

Abstract
Inhibitor of nuclear factor kappa-B kinase subunit beta (IKKβ) is a key regulator of the cannonical NF-κB pathway. IKKβ has been validated as a drug target for pathological conditions, which include chronic inflammatory diseases and cancer. Pharmacological studies revealed that chronic administration of ATP-competitive IKKβ inhibitors resulted in unexpected toxicity. We previously reported the discovery of 13–197 as a non-toxic IKKβ inhibitor that reduced tumor growth. Here, we show that 13–197 inhibits IKKβ in a ATP non-competitive manner and an allosteric pocket at the interface of the kinase and ubiquitin like domains was identified as the potential binding site.
Graphical Abstract
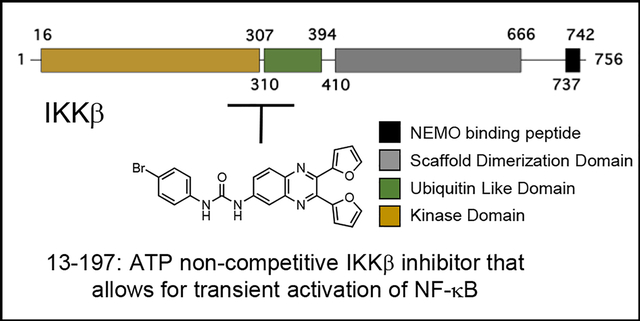
Tumour necrosis factor (TNF) α has been implicated as a driver of chronic inflammatory diseases (CIDs) and is found in ~50% of the tumour microenvironment of surgically resected tumour samples.1, 2 TNFα induced IKKβ phosphorylation results in the activation of the canonical nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway.3 This led to an extensive search for viable IKKβ inhibitors as CID and cancer therapeutics.4 A major complication with the use of ATP-competitive IKKβ inhibitors as therapeutics was elucidated using ML-120B, an ATP-competitive IKKβ inhibitor developed by Millennium pharmaceuticals, Inc.5 ML-120B administration led to granulocytosis,6 and prolonged treatment resulted in increased susceptibility to endotoxin mediated death.7 Another ATP-competitive IKKβ inhibitor, TPCA1, developed by GlaxoSmithKline (GSK),8 exhibited unanticipated toxicity, although it also briefly prolonged the survival of mice with lung cancer.9 These studies clearly demonstrate that IKKβ is an attractive therapeutic target, however, the ATP binding site is not optimal for inhibitor design.
IKKβ is 756 amino acids (aa) in length with aa 16–309 adopting a classical kinase domain (KD), aa 310–404 adopting a ubiquitin-like domain (ULD), and aa 410–664 forming a scaffold dimerization domain (SDD) that has a helical blade structure. To date, only 3 groups have reported the structures of IKKβ that extend through the SDD.10–12 The human IKKβ (pdb id: 4KIK) structure (1–664) is a dimer in which the activation loop (S177 and S181) is phosphorylated in protomer B but not in protomer A.12 The kinase domains (KD) of the two protomers are otherwise identical suggesting that inhibitors that target the ATP binding site are not likely to distinguish between inactive and active IKKβ. However, the protomer conformations differ outside the KD, such as a ~120 Å2 pocket at the KD-SDD interface that is open in protomer B (active IKKβ) but partially closed in protomer A (inactive IKKβ), which offers unique pockets for the design of selective inhibitors.
We previously reported the discovery of a small molecule, 13–197, that inhibited cancer cell growth by perturbing the levels of the anti-apoptotic protein Mcl1.13, 14 Subsequently we showed that 13–197 was orally bioavailable, reduced tumour growth and metastasis in an orthotopic pancreatic cancer model, and nearly doubled the median survival of mice in a mantle cell lymphoma model.15–17 Kinome profiling identified IKKβ as the target of 13–197, however, 13–197 did not share the toxicity profile of ML-120B or TPCA1.16, 18 We hypothesized that the differential toxicity profile of 13–197 was due to a different mechanism of action, i.e., 13–197 is an ATP non-competitive inhibitor.
To test this, we assembled a small panel of IKKβ inhibitors with different core structures for head-to-head comparison studies with 13–197 (Figure 1A). These include ATP-competitive IKKβ inhibitors PS-1145 and ML-120B which are β-carboline derivatives,19 Sc-514 an analogue of TPCA1 with a thiophene-2-carboxamide core,20 the allosteric IKKβ inhibitor BMS345541, which is competitive with IKKβ substrate IκBα,21 and the non-selective kinase inhibitor staurosporine.
Figure 1:
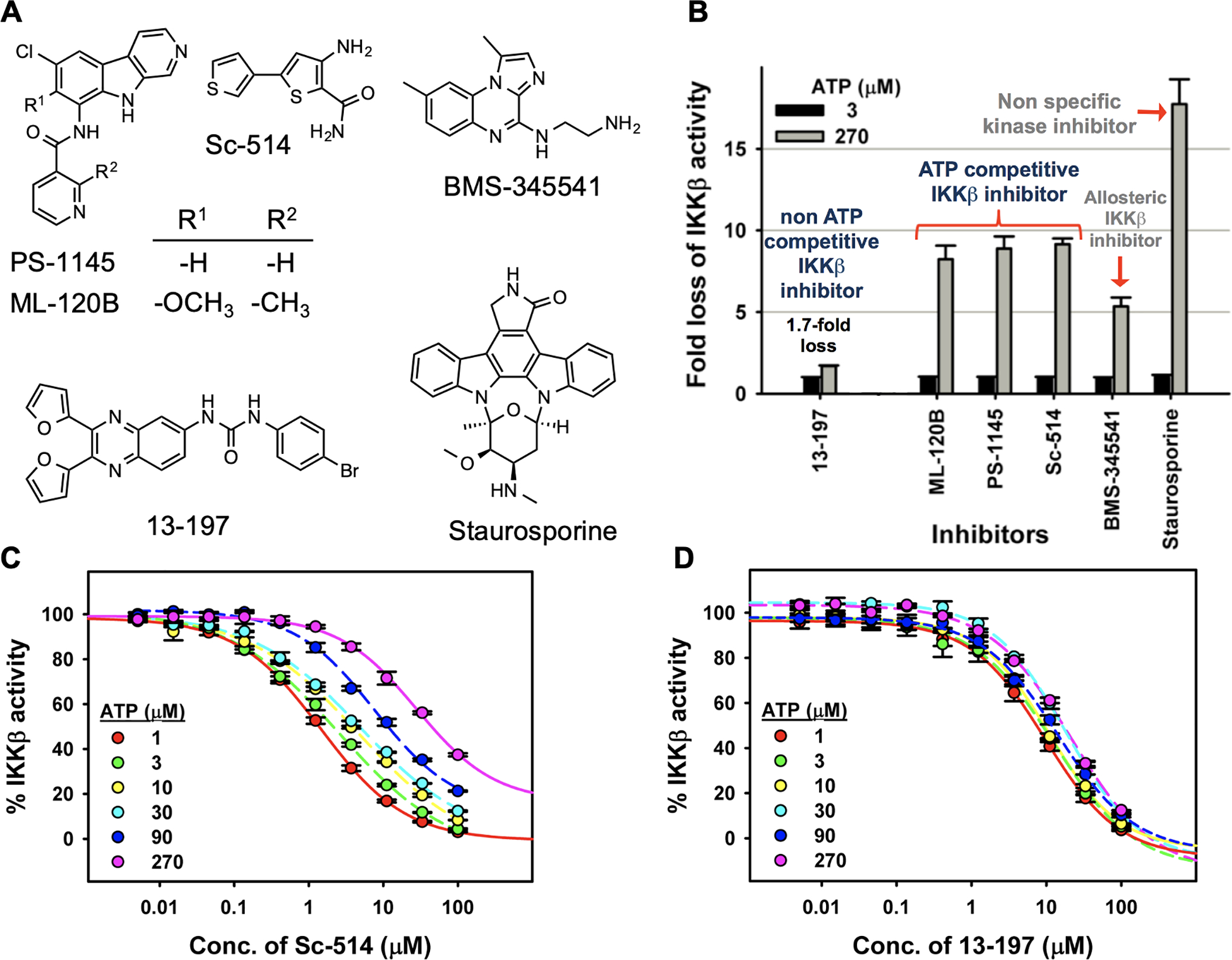
(A) Structures of IKKβ inhibitors. (B) Fold-change in activity of IKKβ inhibitors due to a 90-fold increase in ATP concentration in in vitro IKKβ kinase assays. (C) Sc-514 dose-response studies with IKKβ and increasing concentration of ATP. (D) 13–197 dose-response studies with IKKβ and increasing concentration of ATP.
We screened IKKβ kinase activity in vitro against the panel of IKKβ inhibitors at two different ATP concentrations (3 μM and 270 μM) (Figure 1B). We observed ~10-fold loss of inhibitory activity with ATP competitive IKKβ inhibitors (PS-1145, ML-120B and Sc-514) when the ATP concentration was increased 90-fold in the kinase assay. Under similar conditions, the allosteric inhibitor BMS-345541 fared a little better with ~5-fold loss in inhibition, while the non-selective kinase inhibitor staurosporine was >15-fold less potent at 270μM ATP concentration. Interestingly, with 13–197 we observed only a ~1.7-fold loss of IKKβ kinase inhibitory activity when the ATP concentration was increased 90-fold (Figure 1B). In a follow up ATP dilution series with the ATP-competitive IKKβ inhibitor Sc-514 we observed that the curve shifted to the right in a dose-dependent manner with increasing concentrations of ATP (Figure 1C). On the other hand, the IC50 value of 13–197 remained largely unchanged with increasing concentrations of ATP (Figure 1D). These results strongly suggested that 13–197 functions an ATP non-competitive IKKβ inhibitor.
Next, in the presence or absence of 13–197, the progress curves for IKKβ reactions were linear for up to 2 hours. For the Michaelis-Menten and double reciprocal plots for 13–197 the slopes (μM/min) from the above progress curves were plotted against ATP concentration at indicated 13–197 concentrations (Figure 2A and 2B). In the Michaelis-Menten plot (Figure 2A), the apparent Km remained unchanged when 13–197 concentration was increased and in the double reciprocal plot (Figure 2B) the lines converged on the X-axis suggesting that 13–197 was non-competitive with respect to ATP against IKKβ. In comparison, a similar study with ML-120B showed that the apparent Km increased with increasing concentrations of ML-120B in the Michaelis-Menten plot (Figure 2C) and in the double reciprocal plot the lines converged on the Y-axis (Figure 2D) indicating that ML-120B is competitive with respect to ATP against IKKβ.
Figure 2:
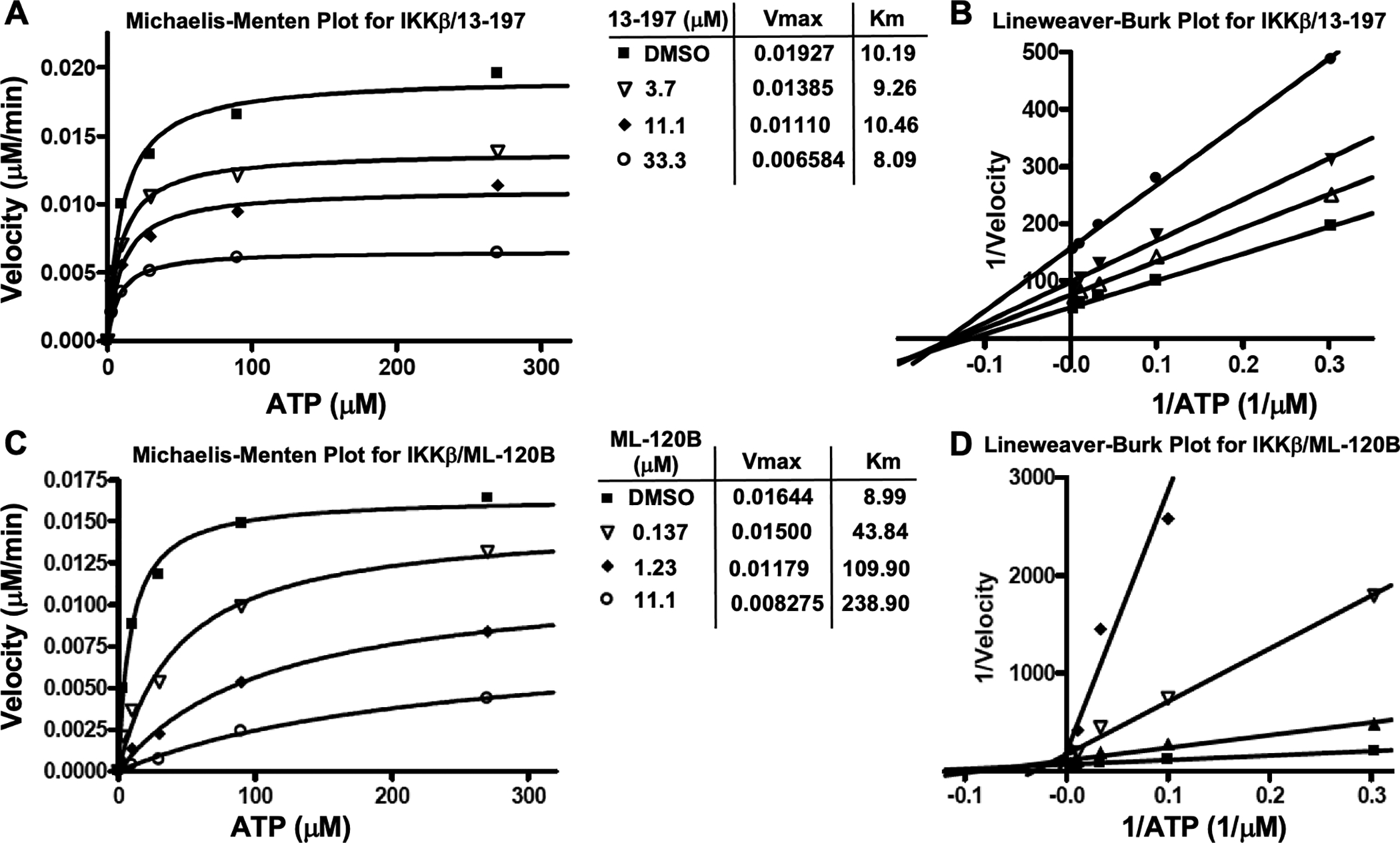
Michaelis-Menten and double reciprocal plots for IKKβ with either 13–197 or ML-120B
To identify the binding site of 13–197 on IKKβ, we adapted the photoaffinity labelling followed by LC-MS/MS approach reported by the Blagg lab to identify the binding site of Novobiocin on HSP90.22 We generated analogue 37–290 in which the bromine atom of 13–197 was replaced with an azide that could be cross linked to target proteins upon exposure to UV light (Figure 3A). Upon exposure to UV light, the aromatic carbon atom that is ortho to the azido group on 37–290 is susceptible to nucleophilic attack by the ε-nitrogen atom on lysine residues in proteins. This results in formation of a 2-amino-azepine ring structure which would yield peptides with a +411 Da signal. Briefly, we incubated IKKβ (20 μg) with 37–290 and one sample was subjected to UV cross linking while the other was used as control. The mixture was then subjected to AspN and trypsin digestion followed by LC-MS/MS analyses (Supplementary Table 1). We identified 9 peptides in the UV crosslinked sample with a +411 Da adduct (Figure 3B). Remarkably, 4/9 peptides had the +411 Da adduct on K310 (Figure 3C). A recently reported virtual screen, identified a small molecule inhibitor (analogue 124, Figure S1A) that occupied the allosteric pocket, which is formed by α-helices from the kinase domain (orange, Figure 3D) and a loop between two β sheets in the ULD (green, Figure 3D), proximal to K310 in IKKβ.23 Consistent with their model, the conformation derived through docking 13–197 using Schrödinger showed that the urea moiety in 13–197 is < 5Å from the imidazole in His380 in IKKβ suggesting potential hydrogen bond interaction (Figure S1B).
Figure 3:
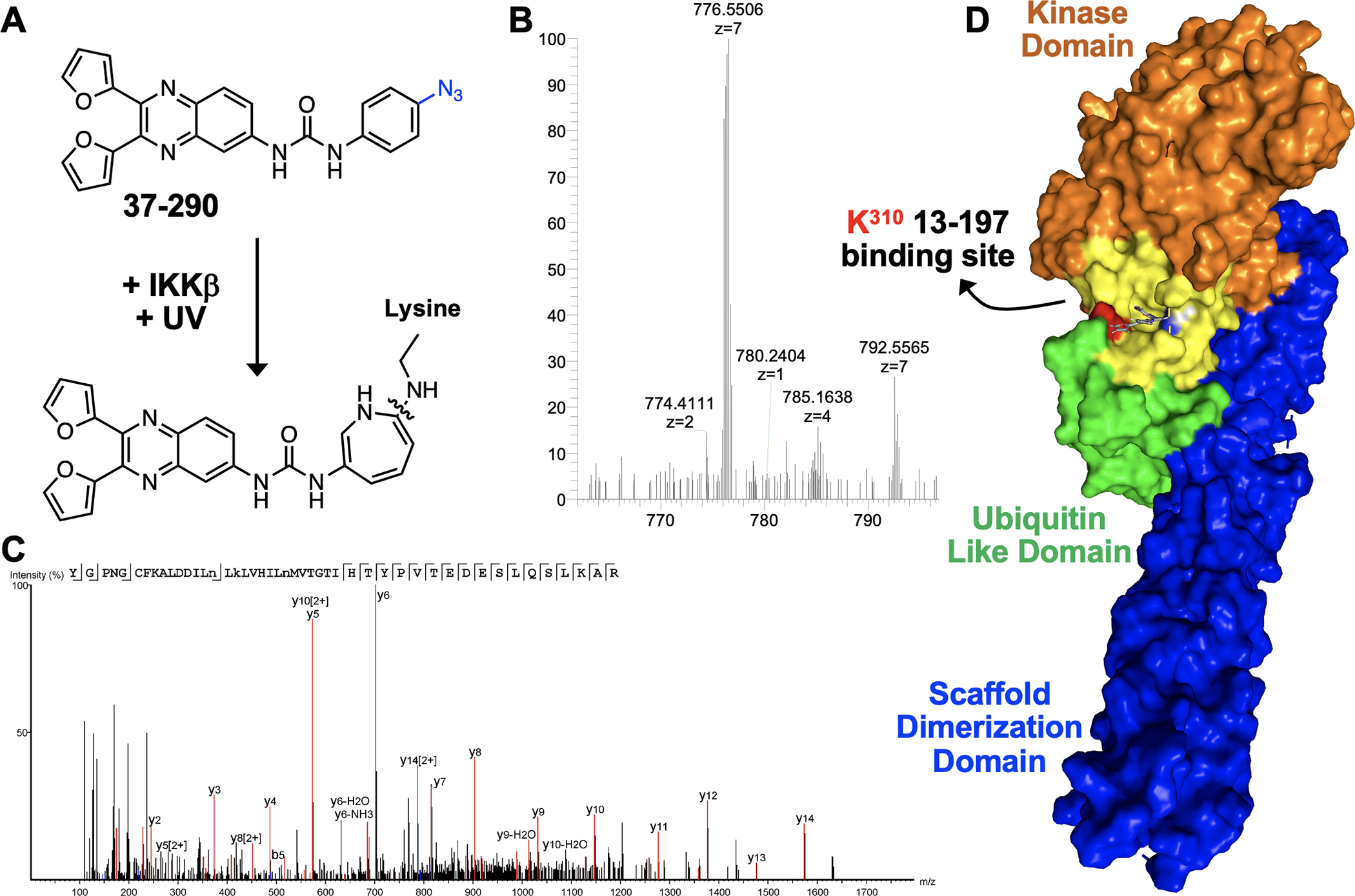
(A) UV crosslinking 37–290 in the presence of IKKβ results in the formation of an amino azapine adduct, (B) Representative 37–290 crosslinked mass spectrum. (C) The MS/MS spectrum associated with the 37–290 crosslinked mass spectrum showing the y-ions. (D) Surface rendering of 13–917 docked into the allosteric pocket adjacent to K310 in IKKβ.
An underappreciated fact associated with NF-κB signalling is the number of feedback inhibitory mechanisms that are in place to ensure that NF-κB activation is transient. An obvious manifestation of innate immunity in vertebrates is the activation of an inflammatory response. TNFα-induced IKKβ- mediated NF-κB activation is a critical signalling pathway that serves as an early inflammatory response. In its latent state, NF-κB is sequestered in the cytoplasm by its endogenous inhibitor IκBα. An array of exogenous stimuli, including TNFα, are known to activate this pathway resulting in the degradation of IκBα and nuclear translocation of NF-κB. Rapid activation of NF-κB mediated gene expression is the cellular response to the stimuli. This results in the resolution of the perceived stress that led to activation of the NF-κB pathway. An early gene that is expressed by NF-κB is its endogenous inhibitor IκBα (under 1h post stimulation), which rapidly dampens the response making the whole process transient. Sustained activation of NF-κB pathway proteins is implicated in a number of disease pathologies including cancer.24, 25 We hypothesized that inhibitors that allow for transient activation of the NF-κB pathway while blocking the sustained activation would serve as viable therapeutics.
To test this hypothesis, we treated HPNE cells with either DMSO or 13–197 and stimulated both sets with TNFα. This resulted in the rapid phosphorylation and degradation of IκBα in both DMSO and 13–197 treated HPNE cells (Figure 4A, compare lanes at 0, 5, 15 and 30 min). Following degradation, IκBα is re-expressed and is phosphorylated due to the continuous presence of TNFα in the media. Interestingly, at the 60 and 120 min time points post-TNFα stimulation we observed reduced phosphorylation of IκBα in the presence of 13–197 when compared to the control (Figure 4A). We also conducted a dose-response study wherein cells were treated with increasing concentrations of 13–197 and stimulated for either 5 min or 120 min with TNFα. The results summarized in Figure 4B show that 13–197 has very little effect on the levels of IκBα phosphorylation at the 5 min time point but inhibited the phosphorylation at the 120 min time point in a dose-dependent manner. Together these results suggests that 13–197 allows for transient NF-κB activation, but blocks the sustained activation of the canonical NF-κB pathway.
Figure 4:
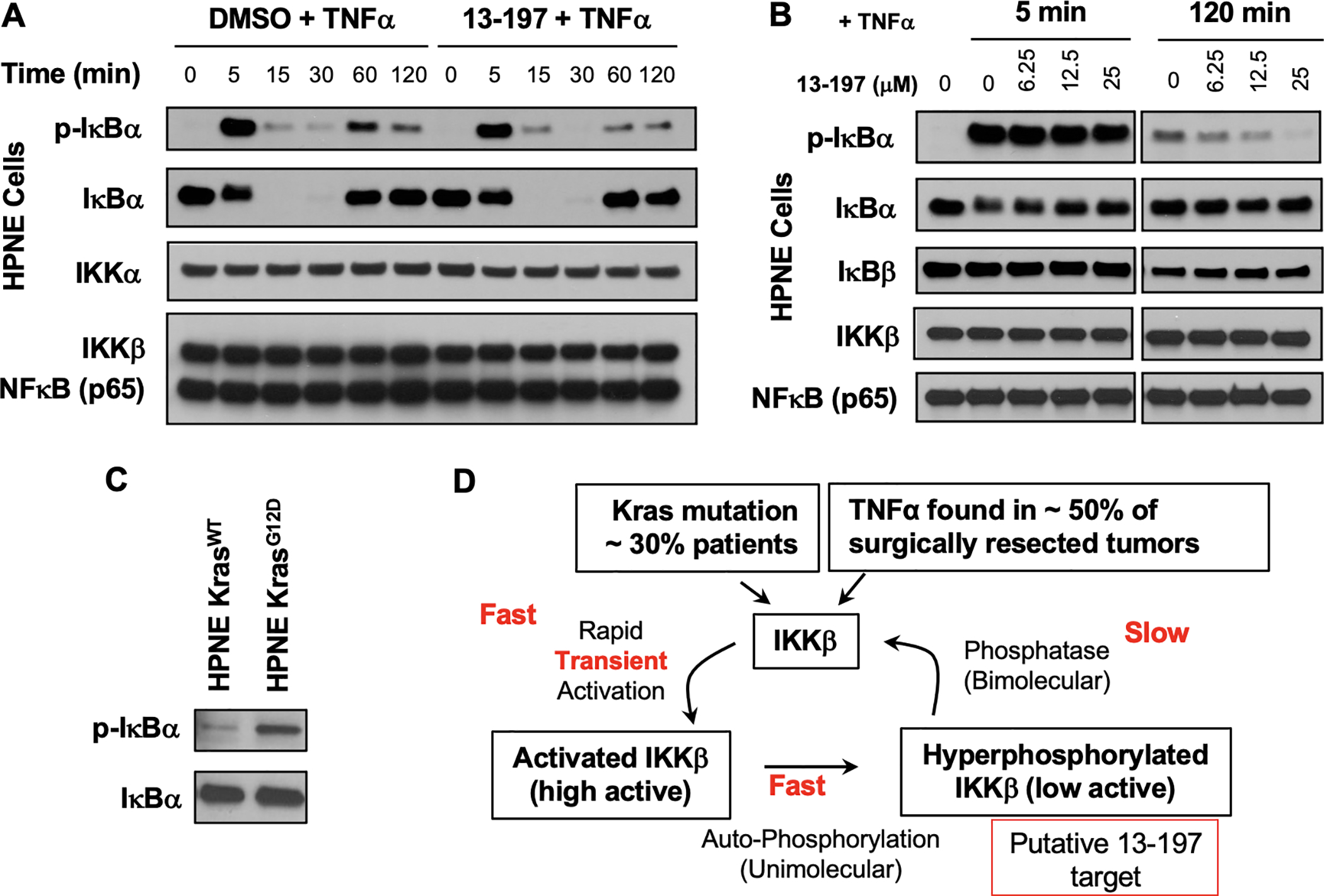
(A - B) Time course and dose-response studies to assess the effect of 13–197 on TNFα induced activation of IKKβ mediated IκBα phosphorylation. (C) Basal levels of IκBα phosphorylation in isogenic cell lines with and without KrasG12D mutation. (D) Our model for IKKβ activation-deactivation and the putative target of 13–197.
Kras mutation is associated with ~30% of all cancers and previous reports have suggested that Kras mutation activates IKKβ.9, 26 Consistently, in Kras mutation driven mouse models concurrent elimination of IKKβ resulted in reduction of tumour growth and increased survival.9, 26 On the other hand, constitutive activation (S177E, S181E) of IKKβ in a Kras mutation-driven model leads to very early onset of tumour growth resulting in a dramatic reduction in survival of mice.27 To determine if Kras mutation contributes to the activation of IKKβ, we subjected lysates from isogenic HPNE cell lines with and without Kras mutation to Western blot analyses (Figure 4C).28, 29 The results reveal elevated basal phosphorylation of IεBα indicating constitutive activation of IKKβ by Kras mutation.
Based on our findings we propose a model for IKKβ regulation and the putative target of 13–197 (Figure 4D). Briefly, in cancers the presence of TNFα in the tumour microenvironment and/or Kras mutation leads to the rapid activation of IKKβ. This is characterized by phosphorylation of serine residues 177 and 181 in the activation loop of IKKβ. Previous reports have shown that pS177,181-IKKβ undergoes autophosphorylation at a stretch of serine residues at the C-terminus over a period of 1–2 h, resulting in a hyperphosphorylated form of IKKβ with reduced kinase activity.30 To return to the resting state, hyperphosphorylated IKKβ associates with phosphatases such as PP5 that will dephosphorylate IKKβ.31 If this pathway is continuously stimulated, as in the case of cancer or CID, we posit that the hyperphosphorylated form of IKKβ will accumulate. Based on our data we propose hyperphosphorylated IKKβ as the putative target of 13–197 (Figure 4D).
In conclusion, here we report the mechanism of action of a non-toxic IKKβ inhibitor 13–197. Our studies show that 13–197 is an ATP non-competitive inhibitor of IKKβ. UV crosslinking using an azido analogue of 13–197 followed by LC-MS/MS implicates an allosteric pocket formed by residues from the kinase and ULD in IKKβ as the potential binding site of 13–197. In cells, 13–197 allows for the transient activation of the NF-κB pathway while inhibiting sustained activation. Our results provide a viable path forward for development of IKKβ inhibitors that lack the toxicity observed previously with ATP-competitive compounds.
Supplementary Material
Acknowledgements
This work was supported in part by NIH grants CA182820, CA197999, CA251151, GM121316 and CA036727. The research resources at UNMC are supported in part by the Nebraska Research Initiative. Biochemistry research at SDSU is supported in part by the California Metabolic Research Foundation. We would like to thank Dr. Koni Stone and the Natarajan lab members for helpful discussions
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
There are no conflicts of interest to declare
References
- 1.Braun J and Sieper J, Expert Opin Biol Ther, 2003, 3, 141–168. [DOI] [PubMed] [Google Scholar]
- 2.Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, Sun HL, Li LY, Ping B, Huang WC, He X, Hung JY, Lai CC, Ding Q, Su JL, Yang JY, Sahin AA, Hortobagyi GN, Tsai FJ, Tsai CH and Hung MC, Cell, 2007, 130, 440–455. [DOI] [PubMed] [Google Scholar]
- 3.Gilmore TD, Oncogene, 2006, 25, 6680–6684. [DOI] [PubMed] [Google Scholar]
- 4.Prescott JA and Cook SJ, Cells, 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wen D, Nong Y, Morgan JG, Gangurde P, Bielecki A, Dasilva J, Keaveney M, Cheng H, Fraser C, Schopf L, Hepperle M, Harriman G, Jaffee BD, Ocain TD and Xu Y, J Pharmacol Exp Ther, 2006, 317, 989–1001. [DOI] [PubMed] [Google Scholar]
- 6.Nagashima K, Sasseville VG, Wen D, Bielecki A, Yang H, Simpson C, Grant E, Hepperle M, Harriman G, Jaffee B, Ocain T, Xu Y and Fraser CC, Blood, 2006, 107, 4266–4273. [DOI] [PubMed] [Google Scholar]
- 7.Greten FR, Arkan MC, Bollrath J, Hsu LC, Goode J, Miething C, Goktuna SI, Neuenhahn M, Fierer J, Paxian S, Van Rooijen N, Xu Y, O’Cain T, Jaffee BB, Busch DH, Duyster J, Schmid RM, Eckmann L and Karin M, Cell, 2007, 130, 918–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Podolin PL, Callahan JF, Bolognese BJ, Li YH, Carlson K, Davis TG, Mellor GW, Evans C and Roshak AK, J Pharmacol Exp Ther, 2005, 312, 373–381. [DOI] [PubMed] [Google Scholar]
- 9.Xia Y, Yeddula N, Leblanc M, Ke E, Zhang Y, Oldfield E, Shaw RJ and Verma IM, Nat Cell Biol, 2012, 14, 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu G, Lo YC, Li Q, Napolitano G, Wu X, Jiang X, Dreano M, Karin M and Wu H, Nature, 2011, 472, 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polley S, Huang DB, Hauenstein AV, Fusco AJ, Zhong X, Vu D, Schrofelbauer B, Kim Y, Hoffmann A, Verma IM, Ghosh G and Huxford T, PLoS biology, 2013, 11, e1001581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu S, Misquitta YR, Olland A, Johnson MA, Kelleher KS, Kriz R, Lin LL, Stahl M and Mosyak L, J Biol Chem, 2013, 288, 22758–22767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Q, Bryant VC, Lopez H, Kelly DL, Luo X and Natarajan A, Bioorg Med Chem Lett, 2011, 21, 1929–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajule R, Bryant VC, Lopez H, Luo X and Natarajan A, Bioorg Med Chem, 2012, 20, 2227–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gautam N, Bathena SP, Chen Q, Natarajan A and Alnouti Y, Biomed Chromatogr, 2013, 27, 900–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Radhakrishnan P, Bryant VC, Blowers EC, Rajule RN, Gautam N, Anwar MM, Mohr AM, Grandgenett PM, Bunt SK, Arnst JL, Lele SM, Alnouti Y, Hollingsworth MA and Natarajan A, Clin Cancer Res, 2013, 19, 2025–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chaturvedi NK, Rajule RN, Shukla A, Radhakrishnan P, Todd GL, Natarajan A, Vose JM and Joshi SS, Mol Cancer Ther, 2013, 12, 2006–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maroni D, Rana S, Mukhopadhyay C, Natarajan A and Naramura M, Immunol Lett, 2015, 168, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, Munshi N, Dang L, Castro A, Palombella V, Adams J and Anderson KC, J Biol Chem, 2002, 277, 16639–16647. [DOI] [PubMed] [Google Scholar]
- 20.Liu Q, Wu H, Chim SM, Zhou L, Zhao J, Feng H, Wei Q, Wang Q, Zheng MH, Tan RX, Gu Q, Xu J, Pavlos N, Tickner J and Xu J, Biochem Pharmacol, 2013, 86, 1775–1783. [DOI] [PubMed] [Google Scholar]
- 21.Burke JR, Pattoli MA, Gregor KR, Brassil PJ, MacMaster JF, McIntyre KW, Yang X, Iotzova VS, Clarke W, Strnad J, Qiu Y and Zusi FC, J Biol Chem, 2003, 278, 1450–1456. [DOI] [PubMed] [Google Scholar]
- 22.Matts RL, Dixit A, Peterson LB, Sun L, Voruganti S, Kalyanaraman P, Hartson SD, Verkhivker GM and Blagg BS, ACS Chem Biol, 2011, 6, 800–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu H, Liang H, Meng H, Deng X, Zhang X and Lai L, Medchemcomm, 2018, 9, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu F, Xia Y, Parker AS and Verma IM, Immunol Rev, 2012, 246, 239–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baltimore D, Nat Immunol, 2011, 12, 683–685. [DOI] [PubMed] [Google Scholar]
- 26.Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, Li J, Peng B, Fleming JB, Wang H, Liu J, Lemischka IR, Hung MC and Chiao PJ, Cancer Cell, 2012, 21, 105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naramura M and Natarajan A, Pancreas, 2018, 47, e27–e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee KM, Nguyen C, Ulrich AB, Pour PM and Ouellette MM, Biochem Biophys Res Commun, 2003, 301, 1038–1044. [DOI] [PubMed] [Google Scholar]
- 29.Purohit A, Varney M, Rachagani S, Ouellette MM, Batra SK and Singh RK, Oncotarget, 2016, 7, 7280–7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delhase M, Hayakawa M, Chen Y and Karin M, Science, 1999, 284, 309–313. [DOI] [PubMed] [Google Scholar]
- 31.Chiang CW, Liu WK, Chiang CW and Chou CK, Biochem J, 2011, 433, 187–196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.