

INTRODUCTION
Diamond-Blackfan anemia (DBA) is a congenital red blood cell aplasia that usually presents during the first year of life. Only 10% of patients are anemic at birth and 80% by 6 months. Although DBA may present at any age, it is considered a disease affecting infants.
The main hematologic features of DBA are severe normochromic and macrocytic anemia, reticulocytopenia, an increase in erythrocyte adenosine deaminase, and erythroid hypoplasia in the bone marrow, with preservation of other lineages. Other developmental abnormalities, most commonly affecting the head, upper limb, kidneys, heart, and eyes, are present in about 40% of cases.
Erythroid failure is characterized by a marked reduction in erythroid precursors and their progenitors, the erythroid burst-forming unit (BFU-E) and colony-forming unit (CFU-E).1 Persistent macrocytosis and increased HbF and erythrocyte “i” antigen expression in patients are features of stress erythropoiesis that is more fetal than adult in nature.2,3 The disease has been associated with point mutations and large deletions in 18 ribosomal protein (RP) genes in about 60% to 65% of patients.4–7 Mutations are also rarely present in two non-RP genes (GATA1, an essential erythroid transcription factor, and TSR2).7
RP haploinsufficiency accounts for most cases of DBA, and an intriguing question is why deficiency of RPs, structural components of ribosomes expressed in all nucleated cells, leads to the specific erythroid and other congenital defects characteristic of DBA. There is good evidence that ribosomal stress leads to free RPs sequestering MDM2, resulting in p53 stabilization and consequent cell cycle arrest or apoptosis, but this does not explain the tissue specificity of disease manifestations. There are two leading hypotheses: the first postulates the importance of specialized (tissue specific) ribosomes that comprise different subsets of RPs that are critical for the translation of specific mRNAs. Support for this theory comes from tissue-specific expression of eL38 in Ts mice that show surprising patterning defects caused by perturbed translation of specific homeobox mRNAs of eL38-deficient tissues. However,8 there is no evidence to date that indicates different ribosome composition in erythroid compared with nonerythroid cells. The second hypothesis posits that ribosome deficiency adversely affects the translation of complex structured mRNAs that have low initiation rates (Fig. 1). Compelling evidence for this theory comes from the demonstration that GATA1 translation is impaired in DBA patients with different RP gene mutations because ribosomes are limiting,9 and that cells in which DBA-associated genes have been knocked down do not have altered ribosome composition compared with their wild-type counterparts, but do have overall decreased levels of ribosomes.10 GATA1 has a complex 5′ UTR that predicts poor translation initiation rates, and such mRNAs are more sensitive to ribosome deficiency than mRNAs with high initiation rates (for an excellent review see11).
Fig. 1.
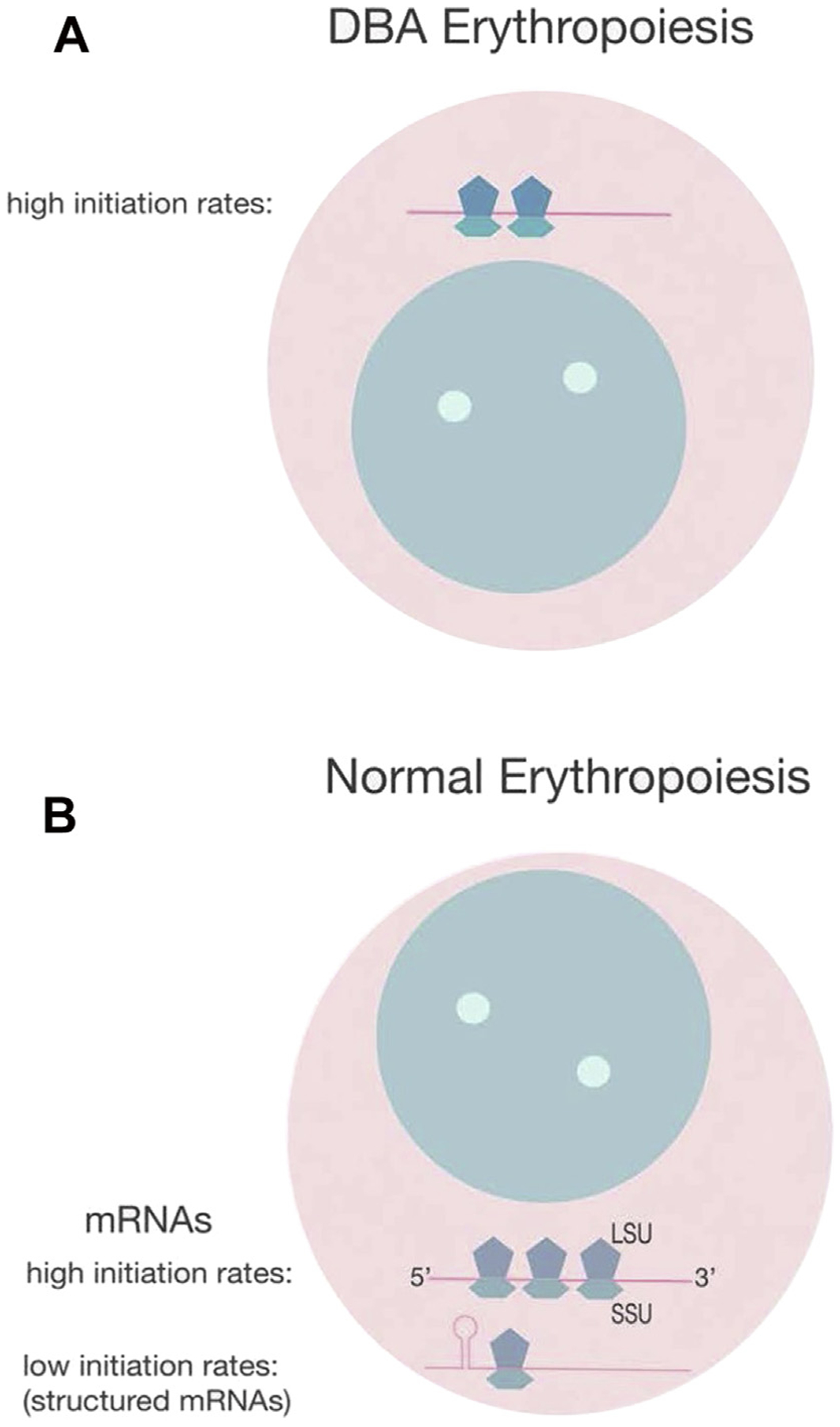
Effect of ribosome availability on mRNA translation. (A) In DBA ribosome structure is normal but ribosomes are reduced (haploinsufficiency), and these cells only translate mRNAs with high initiation rates, such as RPs and hemoglobin, but not mRNAs with low initiation rates, such as those with a structured 5′ open reading frame, such as GATA1, an upstream AUG, or an internal ribosome entry site. LSU, large subunit; SSU, small subunit. (B) Normal erythroid progenitors with a full complement of ribosomes can translate mRNAs with high and low initiation rates (kis).
This article focuses on current issues in the management of patients with DBA and on prospects for novel treatment approaches.
STEROIDS
It is sobering to realize that treatment of DBA with corticosteroids, observed by Gasser in 1951,12 is still the only medication effective in clinical practice. Approximately 75% of patients respond to treatment with an increase in reticulocytes and hemoglobin. Although some patients remain steroid dependent and can be maintained on a tolerably low dose (usually a maximum of 0.5 mg/kg/d prednisone), others require too high a dose of steroids and have to be maintained on regular blood transfusions to avoid steroid toxicity, which includes pathologic fractures, avascular necrosis, cataracts, growth retardation, hypertension, and diabetes. Close monitoring is necessary to avoid these complications because their detection requires a steroid taper and switch to transfusion therapy.13 Although the molecular mechanism of action of corticosteroids in DBA patients has not been completely elucidated, studies of normal mouse erythroid development demonstrate that corticosteroids are able to increase BFU-E cell numbers by stimulating self-renewal, thereby expanding the progenitor cell population first lost in DBA.14 Approximately 40% of patients remain steroid responsive; 40% become steroid refractory, require blood transfusions and are eligible for a hematopoietic stem cell transplant (HSCT); and 20% remit and do not require medication.15
HEMATOPOIETIC STEM CELL TRANSPLANTATION
The only cure for DBA is stem cell transplantation, and excellent results have been obtained for patients aged less than 10 years with HLA-matched allogeneic sibling donors (>90% survival).16 Published results for unrelated HLA-matched transplants have not been as good, in particular for patients transplanted before 2000 and for those older than 10 years.17,18 However, advances in molecular tissue typing and transplantation of young patients to avoid the deleterious effects of iron overload have led to improved results. Table 1 shows several recently published single center studies. Although numbers are still low and length of follow-up limited, all patients were alive at the time of reporting after either myeloablative or reduced intensity conditioning without undue infectious, graft-versus-host disease complications, or failure of engraftment.
Table 1.
Unrelated donor transplantation for Diamond-Blackfan anemia
| # | Age (mo) | Conditioning | HSCT Source | Acute GVHD | Chronic GVHD | Cx | FU (mo) | Ref |
|---|---|---|---|---|---|---|---|---|
| MAC | ||||||||
| 1.1 | 24 | BU/CY/ATG | MUD/PBSC | II | No | CMV react | A,7 | Li et al,45 2017 |
| 1.2 | 42 | “ | MMSD-9/10/PBSC | No | Ltd | Infection | A,14 | |
| 1.3 | 72 | “ | MUD/PBSC | No | No | HC, VOD | A,31 | |
| 1.4 | 10 | “ | UCB-6/6 | II | No | VOD | A,31 | |
| 1.5 | 18 | “ | UCB-5/6 | II | No | No | A,33 | |
| 1.6 | 18 | “ | UCB-6/6 | II | No | No | A,76 | |
| 1.7 | 20 | “ | MUD/PBSC | III | Ltd | ICH, OVD | A,130 | |
| RIC | ||||||||
| 2.1 | 74 | Treo/Flu/rATG | MUD/BM | + | + | AIHA | A,54 | Burroughs et al,46 2017 |
| 2.2 | 146 | “ | MUD/BM | + | No | A,48 | ||
| 2.3 | 263 | “ | MUD/BM | No | No | A,36 | ||
| 2.4 | 27 | “ | MUD/BM | + | No | A,3 | ||
| 3.1 | 9 | Treo/Flu/Thio | MSD | II + 12 | No | A,48 | Crazzolara et al,47 2016 | |
| 3.2 | 7 | “ | MUD | I | A,37 | |||
| 3.3 | 13 | Flu/Thio | MUD | A,124 | ||||
| 4 | 14 | Al/Flu/M | MUD | I | A,21 | Asquith et al,48 2015 |
Abbreviations: A, alive; AIHA, autoimmune hemolytic anemia; BM, bone marrow; CMV, cytomegalovirus; Cx, Complications; FU, follow-up; GVHD, graft-versus-host disease; HC, hemorrhagic cystitis; ICH, intracranial hemorrhage; Ltd, Limited; MUD, matched unrelated donor; MMSD, mismatched sibling donor; MSD, matched sibling donor; OVD, obstructive ventilation disorder; PBSC, peripheral blood stem cells; UCB, umbilical cord blood; VOD, veno-occlusive disease.
Myeloablative Conditioning (MAC): Busufan (BU) 16 mg/kg, Cyclophosphamide 200 mg/kg, rabbit antithymocyte globulin (rATG) 10 mg/kg. Reduced Intensity Conditioning (RIC): Treosulfan 42 mg/m2 (Treo), Fludarabine (Flu) 150 mg/m2, rabbit Antithymocyte Globulin (rATG) 6 mg/kg, Treosulfan (Treo) 56 g/m2, Flu 8mg/m2, Thiotepa (Thio) 160 mg/m2, Alemtuzumab (Al) 45 mg, Flu 150 mg/m2, Melphalan (M) 140 mg/m.
TRANSFUSIONAL HEMOSIDEROSIS
One critical and controversial question is whether DBA patients are more susceptible to early and more severe iron toxicity than other chronically transfused patients. Most patients with DBA are diagnosed during the first few months of life. They frequently present with severe anemia and require an immediate blood transfusion after diagnostic tests have been obtained. The standard of care is to continue blood transfusions for 1 year, because the early use of steroids can impair growth, which is already a concern in patients with DBA. Although 70% to 80% of patients respond to steroids, only half are maintained long term on sufficiently low doses to avoid toxicity. Patients who fail to respond and those who become resistant to steroids require monthly blood transfusions. They are eligible for HSCT if a suitable donor is available and parents choose to pursue this path.
Few studies have compared the effects of chronic transfusions in DBA with other transfused patients, such as those with β-thalassemia major (TM) and sickle cell disease (SCD). A case control study of 31 transfusion-dependent DBA patients and a comparable group of β-thalassemia patients on deferoxamine showed a significantly increased liver iron concentration with a high rate of organ toxicity in DBA, and raised the possibility that DBA patients may be more susceptible to organ toxicity than those with thalassemia.3 However, compliance with the chelation regime was poor, so it was impossible to determine whether these results might have been caused by a difference in biology or by poor compliance. To evaluate the rate of iron loading in 124 children with transfusion-dependent anemias including DBA, TM, congenital dyserythropoietic anemia (CDA), and SCD, Berdoukas and colleagues19 retrospectively evaluated initial MRIs, done before or soon after the start of chelation, and subsequent liver, cardiac, and pancreas R2* MRIs. All initial MRI R2* values for liver iron concentration in patients younger than 10 years were increased and showed no significant differences among diseases. Strikingly, initial and subsequent pancreatic MRIs showed that 50% of subjects with DBA developed pancreatic R2* greater than 100 Hz, compared with 25% CDA or TM and 2.5% with SCD, predicting a risk of glucose intolerance. Similarly, 25% of DBA and CDA patients developed increased cardiac iron (R2* >50 Hz), compared with 10% of those with TM; 2 of the 17 DBA children in the study showed the increase at the young ages of 2.5 and 3.7 years.
Extrahepatic iron deposition is related to an increase in nontransferrin bound iron (NTBI), which includes the toxic redox reactive labile plasma iron component. NTBI levels increase when transferrin saturation exceeds 60% to 70%. Mechanisms of NTBI generation were examined in DBA, TM, and SCD patients with matched transfusion histories by comparing the key parameters of iron metabolism.20 A comparison of hepcidin levels in transfusion-dependent DBA, TM, and SCD shows that hepcidin levels are high in DBA. Does the regulation of hepcidin play a critical role in the extrahepatic iron deposition seen at an early age in DBA?
Although there is no physiologic mechanism for excreting the excess iron acquired by repeated red cell transfusions, hepcidin plays a critical role in iron transport regulation by controlling iron absorption and recirculation through binding to ferroportin, which exports iron into plasma from the basolateral side of duodenal enterocytes and from macrophages and hepatocytes. This leads to ferroportin ubiquitination and degradation. Hepcidin synthesis is regulated by three pathways, iron availability, inflammation, and erythropoiesis:
Iron availability: when iron levels are high, excess transferrin-bound iron (Tf-Fe2) displaces the hemochromatosis protein HFE from the high affinity TFR1 to a complex with the lower affinity TFR2 and hemojuvelin. This complex mediates BMP6 signaling through ALK3 and BMPR2 to activate R-SMAD, SMAD4, and an increase in hepcidin transcription (Fig. 2). Ferroportin is also important for cells that do not contribute to plasma iron, such as cardiomyocytes.8
Inflammation is characterized by increased levels of cytokines, such as interleukin-6 and activin B. Interleukin-6 leads to high hepcidin expression through the interleukin-6 receptor, which activates JAK and STAT3 to increase hepcidin transcription. Activin B can stimulate hepcidin through the BMP6 pathway (see Fig. 2).
When erythropoiesis is increased the requirement for iron is met by an increase in dietary absorption from enterocytes and release of iron from hepatic and reticulo-endothelial stores through inhibition of hepcidin transcription and stabilization of ferroportin. Hepcidin repression depends on erythropoietin (EPO) and a functional bone marrow.21 In mice there is good evidence that EPO induces erythroid cells via JAK2/STAT5 to secrete a long sought erythroid regulator named erythroferrone, because Erf−/−animals, in contrast to wild-type, fail to inhibit hepcidin in response to hemorrhage or EPO injections.22 Ablation of Erfe in thalassemic mice leads to restoration of hepcidin mRNA and reduction in liver and spleen iron content.23 The role of erythroferrone in human erythropoiesis is uncertain, as is the signaling pathway. Growth differentiation factor-15 has also been implicated as a protein secreted by erythroblasts in the ineffective erythropoiesis that characterizes TM24 and CDA type 1,25 although these data are controversial.26
Fig. 2.
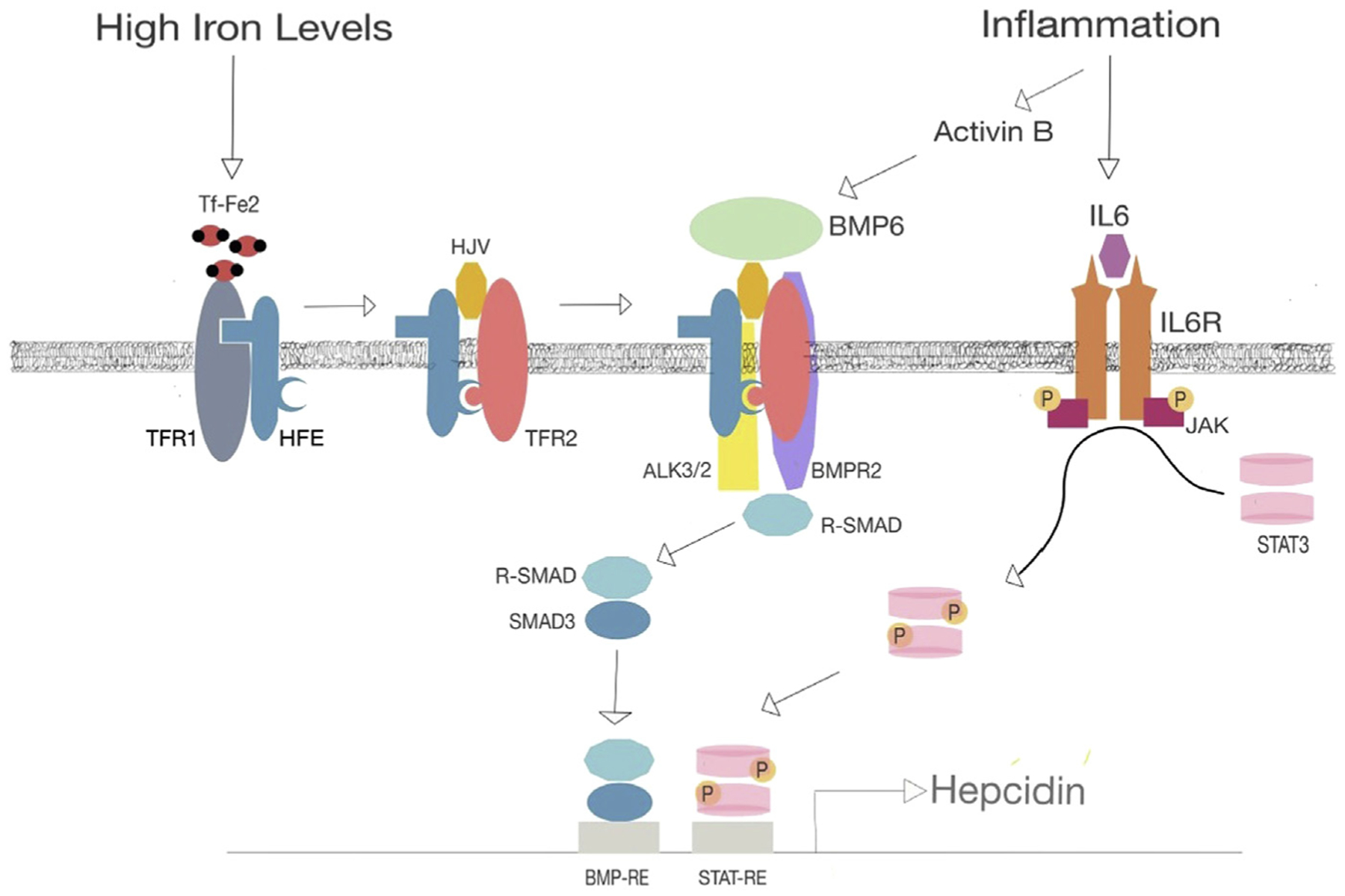
Regulation of hepcidin transcription. DBA patients frequently have high hepcidin levels, higher than other transfusion-dependent anemias, and can load iron rapidly, including the liver, heart, and endocrine organs, such as the pancreas. Increased hepcidin transcription occurs through high concentrations of Tf-Fe2, which displaces HFE from TFR1. HFE forms a complex TFR2 and hemojuvelin, which augments BMP6 activation of serine threonine kinase receptors ALK3/2 and BMPR2 leading to phosphorylation of receptor-activated SMAD (R-SMAD) and hepcidin transcription. Inflammation can lead to increased activin B mediated BMP6 signaling and interleukin (IL)-6 expression, activation of the IL-6 receptor (IL6R), and recruitment/phosphorylation of JAK tyrosine kinase, which in turn phosphorylates STAT3 to augment hepcidin transcription. Hepcidin binds to ferroportin, leading to ubiquitination and degradation of this iron transporter expressed on hepatocytes, macrophages, and cardiomyocytes.
In DBA, hepcidin synthesis is most likely increased through excessive Tf-Fe2 stimulation of the BMP6 signaling pathway, but the downregulation of ferroportin is insufficient to significantly limit transfusional iron overload. High hepcidin levels could also decrease cardiomyocyte expression of ferroportin, thus trapping iron within these cells. Indeed, conditional deletion of ferroportin in cardiomyocytes leads to severe compromise of cardiac function caused by iron accumulation in these cells.8 Although EPO levels are high, suppression of hepcidin does not occur in the absence of erythropoiesis. Most importantly, the absence of erythropoieisis results in lack of iron uptake by erythroblasts, thus accounting for the high transferrin saturation and high levels of NTBI and labile plasma iron. This is most likely the explanation for the increased iron toxicity in DBA.
With respect to clinical practice, chronic transfusion alone in DBA is not a satisfactory long-term management strategy and it is essential that chelation is included early in the treatment plan. DBA patients tolerate deferasirox (Exjade, Jaydenu) and deferoxamine (Desferal) well; however, prolonged subcutaneous administration of deferoxamine is more difficult to manage and to achieve good compliance, especially in older children. DBA patients may be uniquely susceptible to the toxic effects of the other oral agent deferiprone (Ferriprox), which is useful in removing cardiac iron. Indeed, the first case of septic agranulocytosis was reported in a DBA patient and others have been reported.27,28 In summary, because of efficacy, safety, and ease of administration, oral deferasirox is usually tried first; in severely overloaded patients deferoxamine is added either subcutaneously or intravenously, and deferiprone is reserved and used cautiously only in patients with cardiac iron overload.
PROSPECTS FOR NOVEL TREATMENT OF DIAMOND-BLACKFAN ANEMIA
Gene Therapy
For those cases of DBA with an identifiable underlying genetic mutation, gene therapy is an attractive potential therapy to cure DBA. Similar to the goal of allogeneic HSCT, gene therapy for DBA aims to replace DBA HSCs, and their diseased erythroid progeny, with HSCs able to produce adequate numbers of number erythroid progenitors that ultimately give rise to normal mature red blood cells. The difference between allogeneic HSCT and gene therapy is that the source of normal HSCs in gene therapy does not come from an outside donor but from the patient’s own HSCs into which a normal copy of the mutated gene has been inserted.
Given that 25% of DBA cases are associated with a mutation in the RPS19 gene,15 there has been significant interest in the development of gene therapy to deliver a normal copy of RPS19 to RPS19-mutated DBA HSCs. In a series of studies, Karlsson and colleagues have demonstrated that retroviral gene therapy vectors carrying the wild-type RPS19 gene can transduce human DBA patient CD34+ cells, the cell population that contains human HSCs. This results in an erythroid engraftment advantage in immunocompromised mice compared with mock-transduced DBA CD34+ cells.29 They have also shown that lentiviral gene therapy vectors carrying wild-type RPS19 can transduce mouse HSCs and correct the anemia found in a mouse model of DBA that depletes RPS19 protein levels using shRNA knockdown.30 Future studies are needed to determine if the newer lentiviral vectors are also able to efficiently transduce human CD34+ cells. Additionally, CRISPR/Cas9-mediated genome editing has recently been shown to be highly efficient in human HSCs.31 This genome editing paradigm physically corrects a disease-causing mutation within the genome, and may mediate cure without the use of an integrating viral vector.
Novel Drug Therapeutics
To find treatment alternatives to corticosteroids, there has been great interest in identifying novel drug candidates for the management of DBA. Although no other effective medications have been discovered for DBA in the last 60 years, the last decade has seen a substantial increase in the number of candidates with therapeutic potential.
After observing that decreased protein synthesis in lymphoid cells from DBA patients could be increased with high concentrations of l-leucine,32 Pospisilova and colleagues33 treated a single DBA patient with high levels of dietary l-leucine and observed an approximately 50% increase in hemoglobin levels 6 months after l-leucine initiation. Subsequently, Payne and colleagues34 and Jaako and colleagues35 demonstrated that high-levels of l-leucine were able to improve the anemia in RP knockdown DBA models in the zebrafish and mouse, respectively. A clinical trial is currently underway to evaluate l-leucine in DBA patients, but there have been no report of efficacy to date.
To address one of the underlying pathophysiologic mechanisms of disease in DBA, Zon and colleagues (unpublished data) identified through a chemical screen in RPS19-deficient zebrafish that calmodulin inhibition rescued hemoglobin levels. In human cell culture models, the calmodulin inhibitor trifluoperazine blocked nuclear accumulation and transcriptional activity of p53 by inhibiting calmodulin-dependent kinase phosphorylation of the p53 c-terminal domain. Trifluoperazine also increased hemoglobin levels in a RPS19 knockdown mouse model of DBA. Although unpublished, this work has been presented in abstract form at the American Society of Hematology annual meeting in 2015, and a clinical trial will begin soon to evaluate calmodulin inhibition in DBA.
Additional preclinical investigations have focused on finding therapeutic targets that may have a synergistic therapeutic effect with corticosteroids, which would ideally allow DBA patients to either maintain the same level of disease control with less steroid toxicity or maintain disease control when patients become less steroid-responsive. Lodish and colleagues14 probed promoter regions of genes activated in mouse BFU-Es that were stimulated with glucocorticoids and found many promoters contained hypoxia-induced factor 1-α binding sites. Because degradation of hypoxia-induced factor 1- α is mediated by intracellular prolyl hydroxylase activity, they then tested the effect of the prolyl hydroxylase inhibitor dimethyloxalylglycine on mouse BFU-E expansion, with the rationale that increased BFU-E expansion would address the initial underlying deficiency in early erythroid progenitor cells in DBA. They found that although dimethyloxalylglycine treatment by itself did not increase BFU-E expansion, treatment with both dimethyloxalylglycine and corticosteroids resulted in increased BFU-E expansion compared with corticosteroids alone. To date, no clinical trial of prolyl hydroxylase inhibitors in DBA patients have been initiated, but there are currently several prolyl hydroxylase inhibitors being evaluated for EPO induction in chronic kidney disease.36 These studies will be invaluable in defining the toxicity profile of prolyl hydroxylase inhibitors before trials in DBA patients can be undertaken.
Another therapeutic target that potentially synergizes with corticosteroids is the transcription factor peroxisome proliferator activated receptor (PPAR)- α. Lee and colleagues37 identified PPAR-α as the nuclear receptor with the greatest increase in expression on treatment of mouse BFU-Es with corticosteroids, and also determined that the glucocorticoid receptor and PPAR-α share similar sites of chromatin occupancy in mouse BFU-Es. Further experiments with mouse and human BFU-Es demonstrated that the addition of either of the PPAR-α agonists GW7647 or fenofibrate to corticosteroid treatment resulted in increased BFU-E expansion in vitro when compared with corticosteroids alone. This could potentially lower the concentration of corticosteroids needed to increase output of red cells from earlier progenitors. Additionally, GW7647 treatment resulted in a less severe hemoglobin nadir in a mouse model of phenylhydrazine-induced acute hemolytic anemia, and an improvement in hemoglobin levels of a mouse model of chronic anemia caused by heterozygous mutation of the transcription factor erythroid Kruppel-like factor. Further work is still under way to evaluate the efficacy of PPAR-α agonists in preclinical models of DBA.
There have also been recent efforts to use unbiased large-scale chemical screens to identify candidate drugs for DBA therapy. Using induced pluripotent stem cells (iPSCs) generated from DBA patients with RPS19 or RPL5 haploinsufficiency, Doulatov and colleagues38 demonstrated DBA iPSCs have multiple erythropoietic defects in vitro, and in vivo. These DBA iPSCs were then used in an unbiased screen that not only identified corticosteroids as a mediator of erythroid phenotype rescue, but also independently identified the compound SMER28, which is a quinazolinamine derivative previously shown to induce autophagy. They then went on to demonstrate SMER28 is able to alleviate anemia in a zebrafish RP knockdown model of DBA and in a mouse model of DBA with inducible single copy loss of the Rpl11 gene. Given the recent reporting of these findings, clinical translation of SMER28 as a potential DBA therapeutic is still pending.
Lastly, several recent lines of evidence have pointed to inhibition of transforming growth factor (TGF)-β signaling as a potential therapy for DBA. Ge and colleagues39 recently found that iPSCs from DBA patients, with RPS19 or RPL5 haploinsufficiency, are characterized by increased TGF-β receptor signaling and increased expression of TGF-b target genes. Gao and colleagues40 also recently found that the type 3 TGF-β receptor increases in expression during the transition from developmentally early to late BFU-Es, and that treatment with a TGF-β inhibitor increases in vitro expansion of mouse and human BFU-Es. Because the type 3 TGF-β receptor does not possess a signaling domain, the hypothesis is that increasing surface expression of the type 3 TGF-β receptor binds TGF-β signaling molecules and sequesters them at the late BFU-E cell surface, allowing greater opportunity for binding the type 1 and 2 TGF-receptors. Type 1 and 2 TGF-β receptor activation may then mediate signaling that results in differentiation of BFU-Es as opposed to continued BFU-E cell proliferation, whereas inhibition of TGF-β signaling may lead to increased output of red blood cells by increasing BFU-E cell proliferation.
Ear and colleagues41 also reported additional evidence for the therapeutic potential of inhibiting signaling of TGF-β superfamily receptors, recently showing that the activin receptor trap RAP-011 improves erythropoiesis in a RPL11 knockdown zebrafish model of DBA. Activin receptor traps were first proposed as therapy for correcting ineffective erythropoiesis associated with β-thalassemia,42 but activin receptor traps, such as RAP-011, or the humanized version ACE-011, additionally have the potential for binding other members of the TGF-β superfamily of ligands, such as growth differentiation factor-11. Interestingly, ACE-011 has been observed to increase hemoglobin in nonanemic postmenopausal women43 and in oncology patients experiencing chemotherapy-induced anemia.44 Neither of these two patient populations were affected by β -thalassemia and did not demonstrate signs of ineffective erythropoiesis. Given the existing clinical experience with ACE-011, there is now an active clinical trial evaluating the safety and efficacy of ACE-011 in DBA.
KEY POINTS.
Diamond-Blackfan anemia is a congenital red cell aplasia caused by ribosomal protein gene and rarely GATA1 mutations.
The erythroid specificity is caused by reduced ribosome numbers that decrease translation of complex structured mRNAs.
Treatment with steroids is successful long term in approximately 40% of patients and those that fail require hematopoietic stem cell transplantation or red cell transfusions.
Treatment-related issues include steroid toxicity, risks associated with transplantation, and transfusion hemosiderosis.
Several novel treatments for DBA are in trial or under preclinical development.
Footnotes
Disclosure Statement: None of the authors have any disclosures to report.
REFERENCES
- 1.Nathan DG, Clarke BJ, Hillman DG, et al. Erythroid precursors in congenital hypoplastic (Diamond-Blackfan) anemia. J Clin Invest 1978;61:489–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xiang J, Wu D-CC, Chen Y, et al. In vitro culture of stress erythroid progenitors identifies distinct progenitor populations and analogous human progenitors. Blood 2015;125(11):1803–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roggero S, Quarello P, Vinciguerra T, et al. Severe iron overload in Blackfan-Diamond anemia: a case-control study. Am J Hematol 2009;84(11):729–32. [DOI] [PubMed] [Google Scholar]
- 4.Mirabello L, Khincha PP, Ellis SR, et al. Novel and known ribosomal causes of Diamond-Blackfan anaemia identified through comprehensive genomic characterisation. J Med Genet 2017;54:417–25. [DOI] [PubMed] [Google Scholar]
- 5.Mirabello L, Macari ER, Jessop L, et al. Whole-exome sequencing and functional studies identify RPS29 as a novel gene mutated in multicase Diamond-Blackfan anemia families. Blood 2014;124(1):24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farrar JE, Quarello P, Fisher R, et al. Exploiting pre-rRNA processing in Diamond Blackfan anemia gene discovery and diagnosis. Am J Hematol 2014;89(10): 985–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gripp KW, Curry C, Olney AH, et al. Diamond-Blackfan anemia with mandibulofacial dystostosis is heterogeneous, including the novel DBA genes TSR2 and RPS28. Am J Med Genet A 2014;164A(9):2240–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lakhal-Littleton S, Wolna M, Carr CA, et al. Cardiac ferroportin regulates cellular iron homeostasis and is important for cardiac function. Proc Natl Acad Sci U S A 2015;112(10):3164–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ludwig LS, Gazda HT, Eng JC, et al. Altered translation of GATA1 in Diamond-Blackfan anemia. Nat Med 2014;20(7):748–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khajuria RK, Munschauer M, Ulirsch JC, et al. Ribosome levels selectively regulate translation and lineage commitment in human hematopoiesis. Cell 2018; 173(1):90–103.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mills EW, Green R. Ribosomopathies: there’s strength in numbers. Science 2017; 358(6363) [pii:eaan2755]. [DOI] [PubMed] [Google Scholar]
- 12.Gasser C. Aplastiche Anamie (chronische Erythroblastophthise) und Cortison. Schweiz Med Wochenschr 1951;81:1241. [PubMed] [Google Scholar]
- 13.Vlachos A, Muir E. How I treat Diamond-Blackfan anemia. Blood 2010;116(19): 3715–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flygare J, Rayon Estrada V, Shin C, et al. HIF1alpha synergizes with glucocorticoids to promote BFU-E progenitor self-renewal. Blood 2011;117(12):3435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vlachos A, Ball S, Dahl N, et al. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol 2008;142(6):859–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fagioli F, Quarello P, Zecca M, et al. Haematopoietic stem cell transplantation for Diamond Blackfan anaemia: a report from the Italian Association of Paediatric Haematology and Oncology Registry. Br J Haematol 2014;165(5):673–81. [DOI] [PubMed] [Google Scholar]
- 17.Lipton JM, Atsidaftos E, Zyskind I, et al. Improving clinical care and elucidating the pathophysiology of Diamond Blackfan anemia: an update from the Diamond Blackfan Anemia Registry. Pediatr Blood Cancer 2006;46(5):558–64. [DOI] [PubMed] [Google Scholar]
- 18.Roy V, Perez WS, Eapen M, et al. Bone marrow transplantation for Diamond-Blackfan anemia. Biol Blood Marrow Transplant 2005;11(8):600–8. [DOI] [PubMed] [Google Scholar]
- 19.Berdoukas V, Nord A, Carson S, et al. Tissue iron evaluation in chronically transfused children shows significant levels of iron loading at a very young age. Am J Hematol 2013;88(11):5. [DOI] [PubMed] [Google Scholar]
- 20.Porter JB, Walter PB, Neumayr LD, et al. Mechanisms of plasma non-transferrin bound iron generation: insights from comparing transfused Diamond Blackfan anaemia with sickle cell and thalassaemia patients. Br J Haematol 2014;167(5): 692–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pak M, Lopez MA, Gabayan V, et al. Suppression of hepcidin during anemia requires erythropoietic activity. Blood 2006;108(12):3730–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kautz L, Jung G, Valore EV, et al. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet 2014;46(7):678–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kautz L, Jung G, Du X, et al. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of b-thalassemia. Blood 2015;126(17): 2031–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tanno T, Bhanu NV, Oneal PA, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med 2007;13(9): 1096–101. [DOI] [PubMed] [Google Scholar]
- 25.Tamary H, Shalev H, Perez-Avraham G, et al. Elevated growth differentiation factor 15 expression in patients with congenital dyserythropoietic anemia type I. Blood 2008;112(13):5241–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Casanovas G, Swinkels DW, Altamura S, et al. Growth differentiation factor 15 in patients with congenital dyserythropoietic anaemia (CDA) type II. J Mol Med 2011;89(8):811–6. [DOI] [PubMed] [Google Scholar]
- 27.Henter J II, Karlén J. Fatal agranulocytosis after deferiprone therapy in a child with Diamond-Blackfan anemia. Blood 2007;109(12):5157–9. [DOI] [PubMed] [Google Scholar]
- 28.Hoffbrand AV, Bartlett AN, Veys PA, et al. Agranulocytosis and thrombocytopenia in patient with Blackfan-Diamond anaemia during oral chelator trial. Lancet 1989; 2(8660):457. [DOI] [PubMed] [Google Scholar]
- 29.Flygare J, Olsson K, Richter J, et al. Gene therapy of Diamond Blackfan anemia CD34(+) cells leads to improved erythroid development and engraftment following transplantation. Exp Hematol 2008;36(11):1428–35. [DOI] [PubMed] [Google Scholar]
- 30.Jaako P, Debnath S, Olsson K, et al. Gene therapy cures the anemia and lethal bone marrow failure in a mouse model of RPS19-deficient Diamond-Blackfan anemia. Haematologica 2014;99(12):1792–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dever DP, Bak RO, Reinisch A, et al. CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature 2016;539(7629):384–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cmejlova J, Dolezalova L, Pospisilova D, et al. Translational efficiency in patients with Diamond-Blackfan anemia. Haematologica 2006;91(11):1456–64. [PubMed] [Google Scholar]
- 33.Pospisilova D, Cmejlova J, Hak J, et al. Successful treatment of a Diamond-Blackfan anemia patient with amino acid leucine. Haematologica 2007;92(5):7. [DOI] [PubMed] [Google Scholar]
- 34.Payne EM, Virgilio M, Narla A, et al. L-Leucine improves the anemia and developmental defects associated with Diamond-Blackfan anemia and del(5q) MDS by activating the mTOR pathway. Blood 2012;120(11):2214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jaako P, Debnath S, Olsson K, et al. Dietary L-leucine improves the anemia in a mouse model for Diamond-Blackfan anemia. Blood 2012;120(11):2225–8. [DOI] [PubMed] [Google Scholar]
- 36.Gupta N, Wish JB. Hypoxia-inducible factor prolyl hydroxylase inhibitors: a potential new treatment for anemia in patients with CKD. Am J Kidney Dis 2017; 69(6):815–26. [DOI] [PubMed] [Google Scholar]
- 37.Lee HY, Gao X, Barrasa MI, et al. PPAR-alpha and glucocorticoid receptor synergize to promote erythroid progenitor self-renewal. Nature 2015;522(7557):474–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doulatov S, Vo LT, Macari ER, et al. Drug discovery for Diamond-Blackfan anemia using reprogrammed hematopoietic progenitors. Sci Transl Med 2017;9(376) [pii: eaah5645]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ge J, Apicella M, Mills JA, et al. Dysregulation of the transforming growth factor beta pathway in induced pluripotent stem cells generated from patients with Diamond Blackfan anemia. PLoS One 2015;10(8):e0134878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao X, Lee HY, da Rocha EL, et al. TGF-beta inhibitors stimulate red blood cell production by enhancing self-renewal of BFU-E erythroid progenitors. Blood 2016;128(23):2637–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ear J, Huang H, Wilson T, et al. RAP-011 improves erythropoiesis in zebrafish model of Diamond-Blackfan anemia through antagonizing lefty1. Blood 2015; 126(7):880–90. [DOI] [PubMed] [Google Scholar]
- 42.Dussiot M, Maciel TT, Fricot A, et al. An activin receptor IIA ligand trap corrects ineffective erythropoiesis in beta-thalassemia. Nat Med 2014;20(4):398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruckle J, Jacobs M, Kramer W, et al. Single-dose, randomized, double-blind, placebo-controlled study of ACE-011 (ActRIIA-IgG1) in postmenopausal women. J Bone Miner Res 2009;24(4):744–52. [DOI] [PubMed] [Google Scholar]
- 44.Raftopoulos H, Laadem A, Hesketh PJ, et al. Sotatercept (ACE-011) for the treatment of chemotherapy-induced anemia in patients with metastatic breast cancer or advanced or metastatic solid tumors treated with platinum-based chemotherapeutic regimens: results from two phase 2 studies. Support Care Cancer 2016; 24(4):1517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Q, Luo C, Luo C, et al. Disease-specific hematopoietic stem cell transplantation in children with inherited bone marrow failure syndromes. Ann Hematol 2017; 96(8):1389–97. [DOI] [PubMed] [Google Scholar]
- 46.Burroughs LM, Shimamura A, Talano J-A, et al. Allogeneic hematopoietic cell transplantation using treosulfan-based conditioning for treatment of marrow failure disorders. Biol Blood Marrow Transpl 2017;23(10):1669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crazzolara R, Kropshofer G, Haas OA, et al. Reduced-intensity conditioning and stem cell transplantation in infants with Diamond Blackfan anemia. Haematologica 2016;102(3):e73–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Asquith JM, Copacia J, Mogul MJ, et al. Successful use of reduced-intensity conditioning and matched-unrelated hematopoietic stem cell transplant in a child with Diamond-Blackfan anemia and cirrhosis. Pediatr Transplant 2015;19(6): E157–9. [DOI] [PubMed] [Google Scholar]