevented the emergence of cognitive deficits in OPR and OPCR usually seen at 7 to 9 weeks in Fmr1 KO rats. Furthermore, even after treatment was terminated at 9 weeks, these cognitive abilities remained intact for at least another 3 months (the last time point tested). This is particularly important because untreated Fmr1 KO rats were unable to show OPCR at any age tested. Lovastatin also prevented the emergence of age-dependent deficits in FMRP-dependent synaptic plasticity in the prefrontal cortex of Fmr1 KO rats.
To investigate the mechanisms underlying the effect of lovastatin treatment, we next examined basal protein synthesis in the hippocampus. The increase in protein synthesis in rat hippocampus at 4 weeks of age (26) persisted until 6 months of age and was normalized by transient lovastatin treatment from 4 to 9 weeks. This extends previous findings from the mouse model of FXS showing that the acute lovastatin application to hippocampal slices corrects deficits in basal protein synthesis (17) by demonstrating that oral lovastatin administration can reverse this deficit once it has appeared. This reversal lasts for 4 months after removal of the lovastatin, suggesting that the transient inhibition of the ERK signaling pathway is sufficient to promote a long-term reset of protein synthesis in FXS. This effect appears to be inconsistent with the idea that increased protein synthesis in the hippocampus of Fmr1 KO rats is a direct consequence of the loss of FMRP binding to their target RNAs (18). An alternative explanation is that the increased basal protein synthesis observed in Fmr1 KO hippocampus actually reflects a compensatory response to the absence of FMRP, and this is no longer needed after appropriate treatment. Such an interpretation would suggest that most of the excess signal in the protein synthesis assay arises from translation of mRNAs that are not direct FMRP targets, which is supported by our recent study (46) using the translating ribosome affinity purification assay that detects mRNAs associated with ribosomes. We found that FMRP-targeted mRNAs were underrepresented in the ribosome-bound pool in Fmr1 KO CA1 hippocampal pyramidal neurons relative to WT controls (46). Furthermore, Chrm4 mRNA, which encodes the muscarinic acetylcholine receptor 4 (M4), showed an increase in ribosome association, and stimulation of M4 normalized many Fmr1 KO phenotypes. These findings suggest that the increase in translation in Fmr1 KO neurons is a compensatory response to the deletion of FMRP rather than a direct result, at least at the ages tested in these studies (46).
Treatment with lovastatin may remove the trigger for these compensatory changes by preventing the developmental emergence of the cellular or circuit dysfunction associated with the loss of FMRP. Cellular and circuit excitability dysregulation has been shown in a range of brain regions and cell types in Fmr1 KO mice (9, 10). Because circuit activity is known to regulate early neural development, lovastatin could be exerting its disease-modifying role through its regulation of mGluR-dependent neuronal excitability. This hypothesis is supported by our previous findings that acute lovastatin application rescues both audiogenic seizures and mGluR-dependent epileptiform activity in acute brain slices in Fmr1 KO mice (17). Furthermore, acute lovastatin treatment rescued the increase in mGluR-dependent LTD in CA1 in Fmr1 KO mice (17).
One potential limitation of this study is that although there was no alteration in weight gain between genotypes or treatment group, we have not been able to measure the dose of lovastatin received by each animal. Furthermore, we have not defined whether a critical period exists for the effectiveness of lovastatin treatment or defined an effective minimum treatment duration and we have focused on associative learning paradigms that rely on an animal’s inherent attraction to novelty. Whether the beneficial effects of lovastatin can be generalized to other ages and other forms of learning is unknown, but we note that previous studies have reported this to be the case (17). Whether these findings will directly translate to clinical outcome for individuals with FXS is unknown. However, in this context, it is important to remember that our treatment regimen was initiated in 1-month-old rats. Although it is difficult to accurately estimate the corresponding age in humans, it is clear that this is much earlier than most individuals with FX enrolled in ongoing trials with lovastatin (NCT02680379, NCT02998151, and NCT02642653).
In summary, using assays of cognitive ability that rely on an animal’s natural exploratory behavior, we have been able to demonstrate that early, brief treatment restores the normal developmental trajectory of associative memory acquisition that persists well into adulthood. This rescue is paralleled by physiological and biochemical rescue in the prefrontal cortex and hippocampus, respectively. The findings provide proof-of-concept evidence that FXS, and perhaps neurodevelopmental disorders more generally, may be amenable to transient, early intervention to permanently restore normal cognitive developmental trajectories.
MATERIALS AND METHODS
Study design
The purpose of this study was to test the hypothesis that early therapeutic intervention can produce long-lasting benefits on cognition in a rat model of FXS. For behavioral studies, four groups of rats were used: WT control, WT treatment, KO control, and KO treatment; n = 12 per group based on power calculations on published data from the laboratory (32) (effect size = 1.34, α = 0.05, and power = 0.85). Littermates were housed in mixed-genotype cages (3 to 4 rats per cage), and cages were randomly assigned to control or treatment conditions [in line with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines (47)]. For the protein synthesis experiments, sample sizes were chosen on the basis of power calculations on published data from the laboratory (17, 26, 42) (effect size = 2.12, α = 0.05, and power = 0.91); WT control, WT treatment, KO control, and KO treatment (n = 6 per group; at least two slices from each rat used to produce the value for each animal) were used for metabolic labeling. In the same fashion, for in vitro electrophysiology, sample sizes were chosen on the basis of power calculations on published data from the laboratory (17, 26, 42) (effect size = 1.45, α = 0.05, and power = 0.82); WT control (n = 9), WT treatment (n = 7), KO control (n = 9), and KO treatment (n = 8) (at least two slices from each rat used to produce the value for each animal) were used as labels. Experimenters were blind to the genotype and the treatment during data collection, scoring of behavioral data, and data analysis. For behavioral testing, we excluded data from trials in which animals show very low object exploration (less than 5 s of exploration per object or 15 s of total object exploration during the sample phase, or less than 15 s of total object exploration in the test phase). No effect of genotype was observed on the number of trials excluded because of insufficient exploration. Testing was always performed during the light phase of the cycle. All animal experiments were approved by the University of Edinburgh veterinary services before their start and were performed in accordance with the guidelines established by European Community Council Directive 2010/63/EU (22 September 2010) and by the Animal Care (Scientific Procedures) Act 1986.
Animals and treatment
Male LE-Fmr1em1/PWC, hereafter referred to as Fmr1 KO, and WT littermates, bred in-house and kept in a 12-hour/12-hour light/dark cycle with ad libitum access to water and food, were used. Colony founders were produced by SAGE (Sigma Advanced Genetic Engineering) Labs using ZFN-mediated disruption of Fmr1 (48) with a targeted construct containing coding sequence for eGFP; resulting founders did not express FMRP or eGFP. Pups were weaned off their dams at P22 and housed in mixed-genotype cages with littermates (three to four animals per cage). Animals were genotyped by PCR. Ad libitum standard laboratory chow was provided until P29. On P29, the diet was changed to either control or lovastatin-enriched (100 mg/kg; Bio-Serv) diet, which was restocked and weighed once daily. At P64, animals were returned to ad libitum standard laboratory chow until the end of experiment (P164). Rats’ weight and consumption per cage were monitored throughout the dosing period (P29 to P64) to ensure that their diet did not have any adverse effects on their growth.
RNA isolation and RT-PCR
Total hippocampal RNA was isolated from 4-month-old rats (three WT and three Fmr1 KO) using the RNeasy Lipid Tissue Kit (Qiagen) as per the manufacturer’s instructions. Two micrograms of total RNA was used for cDNA synthesis using SuperScript III (Invitrogen) with oligo(dT) and random hexamers. PCR was performed using the GoTaq Green Master Mix (Promega). Fmr1 primers span exons 1 to 4 [Fmr1_e1F: CGA GGA AGG ACG AGA AGA TG and Fmr1_e4R: CAC CCT TTA TCA TCC TCA C; amplicon, 284 base pair (bp)]. Primers to GFP (GFP_F: ACG TAA ACG GCC ACA AGT TC and GFP_R: ATG CCG TTC TTC TGC TTG TC; amplicon, 421 bp) and 18S (18S_F: GTG GAG CGA TTT GTC TGG TT and 18S_R: CAA GCT TAT GAC CCG CAC TT; amplicon, 321 bp) and cDNA from a GFP transgenic mouse were used as positive controls.
Immunoblotting
Hippocampal extracts from Fmr1 KO rats and controls (n = 3 per age for developmental expression; n = 3 per genotype at P14 to verify the loss of expression) were prepared in radioimmunoprecipitation assay buffer containing protease inhibitors (cOmplete EDTA-free), immunoblotted using primary antibodies raised to the C-terminal half of FMRP (1:5000; ab69815, AbCAM), GFP (1:5000; ab6673, AbCAM), and β-actin (1:10,000; AC-74, Sigma-Aldrich) and were imaged as previously described in (36).
Immunohistochemistry
Histology was performed as previously described (49). Coronal sections were reacted with an antibody raised to the N-terminal half of FMRP (1:1500; MAB2160, Millipore).
Spontaneous object exploration tasks
Apparatus
Animals were tested in a rectangular polycarbonate testing box (76 cm long by 45 cm wide by 60 cm tall) with removable walls and floor inserts that could conform to two contexts. In context 1, white textured wallpaper and wood-effect linoleum floor were used. Two 3M Dual-Lock resealable fasteners were attached to the floor, 9 cm from the box walls at north-east and north-west locations, used to keep the two objects firmly attached to the floor in the same locations for every trial. In context 2, matte blue–painted walls and black rubber-textured floor insert were used, with holes cut to gain access to the Dual-Lock resealable fasteners where the objects were attached. The testing box was placed on a table surrounded on three sides by a black curtain, with one opening at the south side of the box (where subjects were placed). A lamp in the north-west corner and a large high contrast poster on the north-east corner were used as external cues, which remained in the same position and orientation throughout the experiments. The external environment was kept as consistent as possible, and a radio on low volume was used to mask potentially distracting noises. A variety of objects were used, which were between 8 cm by 8 cm by 8 cm and 11 cm by 11 cm by 11 cm, were nonporous, and were easily cleaned. Each object was used only once per animal. Objects were cleaned between trials with 70% ethanol solution and unscented baby wipes (Huggies).
Handling and habituation
Starting from P21 (a day before weaning), animals were handled daily in the animal house and experimental room for 7 days before experiments. Task-specific habituation was performed 2 days before experiments (P26 to P27) within the experimental apparatus box to familiarize the animals to the apparatus and type of objects. On P26, the animals were habituated to both context configurations in cage groups (30 min per context) in the morning and individually (10 min per context) in the afternoon. Between exposures to contexts, rats were placed in an opaque holding bucket. On P27, animals were individually habituated twice (morning and afternoon both context configurations each time), but this time, two different objects were fixed in the positions where they would encounter objects during testing (10 min per context, with objects). These objects were not used again during testing. During the habituation sessions, subjects were left undisturbed to explore.
Testing: General procedures
Rats were tested on four different object exploration tasks (OR, OCR, OPR, and OPCR) over a 2-day period, and testing was repeated in the same rats at different ages (P28 and P29, P35 and P36, P42 and P43, P49 and P50, P56 and P57, P63 and P64, P70 and P71, P77 and P78, P98 to P105, and P164 and P165). The general procedures for each task were the same.
For each phase of every task, the experimenter prepared the appropriate context configuration and attached two cleaned appropriate objects to the appropriate locations within the box using the Dual-Lock resealable fasteners. Each animal was then removed from the home cage and placed in the box, facing the south wall of the apparatus. The sample phases and test phases were each 3 min long, during which the animals were free to explore. An overhead black and white camera (Panasonic) was used to monitor the exploring rat around the testing box. The video signal was fed into a DVD recorder and a computer on the desk of the experimenter, which was 2 m away from the testing box. The computer ran an in-house timing software (National Instruments, LabVIEW), whereby the press of a key on the computer mouse would activate a timer. This was performed manually by the experimenter who observed the behavior of the rat via the computer screen and recorded the amount of time that the rat was engaged in exploration of each object. After 3 min, the animal was either placed in the opaque holding bucket (30 cm in diameter, containing standard bedding) for 2 min (after a sample phase) or returned to the home cage (after a test phase). Exploration was defined as the animal actively exploring an object with its snout within 2 cm of the object and performing actions such as sniffing and whisking. Exploration was not scored when the animal was not actively exploring object (i.e., climbing or resting on an object). Novel object positions, test phase contexts, context order in OCR and OPCR, and objects were counterbalanced across genotypes, tasks, and time points to ensure that the final results were as unbiased as possible.
Object recognition
OR (Fig. 1A) is a two-phase nonassociative recognition task. In the sample phase, two identical objects are available in either context 1 or context 2. In the test phase, two objects (one identical to the objects from the sample phase and one novel object) are available in the same context as the sample phase. This task is used to test whether the animal can detect object novelty and discriminate between two nonidentical objects. Higher exploration of the novel than the familiar object is indicative of memory for the familiar object.
Object-place recognition
OPR (Fig. 1A) is a two-phase associative recognition task. In the sample phase, two nonidentical objects are available in either context 1 or context 2. In the test phase, two objects (both identical to one object from the sample phase) are available in the same context as in the sample phase. This task is used to test whether an animal can associate a specific object with a location in space. Higher exploration of the object that is in a different location than it was experienced in the sample phase is indicative of OPR memory.
Object-context recognition
OCR (Fig. 1A) is a three-phase associative recognition task. In sample phase 1, two identical objects are available in either context 1 or context 2. In sample phase 2, a different pair of identical objects is explored in the other context. In the test phase, two objects (one identical to the objects from sample phase 1 and the other identical to the objects from sample phase 2) are available in either context 1 or context 2. This task is used to test whether an animal can associate an object with a surrounding context. Higher exploration of the object, which is in a different context than it was experienced in the sample phase, is indicative of OCR memory.
Object-place-context recognition
OPCR (Fig. 1A) is a three-phase associative recognition task. In sample phase 1, two nonidentical objects are available in either context 1 or context 2. In sample phase 2, objects identical to those in sample phase 1 are available, but the objects have swapped locations and are in the other context. In the test phase, two identical objects (identical to one of the objects in sample phases 1 and 2) are available in one of the two contexts. This task is used to test whether the animal can associate an object to a location in a specific surrounding context (episodic-like memory). Higher exploration of the object, which is in a different location in that context than it was in the sample phase, is indicative of OPCR memory.
In vitro electrophysiology
Medial prefrontal cortex long-term potentiation
Subjects were anesthetized using isoflurane and decapitated. The brain was quickly dissected out and placed in ice-cold (<4°C) modified slicing artificial cerebrospinal fluid (ACSF) solution (NaCl, 86 mM; NaH2PO4, 1.2 mM; KCl, 2.5 mM; NaHCO3, 25 mM; d-glucose, 24 mM; sucrose, 75 mM; CaCl2, 0.5 mM; and MgCl2, 7 mM). Coronal slices (300 μm) containing prelimbic mPFC were cut in ice-cold modified slicing ACSF solution using a vibratome and transferred to a holding chamber containing warmed recording ACSF solution (NaCl, 124 mM; NaH2PO4, 1.2 mM; KCl, 2.5 mM; NaHCO3, 25 mM; d-glucose, 20 mM; CaCl2, 2 mM; and MgCl2, 1 mM), where they were maintained at 35°C for 30 min. Slices were left to recover for a further 30 min at room temperature before the start of experimentation.
Slices were then placed in a submerged recording chamber heated to 31°C and perfused with prewarmed carbogenated recording ACSF solution at a rate of 4 to 5 ml/min. A recording electrode was placed in layer 5 of the prelimbic mPFC, and stimulating electrode was placed in either layer 2/layer 3. Stimulating and recording electrodes were staggered to prevent direct antidromic stimulation. Synaptic responses were evoked every 30 s using a bipolar nichrome stimulating wire attached to a constant current stimulus isolator, delivering a 200-μs pulse. After the acquisition of a 20-min stable baseline, LTP was induced using 5× 500-ms trains of 300-Hz stimulation at 3-min intervals. Responses were then recorded for 60 min after tetanization.
Signal waveforms were amplified 1000×, low pass–filtered at 4 kHz, and digitized at 20 kHz. fEPSP slopes were normalized to baseline values. Magnitude of LTP was calculated from 40 to 60 min after tetanus time points. Normalized data were averaged across experimental groups and reported as means ± SE.
DHPG-induced LTD
Horizontal hippocampal slices (400 μm) prepared from P21 to P32 animals were incubated in oxygenated ACSF at 31°C for 30 min and then stored at room temperature until recording. An incision was made through CA3 before recording. Slices were continuously perfused in an interface chamber with 30° ± 1°C ACSF saturated with 95% O2 and 5% CO2 at 4 to 5 ml/min. mGluR-LTD was induced using S-DHPG (50 μM) for 5 min. LTD magnitude was calculated by dividing the average fEPSP slope from 50 to 60 min after DHPG application by the average fEPSP slope during the 20-min baseline before DHPG application.
Metabolic labeling
Hippocampal slices were prepared from age-matched male WT and Fmr1 KO rats in an interleaved fashion as previously described (17, 26, 42). Briefly, hippocampi were rapidly isolated, and 500-μm slices were prepared from the dorsal half using a Stoelting tissue slicer. Slices were recovered for 4 hours in 32.5°C ACSF (NaCl, 124 mM; KCl, 3 mM; NaH2PO4, 1.25 mM; NaHCO3, 26 mM; dextrose, 10 mM; MgCl2, 1 mM; and CaCl2, 2 mM, saturated with 95% O2 and 5% CO2) and then incubated for 30 min with 25 μM ActD to block transcription. To measure protein synthesis, slices were then transferred to fresh ACSF containing 35S-Met/Cys (10 μCi/ml; PerkinElmer) and incubated for 30 min. After labeling, slices were homogenized, and labeled proteins were isolated by trichloroacetic acid precipitation. Samples were read with a scintillation counter and subjected to a protein concentration assay (Bio-Rad). Final data were expressed as counts per minute per microgram of protein, normalized to the 35S-Met/Cys ACSF used for incubation, and the average incorporation of all samples was analyzed in that experiment (eight samples per experiment: four WT and four Fmr1 KO). All aspects of the experiments were performed blinded to genotype.
Statistical analysis
For all experiments, the researchers conducting the experiments and scoring and analyzing the data were blinded to the genotype of the rats and to treatment group. For the lovastatin dosing experiments, mixed-genotype cages were randomly allocated to lovastatin and control diet groups. Statistical analysis was performed using GraphPad (Prism 6.0), SPSS 16.0 software, or R v.3.4.4 (R Core Team, 2018); scripts were written and run using RStudio 1.0.153 (RStudio Team, 2016). Because of missing data due to our exclusion criteria, LMEs were fitted to our longitudinal behavioral data using the R package lme4 v1.1–17 (50). Animal identity was included in models as a random effect, and the variables of interest were included as fixed effects. To evaluate the significance of effects using LMEs, the model without the variable of interest (a reduced/null model) was compared to the model with the variable of interest using a likelihood ratio test, where the appropriate statistical significance was assessed using either two-or three-way ANOVA. Unpaired two-sample t tests followed by Bonferroni correction were used to compare differences between groups. One-sample t tests were used to compare DIs against chance (DI = 0) controlled for the false discovery rate using the Benjamini-Hochberg procedure (51). Rats were used as an experimental unit throughout the manuscript. In electrophysiological experiments, where multiple slices were used from each animal (LTP/LTD experiments, protein synthesis), the average value for each animal was used in the analysis. Results are presented as means ± SEM. Probabilities of P < 0.05 were considered as significant. The raw data are provided in data file S1.
Supplementary Material
Fig. S1. Adult Fmr1 KO LEH rats exhibit deficits in OPCR.
Fig. S2. Object exploration time during the test phase of spontaneous recognition tasks does not differ between genotypes.
Fig. S3. Rationale of lovastatin treatment in FXS.
Fig. S4. Lovastatin has negligible effect on the weight gain of rats.
Fig. S5. Object exploration time during the test phase of spontaneous recognition tasks does not differ between genotypes with or without lovastatin treatment.
Fig. S6. WT and Fmr1 KO rats perform equally well in OR and OCR tasks throughout development with control and lovastatin diets.
Fig. S7. Fmr1 KO LEH rats exhibit age-specific increase of group I mGluR-LTD in CA1 of the hippocampus.
Table S1. Statistical results from one-sample t tests and post hoc two-sample t tests for object exploration tasks throughout development in WT and Fmr1 KO rats.
Table S2. Statistical results from two-way ANOVA of exploration times in object exploration tasks throughout development in WT and Fmr1 KO rats.
Table S3. Statistical results from one-sample t tests for object exploration tasks throughout development in WT and Fmr1 KO rats with or without lovastatin treatment.
Table S4. Statistical results from post hoc two-sample t tests for object exploration tasks throughout development in WT and Fmr1 KO rats with or without lovastatin treatment.
Table S5. Statistical results from two-way ANOVA of exploration times in object exploration tasks throughout development in WT and Fmr1 KO rats with or without lovastatin treatment.
Table S6. Statistical results from adult object exploration tasks, effect of lovastatin on food intake/weight gain, hippocampal basal protein synthesis, and synaptic plasticity data.
Table S7. LME model distribution tests of behavioral data.
Table S8. LME modeling results of WT and Fmr1 KO object exploration tasks throughout development.
Table S9. LME modeling results of WT and Fmr1 KO object exploration tasks throughout development with or without lovastatin treatment.
Acknowledgments:
We would like to thank A. Aruldass, K. Reed, and C. Neill-Edwards for assistance in behavioral data collection and colleagues in the Wood and Kind groups and the Simons Initiative for the Developing Brain for constructive discussions during the course of this study.
Funding:
A.A. was the recipient of a fellowship from the Greek State Scholarship Foundation IKY (Maria Zaousi bequest). This study was supported by grants from SFARI, The Shirley Foundation, Medical Research Council UK (MR/P006213/1), FRAXA Research Foundation, Wadhwani Foundation, The Patrick Wild Centre, Indian Department of Biotechnology, and RS Macdonald Charitable Trust. This study was also supported by the Wellcome Trust/Royal Society (Sir Henry Dale fellowship 104116/Z/14/Z) and Medical Research Council (MRC MR/M006336/1). This research was also supported, in part, by grants to M.F.B. from NIMH (R01MH106469) and the Simons Foundation (SFARI #240559).
Footnotes
Competing interests: M.F.B. holds a patent titled “Methods of treating disorders with group I mGluR antagonists” (US6890931B2) and has served as a paid consultant to Q-State Biosciences, Vertex Pharmaceuticals, and Sunovion Pharmaceuticals.
Data and materials availability: The LE-Fmr1em1/PWC rat line is available from P.C.K. under a material transfer agreement with the University of Edinburgh. All data associated with this study are present in the paper or Supplementary Materials.
SUPPLEMENTARY MATERIALS
stm.sciencemag.org/content/suppl/2019/05/24/11.494.eaao0498.DC1
Data file S1. Raw data (provided as separate Excel file).
REFERENCES AND NOTES
- 1.Kaufmann WE, Kidd SA, Andrews HF, Budimirovic DB, Esler A, Haas-Givler B, Stackhouse T, Riley C, Peacock G, Sherman SL, Brown WT, Berry-Kravis E, Autism spectrum disorder in fragile X syndrome: Cooccurring conditions and current treatment. Pediatrics 139, S194–S206 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hagerman RJ, Berry-Kravis E, Hazlett HC, Bailey DB Jr., Moine H, Kooy RF, Tassone F, Gantois I, Sonenberg N, Mandel JL, Hagerman PJ, Fragile X syndrome. Nat. Rev. Dis. Primers 3, 17065 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Harlow EG, Till SM, Russell TA, Wijetunge LS, Kind P, Contractor A, Critical period plasticity is disrupted in the barrel cortex of Fmr1 knockout mice. Neuron 65, 385–398 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He Q, Nomura T, Xu J, Contractor A, The developmental switch in GABA polarity is delayed in fragile X mice. J. Neurosci 34, 446–450 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verkerk AJMH, Pieretti M, Sutcliffe JS, Fu Y-H, Kuhl DPA, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang F, Eussen BE, van Ommen G-JB, Blonden LAJ, Riggins GJ, Chastain JL, Kunst CB, Galjaard H, Thomas Caskey C, Nelson DL, Oostra BA, Warran ST, Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914 (1991). [DOI] [PubMed] [Google Scholar]
- 6.De Boulle K, Verkerk AJMH, Reyniers E, Vits L, Hendrickx J, Van Roy B, Van Den Bos F, de Graaff E, Oostra BA, Willems PJ, A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat. Genet 3, 31–35 (1993). [DOI] [PubMed] [Google Scholar]
- 7.Zang JB, Nosyreva ED, Spencer CM, Volk LJ, Musunuru K, Zhong R, Stone EF, Yuva-Paylor LA, Huber KM, Paylor R, Darnell JC, Darnell RB, A mouse model of the human fragile X syndrome I304N mutation. PLOS Genet 5, e1000758 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sidorov MS, Auerbach BD, Bear MF, Fragile X mental retardation protein and synaptic plasticity. Mol. Brain 6, 15 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Contractor A, Klyachko VA, Portera-Cailliau C, Altered neuronal and circuit excitability in fragile X syndrome. Neuron 87, 699–715 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wijetunge LS, Chattarji S, Wyllie DJA, Kind PC, Fragile X syndrome: From targets to treatments. Neuropharmacology 68, 83–96 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Werker JF, Hensch TK, Critical periods in speech perception: New directions. Annu. Rev. Psychol 66, 173–196 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Sengpiel F, Kind PC, The role of activity in development of the visual system. Curr. Biol 12, R818–R826 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Meredith RM, Sensitive and critical periods during neurotypical and aberrant neurodevelopment: A framework for neurodevelopmental disorders. Neurosci. Biobehav. Rev 50, 180–188 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Berry-Kravis EM, Lindemann L, Jønch AE, Apostol G, Bear MF, Carpenter RL, Crawley JN, Curie A, Des Portes V, Hossain F, Gasparini F, Gomez-Mancilla B, Hessl D, Loth E, Scharf SH, Wang PP, Von Raison F, Hagerman R, Spooren W, Jacquemont S, Drug development for neurodevelopmental disorders: Lessons learned from fragile X syndrome. Nat. Rev. Drug Discov 17, 280–299 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tranfaglia MR, Thibodeaux C, Mason DJ, Brown D, Roberts I, Smith R, Guilliams T, Cogram P, Repurposing available drugs for neurodevelopmental disorders: The fragile X experience. Neuropharmacology 147, 74–86 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Henderson C, Wijetunge L, Kinoshita MN, Shumway M, Hammond RS, Postma FR, Brynczka C, Rush R, Thomas A, Paylor R, Warren ST, Vanderklish PW, Kind PC, Carpenter RL, Bear MF, Healy AM, Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci. Transl. Med 19, 152ra128 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Osterweil EK, Chuang S-C, Chubykin AA, Sidorov M, Bianchi R, Wong RKS, Bear MF, Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron 77, 243–250 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bear MF, Huber KM, Warren ST, The mGluR theory of fragile X mental retardation. Trends Neurosci 27, 370–377 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Berry-Kravis E, Hagerman R, Visootsak J, Budimirovic D, Kaufmann WE, Cherubini M, Zarevics P, Walton-Bowen K, Wang P, Bear MF, Carpenter RL, Arbaclofen in fragile X syndrome: Results of phase 3 trials. J. Neurodev. Disord 9, 3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Çaku A, Pellerin D, Bouvier P, Riou E, Corbin F, Effect of lovastatin on behavior in children and adults with fragile X syndrome: An open-label study. Am. J. Med. Genet. A 164, 2834–2842 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Berlyne DE, Novelty and curiosity as determinants of exploratory behaviour. Br. J. Psychol. Gen. Sect 41, 68–80 (1950). [Google Scholar]
- 22.Ennaceur A, Delacour J, A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav. Brain Res 31, 47–59 (1988). [DOI] [PubMed] [Google Scholar]
- 23.Dix SL, Aggleton JP, Extending the spontaneous preference test of recognition: Evidence of object-location and object-context recognition. Behav. Brain Res 99, 191–200 (1999). [DOI] [PubMed] [Google Scholar]
- 24.Norman G, Eacott MJ, Dissociable effects of lesions to the perirhinal cortex and the postrhinal cortex on memory for context and objects in rats. Behav. Neurosci 119, 557–566 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Eacott MJ, Norman G, Integrated memory for object, place, and context in rats: A possible model of episodic-like memory? J. Neurosci 24, 1948–1953 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Till SM, Asiminas A, Jackson AD, Katsanevaki D, Barnes SA, Osterweil EK, Bear MF, Chattarji S, Wood ER, Wyllie DJA, Kind PC, Conserved hippocampal cellular pathophysiology but distinct behavioural deficits in a new rat model of FXS. Hum. Mol. Genet 24, 5977–5984 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Westbrook SR, Brennan LE, Stanton ME, Ontogeny of object versus location recognition in the rat: Acquisition and retention effects. Dev. Psychobiol 56, 1492–1506 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramsaran AI, Westbrook SR, Stanton ME, Ontogeny of object-in-context recognition in the rat. Behav. Brain Res 298, 37–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ainge JA, Langston RF, Ontogeny of neural circuits underlying spatial memory in the rat. Front. Neural Circuits 6, 8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramsaran AI, Sanders HR, Stanton ME, Determinants of object-in-context and object-place-context recognition in the developing rat. Dev. Psychobiol 58, 883–895 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Brown MW, Aggleton JP, Recognition memory: What are the roles of the perirhinal cortex and hippocampus? Nat. Rev. Neurosci 2, 51–61 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Langston RF, Wood ER, Associative recognition and the hippocampus: Differential effects of hippocampal lesions on object-place, object-context and object-place-context memory. Hippocampus 20, 1139–1153 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Chao OY, Huston JP, Li J-S, Wang A-L, de Souza Silva MA, The medial prefrontal cortex—Lateral entorhinal cortex circuit is essential for episodic-like memory and associative object-recognition. Hippocampus 26, 633–645 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Wilson DIG, Watanabe S, Milner H, Ainge JA, Lateral entorhinal cortex is necessary for associative but not nonassociative recognition memory. Hippocampus 23, 1280–1290 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilson DIG, Langston RF, Schlesiger MI, Wagner M, Watanabe S, Ainge JA, Lateral entorhinal cortex is critical for novel object-context recognition. Hippocampus 23, 352–366 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bostrom C, Yau S. -y., Majaess N, Vetrici M, Gil-Mohapel J, Christie BR, Hippocampal dysfunction and cognitive impairment in Fragile-X Syndrome. Neurosci. Biobehav. Rev 68, 563–574 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Hooper SR, Hatton D, Sideris J, Sullivan K, Ornstein PA, Bailey DB Jr., Developmental trajectories of executive functions in young males with fragile X syndrome. Res. Dev. Disabil 81, 73–88 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG, Jaeschke G, Bear MF, Lindemann L, Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74, 49–56 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lambert M, Lupien P-J, Gagné C, Lévy E, Blaichman S, Langlois S, Hayden M, Rose V, Clarke JTR, Wolfe BMJ, Clarson C, Parsons H, Stephure DK, Potvin D, Lambert J, Canadian Lovastatin in Children Study Group, Treatment of familial hypercholesterolemia in children and adolescents: Effect of lovastatin. Pediatrics 97, 619–628 (1996). [PubMed] [Google Scholar]
- 40.Dölen G, Osterweil E, Rao BSS, Smith GB, Auerbach BD, Chattarji S, Bear MF, Correction of fragile X syndrome in mice. Neuron 56, 955–962 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Osterweil EK, Krueger DD, Reinhold K, Bear MF, Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J. Neurosci 30, 15616–15627 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barnes SA, Wijetunge LS, Jackson AD, Katsanevaki D, Osterweil EK, Komiyama NH, Grant SGN, Bear MF, Nägerl UV, Kind PC, Wyllie DJA, Convergence of hippocampal pathophysiology in Syngap+/− and Fmr1−/y mice. J. Neurosci 35, 15073–15081 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martin HGS, Lassalle O, Brown JT, Manzoni OJ, Age-dependent long-term potentiation deficits in the prefrontal cortex of the Fmr1 knockout mouse model of fragile X syndrome. Cereb. Cortex 26, 2084–2092 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Lai JKY, Sobala-Drozdowski M, Zhou L, Doering LC, Faure PA, Foster JA, Temporal and spectral differences in the ultrasonic vocalizations of fragile X knock out mice during postnatal development. Behav. Brain Res 259, 119–130 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Yun S-W, Platholi J, Flaherty MS, Fu W, Kottmann AH, Toth M, Fmrp is required for the establishment of the startle response during the critical period of auditory development. Brain Res 1110, 159–165 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Thomson SR, Seo SS, Barnes SA, Louros SR, Muscas M, Dando O, Kirby C, Wyllie DJA, Hardingham GE, Kind PC, Osterweil EK, Cell-type-specific translation profiling reveals a novel strategy for treating fragile X syndrome. Neuron 95, 550–563.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br. J. Pharmacol 160, 1577–1579 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM, Jenkins SS, Wood A, Cui X, Meng X, Vincent A, Lam S, Michalkiewicz M, Schilling R, Foeckler J, Kalloway S, Weiler H, Ménoret S, Anegon I, Davis GD, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Jacob HJ, Buelow R, Knockout rats via embryo microinjection of zinc-finger nucleases. Science 325, 433 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Till SM, Wijetunge LS, Seidel VG, Harlow E, Wright AK, Bagni C, Contractor A, Gillingwater TH, Kind PC, Altered maturation of the primary somatosensory cortex in a mouse model of fragile X syndrome. Hum. Mol. Genet 21, 2143–2156 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Bates D, Mächler M, Bolker B, Walker S, Fitting linear mixed-effects models using lme4. J. Stat. Softw 67, 1–48 (2015). [Google Scholar]
- 51.Benjamini Y, Hochberg Y, Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B 57, 289–300 (1995). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Adult Fmr1 KO LEH rats exhibit deficits in OPCR.
Fig. S2. Object exploration time during the test phase of spontaneous recognition tasks does not differ between genotypes.
Fig. S3. Rationale of lovastatin treatment in FXS.
Fig. S4. Lovastatin has negligible effect on the weight gain of rats.
Fig. S5. Object exploration time during the test phase of spontaneous recognition tasks does not differ between genotypes with or without lovastatin treatment.
Fig. S6. WT and Fmr1 KO rats perform equally well in OR and OCR tasks throughout development with control and lovastatin diets.
Fig. S7. Fmr1 KO LEH rats exhibit age-specific increase of group I mGluR-LTD in CA1 of the hippocampus.
Table S1. Statistical results from one-sample t tests and post hoc two-sample t tests for object exploration tasks throughout development in WT and Fmr1 KO rats.
Table S2. Statistical results from two-way ANOVA of exploration times in object exploration tasks throughout development in WT and Fmr1 KO rats.
Table S3. Statistical results from one-sample t tests for object exploration tasks throughout development in WT and Fmr1 KO rats with or without lovastatin treatment.
Table S4. Statistical results from post hoc two-sample t tests for object exploration tasks throughout development in WT and Fmr1 KO rats with or without lovastatin treatment.
Table S5. Statistical results from two-way ANOVA of exploration times in object exploration tasks throughout development in WT and Fmr1 KO rats with or without lovastatin treatment.
Table S6. Statistical results from adult object exploration tasks, effect of lovastatin on food intake/weight gain, hippocampal basal protein synthesis, and synaptic plasticity data.
Table S7. LME model distribution tests of behavioral data.
Table S8. LME modeling results of WT and Fmr1 KO object exploration tasks throughout development.
Table S9. LME modeling results of WT and Fmr1 KO object exploration tasks throughout development with or without lovastatin treatment.