

Abstract
Antibiotics profoundly reduced worldwide mortality. However, the emergence of resistance to the growth inhibiting effects of these drugs occurred. New approaches to treat infectious disease that reduce the likelihood for resistance are needed. In bacterial pathogens, complex signaling networks regulate virulence. Anti-virulence therapies aim to disrupt these networks to attenuate virulence without affecting growth. Quorum-sensing, a cell-to-cell communication system, represents an attractive anti-virulence target because it often activates virulence. The challenge is to identify druggable targets that inhibit virulence, while also minimizing the likelihood of mutations promoting resistance. Moreover, given the ubiquity of quorum-sensing systems in commensals, any potential effects of anti-virulence therapies on microbiome function should also be considered. Here we highlight the efficacy and drawbacks of anti-virulence approaches.
Introduction
Over the past century, antibiotics have served as the mainstay for treating bacterial infections. Antibiotics are small molecules that inhibit bacterial growth and, therefore, exert strong selective pressures on bacterial populations, which favor the emergence of antibiotic resistant (AR) clones. Indeed, AR isolates continue to emerge with each new class of antibiotics [1]. Approximately 2.8 million infections by AR pathogens occur annually in the United States, resulting in at least 35 000 deaths [2]. These alarming trends highlight the pressing need for alternative strategies to treat bacterial infections that also reduce the likelihood of resistance.
Anti-virulence therapies represent an alternative approach for treating bacterial infections. In contrast to antibiotics, anti-virulence agents do not directly inhibit bacterial growth [3], which is hypothesized to reduce the selective pressures exerted on pathogen populations. Instead, anti-virulence agents are designed to disrupt intercellular signaling networks essential for host colonization and induction of disease [4]. The challenge is to identify targets within these complex networks that inhibit virulence, while also minimizing the emergence of mutations or compensatory functions that enable pathogen circumvention of the anti-virulence therapy. Secondly, given the ubiquity of cross-species signaling within bacterial communities, another challenge is to inhibit pathogen virulence while avoiding detrimental effects to microbiome function. Herein, we review the implementation of anti-virulence strategies that target quorum sensing in pre-clinical models of infection, with a particular focus on Staphylococcus aureus and Escherichia coli, frequent members of human microbiomes that can also become dangerous opportunistic AR pathogens. The implementation of such strategies with Pseudomonas aeruginosa, another clinically important AR opportunistic pathogen, has been recently reviewed elsewhere [5–8].
Bacterial intercellular signaling through quorum sensing (QS)
Bacteria employ numerous strategies to sense their local environment and integrate this spatial information to regulate their growth, physiology, and behaviors. QS is a form of cell-to-cell communication that enables bacterial populations to coordinate their behaviors in an environmental and cell density-dependent manner [4,9]. The primary components of QS systems include: (1) biosynthetic machinery that generates the signal (autoinducer); and (2) cognate sensory machinery that recognizes and responds to the signal (Figure 1). QS systems represent attractive anti-virulence targets because they often activate virulence-associated functions essential for establishing infection [4]. Moreover, QS can mediate the switch from commensal to opportunistic pathogen and serve as bacterial sensors for host signals that enable appropriate deployment of pathogen virulence programs [10,11]. Thus, much effort has been dedicated toward discovering small molecules that inhibit QS systems [3,5,12,13] (Figure 1).
Figure 1.
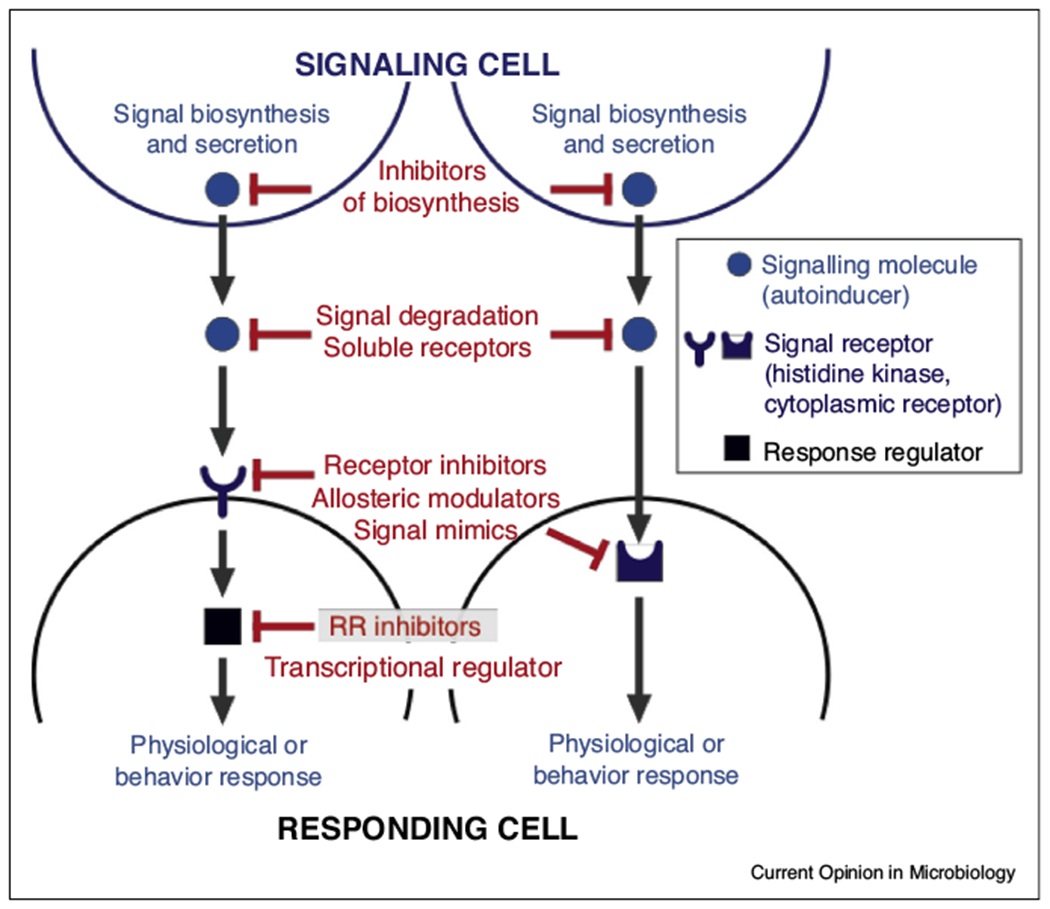
Basic structure of quorum sensing systems in bacteria. Quorum sensing (QS) is a form of cell-to-cell communication that enables bacterial populations to coordinate their behaviors in an environmental and cell density-dependent manner. The primary components of QS systems include the biosynthetic machinery that generates the QS signal and its cognate sensory machinery that recognizes and responds to the signal. The latter is usually a cytoplasmic receptor or histidine sensor kinase (HK), which upon its activation phosphorylates its cognate response regulators (RR). However, other transcription factors can also be inhibited. The QS cytoplasmic receptors and RRs then modulate bacterial behaviors and functions through their functions as transcription factors.
Targeting QS systems in S. aureus
S. aureus is a skin and/or nasal commensal in approximately 30% of the human population that, with inappropriate access to deeper tissues, can become an opportunistic pathogen responsible for potentially lethal infections [14–16,17•]. In S. aureus, the transition from commensal to pathogen is mediated by the accessory gene regulation (agr) locus, which encodes a QS system that activates numerous virulence factors and represses cell surface proteins that contribute to asymptomatic colonization of the nasal cavity and skin. Noticeably, the Agr system cross talks with other regulatory systems, such as SarA/SarR. SarA activates expression of the agr locus, and transcription of sarA is induced by SarA itself, while it is repressed by SarR [18,19] (Figure 2). The importance of agr in S. aureus infections is well established in various rodent models [20–23]. The agr locus encodes the machinery that synthesizes and secretes the QS signal autoinducing peptide (AIP), its receptor AgrC, its cognate response regulator (RR) AgrA, the RNAIII regulatory RNA and δ-toxin. AIP is encoded by the agrD gene. After translation, the AgrD propeptide is processed into AIP by AgrB [19,24] (Figure 2). AgrA and RNAIII together regulate about 200 genes in the S. aureus genome, including virulence-associated functions such as toxins, phenol soluble modulins and proteases [19,25]. Thus, the agr locus represents an attractive therapeutic target for treating S. aureus infections and preventing loss of commensalism in S. aureus carriers.
Figure 2.
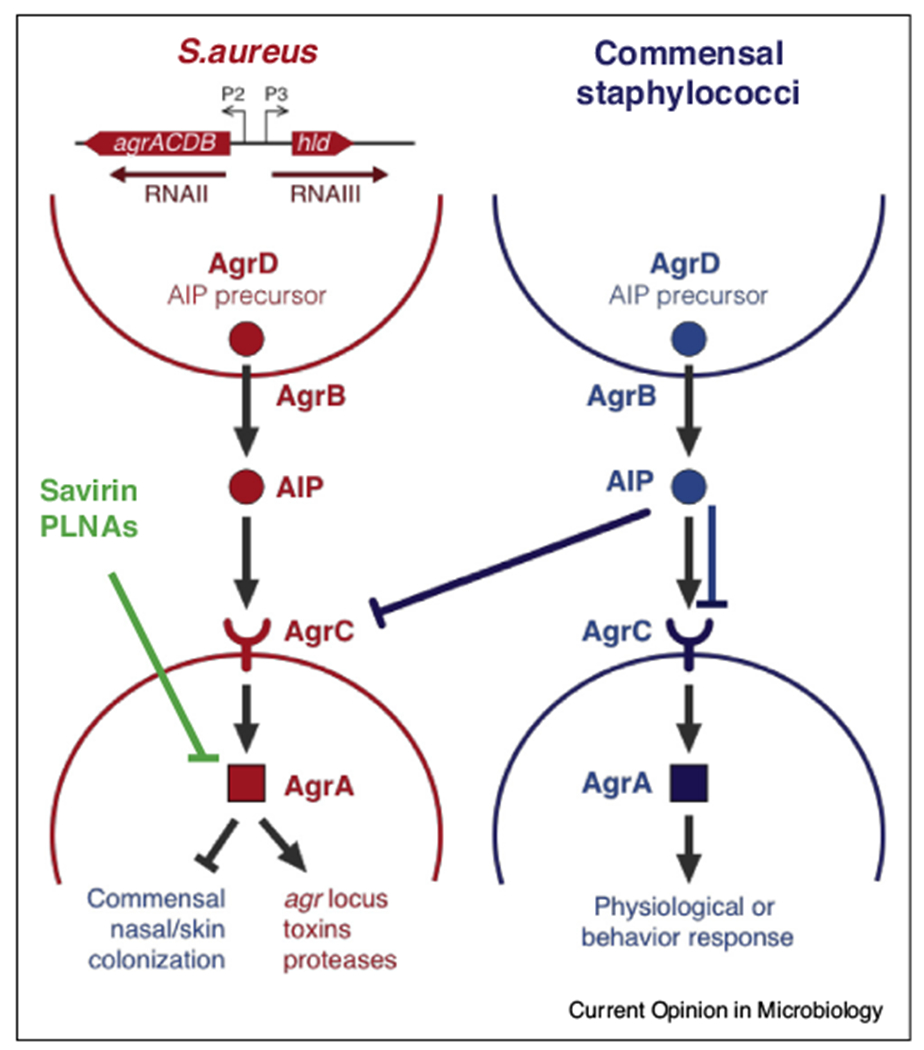
The agr quorum sensing system in S. aureus. The agr locus consists of two divergently encoded operons, agrACDB and RNAIII, which are activated by the transcription factor AgrA. The agrACDB operon encodes AgrD, which is the precursor for the QS signal autoinducing peptide (AIP); AgrB, which is the machinery that processes and secretes AIP; and the AgrAC two-component system comprises the AIP receptor AgrC and its cognate response regulator AgrA. The RNAIII operon consists of the regulatory RNA, RNAIII, and the δ-toxin encoded by hld. AgrA and RNAIII together activate virulence-associated functions such as toxin production, secretion of phenol soluble modulins (PSMs) and protease activity and inhibit the expression of cell surface proteins associated with commensalism. Coagulase negative staphylococcal commensals produce AIPs that inhibit the AgrC receptor in S. aureus, thus attenuating its virulence, they may also inhibit AgrCs from other commensals. Additionally Savirin and PNLAs also inhibit Agr signaling.
Staphylococci exhibit subtle strain-level differences in the chemical structures of their AIPs [26]. S. aureus secretes one of four AIP types that activate their cognate AgrC receptor and inhibit other AgrC alleles [23]. AIPs range from seven to nine aminoacids, and their types stems from variations in the agrD gene. Structural differences in AIP, combined with differences in the N-terminal region of AgrC, which binds this ligand, dictate the outcomes of activation or deactivation of this signaling cascade [19]. Identifying or synthesizing competing analogs to AIPs that inhibit AgrC has been an anti-virulence strategy to treat S. aureus infections [23]. Interspecies QS crosstalk also occurs between S. aureus and staphylococcal skin commensals. For example, Staphylococcus epidermidis’s AIP is able to inhibit the Agr system of most S. aureus, except for one subgroup (subgroup 4), which in turns inhibits the Agr response of S. epidermidis. The AIP produced by Staphylococcus caprae was able to inhibit the Agr system of all classes of S. aureus [27,28,29•,30,31••] (Figure 2). Thus, candidate anti-virulence agents should ideally inhibit all four AIP types in S. aureus, while also avoiding harmful effects to the skin microbiome. Another strategy to inhibit agr activation is to target AgrA, which is more conserved among S. aureus strains. For example, savirin is a QS inhibitor that binds residues uniquely present in the DNA-binding domain of S. aureus AgrA and thus exhibits minimal effects on agr activation in the skin commensal S. epidermidis. Through inhibition of AgrA, savirin also inhibits transcription of RNAIII, which is activated by AgrA [25]. In acute models of skin infection, savirin treatment attenuates pathology as well as local and systemic burdens of S. aureus [25]. Importantly, in contrast to repeated antibiotic treatment, continuous in vivo passaging of S. aureus together with savarin coadministration did not produce savarin-resistant isolates [25]. Another strategy developed to inhibit AgrA and RNAIII was the development of peptide conjugated nucleic acids (PLNAs) that are modified antisense RNAs conjugated to a cell-penetrating peptide [32]. Interestingly, during acute lung infection, nitric oxide (NO) produced by the innate immune system also targets AgrA through a similar mechanism. NO inactivates AgrA through the S-nitrosylation of residues that also interfere with DNA binding and agr activation, thus resulting in attenuated disease. However, one has to be cautious of this approach given that NO production may engender changes in multiple protein targets and have off target pleiotropic effects [71••]. These findings, together with other studies [32–34], highlight the potential of targeting QS systems as an effective strategy for treating acute infections caused by S. aureus.
Commensal microorganisms also produce signaling molecules that interfere with pathogen QS systems to compete for limited nutrients and space. For example, some staphylococcal skin commensals produce AIPs that inhibit the AgrC receptor in S. aureus [27,28,29•, 30,31••,56,57] (Figure 2). Similarly, nasal commensals such as Corynebacterium spp. inhibit virulence-associated functions in S. aureus by blocking QS activation through an undefined mechanism [58,59•]. This antagonistic relationship can be potentially exploited as another antivirulence strategy to treat infections. Indeed, simultaneous administration of live commensal staphylococci or Corynebacterium attenuates S. aureus agr activation, abscess colonization and skin lesion formation [29•,31••,58]. Similarly, therapeutic administration of purified AIP from S. caprae reduces pathogen burdens and accelerates resolution of skin pathology [29•,31••]. Together, these studies demonstrate the potential for developing probiotic treatments that augment the inhibitory effects of commensals on pathogen QS systems. More broadly, studying commensal-pathogen interactions within naturally occurring microbial communities could enable further discovery of antagonistic relationships that can be repurposed for treating infection and maintaining microbiome homeostasis.
Targeting QS systems in Enterobacteriaceae
E. coli encompasses genetically and functionally diverse species that range from benign commensal to professional pathogen. Commensal E. coli are usually present in the gut microbiota [14]. Like S. aureus, endogenous E. coli can become opportunistic pathogens with inappropriate access to extra-intestinal niches and cause diseases such as meningitis, sepsis and urinary tract infections [35–37]. The outgrowth of commensal E. coli within the gut has also been linked to chronic, immune-mediated diseases such as inflammatory bowel disease (IBD) [38]. In contrast, pathogenic E. coli reside in environmental reservoirs such as ruminants and cause human diarrheal diseases upon ingestion of contaminated food and water sources [39,92].
The QseC signaling cascade is a highly conserved QS response system in Gamma-Proteobacteria that consists of the histidine sensor kinase (HK) QseC and its cognate RR QseB [40] (Figure 3). QseC senses the bacterial signal autoinducer-3 (AI-3), which encompass a family of pirazynone molecules that are derived from products from threonine dehydrogenase (Tdh) combined with abortive tRNA synthase reactions. The AI-3 family is synthesized by a variety of bacterial species. Importantly the most active molecule in this AI-3 family is a new pyrazinonetype of metabolite, with very potent activity [94]. QseC and a second HK QseE also sense the host neurotransmitters epinephrine and norepinephrine [41,42], thus enabling E. coli to coordinate population-level behaviors in direct response to the host. QseC activates QseB, thus stimulating flagella biosynthesis [40]. The kinase activity of QseC is promiscuous and also activates the non-cognate RRs, KdpE and QseF, which substantially expands the QseC regulon [43] (Figure 3).
Figure 3.
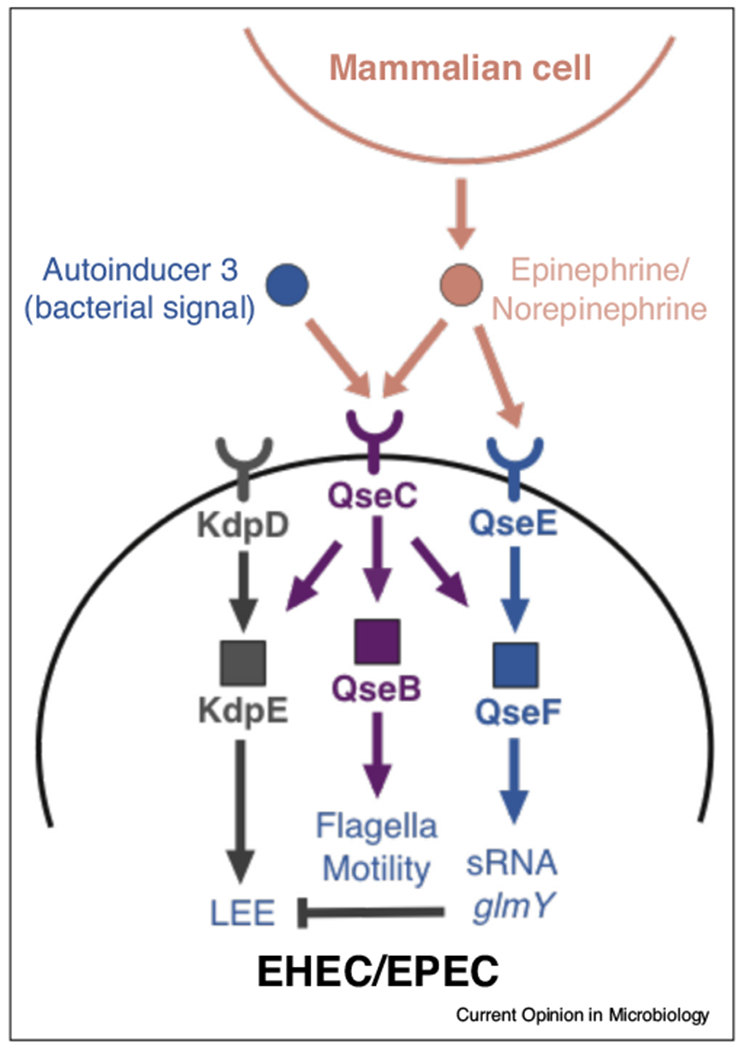
The QseC quorum sensing system in pathogenic Escherichia coli. A major quorum sensing response system in E. coli consists of a two-component system comprises the histidine sensor kinase (HK) QseC and its cognate response regulator (RR) QseB. The QseC receptor is autophosphorylated upon engagement with the QS signal autoinducer-3 and then transfers the phosphate to QseB, which transcriptionally stimulates flagella biosynthesis. QseC, along with a second HK QseE, also sense the host neurotransmitters epinephrine and norepinephrine. Kinase activity in QseC is promiscuous and can activate two additional non-cognate RRs, KdpE and QseF. In enterohemorrhagic and enteropathogenic E. coli (EHEC, EPEC), QseC also regulates the locus of enterocyte effacement (LEE), which is essential for causing intestinal disease. In EHEC and EPEC, QseC activates the LEE through KdpE, a positive regulator of the LEE, and inhibits the LEE through sRNAs that are modulated by QseF.
Genetic studies have demonstrated that QseC is an important activator of virulence in pathogenic E. coli and in the closely related murine pathogen Citrobacter rodentium [44–46,90]. In enterohemorrhagic and enteropathogenic E. coli, QseC regulates the locus of enterocyte effacement (LEE), a pathogenicity island that is essential for causing disease [44,45,90]. QseC activates the LEE through KdpE and QseF-regulated sRNAs [43,47] (Figure 3). Despite employing divergent mechanisms for causing disease, the QseC regulon also modulates virulence-associated functions in other E. coli pathovars such as UPEC and in pathogens such as Francisella tularensis and Salmonella enterica [48–52,90,91]. Indeed, the integration of QseC into the intracellular signaling cascades that modulate virulence is wired differently between bacterial strains [49,53–55,91]. Importantly, the QseC signaling cascade is exploited by many Gram-negative bacterial pathogens to promote virulence. QseC activates expression of a multi and varied array of virulence genes in these pathogens, such as expression of type three secretion systems (T3SS), LPS modification enzymes that promote resistance to stress and antimicrobial peptides, flagella and motility genes, promotion of biofilm formation, expression of multiple iron uptake systems, pili/fimbriae and adhesins, and toxins [49,51,53–55,91].
Consequently, pharmacological inhibition of QseC with LED209 is effective in attenuating pathogen virulence and disease in diverse models of acute infection [46,49,51,90], thus demonstrating the potential efficacy of QseC as an anti-virulence target in numerous pathogens. LED209 was identified through a screen of a small molecule chemical library. Extensive structure activity relationship (SAR) studies revealed that LED209 is a potent pro-drug that is highly selective for QseC. Its warhead allosterically modifies lysines that are only present in QseC, impairing its function, and preventing the activation of the virulence program of several Gram-negative pathogens both in vitro and during murine infection. LED209 does not interfere with pathogen growth, possibly leading to a milder evolutionary pressure toward drug resistance. LED209 has desirable pharmokinetics and does not present toxicity in vitro and in rodents [51,90].
However, given that QseC homologues are also expressed by gut commensals, including endogenous E. coli, studies investigating the effects of QseC inhibition on the human microbiome and on commensal-to-pathogen transitions are important next steps to assess the safety and specificity of this anti-virulence target in pre-clinical models of infection.
Targeting QS systems in chronic infection and disease
While anti-virulence strategies can be effective in models of acute infection, the implementation of such therapies to treat chronic infections may be challenging because of selective pressures continuously exerted by the host [60,61]. Whole genome sequencing has revealed that isolates recovered from chronic disease patients accumulate mutations that fundamentally alter the function of the pathogen [7,16,62–64]. Moreover, in chronic infections that are likely caused by commensal-turned-opportunists, infection-associated isolates harbor mutations that functionally distinguishes the pathogen from its putative commensal ancestor [16,17•,63].
Chronic infections
Chronic S. aureus infections are associated with loss-of-function agr mutations in a subset of patients, which correlates with poorer prognosis and increased mortality [16,62,63,65,66]. The AgrC receptor in chronic infection isolates frequently acquire mutations that increase the AgrC activation threshold or completely inactivates the receptor [17•,62,67••,68•]. These agr dysfunctional isolates generally exhibit decreased virulence potential in vitro and in animal infection models [17•,62,67••,68•]. However, one study reported that an agr dysfunctional bloodstream isolate retained its virulence in an intravenous infection model, which corresponded with the upregulation of agr-regulated virulence factors [17•]. In contrast, agr inactivation in a closely related S. aureus nasal strain from the same patient attenuated its virulence during bloodstream infection [17•]. Moreover, within a population of agr dysfunctional isolates, a subset of cells revert back to an agr+ phenotype through phase variation [69••]. Together, these studies suggest that chronic infection isolates can acquire mutations or functions that bypass the need for agr-dependent activation of its virulence programs, an outcome that could also arise with chronic administration of anti-agr agents.
Another common characteristic of agr dysfunctional mutants is the production of robust biofilms, a functional characteristic that can be recapitulated in isogenic agr mutants [62, 67••,70]. Agr dysfunctional mutants exhibit increased fitness in biofilm infection models compared to agr+ strains, which corresponds with the formation of impenetrable in vivo biofilms that protect the pathogen from leukocyte-mediated killing [67••]. Indeed, agr dysfunctional mutants are detected as early as one week following S. aureus biofilm infection, which is consistent with strong host pressures that select for agr dysfunction to avoid immune-mediated clearance [67••]. In contrast, agr+ isolates exhibit greater fitness and are more virulent during acute skin infections [67••]. Similarly, QS inactivating mutations have also been reported in P. aeruginosa clinical isolates associated with chronic disease [61,72–74]. However, there is a debate on whether these QS mutants in biofilms may be eventually selected out of the population. Taken together, these findings suggest that anti-virulence treatments are likely to be challenging in chronic infections because of the microevolutionary processes that result in loss-of-function mutations in pathogen QS systems. We still need to better understand the arms race between QS proficient and deficient strains within these environments. Another consideration is whether a multi-pronged approach targeting signaling cascades at several levels would work better than a specific target.
Chronic inflammatory diseases
Chronic inflammatory diseases are often associated with compositional changes to the human microbiome that contribute to disease pathogenesis — a state known as dysbiosis. Atopic dermatitis (AD), a chronic skin condition, is associated with dysbiosis of the skin microbiome, which is often characterized by the expansion of commensal S. aureus and a decrease in S. epidermidis [75,76]. The severity of AD corresponds with an elevated S. aureus to S. epidermidis ratio [77]. In a murine model of skin infection, co-administration of the skin commensal Staphylococcus hominis, or its purified AIP, with S. aureus infection attenuates agr activity, inflammation and lesion formation, which corresponds to decreased activation of host proteases that damage the epidermis to cause disease [31••]. Interestingly, the efficacy of S. hominis treatment was lost when co-administered with S. aureus at a 1:10 ratio, which is representative of the dysbiosis observed in AD patients [31••]. Thus, this study demonstrates the potential of utilizing commensal QS systems to treat chronic skin diseases and restore homeostasis within the skin microbiome.
Chronic gastrointestinal diseases such as IBD are also associated with a dysbiotic gut microbiota. In Crohn’s disease, dysbiosis is often characterized by the expansion of commensal Proteobacteria such as E. coli, which includes increased mucosal colonization of a functionally distinct subset known as adherent-invasive E. coli (AIEC) [78,79]. AIEC strains utilize type 1 pili to colonize the intestinal epithelium, which enables aberrant stimulation of mucosal inflammatory responses mediated by AIEC expression of flagellin [80–83,93]. Chemical inhibition (using LED209) of QseC, which stimulates flagella and type 1 pili biosynthesis, attenuates colitis induction by AIEC and inhibits Enterobacteriaceae outgrowth in several experimental models of IBD [40,84•]. Thus, targeting the QseC system in AIEC strains may help alleviate overstimulation of the mucosal immune system while also restoring homeostasis within the gut microbiota of IBD patients. However, it remains unknown whether QS components such as QseC in commensal E. coli are vulnerable to the same selective pressures as S. aureus or P. aeruginosa during chronic disease. A recent study reported the emergence of point mutations in qseC in the aminoacid residues S8R and I283L in Klebsiella pneumoniae that contributed to the development of polymixin resistance following repeated antibiotic exposure. However, the only mutations that inactivate QseC function, which are the direct targets of LED209 are in K256 and K427 [85,86]. Taken together, futures studies are clearly needed to assess the microevolution of QS systems in human commensals and opportunistic pathogens and to define the implications of these genetic events on bacterial function, host response and treating disease.
Concluding remarks and future considerations
Over the past decade, numerous studies have demonstrated the promising potential of anti-virulence agents in preclinical models of bacterial infections. However, the efficacy of targeting QS systems seems to be dependent on the type of bacterial infection (acute versus chronic) and the site of infection. Chronic disease can lead to the emergence of QS dysfunctional mutants and functionally heterogeneous pathogen populations that can render the administration of QS inhibitors challenging. Moreover, given the ubiquity of QS systems in commensals and the potential for crosstalk within the microbiota and between commensal and pathogen [87–89], further studies investigating the contribution of QS in modulating microbiome function and commensal-pathogen interactions are needed in order to evaluate the safety and long-term efficacy of these agents in preclinical models of human disease.
Acknowledgements
This study was supported by the NIH grants: AI053067, AI05135, AI077613, AI114511 to VS.
Footnotes
Conflict of interest statement
Nothing declared.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Khan SN, Khan AU: Breaking the spell: combating multidrug resistant’ superbugs’. Front Microbiol 2016, 7:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.U.S. Centers for Disease Control and Prevention: Antibiotic Resistance Threats in the United States, 2019. 2019. [no volume]. [Google Scholar]
- 3.Njoroge J, Sperandio V: Jamming bacterial communication: new approaches for the treatment of infectious diseases. EMBO Mol Med 2009, 1:201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.LaSarre B, Federle MJ: Exploiting quorum sensing to confuse bacterial pathogens. Microbiol Mol Biol Rev 2013, 77:73–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Welsh MA, Blackwell HE: Chemical probes of quorum sensing: from compound development to biological discovery. FEMS Microbiol Rev 2016, 40:774–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maura D, Ballok AE, Rahme LG: Considerations and caveats in anti-virulence drug development. Curr Opin Microbiol 2016, 33:41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scoffone VC, Trespidi G, Chiarelli LR, Barbieri G, Buroni S: Quorum sensing as antivirulence target in cystic fibrosis pathogens. Int J Mol Sci 2019, 20:1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahator SD, Zhang L: Small is mighty-chemical communication systems in Pseudomonas aeruginosa. Annu Rev Microbiol 2019, 73:559–578. [DOI] [PubMed] [Google Scholar]
- 9.Mukherjee S, Bassler BL: Bacterial quorum sensing in complex and dynamically changing environments. Nat Rev Microbiol 2019, 17:371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krismer B, Weidenmaier C, Zipperer A, Peschel A: The commensal lifestyle of Staphylococcus aureus and its interactions with the nasal microbiota. Nat Rev Microbiol 2017, 15:675–687. [DOI] [PubMed] [Google Scholar]
- 11.Kim E-K, Lee K-A, Hyeon DY, Kyung M, Jun K-Y, Seo SH, Hwang D, Kwon Y, Lee W-J: Bacterial nucleoside catabolism controls quorum sensing and commensal-to-pathogen transition in the drosophila gut. Cell Host Microbe 2020, 27:345– 357. [DOI] [PubMed] [Google Scholar]
- 12.LaSarre B, Federle MJ: Exploiting quorum sensing to confuse bacterial pathogens. Microbiol Mol Biol Rev 2013, 77:73–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu S, Liu J, Liu C, Yang A, Qiao J: Quorum sensing for population-level control of bacteria and potential therapeutic applications. Cell Mol Life Sci 2019, 19 849–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Human Microbiome Project Consortium: Structure, function and diversity of the healthy human microbiome. Nature 2012, 486:207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lakhundi S, Zhang K: Methicillin-resistant Staphylococcus aureus: molecular characterization, evolution, and epidemiology. Clin Microbiol Rev 2018, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young BC, Wu C- H, Gordon NC, Cole K, Price JR, Liu E, Sheppard AE, Perera S, Charlesworth J, Golubchik T et al. : Severe infections emerge from commensal bacteria by adaptive evolution. eLife 2017, 6:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altman DR, Sullivan MJ, Chacko KI, Balasubramanian D, Pak TR, Sause WE, Kumar K, Sebra R, Deikus G, Attie O et al. : Genome plasticity of agr-defective Staphylococcus aureus during clinical infection. Infect Immun 2018, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study identified an agr dysfunctional isolate from a S. aureus bloodstream infection that unexpectedly retained its virulence in a mouse model of intravenous infection. This corresponded with the in vivo upregulation of agr-regulated virulence factors. This finding suggests that chronic infection isolates can acquire mutations that bypass the need for agr-dependent activation of virulence programs in S. aureus, with important implications for anti-virulence therapies that target the agr system.
- 18.Ji G, Beavis RC, Novick RP: Cell density control of staphylococcal virulence mediated by an octapeptide pheromone. Proc Natl Acad Sci U S A 1995, 92:12055–12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jenul C, Horswill AR: Regulation of Staphylococcus aureus virulence. Microbiol Spectr 2018, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abdelnour A, Arvidson S, Bremell T, Rydé n C, Tarkowski A: The accessory gene regulator (agr) controls Staphylococcus aureus virulence in a murine arthritis model. Infect Immun 1993, 61:3879–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheung AL, Eberhardt KJ, Chung E, Yeaman MR, Sullam PM, Ramos M, Bayer AS: Diminished virulence of a sar−/agr− mutant of Staphylococcus aureus in the rabbit model of endocarditis. J Clin Invest 1994, 94:1815–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gillaspy AF, Hickmon SG, Skinner RA, Thomas JR, Nelson CL, Smeltzer MS: Role of the accessory gene regulator (agr) in pathogenesis of staphylococcal osteomyelitis. Infect Immun 1995, 63:3373–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wright JS, Jin R, Novick RP: Transient interference with staphylococcal quorum sensing blocks abscess formation. Proc Natl Acad Sci U S A 2005, 102:1691–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Novick RP: Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol Microbiol 2003, 48:1429–1449. [DOI] [PubMed] [Google Scholar]
- 25.Sully EK, Malachowa N, Elmore BO, Alexander SM, Femling JK, Gray BM, DeLeo FR, Otto M, Cheung AL, Edwards BS et al. : Selective chemical inhibition of agr quorum sensing in Staphylococcus aureus promotes host defense with minimal impact on resistance. PLoS Pathog 2014, 10:e1004174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le KY, Otto M: Quorum-sensing regulation in staphylococci-an overview. Front Microbiol 2015, 6:1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otto M, Echner H, Voelter W, Gotz F: Pheromone cross-inhibition between Staphylococcus aureus and Staphylococcus epidermidis. Infect Immun 2001, 69:1957–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Canovas J, Baldry M, Bojer MS, Andersen PS, Grzeskowiak PK, Stegger M, Damborg P, Olsen CA, Ingmer H: Cross-talk between Staphylococcus aureus and other Staphylococcal species via the agr quorum sensing system. Front Microbiol 2016, 7:1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paharik AE, Parlet CP, Chung N, Todd DA, Rodriguez EI, Van Dyke MJ, Cech NB, Horswill AR: Coagulase-negative staphylococcal strain prevents Staphylococcus aureus colonization and skin infection by blocking quorum sensing. Cell Host Microbe 2017, 22:746–756.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study demonstrates that AIP from the human skin commensal S. caprae inhibits the agr system in S. aureus. Moreover, co-administration of S. caprae and S. aureus attenuated disease in skin infection models, thus highlighting the potential for exploiting commensal QS systems as a strategy to treat S. aureus skin infections.
- 30.Peng P, Baldry M, Gless BH, Bojer MS, Espinosa-Gongora C, Baig SJ, Andersen PS, Olsen CA, Ingmer H: Effect of co-inhabiting coagulase negative staphylococci on S. aureus agr quorum sensing, host factor binding, and biofilm formation. Front Microbiol 2019, 10:2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williams MR, Costa SK, Zaramela LS, Khalil S, Todd DA, Winter HL, Sanford JA, O’Neill AM, Liggins MC, Nakatsuji T et al. : Quorum sensing between bacterial species on the skin protects against epidermal injury in atopic dermatitis. Sci Transl Med 2019, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study screened a collection of human skin commensals to search for strains that inhibit the S. aureus agr system while not directly affecting its growth. In a murine model of atopic dermatitis, co-administration of S. aureus with S. hominis — a hit from the screen — or its purified AIP attenuated inflammation, skin lesion formation and agr system activation in S. aureus. This study demonstrates the potential of utilizing commensal QS systems to treat chronic skin diseases and restore homeostasis within the skin microbiome.
- 32.Salam AM, Quave CL: Targeting virulence in Staphylococcus aureus by chemical inhibition of the accessory gene regulator system in vivo. mSphere 2018, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou Y, Niu C, Ma B, Xue X, Li Z, Chen Z, Li F, Zhou S, Luo X, Hou Z: Inhibiting PSMα-induced neutrophil necroptosis protects mice with MRSA pneumonia by blocking the agr system. Cell Death Dis 2018, 9:362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parlet CP, Kavanaugh JS, Crosby HA, Raja HA, El-Elimat T, Todd DA, Pearce CJ, Cech NB, Oberlies NH, Horswill AR: Apicidin attenuates MRSA virulence through quorum-sensing inhibition and enhanced host defense. Cell Rep 2019, 27:187–198.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen SL, Wu M, Henderson JP, Hooton TM, Hibbing ME, Hultgren SJ, Gordon JI: Genomic diversity and fitness of E. coli strains recovered from the intestinal and urinary tracts of women with recurrent urinary tract infection. Sci Transl Med 2013, 5:184ra. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thä nert R, Reske KA, Hink T, Wallace MA, Wang B, Schwartz DJ, Seiler S, Cass C, Burnham CA, Dubberke ER et al. : Comparative genomics of antibiotic-resistant uropathogens implicates three routes for recurrence of urinary tract infections. mBio 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sarowska J, Futoma-Koloch B, Jama-Kmiecik A, Frej-Madrzak M, Ksiazczyk M, Bugla-Ploskonska G, Choroszy-Krol I: Virulence factors, prevalence and potential transmission of extraintestinal pathogenic Escherichia coli isolated from different sources: recent reports. Gut Pathog 2019, 11:10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sartor RB, Wu GD: Roles for intestinal bacteria, viruses, and fungi in pathogenesis of inflammatory bowel diseases and therapeutic approaches. Gastroenterology 2017, 152:327–339.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen Y, Sperandio V: Enterohemorrhagic E. coli (EHEC) pathogenesis. Front Cell Infect Microbiol 2012, 2:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sperandio V, Torres AG, Kaper JB: Quorum sensing Escherichia coli regulators B and C (QseBC): a novel two-component regulatory system involved in the regulation of flagella and motility by quorum sensing in E. coli. Mol Microbiol 2002, 43:809–821. [DOI] [PubMed] [Google Scholar]
- 41.Clarke MB, Hughes DT, Zhu C, Boedeker EC, Sperandio V: The QseC sensor kinase: a bacterial adrenergic receptor. Proc Natl Acad Sci U S A 2006, 103:10420–10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reading NC, Rasko DA, Torres AG, Sperandio V: The two-component system QseEF and the membrane protein QseG link adrenergic and stress sensing to bacterial pathogenesis. Proc Natl Acad Sci U S A 2009, 106:5889–5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hughes DT, Clarke MB, Yamamoto K, Rasko DA, Sperandio V: The QseC adrenergic signaling cascade in enterohemorrhagic E. coli (EHEC). PLoS Pathog 2009, 5 e1000553–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Njoroge J, Sperandio V: Enterohemorrhagic Escherichia coli virulence regulation by two bacterial adrenergic kinases, QseC and QseE. Infect Immun 2012, 80:688–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moreira CG, Russell R, Mishra AA, Narayanan S, Ritchie JM, Waldor MK, Curtis MM, Winter SE, Weinshenker D, Sperandio V: Bacterial adrenergic sensors regulate virulence of enteric pathogens in the gut. mBio 2016, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Machado Ribeiro TR, Cardinali Lustri B, Elias WP, Moreira CG: QseC signaling in the outbreak O104:H4 Escherichia coli strain combines multiple factors during infection. J Bacteriol 2019, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gruber CC, Sperandio V: Posttranscriptional control of microbe-induced rearrangement of host cell actin. mBio 2014, 5 e01025–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kostakioti M, Hadjifrangiskou M, Pinkner JS, Hultgren SJ: QseC-mediated dephosphorylation of QseB is required for expression of genes associated with virulence in uropathogenic Escherichia coli. Mol Microbiol 2009, 73:1020– 1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moreira CG, Weinshenker D, Sperandio V: QseC mediates Salmonella enterica serovar typhimurium virulence in vitro and in vivo. Infect Immun 2010, 78:914–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kostakioti M, Hadjifrangiskou M, Cusumano CK, Hannan TJ, Janetka JW, Hultgren SJ: Distinguishing the contribution of type 1 pili from that of other QseB-misregulated factors when QseC is absent during urinary tract infection. Infect Immun 2012, 80:2826–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Curtis MM, Russell R, Moreira CG, Adebesin AM, Wang C, Williams NS, Taussig R, Stewart D, Zimmern P, Lu B et al. : QseC inhibitors as an antivirulence approach for Gram-negative pathogens. mBio 2014, 5:e02165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moreira CG, Sperandio V: Interplay between the QseC and QseE bacterial adrenergic sensor kinases in Salmonella enterica serovar typhimurium pathogenesis. Infect Immun 2012, 80:4344–4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guckes KR, Kostakioti M, Breland EJ, Gu AP, Shaffer CL, Martinez CR, Hultgren SJ, Hadjifrangiskou M: Strong crosssystem interactions drive the activation of the QseB response regulator in the absence of its cognate sensor. Proc Natl Acad Sci U S A 2013, 110:16592–16597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Breland EJ, Zhang EW, Bermudez T, Martinez CR, Hadjifrangiskou M: The histidine residue of QseC is required for canonical signaling between QseB and PmrB in uropathogenic Escherichia coli. J Bacteriol 2017, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guckes KR, Breland EJ, Zhang EW, Hanks SC, Gill NK, Algood HMS, Schmitz JE, Stratton CW, Hadjifrangiskou M: Signaling by two-component system noncognate partners promotes intrinsic tolerance to polymyxin B in uropathogenic Escherichia coli. Sci Signal 2017, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ji G, Beavis R, Novick RP: Bacterial interference caused by autoinducing peptide variants. Science 1997, 276:2027–2030. [DOI] [PubMed] [Google Scholar]
- 57.Olson ME, Todd DA, Schaeffer CR, Paharik AE, Van Dyke MJ, Bü ttner H, Dunman PM, Rohde H, Cech NB, Fey PD et al. : Staphylococcus epidermidis agr quorum-sensing system: signal identification, cross talk, and importance in colonization. J Bacteriol 2014, 196:3482–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramsey MM, Freire MO, Gabrilska RA, Rumbaugh KP, Lemon KP: Staphylococcus aureus shifts toward commensalism in response to corynebacterium species. Front Microbiol 2016, 7:1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hardy BL, Dickey SW, Plaut RD, Riggins DP, Stibitz S, Otto M, Merrell DS: Corynebacterium pseudodiphtheriticum exploits Staphylococcus aureus virulence components in a novel polymicrobial defense strategy. mBio 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study demonstrates that the human nasal commensal Corynebacterium pseudodiphtheriticum inhibits virulence-associated functions in S. aureus by blocking QS activation.
- 60.Lieberman TD, Flett KB, Yelin I, Martin TR, McAdam AJ, Priebe GP, Kishony R: Genetic variation of a bacterial pathogen within individuals with cystic fibrosis provides a record of selective pressures. Nat Genet 2014, 46:82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Markussen T, Marvig RL, Gómez-Lozano M, Aanæs K, Burleigh AE, Høiby N, Johansen HK, Molin S, Jelsbak L: Environmental heterogeneity drives within-host diversification and evolution of Pseudomonas aeruginosa. mBio 2014, 5 e01592–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Suligoy CM, Lattar SM, Noto Llana M, Gonzá lez CD, Alvarez LP, Robinson DA, Gó mez MI, Buzzola FR, Sordelli DO: Mutation of Agr is associated with the adaptation of Staphylococcus aureus to the host during chronic osteomyelitis. Front Cell Infect Microbiol 2018, 8:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Read TD, Petit RA, Yin Z, Montgomery T, McNulty MC, David MZ: USA300 Staphylococcus aureus persists on multiple body sites following an infection. BMC Microbiol 2018, 18:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dreux N, Denizot J, Martinez-Medina M, Mellmann A, Billig M, Kisiela D, Chattopadhyay S, Sokurenko E, Neut C, Gower-Rousseau C et al. : Point mutations in FimH adhesin of Crohn’s disease-associated adherent-invasive Escherichia coli enhance intestinal inflammatory response. PLoS Pathog 2013, 9:e1003141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Traber KE, Lee E, Benson S, Corrigan R, Cantera M, Shopsin B, Novick RP: agr function in clinical Staphylococcus aureus isolates. Microbiology 2008, 154:2265–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schweizer ML, Furuno JP, Sakoulas G, Johnson JK, Harris AD, Shardell MD, McGregor JC, Thom KA, Perencevich EN: Increased mortality with accessory gene regulator (agr) dysfunction in Staphylococcus aureus among bacteremic patients. Antimicrob Agents Chemother 2011, 55:1082–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.He L, Le KY, Khan BA, Nguyen TH, Hunt RL, Bae JS, Kabat J, Zheng Y, Cheung GYC, Li M et al. : Resistance to leukocytes ties benefits of quorum sensing dysfunctionality to biofilm infection. Nat Microbiol 2019, 4:1114–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study demonstrates that S. aureus agr dysfunctional mutants exhibit increased fitness in biofilm infection models compared to agr+ strains. This corresponded with the formation of impenetrable in vivo biofilms that protected the pathogen from leukocyte-mediated killing. Moreover, agr dysfunctional mutants are detected as early as one week following S. aureus biofilm infection, whereas no such mutants emerged following skin infection. This suggests that in certain types of S. aureus infections, strong host pressures select for agr dysfunction to the benefit of the pathogen.
- 68.Sloan TJ, Murray E, Yokoyama M, Massey RC, Chan WC, Bonev BB, Williams P: Timing is everything: impact of naturally occurring Staphylococcus aureus AgrC cytoplasmic domain adaptive mutations on autoinduction. J Bacteriol 2019, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study characterized the functional effects of specific genetic mutations that are present in agr dysfunctional clinical isolates.
- 69.Gor V, Takemura AJ, Nishitani M, Higashide M, Medrano Romero V, Ohniwa RL, Morikawa K: Finding of Agr phase variants in Staphylococcus aureus. mBio 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study demonstrates that within a population of S. aureus agr dysfunctional isolates, a subset of cells phenotypically and genotypically revert back to an agr+ phenotype. The rates of reversion vary between clinical isolates. This study introduces another challenge with targeting the agr system as an anti-virulence strategy — the fluctuating heterogeneity of agr functionality within a pathogen population.
- 70.Periasamy S, Joo H-S, Duong AC, Bach T-HL, Tan VY, Chatterjee SS, Cheung GYC, Otto M: How Staphylococcus aureus biofilms develop their characteristic structure. Proc Natl Acad Sci U S A 2012, 109:1281–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Urbano R, Karlinsey JE, Libby SJ, Doulias P-T, Ischiropoulos H, Warheit-Niemi HI, Liggitt DH, Horswill AR, Fang FC: Host nitric oxide disrupts microbial cell-to-cell communication to inhibit Staphylococcal virulence. Cell Host Microbe 2018, 23:594–606.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study demonstrates that host NO production results in the S-nitrosylation of AgrA and consequently disrupts AgrA activation of the agr system in S. aureus. Thus, the host innate immune system targets the agr system during acute infection with S. aureus to minimize disease. The study identified other putative targets for S-nitrosylation in other bacteria, which could likely expand the effects of host NO production on bacterial function.
- 72.Feltner JB, Wolter DJ, Pope CE, Groleau M- C, Smalley NE, Greenberg EP, Mayer-Hamblett N, Burns J, Dé ziel E, Hoffman LR et al.: LasR variant cystic fibrosis isolates reveal an adaptable quorum-sensing hierarchy in Pseudomonas aeruginosa. mBio 2016, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Y, Gao L, Rao X, Wang J, Yu H, Jiang J, Zhou W, Wang J, Xiao Y, Li M et al. : Characterization of lasR-deficient clinical isolates of Pseudomonas aeruginosa. Sci Rep 2018, 8 13344–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kostylev M, Kim DY, Smalley NE, Salukhe I, Greenberg EP, Dandekar AA: Evolution of the Pseudomonas aeruginosa quorum-sensing hierarchy. Proc Natl Acad Sci U S A 2019, 116:7027–7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Meylan P, Lang C, Mermoud S, Johannsen A, Norrenberg S, Hohl D, Vial Y, Prod’hom G, Greub G, Kypriotou M et al. : Skin colonization by Staphylococcus aureus precedes the clinical diagnosis of atopic dermatitis in infancy. J Invest Dermatol 2017, 137:2497–2504. [DOI] [PubMed] [Google Scholar]
- 76.Kennedy EA, Connolly J, Hourihane JO, Fallon PG, McLean WHI, Murray D, Jo J-H, Segre JA, Kong HH, Irvine AD: Skin microbiome before development of atopic dermatitis: early colonization with commensal staphylococci at 2 months is associated with a lower risk of atopic dermatitis at 1 year. J Allergy Clin Immunol 2017, 139:166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alsterholm M, Strö mbeck L, Ljung A, Karami N, Widjestam J, Gillstedt M, Åhren C, Faergemann J: Variation in Staphylococcus aureus colonization in relation to disease severity in adults with atopic dermatitis during a five-month follow-up. Acta Derm Venereol 2017, 97:802–807. [DOI] [PubMed] [Google Scholar]
- 78.Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser A-L, Barnich N, Bringer M-A, Swidsinski A, Beaugerie L, Colombel J-F: High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 2004, 127:412–421. [DOI] [PubMed] [Google Scholar]
- 79.Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M et al. : The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15:382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Boudeau J, Glasser AL, Masseret E, Joly B, Darfeuille-Michaud A: Invasive ability of an Escherichia coli strain isolated from the ileal mucosa of a patient with Crohn’s disease. Infect Immun 1999, 67:4499–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Boudeau J, Barnich N, Darfeuille-Michaud A: Type 1 pili-mediated adherence of Escherichia coli strain LF82 isolated from Crohn’s disease is involved in bacterial invasion of intestinal epithelial cells. Mol Microbiol 2001, 39:1272–1284. [DOI] [PubMed] [Google Scholar]
- 82.Barnich N, Carvalho FA, Glasser A-L, Darcha C, Jantscheff P, Allez M, Peeters H, Bommelaer G, Desreumaux P, Colombel J-F et al. : CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J Clin Invest 2007, 117:1566–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chassaing B, Koren O, Carvalho FA, Ley RE, Gewirtz AT: AIEC pathobiont instigates chronic colitis in susceptible hosts by altering microbiota composition. Gut 2014, 63:1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rooks MG, Veiga P, Reeves AZ, Lavoie S, Yasuda K, Asano Y, Yoshihara K, Michaud M, Wardwell-Scott L, Gallini CA et al. : QseC inhibition as an antivirulence approach for colitis-associated bacteria. Proc Natl Acad Sci U S A 2017, 114:142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study demonstrates that targeting QS systems in pathobionts associated with Crohn’s disease can attenuate colitis and limit dysbiosis that is characterized by increased abundance of E. coli in several mouse models of IBD.
- 85.Parker CT, Russell R, Njoroge JW, Jimenez AG, Taussig R, Sperandio V: Genetic and mechanistic analyses of the periplasmic domain of the enterohemorrhagic Escherichia coli QseC histidine sensor kinase. J Bacteriol 2017, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pitt ME, Cao MD, Butler MS, Ramu S, Ganesamoorthy D, Blaskovich MAT, Coin LJM, Cooper MA: Octapeptin C4 and polymyxin resistance occur via distinct pathways in an epidemic XDR Klebsiella pneumoniae ST258 isolate. J Antimicrob Chemother 2019, 74:582–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wellington S, Greenberg EP: Quorum sensing signal selectivity and the potential for interspecies cross talk. mBio 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hsiao A, Ahmed AMS, Subramanian S, Griffin NW, Drewry LL, Petri WA, Haque R, Ahmed T, Gordon JI: Members of the human gut microbiota involved in recovery from Vibrio cholerae infection. Nature 2014, 515:423–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Thompson JA, Oliveira RA, Djukovic A, Ubeda C, Xavier KB: Manipulation of the quorum sensing signal AI-2 affects the antibiotic-treated gut microbiota. Cell Rep 2015, 10:1861–1871. [DOI] [PubMed] [Google Scholar]
- 90.Rasko D, Moreira CG, Li DR, Reading NC, Ritchie JM, Waldor MK, Williams N, Taussig R, Wei S, Roth M et al. : Targeting QseC signaling and virulence for antibiotic development. Science 2008, 321:1078–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bearson BL, Bearson SMD: The role of the QseC quorum-sensing sensor kinase in colonization and norepinephrine-enhanced motility in Salmonella enterica serovar Typhimurium. Microb Pathog 2008, 44:271–278. [DOI] [PubMed] [Google Scholar]
- 92.Kaper JB, Nataro JP, Mobley HL: Pathogenic Escherichia coli. Nat Rev Microbiol 2004, 2:123–140. [DOI] [PubMed] [Google Scholar]
- 93.Carvalho FA, Barnich N, Sauvanet P, Darcha C, Gelot A, Darfeuille-Michaud A: Crohn’s disease-associated Escherichia coli LF82 aggravates colitis in injured mouse colon via signaling by flagellin. Inflamm Bowel Dis 2008, 14:1051–1060. [DOI] [PubMed] [Google Scholar]
- 94.Kim CS, Gatsios A, Cuesta S, Lam YC, Wei Z, Chen H, Russell RM, Shine EE, Wang R, Wyche TP et al. : Characterization of autoinducer-3structure and biosynthesis in E coli. ACS Cent Sci 2020, 6:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]