

Abstract
De novo, heterozygous, loss of function variants were identified in Pou domain, class 4, transcription factor 1 (POU4F1) via whole exome sequencing in four independent probands presenting with ataxia, intention tremor, and hypotonia. POU4F1 is expressed in the developing nervous system, and mice homozygous for null alleles of Pou4f1 exhibit uncoordinated movements with newborns being unable to successfully right themselves to feed. Head magnetic resonance imaging of the four probands was reviewed and multiple abnormalities were noted including significant cerebellar vermian atrophy and hypertrophic olivary degeneration in one proband. Transcriptional activation of the POU4F1 p.Gln306Arg protein was noted to be decreased when compared to wild-type. These findings suggest that heterozygous, loss-of-function variants in POU4F1 are causative of a novel ataxia syndrome.
Keywords: POU4F1, ataxia, intention tremor, paroxysmal tonic upgaze
Introduction
Hereditary ataxias are a highly heterogeneous group of disorders with symptoms manifesting as slowly progressive incoordination of movement and gait (Jayadev & Bird, 2013). While more than 150 genes have been identified as causative of hereditary ataxias, including genes with autosomal recessive, autosomal dominant, X-linked, and mitochondrial inheritance, many causative disease genes have yet to be recognized and many affected patients remain without an underlying molecular diagnosis (Parodi, Coarelli, Stevanin, Brice, & Durr, 2018).
POU4F1, alternatively known as BRN3A, encodes a class IV POU domain-containing transcription factor expressed in the developing nervous system. The POU domain is a bipartite domain composed of two subunits, a POU-specific domain and a POU-homeodomain, separated by a non-conserved region of 15 to 55 amino acids (Herr et al., 1988). The class IV POU domain group includes C. elegans Unc-86, which is expressed solely in neuroblasts and neurons, and human POU4F1, POU4F2, and POU4F3 genes, which are expressed in the brainstem, retina, dorsal root ganglia, and trigeminal ganglia (Xiang, Gan, Zhou, Klein, & Nathans, 1996). POU4F1 expression is present in the medial habenula, red nucleus, inferior olivary nucleus, and superior colliculus. Targeted disruption of Pou4f1 leads to a specific phenotype in Pou4f1−/− mice where newborns are noted to have decreased suckling with absence of rhythmic jaw opening as well as uncoordinated limb and truncal movements leading to impaired righting and death before 24 hours of age. Pou4f1+/− mice are indistinguishable from wild-type mice in growth and behavior (Xiang et al., 1996). Additionally, Pou4f1−/− mice have loss of neurons in the trigeminal ganglia, medial habenula, red nucleus, inferior olivary nucleus, and nucleus ambiguus (McEvilly et al., 1996; Serrano-Saiz, Leyva-Diaz, De La Cruz, & Hobert, 2018; Xiang et al., 1996)
Here we report four probands from separate families who presented with an unknown ataxia syndrome and who were found to have heterozygous, de novo pathogenic variants in POU4F1. This work expands the known genetic etiologies of hereditary ataxia syndromes.
Methods
Patients, families, and clinical evaluation.
This study was conducted in accordance with the Declaration of Helsinki. The families were enrolled in a research protocol approved by the Institutional Review Board at the Icahn School of Medicine at Mount Sinai. Adult participants and guardians of children provided written informed consent for participation. Medical records from subspecialists including pediatric neurologists, pediatric ophthalmologists, and clinical geneticists were reviewed in detail. Detailed medical histories were taken. Head magnetic resonance imaging (MRI) was obtained for review. Patient videos, including videos showing ataxia and ocular motility, were obtained and reviewed.
Whole exome sequencing.
The four probands received whole exome sequencing through a clinical laboratory (GeneDx, Gaithersburg, MD). Using genomic DNA from the proband and parents, the exonic regions and flanking splice junctions of the genome were captured using the IDT xGen Exome Research Panel v1.0. Massively parallel sequencing was done on an Illumina system with 100bp or greater paired-end reads. Reads were aligned to human genome build GRCh37/UCSC hg19, and analyzed for sequence variants using a custom-developed analysis tool. Additional sequencing technology and variant interpretation protocol has been previously described (Retterer et al., 2016).
Analysis of brain MRI data.
We reviewed head magnetic resonance (MR) imaging from the four probands. Additionally, for each available MR scan the cross-sectional areas of the vermis were measured on the mid-sagittal images as pixel counts within manually drawn contours using the GNU Image Manipulation Program (Open Source, version 2.10.20) (Supp. Tables S1–4, Supp. Figures S2–3). The pixel counts were then normalized to cm2 areas using the intrinsic cm marking strip included within each MR image. Standards for normal mid-sagittal areas of the vermis were as defined by Hayakawa et al. in 92 patients aged newborn to 15 years (Hayakawa et al., 1989).
Measurement of transcriptional activity of POU4F1 p.Gln306Arg variant.
The relative transcriptional activity (Firefly luciferase/Renilla luciferase) for POU4F1 wild-type (POU4F1-wt) and mutant (POU4F1-Q306R) was measured. Transfections were carried out with 60 ng of POU4F1-wt or POU4F1-Q306R, 200 ng of pGL4.23-luc2-RUNX3 or pGL4.23-luc2-minP (minimal promoter), and 50 ng of pRL-null using Lipofectamine3000 (Invitrogen) in HEK293 cells grown in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal calf serum. Cells were plated in a 24 well plate such that the cells were approximately 75% confluent at the time of transfection. The experiment was completed in triplicate, and the luciferase activities were measured 48 hours post-transfection.
Results
Clinical findings.
Proband 1 is a 3-year-old female who was diagnosed with ataxia and intention tremor by age 2 years. She has received strabismus surgery for left eye esotropia and has been diagnosed with paroxysmal tonic upgaze. She has global developmental delay, and has received physical, occupational, and speech therapies. She walked at age 2 years. There has been no regression of developmental milestones. On exam, she is grossly nondysmorphic is hypotonic and displays ataxia.
Proband 2 is a 3-year-old male who was diagnosed with ataxia and intention tremor by age 3 years. He has no history of strabismus. He has been diagnosed with paroxysmal tonic upgaze. He has global developmental delay and has received physical, occupational, and speech therapies. He was able to walk independently at age 3 years 1 month. There has been no regression of developmental milestones. On exam, he is grossly nondysmorphic and shows generalized mild to moderate hypotonia. He has a slight head tremor when sitting. He is mildly unsteady with walking and has a stiff-legged gait. Ataxia is present.
Proband 3 is a 4-year-old male who was noted to have significant truncal hypotonia by age 4 months. He was diagnosed with ataxia and intention tremor by age 2 years. He has a history of a persistent head tremor and underwent strabismus surgery left eye esotropia at 14 months and 39 months. He has global developmental delay and has received physical, occupational, and speech therapies. There has been no regression of developmental milestones. At 4 years of age, he is able to walk with the assistance of a gait trainer. On exam, he is grossly nondysmorphic, and has truncal hypotonia and ataxia.
Proband 4 is a 22-year-old female with ataxia, hypotonia, and intention tremor. Hypotonia was noted from birth. Ataxia was noted as her motor skills developed. Significant motor delays were noted from an early age, and she has received physical therapy from a young age. She rolled over at 18 months, sat without support at 3 years, and walked at age 8 years. She has a mild learning disability diagnosed in grade school. There has been no regression of milestones. She was diagnosed with esotropia and received corrective surgery at age 2 years. A video from age 3 years revealed paroxysmal tonic upgaze, no longer seen in later years. Neuro-ophthalmologic exam at age 11 years 8 months revealed an alternating 12 diopter esotropia with full range of eye movements. On slit lamp examination, she was noted to have a slow vertical-torsional nystagmus. At age 22 years, the proband’s family reports that they have noticed a gradual overall decrease in speech, fine motor skills, coordination, and ambulation in recent years. On exam at age 22 years, she was grossly nondysmorphic. She had decreased tone, hyporeflexia, mild end-task tremor, dysarthria, left greater than right dysmetria of bilateral upper and lower extremities, left greater than right dysdiadochokinesia, and convergence insufficiency. Eye movement range remained full. She was able to achieve full elevation of eyes, but spontaneous drifts of the eyes further up were superimposed on attempts to hold the eyes in elevated position. Saccades were fast, though slightly dysmetric; smooth pursuit was saccadic vertically. She had difficulty maintaining extreme elevation of the eyes in upgaze and convergence insufficiency. Ocular oscillations and palatal tremor were not present.
Each of the four probands were noted to have ataxia, hypotonia, and intention tremor (Table 1). All had significant motor delays. Three of four probands had strabismus, and three of four probands had history of paroxysmal tonic upgaze.
Table 1.
Four probands identified with heteroyzgous variants in POU4F1.
| Proband 1 | Proband 2 | Proband 3 | Proband 4 | |
|---|---|---|---|---|
| Variant | de novo, heterozygous | de novo, heterozygous | de novo, heterozygous | de novo, heterozygous |
| c.917A>G; p.Gln306Arg | c.158_161dup;p.Leu55AlafsTer295 | c.283_290del; p.Thr95SerfsTer251 | c.271_281del;p.Thr91HisfsTer254 | |
| Sex | Female | Male | Male | Female |
| Current Age | 3 yrs 5 months | 3 yrs, 10 months | 4 yrs, 2 months | 22 years old |
| Birth History | Born at 39 weeks via C-section | Born at 38 weeks via vaginal delivery | Born at 41 weeks by induced vaginal delivery | Born at 39 weeks gestation via vaginal delivery |
| Birth wt: 8lbs 1 oz | Birth wt: 7 lbs 7 oz | Birth wt: 8 lb 10 oz | Birth wt: 8 lbs 0.8 oz | |
| Had initial oxygen requirements, but went to the floor with mother | Uneventful birth history | Uneventful birth history | Uneventful birth history | |
| Respiratory system | Concern for aspiration with thin liquids at age 2 years | No issues | No issues | No issues |
| Cardiovascular system | No issues | No issues | No issues | No issues |
| Gastrointestinal systetm | Had early swallowing difficulties; correction of ankyloglossia | No issues | Chronic constipation, better since bowel cleanout | No issues |
| Renal system | No issues | No issues | No issues | No issues |
| Musculoskeletal system | Scoliosis (dextroconvex curvature from T5 to L3); prominent thoracic kyphosis with borderline upper limits of normal lumbar lordosis | No known issues | No known issues | Scoliosis and Scheuermann’s disease |
| Hematologic system | Excessive bleeding after ankyloglossia surgery; dx with von Willebrand’s disease | No issues | No issues | No issues |
| Ophthalmologic system | ||||
| strabismus | Left eye esotropia s/p strabismus surgery | None appreciated | Left eye esotropia s/p surgery at 14 months and 39 months | Bilateral esotropia s/p surgery |
| nystagmus | None appreciated | None appreciated | None appreciated | Slow vertical torsional nystagmus noted on slit-lamp examination |
| other abnormal movements | None appreciated | None appreciated | None appreciated | Spontaneous drifts of the eyes upwards superimposed on tonically held upgaze (after resolution of paroxysmal tonic upgaze) |
| ophthalmoplegia | None appreciated | None appreciated | None appreciated | None appreciated |
| other | Paroxysmal tonic upgaze; wears glasses | Paroxysmal tonic upgaze | Paroxysmal tonic upgaze, later convergence insufficiency | |
| Audiology | Normal hearing | Normal hearing | Normal hearing | Normal hearing |
| Neurological system | ||||
| Intention tremor | Present | Present | Present (particularly with right upper extremity) | Present |
| Ataxia | Present (falls frequently, poor balance and coordination) | Present (falls frequently, poor balance and coordination) | Present | Present |
| Hypotonia | Present | Present | Present (low truncal tone most pronounced) | Present (from infancy; central and peripheral; currently cannot lift anything >10 lbs) |
| Developmental delay/intellectual disability | Global developmental delay | Global developmental delay | Global developmental delay | Isolated motor delays from infancy; learning disability/mild cognitive delays noted at older age |
| Seizures | None | None | None | None |
| Developmental Regression | None | None | None | None |
| Trigeminal involvement | Possible difficulty clenching mouth | None appreciated | Denies difficulty chewing | None |
| Deep tendon reflexe | Patellar 2+ reflexes | Patellar 1+ reflexes | Decreased DTRs (1+ for biceps, tricpes, brachioradialis, and patella bilaterally; 2+ for Achilles) | Biceps 0/4 bilaterally; Triceps and brachioradialis 1/4 bilaterally; Patellar 2/4 bilaterally, Achilles 2+ bilaterally |
| Other | Nerve conduction studies normal at 9 months | Dysmetria, dysarthria, speech apraxia. EEG normal at age 2 years. | ||
| Developmental Milestones | Rolled over at 8 months in both directions; sat at 10 months; crawled at 14 months; walked at age 2 years. | Walked at 3 years 1 month. | Rolled over at 9 months in both directions; sat at 9 months; crawled at 2.5 years; at 4 years is able to walk with a gait trainer | Rolled over in both directions at 18 months; sat at 3 years; walked independently at 8–10 years; received Special Education throughout school. |
| Growth Parameters | ||||
| Height | 64th% at age 2 years | 24th% at 3 years 6 months | 15th% at 2 years 4 months; 16th% at 4 years 4 months | 5th% at 3 years 6 months |
| Weight | 62nd% at age 2 years | 48th% at 3 years 6 months | 25th% at 2 years 4 months; 28th% at 4 years 4 months | 5th% at 3 years 6 months |
| HC | 45th% at age 2 years | 20th% at 3 years 6 months | 33rd% at 2 years 4 months; 17th% at 4 years 4 months | 10th% at 3 years 6 months |
Whole exome sequencing.
Heterozygous, de novo, loss of function variants in POU4F1 were identified in all probands (Table 1). Three of the four probands had frameshift variants, while one proband had a missense change (POU4F1 c.917A>G;p.Gln306Arg; NM_006237.3). POU4F1 is noted to have a high pLI score of 0.89, suggesting intolerance to haploinsufficiency. Variant p.Gln306Arg in POU4F1 gene is not seen in gnomAD, and it is predicted to be deleterious by SIFT and PolyPhen2. Glutamine at codon 306 is highly conserved through a variety of species from human to zebrafish. According to UniProt (www.uniprot.org), POU4F1 Gln306 is located in a POU-specific domain that spans amino acids 261 to 338. A homology model of human POU4F1 is available from the Swiss Model repository, and the position of POU4F1 Gln306 is highlighted (Supp. Figure S1) (http://swissmodel.expasy.org).
Review and analysis of brain MR imaging.
MR images were obtained for each proband and reviewed by an experienced pediatric neuroradiologist. In proband 4, serial MR scans from age 11 months to age 22 years 11 months revealed initially progressive, symmetrical, T2-weighted hyperintensity of the inferior olivary nuclei, which later resolved, reminiscent of hypertrophic olivary degeneration (Fig. 1 A–C) (Goyal et al., 2000). The serial MR scans also documented progressive atrophy of the cerebellar vermis and hemispheres (Fig. 1 D–E; Fig. 2 A–D). MR studies were normal in Proband 1 at ages 9 months, 21 months and 43 months (the first two with contrast enhancement); in proband 2 at age 3 years 2 months; and in proband 3 at age 25 weeks.
Figure 1. Serial MRI studies of Proband 4.
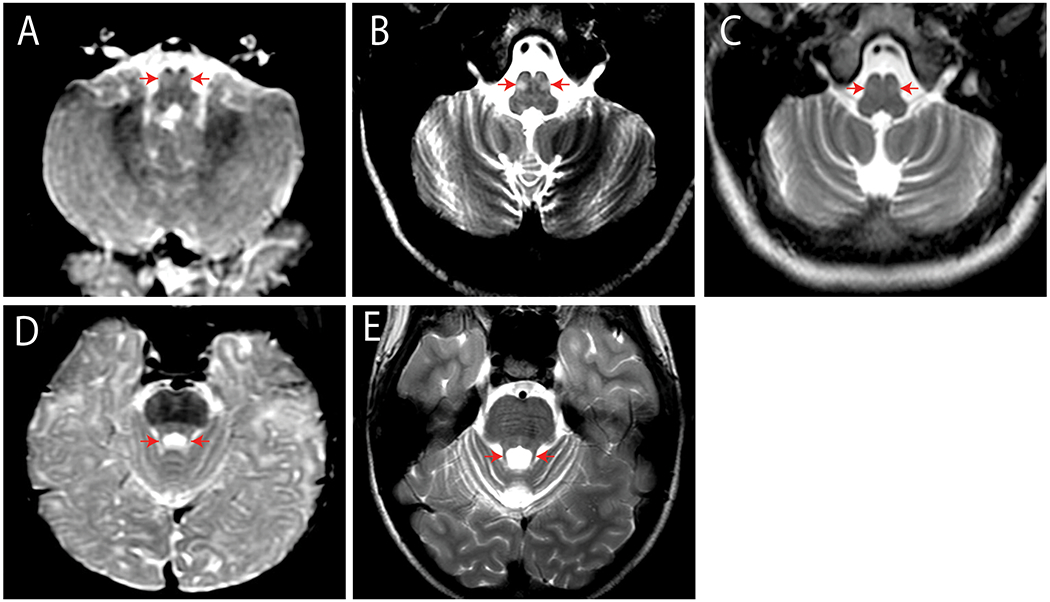
(A-C). Axial T2-Weighted (T2W) MRI sections through the caudal medulla. The inferior olivary nuclei show prominent symmetrical signal increase at age 11.2 months (A) and age 11.0 years (B) that fades by age 16.4 years (C). Progressive enlargement of the cerebellar fissures indicates concurrent atrophy of both inferior cerebellar hemispheres. D-E. Axial T2W sections through the superior cerebellum at age 11.2 months (D) and 13 years (E) show symmetrical thin caliber of the superior cerebellar peduncles and increased size of the superior cerebellar fissures confirming bilateral superior cerebellar atrophy.
Figure 2. Serial mid-sagittal T1-W MRI through the vermis of Proband 4.

(A). At age 11.2 months, the anterior lobe of the vermis is normal. (B-D). T1-WMRI at ages 11 years (B), 13 years (C), and 22 years 11 months (D) show progressive vermian atrophy. See Supp. Figure S3 and Supp.Table S2.
Hayakawa et al. previously found that the growth pattern of the vermis was exponential in healthy individuals, with a growth spurt in the first four years and attainment of adult size in the 6-9 year age range (Hayakawa et al., 1989). In this series of patients with pathogenic POU4F1 variants, Proband 1 showed a 21.2% increase in total vermian cross-sectional area from age 8 months to 21 months and a small additional increase to 23.4% by age 43 months (Supp. Table S1, Supp. Fig. S2). Proband 4 showed a 14.2% loss of the total vermian cross-sectional area from age 11 months to age 11 years, and prominent additional loss to a total loss of 30.2% by age 22 years 11 months (Supp. Table S2, Supp. Fig. S3). It is noteworthy that in both Proband 1 and Proband 4 the changes in vermian area appear to have affected all lobes of the vermis equally, since the ratio of the cross-sectional area of the anterior lobe to the total vermis and the ratio of the anterior lobe to the posterior lobe plus nodulus remain nearly constant over time. Further, these same area ratios are seen in Proband 2 at age 38 months, and Proband 3 at age 5 months. In Proband 4, the serial MRI studies also disclosed symmetric atrophy of the cerebellar hemispheres.
Transcriptional activity of POU4F1 Gln306Arg variant.
POU4F1 has previously been demonstrated to bind a conserved enhancer region 94 kb upstream of the Runx3 locus within histone H3-acetylated chromatin. This binding leads to increases in Runx3 in mRNA and functions to specify neuronal subtype in the trigeminal ganglion (Dykes, Lanier, Eng, & Turner, 2010). We demonstrate that overexpression of POU4F1 p.Q306R leads to stable protein in vitro (Supp. Figure S4). Additionally, POU4F1 p.Q306R has decreased transcriptional activation of the Runx3 locus when compared to wild-type POU4F1 (Figure 3).
Figure 3. Transactivation of POU4F1 p.Gln306Arg is reduced compared to wild-type.

The relative transcriptional activity (Firefly luciferase/Renilla luciferase) for POU4F1 wild-type (POU4F1-wt) and mutant (POU4F1-Q306R) was measured as described. Means ± standard deviations are shown. Significance was determined by a two tailed student t-test, ***p<0.005.
Discussion
To our knowledge, this is the first report of pathogenic variants in POU4F1 causing disease in humans. Pou4f1−/− mice are known to have uncoordinated movements consistent with the ataxia phenotype seen in this patient cohort. All four probands identified in this study had heterozygous, loss-of-function variants in POU4F1 and exhibited ataxia, intention tremor, and hypotonia.
Interestingly, three of the four probands exhibited paroxysmal tonic upgaze in early childhood. In the oldest proband, this resolved over time, but even at age 22 years spontaneous upward drifts of the eyes still occurred when the eyes were held in an elevated position. Paroxymal tonic upgaze is characterized by sudden unprovoked upward deviation of the eyes with preserved consciousness. Episodes are typically brief and often accompanied by a change in head position with chin lowering. This eye movement disorder sometimes occurs as a transient benign phenomenon in healthy infants, but it has also been reported in disorders of neurotransmitter depletion and genetic disorders. It is of particular interest that the few gene mutations with which this finding has been associated (CACNA1A, GRID2, and SEPSECS) all involve proteins related to normal cerebellar function and ataxia typically accompanies the paroxysmal tonic upgaze, similar to the probands in this study (Agamy et al., 2010; Blumkin et al., 2015; Hills et al., 2013; Makrythanasis et al., 2014; Quade et al., 2020). The pathomechanism of this eye movement finding is unclear, but these genetic associations raise the possibility that it may originate from cerebellar dysfunction and may be related to the normal cerebellar flocculus inhibition of upward eye movements and firing bias towards downward eye movements that underly downbeat nystagmus (Baloh & Spooner, 1981; Marti, Straumann, & Glasauer, 2005; Zee, Yamazaki, Butler, & Gucer, 1981). Cerebellar dysfunction would lead to an up-down asymmetry which may facilitate the development of paroxysmal tonic upgaze; indeed, some individuals with paroxysmal tonic upgaze also exhibit downbeat nystagmus with attempts to look down during episodes.
While the phenotype of the four probands is highly consistent, the MR features of the four probands are variable. In proband 4, the sequence of MR findings involving the olivary nucleus is similar to that seen with hypertrophic olivary degeneration. Hypertrophic olivary degeneration is a form of denervation atrophy that typically arises when acute stroke or demyelination disrupts the fibers of the rubro-olivo-cerebellar circuit (triangle of Guillain and Mollaret). Classically, on each side, this circuit is said to extend from the red nucleus to the ipsilateral inferior olivary nucleus, and from there to the contralateral dentate nucleus before returning back through the superior cerebellar peduncle and decussation of the superior cerebellar peduncle to the original red nucleus to complete the triangle (Goyal et al., 2000). More modern explanations of hypertrophic olivary degeneration exclude the red nucleus from relevant circuitry (Shaikh et al., 2010).
Clinically, hypertrophic olivary degeneration is associated with oculopalatal tremor that manifests as pendular vertical-torsional nystagmus and rhythmic involuntary movements of the soft palate, uvula, pharynx, larynx, and upper extremity. In rare instances, including in proband 4, hypertrophic olivary degeneration and oculopalatal tremor may occur in the absence of an acute brainstem event and may be accompanied by progressive ataxia. Such progressive ataxia with palatal tremor has been reported with other genetic diseases, such as Alexander disease and Sandhoff disease (Pretegiani et al., 2015; Schwankhaus, Parisi, Gulledge, Chin, & Currier, 1995). Though proband 4 did not have notable nystagmus or any abnormal palatal movements on her most recent examination at age 22, she was documented to have had vertical-torsional nystagmus at age 11. Particularly unusual is the fact that the hypertrophic olivary degenerative changes on MRI improved over time. The relationship between the clinical and MR features will be the subject of future investigations.
Additional probands will likely be identified in the future as whole exome sequencing is more widely applied. Identification of additional affected individuals will confirm causality of POU4F1 and enable the clinical spectrum of disease due to POU4F1 haploinsufficiency to be further defined.
Supplementary Material
Supplemental Figure S1. Model of Homo sapiens POU4F1 from Swiss-Model repository. This homology model was created using human POU6F1 as the template. A cartoon of POU4F1 is shown with the amino acid Gln306 highlighted.
Supplemental Figure S2. Proband 1. Serial T1-weighted mid-sagittal MR images at ages A. 8 months, B. 1 year 9 months and C. 3 years 7 months.
At each age, the yellow outlines identify the cross-sectional areas of the anterior lobe of the vermis vs. the posterior lobe plus nodulus. The total cross-sectional area of the vermis increases by 23.4% from age 8 months to age 43 months. See Supp. Table S1.
Supplemental Figure S3. Proband 4. Serial T1-weighted mid-sagittal MR images at ages A. 11 months, B. 11 years, C. 13 years, D. 16 years, and E. 22 years.
At each age, the yellow outlines identify the cross-sectional areas of the anterior lobe of the vermis vs. the posterior lobe plus nodulus. The total cross-sectional area of the vermis decreases by 30.2% from age 11 months to age 22 years 11 months. See Supp. Table S2.
Supplemental Figure S4. Expression of POU4F1 in vitro. Wild-type POU4F1 and Gln306Arg POU4F1 were over-expressed in HEK293 cells and compared to a control HEK293 sample. POU4F1 p.Gln306Arg is stable.
Acknowledgements
We are grateful to the families who graciously consented to participate in our research study. Dr. Webb is supported by NIH K08 HD086827.
Footnotes
Conflict of Interest Statement:
LBH, FM, and YS are employees of GeneDx, Inc. EES is an employee of Sema4, and BDW is a consultant for Sema4. The remainder of the authors have no conflicts to disclose.
Web Resources:
Leiden Open Variation Database (LOVD): www.lovd.nl
Swiss-Model repository: http://swissmodel.expasy.org
UniProt: www.uniprot.org
Data Availability Statement:
The data that support the findings of this study are available on request from the corresponding author. The whole exome sequencing data is not publicly available due to privacy or ethical restrictions. The POU4F1 variant information has been submitted to the Leiden Open Variation Database (LOVD) (www.lovd.nl).
References
- Agamy O, Ben Zeev B, Lev D, Marcus B, Fine D, Su D, … Birk OS (2010). Mutations disrupting selenocysteine formation cause progressive cerebello-cerebral atrophy. Am J Hum Genet, 87(4), 538–544. doi: 10.1016/j.ajhg.2010.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baloh RW, & Spooner JW (1981). Downbeat nystagmus: a type of central vestibular nystagmus. Neurology, 31(3), 304–310. doi: 10.1212/wnl.31.3.304 [DOI] [PubMed] [Google Scholar]
- Blumkin L, Leshinsky-Silver E, Michelson M, Zerem A, Kivity S, Lev D, & Lerman-Sagie T (2015). Paroxysmal tonic upward gaze as a presentation of de-novo mutations in CACNA1A. Eur J Paediatr Neurol, 19(3), 292–297. doi: 10.1016/j.ejpn.2014.12.018 [DOI] [PubMed] [Google Scholar]
- Dykes IM, Lanier J, Eng SR, & Turner EE (2010). Brn3a regulates neuronal subtype specification in the trigeminal ganglion by promoting Runx expression during sensory differentiation. Neural Dev, 5, 3. doi: 10.1186/1749-8104-5-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal M, Versnick E, Tuite P, Cyr JS, Kucharczyk W, Montanera W, … Mikulis D (2000). Hypertrophic olivary degeneration: metaanalysis of the temporal evolution of MR findings. AJNR Am J Neuroradiol, 21(6), 1073–1077. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/10871017 [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Konishi Y, Matsuda T, Kuriyama M, Konishi K, Yamashita K, … Hamanaka D (1989). Development and aging of brain midline structures: assessment with MR imaging. Radiology, 172(1), 171–177. doi: 10.1148/radiology.172.1.2740500 [DOI] [PubMed] [Google Scholar]
- Herr W, Sturm RA, Clerc RG, Corcoran LM, Baltimore D, Sharp PA, … et al. (1988). The POU domain: a large conserved region in the mammalian pit-1, oct-1, oct-2, and Caenorhabditis elegans unc-86 gene products. Genes Dev, 2(12A), 1513–1516. doi: 10.1101/gad.2.12a.1513 [DOI] [PubMed] [Google Scholar]
- Hills LB, Masri A, Konno K, Kakegawa W, Lam AT, Lim-Melia E, … Mochida GH (2013). Deletions in GRID2 lead to a recessive syndrome of cerebellar ataxia and tonic upgaze in humans. Neurology, 81(16), 1378–1386. doi: 10.1212/WNL.0b013e3182a841a3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayadev S, & Bird TD (2013). Hereditary ataxias: overview. Genet Med, 15(9), 673–683. doi: 10.1038/gim.2013.28 [DOI] [PubMed] [Google Scholar]
- Makrythanasis P, Nelis M, Santoni FA, Guipponi M, Vannier A, Bena F, … Antonarakis SE (2014). Diagnostic exome sequencing to elucidate the genetic basis of likely recessive disorders in consanguineous families. Hum Mutat, 35(10), 1203–1210. doi: 10.1002/humu.22617 [DOI] [PubMed] [Google Scholar]
- Marti S, Straumann D, & Glasauer S (2005). The origin of downbeat nystagmus: an asymmetry in the distribution of on-directions of vertical gaze-velocity Purkinje cells. Ann N Y Acad Sci, 1039, 548–553. doi: 10.1196/annals.1325.065 [DOI] [PubMed] [Google Scholar]
- McEvilly RJ, Erkman L, Luo L, Sawchenko PE, Ryan AF, & Rosenfeld MG (1996). Requirement for Brn-3.0 in differentiation and survival of sensory and motor neurons. Nature, 384(6609), 574–577. doi: 10.1038/384574a0 [DOI] [PubMed] [Google Scholar]
- Parodi L, Coarelli G, Stevanin G, Brice A, & Durr A (2018). Hereditary ataxias and paraparesias: clinical and genetic update. Curr Opin Neurol, 31(4), 462–471. doi: 10.1097/WCO.0000000000000585 [DOI] [PubMed] [Google Scholar]
- Pretegiani E, Rosini F, Federighi P, Cerase A, Dotti MT, & Rufa A (2015). Pendular nystagmus, palatal tremor and progressive ataxia in GM2-gangliosidosis. Eur J Neurol, 22(6), e67–69. doi: 10.1111/ene.12661 [DOI] [PubMed] [Google Scholar]
- Quade A, Thiel A, Kurth I, Holtgrewe M, Elbracht M, Beule D, … Hausler M (2020). Paroxysmal tonic upgaze: A heterogeneous clinical condition responsive to carbonic anhydrase inhibition. Eur J Paediatr Neurol, 25, 181–186. doi: 10.1016/j.ejpn.2019.11.002 [DOI] [PubMed] [Google Scholar]
- Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, … Bale S (2016). Clinical application of whole-exome sequencing across clinical indications. Genet Med, 18(7), 696–704. doi: 10.1038/gim.2015.148 [DOI] [PubMed] [Google Scholar]
- Schwankhaus JD, Parisi JE, Gulledge WR, Chin L, & Currier RD (1995). Hereditary adult-onset Alexander’s disease with palatal myoclonus, spastic paraparesis, and cerebellar ataxia. Neurology, 45(12), 2266–2271. doi: 10.1212/wnl.45.12.2266 [DOI] [PubMed] [Google Scholar]
- Serrano-Saiz E, Leyva-Diaz E, De La Cruz E, & Hobert O (2018). BRN3-type POU Homeobox Genes Maintain the Identity of Mature Postmitotic Neurons in Nematodes and Mice. Curr Biol, 28(17), 2813–2823 e2812. doi: 10.1016/j.cub.2018.06.045 [DOI] [PubMed] [Google Scholar]
- Shaikh AG, Hong S, Liao K, Tian J, Solomon D, Zee DS, … Optican LM (2010). Oculopalatal tremor explained by a model of inferior olivary hypertrophy and cerebellar plasticity. Brain, 133(Pt 3), 923–940. doi: 10.1093/brain/awp323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang M, Gan L, Zhou L, Klein WH, & Nathans J (1996). Targeted deletion of the mouse POU domain gene Brn-3a causes selective loss of neurons in the brainstem and trigeminal ganglion, uncoordinated limb movement, and impaired suckling. Proc Natl Acad Sci U S A, 93(21), 11950–11955. doi: 10.1073/pnas.93.21.11950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zee DS, Yamazaki A, Butler PH, & Gucer G (1981). Effects of ablation of flocculus and paraflocculus of eye movements in primate. J Neurophysiol, 46(4), 878–899. doi: 10.1152/jn.1981.46.4.878 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. Model of Homo sapiens POU4F1 from Swiss-Model repository. This homology model was created using human POU6F1 as the template. A cartoon of POU4F1 is shown with the amino acid Gln306 highlighted.
Supplemental Figure S2. Proband 1. Serial T1-weighted mid-sagittal MR images at ages A. 8 months, B. 1 year 9 months and C. 3 years 7 months.
At each age, the yellow outlines identify the cross-sectional areas of the anterior lobe of the vermis vs. the posterior lobe plus nodulus. The total cross-sectional area of the vermis increases by 23.4% from age 8 months to age 43 months. See Supp. Table S1.
Supplemental Figure S3. Proband 4. Serial T1-weighted mid-sagittal MR images at ages A. 11 months, B. 11 years, C. 13 years, D. 16 years, and E. 22 years.
At each age, the yellow outlines identify the cross-sectional areas of the anterior lobe of the vermis vs. the posterior lobe plus nodulus. The total cross-sectional area of the vermis decreases by 30.2% from age 11 months to age 22 years 11 months. See Supp. Table S2.
Supplemental Figure S4. Expression of POU4F1 in vitro. Wild-type POU4F1 and Gln306Arg POU4F1 were over-expressed in HEK293 cells and compared to a control HEK293 sample. POU4F1 p.Gln306Arg is stable.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The whole exome sequencing data is not publicly available due to privacy or ethical restrictions. The POU4F1 variant information has been submitted to the Leiden Open Variation Database (LOVD) (www.lovd.nl).