

Abstract
Most disease-associated variants, although located in putatively regulatory regions, do not have detectable effects on gene expression. One explanation could be that we have not examined gene expression in the cell types or conditions that are most relevant for disease. Even large-scale efforts to study gene expression across tissues are limited to human samples obtained opportunistically or post-mortem, mostly from adults. In this review, we evaluate recent findings and suggest an alternative strategy, drawing on the dynamic and highly context-specific nature of gene regulation. We discuss new technologies that can be used to extend the standard regulatory mapping framework to more diverse, disease-relevant cell types and states.
Molecular QTL annotation as a strategy for interpreting disease genetics
A vast majority of the thousands of genetic loci associated with human disease are located outside of coding regions, putatively in genomic regions that participate in gene regulation [1–3]. In most cases, it remains unclear how disease-associated genetic variants in non-coding regions function to alter disease risk between individuals. Indeed, while the biochemical consequences of mutations within protein-coding sequences can sometimes be predicted from the genetic code and protein structure, predicting the effects of mutations in regulatory DNA remains highly challenging. In fact, without additional functional data it is challenging to even predict which genes are regulated by any given non-coding regulatory mutation. Hence, if most disease-associated variants influence pathogenic risk by altering gene regulation, complementary functional information is needed to interpret the impact of such mutations.
Regulatory quantitative trait locus (QTL) mapping is a powerful approach that can connect disease-associated variants to gene regulatory mechanisms (see Box 1). As gene expression is an intermediate link between DNA sequence and organismal phenotype (the central dogma of molecular biology), regulatory associations may provide key insight into the function of disease-associated variants by pointing to the genes and networks they affect. Regulatory associations are particularly critical when the target gene regulated by a disease locus is not the closest gene [4]. For example, mutations strongly associated with obesity are located in the first intron of the FTO gene, but their pathogenic mechanism does not appear to involve the FTO protein. Instead, chromatin conformation and regulatory QTL mapping data have demonstrated that this intronic locus harbors enhancers that regulate the expression of the transcription factor gene IRX3, which is located several megabases away [5]. In other words, genetic variants associated with obesity in FTO are regulatory QTLs that are associated with IRX3 expression. Mouse experiments have confirmed that deletion of Irx3 in the hypothalamus, and in developing adipocytes, protects against obesity, pointing to a causal relationship between regulatory variation and disease [5,6]. Thus, regulatory QTL mapping can enable efforts to understand the biological mechanisms of disease alleles (Figure 1). In addition, identifying the regulatory targets of mutations can shorten the list of candidate genes requiring functional follow-up. Regulatory associations can also help uncover shared mechanisms underlying diseases with different associated disease variants [7,8].
Box 1: regulatory QTL mapping and enrichment for GWAS signals.
Regulatory QTL mapping tests for the association between individual-level genotypes and quantitative molecular phenotypes that are mechanistically related to gene expression. While the majority of reported regulatory QTL studies have focused on mRNA levels (expression QTL or eQTL), other biochemical signatures of gene regulation, including DNA methylation, histone modifications, chromatin accessibility, mRNA synthesis and decay rates, protein levels, or isoform abundance are equally amenable to QTL mapping and provide complementary information about regulatory mechanisms [70]. Considered individually, each class of regulatory QTLs is enriched among disease-associated mutations. This observation is often explained by the notion that each step of regulation influences downstream steps to ultimately determine gene and protein expression levels and thus complex phenotypes such as disease. This notion appears to be partially accurate, as most chromatin-level QTLs seem to influence phenotype via their effects on transcript levels [109,110]. However, protein levels seem to be at least partly buffered with respect to eQTL effects. More generally, derived annotations that integrate multiple forms of regulatory QTL data capture a greater proportion of trait-associated variants [111]. This observation implies that some mutations can make unique contributions at distinct levels of regulation. For instance, variants that alter splicing appear to influence disease risk roughly as much as eQTLs, but act largely independently [59,109,112,113]. A related strategy for detecting cis regulatory effects quantifies allele-specific expression (ASE) across all heterozygous genes, considering the action of a cis-eQTL through its effect on a nearby allele. At heterozygous loci, biased usage of one allele is an internally-controlled indicator of a cis-regulatory difference, an implied eQTL, as both alleles share a common environment in trans [114].
Figure 1:
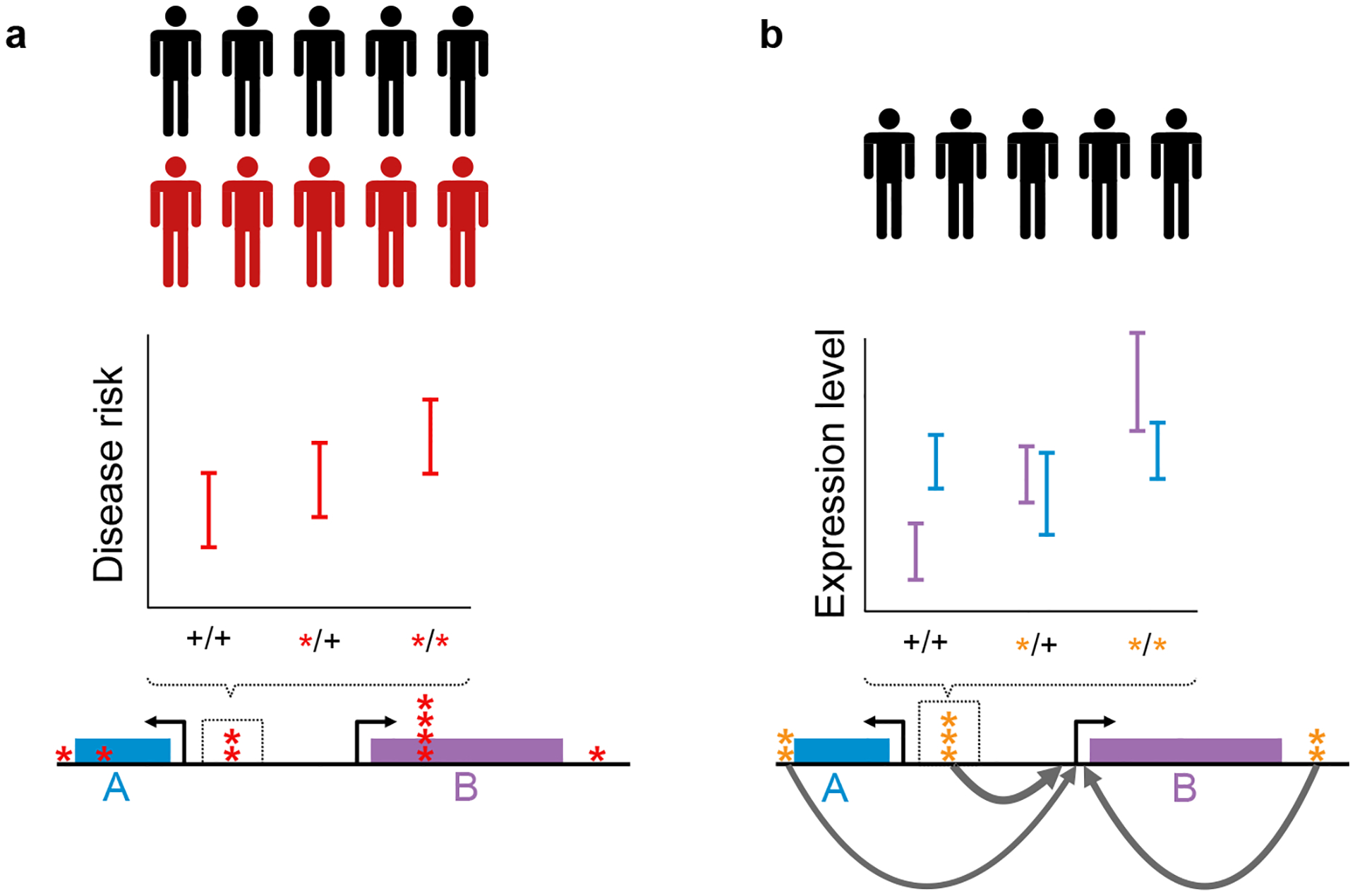
Combining GWAS and eQTL data. (a) By comparing affected and control individuals, disease risk can be associated with genotype at many loci, indicated by red asterisks. Even in large studies, many loci show weak evidence of association, represented by short accumulations of asterisks, and the function of intergenic mutations is unclear. (b) Correlating genotypes with expression levels in healthy subjects can reveal the targets of eQTL variants (orange asterisks). Here, mutations from (a) correlate with expression changes in gene B but not gene A, suggesting possible involvement in disease.
Combining eQTL and GWAS data
Integrating genome-wide association study (GWAS) data with regulatory associations also has the potential to enhance searches for unidentified disease loci. To date, SNPs identified by GWAS account for only a small proportion of the heritability of any given trait. Much of the missing heritability can likely be accounted for by the combined contribution of loci whose individual effects on disease risk are modest [9,10]. However, by treating all SNPs as equal candidates, even well powered studies struggle to identify all disease-associated loci. Because regulatory QTLs have known functional effects, this information can be included as a prior expectation in the analysis of GWAS data.
For example, Cusanovich et al. combined data from GWAS for lymphocyte counts with expression QTL (eQTL) mapping data from immortalized lymphocytes. They found a significant and replicated enrichment of GWAS associations among the top-ranked eQTLs, despite the fact that no single variant achieved genome-wide significance in the original GWAS [11]. In this study, eQTL and GWAS data were not combined to merely propose mechanisms for disease-associated loci, but to identify additional promising candidate disease-associated loci.
Cusanovich et al. did not proceed to demonstrate that a causal relationship exists between the GWAS loci they identified and the regulatory effect they observed. This is an important challenge to address because not all identified QTLs have direct and independent functional roles. For example, linkage disequilibrium (LD) among eQTLs can cause multiple variants or multiple regulated genes to appear to contribute to a phenotype simply due to correlation with a causal site. This challenge is compounded when the LD structure differs between populations from which related datasets are derived, such as the GWAS and gene expression results in Cusanovich et al. As a result, accurate interpretation of QTL data requires a means to disentangle causal variants from other correlated findings.
In the last few years, different methods to jointly evaluate eQTL and GWAS data have been developed. One class of approaches, broadly termed colocalization, focuses on the associated loci themselves. Colocalization approaches test the hypothesis that a shared variant causally impacts both the disease and gene expression [12–15], providing stronger evidence that the regulatory effect underlies the disease mechanism. However, colocalization alone cannot distinguish between a variant that influences expression and disease separately (‘pleiotropy’) and one that affects disease directly via a regulatory effect (‘mediation’) [16]. A complementary set of methods focuses on the associated genes instead of the specific loci. These approaches test the association between genetically influenced gene expression levels and disease, using eQTL data to construct a predictor of gene expression that can be evaluated for association with disease in GWAS studies. In essence, reference eQTL data, relating genotypes to expression, are used to interpret GWAS, which relate genotypes to disease but have no direct measurements of gene expression. Notable examples include PrediXcan and TWAS, which compare predicted expression to disease status in order to prioritize disease genes, and Mendelian Randomization, which uses eQTL data as an instrumental variable for two-step least-squares regression to evaluate the effect of gene expression on disease [17–20].
Gene-based approaches complement, rather than replace, colocalization-based methods [17] and have been particularly successful in identifying new disease associations while narrowing the focus of GWAS results to a tractable number of genes [21]. In turn, a third set of network-based approaches uses regulatory data to infer the structure of gene networks. Thus, rather than implicating individual genes, these approaches can be applied to identify regulatory modules that are perturbed by disease mutations [22–24].
Regardless of the approach used, the ability to predict disease risk based on gene expression data enables construction of individualized transcriptional risk scores, resulting in improved classification of disease risk over genotype information alone [25]. Like colocalization methods, however, the predictive power of gene- and network-level association testing depends on the quality and functional relevance of available gene expression data to the disease in question. Large-scale efforts to comprehensively map regulatory variation in human tissues are therefore instrumental to the efficacy of these approaches.
Does regulatory variation explain GWAS findings?
Even a decade ago, when functional variants in the genome were vastly undercounted, it was already clear that eQTLs are enriched among disease-associated loci [26]. Early efforts to map eQTLs in whole blood and immortalized B cells have found that common genetic variants explain, on average, less than 10% of gene expression variation, but are highly enriched for associations with disease risk [27]. Depending on the traits under consideration, between one sixth and one third of known disease-associated loci were found to be in linkage disequilibrium with an eQTL in blood, and a further 8% of disease-associated loci are near other types of regulatory QTLs that affect isoform usage [1,27,28].
Of course, many disease processes may be specific to tissues other than blood. In recent years, a number of large-scale resources have been generated with the goal of identifying eQTLs in diverse tissue types. The Genotype-Tissue Expression (GTEx) study is the broadest multi-tissue eQTL catalog to date. The GTEx consortium collected data from over 15,000 post-mortem tissue specimens from 838 genotyped donors, representing 49 tissues [29]. This effort is emblematic of what we term ‘standard’ eQTL surveys, which collect data from non-diseased adult tissues in an unperturbed state, using bulk sequencing. The data from GTEx have dramatically increased the number of known eQTLs and regulated genes; the levels of virtually all expressed genes are now known to vary according to at least one nearby (putatively ‘cis’) SNP, and splicing QTLs have been found for about two-thirds of protein-coding genes. As the power to detect eQTLs has increased, so has the proportion of disease variants that are also associated with mapped regulatory variants [4,29].
However, disease-associated loci and eQTLs may be found in LD by chance. In particular, since at least one cis eQTL for practically every known human gene has been catalogued, it would be easy to mistake chance proximity between loci associated with disease and gene expression for a real regulatory mechanism underlying disease. For example, over 95% of catalogued autoimmune disease-associated variants can be found within 100 kb of an eQTL in an immune cell, but recent analysis suggested that expression and disease risk colocalize (that is, map to the same causal site) in only 25% of cases [30]. While colocalization methods are imperfect, and therefore unlikely to identify all cases of sharing between regulatory and disease mutations, this gap is striking. Across all GTEx tissues, 43% of disease-associated loci colocalize with a known eQTL [29]. Again, it is important to acknowledge that given the incomplete power to map and localize eQTLs, and the potential role for other mechanisms, we do not expect a complete overlap between disease-associated loci and inferred casual eQTLs. Still, the relatively low overlap requires us to consider whether the best path forward is to perform even larger standard eQTL studies, or to seek an alternative approach.
The colocalization of disease-associated loci with eQTLs provides important insight into disease mechanisms and the tissues they impact. The ultimate aim would be to increase the proportion of colocalized eQTLs and GWAS hits and provide regulatory annotations for practically all disease-associated loci. Unfortunately, despite the inclusion of nearly twice as many subjects and five additional tissue types relative to previous GTEx analyses [4,31], the proportion of eQTLs that colocalize with disease-associated variants has not changed substantially, even as the total number of identified eQTL variants has surpassed 4.2 million. In part, this may reflect saturation of cis eQTL discovery in bulk, healthy, adult tissue samples; beyond about 600 samples of any tissue, the proportion of genes with an identified eQTL appears to plateau within the GTEx dataset [29]. Larger sample sizes increase power to detect very small eQTL effects and additional independent regulatory associations for the same genes, but it is not clear that these weak regulatory variants correspond to disease risk. Moreover, colocalized eQTLs are not necessarily causal mediators of disease: by using a model that explicitly distinguished cis-mediated from non-mediated (e.g., pleiotropic, trans, or linked) effects of genotypes on traits, Yao et al. estimated that on average, only 11% of trait heritability could be ascribed to GTEx cis-eQTLs [32]. Thus, while a meaningful fraction of genetic disease risk can be attributed to effects on gene regulation, the majority of non-coding disease mutations have yet to be explained.
It should be noted that cis eQTLs explain only a small proportion of expression heritability. It is estimated that up to 70% of inter-individual variance in gene expression may be attributable to trans mechanisms [33,34], which would likely be mediated by other genes in cell-type specific regulatory networks. It is therefore possible that many—and perhaps even all—genes expressed in a relevant cell type collaborate to affect complex diseases via a core set of genes [34,35]. Indeed, preliminary studies suggest distal (‘trans’) eQTLs are particularly cell type-specific and enriched for disease associations [36,37]. However, most eQTL studies have focused almost exclusively on the direct contribution of standard cis eQTLs because the role of trans regulatory mutations has been more difficult to assess, even for large efforts like GTEx. Full detection of trans-eQTL relationships may ultimately require sample sizes of tens or hundreds of thousands of individuals, which are currently only available for blood [38].
It would be tempting to speculate that many disease-associated loci are trans-eQTLs and that with much larger sample sizes we might be able to identify these loci as such. However, the eQTL variants that act in trans regulatory networks frequently also display cis regulatory effects, which often mediate their distal effects [39]. For example, cis-eQTLs for transcription factors, signaling molecules, or receptors can indirectly affect the expression of distal genes in trans via their effects on their local gene products. In fact, common mechanisms through which trans-eQTLs could arise without affecting the structure or expression of a nearby gene in cis are not well characterized. Thus, even without a full accounting of trans regulatory relationships, a detailed catalog of cis-eQTLs would likely include many of the sought-after trans variants even as the full scope of their effects remain underestimated.
Shortcomings of standard eQTL comparisons
In our search to find disease mechanisms and provide functional annotations for disease-associated loci, it is worth considering whether standard regulatory QTL mapping approaches are likely to yield valuable insights. On the one hand, even if all disease-associated loci were also cis eQTLs, one would not expect a 100% recall using colocalization approaches that are applied to current datasets because of incomplete power, allelic heterogeneity when more than one variant affects a gene or the disease, and the fact that linkage disequilibrium is not identical in the GWAS and eQTL study populations [12,14,17]. We may be able to overcome these limitations by performing even larger studies. On the other hand, it is important to ask to what extent we should continue to increase sample sizes and obtain more tissue types in order to discover additional cis eQTLs in steady-state adult samples. Is this the most effective way forward? Are these results likely to account for unexplained disease risk loci? One way to evaluate the expected suitability of this framework is to consider the properties of standard eQTLs compared to our theoretical expectations about functional mutations that cause disease.
Most standard eQTLs do not have specific effects
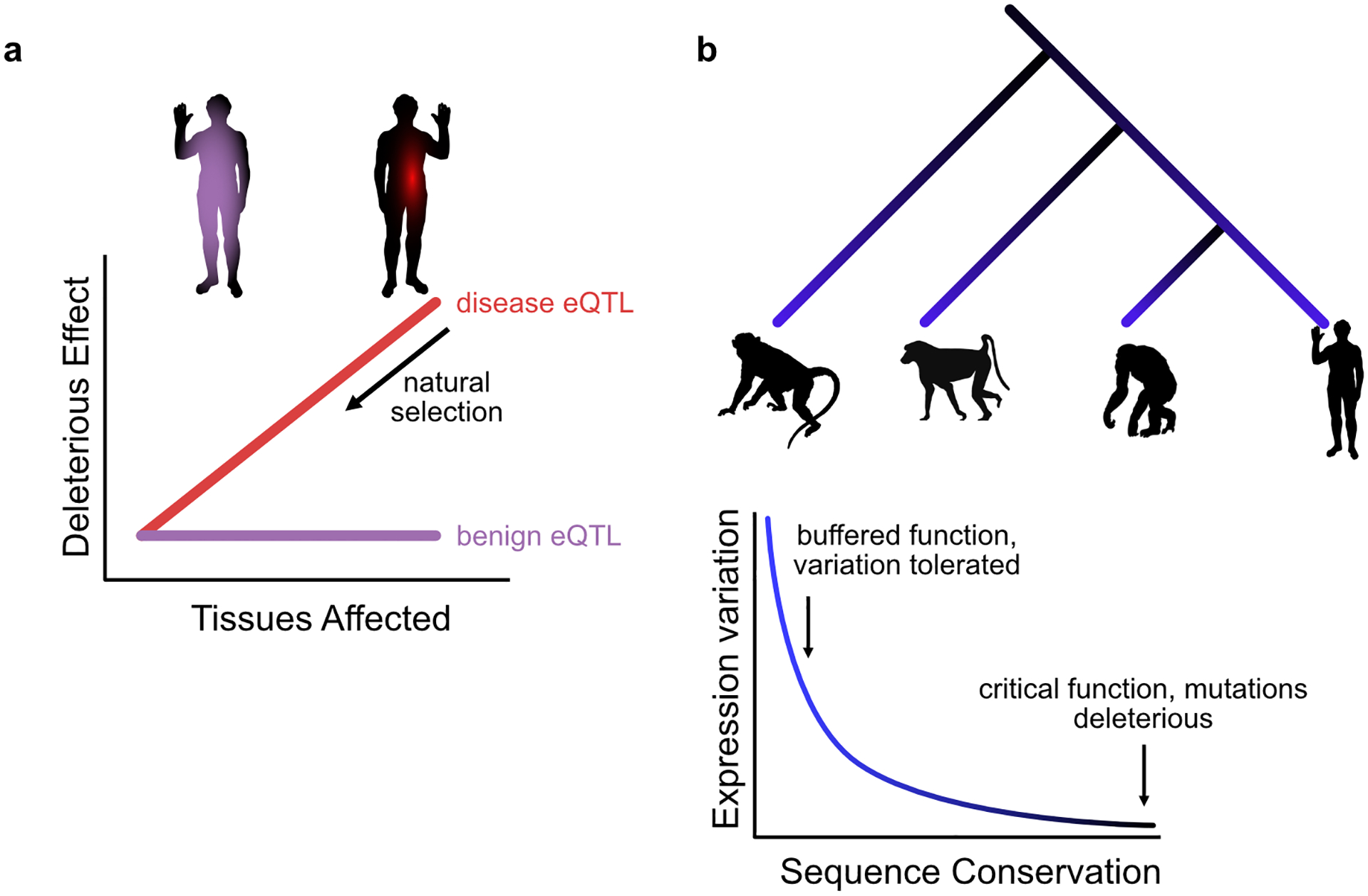
It has long been hypothesized that regulatory changes play an important role in adaptation and speciation. Unlike mutations to protein-coding sequences, which will impact function whenever the protein is expressed, regulatory mutations can have highly dynamic and context-specific effects [40–42]. Regulatory mutations can therefore escape the pleotropic deleterious impacts that would be expected for most changes to structural proteins (see Box 2). Consistent with this notion, nonsynonymous mutations in protein-coding sequences are more rare in the population and more frequently implicated in highly penetrant diseases [43] than regulatory mutations. If most standard eQTLs represent functional regulatory variation, one might expect that their phenotypic impact will be quite specific (Figure 2a).
Box 2: Changes in gene regulation can underlie speciation and adaptation.
Speciation and adaptation require phenotypic variability. Variability in phenotypes can only be tolerated if it does not have a deleterious effect on fitness. Mutations that result in a fitness cost would quickly be purged from the population by natural selection. Mutations that affect structural proteins would have a functional impact everywhere the protein is expressed. Thus, even if a mutation to a protein confers some fitness advantage in a particular functional context, it is likely that it will also have pleiotropic deleterious effects in other contexts. For that reason, most mutations to proteins are overall deleterious and are selected against. In contrast to mutations to structural proteins, regulatory mutations can have circumscribed dynamic and context-specific effects that can allow them to have a fitness benefit in a single context without having otherwise deleterious impacts. Gene expression is controlled by a combination of trans elements, such as transcription factors and small RNA types, binding to cis regulatory elements, including enhancers and promoters, which encode the conditions for transcriptional activation [115]. Cis regulatory elements, such as enhancers, are frequently redundant. Mutations in cis regulatory elements can introduce a new regulatory function while being buffered from causing deleterious effects. Moreover, the modular nature of individual enhancers, which can have spatially and temporally specific dynamics, means that new functional outcomes can be highly specific. Regulatory activity can be gained or lost through single-nucleotide changes at transcription factor binding sites, and transposition of an enhancer can bring new genes under its regulatory control. These changes can all affect the timing, magnitude, and tissue specificity of gene expression but, importantly, do not typically alter the biochemical properties of the gene product. As a result, evolution through changes in gene regulation can lead to phenotypic adaptation without risking deleterious consequences [116–118].
Figure 2:
Features of standard eQTLs are inconsistent with roles in disease. (a) Truly deleterious alleles (red) are unlikely to affect many tissues without encountering strong selective pressures, limiting their persistence in the population. Alleles wide effects are more likely benign (purple). (b) Standard eQTL genes (blue) are shared across primates. These genes show lower levels of sequence conservation, indicating a less sensitive role in physiology.
In fact, many standard eQTLs, including those identified by GTEx, are shared across many tissues [29,37,44,45]. It was estimated that 77% of eQTLs in LD with a disease mutation are not specific to a single tissue [4], although these results are not based on colocalization (see Box 3). This suggests that expanding the scope of standard eQTL surveys to include additional tissues is unlikely to reveal novel findings, instead recapitulating the shared effects that have already been discovered. Moreover, the extensive sharing of eQTLs across tissues is at odds with the expectation that heritable functional mutations would exhibit narrowly tailored effects. The subset of standard eQTLs that do exhibit tissue-specific effects seem to be particularly relevant for disease. For example, eQTLs ascertained specifically in blood cells have greater relevance for immune-related phenotypes [28,46].
Box 3: Colocalization may indicate less sharing of eQTLs across contexts.
The level of eQTL sharing across tissues, cell types, and contexts has been widely studied using statistical methods including meta-analysis, joint regression, and replication studies [119–122]. These studies have indicated high levels of sharing across tissues, with clearly tissue-specific effects being in the minority. Tissue-specific effects have largely been identified in tissues with large sample sizes and strong power to detect small effects not detected in other tissues. However, just as GWAS loci may coincidentally lie in the same region as eQTLs, eQTLs in different tissues may also appear to be shared due to LD, without reflecting the same causal signal. This occurrence may be common, particularly for eQTLs lying in enhancer regions, which tend to be more tissue-specific than promoter elements [123,124]. One consequence is that the relative frequency of truly ‘universal’ eQTLs has likely been overestimated in many cases. Furthermore, allelic heterogeneity, where multiple causal alleles in the same region affect a given gene even within a tissue, also complicates robust identification of shared signals across tissues [12,14]. As a result, although standard eQTLs are shared broadly across tissues, further analysis, such as multi-tissue colocalization, is needed to reliably quantify the extent of causal allele sharing.
It is important to note that almost all GTEx tissues consist of mixtures of different cell types, which have distinct physiological functions. How far gene regulatory patterns ascertained at the level of tissues extend to their constituent cell types remains to be determined, although it is notable that 59–87% of eQTLs found in individual immune cell populations are shared across cell types [23,47]. Computational efforts to deconvolve bulk gene expression data do enable some cell-type-specific mapping, but accuracy is only modest and almost entirely depends on precise cell type definitions, which for the most part are not yet available [29,48]. Emerging efforts to systematically map eQTLs at the single-cell level begin to address this crucial gap in our understanding [49]. For example, it is possible that regulatory variants affecting rare cell types play important functional roles but are not detectable in whole-tissue bulk samples, as has been reported recently for human retinal diseases [50].
Common eQTL variants are very weakly selected
Mutations that increase disease risk generally impose a fitness cost that tends to limit their transmission and their frequency in the population. Mutations that are associated with high risk for disease are indeed rare [51], and most common human diseases are driven by a combination of many loci whose individual effects on disease risk are small. Selection against any single constituent mutation may thus be relatively weak. Moreover, alleles that cause diseases which manifest late in life, beyond reproductive age, are likely to persist in a population [52].
If standard eQTLs are functional but broadly shared across tissues and cell states, they cannot escape pleiotropic deleterious effects. In that case, one might expect that the evolution of such eQTLs will be subject to natural selection in a way that may be similar to the selective pressures that shape the evolution of mutations in structural proteins; at the very least, we might expect the type of selection signature similar to that observed for disease-associated mutations.
Generally speaking, however, efforts to identify loci that evolved under natural selection in humans, without regard to phenotype, have turned up conspicuously few eQTLs [53,54]. Though mutations that are associated with large regulatory effects are extremely rare, the frequency of mutations that underlie standard eQTLs is mostly consistent with neutrality [55–57]. It should be noted that a caveat of this finding is that standard eQTL mapping approaches have limited power to estimate the regulatory effects of truly rare alleles. Yet, even when efforts are made to address this disparity by subsampling, the correlation between allele frequency and regulatory effect size remains modest [27]. Overall, the distribution of regulatory effect sizes and the allele frequency spectrum of standard eQTLs shows some evidence of selection, but its inferred effect is generally quite weak [58].
Consistent with the observation that standard eQTLs generally evolve neutrally or under weak selection, genes that are highly conserved across species, transcription factors, genes that are found to be network hubs, and genes that respond to stress, are all notably depleted for eQTLs. We also rarely find standard eQTLs associated with genes that are intolerant to loss of function mutations in the general population [27,43,55,59,60]. In contrast, highly conserved genes and loss of function intolerant genes are enriched for associations with disease.
The few studies that collected both steady-state mRNA and protein levels have found that the effects of transcriptional variation are strongly buffered, raising the possibility of an attenuated link between most standard eQTLs and baseline phenotypic differences [61–64]. Whether this is a general explanation for the apparent neutrality of many regulatory mutations or not, the overall findings suggest that standard eQTLs generally impact non-essential genes or do not have a major functional consequence.
Common eQTLs are shared across primates
Comparative studies of gene regulation have provided further evidence to support the notion that standard eQTLs may generally be evolving neutrally [65]. Comparing gene expression within and between species can help us identify genes whose regulation evolves under natural selection, as well as genes whose expression pattern is consistent with relaxation of evolutionary constraint (Figure 2b). The observations we discussed above suggest that standard eQTLs are generally found in genes whose expression levels can fluctuate without resulting in deleterious effects. In that case, one might hypothesize that the regulation of orthologs of such genes in closely related species may also evolve under similar evolutionary constraint. In other words, if standard eQTLs are generally neutral, orthologous genes across species are expected to be associated with eQTLs (that is, designated as eGenes) more often than expected by chance.
Indeed, eGenes ascertained from seven tissue types in vervet monkeys were all also identified as eGenes in GTEx, and genes associated with eQTLs in chimpanzees or baboons were significantly more likely to also be associated with eQTLs in humans [66–68]. The shared eGenes tend to display elevated coding sequence divergence, indicative of weaker evolutionary constraint [69]. This observation is consistent with the depletion of eQTLs in functionally important and highly conserved genes in humans, as noted above. Taken together, these observations suggest that a large number of genes regulated by standard eQTLs can vary in expression level, and even protein coding sequence, without incurring a fitness penalty. Put differently, standard eQTLs that operate in adult tissues affect genes whose expression variation has small functional consequences at steady state.
Dynamic gene regulation
The body of work we reviewed above suggests that most standard eQTLs are associated with regulatory changes that have minimal phenotypic impact. If most standard eQTLs are generally benign, increasing sample size and adding more tissue types in an effort to identify yet more standard eQTLs may not help us explain many more disease risk mutations. However, this does not mean that regulatory variation does not play a key role in disease. There are other places to look for this variation by considering the previously mentioned trans eQTLs or eQTLs identified in isolated, as-yet unsampled cell populations, as well as other types of regulatory QTLs that affect different mechanisms, such as protein QTLs, or co-transcriptional QTLs [70]. In this review, however, we would like to focus on the relatively unexplored space of dynamic eQTLs. We highlight two kinds of dynamic eQTL studies, aimed at perturbing gene expression and tracking its development over time.
Response QTLs connect disease-relevant treatments to genetic variation
One alternative screening strategy for regulatory QTLs would apply an extrinsic stimulus relevant to disease pathology in order to prioritize contextually functional regulatory variants, such as exposure to pathogens, stress, or chemical perturbations. This approach emphasizes the importance of regulatory differences that are ordinarily cryptic and sheltered from constitutive selective pressures that might limit their phenotypic effects (see Box 2) [71]. Loci that are associated with inter-individual differences in expression before or after the treatment, but not in both conditions, are termed ‘response eQTLs’. These variants show significant gene-environment interaction effects, where the ‘environment’ is an applied treatment.
The response QTL framework has been particularly well-suited to studies of immune response, as blood can be sampled with low risk to subjects, and stimulated ex vivo. This latter feature is important, as it allows for a more controlled environmental stimulus than can be achieved by sampling from patients in diverse clinical states [72]. Moreover, immune cell activation entails dramatic changes to chromatin accessibility and transcriptional activity [73,74], resulting in considerable power to detect response eQTLs even with modest sample sizes. Many immune response-regulated genes cannot be identified in the absence of stimulation and most regulatory variants implicated in inflammation and infection responses by GWAS manifest uniquely when immune cell types are in their stimulated state [47,73,75–77]. Interestingly, immune cell activation reveals a large number of trans-acting response eQTLs, which are highly cell type-specific [36,75,78]. Responses to pathogens have been under continuous selection throughout human history as changing threats have arisen. In contrast to standard eQTLs identified in blood samples, response eQTLs identified in the context of pathogen sensing can have large effect sizes and show evidence of recent selection [77,79–82]. These immune response eQTLs are therefore critical for understanding disease loci not explained by standard eQTLs.
Model systems unlock a treasure trove of dynamic QTL study possibilities
Response eQTL studies of the immune system are unusual in that they can be pursued in primary human tissues (albeit, in vitro). Despite the obvious physiological relevance of studies in primary tissues, practical and ethical barriers preclude response QTL experiments in most non-blood primary human cell types. In addition, challenges associated with serial sampling mean that time-evolving regulatory processes, such as disease progression, need to be studied ex vivo [82]. For these reasons, experiments in model organisms and in vitro systems play important complementary roles. Laboratory animal studies offer the opportunity to apply treatments to intact tissues composed of mixed cell types while controlling the living environment. Importantly, this allows the application of organism-level treatments that cannot be adequately recapitulated in vitro such as social, behavioral, or dietary manipulations. However, laboratory animals, in particular mice, have often been specifically bred to remove the genetic diversity that powers regulatory QTL studies [83–87]. One notable alternative has been to study captive primate populations [88–91]. These systems have been crucial for disentangling the genetic factors mediating immunological impacts of social stress. However, many of the obstacles to invasive or noxious sampling and treatment procedures in humans similarly limit the scope of studies possible in primates.
The shared limitations of dynamic regulatory experiments in humans and model organisms provide the motivation for in vitro models. The combination of uniform culture conditions, temporally dense sampling, and the possibility to apply pathogenic perturbations make in vitro models an extremely powerful platform for disease-relevant QTL mapping. Some of the earliest response eQTL experiments examined in vitro drug responses in humans, challenging cell lines with steroids, statins, and a range of other chemicals that play roles in disease risk or treatment. By comparing response eQTLs with variants already identified in GTEx and GWAS risk loci, these studies give insight into gene-environment interactions that may play a causal role in disease [92–94]. For example, Mangravite et al. treated lymphoblastoid cell lines with simvastatin and identified response eQTLs for the gene GATM, one of which increases the risk of statin drug toxicity in humans [93]. The authors also found that knocking down GATM expression in hepatocyte-related cell lines reduced secretion of an assayed LDL precursor.
Pushing the limits of the response eQTL study design paradigm, Moyerbrailean et al. exploited the sensitivity of allele-specific expression comparisons for small sample sizes, examining responses of a handful of primary human cell types to 250 environmental exposures [95]. Comparing their response eQTLs to known GWAS variants, they found a dramatic enrichment of SNP heritability among conditionally regulated genes, suggesting that experimentally modulating the cellular environment can have a large payoff in our attempt to explain the mechanisms of disease-associated loci.
The utility of an in vitro response eQTL platform is limited somewhat by the narrow range of available cell lines. In principle, a wide range of human cell types can be accessed through directed differentiation of induced pluripotent stem cell (iPSC) lines, complementing the tissue collections in GTEx with pure cell cultures. That said, many in vitro differentiation protocols yield cells that are essentially juvenile, if fully specified, and may fail to recapitulate important effects of aging on gene regulation [96,97].
Notwithstanding these caveats, the use of in vitro differentiated human cells to study gene regulation has proven to be a powerful approach and is only in its early stages. For example, induced cardiomyocytes have been used to identify genetic variants governing the transcriptional and splicing responses to doxorubicin chemotherapy, which are in turn predictive of toxicity [98]. While drug administration or alteration of a single environmental factor are straightforward treatments, they demonstrate the potential of iPSC systems for regulatory mapping, and we expect that future studies will dramatically extend their application with the use of additional cell types and novel in vitro stress treatments.
Dynamic gene regulation during development
A distinct advantage of iPSC models is the opportunity to track regulatory variation during a developmental process, essentially using time as the context of interest. Despite the importance of early development to disease, studies of prenatal tissue are rare. Post-mortem analysis of prenatal human brain samples, for example, has found a wealth of eQTLs that overlap with psychiatric disease mutations and are not prioritized in standard adult surveys [59,99]. Nonetheless, the inaccessibility of prenatal tissue makes it nearly impossible to study early gene regulatory events in vivo. Many important cell types and developmental events are transient or rare, making an in vitro platform especially useful for identifying regulatory variants that may manifest effects only fleetingly. Mutations of this sort have been identified recently by profiling expression during in vitro differentiation, uncovering eQTLs that are undetectable in the terminal cell type [100,101].
Transient developmental cis regulatory effects could alter gene expression in adult tissues, even as the cis effect diminishes, and could therefore appear later to act in trans (Figure 3c). For instance, a transient cis eQTL affecting SLC29A8 expression in human monocytes shortly after stimulation was recently linked to metallothionein gene expression hours later, appearing only as a single trans effect at the end timepoint [102]. Alternatively, early developmental cis eQTLs can impact adult tissues through mechanisms acting on chromatin state or by altering cell type composition. Cis-acting variants that affect cell type determinants could have even more wide-ranging apparent trans effects, highlighting the value of accessible in vitro systems for developmental studies.
Figure 3:
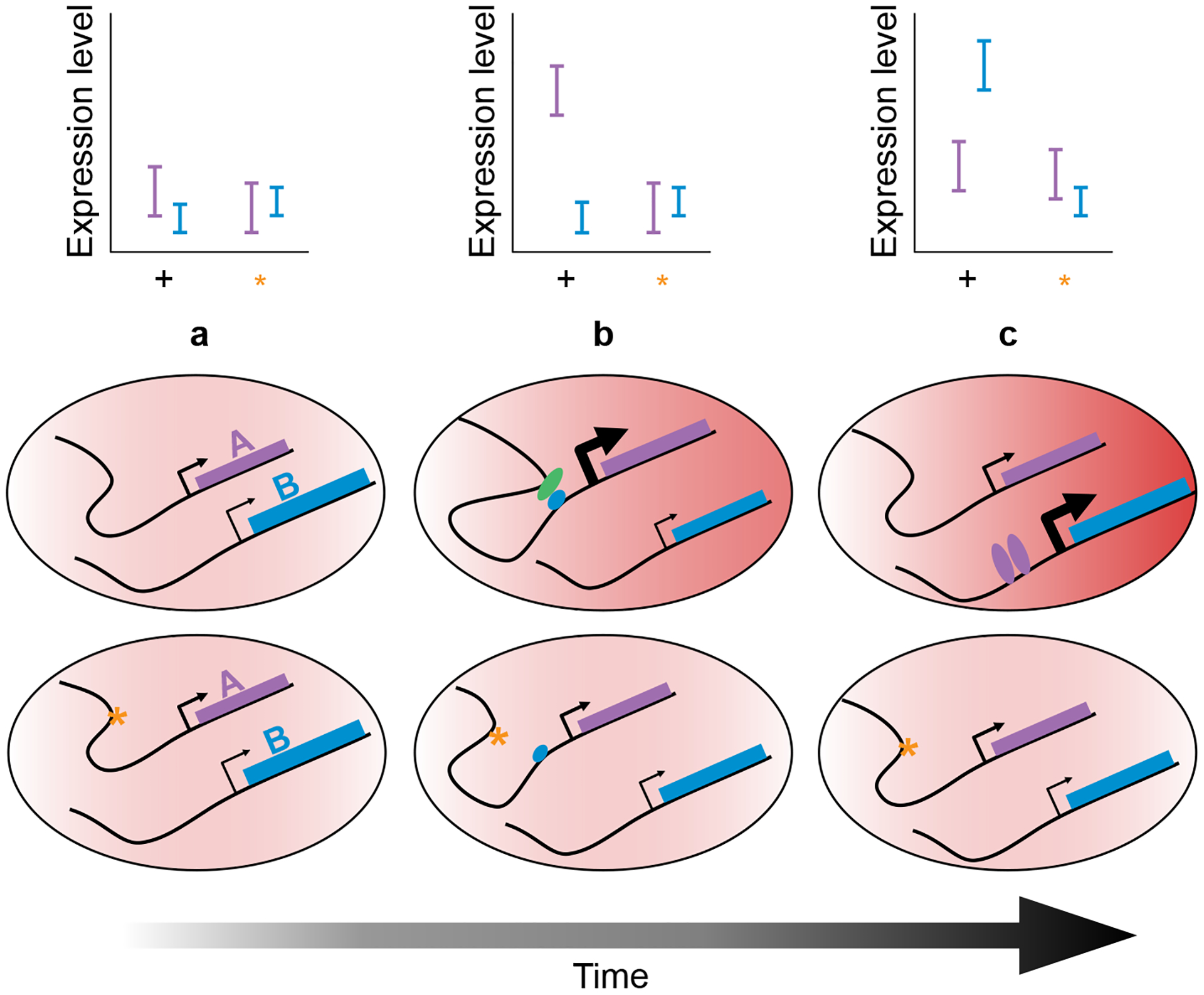
Hypothetical relationship revealed by dynamic eQTL mapping. At time (a), genotype has no detectable effect on expression. In response to changing cellular conditions, transcription of gene A is induced, but attenuated by a cis-eQTL (orange asterisk, b). At time (c), the product of gene A regulates a distal locus, leading to phenotypic divergence of the two cells. Importantly, the cis eQTL is no longer detectable and appears only as a trans effect.
Taken together, iPSC-based strategies offer tremendous potential for studying dynamic gene regulation that has yet to be fully realized. While in vitro experiments have a number of technical advantages, the number of cell types that can be obtained through directed differentiation is still small and their fidelity to primary human cells is often unclear. These constitute potential obstacles to in vitro studies of physiology but not gene regulation: the regulatory QTLs identified in vitro demonstrate their value by explaining features of adult tissues, not the model system itself. As the range of cell types that can be generated in vitro expands, the test of their usefulness will lie in the predictive power of the genotypes that they allow us to identify, and the added power that this information lends to GWAS.
Single-cell technologies empower new QTL study designs
Single-cell sequencing techniques now make it possible to detect cell type-specific changes in a heterogeneous culture. This technological breakthrough has opened the door to studies in developmental systems that yield mixed cell types, such as neurons [103]. By sampling across the duration of the developmental time course, we can now access the full range of intermediate transitional states encountered during lineage decisions and identify regulatory effects reaching across cell types. In one notable first step in this direction, single-cell QTL mapping was applied to a three-day developmental course from human iPSC to definitive endoderm, uncovering nearly 900 dynamic eQTLs [101]. Importantly, single-cell resolution allowed the authors to define developmental stages based on the pseudo-temporal ordering of individual transcriptomes, rather than relying on the fixed schedule of sample collection as a proxy. This result also demonstrates the value that might be expected from applying single-cell expression profiling in order to finely resolve cell types from complex primary tissues (Figure 4a).
Figure 4:
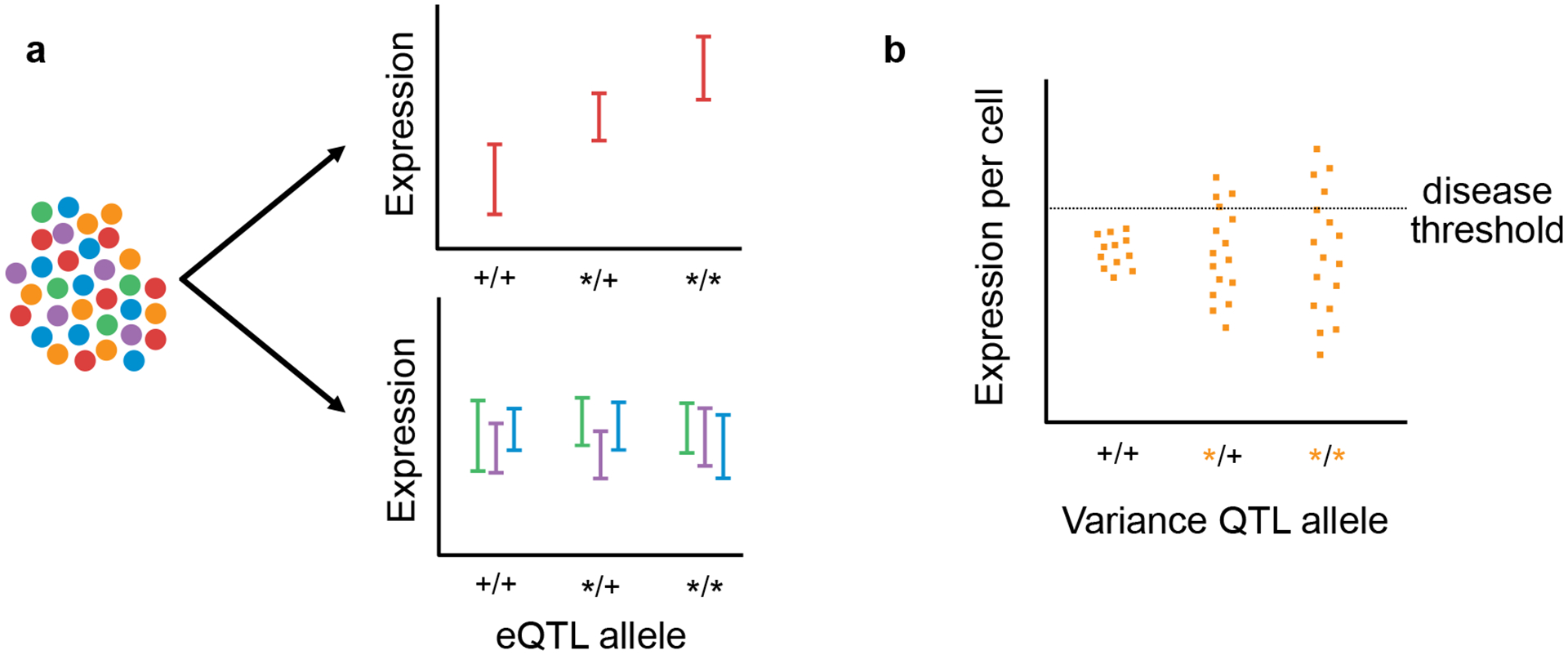
Single-cell profiling can uncover unique regulatory relationships. (a) Single-cell resolution can help uncover eQTLs affecting rare cell types or cell states in primary tissue or mixed cultures. (b) Identifying genetic variants that affect expression dispersion, rather than mean expression levels, may help explain incompletely penetrant phenotypes.
An important next step will be to combine these approaches to efficiently identify response eQTLs particular to a given set of environmental and developmental conditions, such as might be experienced by stress during a critical developmental window. Recently, Jerber et al. provided a first step in this direction as they characterized gene expression in 215 iPSC lines during a 52-day neuronal differentiation procedure while challenging cells with an oxidative stressor [104]. Although the effects of stress at different stages of differentiation were not assessed, the majority of novel eQTLs that colocalized with GWAS variants were identified in late stages of differentiation or under the oxidative stress condition. As such, these results are consistent with the notion that many phenotypically important variants may have limited scopes of action.
In the future, single-cell profiling will be critical for studies of QTLs that affect not only the magnitude of dynamic expression responses, but also their rate and timing. Furthermore, single-cell resolution enables measurement of expression variance within individuals, allowing us to begin studying QTLs that impact the magnitude of biological regulatory noise. Highly variable expression levels may play a key role in disease by allowing a subset of cells to cross a pathological expression threshold while the majority remain healthy. Thus, variance QTLs could lead to incompletely penetrant phenotypes without affecting mean expression levels (Figure 4b). Such breakdowns in robustness during key moments in development or disease progression could have significant consequences. At present, however, variance QTLs are difficult to detect. Pilot experiments using single-cell gene expression data have indicated that thousands of subjects would be required for adequately powered studies, in large part owing to the dominance of non-genetic influences on expression variance [105] (but see also [106] for a discussion of how trans-eQTL effects may impact the power analysis). Future experimental systems designed to accommodate the limitations of single-cell profiling technologies may, therefore, open new frontiers for studying disease.
Concluding remarks
Standard eQTL studies have generated valuable resources for understanding human gene regulation. Just as importantly, these studies have provided the data necessary to critically consider our expectations regarding the connection between regulatory variation and human disease, and to frame the next important questions (see Outstanding Questions). Despite rigorous efforts, standard eQTLs do not, on the whole, provide the missing mechanistic link to the majority of disease-associated genetic variants. Mutations associated with disease may act by altering gene expression under specific conditions, requiring a different experimental framework to prioritize these effects. The dramatic enrichment of disease-associated SNPs among eQTLs identified in such dynamic regulatory experiments suggests that this is an important way forward.
Outstanding Questions.
What is the theoretical limit of the expected overlap between true causal disease-associated mutations and regulatory QTLs?
Will single-cell gene expression studies result in the identification of eQTLs with qualitatively different properties than standard QTLs? Furthermore, can a cell type-specific eQTL reference set accurately predict gene expression in unsampled tissues currently omitted from GTEx?
We highlight two broad classes of dynamic regulatory studies, which use either developmental time or exogenous treatments to unmask response QTLs. How can these and other classes of dynamic manipulations be combined in efficient study designs to comprehensively map regulation under diverse combinations of conditions?
Disease-relevant perturbations, such as immune cell stimulation or drug treatment, have been useful for identifying response QTLs in vitro. The realm of possibilities is almost limitless. Will the most informative manipulations engage core cellular response pathways, or processes specific to a given cell type’s function?
Our discussion has focused primarily on expression QTLs, both because of the rich efforts that have been dedicated to their study and the fact that transcription is a meaningful summary readout of many chromatin-level molecular features. However, the dynamic regulatory mapping framework is equally applicable to other molecular QTLs, and integrative analysis of different regulatory phenotypes will allow for deeper mechanistic insights [107]. New techniques to simultaneously measure multiple molecular features in individual cells will doubtless advance this effort as regulatory QTL studies continue to move to the single-cell level [108]. Indeed, the convergence of iPSC and single-cell profiling technologies offers especially timely opportunities for extending regulatory QTL experiments to study dynamic processes and responses to perturbations. Fully capitalizing on these new opportunities promises to reveal further insights about disease risk mechanisms.
Highlights.
Mapping regulatory QTLs has emerged as a powerful tool to functionally annotate non-coding DNA variants that are associated with disease risk.
Large surveys of gene expression variation in healthy, adult, steady-state tissues have discovered at least one cis eQTL for nearly every human gene.
Standard eQTLs have properties that may be inconsistent with mutations that are associated with a fitness cost, as might be expected for mutations associated with disease.
Regulatory QTL mapping during dynamic cellular processes such as differentiation and perturbation response can reveal otherwise-hidden regulatory variation that may be especially relevant for disease.
New platform technologies, including in vitro differentiated cell types and single-cell profiling, extend the scope of dynamic eQTL studies.
Acknowledgments
We thank Natalia Gonzales, Jonathan Pritchard, Nicholas Banovich, Gabriella Haddad, and Michelle Ward for useful discussions, comments, and edits to the manuscript. AB is supported by NIH grant R01GM120167; YG is supported by NIH grant R35GM131726
Glossary
- GWAS
Genome-wide Association Study. A method for measuring the correlation between genetic variants and phenotypes, such as disease status.
- SNP
single-nucleotide polymorphism. A class of genetic variant consisting of single-letter changes in a DNA sequence.
- Mendelian Randomization
A form of instrumental variable regression used to test the causal effect of an environmental exposure on a phenotype. Genetic variants that cause measurable changes in the environmental exposure (e.g., QTLs) are compared to phenotypes, helping to avoid complications of reverse causality. Mendelian Randomization can be performed using summary-level GWAS data.
- Linkage disequilibrium (LD)
Correlated occurrence of alleles at different loci in a population. Strong LD can allow an assayed allele to “tag” or serve as a proxy for other unspecified alleles.
- Nonsynonymous mutation
A mutation within the protein-coding portion of a gene that changes the encoded amino acid sequence.
- Induced pluripotent stem cells (iPSCs)
Stem cell lines that have been obtained through the in vitro reprogramming of adult somatic cells to a pluripotent state. Through in vitro differentiation procedures, iPSCs serve as renewable sources for obtaining isogenic cell types that might not otherwise be available for study.
- Standard eQTL
A genetic variant (generally a SNP) associated with the expression level of a nearby gene under steady-state conditions. Standard eQTLs are generally assayed in adult tissues or cell lines.
- Tissue-specific eQTL
A genetic variant associated with the steady-state expression level of a nearby gene only in a particular tissue. Tissue-specific eQTLs may derive their specificity by affecting the binding of a tissue-specific transcription factor or chromatin-modifying enzyme, or by other mechanisms that confine the scope of their effect. An eQTL’s designation as “tissue-specific” is not subject to a single standard, and different investigators have set the threshold anywhere from the level of whole organs to molecularly defined cell types.
- Dynamic eQTL
An eQTL ascertained under time-evolving conditions. We use this term to encompass eQTLs revealed by the application of an extrinsic perturbation, sometimes termed “response eQTLs,” as well as those that manifest transiently during the course of a developmental process.
References
- 1.Farh KK-H et al. (2015) Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manolio TA et al. (2009) Finding the missing heritability of complex diseases. Nature 461, 747–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maurano MT et al. (2012) Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gamazon ER et al. (2018) Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat Genet 50, 956–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smemo S et al. (2014) Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507, 371–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Claussnitzer M et al. (2015) FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N Engl J Med 373, 895–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cross-Disorder Group of the Psychiatric Genomics Consortium (2019) Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders. Cell 179, 1469–1482.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fadason T et al. (2018) Chromatin interactions and expression quantitative trait loci reveal genetic drivers of multimorbidities. Nat Commun 9, 5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang J et al. (2010) Common SNPs explain a large proportion of the heritability for human height. Nat Genet 42, 565–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi H et al. (2016) Contrasting the Genetic Architecture of 30 Complex Traits from Summary Association Data. Am J Hum Genet 99, 139–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cusanovich DA et al. (2012) The combination of a genome-wide association study of lymphocyte count and analysis of gene expression data reveals novel asthma candidate genes. Human Molecular Genetics 21, 2111–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hormozdiari F et al. (2016) Colocalization of GWAS and eQTL Signals Detects Target Genes. Am J Hum Genet 99, 1245–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hormozdiari F et al. (2014) Identifying causal variants at loci with multiple signals of association. Genetics 198, 497–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giambartolomei C et al. (2014) Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 10, e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He X et al. (2013) Sherlock: detecting gene-disease associations by matching patterns of expression QTL and GWAS. Am J Hum Genet 92, 667–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeng B et al. (2019) Comprehensive Multiple eQTL Detection and Its Application to GWAS Interpretation. Genetics 212, 905–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barbeira AN et al. (2018) Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat Commun 9, 1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gusev A et al. (2016) Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet 48, 245–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gamazon ER et al. (2015) A gene-based association method for mapping traits using reference transcriptome data. Nat Genet 47, 1091–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu Z et al. (2016) Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 48, 481–487 [DOI] [PubMed] [Google Scholar]
- 21.Gamazon ER et al. (2019) Multi-tissue transcriptome analyses identify genetic mechanisms underlying neuropsychiatric traits. Nat Genet 51, 933–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marbach D et al. (2016) Tissue-specific regulatory circuits reveal variable modular perturbations across complex diseases. Nat Methods 13, 366–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van der Wijst MGP et al. (2018) Single-cell RNA sequencing identifies celltype-specific cis-eQTLs and co-expression QTLs. Nat Genet 50, 493–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saha A et al. (2017) Co-expression networks reveal the tissue-specific regulation of transcription and splicing. Genome Res 27, 1843–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marigorta UM et al. (2017) Transcriptional risk scores link GWAS to eQTLs and predict complications in Crohn’s disease. Nat Genet 49, 1517–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicolae DL et al. (2010) Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet 6, e1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Battle A et al. (2014) Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res 24, 14–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhernakova DV et al. (2017) Identification of context-dependent expression quantitative trait loci in whole blood. Nat Genet 49, 139–145 [DOI] [PubMed] [Google Scholar]
- 29.Aguet F et al. (2019) The GTEx Consortium atlas of genetic regulatory effects across human tissues. bioRxiv DOI: 10.1101/787903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chun S et al. (2017) Limited statistical evidence for shared genetic effects of eQTLs and autoimmune-disease-associated loci in three major immune-cell types. Nat Genet 49, 600–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Consortium GTEx et al. (2017) Genetic effects on gene expression across human tissues. Nature 550, 204–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yao DW et al. (2020) Quantifying genetic effects on disease mediated by assayed gene expression levels. Nat Genet 337, 1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grundberg E et al. (2012) Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat Genet 44, 1084–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu X et al. (2019) Trans Effects on Gene Expression Can Drive Omnigenic Inheritance. Cell 177, 1022–1034.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boyle EA et al. (2017) An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 169, 1177–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Westra H-J et al. (2013) Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet 45, 1238–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mortlock S et al. (2020) Tissue specific regulation of transcription in endometrium and association with disease. Hum. Reprod 35, 377–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vosa U et al. (2018) Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv DOI: 10.1101/447367 [DOI] [Google Scholar]
- 39.Yang F et al. (2017) Identifying cis-mediators for trans-eQTLs across many human tissues using genomic mediation analysis. Genome Res 27, 1859–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wray GA (2007) The evolutionary significance of cis-regulatory mutations. Nat Rev Genet 8, 206–216 [DOI] [PubMed] [Google Scholar]
- 41.Carroll SB (2000) Endless forms: the evolution of gene regulation and morphological diversity. Cell 101, 577–580 [DOI] [PubMed] [Google Scholar]
- 42.Halachev M et al. (2019) Increased ultra-rare variant load in an isolated Scottish population impacts exonic and regulatory regions. PLoS Genet 15, e1008480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lek M et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fu J et al. (2012) Unraveling the regulatory mechanisms underlying tissue-dependent genetic variation of gene expression. PLoS Genet 8, e1002431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He Y et al. (2019) Mechanisms of tissue-specific genetic regulation revealed by latent factors across eQTLs. bioRxiv DOI: 10.1101/785584 [DOI] [Google Scholar]
- 46.Westra H-J et al. (2015) Cell Specific eQTL Analysis without Sorting Cells. PLoS Genet 11, e1005223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmiedel BJ et al. (2018) Impact of Genetic Polymorphisms on Human Immune Cell Gene Expression. Cell 175, 1701–1715.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim-Hellmuth S et al. (2019) Cell type specific genetic regulation of gene expression across human tissues. bioRxiv DOI: 10.1101/806117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van der Wijst M et al. (2020) The single-cell eQTLGen consortium. Elife 9, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Orozco LD et al. (2020) Integration of eQTL and a Single-Cell Atlas in the Human Eye Identifies Causal Genes for Age-Related Macular Degeneration. Cell Rep 30, 1246–1259.e6 [DOI] [PubMed] [Google Scholar]
- 51.UK10K Consortium et al. (2015) The UK10K project identifies rare variants in health and disease. Nature 526, 82–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maher MC et al. (2012) Population genetics of rare variants and complex diseases. Hum Hered 74, 118–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grossman SR et al. (2013) Identifying recent adaptations in large-scale genomic data. Cell 152, 703–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kudaravalli S et al. (2009) Gene expression levels are a target of recent natural selection in the human genome. Molecular Biology and Evolution 26, 649–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li X et al. (2017) The impact of rare variation on gene expression across tissues. Nature Publishing Group 550, 239–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hernandez RD et al. (2019) Ultrarare variants drive substantial cis heritability of human gene expression. Nat Genet 51, 1349–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zeng Y et al. (2015) Aberrant gene expression in humans. PLoS Genet 11, e1004942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Glassberg EC et al. (2019) Evidence for Weak Selective Constraint on Human Gene Expression. Genetics 211, 757–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walker RL et al. (2019) Genetic Control of Expression and Splicing in Developing Human Brain Informs Disease Mechanisms. Cell 179, 750–771.e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cummings BB et al. (2020) Transcript expression-aware annotation improves rare variant discovery and interpretation. 7, 10370–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khan Z et al. (2013) Primate transcript and protein expression levels evolve under compensatory selection pressures. Science 342, 1100–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Battle A et al. (2015) Genomic variation. Impact of regulatory variation from RNA to protein. Science 347, 664–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu L et al. (2013) Variation and genetic control of protein abundance in humans. Nature 499, 79–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Buccitelli C and Selbach M (2020) mRNAs, proteins and the emerging principles of gene expression control. Nat Rev Genet 204, 407. [DOI] [PubMed] [Google Scholar]
- 65.Romero IG et al. (2012) Comparative studies of gene expression and the evolution of gene regulation. Nat Rev Genet 13, 505–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fair BJ et al. (2020) Gene expression variability in human and chimpanzee populations share common determinants. bioRxiv DOI: 10.1101/2020.06.11.146142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jasinska AJ et al. (2017) Genetic variation and gene expression across multiple tissues and developmental stages in a nonhuman primate. Nat Genet 49, 1714–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tung J et al. (2015) The genetic architecture of gene expression levels in wild baboons. Elife 4, 1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tanaka T and Nei M (1989) Positive darwinian selection observed at the variable-region genes of immunoglobulins. Molecular Biology and Evolution 6, 447–459 [DOI] [PubMed] [Google Scholar]
- 70.Ye Y et al. (2020) A Multi-Omics Perspective of Quantitative Trait Loci in Precision Medicine. Trends Genet 36, 318–336 [DOI] [PubMed] [Google Scholar]
- 71.O’Connor LJ et al. (2019) Extreme Polygenicity of Complex Traits Is Explained by Negative Selection. Am J Hum Genet 105, 456–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Taylor DL et al. (2018) Interactions between genetic variation and cellular environment in skeletal muscle gene expression. PLoS ONE 13, e0195788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alasoo K et al. (2018) Shared genetic effects on chromatin and gene expression indicate a role for enhancer priming in immune response. Nat Genet 50, 424–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Calderon D et al. (2019) Landscape of stimulation-responsive chromatin across diverse human immune cells. Nat Genet 51, 1494–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fairfax BP et al. (2014) Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 343, 1246949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gutierrez-Arcelus M et al. (2020) Allele-specific expression changes dynamically during T cell activation in HLA and other autoimmune loci. Nat Genet 52, 247–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim-Hellmuth S et al. (2017) Genetic regulatory effects modified by immune activation contribute to autoimmune disease associations. Nat Commun 8, 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fairfax BP et al. (2012) Genetics of gene expression in primary immune cells identifies cell type-specific master regulators and roles of HLA alleles. Nat Genet 44, 502–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Barreiro LB et al. (2012) Deciphering the genetic architecture of variation in the immune response to Mycobacterium tuberculosis infection. Proc Natl Acad Sci USA DOI: 10.1073/pnas.1115761109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nédélec Y et al. (2016) Genetic Ancestry and Natural Selection Drive Population Differences in Immune Responses to Pathogens. Cell 167, 657–669.e21 [DOI] [PubMed] [Google Scholar]
- 81.Lee MN et al. (2014) Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science 343, 1246980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ye CJ et al. (2014) Intersection of population variation and autoimmunity genetics in human T cell activation. Science 345, 1254665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chick JM et al. (2016) Defining the consequences of genetic variation on a proteome-wide scale. Nature 534, 500–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Keele GR et al. (2020) Integrative QTL analysis of gene expression and chromatin accessibility identifies multi-tissue patterns of genetic regulation. PLoS Genet 16, e1008537–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Keele GR et al. (2019) Determinants of QTL Mapping Power in the Realized Collaborative Cross. G3 9, 1707–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lusis AJ et al. (2016) The Hybrid Mouse Diversity Panel: a resource for systems genetics analyses of metabolic and cardiovascular traits. J. Lipid Res 57, 925–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hasin-Brumshtein Y et al. (2016) Hypothalamic transcriptomes of 99 mouse strains reveal trans eQTL hotspots, splicing QTLs and novel non-coding genes. Elife 5, 3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sanz J et al. (2019) Social history and exposure to pathogen signals modulate social status effects on gene regulation in rhesus macaques. Proceedings of the National Academy of Sciences 186, 201820846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Snyder-Mackler N et al. (2019) Social status alters chromatin accessibility and the gene regulatory response to glucocorticoid stimulation in rhesus macaques. Proceedings of the National Academy of Sciences 116, 1219–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Snyder-Mackler N et al. (2016) Social status alters immune regulation and response to infection in macaques. Science 354, 1041–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tung J et al. (2012) Social environment is associated with gene regulatory variation in the rhesus macaque immune system. Proceedings of the National Academy of Sciences 109, 6490–6495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Maranville JC et al. (2011) Interactions between glucocorticoid treatment and cis-regulatory polymorphisms contribute to cellular response phenotypes. PLoS Genet 7, e1002162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mangravite LM et al. (2013) A statin-dependent QTL for GATM expression is associated with statin-induced myopathy. Nature 502, 377–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Findley AS et al. (2019) Interpreting Coronary Artery Disease Risk Through Gene-Environment Interactions in Gene Regulation. Genetics 213, 651–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Moyerbrailean GA et al. (2016) High-throughput allele-specific expression across 250 environmental conditions. Genome Res 26, 1627–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Studer L et al. (2015) Programming and Reprogramming Cellular Age in the Era of Induced Pluripotency. Cell Stem Cell 16, 591–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Balliu B et al. (2019) Genetic regulation of gene expression and splicing during a 10-year period of human aging. Genome Biology 20, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Knowles DA et al. (2018) Determining the genetic basis of anthracycline-cardiotoxicity by molecular response QTL mapping in induced cardiomyocytes. Elife 7, 1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.la Torre-Ubieta de, L. et al. (2018) The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis. Cell 172, 289–295.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Strober BJ et al. (2019) Dynamic genetic regulation of gene expression during cellular differentiation. Science 364, 1287–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cuomo ASE et al. (2020) Single-cell RNA-sequencing of differentiating iPS cells reveals dynamic genetic effects on gene expression. Nat Commun 11, 810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kolberg L et al. (2020) Co-expression analysis reveals interpretable gene modules controlled by trans-acting genetic variants. 51, 592–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schwartzentruber J et al. (2018) Molecular and functional variation in iPSC-derived sensory neurons. Nat Genet 50, 54–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jerber J et al. (2020) Population-scale single-cell RNA-seq profiling across dopaminergic neuron differentiation. bioRxiv DOI: 10.1101/2020.05.21.103820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sarkar AK et al. (2019) Discovery and characterization of variance QTLs in human induced pluripotent stem cells. PLoS Genet 15, e1008045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Morgan MD et al. (2020) Quantitative genetic analysis deciphers the impact of cis and trans regulation on cell-to-cell variability in protein expression levels. PLoS Genet 16, e1008686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Husquin LT et al. (2018) Exploring the genetic basis of human population differences in DNA methylation and their causal impact on immune gene regulation. Genome Biology 19, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ma S et al. (2020) Chromatin potential identified by shared single cell profiling of RNA and chromatin. bioRxiv DOI: 10.1101/2020.06.17.156943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li YI et al. (2016) RNA splicing is a primary link between genetic variation and disease. Science 352, 600–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wu Y et al. (2018) Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat Commun 9, 918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hormozdiari F et al. (2018) Leveraging molecular quantitative trait loci to understand the genetic architecture of diseases and complex traits. Nat Genet 50, 1041–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lappalainen T et al. (2013) Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Alasoo K et al. (2019) Genetic effects on promoter usage are highly context-specific and contribute to complex traits. Elife 8, 12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Knowles DA et al. (2017) Allele-specific expression reveals interactions between genetic variation and environment. Nat Methods 14, 699–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wilson MD et al. (2008) Species-specific transcription in mice carrying human chromosome 21. Science 322, 434–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Long HK et al. (2016) Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 167, 1170–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Indjeian VB et al. (2016) Evolving New Skeletal Traits by cis-Regulatory Changes in Bone Morphogenetic Proteins. Cell 164, 45–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jones FC et al. (2012) The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484, 55–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nica AC et al. (2011) The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS Genet 7, e1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Flutre T et al. (2013) A statistical framework for joint eQTL analysis in multiple tissues. PLoS Genet 9, e1003486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sul JH et al. (2013) Effectively identifying eQTLs from multiple tissues by combining mixed model and meta-analytic approaches. PLoS Genet 9, e1003491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li G et al. (2018) An empirical Bayes approach for multiple tissue eQTL analysis. Biostatistics 19, 391–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.ENCODE Project Consortium et al. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Roadmap Epigenomics Consortium et al. (2015) Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 [DOI] [PMC free article] [PubMed] [Google Scholar]