

Abstract
Background/Aims
Diabetic nephropathy (DN) is one of the main causes of end-stage kidney disease worldwide. Emerging studies have suggested that its pathogenesis is distinct from nondiabetic renal diseases in many aspects. However, it still lacks a comprehensive understanding of the unique molecular mechanism of DN.
Methods
A total of 255 Affymetrix U133 microarray datasets (Affymetrix, Santa Calra, CA, USA) of human glomerular and tubulointerstitial tissues were collected. The 22 215 Affymetrix identifiers shared by the Human Genome U133 Plus 2.0 and U133A Array were extracted to facilitate dataset pooling. Next, a linear model was constructed and the empirical Bayes method was used to select the differentially expressed genes (DEGs) of each kidney disease. Based on these DEG sets, the unique DEGs of DN were identified and further analyzed using gene ontology and pathway enrichment analysis. Finally, the protein–protein interaction networks (PINs) were constructed and hub genes were selected to further refine the results.
Results
A total of 129 and 1251 unique DEGs were identified in the diabetic glomerulus (upregulated n = 83 and downregulated n = 203) and the diabetic tubulointerstitium (upregulated n = 399 and downregulated n = 874), respectively. Enrichment analysis revealed that the DEGs in the diabetic glomerulus were significantly associated with the extracellular matrix, cell growth, regulation of blood coagulation, cholesterol homeostasis, intrinsic apoptotic signaling pathway and renal filtration cell differentiation. In the diabetic tubulointerstitium, the significantly enriched biological processes and pathways included metabolism, the advanced glycation end products–receptor for advanced glycation end products signaling pathway in diabetic complications, the epidermal growth factor receptor (EGFR) signaling pathway, the FoxO signaling pathway, autophagy and ferroptosis. By constructing PINs, several nodes, such as AGR2, CSNK2A1, EGFR and HSPD1, were identified as hub genes, which might play key roles in regulating the development of DN.
Conclusions
Our study not only reveals the unique molecular mechanism of DN but also provides a valuable resource for biomarker and therapeutic target discovery. Some of our findings are promising and should be explored in future work.
Keywords: bioinformatics, diabetic nephropathy, microarray, molecular mechanisms, transcriptome
INTRODUCTION
Owing to the increasing prevalence of diabetes mellitus, diabetic nephropathy (DN), which is one of its major microvascular complications, has become a worldwide public health problem [1]. DN is associated with more rapid progression, higher risk of cardiovascular disease and higher mortality rates compared with nondiabetic renal diseases (NDRDs). However, only a few antidiabetic drugs, including renin–angiotensin system (RAS) inhibitors and sodium–glucose co-transporter-2 (SGLT2) inhibitors, have been demonstrated to be beneficial to survival. The onset of microalbuminuria (MA) is regarded as an early marker of kidney injury in DN. However, emerging evidence has shown that MA is not an ideal diagnostic tool. As suggested by a recent biopsy study, comorbidity of DN and NDRD is observed in nearly 50% of patients with DN [2]. Therefore a comprehensive understanding of the unique molecular mechanism of DN is urgently needed to improve the diagnosis and treatment.
Numerous animal studies have suggested that the pathogenesis of DN is distinct from NDRDs in many aspects. These studies constructed our knowledge of the molecular mechanism of DN by steadily laying ‘stone on stone’. However, there is still a lack of large-scale, parallel and nonbiased comparisons regarding the molecular differences between DN and NDRD.
For the past 20 years, high-throughput technologies and bioinformatics have emerged as powerful tools to identify the molecular signatures of diseases. For example, using gene set enrichment analysis, a recent study identified 20 hub genes, including albumin (ALB), epidermal growth factor (EGF) and collagen type IV alpha 1 chain (COL4A1), in diabetic tubulointerstitial tissues [3]. Another RNA sequencing study found a set of unique genes for early DN by comparing its transcriptomic profile with advanced DN [4]. However, these studies did not include other chronic kidney diseases (CKDs) and only contained several datasets. To this end, we aim to provide integrative insight into the unique molecular mechanism of DN using a bioinformatics analysis of >250 microarray datasets.
MATERIALS AND METHODS
Dataset collection
The overall study design is presented in Figure 1. First, human microarray datasets including DN, NDRDs [hypertensive nephropathy (HN) and three types of glomerulonephritis (GN) including membranous nephropathy (MN), focal segmental glomerular sclerosis (FSGS) and immunoglobulin A nephropathy (IgAN)] and healthy controls were collected from gene expression omnibus (https://www.ncbi.nlm.nih.gov/geo/) in June 2019 according to the search criteria described in the supplemental documentation. To minimize the platform variation, all datasets were generated by either the Affymetrix U133 Plus 2.0 or U133 Array (Affymetrix, Santa Clara, CA, USA) , which both belong to the Affymetrix Human Genome U133 platform. The summary and sample description of each dataset were carefully evaluated by two investigators (L.T. Z. and Y.S.Z.) before being included.
FIGURE 1:

Overall study design and bioinformatics analysis workflow. The methods used in corresponding processes were presented in bold type in the figure. Quality control: QC stats in the simplify package and RNA degradation analysis in the affy package; data preprocessing: the RMA method in the affy package; DEG selection: the empirical Bayes method in the limma package and RMA: robust multiarray average.
Microarray dataset processing and DEG identification
The bioinformatics analysis was performed using R (version 3.6.0; R Foundation for Statistical Computing, Vienna, Austria) according to the systematic workflow described in Figure 1 [5]. Briefly, the quality of each dataset was examined using the general quality control stats in the simplify package and the RNA degradation analysis in the affy package. Then the 22 215 shared Affymetrix identifiers of the Human Genome U133 platform were extracted and datasets of the same category were pooled to expand the sample size. In particular, the healthy controls were pooled from all studies regardless of kidney diseases. The relative log expression graph was used to evaluate the consistency among datasets and those with significant bias were discarded. Then the robust multiarray average method in the affy package was used to preprocess the original data. Lastly, the empirical Bayes method in the limma package was used to select differentially expressed genes (DEGs). Statistically significant DEGs were defined as those with a P-value <0.05 after adjustment by the Benjamini–Hochberg method and fold changes >1.5.
Then we defined the unique DEGs of DN as those dysregulated in DN but not in the other types of kidney diseases. Mathematically, it can be written as uniDN = DN − (IgAN∪MN∪FSGS∪HN), where uniDN denotes the unque DEG set of DN and ∪ denotes the union operation. Let GN = (IgAN∪MN∪FSGS) and the final equation becomes uniDN = DN − (GN∪HN)’, from which the unique DEGs of DN were identified.
GO and pathway enrichment analysis
The enrichment analysis was performed using the cluster Profiler package (version 3.12.0) in R [6]. Specifically, gene ontology (GO) annotation (http://geneontology.org/), the Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway (http://www.genome.jp/kegg) and the Reactome Pathway (http://www.reactome.org) query were realized via Annotation Hub, the KEGG Pathway Module and ReactomePA, respectively. The ggplot2 and enrich plot packages were used for visualization. A hypergeometric test was applied for enrichment analysis. P-values <0.05 after adjustment by the Benjamini–Hochberg method were regarded as statistically significant.
Construction of the protein–protein interaction network (PIN), recognition of hub genes and cluster analysis
The PIN analysis was performed based on three databases:the Biological General Repository for Interaction Datasets (BioGRID;http://thebiogrid.org/), the Database of Interacting Proteins (DIP; http://dip.doe-mbi.ucla.edu/) and the Human Protein Reference Database (HPRD; http://www.hprd.org/). Cytoscape software (version 3.6.0) was utilized to visualize PINs and analyze the characteristics of each node. The degree method in the CytoHubba plug-in was used to identify hub genes, defined as the number of edges incident to each node [7].
RESULTS
Characteristics of datasets
As presented in Table 1, a total of 255 Affymetrix U133 microarray datasets including biopsy-proven CKDs (DN tubulointerstitium n = 11, DN glomeruli n = 7; HN tubulointerstitium n = 21, HN glomeruli n = 15, IgAN tubulointerstitium n = 25, IgAN glomeruli n = 43; MN tubulointerstitium n = 18, MN glomeruli n = 18; FSGS tubulointerstitium n = 12, FSGS glomeruli n = 23) and healthy controls (normal tubulointerstitium n = 22, normal glomeruli n = 40) were collected. Healthy controls were either patients after tumor nephrectomy or pretransplant living donors. The resources of the datasets include GSE37463 [8], GSE47185 [9], GSE35489 [10], GSE21785 [11], GSE20602 [12], GSE50469 [13] and GSE69438 [14]. The baseline clinical characteristics of each included cohort can be found in the supplemental documentation.
Table 1.
Datasets included in the study after QC
| Sample names | Cases, n | Controls, n | Resources | Platforms |
|---|---|---|---|---|
| DN glomeruli | 7 | 18 | GSE37463 [8], GSE47185 [9] | Affymetrix U133 Plus 2.0 |
| DN tubulointerstitium | 11 | 22 | GSE35489 [10], GSE47185 [9] | Affymetrix U133 A |
| HN glomeruli | 15 | 22 | Affymetrix U133 A | |
| HN tubulointerstitium | 21 | 22 | GSE47185 [9], GSE37463 [8] | Affymetrix U133 A |
| IgAN glomeruli | 43 | 22 | Affymetrix U133 A | |
| IgAN tubulointerstitium | 25 | 22 | GSE35489 [10], GSE47185 [9] | Affymetrix U133 A |
| MN glomeruli | 18 | 22 | Affymetrix U133 A | |
| MN tubulointerstitium | 18 | 22 | Affymetrix U133 A | |
| FSGS glomeruli | 23 | 40 | GSE37463 [8], GSE47185 [9], GSE20602 [12], GSE21785 [11] |
Affymetrix U133 A Affymetrix U133 Plus 2.0 |
| FSGS tubulointerstitium | 12 | 22 | GSE35489 [10], GSE47185 [9] | Affymetrix U133 A |
Unique DEGs of DN
First, we constructed the gene expression patterns of all CKDs (Figure 2). As shown in Figure 3, we identified 417 DEGs (upregulated n = 303, downregulated n = 114) in the diabetic glomerulus, among which 129 DEGs were unique to DN (30.9%; upregulated n = 83, downregulated n = 46). In comparison, many more transcripts (upregulated n = 1291, downregulated n = 926) were dysregulated in the glomerular compartment of GN (Figure 2).
FIGURE 2:
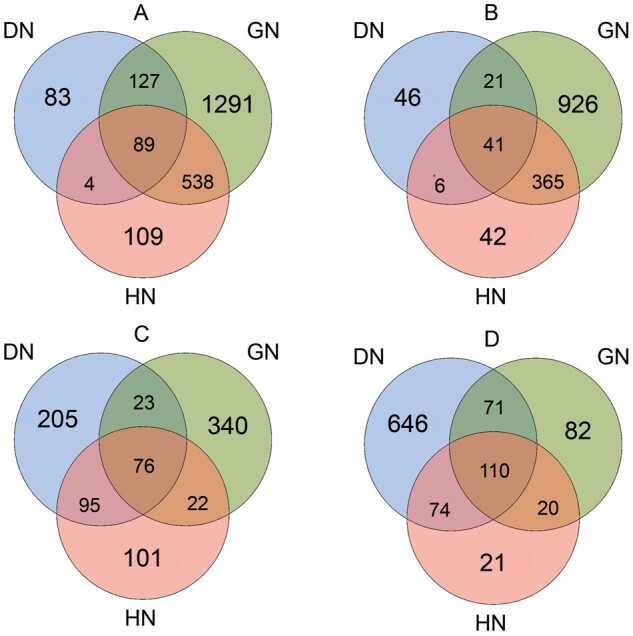
Venn plots for identification of the unique DEGs in DN. Different colors represent different primary diseases. Overlapping areas represent shared DEGs. (A) Upregulated genes in glomerular compartments in various types of CKD; (B) downregulated genes in glomerular compartments in various types of CKD; (C) upregulated genes in tubulointerstitial compartments in various types of CKD and (D) downregulated genes in tubulointerstitial compartments in various types of CKD.
FIGURE 3:

Volcano plots showing the unique DEGs of the diabetic (A) glomerulus and (B) tubulointerstitium.
In the diabetic tubulointerstitium, 1251 DEGs were identified (Figure 2; upregulated n = 399, downregulated n = 874). Among these, 851 DEGs were DN-specific (68.0%; upregulated n = 205, downregulated n = 646) (Figure 3). Complete lists of the DEGs were presented in the Supplementary data, Table S1.
GO enrichment analysis of unique DEGs
The glomerular DEG set of DN was significantly enriched in 58 GO terms [Supplementary data, Table S2; molecular function (MF) n = 6, cellular component (CC) n = 4, biological process (BP) n = 48]. The 30 most enriched terms and involved genes were presented in Figure 4 and Table 2, respectively. Notably, the extracellular matrix, a CC term, was identified as the most enriched term. Further pathway analysis indicated that the involved DEGs were most related to the Wnt signaling pathway, the formation of fibrin clot (clotting cascade) and diseases associated with glycosaminoglycan metabolism (Supplementary data, Table S4). The enriched BP and MF terms include kidney epithelium development (BP), regulation of cell growth (BP), cholesterol homeostasis (BP) and cell adhesion molecule binding (MF).
FIGURE 4:

Significantly enriched GO terms of DEGs in the diabetic (A) glomerulus and (B) tubulointerstitium.
Table 2.
Enriched GO terms and involved genes
| ID | Description | Genes |
|---|---|---|
| Diabetic glomerular compartment | ||
|
| ||
| GO: 0031012 | Extracellular matrix | MMP7, LUM, SLPI, SFRP1, CXCL12, SERPINE2, ANXA2, SERPING1, GPC4, DCN, MXRA7, ANXA4, F3, CCN2, FGF1, NDNF |
| GO: 0062023 | Collagen-containing extracellular matrix | LUM, SLPI, SFRP1, CXCL12, SERPINE2, ANXA2, SERPING1, GPC4, DCN, MXRA7, ANXA4, F3, CCN2 |
| GO: 0072073 | Kidney epithelium development | PROM1, SFRP1, TFAP2B, CD24, SMAD5, EPHA7, KLHL3, FGF1, NPHS2 |
| GO: 0002237 | Response to molecule of bacterial origin | SLPI, VIM, CD24, ABCA1, HMGB2, DCN, LIAS, THBD, CEBPB, GCH1, NFKBIA, ADM |
| GO: 0098552 | Side of membrane | VTCN1, SERPINE2, ABCA1, GPC4, LDLR, IL1RL1, F3, HLA-DRB4, CD83, NPHS2, RGS2 |
| GO: 0009897 | External side of plasma membrane | VTCN1, SERPINE2, ABCA1, GPC4, LDLR, IL1RL1, F3, CD83 |
| GO: 0032496 | Response to lipopolysaccharide | SLPI, VIM, ABCA1, HMGB2, DCN, LIAS, THBD, CEBPB, GCH1, NFKBIA, ADM |
| GO: 0001655 | Urogenital system development | PROM1, SFRP1, TFAP2B, CD24, SMARCC1, DCN, SMAD5, EPHA7, KLHL3, FGF1, NPHS2 |
| GO: 0001822 | Kidney development | PROM1, SFRP1, TFAP2B, CD24, DCN, SMAD5, EPHA7, KLHL3, FGF1, NPHS2 |
| GO: 0072001 | Renal system development | PROM1, SFRP1, TFAP2B, CD24, DCN, SMAD5, EPHA7, KLHL3, FGF1, NPHS2 |
| GO: 0032103 | Positive regulation of response to external stimulus | CXCL12, STX3, NUPR1, CCL4, LDLR, THBD, IL1RL1, F3, CEBPB, NFKBIA |
| GO: 0045861 | Negative regulation of proteolysis | SLPI, WFDC2, SERPINE2, TFAP2B, SERPING1, SMARCC1, CSNK2A1, CAMLG, PLAUR, KLF4 |
| GO: 0016049 | Cell growth | SFRP1, CXCL12, SERPINE2, SORBS2, CSNK2A1, PAPPA2, TAF9B, EPHA7, IFRD1, SOCS2, SGK1, RGS2 |
| GO: 0001558 | Regulation of cell growth | SFRP1, CXCL12, SERPINE2, CSNK2A1, PAPPA2, TAF9B, EPHA7, IFRD1, SOCS2, SGK1, RGS2 |
| GO: 0001101 | Response to acid chemical | SFRP1, BCHE, UFL1, ABCA1, FABP3, LAMTOR3, LDLR, KLF4, CEBPB, CCN2 |
| GO: 0072009 | Nephron epithelium development | PROM1, TFAP2B, CD24, KLHL3, FGF1, NPHS2 |
| GO: 0032365 | Intracellular lipid transport | ANXA2, ABCA1, FABP3, LDLR |
| GO: 0045823 | Positive regulation of heart contraction | GCH1, CCN2, ADM, RGS2 |
| GO: 0030193 | Regulation of blood coagulation | SERPINE2, SERPING1, PLAUR, THBD, F3 |
| GO: 1900046 | Regulation of hemostasis | SERPINE2, SERPING1, PLAUR, THBD, F3 |
| GO : 0061318 | Renal filtration cell differentiation | PROM1, CD24, NPHS2 |
| GO: 0072112 | Glomerular visceral epithelial cell differentiation | PROM1, CD24, NPHS2 |
| GO: 0050818 | Regulation of coagulation | SERPINE2, SERPING1, PLAUR, THBD, F3 |
| GO: 0072311 | Glomerular epithelial cell differentiation | PROM1, CD24, NPHS2 |
| GO: 0030195 | Negative regulation of blood coagulation | SERPINE2, SERPING1, PLAUR, THBD |
| GO: 1900047 | Negative regulation of hemostasis | SERPINE2, SERPING1, PLAUR, THBD |
| GO : 0032373 | Positive regulation of sterol transport | ANXA2, ABCA1, NFKBIA |
| GO: 0032376 | Positive regulation of cholesterol Transport | ANXA2, ABCA1, NFKBIA |
| GO: 0072010 | Glomerular epithelium development | PROM1, CD24, NPHS2 |
| GO: 0052547 | Regulation of peptidase activity | SLPI, WFDC2, TFAP2B, CSNK2A1, EPHA7, PLAUR, KLF4, F3, PCOLCE2, CCN2 |
| Diabetic tubulointerstitial compartment | ||
|
| ||
| GO: 0044282 | Small molecule catabolic process | ENTPD1, IDUA, HYAL1, TST, IVD, GLS, ENPP4, ACOX1, MTRR, MCCC2, HADH, ENO1, ENTPD5, AHCY, HNMT, ECI2, PFKFB2, EHHADH, PEX7, ALDH7A1, ETFDH, DBT, ACADSB, CRYM, ACADM, PNP, IMPA1, ACADL, HAGH, KYNU, DECR2, MMUT, FUT6, KHK, RIDA, BCKDHB, MCCC1, GLUD2, AUH, GOT1, ALDH8A1, CRYL1, FAH, CTH, ALDH3A2, DERA, QDPR, ACAA1, HGD, UPB1, ALDH1L1, ALDH1B1, GK, KMO, NUDT3, ALDH6A1, QPRT, SORD, HADHA, ABAT, ACOX2, PRODH2, NPL, DPYS, FABP1 |
| GO: 0016054 | Organic acid catabolic process | IDUA, HYAL1, TST, IVD, GLS, ACOX1, MTRR, MCCC2, HADH, AHCY, HNMT, ECI2, EHHADH, PEX7, ALDH7A1, ETFDH, DBT, ACADSB, CRYM, ACADM, ACADL, KYNU, DECR2, MMUT, RIDA, BCKDHB, MCCC1, GLUD2, AUH, GOT1, ALDH8A1, CRYL1, FAH, CTH, ALDH3A2, QDPR, ACAA1, HGD, ALDH1L1, KMO, ALDH6A1, QPRT, SORD, HADHA, ABAT, ACOX2, PRODH2, NPL, FABP1 |
| GO: 0046395 | Carboxylic acid catabolic process | IDUA, HYAL1, TST, IVD, GLS, ACOX1, MTRR, MCCC2, HADH, AHCY, HNMT, ECI2, EHHADH, PEX7, ALDH7A1, ETFDH, DBT, ACADSB, CRYM, ACADM, ACADL, KYNU, DECR2, MMUT, RIDA, BCKDHB, MCCC1, GLUD2, AUH, GOT1, ALDH8A1, CRYL1, FAH, CTH, ALDH3A2, QDPR, ACAA1, HGD, ALDH1L1, KMO, ALDH6A1, QPRT, SORD, HADHA, ABAT, ACOX2, PRODH2, NPL, FABP1 |
| GO: 0050662 | Coenzyme binding | QSOX1, CYBB, MICAL2, IVD, IDH3G, ACOX1, MTRR, HADH, IDH2, AHCY, ECI2, GRHPR, PNPLA3, CAT, ETFDH, ACADSB, IDH3B, CRYM, MAOB, ACADM, ACADL, KYNU, AIFM1, P4HA2, CRYZL1, MARC2, MCCC1, GPHN, GOT1, CRYL1, HPGD, NOX4, CTH, IDH3A, DDC, AOX1, ALDH1B1, NQO2, KMO, ALDH6A1, SORD, GPD1, HADHA, ABAT, ACOT7, ACOX2, DHTKD1 |
| GO: 0048037 | Cofactor binding | LCN2, QSOX1, CYBB, PTGES, MICAL2, IVD, IDH3G, FDX1, ACLY, ACOX1, ACO2, MTRR, HADH, IDH2, AHCY, ECI2, GRHPR, PNPLA3, CAT, ETFDH, ACADSB, IDH3B, CRYM, MAOB, CYP11B2, ACADM, CYB5A, CYP20A1, ACADL, KYNU, AIFM1, P4HA2, MMUT, CRYZL1, ACO1, MARC2, MCCC1, GPHN, GOT1, CRYL1, HPGD, NOX4, CTH, SDHC, SDHB, IDH3A, DDC, AOX1, ALDH1B1, NQO2, KMO, ALDH6A1, GSTM4, SORD, GPD1, HADHA, ABAT, ACOT7, ACOX2, CUBN, DHTKD1, AOC1 |
| GO: 0006520 | Cellular amino acid metabolic process | TST, IVD, GLS, MTRR, MCCC2, AHCY, HNMT, NARS2, PSMA3, PSMD1, PSMF1, ALDH7A1, DBT, PSMD12, ACADSB, CRYM, PEPD, ADI1, EPRS, KYNU, MMUT, PSMD11, RIDA, BCKDHB, MCCC1, GLUD2, AUH, PSMC2, GOT1, NR1H4, ALDH8A1, NOX4, FAH, CTH, GLYAT, ASS1, DDC, BPHL, QDPR, HGD, UPB1, KMO, ALDH6A1, GCLM, DPEP1, ABAT, FOLH1B, BHMT2, PRODH2, PSAT1, DPYS, SLC7A7 |
| GO: 0009063 | Cellular amino acid catabolic process | TST, IVD, GLS, MTRR, MCCC2, AHCY, HNMT, ALDH7A1, DBT, ACADSB, CRYM, KYNU, RIDA, BCKDHB, MCCC1, GLUD2, AUH, GOT1, ALDH8A1, FAH, CTH, QDPR, HGD, KMO, ALDH6A1, ABAT, PRODH2 |
| GO: 0010038 | Response to metal ion | ITPR3, CAV1, CALR, FABP4, CYBB, B2M, MEF2C, LOXL2, HNRNPD, PTGES, ABCC2, HOMER1, NEDD4L, CREB1, PRKAA2, SLC30A5, SLC11A2, BNIP3, CAT, MT1E, MAOB, MT1X, CYP11B2, CYB5A, MT1H, ALAD, MT1G, MT1F, LGMN, ACO1, GPHN, GOT1, EGFR, GGH, ASS1, QDPR, PRNP, CLIC4, DPEP1, CA2, SORD, CPNE3, ABAT, AOC1 |
| GO: 1901605 | Alpha-amino acid metabolic process | IVD, GLS, MTRR, MCCC2, AHCY, HNMT, ALDH7A1, CRYM, ADI1, KYNU, MMUT, RIDA, MCCC1, GLUD2, AUH, GOT1, NR1H4, ALDH8A1, NOX4, FAH, CTH, GLYAT, ASS1, QDPR, HGD, KMO, ALDH6A1, GCLM, DPEP1, BHMT2, PRODH2, PSAT1 |
| GO: 0005777 | Peroxisome | ACSL4, PEX19, ACOX1, ACSL3, ISOC1, IDH2, ECI2, GRHPR, PLAAT3, EHHADH, PEX7, CAT, ACSL1, CRYM, DECR2, MARC2, RIDA, ALDH3A2, EPHX2, ACAA1, HSDL2, ACOX2, AOC1, FABP1 |
| GO: 0042579 | Microbody | ACSL4, PEX19, ACOX1, ACSL3, ISOC1, IDH2, ECI2, GRHPR, PLAAT3, EHHADH, PEX7, CAT, ACSL1, CRYM, DECR2, MARC2, RIDA, ALDH3A2, EPHX2, ACAA1, HSDL2, ACOX2, AOC1, FABP1 |
| GO: 0005759 | Mitochondrial matrix | ERBB4, ISCA1, TST, GRSF1, IVD, IDH3G, GLS, PDP1, FDX1, HSPD1, ACO2, MCCC2, HADH, IDH2, CREB1, NARS2, ACSS3, MRPL42, GRPEL1, ALDH7A1, ETFDH, DBT, MRPL22, MAIP1, ACADSB, IDH3B, VDAC1, ACADM, ACADL, LRPPRC, HAGH, MMUT, RIDA, BCKDHB, MCCC1, MRPS18B, AUH, FOXO3, GLYAT, IDH3A, DLAT, ALDH1B1, ALDH6A1, LACTB2, NR3C1, ACSF2, HADHA, ABAT, DHTKD1 |
| GO: 1901606 | Alpha-amino acid catabolic process | IVD, GLS, MTRR, MCCC2, AHCY, HNMT, ALDH7A1, CRYM, KYNU, RIDA, MCCC1, GLUD2, AUH, GOT1, ALDH8A1, FAH, QDPR, HGD, KMO, ALDH6A1, PRODH2 |
| GO: 0006732 | Coenzyme metabolic process | NNMT, PFKFB3, ACSL4, NUP214, FOLR2, CD38, NUP50, PDP1, ACLY, ACSL3, MTRR, MCCC2, IDH2, ENO1, ENTPD5, PFKFB2, MAT2A, PRKAA2, RFK, ZBTB7A, ACSL1, STAT3, PNP, CYB5A, KYNU, MOCS2, MCCC1, GPHN, AMD1, GLYAT, DERA, DLAT, ALDH1L1, KMO, ACOT13, ACSF2, QPRT, GPD1, ACOT7, AASDHPPT, BHMT2, DHTKD1, INSR |
| GO: 0006790 | Sulfur compound metabolic process | ACPP, ACSL4, CHST15, IDUA, BGN, MICAL2, HYAL1, TST, PDP1, ABCC2, ACLY, CIAO1, ACSL3, ARSB, ST3GAL1, MTRR, MCCC2, SULT1C2, AHCY, PAX8, MAT2A, ACSL1, ADI1, HAGH, KYNU, MMUT, MCCC1, NOX4, CTH, AMD1, GLYAT, DLAT, ACOT13, GNS, GSTM4, GCLM, ACSF2, DPEP1, ACOT7, BHMT2, SLC35D1 |
| GO: 0002283 | Neutrophil activation involved in immune response | LTF, C3, LCN2, ACPP, CD93, CFD, QSOX1, RNASET2, CYBB, B2M, LAIR1, RAB31, FCER1G, FCN1, MNDA, DNASE1L3, ATP6V1D, PGRMC1, ENPP4, RAB5C, ACLY, ARSB, CTSA, ILF2, CPPED1, DEGS1, PSMD1, CAT, IGF2R, CTSB, PSMD12, HMOX2, PNP, RAB14, ALAD, PSMD11, NRAS, PSMC2, METTL7A, RAB7A, GGH, LAMP2, DERA, MAGT1, ACAA1, SLC2A5, CMTM6, IQGAP2, GNS, CPNE3, AOC1 |
| GO: 0002446 | Neutrophil mediated immunity | LTF, C3, LCN2, ACPP, CD93, CFD, QSOX1, RNASET2, CYBB, B2M, LAIR1, RAB31, FCER1G, FCN1, MNDA, DNASE1L3, ATP6V1D, PGRMC1, ENPP4, RAB5C, ACLY, ARSB, CTSA, ILF2, CPPED1, DEGS1, PSMD1, CAT, IGF2R, CTSB, PSMD12, HMOX2, PNP, RAB14, ALAD, PSMD11, NRAS, PSMC2, METTL7A, RAB7A, GGH, LAMP2, DERA, MAGT1, ACAA1, SLC2A5, CMTM6, IQGAP2, GNS, CPNE3, AOC1 |
| GO: 0042119 | Neutrophil activation | LTF, C3, LCN2, ACPP, CD93, CFD, QSOX1, RNASET2, CYBB, B2M, LAIR1, RAB31, FCER1G, FCN1, MNDA, DNASE1L3, ATP6V1D, PGRMC1, ENPP4, RAB5C, ACLY, ARSB, CTSA, ILF2, CPPED1, DEGS1, PSMD1, CAT, IGF2R, CTSB, PSMD12, HMOX2, PNP, RAB14, ALAD, PSMD11, NRAS, PSMC2, METTL7A, RAB7A, GGH, LAMP2, DERA, MAGT1, ACAA1, SLC2A5, CMTM6, IQGAP2, GNS, CPNE3, AOC1 |
| GO: 0043312 | Neutrophil degranulation | LTF, C3, LCN2, ACPP, CD93, CFD, QSOX1, RNASET2, CYBB, B2M, LAIR1, RAB31, FCER1G, FCN1, MNDA, ATP6V1D, PGRMC1, ENPP4, RAB5C, ACLY, ARSB, CTSA, ILF2, CPPED1, DEGS1, PSMD1, CAT, IGF2R, CTSB, PSMD12, HMOX2, PNP, RAB14, ALAD, PSMD11, NRAS, PSMC2, METTL7A, RAB7A, GGH, LAMP2, DERA, MAGT1, ACAA1, SLC2A5, CMTM6, IQGAP2, GNS, CPNE3, AOC1 |
| GO: 0051287 | Nicotinamide adenine dinucleotide (NAD) binding | IDH3G, HADH, IDH2, AHCY, GRHPR, IDH3B, CRYL1, HPGD, IDH3A, AOX1, ALDH1B1, SORD, GPD1, HADHA |
| GO: 0050660 | Flavin adenine dinucleotide binding | QSOX1, CYBB, MICAL2, IVD, ACOX1, MTRR, ETFDH, ACADSB, MAOB, ACADM, AIFM1, NOX4, AOX1, NQO2, KMO, ACOX2 |
| GO: 0006631 | Fatty acid metabolic process | C3, ACSL4, CAV1, CD74, PTGES, RGN, IVD, PDP1, ACOX1, ACSL3, HADH, ECI2, PRKAA2, EHHADH, DEGS1, PNPLA3, PEX7, ETFDH, ACSL1, ACADSB, ACADM, ACADL, DECR2, MMUT, LYPLA1, CRYL1, HPGD, CES2, ALDH3A2, EPHX2, ACAA1, MSMO1, GSTM4, ACSF2, HADHA, ACOT7, ACOX2, QKI, FABP1 |
| GO: 0043202 | Lysosomal lumen | AGRN, RNASET2, CD74, IDUA, BGN, SGSH, HYAL1, CTSL, ARSB, CTSA, CTSB, TPP1, LGMN, GPC3, LAMP2, GNS, CUBN, CTSV |
| GO: 0016999 | Antibiotic metabolic process | CYBB, IDH3G, ABCC2, ADH5, ACLY, ST3GAL1, ACO2, IDH2, GPX3, CAT, STAT3, IDH3B, MAOB, ACO1, EGFR, SDHC, SDHB, IDH3A, DLAT, ALDH1B1, DPEP1, NPL |
| GO: 0044438 | Microbody part | ACSL4, PEX19, ACOX1, ACSL3, ECI2, GRHPR, PLAAT3, EHHADH, PEX7, CAT, ACSL1, CRYM, DECR2, ALDH3A2, EPHX2, ACAA1, ACOX2, FABP1 |
| GO: 0044439 | Peroxisomal part | ACSL4, PEX19, ACOX1, ACSL3, ECI2, GRHPR, PLAAT3, EHHADH, PEX7, CAT, ACSL1, CRYM, DECR2, ALDH3A2, EPHX2, ACAA1, ACOX2, FABP1 |
| GO: 0045177 | Apical part of cell | ANXA1, ITPR3, OXTR, PLAT, ABCC2, HOMER1, USH1C, SORBS2, SLC30A5, SLC11A2, PDZK1, SLC3A2, LRP2, ATP6V1C1, LGMN, ERBB3, STX3, NOX4, ABCC6, EZR, EGFR, SLC2A5, VAMP3, CLCN5, TUBG1, CLIC4, SLC22A4, SLC17A1, DPEP1, CA2, PRKCI, KL, SLC7A9, THY1, PODXL, CUBN, MTDH, CTSV, FABP1 |
| GO: 0031983 | Vesicle lumen | LTF, C3, LCN2, CFD, QSOX1, CALR, RNASET2, B2M, TOR4A, HGF, FCN1, MNDA, VEGFC, SERPINF2, ACLY, ARSB, CTSA, ILF2, CPPED1, PSMD1, CAT, PSMD12, PNP, ALAD, PSMD11, VEGFA, PSMC2, EGFR, GGH, DERA, ACAA1, KNG1, GNS, APOE, APOH, PLG, AOC1 |
| GO: 0072350 | Tricarboxylic acid metabolic process | IDH3G, ACLY, ACO2, IDH2, IDH3B, ACO1, SDHC, SDHB, IDH3A, ASS1, DLAT |
| GO: 0005775 | Vacuolar lumen | C3, AGRN, RNASET2, CD74, IDUA, BGN, MNDA, SGSH, HYAL1, CTSL, ACLY, ARSB, CTSA, CPPED1, PSMD1, CTSB, TPP1, LGMN, GPC3, GGH, LAMP2, GNS, CUBN, CTSV |
In comparison, many more enriched terms were identified in the tubulointerstitial DEG set of DN (Supplementary data, Table S3; N = 467, MF n = 32, CC n = 65, BP n = 370). Dysregulation of a wide range of small molecule catabolic processes, including organic acid, carboxylic acid, amino acid and fatty acid, was suggested to be involved in DN (Figure 4). Moreover, neutrophil-mediated immunity, oxidative stress, aging and platelet activation might also participate in the development of DN (Figure 4). We also performed GO enrichment analysis in upregulated and downregulated DEG sets separately. The results can be found in the Supplementary data, Table S4.
Pathway enrichment analysis
Two databases (KEGG pathway, Reactome) were used for pathway enrichment analysis. After analyzing the whole DEG set, there is no evidence of an enriched term within the glomerular compartment, and the same holds true for the upregulated and downregulated DEG sets.
In contrast, 75 terms were found enriched in the DEG set of the tubulointerstitial compartment (Supplementary data, Table S5; KEGG pathway n = 27, Reactome pathway n = 48). Similar to the results of the GO enrichment analysis, pathway enrichment analysis revealed a wide range of metabolism disorders. Notably, valine, leucine and isoleucine degradation was identified as the term with the highest enrichment (q value = 3.86E-11). Other enriched terms include the peroxisome proliferator-activated receptor (PPAR) signaling pathway, epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor resistance, FoxO signaling pathway, neutrophil degranulation and autophagy (Figures 5 and 6). Moreover, the upregulated genes were mainly related to hemostasis, signaling by receptor tyrosine kinases, platelet activation and extracellular matrix organization (Figure 5 and Supplementary data, Table S5). The downregulated genes, not surprisingly, were mainly associated with metabolism disorders, FoxO signaling pathway, PPAR signaling pathway, etc., most of which can also be found in the list of the whole set enrichment terms (Figure 5 and Supplementary data, Table S5). The enrichment map for the KEGG enrichment analysis of the diabetic tubulointerstitium further revealed the inner relationship between different enriched pathways (Figure 6). Most metabolism-related pathways have shared genes and constitute a pathological network. It is also noteworthy that ferroptosis might also contribute to renal tubular cell death as well as autophagy.
FIGURE 5:

Significantly enriched pathways for (A) dysregulated DEGs, (B) upregulated DEGs and (C) downregulated DEGs in the diabetic tubulointerstitium.
FIGURE 6:

Enrichment map for the KEGG enrichment analysis of the diabetic tubulointerstitium. Overlapping gene sets tend to cluster together by edges.
PIN reconstruction and hub gene identification
To further reveal the relationship of the DEGs, PINs with physical interaction were reconstructed (Figure 7). Each edge represents a physical interaction established by a given method, such as mass spectrometry or two-hybrid assay. While calculating topological scores, the edges between two nodes were regarded as one. Compared with those of the glomerular compartment, tubulointerstitial DEGs formed a much more complex network (glomerular PIN: 36 nodes, 30 edges; tubulointerstitial PIN: 597 nodes, 1870 edges). As presented in Tables 3 and 4, nodes with the highest degrees were recognized as hub genes for each network. Hub genes of the glomerular PIN include AGR2, VIM, CSNK2A1, ANXA2 and TERF1, whereas 10 genes, including EGFR, LMNA and HSPD, were identified as hub genes in the tubulointerstitial PIN.
FIGURE 7:

PIN reconstruction of the diabetic (A) glomerulus and (B) tubulointerstitium. Red, upregulated nodes; green, downregulated nodes.
Table 3.
Hub genes identified in the glomerular PIN
| Rank | Gene symbol | Full name | Description | Degrees |
|---|---|---|---|---|
| 1 | AGR2 | Anterior gradient 2, protein disulfide isomerase family member | This gene encodes a member of the disulfide isomerase family of ER proteins that catalyze protein folding and thiol-disulfide interchange reactions. The encoded protein has an N-terminal ER-signal sequence, a catalytically active thioredoxin domain and a C-terminal ER-retention sequence. This protein plays a role in cell migration, cellular transformation and metastasis and is as a p53 inhibitor. As an ER-localized molecular chaperone, it plays a role in the folding, trafficking and assembly of cysteine-rich transmembrane receptors and the cysteine-rich intestinal gylcoprotein mucin. This gene has been implicated in inflammatory bowel disease and cancer progression | 10 |
| 2 | VIM | Vimentin | This gene encodes a type III intermediate filament protein. Intermediate filaments, along with microtubules and actin microfilaments, make up the cytoskeleton. The encoded protein is responsible for maintaining cell shape and integrity of the cytoplasm and stabilizing cytoskeletal interactions. This protein is involved in neuritogenesis and cholesterol transport and functions as an organizer of a number of other critical proteins involved in cell attachment, migration and signaling. Bacterial and viral pathogens have been shown to attach to this protein on the host cell surface. Mutations in this gene are associated with congenital cataracts in human patients | 4 |
| 3 | CSNK2A1 | Casein kinase 2 alpha 1 | Casein kinase II is a serine/threonine-protein kinase that phosphorylates acidic proteins such as casein. It is involved in various cellular processes, including cell cycle control, apoptosis and circadian rhythm | 4 |
| 4 | ANXA2 | Annexin A2 | This gene encodes a member of the annexin family. Members of this calcium-dependent phospholipid-binding protein family play a role in the regulation of cellular growth and in signal transduction pathways. This protein functions as an autocrine factor, which heightens osteoclast formation and bone resorption | 4 |
| 5 | TERF1 | Telomeric repeat binding factor 1 | This gene encodes a telomere- specific protein that is a component of the telomere nucleoprotein complex. This protein is present at telomeres throughout the cell cycle and functions as an inhibitor of telomerase, acting in cis to limit the elongation of individual chromosome ends | 3 |
ER: endoplasmic reticulum. Descriptions of the genes were abstracted from the RefSeq database.
Table 4.
Hub genes identified in the tubulointerstitial PIN
| Rank | Gene symbol | Full name | Description | Degrees |
|---|---|---|---|---|
| 1 | EGFR | Epidermal growth factor receptor | The protein encoded by this gene is a transmembrane glycoprotein that is a member of the protein kinase superfamily. This protein is a receptor for members of the epidermal growth factor family. EGFR is a cell surface protein that binds to epidermal growth factor. Binding of the protein to a ligand induces receptor dimerization and tyrosine autophosphorylation and leads to cell proliferation | 99 |
| 2 | LMNA | Lamin A/C | The nuclear lamina consists of a two-dimensional matrix of proteins located next to the inner nuclear membrane. The lamin family of proteins make up the matrix and are highly conserved in evolution. During mitosis, the lamina matrix is reversibly disassembled as the lamin proteins are phosphorylated. Lamin proteins are thought to be involved in nuclear stability, chromatin structure and gene expression. Vertebrate lamins consist of two types, A and B. Alternative splicing results in multiple transcript variants. Mutations in this gene lead to several diseases: Emery–Dreifuss muscular dystrophy, familial partial lipodystrophy, limb–girdle muscular dystrophy, dilated cardiomyopathy, Charcot–Marie–Tooth disease and Hutchinson–Gilford progeria syndrome | 48 |
| 3 | NRAS | NRAS proto-oncogene, GTPase | This is an N-ras oncogene encoding a membrane protein that shuttles between the Golgi apparatus and the plasma membrane. This shuttling is regulated through palmitoylation and depalmitoylation by the ZDHHC9-GOLGA7 complex. The encoded protein, which has intrinsic GTPase activity, is activated by a guanine nucleotide-exchange factor and inactivated by a GTPase activating protein. Mutations in this gene have been associated with somatic rectal cancer, follicular thyroid cancer, autoimmune lymphoproliferative syndrome, Noonan syndrome and juvenile myelomonocytic leukemia | 47 |
| 4 | HSPD1 | Heat shock protein family D (Hsp60) member 1 | This gene encodes a member of the chaperonin family. The encoded mitochondrial protein may function as a signaling molecule in the innate immune system. This protein is essential for the folding and assembly of newly imported proteins in the mitochondria. This gene is adjacent to a related family member and the region between the two genes functions as a bidirectional promoter. Several pseudogenes have been associated with this gene. Two transcript variants encoding the same protein have been identified for this gene. Mutations associated with this gene cause autosomal recessive spastic paraplegia 13 | 46 |
| 5 | HEXIM1 | HEXIM P-TEFb complex subunit 1 | Expression of this gene is induced by hexamethylene-bis-acetamide in vascular smooth muscle cells | 43 |
| 6 | TGOLN2 | Trans-golgi network protein 2 | This gene encodes a type I integral membrane protein that is localized to the trans-Golgi network, a major sorting station for secretory and membrane proteins. The encoded protein cycles between early endosomes and the trans-Golgi network and may play a role in exocytic vesicle formation | 42 |
| 7 | MAPK6 | Mitogen-activated protein kinase 6 | The protein encoded by this gene is a member of the Ser/Thr protein kinase family and is most closely related to MAP kinases. MAP kinases also known as extracellular signal-regulated kinases, are activated through protein phosphorylation cascades and act as integration points for multiple biochemical signals. This kinase is localized in the nucleus and has been reported to be activated in fibroblasts upon treatment with serum or phorbol esters | 40 |
| 8 | RC3H2 | Ring finger and C-x8-C-x5-C-x3-H (CCCH)-type domains 2 | RC3H2 is a protein coding gene. GO annotations related to this gene include mRNA binding | 36 |
| 9 | CHD3 | Chromodomain helicase DNA binding protein 3 | This gene encodes a member of the CHD family of proteins which are characterized by the presence of chromo (chromatin organization modifier) domains and SNF2-related helicase/ATPase domains. This protein is one of the components of a histone deacetylase complex referred to as the Mi-2/nucleosome remodeling and deacetylase(NuRD) complex which participates in the remodeling of chromatin by deacetylating histones. Chromatin remodeling is essential for many processes including transcription. Autoantibodies against this protein are found in a subset of patients with dermatomyositis | 35 |
| 10 | SUZ12 | SUZ12 polycomb repressive complex 2 subunit | This zinc finger gene has been identified at the breakpoints of a recurrent chromosomal translocation reported in endometrial stromal sarcoma. Recombination of these breakpoints results in the fusion of this gene and juxtaposed with another zinc finger protein 1 (JAZF1) | 34 |
Descriptions of the genes were abstracted from the RefSeq database. As no summary was available for RC3H2 in Refseq, the GeneCards summary was used instead.
DISCUSSION
DN is the leading cause of end-stage kidney disease worldwide. Its pathogenesis is distinct from other CKDs such as GN and HN. In this study we reported a unique transcriptomic feature of DN based on bioinformatics analysis of >250 microarray datasets. To our knowledge, this is the largest comparative bioinformatics study on this topic.
Our study found more DEGs in the diabetic tubulointerstitium than in the glomerulus. Moreover, a larger proportion of DEGs were DN-specific in the tubulointerstitium (68.0% versus 30.9%). As a result, the number of unique DEGs identified in diabetic tubulointerstitium was 6.59 times as many as in the glomerulus, despite the fact that more tubulointerstitial samples were included in the analysis. In addition, the tubular DEG formed a much more complex PIN. These data provide compelling evidence for the presence of diabetic tubulopathy and support the tubulocentric view of DN.
According to the GO enrichment analysis, the unique DEGs of the glomerular compartment were related to the extracellular matrix, cell growth, regulation of blood coagulation, cholesterol homeostasis, intrinsic apoptotic signaling pathway and renal filtration cell differentiation. However, we did not find enriched terms in either the Reactome or KEGG pathway database using the cluster profile package in R, which might be due to the conservative algorithm design (fewer background genes in hypergeometric test) of this package. Consistent with the notion that glomerulosclerosis is a key pathological feature of DN, we observed that the extracellular matrix was the most significantly enriched GO term in the diabetic glomerulus. Further pathway analysis revealed that the related DEGs were enriched in the wnt signaling pathway, the formation of fibrin clot (clotting cascade) and diseases associated with glycosaminoglycan metabolism. Dysregulation of glomerular wnt/β-catenin signaling has been documented to increase the deposition of fibrous tissue in DN, despite the fact that its mechanism remains to be elucidated [15]. Interestingly, most of the enriched terms (extracellular matrix, cell growth, regulation of blood coagulation and apoptosis) fell into the broader BPs of wound healing. A recent bioinformatics analysis using urinary proteomics also showed that wound healing is a hallmark in both uncomplicated diabetes and incipient DN [16]. Experimental studies have shown that activation of coagulation, one of the early processes in wound healing, can facilitate immune cell infiltration, impair glomerular permselectivity and induce apoptosis of podocytes and endothelial cells. On the other hand, chronic injury induces progressive accumulation of extracellular matrix and improper tissue development, which contributes to the development of renal lesions in DN. Moreover, our analysis also observed a disturbance of cholesterol homeostasis pathways in the diabetic glomerulus. Interestingly, the altered expression of related DEGs, including ABCA1 and low-density lipoprotein receptor (LDLR), has been shown to promote cholesterol accumulation by affecting the efflux and influx of LDL, which aggravates glomerulopathy in DN [17, 18]. We also identified five hub genes of the glomerular PIN, including AGR2, VIM, CSNK2A1, ANXA2 and TERF1. Prior work has documented some of their roles in the development of DN. For example, Huang et al. [19] reported that CK2α (encoded by CSNK2As) can regulate the process of fibrosis in the diabetic kidney via the nuclear factor κB (NF-κB) pathway. In addition, a recent study reported a significant upregulation of AGR2, Tff2 and Gkn3 in the diabetic mouse model after administration of dapagliflozin [20]. However, their roles in the development of diabetic tubulopathy remain to be elucidated.
In the diabetic tubulointerstitium, the significantly enriched BPs and pathways included metabolism, the advanced glycation end products–receptor for advanced glycation end products signaling pathway in diabetic complications, the EGFR signaling pathway, the FoxO signaling pathway, autophagy and ferroptosis. In contrast to the diabetic glomerulus, a wide range of metabolism disorders emerged as the top pathological processes in diabetic tubulopathy. It is widely accepted that diabetes is associated with altered cellular metabolism. By analyzing diabetic kidney transcriptomic data from a cohort of southwestern American Indians, Sas et al. [21] reported dysregulation of glycolysis, fatty acid and amino acid metabolism pathways in the human kidney. Our analysis highlighted that these changes are characteristic in diabetic tubulointerstitium, and the PPAR signaling pathway might play important roles in regulating renal tubular metabolism (especially in fatty acid metabolism) in DN. We observed a significant dysregulation of downstream target genes of PPARs that participate in the maintenance of renal metabolic homeostases, such as glycerol kinase (GK), 3-hydroxy-3-methylglutaryl-coenzyme A synthase 2 (HMGCS2) and acyl-coenzyme A dehydrogenase long chain (ACADL). We have also observed a significant downregulation of the pathway for branched-chain amino acids (BCAAs; including valine, leucine and isoleucine) degradation. Urinary BCAA catabolites are decreased in diabetic subjects [22]. Elevations of plasm BCAAs are independently associated with an increased risk of type 2 diabetes and insulin resistance [23]. Whether urinary BCAAs could be used as an early biomarker of diabetic tubulopathy remains to be investigated.
Our study also showed ferroptosis might be involved in renal tubular cell death in DN. Ferroptosis is an iron-dependent form of cell death resulting from lipid-based reactive oxygen species, which has implications in tumors, nervous system diseases and kidney diseases such as polycystic kidney disease and acute kidney injury [24]. A recent study reported that inhibition of ferroptosis can reduce myocardial ischemia–reperfusion injury in diabetic models [25]. Considering there is still a lack of literature, it is noteworthy for future studies to investigate the role of ferroptosis in DN.
Ten genes with the highest degrees, including EGFR, LMNA and HSPD1, were identified as hub genes in the tubulointerstitial PIN. Interestingly, some of these genes, such as EGFR and HSPD1, have been gaining increasing research attention in recent years [26, 27]. Using global protein network analysis and subsequent functional validation, Aluksanasuwan et al. [27] reported HSPD1 was involved in diabetic tubular cell dysfunction probably via regulation of intracellular adenosine triphosphate (ATP) production. Here we only discussed several terms or genes according to their importance given by statistics tests. However, it is rather common that those with less statistical significance have more important roles in real circumstances. Therefore readers are encouraged to explore the supplementary materials for further information.
However, some limitations are worth noting. First, although >250 datasets were included, only transcriptomics data were analyzed in our study. Future work should therefore include proteomic and metabolomic data to provide a more comprehensive understanding of the molecular mechanism. Second, expression of mRNAs, rather than proteins, was used to reconstruct PINs, which might not accurately reflect the real situation. Third, to increase the specificity of unique DEG identification, the union but not the intersection operation was used to generate the DEG set of GN. Despite its logical reasonability, this operation might miss some potential genes that play important roles in DN. Last but not least, several resources of heterogeneity, including different genetic and clinical backgrounds of the study populations, might also introduce bias to our results.
In conclusion, our study not only reveals the unique molecular mechanism of DN, but also provides a valuable resource for biomarker and therapeutic target discovery. Some of our findings are promising and should be explored by future work.
SUPPLEMENTARY DATA
Supplementary data are available at ckj online.
FUNDING
Funding was provided by the National Natural Science Foundation of China (Youth Program 81900698). This project was supported by the Scientific Research Projects of Wuxi Municipal Health and Family Planning Commission (Q201761).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
Supplementary Material
REFERENCES
- 1. Webster AC, Nagler EV, Morton RL. et al. Chronic kidney disease. Lancet 2017; 389: 1238–1252 [DOI] [PubMed] [Google Scholar]
- 2. Kritmetapak K, Anutrakulchai S, Pongchaiyakul C. et al. Clinical and pathological characteristics of non-diabetic renal disease in type 2 diabetes patients. Clin Kidney J 2018; 11: 342–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zeng M, Liu J, Yang W. et al. Multiple-microarray analysis for identification of hub genes involved in tubulointerstial injury in diabetic nephropathy. J Cell Physiol 2019; 234: 16447–16462 [DOI] [PubMed] [Google Scholar]
- 4. Fan Y, Yi Z, D’Agati VD. et al. Comparison of kidney transcriptomic profiles of early and advanced diabetic nephropathy reveals potential new mechanisms for disease progression. Diabetes 2019; 68: 2301–2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou LT, Qiu S, Lv LL. et al. Integrative bioinformatics analysis provides insight into the molecular mechanisms of chronic kidney disease. Kidney Blood Press Res 2018; 43: 568–581 [DOI] [PubMed] [Google Scholar]
- 6. Yu G, Wang LG, Han Y. et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012; 16: 284–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chin CH, Chen SH, Wu HH. et al. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol 2014; 8: S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Berthier CC, Bethunaickan R, Gonzalez-Rivera T. et al. Cross-species transcriptional network analysis defines shared inflammatory responses in murine and human lupus nephritis. J Immunol 2012; 189: 988–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martini S, Nair V, Keller BJ. et al. Integrative biology identifies shared transcriptional networks in CKD. J Am Soc Nephrol 2014; 25: 2559–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reich HN, Tritchler D, Cattran DC. et al. A molecular signature of proteinuria in glomerulonephritis. PLoS One 2010; 5: e13451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lindenmeyer MT, Eichinger F, Sen K. et al. Systematic analysis of a novel human renal glomerulus-enriched gene expression dataset. PLoS One 2010; 5: e11545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Neusser MA, Lindenmeyer MT, Moll AG. et al. Human nephrosclerosis triggers a hypoxia-related glomerulopathy. Am J Pathol 2010; 176: 594–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hodgin JB, Berthier CC, John R. et al. The molecular phenotype of endocapillary proliferation: novel therapeutic targets for IgA nephropathy. PLoS One 2014; 9: e103413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ju WJ, Nair V, Smith S. et al. Tissue transcriptome-driven identification of epidermal growth factor as a chronic kidney disease biomarker. Sci Transl Med 2015; 7: 316ra193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guo Q, Zhong W, Duan A. et al. Protective or deleterious role of Wnt/beta-catenin signaling in diabetic nephropathy: an unresolved issue. Pharmacol Res 2019; 144: 151–157 [DOI] [PubMed] [Google Scholar]
- 16. Van JAD, Scholey JW, Konvalinka A.. Insights into diabetic kidney disease using urinary proteomics and bioinformatics. J Am Soc Nephrol 2017; 28: 1050–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang Y, Ma KL, Liu J. et al. Dysregulation of low-density lipoprotein receptor contributes to podocyte injuries in diabetic nephropathy. Am J Physiol Endocrinol Metab 2015; 308: E1140–E1148 [DOI] [PubMed] [Google Scholar]
- 18. Yin Q-H, Zhang R, Li L. et al. Exendin-4 ameliorates lipotoxicity-induced glomerular endothelial cell injury by improving ABC transporter A1-mediated cholesterol efflux in diabetic apoE knockout mice. J Biol Chem 2016; 291: 26487–26501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang J, Chen Z, Li J. et al. Protein kinase CK2α catalytic subunit ameliorates diabetic renal inflammatory fibrosis via NF-κB signaling pathway. Biochem Pharmacol 2017; 132: 102–117 [DOI] [PubMed] [Google Scholar]
- 20. Kanno A, Asahara S-I, Kawamura M. et al. Early administration of dapagliflozin preserves pancreatic β-cell mass through a legacy effect in a mouse model of type 2 diabetes. J Diabetes Investig 2019; 10: 577–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sas KM, Kayampilly P, Byun J. et al. Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight 2016; 1: e86976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Connor SC, Hansen MK, Corner A. et al. Integration of metabolomics and transcriptomics data to aid biomarker discovery in type 2 diabetes. Mol Biosyst 2010; 6: 909–921 [DOI] [PubMed] [Google Scholar]
- 23. Flores-Guerrero JL, Osté MCJ, Kieneker LM. et al. Plasma branched-chain amino acids and risk of incident type 2 diabetes: results from the PREVEND prospective cohort study. J Clin Med 2018; 7: 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li J, Cao F, Yin H-L. et al. Ferroptosis: past, present and future. Cell Death Dis 2020; 11: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li W, Li W, Leng Y. et al. Ferroptosis is involved in diabetes myocardial ischemia/reperfusion injury through endoplasmic reticulum stress. DNA Cell Biol 2020; 39: 210–225 [DOI] [PubMed] [Google Scholar]
- 26. Li Z, Li Y, Overstreet JM. et al. Inhibition of epidermal growth factor receptor activation is associated with improved diabetic nephropathy and insulin resistance in type 2 diabetes. Diabetes 2018; 67: 1847–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aluksanasuwan S, Sueksakit K, Fong-Ngern K. et al. Role of HSP60 (HSPD1) in diabetes-induced renal tubular dysfunction: regulation of intracellular protein aggregation, ATP production, and oxidative stress. FASEB J 2017; 31: 2157–2167 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.