

Abstract
Much has been written about the validity of mice as a preclinical model for brain disorders. Critics cite numerous examples of apparently effective treatments in mouse models that failed in human clinical trials, raising the possibility that the two species’ neurobiological differences could explain the high translational failure rate in psychiatry and neurology (neuropsychiatry). However, every stage of translation is plagued by complex problems unrelated to neurobiological conservation. Therefore, although these case studies are intriguing, they cannot alone determine whether these differences observed account for translation failures. Our analysis of the literature indicates that most neuropsychiatric treatments used in humans are at least partially effective in mouse models, suggesting that neurobiological differences are unlikely to be the main cause of neuropsychiatric translation failures.
We are in the midst of a crisis in neuropsychiatric drug development. Only 9% of neuropsychiatric drugs entering clinical trials succeed and go on to market, one of the lowest rates across all therapeutic areas [1]. Although attrition occurs at all phases of clinical trials, the majority take place during Phases II and III, where failure is costliest in terms of both time and money [1,2]. The low success rate in the development of neuropsychiatric drugs is partially due to one of the highest failure rates in Phase III clinical trials [1]. Thus, even within human studies, many initial findings do not extrapolate to larger, more complex cohorts. A combination of high cost and high risk makes investment unappealing, and multiple pharmaceutical companies have downsized or altogether abandoned neuropsychiatric drug development [3].
All aspects of neuropsychiatric drug development have attracted heavy scrutiny, but preclinical trials, in which use of mouse disease models is ubiquitous, have received the greatest attention. Numerous problems have been identified in preclinical studies (Table 1). These include a low standard of evidence, a reliance on inappropriate statistics, conflation of confirmatory and exploratory designs, poor reproducibility (partly from low animal numbers, resulting in underpowered studies), inadequate consideration of pharmacokinetic/pharmacodynamic issues, improper modeling, and the adoption of flawed methods [4–6]. Fundamental differences between the neurobiology of mice and humans have also been proposed to account for translation failures in neuropsychiatry [3]. Effective mouse models should have construct validity (a similar neurobiological cause, such as a genetic variant or neuroanatomical abnormality, to the human disorder) and predictive validity (similar responses to treatments that prevent or reverse the human disorder) [7]. Ideally, they should also have face validity (compelling similarities to the endophenotypes of the human condition), though behavioral effects of a shared disease etiology may differ across species.
Table 1.
Sources of low predictive validity in preclinical study and proposed solutions.
| Sources of low predictive validity | Solutions |
|---|---|
| Clinically non-predictive populations | Use multiple background strains |
| Use multiple genetic models for the same disease | |
| Outbreed mouse population | |
| Develop humanized mouse models | |
| Clinically non-predictive behavior | Use multiple complementary behavioral assays |
| Validate assays with both clinically successful and failed compounds | |
| Develop new assays to model neglected disease symptoms | |
| Low construct validity | Preclinically test pharmacokinetics/pharmacodynamics |
| Develop behavioral assays that work similarly for humans and mice | |
| Discover predictive clinical biomarkers shared by humans and mice | |
| Identify human biomarkers that better recapitulate drug efficacy | |
| Low preclinical statistical power | Pre-register behavioral assays and metrics |
| Increase baseline sample sizes | |
| Independently replicate all successful tests before clinical trials |
The predictive validity of a preclinical model reflects how well assays predict a compound’s future therapeutic performance in the clinic, and it has two core components [7]. The first is positive predictive value: the probability of a positive preclinical finding resulting in a successful clinical trial (a true positive) [8]. The second is its inverse, the false discovery rate: the probability of a positive preclinical finding resulting in a negative or a failed clinical trial (a false positive). In recent years, a growing number of investigators have called for a two-way dynamic interaction between preclinical and clinical studies, where — besides preclinical work informing clinical studies — clinical studies would inform further preclinical work [9]. These opposite, complementary approaches are known as translational and ‘back-translational’ frameworks, respectively (Figure 1). Back-translation is not unlike the practice of hindcasting in environmental science, where investigators refine predictive models by examining their performance on known historical data. Here, we determined whether preclinical mouse models and assays in the literature could detect the therapeutic effect of a representative set of known clinically effective neuropsychiatric drugs, independent of whether they originally were developed using mouse behavioral models or assays.
Figure 1. Analysis of conservation requires both translation and back-translation.
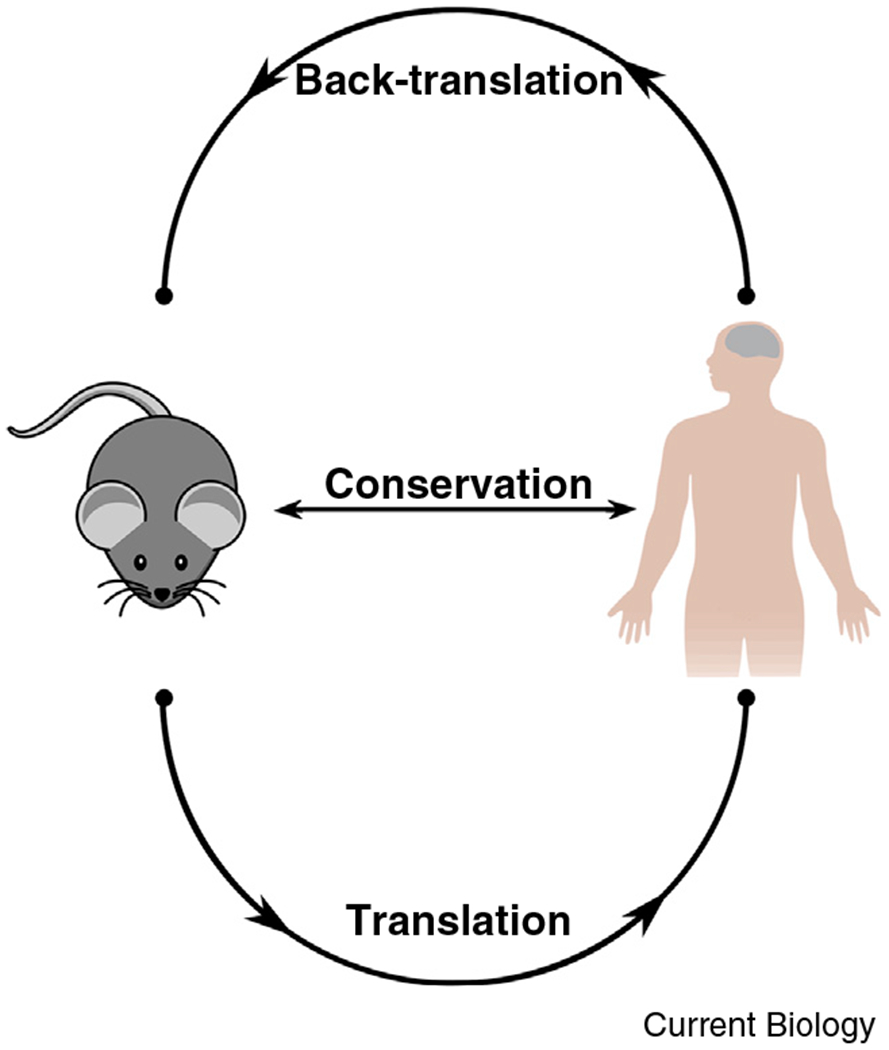
Translation and back-translation complement each other to uncover conservation. Evolutionary conservation is a bidirectional relationship between two species (in this case, mice and humans) characterized by their mutual similarity. A symmetrical approach is required to reveal it: translational approaches for what mice share with humans, and back-translation for what humans share with mice.
All surveyed neuropsychiatric drugs back-translate to mice
We performed a representative, but not exhaustive, review to determine whether clinically effective neuropsychiatric drugs were similarly effective in preclinical mouse models (see Supplemental Information, published with this article online for full details). We selected the 40 top-selling neuropsychiatric drugs and reviewed their efficacy in mouse models of all neuropsychiatric disorders indicated for treatment in humans by the Food and Drug Administration (FDA), for a total of 66 drug–indication combinations. For this purpose, we reviewed each compound’s most impactful publications in which investigators used relevant behavioral assays to test its effects in mouse models of its indicated neuropsychiatric disease, as well as relevant patents, to determine whether mouse behavioral assays were used during the compound’s preclinical development. We summarize our results in Table S1 (see Supplemental Information).
To determine whether drugs successfully back-translated from humans to mice, we examined whether their therapeutic effect had been tested in at least one behavioral assay in a relevant neuropsychiatric disease mouse model. Even when we excluded results from behavioral assays originally validated by compounds of interest (e.g., amitriptyline in the forced swim and tail suspension tests and diazepam in the elevated plus maze), all compounds still back-translated in at least one behavioral assay and most in a highly reliable manner. The genetic background, specific disease model, variation in dosage, and route of administration did not appear to affect the back-translation potential of a drug–indication combination overall. When a drug’s effect replicated consistently in a behavioral assay for an indication, it was remarkably robust, consistently retaining efficacy across numerous combinations of various levels of many factors. Interestingly, <20% (13/66) of all reviewed drugs were developed using mouse behavioral assays: all others either used other species in behavioral assays or did not report preclinical behavioral testing. Nevertheless, when mouse behavioral assays were used in preclinical testing, all compounds back-translated in assays that had not been used for their initial development.
Criticisms of the predictive validity of mouse models have focused on potentially insufficient conservation between mice and men, speculating that these differences may constrain construct validity so much that a drug’s activity in mouse neuropsychiatric disease models cannot reliably inform future activity in humans [3]. However, more than 99% of mouse genes have human homologs, including most known proteins involved in synaptic transmission, such as neurotransmitters, receptors, and ion channels [10]. Many neuropsychiatric drugs tested to date function primarily by modulating synaptic transmission. Therefore, the molecular target of a drug developed in mice is unlikely to be absent in humans, and many molecular pathways of interest in humans are likely conserved in mice. Indeed, we consider it unlikely that genomic differences between humans and mice account for most translation failures, at least when discussing most current mechanisms of action in neuropsychiatry. The two species’ transcriptomes are more divergent than their genetic codes, however, and these differences in gene expression could affect neuropsychiatric disease manifestations and treatment outcomes [11]. Further, if most future neuropsychiatric drugs in development had mechanisms of action directly targeting gene expression (e.g. via transcription factors, epigenetics, or post-transcriptional modifications), these differences would be more likely to become salient. The relationship between variations in transcriptomes and neuropsychiatric drug efficacy has not yet been extensively explored, and could be a fruitful avenue for further research.
Species differences great enough to impact construct validity would be unlikely to homogenously impact the validity of mouse models of all neuropsychiatric diseases. Instead, they would be more likely to affect specific models of neuropsychiatric disorders designed to simulate higher-order cognitive defects arising from changes in neocortical pathways, such as those involved in schizophrenia [12]. Indeed, there are major differences between humans and mice in nearly all cognitive domains. A large number of them result from the increased size and complexity of the human connectome, as well as its relatively unique organization, especially in the neocortex [12]. Differences in neural gene expression between mice and primates are most pronounced in the neocortex, particularly among genes regulating its development and organization [12].
Although neurotransmitters and neurotransmission-associated molecules may not differ much between species, density and distribution differences in certain pathways do exist, but their impact on predictive validity is unknown. Short of in vivo studies in primates, which have their own associated concerns, it is unclear if phenotypes in other commonly used model species would be significantly more similar to those in humans [13]. Humanized mice are an interesting new model that may be able to bridge some gaps between humans and mice. However, to our knowledge, there are few (if any) published studies comparing side-by-side the effect of neuropsychiatric drugs in behavioral assays in humanized versus non-humanized mouse models. Humanized mice or mice engineered to contain human induced pluripotent stem cell-derived cerebral organoids could conceivably improve the construct validity of mouse models, but sufficient evidence is not yet available to determine whether these changes will result in tangible increases in predictive validity.
Species differences in drug bioavailability, metabolism and toxicity can also complicate translation from experimental models to humans. In many clinical trials, the maximally tolerated human dose is lower than the effective dose used in preclinical mouse models, which may contribute to the low clinical success rate in neuropsychiatry. However, this is by no means unique to research involving mice. Few, if any, commonly-used preclinical animal models, including non-human primates, can reliably predict human oral bioavailability [14]. Non-animal models have probably even lower predictive power for dosing. Neuropsychiatric drug efficacy is highly multivariate and can be affected by seemingly minor differences in many non-neuronal properties (e.g. blood–brain barrier permeability, sexual dimorphism, and glia-specific effects). For these and other reasons, the pharmaceutical industry is beginning to pay much more attention to the influence of complex biological systems in pharmacology. Even if aspects of these complex biological systems differ between species, models possessing imperfectly conserved versions of these systems will likely possess higher predictive validity for dosing than one lacking them, especially in neuropsychiatry, where the blood–brain barrier still poses a unique challenge to delivery.
Mouse behavioral assays are variably effective in detecting clinically effective neuropsychiatric drugs
As part of our analysis, we assigned rudimentary measures of confidence for the ability of each behavioral assay to detect clinically effective neuropsychiatric drugs, based on the reliability of the assay (see Supplemental Information for details). Of the 66 reviewed drug–indication combinations, 46 used at least one behavioral assay we classified as ‘high confidence’. Of the remaining 20 combinations, 6 used at least one assay we classified as ‘moderate confidence’, and the remaining 14, including all 6 adjuncts, only used assays we classified as ‘low confidence’, and we subsequently rated the back-translation of this last group as ‘inconclusive’. We generally underestimated confidence in the back-translation of adjuncts because three factors (adjunct compound, main compound, and assay) needed to replicate across studies, not just two (compound and assay), as for primary drugs. This additional requirement generally reduced the number of times each combination would replicate. Thus, low confidence in adjunct back-translation may stem, at least in part, from the classification criteria used.
The specific ability of particular assays to detect true positive results varied considerably, mostly based on indication. By far, drugs indicated to treat pain or seizures back-translated best to mouse behavioral assays. For example, drugs to treat pain back-translated with high confidence in multiple assays, and few assays yielded negative results. In contrast, no individual assays could reliably detect all clinically effective antidepressants or anxiolytics with even moderate confidence. Interestingly, when we compared our analysis to more focused reviews of the preclinical efficacy of these conditions, our conservative methods actually arrived at lower estimates of preclinical efficacy than most other systematic studies (see Supplemental Information for full information of validation). Depression and anxiety both manifest as complex sets of multivariate symptoms, increasing the difficulties involved in modeling and assaying, as compared to seizures and pain (which we acknowledge are not necessarily easy to treat either). All together, these results support using multiple appropriate behavioral assays in preclinical drug development.
For other indications, selective use of individual behavioral assays complicated efficacy judgments. For example, the assessment of drugs indicated for obsessive-compulsive disorder (OCD) or attention deficit hyperactivity disorder in mice overwhelmingly rely on individual assays (marble burying and hyperlocomotion, respectively), which in our analysis were performed more than all other assays combined for their related indications. This uniformity could magnify potential disparities between a disorder and its behavioral model.
Multiple compounds studied preclinically in mice may effectively treat a single symptom of a complex human condition corresponding to the single behavior examined, but they may not treat other symptoms not corresponding to the consensus assay. This would clearly impact predictive validity because reliance on a few assays could return both false positives and false negatives: compounds could alter the behavior(s) tested without altering the underlying pathology, or the paucity of behavioral assays available could miss compounds that improve un-modeled symptoms. Modeling OCD nearly exclusively via marble burying serves as an example of these pitfalls. The specific aspect of OCD assayed by marble burying and even its correspondence to OCD or anxiety are perennially debated; likewise, some anxiolytic compounds with no human anti-compulsive effects consistently pass this assay while some known anti-compulsive drugs consistently fail [15].
Because bipolar disorder and schizophrenia are both characterized by a diverse set of symptoms and manifestations, reliance on even a few assays modeling specific behaviors can be overly reductive [7]. Even though drugs indicated for schizophrenia and bipolar disorder succeed in multiple mouse assays, experimenters converge on a subset of these assays, such as prepulse inhibition and hyperlocomotion, which are very well-characterized, but at best, they only partially model these diseases’ behavioral complexity. Other behavioral assays, such as socialization and Y-maze alternation, represent different symptoms than the more canonical assays, but are less common. Thus, during testing, these highly multifactorial diseases often become reduced to a small subset of their overall symptoms: preclinical studies will be much less likely to identify drugs treating symptoms exclusively modeled by less popular assays, because canonical assays are most likely to be used when quickly screening candidate drugs, biasing drug development towards their associated subset of modeled symptoms [7].
These less popular assays represent an opportunity. Drugs succeeding in unconventional assays via unconventional mechanisms face a less crowded development pipeline and patient population than those acting on well-characterized mechanisms and succeeding in the disease’s most common behavioral assays. For example, the highly successful antiepileptic levetiracetam preclinically failed in the pentylenetetrazole and maximal electroshock tests, the two most commonly used preclinical seizure models in our analysis, but succeeded in the less common audiogenic and kindled seizure models [16]. Had levetiracetam been tested in only the two orthodox models, it would likely never have been discovered. Instead, it required a more thorough investigation, using a wide spectrum of behavioral models, not just the canonical ones.
Improving preclinical trial payoffs
Neuropsychiatric drugs have the lowest aggregate clinical success rate, which — combined with their high development costs — has strongly discouraged industrial research and development. The vast majority of resource and time expenditures during drug development are incurred during mid- to late-stage clinical trials. Therefore, optimizing early-stage drug development (e.g., target screening and preclinical studies) is a sensible way to reduce costs and attrition in late-stage clinical trials, where failure is most expensive [2]. However, neuropsychiatric preclinical testing has an intolerably high false discovery rate and correspondingly low payoffs. But are experimental models responsible?
Construct validity likely affects predictive validity, wherein a model with underlying mechanisms closely related to the disease of interest should better predict the effect of a given therapeutic on the disease’s phenotype. However, some upstream construct differences may not necessarily impair predictive validity, so long as the model and the human condition share downstream neural alterations that similarly induce the disease’s phenotype and respond to treatment.
Correspondingly, species differences between mice and humans may not be great enough to fully explain the discrepancy between observed preclinical and clinical results. Such differences should yield not only drugs effective in mice and ineffective in humans, but also drugs effective in humans and ineffective in mice. If the two species are sufficiently divergent to prevent adequate translation, then mouse studies would not only consistently return false positives but also false negatives, regardless of model or assay. However, we find that mouse studies can confirm the efficacy of every examined drug effective in human neuropsychiatric disease, which indicates that mouse models can sufficiently capture critical aspects of human neuropsychiatry to detect true positive results. Detecting false positives and true positives, but not false negatives, would suggest a process that solely amplifies effects of all neuropsychiatric drugs in mice. Such a process would need to amplify the effects of diverse drugs working via numerous unrelated mechanisms and targets in separate parts of the brain, without inducing any changes that reduce their effectiveness, which seems very unlikely. Overall, our findings do not support the claim that intrinsic biological differences between the two species explain most observed differences in preclinical and clinical outcomes.
Nevertheless, there is plenty of room for improvement in the current use of mouse models, starting with a need for more research on the cause of neuropsychiatric diseases (Table 1). Without this information it will be impossible to properly model them in any system, including mice. Additionally, we need to enforce a higher standard of evidence in preclinical trials, such as requiring success in multiple behavioral assays for each relevant phenotype of interest, with more rigorous methodologies and using multiple background strains and/or genetic models before advancing to clinical trials, and ensuring that the concentration of drug found effective in an animal model is the actual concentration tested in humans after accounting for species differences in ADME/PK (absorption, distribution, metabolism, excretion/pharmacokinetics) [17]. So long as the behavioral assays model different aspects of a neuropsychiatric disease (e.g. forced swim, sucrose preference, and splash tests in depression), the increase in predictive validity will almost certainly offset the decrease in throughput from more thorough testing, reducing costs and increasing proportional FDA approval by moving fewer compounds forward that are more likely to succeed in clinical trials [8]. Choosing behavioral assays that interrogate different aspects of the disease is also key. Of course, these should not be the only steps taken. More study should also be undertaken to improve the translational potential and reproducibility of individual preclinical models/assays, and many such approaches have been proposed and discussed elsewhere [9,17,18].
Identifying potentially translatable biomarkers correlated with target engagement and/or therapeutic response could also increase the positive predictive value of mouse models in neuropsychiatric drug discovery. Whether based in genetics, biochemistry, electrophysiology or neuroimaging, such biomarkers, and resulting mechanistic insights could potentially be critical for determining which preclinical candidates should go forward into clinical trials. Identifying validated and quantifiable outcome measures in clinical trials for neuropsychiatric diseases is also essential [19]. Clinical failures could occur for reasons unrelated to compound efficacy or target mechanism. Even a perfect preclinical program could not make up for the problems caused by a lack of appropriate outcome measures. A comprehensive approach including improved mouse behavioral assay predictive validity, optimized clinical biomarkers, in-depth validation of drug mechanisms in humans, and better outcome measures may be needed to solve the present crisis in neuropsychiatry.
Despite the current suboptimal state of preclinical mouse studies in neuropsychiatry, no clear and viable alternatives seem to exist, though many new promising technologies may lie just beyond the horizon. Target-based screening only discovered one neuropsychiatric drug approved between 1999 and 2008, whereas preclinical phenotypic screening, which includes behavioral assays, discovered seven, despite a throughput orders of magnitude lower [20]. Further, it is uncertain whether other proposed screening methods (e.g. murine or human cell culture models) actually possess greater construct validity. Although such assays do have obvious advantages, (e.g., greater ease of manipulation and higher throughput capacity), defining truly disease-relevant outcome measures could be even harder in cell and tissue culture models than in animal models, particularly in regard to neuropsychiatric diseases that affect or depend on complex network properties and/or aging, two features difficult to reliably simulate in vitro.
Once these technologies mature, however, it is quite easy to envision a preclinical pipeline where these techniques complement each other for more sophisticated predictions. One phenomenon that comes to mind is pharmacokinetics/pharmacodynamics, which involves both complex and species-specific drug responses. While most known aspects of the synapse are conserved between mice and humans, some highly specific differences do still exist, such as mutations in analogous synaptic receptors expressed by different species. Human stem cell-derived brain cells or organoids could be used to model the molecular dynamics of different doses in human neural tissues. The mouse could then model the drug’s effects on complex neural networks, although doses may have to be adjusted for species differences in bioavailability and metabolism. Thus, these different model paradigms can co-exist and compensate for each other’s trade-offs.
The current neuropsychiatric drug development crisis has called into question many previously held assumptions. One such assumption is that the mouse itself is a sufficiently valid model for neuropsychiatric drug discovery. Our review of the literature presented here indicates that preclinical mouse behavioral assays can detect the entirety of a representative set of clinically effective neuropsychiatric drugs, even for drugs not originally developed using mice. It will also be important to better understand why so many phase II trials in humans do not extrapolate to phase III trials, and to explore whether similar phenomena could contribute to the false discovery rate in animal models. Every stage of the drug development pipeline urgently needs improvement, and it is even possible that we may need to fundamentally re-invent how we take discoveries from the laboratory to the clinic.
Supplementary Material
ACKNOWLEDGMENTS
S.A.L. is supported by NIH grants R01AG056259, R01NS086890, DP1DA041722, and RF1AG057409.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information contains methods and one table, and can found with this article online at https://doi.org/10.1016/j.cub.2018.07.046.
DECLARATION OF INTERESTS
M.F.B. is a consultant for Q-State Biosciences and a member of the scientific advisory board of BioAxone Biosciences. S.A.L. is the inventor on worldwide patents for the use of the drug memantine in neurodegenerative and neuropsychiatric disorders. Per Harvard University guidelines, S.A.L. participates in a royalty-sharing agreement with his former institution Boston Children’s Hospital/Harvard Medical School, which licensed memantine (Namenda®) to Forest Laboratories, Inc./Actavis/Allergan, Inc. S.A.L. also declares that in addition to his affiliations on the masthead, he is also an Adjunct Professor of Neurology at the Yale School of Medicine. L.M. serves pro bono on the Research Management Committee of Cure Network Dolby Acceleration Partners, LLC, and is also a member of the External Advisory Board of the NIH-sponsored Alzheimer’s Disease Precision Models Center of Indiana University and The Jackson Laboratory. M.S. has received grants and consulting fees from Roche, grants from Novartis, Pfizer, LAM Therapeutics, Ibsen, Navitor, Rugen and Neuren outside the submitted work. He also has served on the Scientific Advisory Board of Sage Therapeutics, PTEN Research Foundation and PTEN Hamartoma Tumor Syndrome Foundation. M.S. also serves on the professional advisory board of Tuberous Sclerosis Alliance and is part of the Preclinical Autism Consortium for Therapeutics (PACT) funded by Autism Speaks.
REFERENCES
- 1.Hay M, Thomas DW, Craighead JL, Economides C, and Rosenthal J (2014). Clinical development success rates for investigational drugs. Nat. Biotechnol. 32, 40–51. [DOI] [PubMed] [Google Scholar]
- 2.Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, and Schacht AL (2010). How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 9, 203–214. [DOI] [PubMed] [Google Scholar]
- 3.Hyman SE (2014). Revitalizing psychiatric therapeutics. Neuropsychopharmacology 39, 220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kimmelman J, Mogil JS, and Dirnagl U (2014). Distinguishing between exploratory and confirmatory preclinical research will improve translation. PLoS Biol. 12, e1001863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kleiman RJ, and Ehlers MD (2016). Data gaps limit the translational potential of preclinical research. Sci. Transl. Med 8, 320ps321. [DOI] [PubMed] [Google Scholar]
- 6.van der Worp HB, Howells DW, Sena ES, Porritt MJ, Rewell S, O’Collins V, and Macleod MR (2010). Can animal models of disease reliably inform human studies? PLoS Med. 7, e1000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nestler EJ, and Hyman SE (2010). Animal models of neuropsychiatric disorders. Nat. Neurosci. 13, 1161–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scannell JW, and Bosley J (2016). When quality beats quantity: decision theory, drug discovery, and the reproducibility crisis. PLoS One 11, e0147215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Markou A, Chiamulera C, Geyer MA, Tricklebank M, and Steckler T (2009). Removing obstacles in neuroscience drug discovery: the future path for animal models. Neuropsychopharmacology 34, 74–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. (2002). Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562. [DOI] [PubMed] [Google Scholar]
- 11.Miller JA, Horvath S, and Geschwind DH (2010). Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc. Natl. Acad. Sci. USA 107, 12698–12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geschwind DH, and Rakic P (2013). Cortical evolution: judge the brain by its cover. Neuron 80, 633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jennings CG, Landman R, Zhou Y, Sharma J, Hyman J, Movshon JA, Qiu Z, Roberts AC, Roe AW, Wang X, et al. (2016). Opportunities and challenges in modeling human brain disorders in transgenic primates. Nat. Neurosci. 19, 1123–1130. [DOI] [PubMed] [Google Scholar]
- 14.Musther H, Olivares-Morales A, Hatley OJD, Liu B, and Hodjegan AH (2014). Animal vs. human oral drug bioavailability: do they correlate? Eur. J. Pharm. Sci. 57, 280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Albelda N, and Joel D (2012). Animal models of obsessive-compulsive disorder: exploring pharmacology and neural substrates. Neurosci. Biobehav. Rev. 38, 47–63. [DOI] [PubMed] [Google Scholar]
- 16.Crepeau AZ, and Treiman DM (2010). Levetiracetam: a comprehensive review. Expert Rev. Neurother. 10, 159–171. [DOI] [PubMed] [Google Scholar]
- 17.Steward O, and Balice-Gordon R (2014). Rigor or mortis: best practices for preclinical research in neuroscience. Neuron 84, 572–581. [DOI] [PubMed] [Google Scholar]
- 18.Institute of Medicine. (2013). Improving the Utility and Translation of Animal Models for Nervous System Disorders: Workshop Summary (Washington, DC: National Academies Press; ). [PubMed] [Google Scholar]
- 19.Jeste SS, and Geschwind DH (2016). Clinical trials for neurodevelopmental disorders: At a therapeutic frontier. Sci. Transl. Med 8, 321fs1. [DOI] [PubMed] [Google Scholar]
- 20.Swinney DC, and Anthony J (2011). How were new medicines discovered? Nat. Rev. Drug Discov. 10, 507–519. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.