

Abstract
Monoterpene indole alkaloids are a large class of natural products derived from a single biosynthetic precursor, strictosidine. We describe a synthetic approach to strictosidine that relies on a key facially selective Diels–Alder reaction between a glucosyl-modified alkene and an enal to set the C15–C20–C21 stereotriad. DFT calculations were used to examine the origin of stereoselectivity in this key step, wherein two of 16 possible isomers are predominantly formed. These calculations suggest the presence of a glucosyl unit, also inherent in the strictosidine structure, guides diastereoselectivity, with the reactive conformation of the vinyl glycoside dienophile being controlled by an exo-anomeric effect. (−)-Strictosidine was subsequently accessed using late-stage synthetic manipulations and an enzymatic Pictet–Spengler reaction. Several new natural product analogs were also accessed, including precursors to two unusual aryne natural product derivatives termed “strictosidyne” and “strictosamidyne”. These studies provide a strategy for accessing glycosylic natural products and a new platform to access monoterpene indole alkaloids and their derivatives.
Graphical Abstract
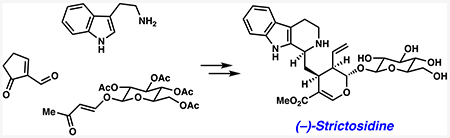
INTRODUCTION
Monoterpene indole alkaloids (MIAs) are a large class of natural products, many of which possess valuable pharmacological properties. To date, more than 3000 MIAs have been identified with diverse structures and bioactivities, which are exemplified by three of the most well-known members: quinine (1), strychnine (2), and vinblastine (3) (Figure 1).1–3 Quinine (1) belongs to the family of Cinchona alkaloids and is an antimalarial drug;4–6 strychnine (2), one of the most complex Strychnos alkaloids, is a potent toxin;7–9 and vinblastine (3), a Vinca alkaloid, is a frontline anticancer therapeutic and one of the most expensive small-molecule, off-patent drugs on the pharmaceutical market.10–13
Figure 1.
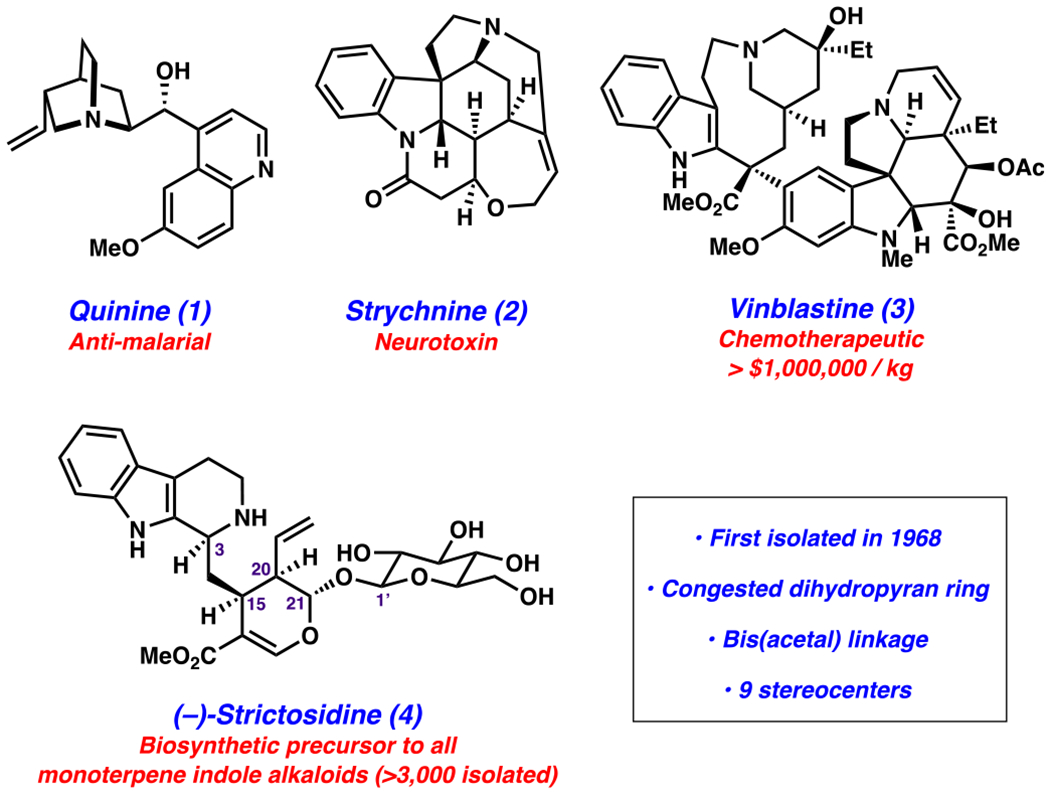
Strictosidine (4) and select natural products biosynthetically derived from 4.
An active area of research is the development of new strategies to access complex MIAs, such as vinblastine (3), through a combination of isolation from natural sources, biosynthesis, and total synthesis.1,14–31 We identified the natural product (−)-strictosidine (4) as an attractive entryway to access MIAs and derivatives.1 (−)-Strictosidine (4) is the last common biosynthetic precursor to all MIAs. It was first isolated in 1968 and contains nine stereocenters, a highly congested dihydropyran ring, a glucosyl moiety, and a bis(acetal) linkage.32 Despite its importance in MIA biosynthesis and being known for over 50 years, (−)-strictosidine (4) has remained challenging to access. Isolation of 4 from natural sources is unreliable, and its complete biosynthesis has proven difficult to engineer.32 Seminal efforts in this field include O’Connor and co-workers’ breakthrough in the biocatalytic production of strictosidine in yeast, albeit with a modest titer,1,33 and our groups’ finding of prevalent shunt pathways in the bioengineering of the early steps in S. cerevisiae.34,35 Furthermore, 4 has been largely overlooked by the synthetic community until very recently. The first total synthesis of strictosidine (4) was published during the course of our studies by Ishikawa and co-workers.36,37
Our laboratories sought to achieve the synthesis of (−)-strictosidine (4) using a blend of synthetic chemistry and biocatalysis and then use our approach as a platform for the preparation of new, unnatural derivatives thereof. Herein, we report: (a) a facially selective Diels–Alder reaction to access the dihydropyran moiety of 4, including the C15–C20–C21 stereotriad, (b) a computational analysis of this key step, (c) access to (−)-strictosidine (4) and an unnatural C15 epimer via enzymatic and nonenzymatic late-stage Pictet–Spengler reactions, and (d) the preparation and interception of “strictosidyne” and “strictosamidyne,” which are aryne derivatives of natural products.
RESULTS AND DISCUSSION
Retrosynthetic Analysis.
Our retrosynthetic analysis of (−)-strictosidine (4) is shown in Scheme 1. We envisioned (−)-strictosidine (4) would be obtained from its biosynthetic precursors (−)-secologanin (6) and tryptamine (5) via an enzymatic Pictet—Spengler reaction.33 This known step would build the tetrahydro-β-carboline ring, establish the stereochemistry at the C3 stereocenter, and provide a platform for the synthesis of unnatural strictosidine analogs. It should be emphasized that secologanin derivatives have been popular synthetic targets, yet only one synthesis of this compound exists, as reported by Ishikawa in 2019.37–45 (−)-Secologanin (6) could be obtained from vinylogous ester 7 through a series of manipulations, including introduction of the terminal olefin and oxidative cleavage of the five-membered ring. In a key step, the dihydropyran of vinylogous ester 7 could be accessed by an inverse electron-demand hetero-Diels–Alder reaction between enal 8 and enol ether 9. Enabled by the presence of the acetylated glucose moiety, this transformation would set three key stereocenters (C15, C20, C21). Whereas enal 8 can be obtained from cyclopentenone using known chemistry,46 we envisioned enol ether 9 to be accessible from glucose.47,48
Scheme 1.
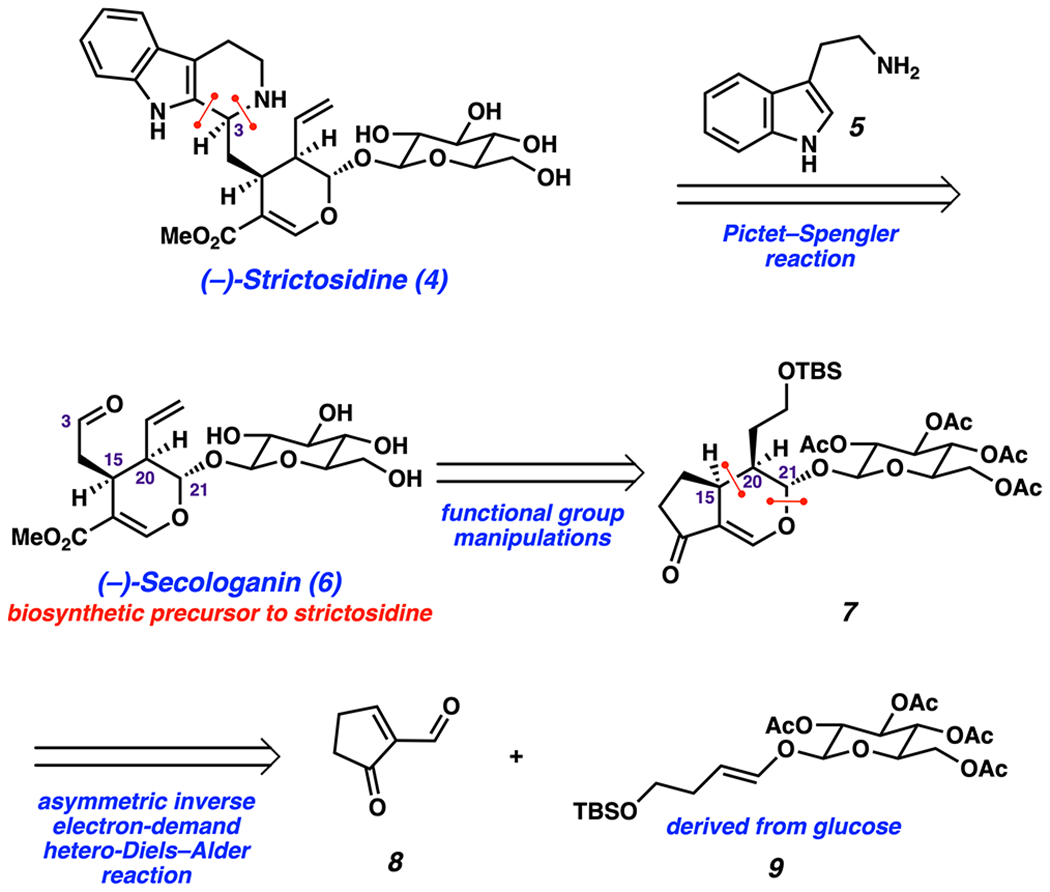
Retrosynthetic Analysis of (−)-Strictosidine (4)
Substrate Synthesis and Experimental and Computational Studies of Facially Selective Diels–Alder Reaction.
To initiate our synthetic effort, we prepared enol ether 9 using the sequence shown in Scheme 2. Known vinylogous ester 1048 underwent silylation/Rubottom oxidation to give an intermediate α-siloxy ketone.49 Subsequent ketone reduction and acetylation50,51 provided allylic acetate 11 as a 1:1 diastereomeric mixture in 56% yield over the two steps. Next, substantial effort was put forth to reductively remove the acetoxy group, which proved quite challenging. Reductions of allylic acetates bearing oxygen on the vinylic carbon are precedented on cyclic systems,52–59 but the corresponding reduction on linear substrates is rare and gives poor E/Z selectivity.60 Ultimately, we optimized nickel-catalyzed allylic reduction conditions reported by Yin, which afforded enol ether 9.60 Of note, the position and E geometry of the olefin was maintained.61,62
Scheme 2.
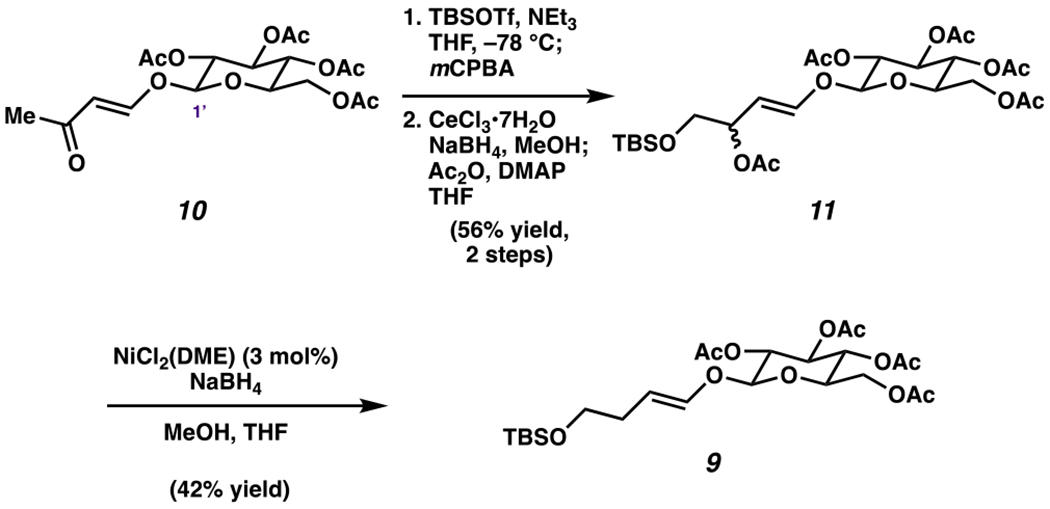
Synthesis of Enol Ether 9
With enol ether 9 in hand, we sought to assess its viability as a dienophile in the key inverse electron-demand hetero-Diels–Alder reaction with known enal 846 (Scheme 3).63,64 Of note, a successful Diels–Alder cycloaddition would lead to the introduction of three new stereocenters, where we hoped selectivity would be guided by the sugar moiety in 9. Moreover, in considering the formation of these stereocenters and regioselectivity possibilities, 16 isomers of the Diels–Alder cycloadduct could arise. After examining a variety of reaction conditions (i.e., solvents, Lewis acids, and temperatures),65 we identified optimal reaction conditions, which involved heating 8 and 9 in hexafluoroisopropanol (HFIP) at 50 °C for 16 h. This gave rise to cycloadducts 7a (desired) and its C15 epimer, 7b, as the major productsin a 1:1 ratio (55% combined yield).66 Sugars have rarely been employed to dictate stereochemistry in intermolecular inverse electron-demand hetero-Diels–Alder reactions, where the sugar resides on the dienophilic component.67–80 Furthermore, in the present example, the sugar is not used as a chiral auxiliary, but it is a component of both (−)-secologanin (6) and (−)-strictosidine (4). Thus, our approach involving early introduction of the sugar to guide stereochemical outcomes represents a useful strategy for accessing single enantiomers of glycosylated natural products.
Scheme 3.

Facially Selective Hetero-Diels–Alder Reaction Affords Cycloadducts 7a (Desired) and 7b out of 16 Possible Isomers
To explore the factors that control selectivity in the Diels–Alder reaction, we undertook density functional calculations (DFT) with the M06–2X functional. This method is known to give reliable energetics of stereoisomeric transition states of Diels–Alder reactions.81–83 Transition states were calculated for stepwise and concerted pathways, and the latter were found to be more favorable. As such, the E geometry in dienophile 9 leads to the trans relationship between C20 and C21 in the products 7a and 7b (see Scheme 3). Four possible stereoisomeric transition states, corresponding to endo/exo pathways and different facial approach, were investigated, with bond formation occurring between C15 and C20 and O17 and C21 of the reactants. These are shown in Figure 2.84–88 TS1(exo) and TS1(endo) were energetically most favorable and correlate to the two major products isolated experimentally, 7a and 7b, respectively. TS2(exo) and TS2(endo) were found to have higher activation barriers, and the corresponding products were not isolated experimentally.
Figure 2.
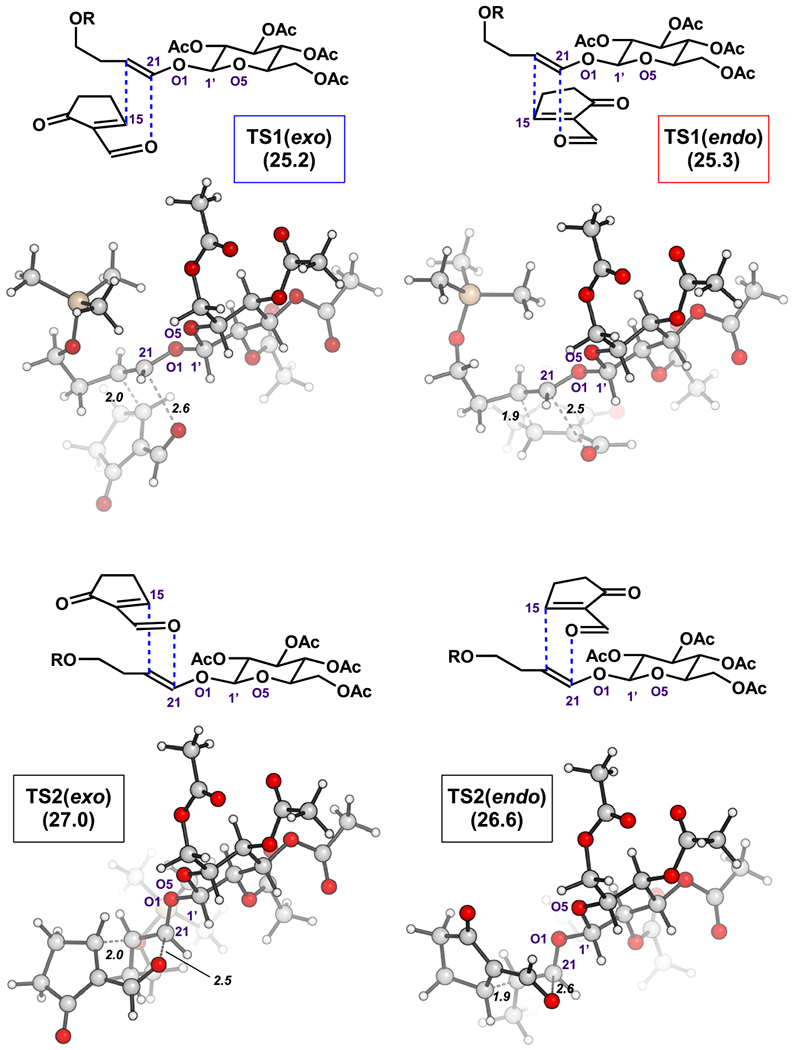
Four stereoisomeric transition states of the hetero-Diels–Alder reaction, with activation energies shown in kcal/mol. TS1(exo) and TS2(endo) correspond to observed products 7a and 7b, respectively. R = TBS in experimental work. R = TMS in calculated structures.
Two key factors that were investigated are the conformation of the glucosyl moiety and the adjacent reactive double bond (Figure 3a). Although the conformation of the glucosyl unit in dienophile 9 was found to be similar to that in all stereoisomeric transition states (i.e., TS1 and TS2), the orientation of the adjacent reactive olefin is more variable and is believed to dictate the stereochemical outcome of the reaction.
Figure 3.
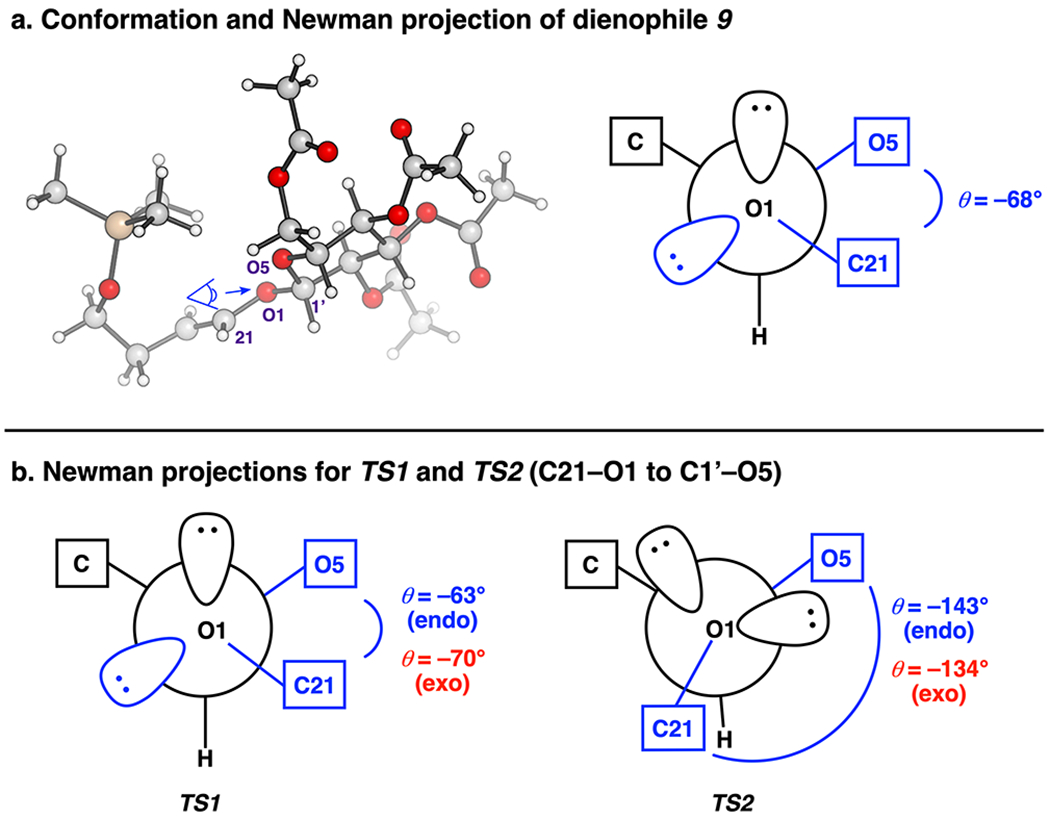
(a) Conformation and Newman projection of dienophile 9. (b) Newman projections for TS1 and TS2.
On dienophile 9, the C21 alkene adopts an exo-anomeric conformation (Figure 3).89 The dihedral angle between C21–O1 and C1’—O5 is −68°. Here, one lone pair on exocyclic oxygen O1 overlaps with the C1’—O5 σ* antibonding orbital and stabilizes itself by negative hyperconjugation as shown in the Newman projection in Figure 3a. The glucosyl enol ether is s-trans in order to avoid steric repulsion of the glucosyl group that would occur in the s-cis conformation that is normally favored for enol ethers.90,91 Each acetate is syn with the C=O aligned with the axial CH of the ring, similar to the XRD structure of an acetylated glucose.67
In each TS1, approach of the heterodiene occurs anti to the face of pyranyl oxygen O5 (re face) (Figure 2). The C21–O1 and C1’–O5 dihedral angles are −63° and −70°, respectively, for TS1(endo) and TS1(exo), indicating exo anomeric preferences in the transition states, similar to the orientation present in dienophile 9 (Figure 3b).92 This stabilizing effect imparted by the glucosyl ring leads to the face anti to pyranyl oxygen O5 (re face) being more accessible to the diene. In TS2(endo) and TS2(exo), involving si facial approach, some rotation around the C1’–O1 bond from the stable exo-anomeric conformation is required (C21–O1 and C1’–O5 dihedral angles are −143° and −134°, respectively). As a result, TS2(endo) and TS2(exo) are 1.3 and 1.8 kcal/mol higher in free energies than the corresponding TS1(endo) and TS1(exo) facial approaches. The computationally predicted activation energies for TS1(endo) and TS1(exo) correlate to the experimentally observed ratio of products 7a and 7b.
This hetero-Diels–Alder reaction is inverse electron-demand, since the LUMO of the heterodiene and the HOMO of the dienophile have a lower energy gap (9.6 eV) than the opposite HOMO–LUMO pair (14.5 eV) as shown in Figure 4a. There is a strong preference for one regioisomer involving the union of the nucleophilic carbon (C20) of the enol ether with the electrophilic carbon (C15) of the α,β-unsaturated aldehyde heterodiene due to a larger HOMO coefficient at C20 than C21. The frontier orbital interactions involving the π orbitals of the enal 8 and enol ether 9 are shown in Figure 4a. Endo/exo selectivity is not observed experimentally. The π lone pair of O1 mixes slightly with the alkene HOMO, but the coefficient is small, and the stabilizing secondary orbital interaction in the endo transition state is small. By contrast in a normal Diels–Alder reaction, such as that of butadiene plus acrolein, the large coefficient on the carbonyl carbon in the LUMO gives strong secondary orbital stabilization of the endo transition state (Figure 4b).
Figure 4.

(a) Frontier orbital interactions in the inverse electron-demand Diels–Alder reaction of 8 with 9. Orbital energies were calculated with HF/6-31G(d,p)/SMD(toluene).93,94 HOMO–LUMO energies are shown with the inverse electron-demand pathway in blue (HOMO–LUMO gap = 9.6 eV) and the normal electron-demand pathway in red (HOMO–LUMO gap = 14.5 eV). (b) Schematic representation of the strong endo-stabilizing secondary orbital interactions in a normal electron-demand Diels–Alder reaction (compared to weak interactions for the inverse electron-demand case studied here).
Elaboration to (−)-Secologanin and (−)-Strictosidine.
As shown in Scheme 4, Diels–Alder adduct 7a was elaborated to (−)-secologanin (6). Deprotection of 7a afforded the corresponding free alcohol, which underwent elimination under standard Grieco-olefination conditions.95 This sequence gave olefin 12 in 93% yield over two steps. Next, 12 was converted to the corresponding TBS enol ether, which set the stage for a Rubottom oxidation. The corresponding α-hydroxy ketone 13 was obtained in 53% yield as a single diastereomer. This intermediate was subjected to lead tetraacetate in methanol36,96 to effect oxidative cleavage97 and introduce the necessary aldehyde and methyl ester groups. Lastly, global acetyl removal gave (−)-secologanin (6) in 65% yield over 2 steps.40 Overall, (−)-secologanin (6) was accessed in nine steps from known materials.
Scheme 4.
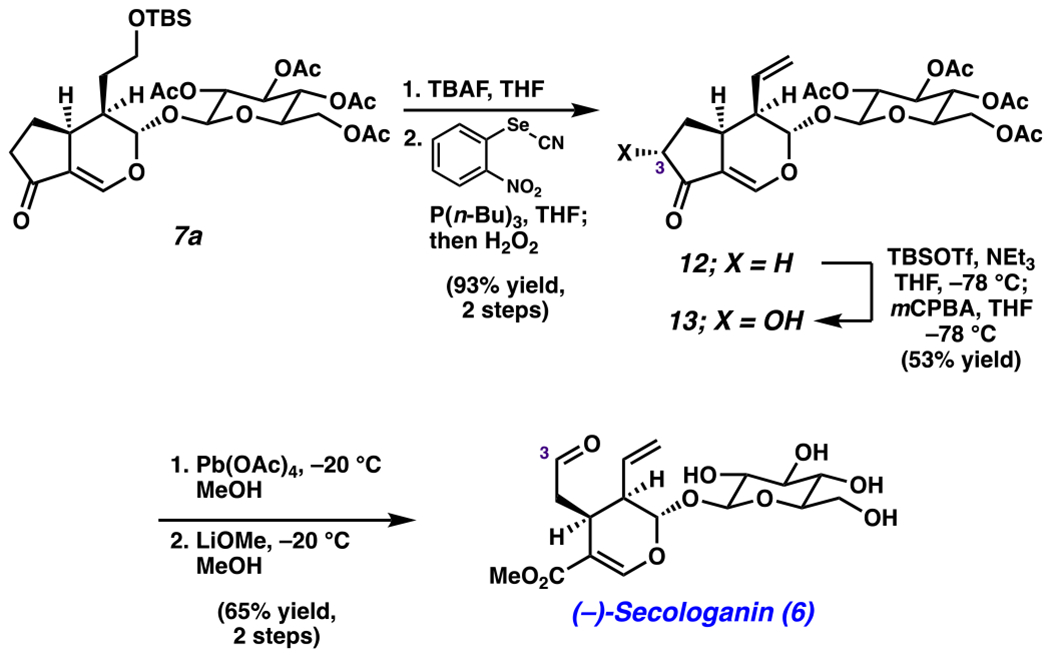
Synthesis of (−)-Secologanin (6)
To access (−)-strictosidine (4), we turned to the late-stage enzymatic Pictet–Spengler reaction between (−)-secologanin (6) and tryptamine (5) (Scheme 5). The natural biocatalyst for this transformation, strictosidine synthase, has previously been used successfully in the laboratory setting to prepare 4.33,98–103 As a practical advance, we sought to use crude cell lysate from an Escherichia coli BL21 overexpressing the strictosidine synthase strain in place of purified enzyme. The lyophilized crude lysate was found to be a stable white powder that could be easily weighed on the benchtop.104 To test the key biocatalytic step, (−)-secologanin (6) and tryptamine (5) were combined in an aqueous phosphate buffer with the crude lysate containing strictosidine synthase. This procedure delivered the natural product, (−)-strictosidine (4), in 82% yield, as a single C3 epimer, for which the spectral data are consistent with the published data.105 Overall, (−)-strictosidine (4) was accessed in 10 steps from known materials, utilizing a blend of chemical synthesis and enzymatic catalysis.
Scheme 5.
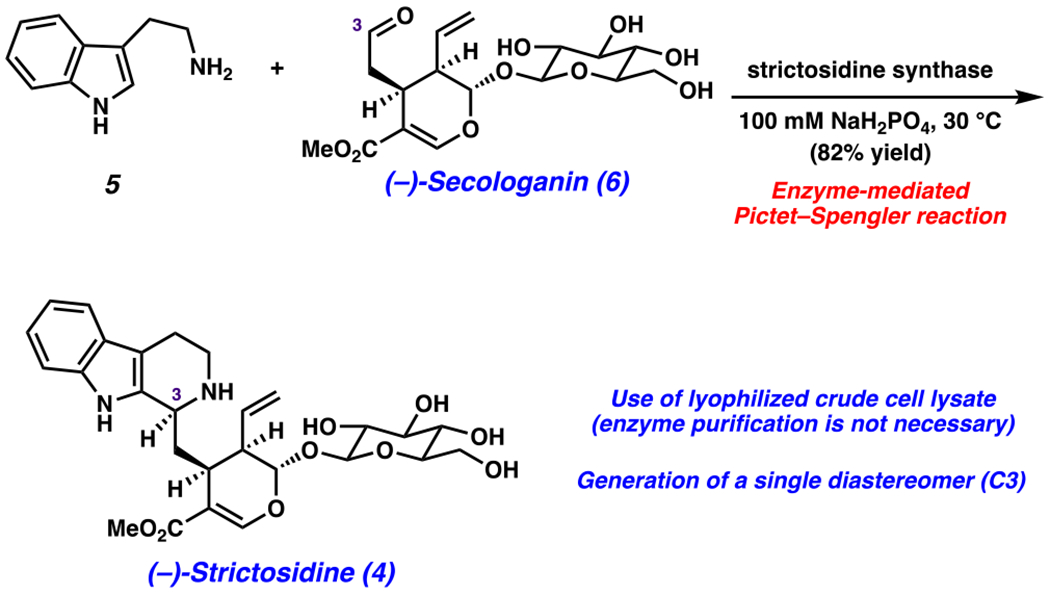
Enzymatic Pictet–Spengler Reaction Provides (−)-Strictosidine (4)
Synthesis of epi-Strictosidine, “Strictosidyne”, and “Strictosamidyne”.
Our next objective was to prepare new, unnatural analogs of strictosidine (4).106–109 This was pursued via two complementary strategies, the first of which is highlighted in Scheme 6 and involves the use of one of our synthetic intermediates that would not be readily accessible by other means. Specifically, 7b, the C15 epimer of the desired product of the Diels–Alder reaction was elaborated to an unnatural secologanin derivative 14 by applying a similar synthetic sequence as that from 7a to 6 (Scheme 3). The enzymatic Pictet–Spengler reaction of 14 with strictosidine synthase was attempted, but unfortunately, it led to the return of starting material, thus highlighting the substrate specificity of the enzyme.110 We were delighted to find that treatment of 14 with TFA and tryptamine (5) generated the desired tetrahydro-beta-carboline ring system (1:1 diastereomeric ratio (dr) with respect to C3).111–113 Subsequent removal of the acetates afforded epi-strictosidine isomers 15. It is worth noting that isomers 15 would not be readily accessible from epimerization of strictosidine (4) or by manipulating the biosynthetic pathway.114
Scheme 6.
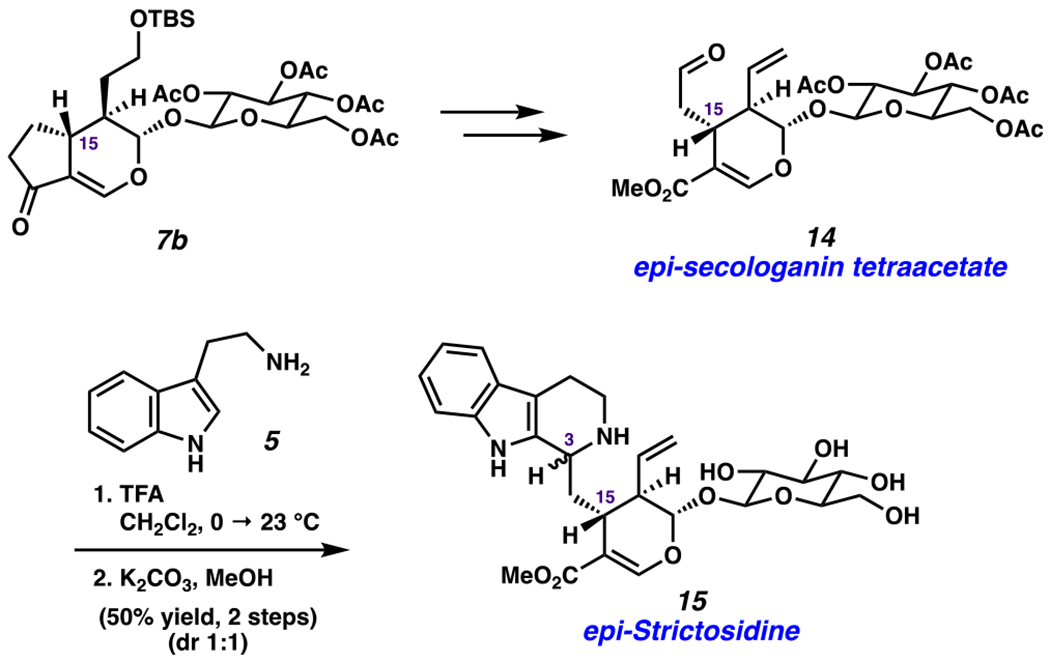
Synthesis of Unnatural Derivative epi-Strictosidine 15
The second strategy we pursued for analog synthesis involved varying the tryptamine fragment using a new and unconventional building block (Figure 5). Specifically, we questioned if tryptamine derivative 17 could be accessible. In turn, 17 could serve as a masked synthetic equivalent of “tryptaminyne” 18, which itself could find use in aryne trapping experiments or, for the purposes of our current study, be used in Pictet–Spengler reactions to make unique strictosidine derivatives. Of note, tryptamine is a prevalent precursor in both biosynthesis and chemical synthesis, 1,115,116 so the previously unknown “tryptaminyne” precursor could prove generally useful. We were delighted to find that commercially available indolyne precursor 16 could be elaborated to silyltriflate 17 in four steps.117
Figure 5.
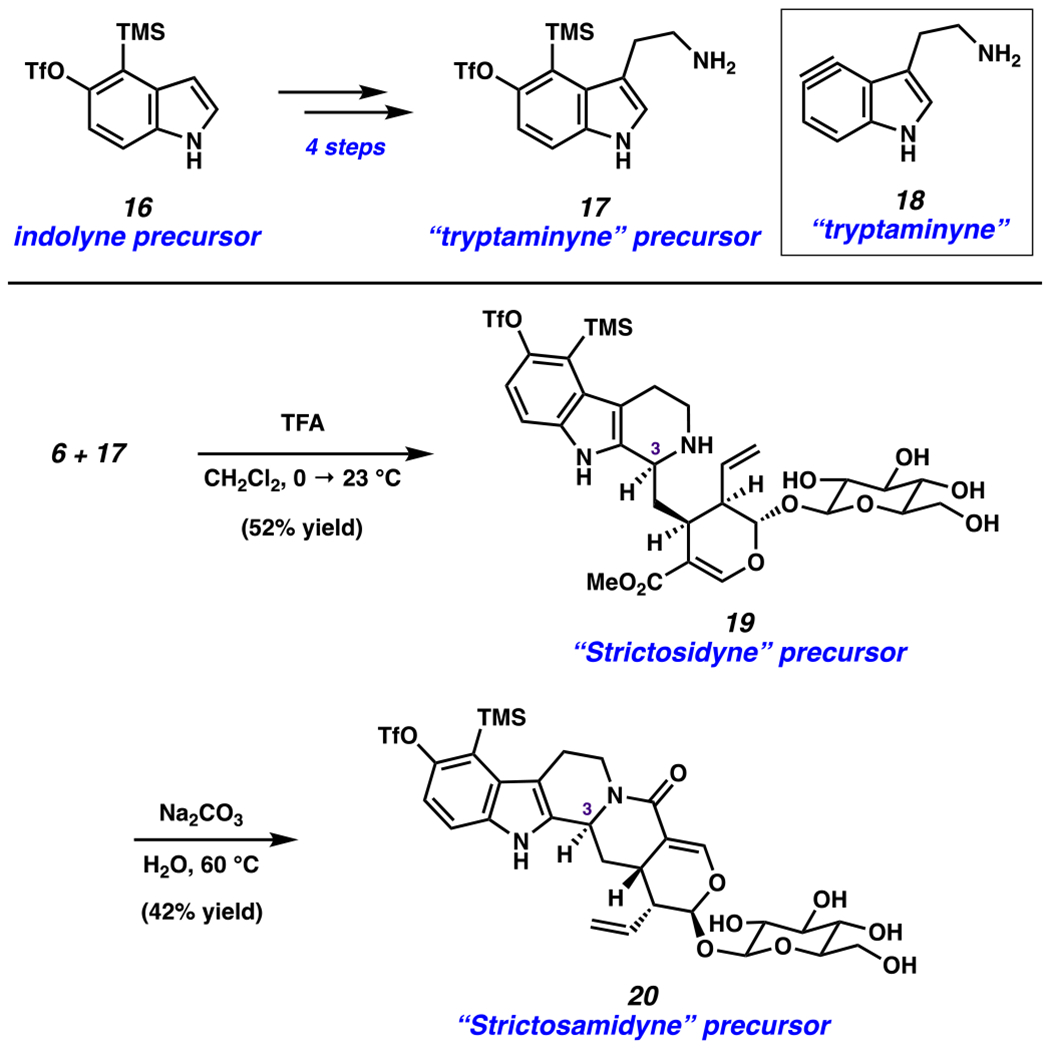
Synthesis of tryptaminyne precursor 17, “strictosidyne” precursor 19, and “strictosamidyne” precursor 20.
With silyl triflate 17 in hand, we attempted the Pictet–Spengler reaction using (−)-secologanin (6). Attempts to promote the desired reaction with strictosidine synthase were unsuccessful and only led to unreacted starting material. However, we found that the use of TFA led to the desired fragment coupling and annulation. “Strictosidyne” precursor 19 was obtained in 52% yield. The C3 epimer was also observed (16% yield, not depicted).118 We also took advantage of the opportunity to make new derivatives of strictosamide, a related natural product.119–121 As such, a single diastereomer of 19 (as depicted) was treated with sodium carbonate to afford 20, which we envisioned serving as a precursor to the aryne derivative of strictosamide we term “strictosamidyne”.
Lastly, we demonstrated that “strictosidyne” (22) and “strictosamidyne” (23) could be generated from precursors 19 and 20, respectively, by performing Diels–Alder trapping experiments (Scheme 7). Each silyltriflate was independently subjected to furan (21) and cesium fluoride in acetonitrile at 50 °C.122 To our delight, this gave cycloadducts 24 and 25 in 66% and 55% yield (both 1:1 dr), respectively. The chemoselectivity in both reactions is noteworthy, given that the highly reactive aryne moieties could be generated and trapped in the presence of nucleophilic groups, such as unprotected amines and the four free alcohols on the glucosyl unit. To our knowledge, 19 and 20 are the first silyl triflate derivatives of complex alkaloids. Likewise, 22 and 23 are the first aryne derivatives of such complex naturally occurring structures.123 We expect the ability to use and intercept aryne derivatives of complex natural products will prove useful in future efforts, especially those geared toward late-stage structural diversification.
Scheme 7.
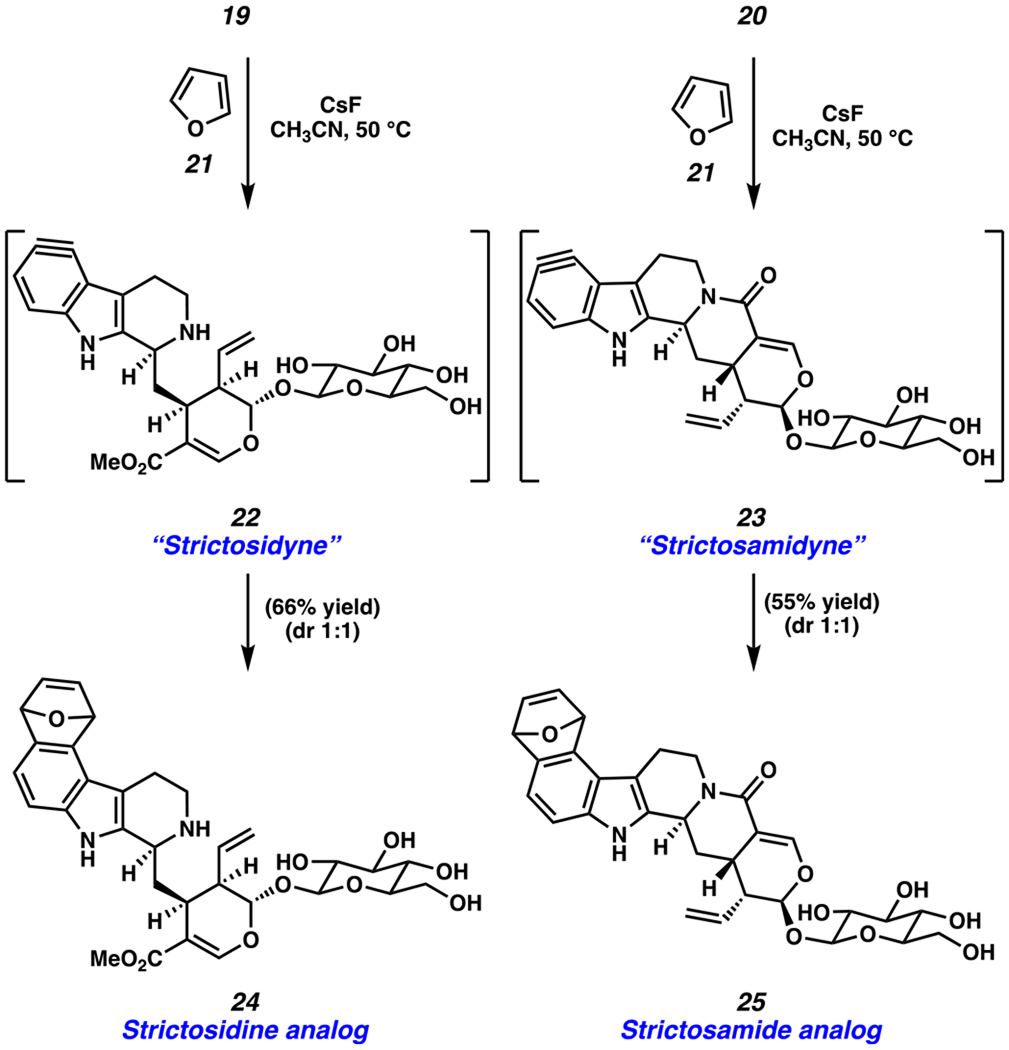
Trapping Experiments of Strictosidyne Precursor 19 and Strictosamidyne Precursor 20
CONCLUSIONS
In summary, we have completed the total synthesis of (−)-strictosidine and several unnatural analogs thereof. Our stereospecific approach features a facially selective Diels–Alder reaction to access the C15–C20–C21 stereotriad. As shown by DFT calculations, stereoselectivity in this key step is ultimately controlled by the glucosyl unit present in both the dienophile and (−)-strictosidine itself as a result of an exo-anomeric effect. This key step permits access to (−)-secolo- ganin and an unnatural derivative, which are subsequently employed in enzymatic or reagent-based Pictet–Spengler reactions, to give (−)-strictosidine and an unnatural epimer. Moreover, by accessing a “tryptaminyne” precursor, two unusual aryne natural product derivatives termed “strictosidyne” and “strictosamidyne” were generated and intercepted in Diels–Alder cycloadditions. These studies not only provide a means to access strictosidine and new derivatives thereof but also showcase the ability of a glucosyl unit to guide stereoselectivity through conformational effects, the synergy between synthetic chemistry, biocatalysis, and computations, and the use of “tryptaminyne” chemistry as a strategy to access derivatives of complex alkaloids.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful to the University of California, Los Angeles for financial support. We are grateful to the NIH-NIGMS (F31-GM121016 to L.A.M. and T32-GM067555 to J.M.B), the NIH-NCCIH (R01AT010001), the Tobacco-Related Disease Research Program of the University of California (T29DT0359 to S.M.A.), the Foote Family (S.M.A. and L.A.M.), the Trueblood Family (N.K.G.), NSF (CHE-1900178 to N.K.G and CHE-1764328 to K.N.H.), and CCRC (CRR-19-584627 to N.K.G). Calculations were performed on the Hoffman2 cluster and the UCLA Institute of Digital Research and Education (IDRE) at UCLA and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation (OCI-1053575). These studies were supported by shared instrumentation grants from the NSF (CHE-1048804) and the National Center for Research Resources (S10RR025631).
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c02004
The Supporting Information. is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c02004.
Detailed experimental procedures and compound characterization data (PDF)
The authors declare no competing financial interest.
Contributor Information
Sarah M. Anthony, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States.
Veronica Tona, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States.
Yike Zou, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States.
Lucas A. Morrill, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States
John M. Billingsley, Department of Chemistry and Biomolecular Engineering, University of California, Los Angeles, California 90095, United States.
Megan Lim, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States.
Yi Tang, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States.
K. N. Houk, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States.
Neil K. Garg, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States.
REFERENCES
- (1).O’Connor SE; Maresh JJ Chemistry and biology of monoterpene indole alkaloid biosynthesis. Nat. Prod. Rep. 2006, 23, 532—547. [DOI] [PubMed] [Google Scholar]
- (2).Pan Q; Mustafa NR; Tang K; Choi YH; Verpoorte R Monoterpenoid indole alkaloids biosynthesis and its regulation in Catharanthus roseus: a literature review from genes to metabolites. Phytochem. Rev. 2016, 15, 221—250. [Google Scholar]
- (3).De Luca V; Salim V; Thamm A; Masada AS; Yu F Making iridoids/secoiridoids and monoterpenoid indole alkaloids: progress on pathway elucidation. Curr. Opin. Plant Biol. 2014, 19, 35—42. [DOI] [PubMed] [Google Scholar]
- (4).Kaufman TS; Rúveda EA The quest for quinine: those who won the battles and those who won the war. Angew. Chem., Int. Ed. 2005, 44, 854–885. [DOI] [PubMed] [Google Scholar]
- (5).Nicolaou KC; Snyder SA Classics in total synthesis II: more targets, strategies, methods; Wiley-VCH: Weinheim, Germany, 2003; Ch. 15. pp 443—462. [Google Scholar]
- (6).Souza KAFD; Porto PA History and epidemiology of science in the classroom: The synthesis of quinine as a proposal. J. Chem. Educ. 2012, 89, 58—63. [Google Scholar]
- (7).Nicolau KC; Sorensen EJ Classics in total synthesis: targets, strategies, methods. VHC: Weinheim & New York, 1996; Ch. 2. pp 22—40. [Google Scholar]
- (8).Cannon JS; Overman LE Is there no end to the total syntheses of strychnine? Lessons learned in strategy and tactics in total synthesis. Angew. Chem., Int. Ed. 2012, 51, 4288—4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).He W; Wang P; Chen J; Xie W Recent progress in the total synthesis of Strychnos alkaloids. Org. Biomol. Chem. 2020, 18, 1046–1056. [DOI] [PubMed] [Google Scholar]
- (10).Ishikawa H; Colby DA; Seto S; Va P; Tam A; Kakei H; Rayl TJ; Hwang I; Boger DL Total synthesis of vinblastine, vincristine, related natural products, and key structural analogues. J. Am. Chem. Soc. 2009, 131, 4904—4916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Datta A; Srivastava PS Variation in vinblastine production by Catharanthus roseus during in vivo and in vitro differentiation. Phytochemistry 1997, 46, 135—137. [Google Scholar]
- (12).Miettinen K; Dong L; Navrot N; Schneider T; Burlat V; Pollier J; Woittiez L; Krol D. v. d; Lugan R; Ilc T;Verpoorte R; Oksman-Caldentry K-M; Martinoia E; Bouwmeester H; Goossens A; Memelink J; WerkReichhart D The seco-iridoid pathway from Catharanthus roseus. Nat. Commun. 2014, 5, 3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mehrotra S; Mishra S; Srivastava V Hairy root cultures for monoterpene indole alkaloid pathway: investigation and biotechnological production; Springer: Singapore, 2018; pp 95–121. [Google Scholar]
- (14).Kries H; O’Connor S Biocatalysts from alkaloid producing plants. Curr. Opin. Chem. Biol. 2016, 31, 22–30. [DOI] [PubMed] [Google Scholar]
- (15).Zhu H; Kerčmar P; Wu F; Rajendran C; Sun L; Wang M; Stöckigt J Using strictosidine synthase to prepare novel alkaloids. Curr. Med. Chem. 2015, 22, 1880–1888. [PubMed] [Google Scholar]
- (16).Leonard E; Runguphan W; O’Connor S; Prather KJ Opportunities in metabolic engineering to facilitate scalable alkaloid production. Nat. Chem. Biol. 2009, 5, 292–300. [DOI] [PubMed] [Google Scholar]
- (17).Caputi L; Franke J; Farrow SC; Chung K; Payne RME; Nguyen T-D; Dang T-TT; Carqueijeiro IST; Koudounas K; de Bernonville TD; Ameyaw B; Jones DM; Vieira IJC; Courdavault V; O’Connor SE Missing enzymes in the biosynthesis of the anticancer drug vinblastine in Madagascar periwinkle. Science 2018, 360, 1235–1239. [DOI] [PubMed] [Google Scholar]
- (18).Tanifuji R; Minami A; Oguri H; Oikawa H Total synthesis of alkaloids using both chemical and biochemical methods. Nat. Prod. Rep. 2020, 37, 1098–1121. [DOI] [PubMed] [Google Scholar]
- (19).Liu X-Y; Qin Y Indole alkaloid synthesis facilitated by photoredox catalytic radical cascade reactions. Acc. Chem. Res. 2019, 52, 1877–1891. [DOI] [PubMed] [Google Scholar]
- (20).Saya JM; Ruijter E; Orru RVA Total synthesis of Aspidosperma and Strychnos alkaloids through indole dearomatization. Chem. - Eur. J. 2019, 25, 8916–8935. [DOI] [PubMed] [Google Scholar]
- (21).Tokuyama H The total synthesis of biosynthetically related monoterpene indole alkaloids. Yuki Gosei Kagaku Kyokaishi 2015, 15, 1120–1129. [Google Scholar]
- (22).Dou Y; Kouklovsky C; Vincent G Bioinspired divergent oxidative cyclization from strictosidine and vincoside derivatives: second-generation total synthesis of (−)-cymoside and access to an original hexacyclic-fused furo[3,2-b]indoline. Chem. - Eur. J. 2020, 26, 17190–17194. [DOI] [PubMed] [Google Scholar]
- (23).Zhang B; Wang X; Li C Enantioselective total synthesis of (+)-corymine and (−)-deformylcorymine. J. Am. Chem. Soc. 2020, 142, 3269–3274. [DOI] [PubMed] [Google Scholar]
- (24).Dou Y; Kouklovsky C; Gandon V; Vincent G Enantioselective total synthesis of cymoside through a bioinspired oxidative cyclization of a strictosidine derivative. Angew. Chem., Int. Ed. 2020, 59, 1527–1531. [DOI] [PubMed] [Google Scholar]
- (25).Jarret M; Turpin V; Tap A; Gallard J-F; Kouklovsky C; Poupon E; Vincent G; Evanno L Bioinspired oxidative cyclization of the geissoschizine skeleton for enantioselective total synthesis of mavacuran alkaloids. Angew. Chem. Int. Ed. 2019, 58, 9861–9865. [DOI] [PubMed] [Google Scholar]
- (26).Liu Y; Wang H Unified enantioselective total syntheses of (−)-scholarisine G, (+)-melodinine E, (−)-leuconoxine and (−)-mersicarpine. Chem. Commun. 2019, 55, 3544–3547. [DOI] [PubMed] [Google Scholar]
- (27).Mason JD; Weinreb SM Total syntheses of the monoterpenoid indole alkaloids (±)-alstoscholarisine B and C. Angew. Chem., Int. Ed. 2017, 56, 16674–16676. [DOI] [PubMed] [Google Scholar]
- (28).Liang X; Jiang S-Z; Wei K; Yang Y-R Enantioselective total synthesis of (−)-alstoscholarisine A. J. Am. Chem. Soc. 2016, 138, 2560–2562. [DOI] [PubMed] [Google Scholar]
- (29).Piemontesi C; Wang QW; Zhu J Enantioselective total synthesis of (−)-terengganensine A. Angew. Chem. Int. Ed. 2016, 55, 6556–6560. [DOI] [PubMed] [Google Scholar]
- (30).Xu Z; Bao X; Wang Q; Zhu J An enantioselective total synthesis of (−)-isoschizogamine. Angew. Chem. Int. Ed. 2015, 54, 14937–14940. [DOI] [PubMed] [Google Scholar]
- (31).Wagnières O; Xu Z; Wang Q; Zhu J Unified strategy to monoterpene indole alkaloids: total syntheses of (±)-goniomitine, (±)-1,2-dehydroaspidospermidine, (±)-aspidospermidine, (±)-vinca-difformine, and (±)-kopsihainanine A. J. Am. Chem. Soc. 2014, 136, 15102–15108. [DOI] [PubMed] [Google Scholar]
- (32).Smith GN Strictosidine: a key intermediate in the biogenesis of indole alkaloids. Chem. Commun. (London) 1968, 912–914. [Google Scholar]
- (33).Brown S; Clastre M; Courdavault V; O’Connor S De novo production of the plant-derived alkaloid strictosidine in yeast. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 3205–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Billingsley JM; DeNicola AB; Barber JS; Tang M-C; Horecka J; Chu A; Garg NK; Tang Y Engineering the biocatalytic selectivity of iridoid production in Saccharomyces cerevisiae. Metab. Eng. 2017, 44, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Yee DA; DeNicola AB; Billingsley JM; Creso JG; Subrahmanyam V; Tang Y Engineered mitochondrial production of monoterpenes in Saccharomyces cerevisiae. Metab. Eng. 2019, 55, 76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ishikawa’s route to strictosidine features an organocatalytic Michael reaction, Fukuyama reduction, cyclization cascade; see: Ishikawa H; Sakamoto J; Umeda Y; Rakumitsu K; Sumimoto M Total syntheses of (−)-strictosidine and related indole alkaloid glycosides. Angew. Chem. Int. Ed. 2020, 59, 13414–13422. [DOI] [PubMed] [Google Scholar]
- (37).For Ishikawa’s route to secologanin; see: Rakumitsu K; Sakamoto J; Ishikawa H Total syntheses of (−)-secologanin, (−)-5-carboxystrictosidine, and (−)-rubenine. Chem. - Eur. J. 2019, 25, 8996–9000. [DOI] [PubMed] [Google Scholar]
- (38).Bernhardt P; O’Connor SE Synthesis and biochemical evaluation of des-vinyl secologanin aglycones with alternate stereochemistry. Tetrahedron Lett. 2009, 50, 7118–7120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bernhardt P; Yerkes N; O’Connor SE Bypassing stereoselectivity in the early steps of alkaloid biosynthesis. Org. Biomol. Chem. 2009, 7, 4166–4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Tietze L-F Secologanin, a biogenetic key compound–synthesis and biogenesis of the iridoid and secoiridoid glycosides. Angew. Chem. Int. Ed. Engl. 1983, 22, 828–841. [Google Scholar]
- (41).Nakane M; Hutchinson CR Stereoselective synthesis of (±)-1-O-methylloganin, 10-hydroxyloganin, secologanin, and sweroside aglucons from a common 1-hydroxy-4a,5,8,8-tetrahydroiso-chromene synthon. J. Org. Chem. 1980, 45, 4233–4236. [Google Scholar]
- (42).Cheng PTW; McLean S A synthetic approach to secologanin: synthesis of a protected form of the secoxyloganin aglucone. Tetrahedron Lett. 1988, 29, 3511–3512. [Google Scholar]
- (43).Isoe S; Katsumura S; Okada T; Yamamoto K; Takemoto T; Inaba H; Han Q; Nakatani K Novel synthesis of (−)-secologanin aglucon-O-silyl ether from (+)-genipin via oxidative fragmentation of γ-hydroxyalkylstannane. Tetrahedron Lett. 1987, 28, 5865–5868. [Google Scholar]
- (44).Chang N-C; Day H-M; Lu W-F Total synthesis of (±)-secologanoside O-methyl ether. J. Org. Chem. 1989, 54, 4083–4088. [Google Scholar]
- (45).Tai H-M; Huang M-H; Yang C-C Formal total synthesis of (±)-dimethyl secologanoside. J. Chin. Chem. Soc. 2003, 50, 441–444. [Google Scholar]
- (46).Adary EM; Chang C-W; D’Auria DT; Nguyen PM; Polewacz K; Reinicke JA; Seo H; Berger GO Improved synthesis of and nucleophilic addition to 2-formyl-2-cyclohexenone. Tetrahedron Lett. 2015, 56, 386–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Frank RL; Varland RH 1,3,5-Triacetylbenzene. Org. Synth. 1947, 27, 91. [Google Scholar]
- (48).Gupta R; Harland FA; Stoodley RJ An efficient enantiocontrolled synthesis of (+)-4-demethoxydaunomycinone. Tetrahedron 1984, 40, 4657–4667. [Google Scholar]
- (49).Craita C; Didier C; Vogel P Short synthesis of the C16-C28 polyketide fragment of apoptolidin A aglycone. Chem. Commun. 2007, 2411–2413. [DOI] [PubMed] [Google Scholar]
- (50).Jung J; Lehnemann BW Preparation of cis-2-cyclopentene-1,4-diol. US Patent US 2009–663763. December 9, 2009. [Google Scholar]
- (51).Nishiyama S; Ikeda Y; Yoshida S-I; Yamamura S Synthetic study on breynin A: synthesis of breynolide sulfone. Tetrahedron Lett. 1989, 30, 105–108. [Google Scholar]
- (52).Clark JS; Romiti F; Sieng B; Paterson LC; Stewart A; Chaudhury S; Thomas LH Synthesis of the A-D ring system of the gambieric acids. Org. Lett. 2015, 17, 4694–4697. [DOI] [PubMed] [Google Scholar]
- (53).Fukumoto S; Ujikawa O; Morimoto S; Asano Y; Mikami S; Tokunaga N; Kori M; Imaeda T; Fukuda K; Nakamura S; Iwanaga K Sulfonamide derivative and use thereof. International Patent WO 2012/137982 A2, October 11, 2012. [Google Scholar]
- (54).Hanessian S; Maianti JP; Matias RD; Feeney LA; Armstrong ES Hybrid aminoglycoside antibiotics via Tsuji palladium-catalyzed allylic deoxygenation. Org. Lett. 2011, 13, 6476–6479. [DOI] [PubMed] [Google Scholar]
- (55).Wyatt PG; Coomber BA; Evans DN; Jack TI; Fulton HE; Wonacott AJ; Colman P; Varghese J Sialidase inhibitors related to zanamivir. Further SAR studies of 4-amino-4H-pyran-2-carboxylic acid-6-propylamides. Bioorg. Med. Chem. Lett. 2001, 11, 669–673. [DOI] [PubMed] [Google Scholar]
- (56).Jung ME; Trilunovich ID Efficient Synthesis of 2′,3′-dideoxynucleosides and C-nucleosides from D-glucosamine. Tetrahedron Lett. 1992, 33, 2921–2924. [Google Scholar]
- (57).Kovács L; Herczegh P; Batta G; Farkas I Thiazole-C-nucleosides IV. An entry to pent-1’-enopyranosylthazole derivatives. Tetrahedron 1991, 47, 5549–5560. [Google Scholar]
- (58).Schreiner E; Zbiral E A convenient approach to 3-deoxy-D-glycero-D-galacto-nonulosonic acid (KDN), 5-Azido-5-deoxy-KDN and 5-deoxy-KDN, and their 4-methylumbelliferyl 2α-glycosides. Liebigs Ann. Chem. 1990, 1990, 581–586. [Google Scholar]
- (59).Greenspoon N; Keinan E Selective deoxygenation of unsaturated carbohydrates with Pd(0)/Ph2SiH2/ZnCl2. J. Org. Chem. 1988, 53, 3723–3731. [Google Scholar]
- (60).Yin B-L; Cai C-B; Lai J-Q; Zhang Z-R; Huang L; Xu L-W; Jiang H-F Sodium borohydride-nickel chloride-methanol catalytic system for regioselective reduction of electron-rich conjugated dienes and reductive cleavage of allyl esters involving π-allylnickel intermediates. Adv. Synth. Catal. 2011, 353, 3319–3324. [Google Scholar]
- (61).Several reductants were surveyed, including silanes and Lewis acid-promoted conditions. No other conditions gave the desired reduction. Inclusion of additional nickel catalyst gave the isomerized olefin and over-reduced products, which we inseparable from enol ether 9. [Google Scholar]
- (62).Other methods to construct the olefin were explored, such as a Wittig reaction, olefin metathesis, and olefin isomerization. Furthermore, constructing the C–O bond were also investigated, but no methodologies gave adequate yield of enol ether 9. [Google Scholar]
- (63).Tietze LF; Meier H; Nutt H Synthesis of (±)-secologanin alucone O-ethyl ether and derivatives by tandem Knoevenagel hetero Diels-Alder reaction. Liebig Ann. Chem. 1990, 1990, 253–260. [Google Scholar]
- (64).Tietze LF; Meier H; Nutt H Inter- and intramolecular hetero Diels-Alder reactions, XXV. The tandem Knoevenagel hetero Diels-Alder reaction with a formylacetic acid equivalent. Synthesis of dihydropyrancarboxylates. Chem. Ber. 1989, 122, 643–650. [Google Scholar]
- (65).See SI for the Diels–Alder reaction optimization details. [Google Scholar]
- (66).Although we cannot rule out the formation of other possible isomers in trace quantities, based on 1H NMR analysis of the crude reaction mixture, we estimate that any given byproduct is produced in less than 5% yield. [Google Scholar]
- (67).For previous examples of glucose-directed inverse-electron demand Diels–Alder reactions see: Choudhury A; Franck RW; Gupta RB Cycloaddition of isoquinolinium salts: homochiral tetralins via dienophiles bearing chiral auxiliaries. Tetrahedron Lett. 1989, 30, 4921–4924. [Google Scholar]
- (68).Normal demand Diels–Alder reactions with a glucosyl moiety are more common, see references70–79 and:Lubineau A; Queneau Y Aqueous cycloadditions using glyco-organic substrates. 1. Stereochemical course of the reaction. J. Org. Chem. 1987, 52, 1001–1007. [Google Scholar]
- (69).Larsen DS; Stoodley RJ Asymmetric Diels–Alder reactions. Part 3. Influence of butadiene structure upon the diastereofacial reactivity of (E)-1-(2’,3′,4’,6’-tetra-O-acetyl-β-D-glucopyranosyloxy)buta-1,3-dienes. J. Chem. Soc., Perkin Trans. 1 1989, 1841–1852. [Google Scholar]
- (70).Beagley B; Curtis ADM; Pritchard RG; Stoodley RJ Asymmetric Diels–Alder reactions. Part 6. Regio- and stereo-selective cycloadditions of 5-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyloxy)-1,4-naphthoquinone. J. Chem. Soc., Perkin Trans. 1 1992, 1981–1991. [Google Scholar]
- (71).Larsen DS; Lins RJ; Stoodley RJ; Trotter NS Studies related to carba-pyranoses: a strategy for the synthesis of β-1,3-glycosidically linked aminomonocarba-disaccharides. J. Chem. Soc. Perkin Trans. 1 2001, 2204–2212. [Google Scholar]
- (72).Alves MJ; Almeida IG; Fortes AG; Freitas AP Stereoselective cycloaddition of 1-glucosyl-1,3-butadienes with tert-butyl 2H-azirine-3-carboxylate, glyoxylates and imines. Tetrahedron Lett. 2003, 44, 6561–6565. [Google Scholar]
- (73).Cousins RPC; Curtis ADM; Ding WC; Stoodley RJ 1,5-asymmetric inductions in the reactions of 2-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyloxy)benzaldehyde with Danishefsky’s diene. Tetrahedron Lett. 1995, 36, 8689–8692. [Google Scholar]
- (74).Pritchard RG; Stoodley RJ; Yuen W-H Studies related to carba-pyranoses: synthesis of acetylated derivatives of 4-amino-2,4-dideoxy-3-O-(β-D-glucopyranosyl)-β-L-(and β-D-) altrocarba-pyranose from a D-glucose template. Org. Biomol. Chem. 2005, 3, 162–171. [DOI] [PubMed] [Google Scholar]
- (75).Luo S-Y; Jang Y-J; Liu J-Y; Chu C-S; Liao C-C; Hung S-C Carbohydrate-templated asymmetric Diels–Alder reactions of masked ortho-benzoquinones for the synthesis of chiral bicyclo[2.2.2]-oct-5-en-2-ones. Angew. Chem., Int. Ed. 2008, 47, 8082–8085. [DOI] [PubMed] [Google Scholar]
- (76).Helliwell M; Phillips IM; Pritchard RG; Stoodley RJ Asymmetric synthesis of (5S)-4-deoxy-5-C-(4-nitrophenyl)-L-threo-pentose and (5R)-5-C-(4-nitrophenyl)-L-arabinose. Tetrahedron Lett. 1999, 40, 8651–8655. [Google Scholar]
- (77).Weng C-H; Hsu D-S; Liao C-C Application of carbohydrate-templated asymmetric Diels–Alder reaction to the syntheses of ent-penicillones A and B. J. Org. Chem. 2016, 81, 11421–11426. [DOI] [PubMed] [Google Scholar]
- (78).Aucagne V; Avera MC; Barattucci A; Bonaccorsi P; Giannetto P; Rollin P; Tatibouet A Sulfenic acids in the carbohydrate field. Synthesis of transient glycosulfenic acids and their addition to unsaturated acceptors. J. Org. Chem. 2002, 67, 6925–6930. [DOI] [PubMed] [Google Scholar]
- (79).Totani K; Takao K.-i.; Tadano K.-i. Sugar as a tool for asymmetric synthesis: some effective approaches. Synlett 2004, 12, 2066–2080. [Google Scholar]
- (80).Kunz H; Ruck K Carbohydrates as chiral auxiliaries in stereoselective synthesis. New synthetic methods. Angew. Chem., Int. Ed. Engl. 1993, 32, 336–358. [Google Scholar]
- (81).Levandowski BJ; Hamlin TA; Helgeson RC; Bickelhaupt FM; Houk KN Origins of the endo and exo selectivities in cyclopropenone, iminocyclopropene, and triafulvene Diels-Alder cycloadditions. J. Org. Chem. 2018, 83, 3164–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Houk KN; Luskus LJ Influence of steric interactions on endo stereoselectivity. J. Am. Chem. Soc. 1971, 93, 4606–4607. [Google Scholar]
- (83).McCarrick MA; Wu YD; Houk KN Hetero-Diels–Alder reaction transition structures: reactivity, stereoselectivity, catalysis, solvent effects, and the exo-lone-pair effect. J. Org. Chem. 1993, 58, 3330–3343. [Google Scholar]
- (84).TMS replaces TBS group in our models to restrict conformational flexibility. Due to the conformational flexibility of the glycosyl enol ether dienophile, we performed extensive conformational searches using metadynamics approaches in Grimme’s program CREST. More than 1700 conformations were generated for the dienophile; in order to find what proved to be the most favored chair 4C1 conformation of the glycosyl group, manual adjustments of the glycosyl group to the lowest energy 4C1 conformation were also required. Low energy conformations were reoptimized with M06-2X. [Google Scholar]
- (85).Grimme S Exploration of chemical compound, conformer, and reaction space with meta-dynamics simulations based on tight-binding quantum chemical calculations. J. Chem. Theory Comput. 2019, 15, 2847–2862. [DOI] [PubMed] [Google Scholar]
- (86).Pracht P; Bohle F; Grimme S Automated exploration of the low-energy chemical space with fast quantum chemical methods. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. [DOI] [PubMed] [Google Scholar]
- (87).Biarnés X; Ardevol A; Planas A; Rovira C; Laio A; Parrinello M The conformational free energy landscape of β-D-glucopyranose. Implications for substrate preactivation in β-glucoside hydrolases. J. Am. Chem. Soc. 2007, 129, 10686–10693. [DOI] [PubMed] [Google Scholar]
- (88).Mayes HB; Broadbelt LJ; Beckham GT How sugars pucker: electronic structure calculations map the kinetic landscape of five biologically paramount monosaccharides and their implications for enzymatic catalysis. J. Am. Chem. Soc. 2014, 136, 1008–1022. [DOI] [PubMed] [Google Scholar]
- (89).Lemieux RU; Koto S; Voisin D, The Exo-Anomeric Effect. In Anomeric Effect; American Chemical Society: 1979; Vol. 87, pp 17–29. [Google Scholar]
- (90).Owen NL; Sheppard N Infra-red spectra and structure of methyl vinyl ether. Trans. Faraday Soc. 1964, 60, 634–645. [Google Scholar]
- (91).Capon B; Siddhanta AK Simple enols. 3. Stereochemistry of simple enols in solution. J. Org. Chem. 1984, 49, 255–257. [Google Scholar]
- (92).The s-cis conformation of the enol ether in 9 was found to be energetically disfavorable both in the ground state and transition state, presumably due to steric repulsion of the glucosyl group. [Google Scholar]
- (93).The reaction conducted in toluene provided the same dr as in HFIP. See the Supporting Information for more details. [Google Scholar]
- (94).TBS group was replaced by TMS group in the orbital calculations. [Google Scholar]
- (95).Grieco PA; Gilman S; Nishizawa M Organoselenium chemistry. A facile one-step synthesis of alkyl aryl selenides from alcohols. J. Org. Chem. 1976, 41, 1485–1486. [Google Scholar]
- (96).Kou KGM; Kulyk S; Marth CJ; Lee JC; Doering NA; Li BX; Gallego GM; Lebold TP; Sarpong R A unifying synthesis approach to the C18-, C19-, and C20-diterpenoid alkaloids. J. Am. Chem. Soc. 2017, 139, 13882–13896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Tietze LF Fragmentation of hydroxyloganin derivatives. Easy access to secologanin type compounds. J. Am. Chem. Soc. 1974, 96, 946–947. [Google Scholar]
- (98).Yang L; Zou H; Zhu H; Ruppert M; Gong J; Stockigt J Improved expression of His(6)-tagged strictosidine synthase cDNA for chemo-enzymatic alkaloid diversification. Chem. Biodiversity 2010, 7, 860–870. [DOI] [PubMed] [Google Scholar]
- (99).Pfitzner U; Zenk MH Immobilization of strictosidine synthase from Catharanthus cell cultures and preparative synthesis of strictosidine. Planta Med. 1982, 46, 10–14. [DOI] [PubMed] [Google Scholar]
- (100).Zou H-B; Zhu H-J; Zhang L; Yang L-Q; Yu Y-P; Stockigt J A facile chemoenzymatic approach: one-step syntheses of monoterpenoid indole alkaloids. Chem. - Asian J. 2010, 5, 2400–2404. [DOI] [PubMed] [Google Scholar]
- (101).Bernhardt P; Usera A R; O’Connor, S. E. Biocatalytic asymmetric formation of tetrahydro-β-carbolines. Tetrahedron Lett. 2010, 51, 4400–4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Pressnitz D; Fischereder E-M; Pletz J; Kofler C; Hammerer L; Hiebler K; Lechner H; Richter N; Eger E; Kroutil W Asymmetric synthesis of (R)-1-alkyl-substituted tetrahydro- -carbolines catalyzed by strictosidine synthases. Angew. Chem., Int. Ed. 2018, 57, 10683–10687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Cai Y; Shao N; Xie H; Futamura Y; Panjikar S; Liu H; Zhu H; Osada H; Zou H Stereocomplementary chemoenzymatic Pictet–Spengler reactions for formation of rare azepino-indole frameworks: discovery of antimalarial compounds. ACS Catal. 2019, 9, 7443–7448. [Google Scholar]
- (104).Standard methods were used to generate the strictosidine synthase E. coli expression strain. The lyophilized crude cell lysate was found to be stable for at least 6 months at 23 °C. See the Supporting Information for more details. [Google Scholar]
- (105).Achenbach H; Benirschke M Confirmation of the absolute configuration of dolichantoside and isodolichantoside by synthesis from (−)-secologanin. Phytochemistry 1997, 44, 1387–1390. [Google Scholar]
- (106).Most prior studies for analog synthesis have involved manipulating secologanin (6) or employing substituted tryptamines. For examples, see: McCoy E; Galan MC; O’Connor SE Substrate specificity of strictosidine synthase. Bioorg. Med. Chem. Lett. 2006, 16, 2475–2478. [DOI] [PubMed] [Google Scholar]
- (107).Most prior studies for analog synthesis have involved manipulating secologanin (6) or employing substituted tryptamines. For examples, references108,109 and: Yerkes N; Wu JX; McCoy E; Galan MC; Chen S; O’Connor SE Substrate specificity and diastereoselectivity of strictosidine glucosidase, a key enzyme in monoterpene indole alkaloid biosynthesis. Bioorg. Med. Chem. Lett. 2008, 18, 3095–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Bernhardt P; McCoy E; O’Connor SE Rapid identification of enzyme variants for reengineered alkaloid biosynthesis in periwinkle. Chem. Biol. 2007, 14, 888–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Lee H-Y; Yerkes N; O’Connor SE Aza-tryptamine substrates in monoterpene indole alkaloid biosynthesis. Chem. Biol. 2009, 16, 1225–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Similarly, the deprotected derivative of 14 also failed to undergo the enzymatic PS reaction. [Google Scholar]
- (111).Patty-Lukats A; Karolyhazy L; Szabo LF; Podanyi B First direct and detailed stereochemical analysis of strictosidine. J. Nat. Prod. 1997, 60, 69–75. [Google Scholar]
- (112).Patty-Lukats A; Kocsis A; Szabo LF; Podanyi B Configurative correlation and conformational analysis of strictosidine and vincoside derivatives. J. Nat. Prod. 1999, 62, 1492–1499. [DOI] [PubMed] [Google Scholar]
- (113).Battersby AR; Burnett AR; Parsons PG Alkaloid biosynthesis. Part XV. Partial synthesis and isolation of vincoside and isovincoside: biosynthesis of the three major classes of indole alkaloids from vincoside. J. Chem. Soc. C 1969, 1193–1200. [Google Scholar]
- (114).The C15 stereocenter is set early in the biosynthesis and is controlled by enzymes and a 5/6 fused ring system. For the full biosynthesis, see ref 33. [Google Scholar]
- (115).For a review on chemical Pictet–Spengler reactions involving tryptamine derivatives, see: Royer J; Bonin M; Micouin L Chiral heterocycles by iminium ion cyclization. Chem. Rev. 2004, 104, 2311–2352. [DOI] [PubMed] [Google Scholar]
- (116).For a review on Pictet–Spenglerase enzymes and their applications in biocatalysis: Roddan R; Ward JM; Keep NH; Hailes HC Pictet–Spenglerases in alkaloid biosynthesis: Future applications in biocatalysis. Curr. Opin. Chem. Biol. 2020, 55, 69–76. [DOI] [PubMed] [Google Scholar]
- (117).See the Supporting Information for more details. [Google Scholar]
- (118).Reagent-based Pictet–Spengler reactions of secologanin derivatives often give 1:1 dr. However, we found that shorter reaction times (i.e., 20 min) led to roughly 2:1 dr with respect to C3 based on 1H NMR analysis of crude reaction mixtures. Similarly, we have observed that the TFA-promoted Pictet–Spengler reaction of 5 and 6 to give strictosidine (4) similarly favors the natural C3 epimer at shorter reaction times. [Google Scholar]
- (119).Strictosamide has recently been investigated for various biological activity. See: Candeias MF; Abreu P; Pereira A; Cruz-Morais J Effects of strictosamide on mouse brain and kidney Na+, K+-ATPase and Mg2+-ATPase activities. J. Ethnopharmacol 2009, 121, 117–122. [DOI] [PubMed] [Google Scholar]
- (120).Strictosamide has recently been investigated for various biological activity. See: Kuete V; Sandjo LP; Mbaveng AR; Seukep JA; Ngadjui BT; Efferth T Cytotoxicity of selected Cameroonian medicinal plants and Nauclea pobeguinii towards multifactorial drug-resistant cancer cells. BMC Complementary Altern. Med. 2015, 15, 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Strictosamide has recently been investigated for various biological activity. See: Yuce I; Agnaniet H; Morlock GE New Antidiabetic and Free-Radical Scavenging Potential of Strictosamide in Sarcocephalus pobeguinii Ground Bark Extract via Effect-Directed Analysis. ACS Omega 2019, 4, 5038–5043. [Google Scholar]
- (122).Silyl triflates 19 and 20 were employed in these experiments as single diastereomers. [Google Scholar]
- (123).For reactions of natural products with arynes, see: Ross SP; Hoye TR Reactions of hexadehydro-Diels–Alder benzynes with structurally complex multifunctional natural products. Nat. Chem. 2017, 9, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.