

Abstract
The idiosyncratic characteristics and severity of acetaminophen (APAP) overdose-induced hepatotoxicity render identifying the predisposing factors and mechanisms of APAP-induced liver toxicity necessary and urgent. Farnesoid X receptor (FXR) controls bile acid homeostasis and modulates the progression of various liver diseases. Although global FXR deficiency in mice enhances APAP intoxication, the mechanism remains elusive. In this study, an increased sensitivity to APAP-induced toxicity was found in global Fxr-null (Fxr−/−) mice, but was not observed in hepatocyte-specific or macrophage-specific Fxr-null mice, suggesting that global FXR deficiency enhances APAP hepatotoxicity via disruption of systematic bile acid homeostasis. Indeed, more bile acid accumulation was found in global Fxr−/− mice, while 2% cholestyramine diet feeding decreased serum bile acids and alleviated APAP hepatotoxicity in global Fxr−/− mice, suggesting that bile acid accumulation contributes to APAP toxicity. Bile acids were suspected to induce macrophage to release tumor necrosis factor-α (TNF-α), which is known to enhance the APAP hepatotoxicity. In vitro, deoxycholic acid (DCA), a secondary bile acid metabolite, significantly induced Tnfa mRNA and dose-dependently enhanced TNF-α release from macrophage, while the same dose of DCA did not directly potentiate APAP toxicity in cultured primary hepatocytes. In vivo, DCA enhanced TNF-α release and potentiated APAP toxicity, both of which were abolished by the specific TNF-α antagonist infliximab. These results reveal an FXR-DCA-TNF-α axis that potentiates APAP hepatotoxicity, which could guide the clinical safe use of APAP.
Keywords: drug-induced liver injury, acute liver failure, acetaminophen, FXR, DCA
Acetaminophen (APAP), also known as paracetamol, is a widely used antipyretic and analgesic in clinic. However, APAP overdosing can cause acute liver failure, responsible for nearly 500 deaths in the United States alone, and remains the leading cause of drug-induced liver injury in several countries (Lee, 2017). Many risk factors, such as gender (Masubuchi et al., 2011), genetic diversity, metabolic disorders (Kučera et al., 2012), viral hepatitis infection (Maddox et al., 2010), and chronic alcohol use (Sato et al., 1981), could affect the propensity of APAP to cause acute liver failure. Identifying the predisposing factors and mechanisms is necessary to understand the pathological progression of APAP hepatotoxicity and for guiding the rational safe use of APAP.
APAP, when metabolized mainly by CYP2E1, produces the reactive metabolite N-acetyl-p-benzoquinone imine, which then causes depletion of hepatic glutathione (GSH) and stimulates macrophage activation to release proinflammatory mediators (Lancaster et al., 2015; Martin-Murphy et al., 2010). Once GSH is depleted, hepatocyte death is initiated, which is further enhanced by tumor necrosis factor-α (TNF-α), particularly under conditions of GSH depletion (Colell et al., 1998; Matsumaru et al., 2003; Yan et al., 2016). Thus, any factors that affect APAP metabolic activation, chemoprevention or TNF-α release could influence the hepatic sensitivity to APAP toxicity. In the APAP-induced liver injury model, farnesoid X receptor (FXR) activation by its agonist 3-(2,6-dichlorophenyl)-4-(3’-carboxy-2-chlorostilben-4-yl) oxymethyl-5-isopropylisoxazole (GW4064) alleviated APAP-induced liver injury, while global Fxr-null mice (Fxr−/−) showed enhanced liver injury (Lee et al., 2010); this effect was suggested to be through inducing expression of xenobiotic metabolism-related genes in wild-type (WT) mice, but not Fxr−/− mice, leading to an increased metabolism of toxic APAP derivatives and reduced GSH levels (Lee et al., 2010). More recently, the FXR agonist GW4064 was found to alleviate APAP-induced liver injury by promoting cell proliferation via pyruvate dehydrogenate kinase 4-controlled metabolic reprogramming (Xie et al., 2016). Although FXR activation is known to ameliorate APAP-induced hepatotoxicity, the mechanism for how FXR deficiency potentiates APAP hepatotoxicity remains largely unknown.
FXR is highly expressed in liver and intestine as the primary regulator of bile acid homeostasis. FXR deficiency causes cholestasis in mice and FXR expression is correlated with cholestasis in humans (Eloranta and Kullak-Ublick, 2008; Sinal et al., 2000). Prospective studies indicate that bile acid level is a prognostic biomarker in APAP-induced acute liver failure in patients (Woolbright et al., 2014). Accumulated toxic bile acids potentiate hepatotoxicity in various liver injury models or metabolic diseases by causing hepatocellular stress and inducing cell death (Gumpricht et al., 2005; Sokol et al., 1995, 1998; Woolbright et al., 2015; Yerushalmi et al., 2001), or activating the macrophage via inducing NLR family, pyrin domain containing 3 (NLRP3) inflammasome activation as inflammatory damage-associated molecular patterns DAMPs (Hao et al., 2017). NLRP3 inflammasome activation was suggested to result in enhanced metainflammation and TNF-α release (Samstad et al., 2014; Yan et al., 2018). However, little is known whether bile acids as potential DAMPs could induce the release of TNF-α, which is known to potentiate hepatocyte death in GSH-depleted hepatocytes when mice are challenged by APAP or other hepatic chemical toxicants (Colell et al., 1998; Matsumaru et al., 2003; Yan et al., 2016). Thus, the question arises whether FXR deficiency enhances APAP hepatotoxicity by increasing bile acid accumulation that in turn stimulates macrophage to release TNF-α, which then increases APAP hepatotoxicity. To explore the role and mechanism of how FXR influences APAP toxicity, global Fxr−/− mice, hepatocyte-specific Fxr-null (FxrΔHep) mice, and macrophage-specific Fxr-null (FxrΔMac) mice, in combination with liquid chromotagraphy-mass spectrometry (LC-MS) analyses of bile acid levels and bile acid-treated macrophage cell cultures were used to investigate whether any liver cell specific-FXR mediated APAP hepatotoxicity and whether FXR deficiency enhanced APAP toxicity via modulating bile acid-induced TNF-α release from macrophage.
MATERIALS AND METHODS
Chemicals and reagents
APAP, GW4064, lipopolysaccharides from Escherichia coli O111: B4, cholestyramine, cholic acid, α-muricholic acid, β-muricholic acid, deoxycholic acid (DCA), ursodeoxycholic acid, hyodeoxycholic acid, lithocholic acid, taurocholic acid (TCA), tauro-α-muricholic acid, tauro-β-muricholic acid, taurodeoxycholic acid, taurochenodeoxycholic acid, tauroursodeoxycholic acid, and taurohyodeoxycholic acid were purchased from Sigma (St Louis, Missouri). Infliximab (INF) was obtained from Zhongda hospital (Nanjing, China). Cell counting kit-8 kit was purchased from Dojindo Molecular Technologies (Rockville, Maryland). Cholestyramine diet (2%) and its matched control chow diet were made in Research Diets (New Brunswick, New Jersey). Deionized water was purified using a Milli-Q system (Millipore, Billerica, Massachusetts). Primers were ordered from Integrated DNA Technologies (Coralville, Iowa,).
Animals and treatments
Fxr −/− mice and strain-matched WT mice, FxrΔHep and Fxr-floxed (Fxrfl/fl) mice on a C57BL/6N background as described previously (Kim et al., 2007). FxrΔMac mice were produced by crossing Fxrfl/fl mice with LysM-Cre on the C57BL/6N background. The LysM-Cre transgenic line was previously described (Brocker et al., 2017). FxrΔHep, FxrΔMac, Fxrfl/fl, Fxr−/−, and matched WT mice were maintained at the National Cancer Institute (Bethesda, Maryland, USA) and allowed free access to food and water until experimental use. The animal vivarium was maintained at 25°C ± 2°C with a 12/12 h light/dark cycle and 50% ± 10% humidity. Six- to eight-week-old, body weight-matched male Fxr−/−, FxrΔHep, FxrΔMac, and Fxrfl/fl mice, were dosed with APAP at 200 mg/kg via IP injection and then killed 12 or 24 h after treatment as indicated. For the GW4064/APAP animal experiment, 6- to 8-week-old age and body weight-matched male C57BL/6N mice were used. The mice were intraperitoneally injected with GW4064 at 30 mg/kg for 5 consecutive days and 30 min after GW4064 dosing, the overnight-fasted mice were intraperitoneally dosed with 300 mg/kg of APAP and refed 2 h after APAP dosing until the end of the experiment. All mice were killed 24 h after APAP dosing. All experiments were performed at the National Cancer Institute with mouse handling in accordance with animal study protocols approved by the National Cancer Institute Animal Care and Use Committee.
For the APAP/DCA dosing experiment, 6- to 8-week-old male C57BL/6 mice were obtained from Shanghai SLAC Laboratory Animal Center (Shanghai, People’s Republic of China). The mice were dosed with APAP at 150 mg/kg via IP injection and DCA at 30 mg/kg dissolved in saline via IP injection at 2 h after APAP dosing. Before IP injection of APAP, the mice were fasted overnight (14 h) with free access to water and refed 2 h after APAP dosing. All mice were killed 24 h after APAP administration. For the INF/APAP/DCA experiment, mice were pretreated with 6 mg/kg INF once daily via IP injection for 3 consecutive days and then treated with 200 mg/kg of APAP 30 min after the last dose of INF, followed by 30 mg/kg of DCA 2 h after APAP treatment. All mice were killed 24 h after APAP treatment. The selection of the doses and duration of APAP, DCA, or INF exposure were based on previous studies (Ferah et al., 2013; Hao et al., 2017; Lee et al., 2010; Saini et al., 2011; Yan et al., 2016; Zhao et al., 2016). All experiments were approved by the animal ethics committee of China Pharmaceutical University and performed in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health.
Serum aminotransferase analysis
Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were quantified using commercial assay kits (Catachem, Bridgeport, Connecticut) or a standard clinical automatic analyzer as described previously (Yan et al., 2016). In brief, serum samples were diluted 10 times using saline and then subjected to analyses.
Bile acid analysis by LC-MS
Bile acids were extracted from mouse serum and analyzed by LC-MS as described previously (Xie et al., 2019). In brief, an aliquot of 25 µl serum was deproteinated with 100 µl of acetonitrile containing 1 µM d5-UDCA as internal standard. After centrifugation for 15 min at 15 000×g, 100 µl of supernatant was further diluted with 100 µl of water containing 0.1% formic acid. A 5 µl aliquot of the supernatants was injected into an Acquity ultrahigh-performance LC/Synapt G2Si quadrupole time-of-flight mass spectrometer (Waters Corporation, Milford, Massachusetts) equipped with an Acquity BEH C18 column (100×2.1mm internal diameter, 1.7mm, Waters Corp.) for chromatographic separation.
Gene expression analysis
Total tissue mRNA was extracted from frozen liver samples using TRIzol reagent (Thermo-Fisher, Waltham, Massachusetts) with a Percellys bead homogenizer (Bertin, Rockville, Maryland) using 1 mm zirconia/silica beads. Total RNA was quantified using a NanoDrop spectrophotometer (NanoDrop Products, Wilmington, Delaware) and 1 µg of RNA was reverse transcribed with cDNA synthesis Super Mix (Quanta Biosciences, Beverly, Massachusetts), followed by a quantitative real-time polymerase chain reaction analysis using an Applied Biosystems QuantStudio 7 Flex Real-time PCR System (Thermo-Fisher). Values calculated using the 2ΔCt method were normalized to Actb mRNA and expressed as fold change versus control group (Primers were listed in Supplementary Table 1).
Histology analysis
Formalin-fixed liver tissues were embedded in paraffin and 5-µm thick sections were cut for hematoxylin and eosin (H&E) staining in the Department of Pathophysiology at the Affiliated Hospital of Nanjing University of Chinese Medicine (Nanjing, Jiangsu, China, Figs. 7 and 8) and at Histoserv Inc. (Germantown, Maryland; for other Figures).
Figure 7.

Deoxycholic acid (DCA) potentiates APAP hepatotoxicity via enhancing tumor necrosis factor-α (TNF-α) release. A, Time scheme of experiment procedure. B–D, Serum alanine aminotransferase, aspartate aminotransferase, and TNF-α levels in DCA/APAP-treated mice. E, Hematoxylin and eosin staining analysis of liver sections DCA/APAP-treated mice; scale bar 100 µm. Data are presented as means ± SEM (n = 6 mice per group). *p < .05, **p < .01, and ***p < .005, compared with acetaminophen (APAP) group or DCA group as indicated. Control, control saline-treated mice; DCA, 30 mg/kg of DCA-treated mice; APAP, 150 mg/kg of APAP-treated mice; APAP+DCA, mice treated with 150 mg/kg of APAP and then treated with 30 mg/kg of DCA.
Enzyme-linked immunosorbent assay
TNF-α enzyme-linked immunosorbent assay kits for mouse TNF-α were purchased from Excel (Shanghai, People’s Republic of China). Fifty microliters of serum was used for analysis.
Cell culture experiments
Raw 264.7 cells were purchased from ATCC (Manassas, Virginia) and cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fatal bovine serum and 1% antibiotics (0.5% penicillin and 0.5% streptomycin) for no more than 10 generations as described previously (Hao et al., 2017). The tested bile acids were used at a concentration of 50–200 μM for the indicated time for cell viability assays in 96-well plates or TNF-α assays in 24-well plates. TNF-α release was determined using the supernatant of DCA-treated RAW264.7 cells by enzyme-linked immunosorbent assays. Human nontumor hepatic LO2 cells were purchased from the Chinese Academy of Sciences (Shanghai, China) and cultured in RPMI 1640 medium with 10% fatal bovine serum and 1% antibiotics (0.5% penicillin and 0.5% streptomycin). APAP was dissolved in culture medium at a dose of 10 mM for 24 h, followed by a cell viability test using the Cell Counting Kit-8 kit per the manual protocol. The doses and duration of APAP or DCA exposure were based on previous studies (Hao et al., 2017; Yan et al., 2016; Zhao et al., 2016).
Kupffer cell isolation
Kupffer cells were isolated as described previously (Matsuda et al., 2018). In brief, mouse hepatic nonparenchymal cells were obtained by the collagenase perfusion method as described (Yagai et al., 2014). Kupffer cells were sorted from the nonparenchymal cells by autoMACS Pro (Miltenyi Biotec, Auburn, California) using fluorescein isothiocyanate antimouse F4/80 antibody (Catalog No. 123108, Biolegend, San Diego, California; RRID: AB_893502) and anti-fluorescein isothiocyanate microbeads (Catalog No. 130-048-701, Miltenyi Biotec; RRID: AB_244371). The sorting was repeated to increase the purity of Kupffer cells. The harvested cells were then subjected to mRNA extraction using TRIzol reagent (Thermo-Fisher).
Statistical analysis
Experimental values are presented as mean ± SEM. Statistical analysis was performed using Prism version 7.05 (GraphPad Software, San Diego, California). Two-tailed t test was used between 2 groups, whereas one-way analysis of variance followed by the Dunnett multiple-comparisons test was used among multiple groups. p Values < .05 were considered statistically significant.
RESULTS
APAP-Induced Hepatotoxicity Is Enhanced in Fxr−/− Mice and Alleviated in GW4064-Treated Mice
To determine whether FXR mediates APAP-induced liver injury in vivo, the specific FXR agonist GW4064 and global Fxr−/− mice were used. Fxr−/− mice and strain-matched C57BL/6N WT mice were subjected to treatment with 200 mg/kg APAP. Biochemical analysis of serum ALT and AST levels demonstrated that APAP significantly increased serum ALT and AST levels in Fxr−/− mice compared with WT mice (Figs. 1A and 1B). H&E staining of livers showed that APAP-treated Fxr−/− mice had significant cellular damage as compared with the similarly treated WT mice (Figure 1C). In non-APAP treated control mice, serum ALT and AST levels were slightly increased in global Fxr−/− mice, and no obvious histological difference was noted between the two genotypes (Supplementary Figs. 1A–C).
Figure 1.

GW4064 protects, while farnesoid X receptor (FXR) deficiency enhances, acetaminophen (APAP) -induced liver injury in mice. A and B, Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST)levels at 3 or 12 h after APAP dosing in global Fxr−/− mice and wild-type (WT) mice. C, Liver hematoxylin and eosin (H&E) staining for WT mice and Fxr−/− mice at 12 h after APAP treatment, scale bar 100 µm. D, Serum ALT and AST levels. E, tumor necrosis factor (TNF)-α levels in control or GW4064-treated mice. F, Liver H&E staining for GW4064/APAP-treated mice at 24 h after APAP dosing, scale bar 50 µm (n = 5 mice per group). Data are presented as means ± SEM. *p < .05 and **p < .01 compared with WT group or GW4064-treated group. ###p < .005 compared with Control group. WT mice treated with 200 mg/kg of APAP. Fxr−/−, Fxr-null mice treated with 200 mg/kg of APAP. Vehicle + APAP, APAP-dosed mice pretreated with vehicle; GW4064, APAP-dosed mice pretreated with GW4064; Control, control saline-treated mice.
Consistent with this finding, FXR activation by a 5-day pretreatment with GW4064 sharply decreased the APAP-induced increase of serum ALT and AST levels, and decreased APAP-induced serum TNF-α release (Figs. 1D and 1E). H&E staining further demonstrated that GW4064 treatment also decreased liver necrosis in APAP-treated mice (Figure 1F). These data demonstrated that global FXR deficiency potentiated APAP toxicity, while FXR activation by GW4064 decreased APAP hepatotoxicity, together revealing that FXR signaling restricts APAP-induced liver injury.
Loss of Hepatocyte FXR Does Not Enhance APAP Toxicity
The mechanism by which hepatocyte FXR influences APAP-induced liver injury was investigated. In WT mice, APAP decreased expression of Fxr mRNA as well as FXR target genes (Supplementary Figs. 1D–F). Consistent with the in vivo data, APAP treatment at 5 and 10 mM markedly decreased Fxr mRNA in primary hepatocytes (Supplementary Figure 1G). Thus, APAP challenge inhibited expression of Fxr mRNA and FXR target genes, which may be due to increased stress from APAP-induced inflammation.
FXR is highly expressed in liver, particularly hepatocytes, while FXR is known to directly act as a hepatocyte protector since hepatic FXR inhibits endoplasmic reticulum stress-induced NLRP3 inflammasome activation (Han et al., 2018), suppresses TNF-α-induced apoptosis in hepatocytes, and ameliorates liver injury (Wang et al., 2018a). Thus, hepatocyte FXR may directly modulate APAP intoxication. To examine whether hepatocyte FXR could directly influence APAP intoxication, FxrΔHep mice and Fxrfl/fl were subjected to APAP treatment. No significant difference was found between Fxrfl/fl and FxrΔHep mice, as revealed by serum ALT and AST levels and H&E staining in livers of APAP-treated mice (Figs. 2A–C); this experiment was repeated three times using both age- and body weight-matched mice, which all yielded the same results that hepatocyte FXR deficiency did not significantly alter the sensitivity of mice to APAP-induced toxicity. Similarly, no significant difference was found between untreated Fxrfl/fl and FxrΔHep mice (Figs. 2D–F). Thus, hepatocyte Fxr disruption itself did not further enhance APAP toxicity.
Figure 2.

Hepatocyte farnesoid X receptor (FXR) does not further potentiate acetaminophen (APAP) toxicity. A and B, serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels of APAP-treated FxrΔHep mice and Fxrfl/fl mice. C, Representative hematoxylin and eosin (H&E) staining of APAP-dosed FxrΔHep mice and its matched Fxrfl/fl mice, scale bar 100 µm. D and E, serum ALT and AST levels of untreated control FxrΔHep mice and Fxrfl/fl mice. F, Representative H&E staining of untreated control FxrΔHep mice and Fxrfl/fl mice, scale bar 100 µm (n = 5 mice per group). Data are presented as means ± SEM.
Loss of Macrophage FXR Does Not Enhance APAP-Induced Toxicity
Since FXR in macrophage was found to directly regulate NLRP3 inflammasome activation (Hao et al., 2017), a role for macrophage FXR was suspected to influence the APAP-induced hepatic toxicity. To examine the direct contribution of macrophage FXR in modulation of APAP-induced hepatotoxicity, FxrΔMac mice were generated (Figs. 3A and 3B). FxrΔMac and age- and body weight-matched Fxrfl/fl littermates were challenged with 200 mg/kg APAP. Mice were killed at 6 or 24 h after APAP dosing. However, no significant difference in extent of toxicity was found between the two genotypes upon APAP treatment at either 6 or 24 h, as revealed by serum ALT and AST levels (Figs. 3C and 3D) and liver H&E staining (Figure 3E). No significant difference in serum ALT, AST levels and liver H&E staining were found between FxrΔMac and Fxrfl/fl mice in the absence of APAP challenge (Figs. 3F–H). These data demonstrate that macrophage-specific FXR deficiency does not enhance APAP toxicity.
Figure 3.

Loss of macrophage farnesoid X receptor (FXR) expression does not further enhance acetaminophen (APAP)-induced toxicity. A, Breeding scheme of FxrΔMac mice. B, mRNA levels of Kupffer cell Fxr in FxrΔMac and Fxrfl/fl mice. C, serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels at 6 h after APAP dosing in FxrΔMac and Fxrfl/fl mice. D, serum ALT and AST levels at 24 h after APAP dosing in FxrΔMac and Fxrfl/fl mice. E, hematoxylin and eosin (H&E) staining of liver sections from APAP-dosed FxrΔMac and Fxrfl/fl mice; scale bar 100 µm. F and G, serum ALT and AST levels of untreated control FxrΔMac and Fxrfl/fl mice. H, H&E staining of liver sections from untreated control FxrΔMac and Fxrfl/fl mice; scale bar 50 µm (n = 4 for Figure 3B, whereas n = 5 mice per group for Figs. 3C–H). Data are presented as means ± SEM. ***p < .005 compared with the Fxrfl/fl group.
Global FXR Deficiency Enhances and GW4064 Decreases APAP-Induced Bile Acid Accumulation
Neither loss of macrophage FXR nor hepatocyte FXR mimicked the phenotype of global FXR deficiency after APAP dosing, suggesting that FXR protein expression itself in liver cells may not provide direct chemoprevention. In previous studies, global FXR deficiency was shown to cause cholestasis that potentiated carbon tetrachloride-induced liver injury by increasing accumulated TCA levels (Takahashi et al., 2017) and to potentiate endotoxin-induced shock by increasing the accumulation of proinflammatory bile acids (Hao et al., 2017). Thus, global FXR deficiency was suspected to cause more aberrant accumulation of bile acids, among which, some toxic bile acids could act as circulated DAMPs to enhance APAP toxicity. To test this hypothesis, serum bile acid levels were measured but were not changed in APAP-treated FxrΔMac and Fxrfl/fl mice (Supplementary Figure 2). Given that hepatocyte FXR is known to modulate bile acid signaling, serum bile acid levels were measured in global Fxr-null mice and FxrΔHep mice revealing that bile acids were increased in FxrΔHep mice compared with Fxrfl/fl mice, while levels of serum bile acids in APAP-dosed Fxr−/− mice accumulated to a much greater extent than that in FxrΔHep mice (Figs. 4A and 4B). Similarly, bile acid levels were also examined in untreated control mice. Global FXR deficiency caused significantly more accumulation of several bile acid metabolites when compared with hepatocyte-specific FXR deficiency, even under basal conditions (Supplementary Figs. 3A and 3B). GW4064 markedly alleviated almost all the taurine-conjugated bile acids and decreased the APAP-induced increase of DCA, a nonconjugated secondary bile acid in serum of APAP-treated mice (Supplementary Figs. 4A and 4B). These data suggest that increased bile acids may contribute to APAP hepatotoxicity.
Figure 4.
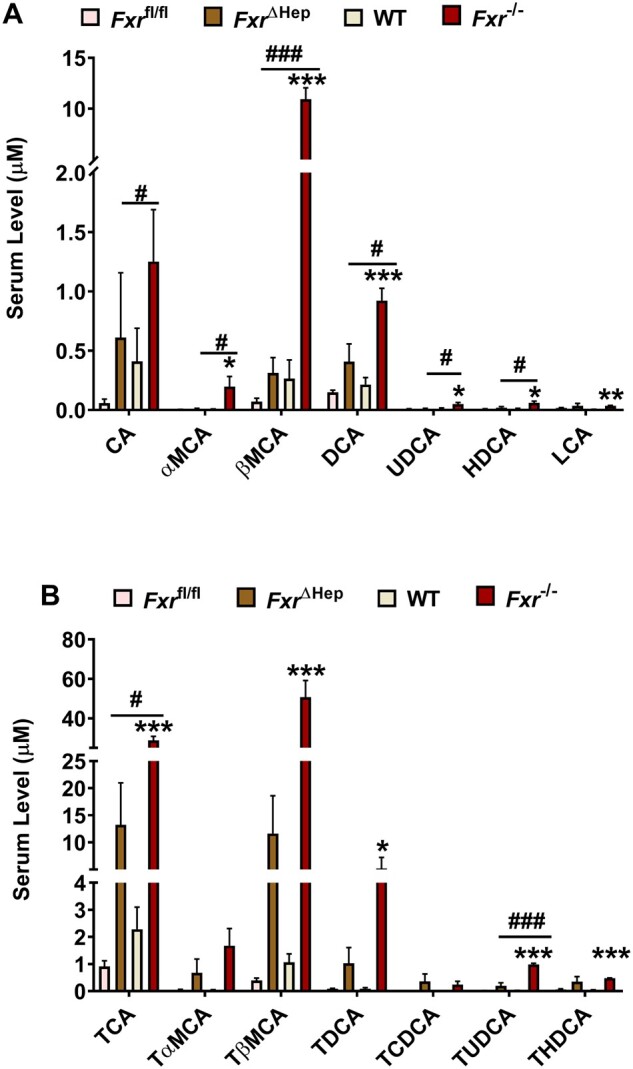
Bile acids accumulates more significantly in global farnesoid X receptor (Fxr)-null mice than FxrΔHep mice after acetaminophen (APAP) dosing. A, serum free bile acids at 24 h after APAP dosing. B, Serum conjugated bile acids at 24 h after APAP dosing (n = 5 mice per group). Data are presented as means ± SEM. *p < .05, **p < .01, and ***p < .005 compared with wild-type group. #p < .05, ##p < .01, and ###p < .005 compared with the FxrΔHep group. Abbreviations: CA, cholic acid; α-MCA, α-muricholic acid; βMCA, β-muricholic acid; DCA, deoxycholic acid; UDCA, ursodeoxycholic acid; HDCA, hyodeoxycholic acid; LCA, lithocholic acid; TCA, taurocholic acid; TαMCA, tauro-α-muricholic acid; TβMCA, tauro-β-muricholic acid; TDCA, taurodeoxycholic acid; TCDCA, taurochenodeoxycholic acid; TUDCA, tauroursodeoxycholic acid; THDCA, taurohyodeoxycholic acid.
Cholestyramine Feeding Rescues APAP Toxicity in Fxr−/− Mice
To determine whether accumulation of bile acids could enhance APAP toxicity in global Fxr−/− mice, the bile acid chelator cholestyramine was employed. Fxr−/− mice were fed a 2% cholestyramine diet or control chow diet for 1 week and then treated with APAP at 200 mg/kg for 24 h (Figure 5A). Cholestyramine diet feeding significantly alleviated APAP-induced serum ALT levels and histological damage (Figs. 5B and 5C), accompanied by decreased serum bile acid levels (Figs. 5D and 5E). These data suggest that bile acid accumulation in APAP-dosed Fxr−/− mice enhanced hepatic toxicity.
Figure 5.

Cholestyramine feeding rescues enhanced acetaminophen toxicity in global Fxr-null mice. A, time scheme of experiment procedure. B, serum alanine aminotransferase levels. C, Hematoxylin and eosin staining analysis, scale bar 100 µm. D, quantitation of serum free bile acids level. E, quantitation of serum conjugated bile acid levels (n = 8 mice per group). Data are presented as means ± SEM. *p < .05, **p < .01, and ***p < .005 compared with chow group. Full names of abbreviated bile acids were same as Figure 4.
DCA Markedly Induces TNF-α Release in Macrophage
Since bile acids accumulate in global Fxr−/− mice and cholestyramine feeding rescued APAP toxicity in these mice, bile acids may influence the sensitivity differences to APAP toxicity between Fxr−/− mice and FxrΔHep mice. Bile acids are known to induce NLRP3 inflammasome activation as proinflammatory DAMPs (Hao et al., 2017), and NLRP3 inflammasome activation was suggested to result in enhanced metainflammation and TNF-α release (Samstad et al., 2014; Yan et al., 2018). When FXR activation by GW4064 decreased TNF-α release in APAP-treated mice (Figure 1E), whether and which bile acid induces macrophage to release TNF-α to potentiate APAP toxicity was investigated. In Raw264.7 cells, 100 µM DCA, among all tested bile acid metabolites, was found to significantly increase Tnfa mRNA levels 12 h after each compound treatment, with lipopolysaccharides used as a positive control (Figs. 6A and 6B). To further validate this result, a dose-dependency experiment was performed with DCA at 0, 20, 50, and 100 µM, and DCA was found to induce TNF-α release dose-dependently in Raw264.7 cells (Figure 6C). A time-course analyses of Tnfa mRNA levels or TNF-α release were performed with 100 µM of DCA, and DCA was further found to markedly activate TNF-α signaling at both 1, 2, 4, and 8 h, as revealed by a marked induction of Tnfa mRNA levels that peaked at 1 h (Figure 6D), and increased TNF-α release that peaked at 8 h (Figure 6E). The increased TNF-α release was not due to induced cell death (Figs. 6F and 6G). Furthermore, the same doses (50–200 uM) of DCA did not directly potentiate APAP-induced hepatocyte damage in LO2 cells (Figure 6H). These data suggest that DCA among the bile acids has the potential to synergize with APAP to cause hepatocyte damage via inducing macrophage to release TNF-α.
Figure 6.

Deoxycholic acid (DCA) induces tumor necrosis factor-α (TNF-α) release in macrophage. A and B, Tnfa mRNA levels induced by free bile acids and taurine-conjugated bile acids in macrophage. C, Dose-dependency of DCA-induced TNF-α protein release at 20, 50, and 100 µM in macrophage. D, Time course of Tnfa mRNA levels after treating with 100 µM of DCA for 1, 2, 4, and 8 h. E, Time course of DCA (100 µM)-induced TNF-α release at 1, 2, 4, and 8 h in macrophage. G and H, Cell viabilities treated with free bile acids and taurine-conjugated bile acids in macrophage. I, DCA at 50–200 µM per se did not directly potentiate acetaminophen toxicity in LO2 cells (n = 6 per group). Data are presented as means ± SEM. #p < .05, ##p < .01, and ###p < .005 compared with the control group. Abbreviation: LPS, lipopolysaccharides. Full name of abbreviated bile acids were same as Figure 4.
DCA Potentiates APAP Hepatotoxicity by Increasing TNF-α Release
To further test whether DCA synergizes with APAP to cause hepatotoxicity by increasing TNF-α release, overnight-fasted C57BL/6N mice were treated with control vehicle or 30 mg/kg DCA 2 h after APAP and then killed to collect blood and livers for further analysis 24 h after APAP administration (Figure 7A). The results demonstrated that 30 mg/kg of DCA significantly potentiated APAP-induced toxicity, as revealed by significant increases of serum ALT and AST levels in APAP/DCA-treated mice as compared with mice treated with DCA alone or APAP alone (Figs. 7B–D). Consistent with the current hypothesis, serum TNF-α release was significantly increased in APAP/DCA-treated mice (Figure 7D). In addition, H&E staining showed data consistent with serum ALT and AST levels (Figure 7E). These results suggest that increased levels of DCA after APAP dosing potentiated APAP toxicity.
To further examine whether increased TNF-α release in DCA/APAP-treated mice contributed to APAP hepatotoxicity, a TNF-α-specific antagonist INF was used. Mice were pretreated with INF for 3 days and a single IP injection of 150 mg/kg APAP followed 2 h later by a single IP injection of 30 mg/kg DCA (Figure 8A). Mice pretreated with INF had much less serum TNF-α release (Figure 8B), markedly decreased serum ALT levels and AST levels (Figs. 8C and 8D), as well as improved liver damage as histologically assessed (Figure 8E), supporting the view that INF efficiently alleviated TNF-α release and associated liver injury. These data support the notion that DCA, when dosing after APAP challenge, potentiated APAP hepatotoxicity by stimulating TNF-α release, which could be a possible explanation why mice are more sensitive to APAP toxicity when FXR is inhibited (Figure 8F).
Figure 8.

Tumor necrosis factor-α (TNF-α) antagonist infliximab (INF) decreases serum TNF-α levels and alleviated deoxycholic acid (DCA)/acetaminophen (APAP)-induced liver injury. A, Time scheme of experiment procedure. B, Serum TNF-α levels. C, Serum alanine aminotransferase levels. D, Serum aspartate aminotransferase levels. E, Hepatic hematoxylin and eosin staining; 100 times scale bar 100 µm. F, Graphic abstract of proposed mechanism (n = 6 mice per group). Data are presented as means ± SEM. Control, control saline-treated mice; APAP+DCA, mice pretreated with saline for three consecutive days, adminstered with APAP + DCA; INF, INF only treated group; INF + APAP/DCA, mice pretreated with INF for three consecutive days, and then treated with APAP+DCA. *p < .05, **p < .01, and ***p < .005, compared with Saline + A/D group. #p < .05, ##p < .01, and ###p < .005 compared with control group.
DISCUSSION
Although global FXR deficiency was documented to sensitize mice to APAP hepatotoxicity, the mechanism and which cell type-specific FXR mediates APAP intoxication. This study demonstrated that loss of global FXR signaling potentiated APAP-induced liver injury; neither hepatocyte-specific nor macrophage-specific Fxr disruption had a significant impact on APAP toxicity. Global Fxr disruption showed enhanced accumulation of circulating serum bile acids, among which DCA was identified to stimulate macrophage to release TNF-α that in turn could synergize with APAP to induce liver injury. The DCA-potentiated APAP hepatotoxicity and TNF-α release was further abolished by the specific TNF-α antagonist INF. These findings reveal an FXR-DCA-TNF-α axis that modulates APAP hepatotoxicity (Figure 8F).
Hepatocyte FXR directly protects hepatocytes against inflammation (Wang et al., 2008) and cell death (Han et al., 2018; Wang et al., 2018a), whereas macrophage FXR modulates NLRP3 inflammasome activation (Hao et al., 2017). However, in this study, neither hepatocyte-specific Fxr nor macrophage-specific Fxr disruption was found to directly influence APAP hepatotoxicity, suggesting that cell-specific FXR could play distinct roles under different conditions. Similar with the present findings, hepatocyte-specific Fxr disruption had no role in modulating the severity of alcoholic liver disease, whereas global loss of FXR significantly enhanced the alcoholic liver disease (Zhang et al., 2018). Fxr−/− mice developed spontaneous liver tumors, while neither hepatocyte-specific Fxr−/− mice nor intestine-specific Fxr−/− mice developed spontaneous liver tumors during aging (Takahashi et al., 2018). In addition, serum bile acid accumulation in global Fxr−/− mice was more significant than that of FxrΔHep mice in APAP-treated mice in the current study. In line with this point, FXR was reported to regulate carbon tetrachloride-induced liver injury and liver tumor development by negatively modulating levels of circulating TCA, which could induce JNK-stress signaling in this model and upregulate MYC expression in Fxr-deficient hepatocytes (Takahashi et al., 2017, 2018). These studies suggest an important pathophysiological role for FXR-bile acids axis in preventing liver injury. In addition, although the FXR-DCA-TNF-α axis was demonstrated to at least partially to mediate APAP intoxication in this study, it remains a possibility that FXR signaling in extrahepatic tissues also plays a coordinated role with liver FXR. Deficiency of fibroblast growth factor 15, encoded by an FXR target gene in intestine, significantly increases susceptibility to APAP-induced liver injury, although to a lesser extent (Huang et al., 2018). Thus, it is reasonable to infer that neither any cell type-specific FXR in liver nor extrahepatic tissues alone, but combined FXR expression from two or more cell types together with the accumulation of bile acids, coordinate to modulate the extent of APAP toxicity.
Global FXR disruption results in an increase in the primary bile acid cholic acid, which could then lead to increased production of almost all bile acids, including the proposed biomarker DCA that could potentiate TNF-α release from macrophage. In previous studies, DCA was found to induce NLRP3 inflammasome activation and potentiate colitis (Zhao et al., 2016) and sepsis (Hao et al., 2017). These data and earlier studies together support the view that DCA could act as one proinflammatory DAMPs to mediate diseases progression.
Increased accumulation of DCA in the global Fxr−/− mice could be due to enhanced production of cholic acid, the precursor of DCA, as a result of increased expression of CYP7A1 that is elevated under FXR deficiency (Eloranta and Kullak-Ublick, 2008; Wang et al., 2018b). The mechanism by which DCA accumulates in global Fxr−/− mice compared with FxrΔHep mice remains undetermined. One possible explanation is that extrahepatic FXR, such as intestinal FXR, plays a coordinated role with hepatocyte FXR to regulate the bile acid homeostasis, particularly for the secondary bile acid DCA, which is produced in the intestine by gut microbiota (Matsubara et al., 2013; Wahlstrom et al., 2016). Indeed, an earlier study demonstrated that regulation of bile acid homeostasis is dependent on liver and intestine FXR signaling and DCA is significantly increased in feces in intestine-specific Fxr-null mice, but not from FxrΔHep mice or Fxr−/− mice (Kim et al., 2007).
In summary, while Fxr−/− mice are more sensitive to APAP toxicity, neither FxrΔHep nor FxrΔMac mice alone show enhanced APAP-induced hepatic toxicity. DCA elevation under global FXR deficiency induces macrophage to release TNF-α that potentiates APAP toxicity. Thus, combined FXR deficiency and accumulation of DCA predict more risk for APAP hepatotoxicity, providing new insights into crosstalk among FXR-mediated metabolic homeostasis, macrophage activation and liver injury. FXR signaling is frequently disrupted in various diseases (Kemper, 2011), such as sepsis (Hao et al., 2017), alcoholic liver injury (Wu et al., 2014), fibrosis or inflammation (Wang et al., 2018a), obesity (Kim et al., 2015), and colorectal cancer (Bayerdorffer et al., 1993), which could be accompanied by cholestasis and DCA accumulation (Bayerdorffer et al., 1993; Hao et al., 2017). In addition, the elderly population also have particularly high DCA levels (van der Werf et al., 1981). The current finding suggests that any group with decreased FXR signaling and elevated DCA needs to be carefully monitored under APAP use.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
AUTHOR CONTRIBUTIONS
Conceptualization: Ti.Y., H.H., G.W., and F.G.; Project management: Ti.Y., H.H., G.W., and F.G.; data acquisition and analyses: Ti.Y., N.Y., H.H., H.W., To.Y., Y.L., S.T., M.Z., and K.W.; Article writing, review, and edit: Ti.Y., F.G., H.H., and S.T.
Supplementary Material
ACKNOWLEDGMENTS
We thank Linda G. Byrd for preparation and submission of the animal protocols. We thank Grace L. Guo, Fei Li, and Cen Xie for expert discussion.
FUNDING
National Cancer Institute Intramural Research Program, Center of Cancer Research, National Institutes of Health; National Natural Science Foundation of China (grants 81720108032, 81930109, 82073928, and 82073926); Overseas Expertise Introduction Project for Discipline Innovation (grant G20582017001); “Double-First Class” Initiative Project (grants CPU2018GF09 and CPU2018GF01); State Key Laboratory of Natural Medicines at China Pharmaceutical University (grants SKLNMZZ202020 and SKLNMZZ202001); Leading technology foundation research project of Jiangsu province (grant BK20192005); the Major State Basic Research Development Program of China (2017YFA0205400); and Sanming Project of Medicine in Shenzhen (SZSM201801060). N.Y. was supported by Natural Science Foundation of China grant (81530098); and the World Discovery Scholarship supported by China Pharmaceutical University.
DECLARATION OF CONFLICTING INTERESTS
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
REFERENCES
- Bayerdorffer E., Mannes G. A., Richter W. O., Ochsenkuhn T., Wiebecke B., Kopcke W., Paumgartner G. (1993). Increased serum deoxycholic acid levels in men with colorectal adenomas. Gastroenterology 104, 145–151. [DOI] [PubMed] [Google Scholar]
- Brocker C. N., Yue J., Kim D., Qu A., Bonzo J. A., Gonzalez F. J. (2017). Hepatocyte-specific PPARA expression exclusively promotes agonist-induced cell proliferation without influence from nonparenchymal cells. Am. J. Physiol. Gastrointest. 312, G283–G299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colell A., Garcia-Ruiz C., Miranda M., Ardite E., Mari M., Morales A., Corrales F., Kaplowitz N., Fernandez-Checa J. C. (1998). Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology 115, 1541–1551. [DOI] [PubMed] [Google Scholar]
- Eloranta J. J., Kullak-Ublick G. A. (2008). The role of FXR in disorders of bile acid homeostasis. Physiology 23, 286–295. [DOI] [PubMed] [Google Scholar]
- Ferah I., Halici Z., Bayir Y., Demirci E., Unal B., Cadirci E. (2013). The role of infliximab on paracetamol-induced hepatotoxicity in rats. Immunopharmacol. Immunotoxicol. 35, 373–381. [DOI] [PubMed] [Google Scholar]
- Gumpricht E., Dahl R., Devereaux M. W., Sokol R. J. (2005). Licorice compounds glycyrrhizin and 18 beta-glycyrrhetinic acid are potent modulators of bile acid-induced cytotoxicity in rat hepatocytes. J. Biol. Chem. 280, 10556–10563. [DOI] [PubMed] [Google Scholar]
- Han C. Y., Rho H. S., Kim A., Kim T. H., Jang K., Jun D. W., Kim J. W., Kim B., Kim S. G. (2018). FXR inhibits endoplasmic reticulum stress-induced NLRP3 inflammasome in hepatocytes and ameliorates liver injury. Cell Rep. 24, 2985–2999. [DOI] [PubMed] [Google Scholar]
- Hao H., Cao L., Jiang C., Che Y., Zhang S., Takahashi S., Wang G., Gonzalez F. J. (2017). Farnesoid X receptor regulation of the nlrp3 inflammasome underlies cholestasis-associated sepsis. Cell Metab. 25, 856–867.e855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M., Williams J., Kong B., Zhu Y., Li G., Zhu Z., Guo G. L. (2018). Fibroblast growth factor 15 deficiency increases susceptibility but does not improve repair to acetaminophen-induced liver injury in mice. Dig. Liver Dis. 50, 175–180. [DOI] [PubMed] [Google Scholar]
- Kemper J. K. (2011). Regulation of FXR transcriptional activity in health and disease: Emerging roles of FXR cofactors and post-translational modifications. Biochim. Biophys. Acta 1812, 842–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. H., Xiao Z., Kwon S., Sun X., Ryerson D., Tkac D., Ma P., Wu S. Y., Chiang C. M., Zhou E., et al. (2015). A dysregulated acetyl/sumo switch of FXR promotes hepatic inflammation in obesity. EMBO J. 34, 184–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I., Ahn S. H., Inagaki T., Choi M., Ito S., Guo G. L., Kliewer S. A., Gonzalez F. J. (2007). Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J. Lipid Res. 48, 2664–2672. [DOI] [PubMed] [Google Scholar]
- Kučera O., Roušar T., Staňková P., Haňáčková L., Lotková H., Podhola M., Cervinková Z. (2012). Susceptibility of rat non-alcoholic fatty liver to the acute toxic effect of acetaminophen. J. Gastroenterol. Hepatol. 27, 323–330. [DOI] [PubMed] [Google Scholar]
- Lancaster E. M., Hiatt J. R., Zarrinpar A. (2015). Acetaminophen hepatotoxicity: An updated review. Arch. Toxicol. 89, 193–199. [DOI] [PubMed] [Google Scholar]
- Lee W. M. (2017). Acetaminophen (APAP) hepatotoxicity-isn’t it time for APAP to go away? J. Hepatol. 67, 1324–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee F. Y., de Aguiar Vallim T. Q., Chong H. K., Zhang Y., Liu Y., Jones S. A., Osborne T. F., Edwards P. A. (2010). Activation of the farnesoid X receptor provides protection against acetaminophen-induced hepatic toxicity. Mol. Endocrinol. 24, 1626–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddox J. F., Amuzie C. J., Li M., Newport S. W., Sparkenbaugh E., Cuff C. F., Pestka J. J., Cantor G. H., Roth R. A., Ganey P. E. (2010). Bacterial- and viral-induced inflammation increases sensitivity to acetaminophen hepatotoxicity. J. Toxicol. Environ. Health 73, 58–73. [DOI] [PubMed] [Google Scholar]
- Martin-Murphy B. V., Holt M. P., Ju C. (2010). The role of damage associated molecular pattern molecules in acetaminophen-induced liver injury in mice. Toxicol. Lett. 192, 387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masubuchi Y., Nakayama J., Watanabe Y. (2011). Sex difference in susceptibility to acetaminophen hepatotoxicity is reversed by buthionine sulfoximine. Toxicology 287, 54–60. [DOI] [PubMed] [Google Scholar]
- Matsubara T., Li F., Gonzalez F. J. (2013). Fxr signaling in the enterohepatic system. Mol. Cell Endocrinol. 368, 17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda M., Tsurusaki S., Miyata N., Saijou E., Okochi H., Miyajima A., Tanaka M. (2018). Oncostatin m causes liver fibrosis by regulating cooperation between hepatic stellate cells and macrophages in mice. Hepatology 67, 296–312. [DOI] [PubMed] [Google Scholar]
- Matsumaru K., Ji C., Kaplowitz N. (2003). Mechanisms for sensitization to TNF-induced apoptosis by acute glutathione depletion in murine hepatocytes. Hepatology 37, 1425–1434. [DOI] [PubMed] [Google Scholar]
- Saini S. P. S., Zhang B., Niu Y., Jiang M., Gao J., Zhai Y., Hoon Lee J., Uppal H., Tian H., Tortorici M. A., et al. (2011). Activation of liver X receptor increases acetaminophen clearance and prevents its toxicity in mice. Hepatology 54, 2208–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samstad E. O., Niyonzima N., Nymo S., Aune M. H., Ryan L., Bakke S. S., Lappegard K. T., Brekke O. L., Lambris J. D., Damas J. K., et al. (2014). Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J. Immunol. 192, 2837–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato C., Matsuda Y., Lieber C. S. (1981). Increased hepatotoxicity of acetaminophen after chronic ethanol consumption in the rat. Gastroenterology 80, 140–148. [PubMed] [Google Scholar]
- Sinal C. J., Tohkin M., Miyata M., Ward J. M., Lambert G., Gonzalez F. J. (2000). Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102, 731–744. [DOI] [PubMed] [Google Scholar]
- Sokol R. J., McKim J. M. Jr, Goff M. C., Ruyle S. Z., Devereaux M. W., Han D., Packer L., Everson G. (1998). Vitamin E reduces oxidant injury to mitochondria and the hepatotoxicity of taurochenodeoxycholic acid in the rat. Gastroenterology 114, 164–174. [DOI] [PubMed] [Google Scholar]
- Sokol R. J., Winklhofer-Roob B. M., Devereaux M. W., McKim J. M. Jr. (1995). Generation of hydroperoxides in isolated rat hepatocytes and hepatic mitochondria exposed to hydrophobic bile acids. Gastroenterology 109, 1249–1256. [DOI] [PubMed] [Google Scholar]
- Takahashi S., Tanaka N., Fukami T., Xie C., Yagai T., Kim D., Velenosi T. J., Yan T., Krausz K. W., Levi M., et al. (2018). Role of farnesoid x receptor and bile acids in hepatic tumor development. Hepatol. Commun. 2, 1567–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S., Tanaka N., Golla S., Fukami T., Krausz K. W., Polunas M. A., Weig B. C., Masuo Y., Xie C., Jiang C. T., et al. (2017). Farnesoid X receptor protects against low-dose carbon tetrachloride-induced liver injury through the taurocholate-JNK pathway. Toxicol. Sci. 158, 334–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Werf S. D., Huijbregts A. W., Lamers H. L., van Berge Henegouwen G. P., van Tongeren J. H. (1981). Age dependent differences in human bile acid metabolism and 7 alpha-dehydroxylation. Eur. J. Clin. Invest. 11, 425–431. [DOI] [PubMed] [Google Scholar]
- Wahlstrom A., Sayin S. I., Marschall H. U., Backhed F. (2016). Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 24, 41–50. [DOI] [PubMed] [Google Scholar]
- Wang H., Ge C., Zhou J., Guo Y., Cui S., Huang N., Yan T., Cao L., Che Y., Zheng Q., et al. (2018a). Noncanonical farnesoid X receptor signaling inhibits apoptosis and impedes liver fibrosis. EBioMedicine 37, 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., He Q., Wang G., Xu X., Hao H. (2018b). Fxr modulators for enterohepatic and metabolic diseases. Expert Opin. Ther. Pat. 28, 765–782. [DOI] [PubMed] [Google Scholar]
- Wang Y. D., Chen W. D., Wang M., Yu D., Forman B. M., Huang W. (2008). Farnesoid X receptor antagonizes nuclear factor κb in hepatic inflammatory response. Hepatology 48, 1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolbright B. L., Dorko K., Antoine D. J., Clarke J. I., Gholami P., Li F., Kumer S. C., Schmitt T. M., Forster J., Fan F., et al. (2015). Bile acid-induced necrosis in primary human hepatocytes and in patients with obstructive cholestasis. Toxicol. Appl. Pharmacol. 283, 168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolbright B. L., McGill M. R., Staggs V. S., Winefield R. D., Gholami P., Olyaee M., Sharpe M. R., Curry S. C., Lee W. M., Jaeschke H.; Acute Liver Failure Study Group. (2014). Glycodeoxycholic acid levels as prognostic biomarker in acetaminophen-induced acute liver failure patients. Toxicol. Sci. 142, 436–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W., Zhu B., Peng X., Zhou M., Jia D., Gu J. (2014). Activation of farnesoid X receptor attenuates hepatic injury in a murine model of alcoholic liver disease. Biochem. Biophys. Res. Commun. 443, 68–73. [DOI] [PubMed] [Google Scholar]
- Xie C., Takahashi S., Brocker C. N., He S., Chen L., Xie G., Jang K., Gao X., Krausz K. W., Qu A., et al. (2019). Hepatocyte peroxisome proliferator-activated receptor alpha regulates bile acid synthesis and transport. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 1864, 1396–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y., Wang H., Cheng X., Wu Y., Cao L., Wu M., Xie W., Wang G., Hao H. (2016). Farnesoid X receptor activation promotes cell proliferation via PDK4-controlled metabolic reprogramming. Sci. Rep. 6, 18751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagai T., Miyajima A., Tanaka M. (2014). Semaphorin 3E secreted by damaged hepatocytes regulates the sinusoidal regeneration and liver fibrosis during liver regeneration. Am. J. Pathol. 184, 2250–2259. [DOI] [PubMed] [Google Scholar]
- Yan T., Wang H., Cao L., Wang Q., Takahashi S., Yagai T., Li G., Krausz K. W., Wang G., Gonzalez F. J., et al. (2018). Glycyrrhizin alleviates nonalcoholic steatohepatitis via modulating bile acids and meta-inflammation. Drug Metab. Dispos. 46, 1310–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan T., Wang H., Zhao M., Yagai T., Chai Y., Krausz K. W., Xie C., Cheng X., Zhang J., Che Y., et al. (2016). Glycyrrhizin protects against acetaminophen-induced acute liver injury via alleviating tumor necrosis factor alpha-mediated apoptosis. Drug Metab. Dispos. 44, 720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yerushalmi B., Dahl R., Devereaux M. W., Gumpricht E., Sokol R. J. (2001). Bile acid-induced rat hepatocyte apoptosis is inhibited by antioxidants and blockers of the mitochondrial permeability transition. Hepatology 33, 616–626. [DOI] [PubMed] [Google Scholar]
- Zhang M., Kong B., Huang M., Wan R., Armstrong L. E., Schumacher J. D., Rizzolo D., Chow M. D., Lee Y. H., Guo G. L. (2018). FXR deletion in hepatocytes does not affect the severity of alcoholic liver disease in mice. Dig. Liver Dis. 50, 1068–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S., Gong Z., Zhou J., Tian C., Gao Y., Xu C., Chen Y., Cai W., Wu J. (2016). Deoxycholic acid triggers NLRP3 inflammasome activation and aggravates DSS-induced colitis in mice. Front. Immunol. 7, 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.