

Fetal BPA exposure, even below safe regulatory limits, disrupts the developing hypothalamus and affects behavior and activity.
Abstract
Critical physiological processes such as sleep and stress that underscore health are regulated by an intimate interplay between the endocrine and nervous systems. Here, we asked how fetal exposure to the endocrine disruptor found in common plastics, bisphenol A (BPA), causes lasting effects on adult animal behaviors. Adult mice exposed to low-dose BPA during gestation displayed notable disruption in circadian activity, social interactions, and associated neural hyperactivity, with some phenotypes maintained transgenerationally. Gestational BPA exposure increased vasopressin+ neurons in the suprachiasmatic nucleus (SCN), the region that regulates circadian rhythms, of F1 and F3 generations. Mechanistically, BPA increased proliferation of hypothalamic neural progenitors ex vivo and caused precocious neurogenesis in vivo. Co-antagonism of both estrogen and androgen receptors was necessary to block BPA’s effects on hypothalamic neural progenitors, illustrating a dual role for these endocrine targets. Together, gestational BPA exposure affects development of circadian centers, with lasting consequences across generations.
INTRODUCTION
Bisphenol A (BPA) is a common plasticizer and controversial endocrine-disrupting chemical that has affinity for all estrogen receptors (ERs) (1–3), as well as androgen (AR), thyroid (TH), and perhaps glucocorticoid receptors (4–7). In humans, BPA is detected in maternal and fetal serum (8, 9) and readily transfers to offspring across the placenta and via breast milk (8, 10). Gestational and early-life BPA exposure correlates with a variety of neurodevelopmental disorders, including attention deficit hyperactivity disorder (ADHD), autism spectrum disorder (ASD), depression, and anxiety (11–13), conditions that often copresent with sleep disorders (14, 15). However, the mechanistic underpinnings linking early-life BPA and behavioral outcomes later in life has lagged behind these findings (10).
BPA is deemed safe at environmental levels by various governmental bodies, including the U.S. Environmental Protection Agency (16), Health Canada (17), and the European Chemicals Agency (18). Historically, the reference dose (RfD) for BPA, considered the acceptable daily human intake, was 50 μg/kg body weight (BW) per day, based on a thousand-fold dilution of the lowest observable adverse effect level (LOAEL) (19). In 2008, Health Canada revised its recommended total daily intake (TDI) to 25 μg/kg BW and maintained this level in an updated guideline in 2012, concluding that “dietary exposure to BPA through food packaging uses is not expected to pose a health risk to the general population, including newborns and young children” (17). Most recently, the European Food and Safety Agency published a scientific opinion that recommended a temporary TDI (t-TDI) of only 4 μg/kg BW while simultaneously reconfirming “that there is no health concern for BPA at the estimated [current] levels of exposure” (20). A “no observable adverse effect level,” the standard measure normally used to determine the RfD, has yet to be calculated for BPA since adverse effects are observed at even the lowest tested doses (19). Animal studies have documented a variety of effects at doses below the LOAEL and/or below the RfD level (21–23).
Gestational BPA exposure in animal models demonstrates that the hypothalamus is a particularly susceptible brain region (10, 24), one that is also responsible for the regulation of neuroendocrine interactions and a variety of behaviors including feeding, social associations, and circadian rhythms (25, 26). The hypothalamus consists of 11 nuclei populated with hypothalamic neurons and glia that arise from neural stem and progenitor cells (NSPCs, also called radial glial cells) that line the third ventricle (25). These NSPCs divide at the third ventricle and differentiated daughter cells then migrate outward to populate the developing hypothalamic parenchyma (25, 26). The hypothalamic suprachiasmatic nucleus (SCN) uniquely consists of the pacemaker cells that rhythmically express clock genes to regulate daily cycles (27). Structurally, the SCN is located in the ventral hypothalamus with two bilateral nuclei surrounding the third ventricle that are classically divided into two subdomains—the core and the shell—that display differential gene expression to underscore their unique roles (28, 29). The core contains the retinal-responsive neurons that express vasoactive intestinal peptide (VIP), as well as calbindin, calretinin, and gastrin-releasing peptide, and it rhythmically oscillates the expression of the clock genes that drives daily rhythms (27). In contrast, the shell primarily expresses arginine vasopressin (AVP) as well as angiotensin II and met-enkephalin to a lesser extent (28, 30, 31), and it predominantly regulates rhythmic signaling to other regions of the brain and body using the input it receives from the core. The shell is also responsible for synchronizing the SCN master clock with peripheral clocks in other brain regions and throughout the body (31). Developmental disruption to either the core (e.g., altering pacemaker performance) or the shell (e.g., compromising the linkages to peripheral pacemakers around the body) disrupts the daily rhythmicity of a variety of bodily functions, most obviously activity and sleep, but also impedes other aspects of body chemistry such as blood pressure, serum cortisol levels, and a variety of complex neurological and physiological processes such as mental focus and feeding, among many others (27).
Given that gestational BPA exposure can have lasting effects on behaviors that are comorbid with sleep disruptions, we asked whether fetal BPA exposure affects SCN development and function. We determined that vasopressin neurons in the SCN are particularly susceptible to gestational BPA exposure and showed that increased hypothalamic neurogenesis had lasting effects on circadian activity, neural hyperactivity, and social interactions that persisted across multiple generations. We further characterized hypothalamic NSPC behavior following both ex vivo and in vivo BPA exposures and determined that BPA caused increased proliferation and decreased stemness. Moreover, we characterized BPA molecular targets and revealed that both ER and AR are required to mediate fetal BPA effects. Our findings point to an unappreciated sensitivity of developing circadian centers to embryonic BPA exposure and, perhaps, other endocrine-disrupting chemicals more broadly.
RESULTS
We first established a mouse paradigm for gestational BPA exposure with the goal of modeling human exposure levels. Although we initially characterized two BPA-laced diets, 50 mg of BPA per kg diet and 50 μg of BPA per kg diet, representing a “high dose” and “low dose,” respectively, nearly identical behavioral and neurogenic results were observed with both diets. High-dose results are shown in fig. S4, including data on high-dose BPA effects on neurogenesis, neurosphere proliferation and self-renewal, and first generation (F1) offspring behavior from control and BPA-fed dams.
Enzyme-linked immunosorbent assays (ELISAs) were used to measure serum BPA in both dams and postnatal day 0 (P0) pups following maternal ingestion of the 50 μg/kg BPA diet. Pups from BPA-fed dams had approximately 25-fold higher BPA (~0.08 ng/ml) than controls (P = 0.006, n = 5 to 7 pools of serum from independent litters of 10 to 14 pups; fig. S1A), whereas dams fed this diet displayed ~134-fold higher BPA levels (~0.4 ng/ml) in their blood than control dams (fig. S1B). We measured the amount of food consumed by pregnant dams fed either control or BPA diet (50 μg/kg) and observed no difference in diet intake by dams from these two groups (fig. S1C). In addition, we observed no obesogenic effects in the offspring from gestational BPA at this dosage, with no significant change in body mass between BPA-exposed F1 mice either as adults (fig. S1D) or during the early growth phase from P5 to P35 (fig. S1E). Using the average daily diet intake, we calculated the approximate dose of BPA to the dams (fig. S1F) as ~2.25 μg/kg BW, which was approximately 22 times lower than the “safe” human RfD of 50 μg/kg BW and lower still than the proposed safe t-TDI of 4 μg/kg BW (20). As far as correlating serum levels, in human studies, BPA in maternal serum can vary considerably (up to ~22 ng/ml) (32), but mean values are generally in the low nanograms per milliliter range, with specific studies reporting 0.46 ng/ml (33), 1.4 ng/ml (9), 4.4 ng/ml (8), and 5.9 ng/ml (32). These values are all higher (1.15- to 14.75-fold) than the ~0.4 ng/ml level (fig. S1A) measured in our BPA-fed dams. Moreover, human fetal serum levels range between 0.62 ng/ml (33), 2.18 ng/ml (34), 2.2 ng/ml (9), and 2.9 ng/ml (8). Again, values are all significantly higher (7.75- to 36.25-fold) than the ~0.08 ng/ml (fig. S1B) we observed in newborn pup serum using our gestational BPA exposure model as measured by ELISA. We confirmed our ELISA measurements via liquid chromatography–mass spectrometry (LC-MS) on a different set of serum samples from newborn pups. These analyses showed that the concentration of BPA in the serum of gestationally exposed F1 pups is ~0.125 ng/ml (fig. S1I), which is ~4.96- to 23.2-fold lower than the reported human fetal serum values noted above. Last, we used LC-MS to measure BPA in newborn pup brains and confirmed that our low-dose BPA diet significantly elevated brain BPA content at birth (fig. S1J). Combined, these data confirm that murine maternal ingestion of 50 μg/kg BPA-laced diet facilitates an exposure to the developing fetus at levels lower than governmental BPA RfDs and reported BPA levels in human adult and fetal serum.
Gestational BPA exposure alters behavior in 12-week-old mice
Next, we asked whether our gestational exposure model led to lasting consequences by using a variety of well-established behavioral tests that inform on phenotypes consistent with neurodevelopmental disruptions. In the open-field test, when compared to control mice, fetal BPA-exposed mice of both sexes (i) spent significantly more time in the center of the testing area (representative tracking images in Fig. 1A and quantification in Fig. 1B; 2.2-fold increase, P = 0.012), (ii) moved more between the designated zones (1.9-fold increase, P = 0.0007; Fig. 1C), and (iii) traveled a significantly greater distance during the testing period (1.2-fold increase, P = 0.022; Fig. 1D). These data indicate higher overall activity and potentially more adventurous and/or less anxious behavior in BPA-exposed offspring relative to controls.
Fig. 1. Gestational BPA exposure alters behaviors.
In open-field testing [representative tracking images in (A)], BPA mice exhibited increased time away from the wall (B), zone transitions (C), and total distance (D). In the elevated plus maze [representative tracking images in (E)], BPA mice spent more time in the open arms (F) and again exhibited increased zone transitions (G). In forced swim testing, time to initial immobility was unchanged (H), but BPA mice exhibited higher total movement time (I). The rotarod test indicates that locomotion is unaffected as measured by time to failure (J) or maximum speed achieved (K). In three-chamber testing, BPA mice interacted longer with a stranger mouse versus an empty cage [representative tracking images in (L) and quantification in (M)] or a littermate (N). Unexposed F3 mice descended from F1 BPA dams also displayed altered behavior, interacting less with a stranger animal versus an empty cage (O) while displaying higher activity (P), although this activity effect was only found in males (Q) as female F3 mice were indistinguishable from controls (R). Similar effects were observed against a littermate. F3 BPA mice entered the stranger zone less (S) but traveled more distance (T), again entirely due to male F3 BPA mice (U) not females (V). WT, wild type.
In the elevated plus maze, designed to test anxiety-like behaviors, gestational BPA exposure caused mice of both sexes to spend significantly more time in the open arms of the maze (representative tracking images in Fig. 1E and quantification in Fig. 1F; 1.9-fold increase, P = 0.0016) and likewise displayed an increased likelihood of crossing from one zone to another (1.6-fold increase, P = 0.002; Fig. 1G). These data support the generalized findings from the open-field test whereby BPA-exposed F1 mice appeared less anxious than control animals.
We next used the forced swim test to assess whether gestational BPA exposure had any effects on depressive tendencies. We observed no BPA effect on the primary measure of this test: the time to initial immobility (Fig. 1H); however, we did detect that over the course of the test, BPA mice of both sexes were more likely to become mobile again and renew efforts to escape the water, leading to a significant increase in total time spent moving (2.4-fold increase, P < 0.0001; Fig. 1l).
As a control for gross motor defects or cerebellar deficiencies that might have inadvertently influenced other behavioral tests, we used the rotarod apparatus to test for errors in spatiomotor control. We observed no significant effects of gestational BPA exposure on either the time that mice were able to walk on the rotating rod as it increased in speed over 4 min (Fig. 1J) or the maximum speed they were able to achieve (Fig. 1K). This finding, along with the qualitative observations of all mice across the various behavioral tests, suggests no defects in cerebellar or locomotor circuits that might have impaired or compromised other tests. In addition, we used the Barnes maze and Morris water maze tasks to assess learning and memory and observed no significant differences in F1 mice as a result of gestational BPA exposure (fig. S2, A and B).
Last, to determine whether gestational BPA exposure impeded social interactions, we placed either a control or gestationally exposed BPA mouse into a three-chamber apparatus that contained a wild-type stranger (never previously encountered) mouse in one chamber and either an empty cage (representative tracking images in Fig. 1L with location of stranger mouse marked with red X; quantification in Fig. 1M) or a littermate (Fig. 1N) in the opposite chamber. In both scenarios, BPA-exposed mice of both sexes spent significantly more time investigating the unknown mouse than control mice, exhibiting a 1.2-fold increase in time spent with the unknown mouse relative to an empty cage (P = 0.021; Fig. 1M) and a 1.4-fold increase in time with the unknown mouse relative to a littermate (P = 0.0002; Fig. 1N), also consistent with the notion that gestational BPA exposure might decrease anxiety.
Gestational BPA exposure has transgenerational effects
Given the alterations in behavior and activity in our F1 mice and the known epigenetic effects of BPA in other organs and systems (35), we examined behavior in F2 and F3 animals. Of note, we consider BPA-induced changes intergenerational for the adult female (F0), first generation of offspring (F1), and second generation of offspring (F2) since the adult, the fetus, and the primordial germ cells were all directly exposed to BPA. We deem any BPA effects transgenerational for the subsequent generations (F3 or later) since BPA exposure is absent. All nonsignificant behavioral changes in F2 and F3 mice are described in fig. S2.
The most significant behavioral effect observed in the F3 mice included alterations in social behavior using both methodologies of the three-chamber task. Specifically, when animals were presented with a stranger mouse in one region and an empty cage control in the other, F3 mice descended from BPA-exposed F1s spent significantly less time investigating the unknown animal than mice from control F1 lineages (0.83-fold change, P = 0.018; Fig. 1O) and traveled more distance in total, investigating the entire apparatus (1.5-fold increase, P = 0.006; Fig. 1P); however, this response was sexually dimorphic whereby this behavioral change increased only in males (1.8-fold increase, P = 0.026; Fig. 1Q) with no significant difference observed in females (Fig. 1R). Similarly, when the mice were given the option of a stranger mouse versus a sex-matched littermate, descendants of BPA-exposed F1 mice entered the stranger zone less than descendants of control F1 mice (0.62-fold change, P = 0.019; Fig. 1S), and these mice also traveled more distance overall than control mice during the testing exposure (1.66-fold increase, P = 0.006; Fig. 1T). Again, when sex was taken into account, this behavioral effect was observed only in male animals (2.5-fold increase, P = 0.007; Fig. 1U) and not female animals (Fig. 1V). Overall, these data demonstrate that gestational BPA exposure may have transgenerational effects on social behaviors.
Prenatal BPA exposure alters circadian rhythms
Given that sleep disruption is comorbid with ADHD and other neurodevelopmental disorders with behavioral phenotypes akin to those observed above (14, 15), we next examined whether gestational BPA exposure alters circadian rhythms. We continuously monitored the wheel-running activity of singly housed F1 mice of both sexes over the course of 2 months in both a 12:12-hour light-dark (LD) environment and in constant darkness (DD), representative actograms are displayed in Fig. 2A. In LD conditions, we observed an increase in the total daily activity of BPA mice (1.6-fold increase, P = 0.008; Fig. 2B) and a longer activity duration in these animals (~90 min longer; 1.14-fold increase, P = 0.036; Fig. 2C) relative to controls. BPA-exposed F1 mice also exhibited an alteration in activity onset, showing less of an anticipatory increase in behavior before the dark period (Fig. 2D). Specifically, control mice became active an average of 15 min ahead of the dark period, while gestational BPA-exposed mice only became active an average of 2 min ahead of the dark period (P = 0.026; Fig. 2D). Last, given the increase in total daily activity in fetal BPA-exposed mice, we plotted the data to yield a 24-hour time course of activity and then compared the activity differences between control and BPA-exposed mice in 4-hour segments. We observed no difference in activity during the first 12 hours of the light period but found a significant increase in activity in BPA mice during both the first (P = 0.03; Fig. 2E) and second 4-hour increments of the dark period (P = 0.027; Fig. 2E) but no significant difference in the final 4-hour portion of the dark period as animals from both groups showed a marked decline in activity. Next, we analyzed circadian patterns in the DD condition to observe the effects of BPA on activity rhythms in the absence of a normalizing light entrainment. As with LD conditions, we observed a significant increase in total daily activity for gestational BPA-exposed F1 mice in total darkness compared to controls (1.35-fold increase, P = 0.007; Fig. 2F). We also examined the free-running circadian period for these animals in the DD condition and observed that gestational BPA-exposed mice displayed a significantly shorter intrinsic day than control animals (~15 min less; P = 0.008; Fig. 2G). Similar to LD conditions, fetal BPA-exposed mice housed in total darkness were active for a longer duration than control animals (more than 2.5-hour-longer average duration; P = 0.02; Fig. 2H), and when data were organized to synchronize each animal’s activity onset [set to circadian time (CT) 12] and then plotted into a 24-hour activity time course, we again observed a significant increase specifically in the first and second sets of 4-hour increments (P = 0.004 and P = 0.018, respectively; Fig. 2I) of activity but no change in the final 4-hour segment. Last, we observed the resultant phase shift in circadian activity after exposure of mice to a 15-min light exposure 4 hours after activity onset (i.e., at CT16) and noted that, gestationally, BPA-exposed mice were less responsive to the light pulse, shifting their activity of, on average, ~2.2 hours compared to a shift of ~3.4 hours for control animals (P = 0.025; Fig. 2J). Last, we analyzed the activity curves of all animals (full data in Table 1) and observed no significant effect of treatment on the activity mesor (P = 0.827), amplitude (P = 0.594), acrophase (P = 0.36), or acrophase amplitude (P = 0.926), as assessed by two-way analysis of variance (ANOVA). Combined, these data demonstrate that gestational BPA exposure affects circadian behaviors.
Fig. 2. Gestational BPA exposure alters circadian rhythms.
Activity was monitored in both 12:12-hour LD and 24-hour DD conditions. Representative actograms are displayed [(A) gray, dark hours; white, light hours; black marks represent activity, each row is 24 hours). In LD conditions, BPA mice exhibited higher total daily activity (B), were active for a longer duration following the start of the dark period (C), and exhibited less anticipatory onset of activity before LD transition (D) than control mice. Increased total daily activity was due to significantly higher activity throughout the dark period (E) (data grouped in 6-hour intervals, n = 6 to 7 mice per treatment). When animals were entrained to 24-hour darkness, BPA-exposed mice still displayed higher total activity (F) as well as a shorter circadian period (G) and longer activity duration (H). When tracked activity was aligned at like circadian times, BPA mice displayed significantly more activity within the first 12 hours (I) (data grouped in 6-hour intervals, n = 6 to 7 mice per treatment). When animals were exposed to two 15-min light pulses, BPA-exposed mice displayed less of a phase shift than control mice in response to the unexpected light cue (J). (No comparison is made across time within a given treatment for time series).
Table 1.
Activity rhythm analyses.
| Animal | Sex | Treatment | Mesor | Amplitude | Acrophase (hours) |
Acrophase amplitude |
| BPA-F1 | F | BPA | 5.7005 | 22.9714 | 16.28702 | 7.688 |
| BPA-F2 | F | BPA | 6.1907 | 20.9857 | 16.97199 | 8.492 |
| BPA-F3 | F | BPA | 6.8206 | 23.5 | 16.63125 | 9.5107 |
| BPA-F4 | F | BPA | 9.2086 | 27.3857 | 17.33631 | 12.7518 |
| BPA-M1 | M | BPA | 5.7212 | 19.7429 | 16.27929 | 8.7901 |
| BPA-M2 | M | BPA | 3.1872 | 17.9143 | 15.80129 | 3.671 |
| BPA-M3 | M | BPA | 4.5932 | 23.2571 | 16.04463 | 5.9749 |
| CON-F1 | F | Control | 7.0091 | 21.8714 | 18.59813 | 8.7794 |
| CON-F2 | F | Control | 4.6302 | 21.5714 | 16.47778 | 5.7203 |
| CON-F3 | F | Control | 6.9877 | 24.3286 | 16.62251 | 10.7644 |
| CON-M1 | M | Control | 5.247 | 18.8857 | 17.21187 | 7.1306 |
| CON-M2 | M | Control | 7.4759 | 21.8714 | 16.50297 | 9.6499 |
| CON-M4 | M | Control | 4.2123 | 19.3286 | 15.53207 | 5.9592 |
BPA accelerates hypothalamic neurogenesis
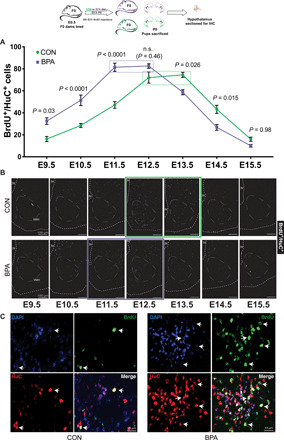
To ascertain the underlying cellular changes conferred by fetal BPA exposure, we next examined whether neurogenesis within the hypothalamus, the brain region that regulates circadian behaviors, was altered. Previously, we showed that hypothalamic neurogenesis commences at approximately embryonic day 9 (E9), peaks at E12 to E13, and ends by ~E15 (36). Here, birthdating revealed that gestational BPA caused precocious neurogenesis in the F1 pups, resulting in the peak of neurogenesis to shift from E12-E13 to E11-E12 (Fig. 3A). This shift in peak neurogenesis is revealed both by quantification (Fig. 3A) and in viewing representative images (Fig. 3B, red boxes; n = 5 to 6 mice from different litters per time point; double-positive cells that cross the threshold after processing are shown; high-magnification representative images in Fig. 3C; see Materials and Methods for details and see fig. S3A for raw color images) from the tuberal hypothalamus. We performed a two-way ANOVA to analyze the data and found statistically significant effects of both time (P < 0.0001) and treatment (P = 0.0001), as well as significant interactions (P < 0.0001). We used Tukey’s post hoc test to compare and identify statistically significant differences between treatments at each time point. Specifically, gestational BPA exposure increased the number of neurons born at E9.5 by 2-fold (P = 0.03), at E10.5 by 1.8-fold (P < 0.0001), at E11.5 by 1.7-fold (P < 0.001), and at E12.5 by 1.15-fold [not significant (n.s.), P = 0.46]. Concomitantly, in the later stages of neurogenesis, pups from BPA-fed dams exhibited a 0.79-fold reduction at E13.5 (P = 0.026), 0.62-fold reduction at E14.5 (P = 0.015), and 0.61-fold reduction at E15.5 (n.s., P = 0.98), suggesting that the progenitor pool was prematurely reduced by the increased neurogenesis during the early phase. In addition, birthdating experiments from the same time points in the neighboring thalamic region showed no significant differences following gestational BPA exposure (fig. S3, B and C). We also confirmed that the accelerated neurogenesis did not result in an alteration of the total number of hypothalamic neurons by counting neuronal nuclei-positive (NeuN+) cells in P0 pup sections from the tuberal hypothalamus as a proportion of 4′,6-diamidino-2-phenylindole–positive (DAPI+) nuclei. We observed no significant change between control (26.2%) and BPA (25.4%) pups (measured by the sum of cells from three tuberal hypothalamic sections each ~100 μm apart; n = 5 animals; P = 0.78, unpaired t test). These data indicate that the overall proportion of neurons in the hypothalamus is not affected by the acceleration in neurogenesis due to BPA exposure.
Fig. 3. Gestational BPA exposure accelerates hypothalamic neurogenesis.
Sections of the tuberal hypothalamus were stained for BrdU+/HuC+ neurons born on each day during the window of hypothalamic neurogenesis (E9 to E15). BPA exposure resulted in more early-born neurons in this window and less late-born neurons. Quantification (A) (two-way ANOVA, P values compare treatments at each time point) and representative images (B) are shown, with peak neurogenesis indicated by green (CON) or purple (BPA) boxes. The third ventricle edge (3V), presumptive ventromedial hypothalamus (VMH), and the pial edge is shown in white dashed outline. Representative high-magnification images of dual-positive cells are shown within the VMH of CON and BPA mice (C). IHC, immunohistochemistry.
Gestational BPA exposure specifically affects progenitors in the developing SCN
Given the effects of fetal BPA exposure on circadian activity, we next asked whether progenitors in the SCN were particularly sensitive to BPA’s effects and, if so, which neuronal phenotypes were affected. We used birthdating to quantify SCN neurogenesis in F1 mice at P0 after labeling from E9.5 to E15.5, as per above. Two-way ANOVA revealed significant effects of both time (P < 0.0001) and treatment (P = 0.0042), as well as significant interactions (P < 0.0001). The early phase of SCN neurogenesis was shifted in BPA-exposed animals, consistent with precocious neurogenesis of early-born neurons, whereas no change in later-born neurons was observed (representative images in Fig. 4A). Specifically, significantly more neurons were born in the SCN of BPA-exposed animal at E10.5 (3.15-fold increase, P = 0.035; Fig. 4B) and E11.5 (1.58-fold increase, P = 0.0044; Fig. 4B) and E12.5 (1.4-fold increase, P = 0.0002; Fig. 4B) than control animals. Very few SCN neurons were born in either group at E9.5 or E15.5, and there were no significant differences observed in neuronal birth at E13.5 or E14.5 (Fig. 4B).
Fig. 4. Changes in neuropeptide expression in the SCN.
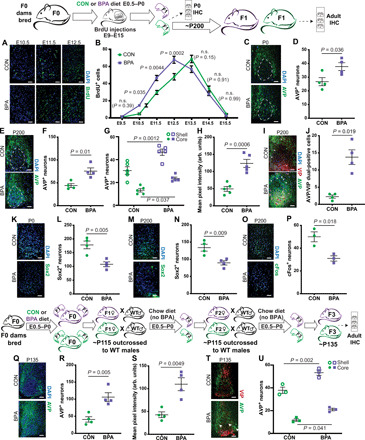
BPA exposure increases early (E10 to E12) SCN neurogenesis [representative images from the significantly changed time points shown in (A) and quantification in (B), two-way ANOVA, P values compare treatments at each time point], increases AVP+ neurons in P0 [representative images in (C) and quantification in (D)], and adult animals relative to controls [representative images in (E) and quantification in (F)]. This was consistent when the shell and core were counted separately (G) (two-way ANOVA), and total AVP expression was also higher in BPA animals (H). AVP and VIP costaining revealed a BPA-mediated increase in AVP/VIP dual-positive neurons [representative images in (I) and quantification in (J), white arrows mark sample shell AVP+ cells, and pink arrows mark dual-positive cells in BPA sections]. SCN Sox2+ cells were reduced in BPA mice at P0 [representative images in (K) and quantification in (L)] and adult mice [representative images in (M) and quantification in (N)]. BPA exposure reduced light-induced cFos expression [representative images in (O) and quantification in (P)]. BPA-exposed F3 mice also had less SCN AVP+ neurons [representative images in (Q) and quantification in (R)] and lower overall expression (S). F3 mice also displayed the same change in region-specific AVP+ neurons [representative images in (T) and quantification in (U), two-way ANOVA]. (Where appropriate, dashed line delineates the SCN core).
To determine the phenotype of these precocious SCN neurons in gestationally exposed pups, we first examined AVP neurons due to their key role in circadian signaling (29, 30, 37, 38). We observed a significant increase in the number of AVP+ neurons both at P0 (representative images are shown in Fig. 4C and quantification in Fig. 4D; 1.4-fold increase, P = 0.036) and in the adult animal (representative images are shown in Fig. 4E and quantification in Fig. 4F; 1.4-fold increase, P = 0.036; 1.7-fold increase, P = 0.01) of gestational BPA-exposed animals relative to controls, suggesting a lasting increase in AVP+ neurons in the SCN following prenatal BPA exposure. We observed a significant increase in AVP+ neurons in the shell (Fig. 4G; 1.5-fold increase, P = 0.0012) and the core (Fig. 4G; 1.75-fold increase, P = 0.037). We also quantified the mean pixel intensity of AVP fluorescence in the adult animal as a measure of the total expression of AVP both in cells and fibers since we observed abundant AVP expression in projections throughout the SCN shell, and found a statistically significant increase in fetal BPA-exposed animals compared to control mice (Fig. 4H; 2.5-fold increase, P = 0.0006).
Moreover, AVP+ neurons were observed in the ventral SCN core, a region normally expressing minimal AVP, which was confirmed by coexpression of the core marker VIP (representative images are shown in Fig. 4I). We quantified AVP+/VIP+ neurons in BPA-exposed animals and observed that BPA increased the total number of dual-positive neurons (Fig. 4J; 4.6-fold increase, P = 0.016), which were generally absent in control animals. Combined, these data indicate an effect of gestational BPA exposure on AVP+ neurons within the SCN.
This increase in AVP+ expression in the SCN core of gestational BPA-exposed mice could occur via two mechanisms (not mutually exclusive): (i) a neuronal change such that core SCN neurons acquired an AVP fate at the expense of other neuronal phenotypes or (ii) AVP expression is ectopic, such that extraneous AVP+ neurons are now localized in the core region. To distinguish between these two possibilities, we quantified the number of Sox2+ cells, a known regulator of core signaling in Period2 neurons (39). We observed a reduction in Sox2 expression in BPA-treated mice at P0 (representative images are shown in Fig. 4K and quantification in Fig. 4L; 0.6-fold change, P = 0.005) and in the adult animal (representative images are shown in Fig. 4M and quantification in Fig. 4N; 0.6-fold change, P = 0.036) relative to controls, perhaps due to core neurons acquiring an AVP fate at the expense of Sox2. We also examined whether this change in neuronal specification would affect SCN neuronal activity as measured by the expression of cFos in the SCN of animals following a brief 72-hour adjustment to DD conditions followed by a 1-hour light exposure at CT16 (to maximize cFos expression). We observed a significant reduction in cFos+ neurons in BPA-exposed animals relative to controls (representative images are shown in Fig. 4O and quantification in Fig. 4P; 0.6-fold change, P = 0.018), indicating that the ability of the SCN to respond to light cues is compromised when an increased number of AVP+ neurons reside in the core.
To determine whether this change in AVP expression persisted transgenerationally, we quantified AVP expression in the SCN of F3 animals. We observed significantly more AVP+ neurons (representative images are shown in Fig. 4Q and quantification in Fig. 4R; 2.6-fold increase, P = 0.005) and mean pixel intensity (Fig. 4S; 2.4-fold increase, P = 0.0049). In addition, AVP+ neurons were increased in both the shell (representative images shown in Fig. 4T, arrowheads, and quantification in Fig. 4U; 1.4-fold increase, P = 0.002) and the core (1.8-fold increase, P = 0.041; Fig. 4U) in the SCN of F3 descendants of BPA-exposed mice than in those from control animals, suggesting that gestational BPA exposure may cause lasting epigenetic changes in hypothalamic neural progenitors.
BPA acts on hypothalamic progenitors
To elucidate the molecular changes by which BPA might direct this increase in AVP+ specification, we used the neurosphere assay that serves as a primary culture model of hypothalamic NSPCs that allows the study of NSPC activation, proliferation, self-renewal (stemness), and differentiation (40). To start, we examined the effects of exogenous BPA on naïve hypothalamic NSPCs. BPA treatment (10 nM) in culture [chosen to approximate the commonly found human fetal serum BPA levels of ~2 ng/ml (8, 9, 34)] significantly increased the number of primary neurospheres observed (representative images in Fig. 5A and quantification in Fig. 5B; 1.6-fold increase, P < 0.0003) relative to vehicle treatment, indicating that BPA increased sphere-forming capacity and/or NSPC activation. BPA treatment also resulted in a higher proportion of large (>200 μM) spheres compared to vehicle only (representative images in Fig. 5A and quantification in Fig. 5C; 2.4-fold increase, P = 0.0022), consistent with an increase in NSPC proliferation by BPA treatment. After dissociation and replating, a decrease was observed in the number of secondary neurospheres that reformed from BPA-treated cells relative to vehicle controls (representative images in Fig. 5D and quantification in Fig. 5E; 0.71-fold reduction, P = 0.022), indicating a loss of self-renewal capacity and a bias toward cell cycle exit in BPA-treated NSPCs. Despite fewer secondary spheres following BPA treatment, those that did form were larger than non–BPA-exposed spheres (representative images in Fig. 5D and quantification in Fig. 5F; 1.87-fold increase, P = 0.0044), further evidence that NSPC proliferation was BPA responsive. To confirm that the loss of self-renewal capacity observed in secondary neurosphere formation was not transient, we performed three more passages and found a significant reduction of sphere-forming cells after the third (Fig. 5G; 0.79-fold reduction, P = 0.021) and fourth (Fig. 5H; 0.7-fold reduction, P = 0.015) passages, but not after the fifth passage (Fig. 5I; 0.72-fold reduction, P = 0.081). To confirm that BPA-treated hypothalamic NSPCs were exiting the cell cycle, we dissociated primary hypothalamic neurospheres and replated the cells in the absence of growth factors [epidermal growth factor (EGF) and fibroblast growth factor (FGF)], which unleashes differentiation programs. BPA treatment resulted in a marked increase in neuronal differentiation, determined by higher numbers of NeuN+ cells (representative images in Fig. 5J and quantification in Fig. 5K; 2.5-fold increase, P = 0.023), and a notable loss of multipotency shown by reduced Sox2+ neural progenitors (representative images in Fig. 5L and quantification in Fig. 5M; 0.56-fold change, P = 0.006) relative to vehicle-treated controls. Given that it is not possible to isolate SCN NSPCs from overall hypothalamic progenitors, we could not access the effects of exogenous BPA treatment on SCN AVP neuronal formation per se. BPA treatment selectively conferred a neuronal fate since no change in astrocyte [i.e., glial fibrillary acidic protein–positive (GFAP+)] or oligodendrocyte [i.e., platelet-derived growth factor receptor α–positive (PDGFRα+)] cell numbers was observed (fig. S5).
Fig. 5. Ex vivo hypothalamic NSPC culture elucidates mechanisms of BPA action.
BPA treatment (10 nM) increased the number [representative images in (A) and quantified in (B)] and size [representative images in (A) and quantified in (C)] of primary neurospheres. BPA (10 nM) decreased the number [representative images in (D) and quantified in (E)] but increased the size [representative images in (D) and quantified in (F)] of secondary neurospheres. We observed significant reduction in sphere-forming cells in tertiary (G) and quaternary (H) cells but not in the fifth culturing (I). Differentiated 10 days in vitro (DIV) BPA-treated cells expressed higher NeuN [representative images in (J) and quantified in (K)] and lower Sox2 [representative images in (L) and quantified in (M)] than controls. Gestational BPA exposure increased primary neurosphere number without direct BPA treatment [graphical experimental explanation in (N) and quantification in (O)] even when BPA diet was only given in the past [quantification in (P)]. Use of fulvestrant (Fulv) and flutamide (Flut) indicates that dual ER/AR antagonism is required for phenotypic rescue of primary sphere number [representative images in (Q) and quantified in (R), ANOVA] and size [representative images in (Q) and quantified in (S), ANOVA]. Combined antagonism also reversed BPA effects on secondary sphere number [representative images in (T) and quantified in (U), ANOVA] and size [representative images in (T) and quantified in (V), ANOVA]. NS, neurospheres; n.s., no significance.
To determine whether hypothalamic NSPCs exposed to BPA in vivo displayed similar properties, we isolated E12.5 fetal hypothalamic NSPCs from dams fed either control or BPA diet (50 μg/kg) under two scenarios. In the first scenario, dams were grouped (and groups never interchanged; see Fig. 5N) and fed either control or BPA diet during gestation for two breeding cycles with 6- to 8-week rest times on regular diet in between breeding rounds. On their third round of breeding, pregnant dams were placed on control or BPA diets, E12.5 pups were collected, and hypothalamic NSPCs were harvested and cultured with no additional BPA treatment. Gestational BPA exposure in vivo increased hypothalamic NSPC sphere-forming capacity (1.65-fold increase, P = 0.0057; Fig. 5O), consistent with exogenous BPA treatment. In the second scenario, dams were grouped and bred as shown (Fig. 5N); except in the third round of breeding, the BPA group was instead kept on the control diet. Perhaps unexpectedly, an increase in sphere-forming capacity was observed in hypothalamic NSPCs from pups never exposed to BPA themselves but collected from dams that received BPA diet during two previous pregnancies (Fig. 5P; 1.45-fold increase, P = 0.0029), perhaps demonstrating that maternal BPA exposure can affect future pregnancies.
Antagonism of both ER and AR is required to block BPA’s effects
To determine the molecular targets of BPA in NSPCs, we focused on the steroid receptors, ER and AR, which are expressed in NSPCs (41, 42). ER and AR are recognized to be agonized and antagonized by BPA, respectively (1–5, 24), although we previously showed that AR can be agonized by BPA in hypothalamic progenitors (24). Using our neurosphere assay, we treated hypothalamic NSPCs with antagonists for ER [i.e., fulvestrant (Fulv); 1 μM] or AR [i.e., flutamide (Flut); 1 μM] or both. Antagonist concentrations were previously established in our laboratory during the development of our hypothalamic neurosphere technique (40) to reliably block receptor activation while testing seeding densities. As per above, BPA treatment increased the number of primary spheres formed (representative images in Fig. 5Q and quantification in Fig. 5R, 1.4-fold above vehicle). Individually, each inhibitor partially blocked the BPA-mediated effects (Fulv, 1.13-fold increase above vehicle; Flut, 1.16-fold increase above vehicle; Fig. 5R), but only dual antagonism of ER and AR was sufficient to fully reverse BPA effects on NSPCs to control levels (1.04-fold increase above vehicle; Fig. 5R). Fulv treatment, while statistically indistinguishable from vehicle (Fig. 5R; P = 0.098), was simultaneously not significantly different from BPA treatment only (P = 0.088), indicating that it was not sufficient to completely rescue the BPA effects. Moreover, the size of primary hypothalamic neurospheres treated with BPA was also reduced to vehicle levels following dual antagonism of ER and AR (1.26-fold increase over vehicle; Fig. 5S). Flut alone reduced the proportion of large spheres to the level of vehicle treatment (P = 0.16) but to a lesser extent than the combined treatment (representative images in Fig. 5Q and quantification in Fig. 5S; 1.7-fold increase over vehicle versus 2.85-fold increase over vehicle) and was not statistically significantly different from BPA treatment alone (P = 0.09).
For secondary hypothalamic neurospheres, only the combined treatment of Fulv and Flut in BPA-treated cultures restored the number of secondary spheres formed to the level of vehicle (representative images in Fig. 5T and quantification in Fig. 5U; 0.98-fold change from vehicle for combined treatment versus 0.72-fold change for BPA treatment only). Concomitantly, dual antagonism of both ER and AR in BPA-treated cultures was the only treatment able to restore hypothalamic NSPC neurosphere size to vehicle levels (1.2-fold increase over vehicle; Fig. 5V), further demonstrating that ER and AR are both required to mediate BPA’s effects on hypothalamic progenitor behaviors.
DISCUSSION
Here, we examine the effects of an environmentally relevant dose of BPA on murine hypothalamic development and associated behaviors and report a particular sensitivity of vasopressin neuronal precursors to this plasticizer. Specifically, by using a BPA dose below what is considered safe for human exposure by the Food and Drug Administration (FDA), Health Canada, and the European Food Safety Authority, we observe accelerated embryonic neurogenesis in the BPA-exposed offspring that manifests as ectopic specification of AVP neurons in the SCN and associated altered circadian activity, among other behavioral abnormalities. Using an ex vivo culture model of hypothalamic NSPCs, we show that BPA treatment increases the activation, proliferation, and neuronal-specific differentiation of these progenitors in an ER and AR codependent manner. Last, we present evidence that AVP ectopic expression and behavioral changes persist for multiple generations despite the lack of further BPA exposure, underscoring the lasting effects of BPA action on hypothalamic NSPCs. Combined, these findings serve as additional evidence that low-dose BPA exposure has transgenerational effects on developing brains and provides further impetus to revisit the current BPA RfD and LOAEL levels, in addition to government-recommended TDIs.
Neurodevelopment is a protracted series of steps influenced by an extensive niche of developmental factors (10, 26) such that premature hypothalamic neuronal birth can lead to aberrant specification, migration, and/or circuit establishment that undermines hypothalamic function. The changes in SCN development described here are a compelling example of this type of disruption. For example, gestational BPA exposure induces precocious SCN neurogenesis specifically in the early neurogenic window (E10 to E12), which we propose influences some SCN progenitors to specify ectopic AVP neurons at the expense of other SCN neuronal phenotypes, such as Sox2+ cells in the SCN core. This misexpression of AVP leads to consequences on SCN-mediated physiologies, including alterations in free-running period and light entrainment. Notably, AVP is a critical regulator of circadian signaling from the SCN shell to other brain regions (29, 31), with expression of the clock gene Bmal1 in AVP neurons regulating aspects of circadian signaling such as light entrainment (38) and synchronizing the pacemaker cells (37, 43), making misexpression of this neuropeptide likely to disrupt SCN function. In addition, spatial AVP distribution within the SCN appears to be critical to clock gene coordination (44), and there is evidence that spatial VIP organization is also needed for clock rhythmicity (45). These linkages indicate that BPA-mediated alterations in SCN neuronal specification and spatial organization are likely causes of the altered daily activity rhythms in BPA-exposed mice. At the same time, however, Sox2 is a transcriptional regulator of the clock gene Per2 in SCN neurons (39), also making it likely that the loss of Sox2+ cells in the SCN results in at least some of the deficiencies observed in the BPA-exposed mice. It is unclear whether the ectopic expression of AVP neurons in the core, the loss of other core neuronal phenotypes, or both ultimately explains the irregular circadian activity observed in BPA-exposed offspring, although the decrease in cFos signaling within core neurons suggests that core AVP+ neurons have not adopted the physiological role of neurons intended for this subdomain.
Beyond circadian activity, gestational BPA exposure also affects a wide range of behaviors in both the offspring and transgenerationally, raising the intriguing notion that defects in SCN-mediated diurnal activity may explain some of the more unusual behavior results in tests such as the elevated plus maze and forced swim tests, where our findings contradict established literature from higher-dose BPA studies. Further, defects in SCN development might lead to an overall more active phenotype across the behavioral tests, confounding the emotional/behavioral factors the test is intended to assess. For example, we observe a robust increase in hyperactivity in F1 BPA-exposed mice as measured by distance traveled in a variety of tests, a lowering of anxiety-like and depressive behaviors, and altered social interaction with a stranger mouse. Hyperactivity is perhaps the most common behavioral phenotype observed in animal BPA studies (10), and a recent meta-analysis of research in both rodents and humans indicated a clear correlation between early BPA exposure and hyperactivity (46). Depressive and anxiety behavior effects are more mixed in animal models, with higher-dose studies indicating an increase in these behaviors with gestational and/or lactational exposure (47), perinatal exposure (48, 49), and chronic exposure in males (50), but no effect of low-dose exposure was found in the primary report from the Consortium Linking Academic and Regulatory Insights on BPA Toxicity (CLARITY-BPA) program (51). A variety of social behaviors are perturbed by BPA exposure, including parental attachment and sociosexual behaviors (52), with one study (using a 100× higher dose) showing a similar effect on behavior in rodent juveniles as we did in adults. Specifically, F1 BPA-exposed juvenile mice are more interested in an unknown animal, an effect that persists to the F3 generation (53), although, instead, we observe less interest in a stranger animal in the F3 adults. Social interactions are complex, and the necessarily simplified traits measured by the three-chamber tasks are difficult to translate to human cohort data. Are our F1 mice more interested in social interactions so they spend more time with the unknown animal, or are they slower to acclimate to an unknown animal due to reduced social ability so they spend more time investigating this strange new mouse? Both social interaction (despite the opposing effect) and related hyperactivity (although restricted to males) persist to the F3 generation in our study, consistent with an epigenetic modification that may be occurring during gestation. BPA causes epigenetic modifications in a variety of studies, so this finding is not particularly unexpected (35), and is supported by other reports using higher BPA dose as well (53). In human studies, BPA levels are correlated with increased incidence of a variety of neurodevelopmental and mood disorders, especially ADHD and ASDs (11–13), making our findings of BPA-mediated hyperactivity consistent. It is also noteworthy that the social behavior phenotypes shifted across the BPA-exposed generations. For example, gestationally exposed BPA F1 animals display increased time with the unknown conspecific, whereas the BPA F2 lineage animals show no social behavior phenotypes and the BPA F3s display decreased time with the unknown conspecific. Transgenerational epigenetic inheritance is one explanation for environmental effects across generations (54, 55); however, one would expect the ensuing phenotypes to be consistent if a conserved epigenetic modification(s) was the underlying cause. Discordant multigenerational behaviors are not entirely novel since maladaptation of the neuroendocrine system can lead to disparate phenotypes. For example, high maternal stress can adversely prime the hypothalamic-pituitary axis during development such that the offspring display hypoactive stress responses later in life (56, 57). In addition, we propose a third consideration that might contribute to this changing transgenerational phenotype: changes to maternal care. Notably, maternal care is mediated at least in part by the vasopressin system (58), which we show here is disrupted by BPA. Moreover, low-dose BPA exposure alters maternal care in both gestationally exposed F1 and unexposed F2 generations (59, 60), and since disrupted maternal behaviors are linked to altered anxiety-like behaviors and social interactions in the offspring (61, 62), it is possible that maternal care is different across the BPA generations, causing this change in social behaviors causing this change in social behaviors in offspring. Future studies are needed to investigate this intriguing notion that low-dose gestational BPA exposure disrupts the development of hypothalamic circuits that underlie maternal care, thereby leading to lasting effects in the offspring that manifest as changes in social behaviors.
Historically, one of the challenges of BPA animal studies is the relevance of exposure dose relative to environmental levels of the average human, and this is especially true for gestational exposure models. Previous challenges to measuring actual BPA urine or serum levels in animals hindered the translation of the findings back to humans. Here, we use ELISAs and LC-MS to determine the concentration of serum BPA in newborn pups exposed gestationally and found levels to be ~0.08 and ~0.125 ng/ml, respectively, which is ~8- to 35-fold and ~5- to 20-fold lower, respectively, than BPA concentrations consistently reported in human fetal cord blood (8, 9, 34). Moreover, we determine the approximate BPA dosage consumed by the pregnant dams ~2.25 μg/kg BW per day, which is nearly 25-fold lower than what is considered the safe TDI in humans in North America. Further, this amount of BPA ingestion elevates serum BPA in the dams to ~0.4 ng/ml (ELISA) and which is 1.15- to 14.75-fold (ELISA) or lower than recorded human blood serum levels of mothers at birth (8, 9, 32, 33), providing additional support to the relevance of our gestational exposure paradigm. Coincidentally, this ~2.25 μg/kg BW per day dietary dose delivered by our BPA diet nearly matches the lowest dose delivered to rats as part of the CLARITY-BPA studies (63), which shows a variety of brain-specific BPA effects including changes in volume of the anteroventral periventricular nucleus of the hypothalamus (64), hypothalamic oxytocin receptor expression (65), and gene expression in the hypothalamus and other brain regions (66, 67). Combined, given the nonmonotonic dose-response curve of BPA action (68), as well as the demonstration that BPA exposure models in animals matches human environmental exposure levels, it is becoming more evident that low-dose BPA affects our bodies and especially developing brains.
Several studies have now demonstrated that BPA can influence neural progenitors to perturb neurogenesis. For example, low-dose waterborne BPA exposure [~8-fold lower than the estimated average surface waters value of 12 μg/liter and up to ~1000-fold lower than high-risk watersheds (69)] causes precocious hypothalamic neurogenesis in zebrafish embryos (24), whereas higher exposures of 20 mg/kg and 200 μg/kg BW BPA doses in mice impair hippocampal (70) and neocortical neurogenesis (71), respectively. To understand the hypothalamic mechanistic pathways disrupted by BPA, we used our neurosphere assay that serves as an ex vivo model to probe hypothalamic NSPC behaviors, including activation, proliferation, differentiation, and self-renewal capacity. We hypothesized that hypothalamic NSPCs must (i) rapidly proliferate to produce daughter neurons, (ii) display reduced self-renewal capacity properties, and (iii) initiate neuronal differentiating programs. We used our neurosphere assay (40) at the window of peak hypothalamic neurogenesis (E12.5), when neurogenic progenitor division is maximized, to test these hypotheses, with our culturing and treatment paradigm designed to mimic the in utero environment whereby these NSPCs would be bathed in constant BPA exposure. Both ex vivo and in vivo BPA treatments of hypothalamic NSPCs induce these behaviors. These findings are in agreement with studies in cultured rat hypothalamic progenitors whereby BPA delivered to pregnant dams via drinking water increases neural proliferation and expression of proneural markers in BPA-treated NSPCs (72). An unexpected finding of our study is that hypothalamic NSPCs display increased proliferative behaviors even when the fetal brain was not exposed to BPA during the current gestational period but, instead, the pregnant dam was exposed during previous pregnancies. These data suggest that perhaps BPA can accumulate in maternal fat to deliver a steady low dose to the fetus even in the absence of consumption of BPA during that isolated pregnancy, which may occur concomitantly with epigenetic changes.
Notably, estrogen signaling can induce neural proliferation and alter the neuron-to-glia ratio of cultured rat embryonic neural stem cells (42), suggesting that BPA is estrogenic in neural progenitors. However, here, BPA effects on hypothalamic NSPCs are blocked only when both ER and AR signaling are pharmacologically inhibited. These findings are perhaps unpredicted because although BPA is classically considered estrogenic, it is also thought to be antiandrogenic (4, 5). However, our previous study found that AR signaling was required for BPA-mediated precocious hypothalamic neurogenesis in zebrafish embryos (24). It is possible that hypothalamic NSPCs are unique in their sensitivity to BPA-mediated AR activation during developmental time points, although further studies in other brain regions are needed. The dual requirement of ER and AR for BPA-induced hypothalamic NSPC proliferation (as measured by primary and secondary neurosphere size) is more robust than its effects on activation (primary number) and self-renewal capacity (secondary number) since the combined antagonist treatment still did not completely rescue the size effects. It is possible that BPA-mediated proliferation involves other unidentified pathways, which is perhaps not unexpected given that BPA can bind TH (6) and potentially glucocorticoid (7) receptors in addition to ER and AR. Together, we propose a model whereby maternal BPA ingestion crosses the placenta and travels via fetal circulation to the developing brain, where it binds ER and AR in the NSPCs and induces changes in developmental programs that cause neural progenitors to induce precocious neurogenesis (Fig. 6).
Fig. 6. Working model of BPA effects on the developing SCN.
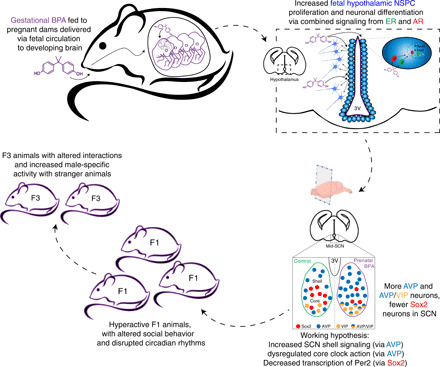
We propose that maternally ingested BPA crosses the placental to enter the developing fetal bloodstream. The hypothalamus is particularly BPA sensitive, and hypothalamic NSPCs respond to BPA with increased proliferation and neuron-specific differentiation via a combined ER- and AR-dependent mechanism. In the SCN, this results in increased overall AVP+, AVP+/VIP+ core, and fewer Sox2+ core neurons. This likely results in a variety of disruptions to the SCN molecular clock, including clock gene expression, as well as altered AVP-dependent shell signaling to other brain regions. Combined, these developmental changes might disrupt circadian activity and result in the other behavioral phenotypes displayed by BPA mice including hyperactivity and altered social interactions.
There is timeliness to our study given the release of the government/academic collaborative National Institute of Environmental Health Sciences/FDA CLARITY-BPA Core Study report, which concluded that no significant effects of BPA are found at levels below recommended safe doses (63). However, data not included in the government’s final analyses have since been published independently by CLARITY-BPA partner academic laboratories and demonstrate many cases of low-dose BPA effects across a variety of tissues, including the brain (22, 64, 66, 67, 73). An arm’s length, secondary analysis of data from both the government and academic laboratories concludes that adverse outcomes from low-dose BPA should not be dismissed and that the brain was the most consistently responsive organ studied (74). Furthermore, a recent study raises the possibility that researchers may be inadvertently underestimating BPA levels in human fluids when indirect analytical methods are used (75) since direct measurement shows a 19-fold higher level of BPA in human fluids, which is 44-fold higher than the most recent mean exposure value cited from the U.S. National Health and Nutrition Examination Survey (76). Thus, our study here is apropos to this larger discussion given our use of a BPA dose that is ~30-fold lower than commonly reported levels in newborn cord blood (8, 9, 34). Our results are in agreement with published reports from CLARITY academic partners and supportive of ongoing examination of safe BPA exposure levels.
In summary, we present a comprehensive study of the neurodevelopmental effects of gestational low-dose BPA that includes behavioral, cellular, and molecular end points. The resulting accelerated hypothalamic neurogenesis and the disruption in timing of neuronal birth likely alter the specification and integration of affected hypothalamic neurons, which is known to be spatiotemporally driven. Our study provides additional evidence that the developing hypothalamus is particularly susceptible to endocrine disruption by BPA, even at very low doses, and may provide mechanistic insights into the established correlations between early-life BPA exposure and neurodevelopmental disorders in humans.
MATERIALS AND METHODS
Experimental design
This study was designed to assess the effects of gestational exposure to environmentally relevant doses of BPA delivered via maternal diet to mimic human exposure levels, on the developing hypothalamus. All animal and experimental protocols were approved by the University of Calgary Animal Care Committee and followed the Guidelines for the Canadian Council of Animal Care. The number of samples was determined on the basis of experimental approach, availability, and feasibility required to obtain definitive results and is specified below in the detailed methods or alongside results. All experiments were carefully controlled. All behavioral and circadian experiments used both male and female animals. Behavioral experiments used, at most, two animals from a given litter; social interactions only involved animals that had previously interacted if specifically described (e.g., a littermate). For neurogenesis and immunohistochemistry experiments, only one animal was analyzed from a given litter. For neurosphere experiments, technical replicates were performed in triplicate within a culturing experiment from pooled fetal hypothalamic tissue. All data were included for analysis, and wherever feasible, all data points are displayed in figures. Researchers were not blinded for either data collection or analysis.
Mouse husbandry and breeding
Mice were housed and maintained at the University of Calgary Animal Resource Centre, fed our laboratory standard chow diet (chow; LabDiet Pico-Vac Lab Rodent Diet, 5061), and watered ad libitum. All material used for housing, feeding, and water were BPA free. Wild-type C57bl6 mice from Charles River were paired and females were monitored daily for breeding success. Pregnant dams were placed on either a 7% corn oil diet (control or CON; Envigo diet code TD.120176) or 500 parts per billion (50 μg/kg) BPA diet (BPA; Envigo diet code TD.160491). Dams were used one to three times for breeding rounds (most dams were bred twice), but never switched feeding groups, and were rested for 6 to 8 weeks postweaning and fed only chow diet before being bred again. Diet intake for the gestational period was measured, and after pups were born, the diet was replaced with chow diet. Pups were weaned to separate cages at P21. A subset of pups were weighed at P5, P10, P15, P21, P28, and P35, and adult mice were weighed at ~6 months old. Serum BPA was measured by ELISA (Abnova), dams were euthanized after giving birth, and blood was collected by cardiac puncture; newborn pups were decapitated, and blood was pooled together from all pups. Blood was collected in heparinized polystyrene tubes and centrifuged at 2000g at 4°C to separate cells. Serum was collected in glass vials and frozen until analysis. ELISA was performed as per the manufacturer’s instructions.
Female F1 mice were outcrossed at ~P115 to wild-type males to generate F2 mice. F2 female mice were again outcrossed at ~P115 to new wild-type males to generate F3 mice for analysis of behavior and SCN neuropeptide expression. F1, F2, and F3 mice were fed only chow diet at all times and were not subjected to any other BPA exposure other than the original diet fed to F0 dams during pregnancy.
Wild-type CD1 mice were housed, fed chow diet only with no BPA diet exposure, and bred as above, but pregnant dams were euthanized on E12.5 and fetal pup brains were microdissected for isolation of the presumptive hypothalamic NSPCs for ex vivo culture in the neurosphere assay, as we have previously established (see below for details) (40).
Quantification of neurogenesis
Pregnant C57 dams fed diets were injected with the thymidine analog 5-bromo-2′-deoxyuridine (BrdU; 10 mg/ml, 200 μl of injection volume, n = 5 to 6 dams per time point) on one of E9.5 to E15.5 to label neurons born on each day. Pups were euthanized at P0; brains were removed and fixed in 4% paraformaldehyde (PFA) for 48 hours, transferred to 20% sucrose for 48 hours, then embedded in optimal cutting temperature (OCT) compound, and frozen at −80°C. Adult brains were fixed and frozen as above, except that the animal was anesthetized and perfused first with saline and then with 4% PFA before the 48-hour whole fixation step. Frozen brains were sliced into 12- to 14-μm sections.
For quantification of BrdU and neuropeptides in P0 brains in the tuberal hypothalamus or the central SCN, sections were washed in phosphate-buffered saline (PBS), PBS with 0.1% Triton X-100 (PBST), and PBS with 1% Triton X-100, incubated with 2 N of HCl at 37°C for 45 min and then blocked in PBST and 5% normal donkey serum for 1 hour. Primary antibodies (rat anti-BrdU, Abcam, 1:200; rabbit anti-HuC, Invitrogen, 1:100; rabbit anti-AVP, Abcam, 1:400; rabbit anti-Sox2, EMD Millipore, 1:400) were incubated for 12 hours overnight at 4°C. The next day, sections were washed in PBST and incubated with secondary antibody (Alexa Fluor 488 donkey anti-mouse, 1:500; Alexa Fluor 555 donkey anti-rabbit, 1:500) for 2 hours at room temperature, washed again in PBST, and lastly stained with DAPI nuclear stain (5 min, room temperature) before final washes and mounting. Slides were imaged with a Zeiss Axioplan 2 fluorescent microscope. Brightness and/or contrast of the entire image was adjusted using Adobe Photoshop CC 2018 where necessary to remove background or improve visibility. For BrdU+/HuC+ double-positive cells, Fiji image processing software was used to threshold for dual-positive cells, the images were then converted to grayscale and cells were counted. Neurogenesis counts were averaged from three sections (each ~100 μm from the others) of the tuberal hypothalamus (as marked by landmarking and confirmed by the presence of the ventromedial nucleus marker FezF1 in adjacent sections). The hypothalamic/thalamic border was defined by the hypothalamic sulcus. Each data point consists of an n of five mice, all from different litters.
Behavior
F1, F2, and F3 pups were raised to ~12 weeks of age and then underwent behavioral testing for 7 to 10 days. All tests were done in accordance with University of Calgary Cumming School of Medicine Optogenetics Facility behavioral protocols. Briefly, mice were acclimated in behavioral rooms for at least 1 hour before testing. For the open-field test, elevated plus maze, and three-chamber tests, mice were monitored via an overhead camera for the duration of the test and evaluated via the Panlab Smart 3.0 Video Tracking software package (F1 and F2 animals) or the ANY-maze software package (F3 animals). The open-field test was 10 min in duration, elevated plus maze was 5 min in duration, and the three-chamber and novel animal tests were 10 min each after a 10-min acclimation period to an empty apparatus (no other mice present). The forced swim test and rotarod test were monitored manually. For the forced swim, mice were placed in a 2-liter beaker filled with ~1.7 liter of 25°C water and were individually monitored for more than 5 min to record the time to immobility and the total movement time. For the rotarod test of motor control, mice were trained on the apparatus for more than 4 days with increasing speed (4, 7, 10, and 12 rpm), followed by a final day of testing where the rod increased speed over time and the mice were monitored for up to 5 min to determine the length of time maintained on the apparatus and the maximum rpm achieved. For all behavioral assays, both male and female mice were used in approximately equal numbers, and no more than two animals were from the same litter.
Circadian testing
A subset of F1 mice aged 4 months old (four males and three females for both control and BPA mice) were monitored for daily activity in a separate, light-controlled facility equipped for constant measurement of wheel-running activity. Mice were singly housed in cages equipped with a wheel, acclimated in a 12:12-hour LD environment for 10 days. After acclimation, 19 days of wheel-running activity was selected for analysis. Animals were then switched to a 24-hour DD environment and acclimated again for 10 days, followed by a subsequent 14 days of activity used for analysis. The ClockLab analysis package (Actimetrics) was then used to quantify total activity, circadian period (dark only), activity duration, activity onset, and a time series of daily activity in each of the lighting conditions. Last, dark-acclimated animals were subjected to a 15-min light pulse at CT16 (CT12 defined as each animal’s daily activity onset), followed by assessment of the shift in their circadian rhythm over the next 4 days.
Neurosphere assay
We have adapted the neurosphere assay for use during the window of peak neurogenesis in the fetal mouse hypothalamus (40). Briefly, pregnant CD1 dams (fed only chow diet at all times) were euthanized on E12.5 and fetuses were removed, and the developing hypothalamic region was microdissected from each fetus and pooled together, triturated, strained, and grown in suspension culture at 5000 cells/ml in 24-well plates for treatment. Cells were treated with vehicle only, 10 nM BPA only, or 10 nM BPA with Fulv, Flut, or a combination of both (1 μM). Cells were incubated for 10 days in vitro (DIV) at 37°C with 5% CO2, with 50% media replenishment at 5 DIV. At 10 DIV, primary neurospheres were counted and imaged; then dissociated in sterile media, counted, and replated at 1000 cells/ml; and cultured again for 10 days (until 20 DIV, media were replenished at 15 DIV) as above for secondary neurosphere formation. For differentiation assays, growth factors (EGF and FGF) were removed, and cells were allowed to differentiate for more than 7 days in culture and then fixed in 2% PFA in NeuroCult media. Differentiation was measured by staining for NeuN, GFAP, and PDGFRα (primary antibodies: rabbit anti-NeuN, Abcam, 1:200; rabbit anti-GFAP, Dako, 1:500; goat anti- PDGFRα, R&D Systems, 1:200; secondary antibody: Alexa Fluor 488 donkey anti-rabbit or donkey anti-goat, 1:500) and Sox2 as noted above for embryonic brain sections.
Adult hypothalamic immunohistochemistry
Adult F1 mice were euthanized at ~P200 and brains were fixed in 4% PFA, cryoprotected in 20% sucrose, and then transferred to OCT before sectioning. Sections (10 μm) were stained for AVP, VIP (1:100; guinea pig anti-VIP, Peninsula Labs), cFos (1:700; rabbit anti-cFos, Synaptic Systems), and Sox2 as noted above for embryonic brain sections. Adult F3 mice were euthanized at ~P135, fixed, sectioned, and stained for AVP and VIP as above. Sections were imaged using a Zeiss Axioplan 2 fluorescent microscope, images were processed in Adobe Photoshop CC software, and mean pixel intensity quantification or cell counts were performed with the FIJI image processing package. In all image subfigures, scale bars on micrograph indicate the same size unless specifically noted. For all analyses, male and female mice were used in approximately equal numbers (two each when n = 4; one extra female when n = 3 or 5).
Statistics
All data are presented as means ± SEM. Data were assessed using the GraphPad Prism software package; statistics were performed on raw data and are described in the figure legends. Briefly, unpaired t tests or ANOVAs (with Tukey’s post hoc test for multiple comparisons) were used to analyze data. Except where specified in the text or figure legends, the statistical comparison was an unpaired t test. For all tests, α = 0.05.
Acknowledgments
We thank N. Klenin for assistance with cryosectioning and J. V. Shankara for assistance with circadian monitoring. Funding: This study was supported by postdoctoral fellowships to D.N. from the Canadian Institutes of Health Research (201611MFE-381713-245522), Alberta Innovates Health Solutions (201610151), and the University of Calgary Eyes High Competition; a summer studentship to K.M.F. from Alberta Innovates Health Solutions; Natural Sciences and Engineering Research Council of Canada’s support to M.C.A.; and a Natural Sciences and Engineering Research Council of Canada Discovery grant (DG386445) to D.M.K. Author contributions: D.N., M.C.A., and D.M.K. conceived of the experiments. D.N. and K.M.F. performed the experiments. D.N., K.M.F., M.C.A., and D.M.K. analyzed the data. D.N. and D.M.K. wrote the manuscript. K.M.F. and M.C.A. reviewed and edited the manuscript. Competing interests: The authors declare that they have no competing interests. D.M.K. is a co-founder of Path Therapeutics, focused on epilepsy drug discovery. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/22/eabd1159/DC1
REFERENCES AND NOTES
- 1.Gould J. C., Leonard L. S., Maness S. C., Wagner B. L., Conner K., Zacharewski T., Safe S., McDonnell D. P., Gaido K. W., Bisphenol A interacts with the estrogen receptor α in a distinct manner from estradiol. Mol. Cell. Endocrinol. 142, 203–214 (1998). [DOI] [PubMed] [Google Scholar]
- 2.Kuiper G. G. J. M., Lemmen J. G., Carlsson B., Corton J. C., Safe S. H., van der Saag P. T., van der Burg B., Gustafsson J.-Å., Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor β. Endocrinology 139, 4252–4263 (1998). [DOI] [PubMed] [Google Scholar]
- 3.Tohmé M., Prud’homme S. M., Boulahtouf A., Samarut E., Brunet F., Bernard L., Bourguet W., Gibert Y., Balaguer P., Laudet V., Estrogen-related receptor γ is an in vivo receptor of bisphenol A. FASEB J. 28, 3124–3133 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Lee H. J., Chattopadhyay S., Gong E. Y., Ahn R. S., Lee K., Antiandrogenic effects of bisphenol A and nonylphenol on the function of androgen receptor. Toxicol. Sci. 75, 40–46 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Teng C., Goodwin B., Shockley K., Xia M., Huang R., Norris J., Merrick B. A., Jetten A. M., Austin C. P., Tice R. R., Bisphenol A affects androgen receptor function via multiple mechanisms. Chem. Biol. Interact. 203, 556–564 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moriyama K., Tagami T., Akamizu T., Usui T., Saijo M., Kanamoto N., Hataya Y., Shimatsu A., Kuzuya H., Nakao K., Thyroid hormone action is disrupted by bisphenol A as an antagonist. J. Clin. Endocrinol. Metab. 87, 5185–5190 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Prasanth G. K., Divya L. M., Sadasivan C., Bisphenol-A can bind to human glucocorticoid receptor as an agonist: An in silico study. J. Appl. Toxicol. 30, 769–774 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Schonfelder G., Wittfoht W., Hopp H., Talsness C. E., Paul M., Chahoud I., Parent bisphenol A accumulation in the human maternal-fetal-placental unit. Environ. Health Perspect. 110, A703–A707 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ikezuki Y., Tsutsumi O., Takai Y., Kamei Y., Taketani Y., Determination of bisphenol A concentrations in human biological fluids reveals significant early prenatal exposure. Hum. Reprod. 17, 2839–2841 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Nesan D., Sewell L. C., Kurrasch D. M., Opening the black box of endocrine disruption of brain development: Lessons from the characterization of Bisphenol A. Horm. Behav. 101, 50–58 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Harley K. G., Gunier R. B., Kogut K., Johnson C., Bradman A., Calafat A. M., Eskenazi B., Prenatal and early childhood bisphenol A concentrations and behavior in school-aged children. Environ. Res. 126, 43–50 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tewar S., Auinger P., Braun J. M., Lanphear B., Yolton K., Epstein J. N., Ehrlich S., Froehlich T. E., Association of bisphenol a exposure and attention-deficit/hyperactivity disorder in a national sample of U.S. children. Environ. Res. 150, 112–118 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Cock M., Maas Y. G., van de Bor M., Does perinatal exposure to endocrine disruptors induce autism spectrum and attention deficit hyperactivity disorders? Review. Acta Paediatr. 101, 811–818 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Robinson-Shelton A., Malow B. A., Sleep disturbances in neurodevelopmental disorders. Curr. Psychiatry Rep. 18, 6 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Hoban T. F., Sleeplessness in children with neurodevelopmental disorders. CNS Drugs 14, 11–22 (2000). [Google Scholar]
- 16.Environmental Protection Agency, “TSCA work plan for chemical assessments: 2014 update,” 2014; www.epa.gov/sites/production/files/2015-01/documents/tsca_work_plan_chemicals_2014_update-final.pdf.
- 17.Health Canada, “Health Canada’s updated assessment of bisphenol A (BPA) exposure from food sources,” 2012; www.canada.ca/content/dam/hc-sc/migration/hc-sc/fn-an/alt_formats/pdf/securit/packag-emball/bpa/bpa_hra-ers-2012-09-eng.pdf.
- 18.European Chemicals Agency, “MSC unanimously agrees that Bisphenol A is an endocrine disruptor,” 2017; https://echa.europa.eu/-/msc-unanimously-agrees-that-bisphenol-a-is-an-endocrine-disruptor.
- 19.Welshons W. V., Thayer K. A., Judy B. M., Taylor J. A., Curran E. M., S F., Large effects from small exposures. I. Mechanisms for endocrine-disrupting chemicals with estrogenic activity. Environ. Health Perspect. 111, 994–1006 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.EFSA Panel on Biological Hazards (BIOHAZ) , Scientific opinion on the risks to public health related to the presence of bisphenol A (BPA) in foodstuffs. EFSA J. 13, 4139 (2015).32313571 [Google Scholar]
- 21.Teeguarden J. G., Hanson-Drury S., A systematic review of Bisphenol A "low dose" studies in the context of human exposure: A case for establishing standards for reporting "low-dose" effects of chemicals. Food Chem. Toxicol. 62, 935–948 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Prins G. S., Patisaul H. B., Belcher S. M., Vandenberg L. N., CLARITY-BPA academic laboratory studies identify consistent low-dose Bisphenol A effects on multiple organ systems. Basic Clin. Pharmacol. Toxicol. 125 ( Suppl 3), 14–31 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vandenberg L. N., Ehrlich S., Belcher S. M., Ben-Jonathan N., Dolinoy D. C., Hugo E. R., Hunt P. A., Newbold R. R., Rubin B. S., Saili K. S., Soto A. M., Wang H.-S., vom Saal F. S., Low dose effects of bisphenol A. Endocr. Disruptors 1, e26490 (2014). [Google Scholar]
- 24.Kinch C. D., Ibhazehiebo K., Jeong J. H., Habibi H. R., Kurrasch D. M., Low-dose exposure to bisphenol A and replacement bisphenol S induces precocious hypothalamic neurogenesis in embryonic zebrafish. Proc. Natl. Acad. Sci. U.S.A. 112, 1475–1480 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nesan D., Kurrasch D. M., Genetic programs of the developing tuberal hypothalamus and potential mechanisms of their disruption by environmental factors. Mol. Cell. Endocrinol. 438, 3–17 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Xie Y., Dorsky R. I., Development of the hypothalamus: Conservation, modification and innovation. Development 144, 1588–1599 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.R. Silver, M. Rainbow, in Neuroscience in the 21st Century, D. W. Pfaff, Ed. (Springer, 2013), chap. 66, pp. 1847–1888. [Google Scholar]
- 28.Abrahamson E. E., Moore R. Y., Suprachiasmatic nucleus in the mouse: Retinal innervation, intrinsic organization and efferent projections. Brain Res. 916, 172–191 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Kriegsfeld L. J., Leak R. K., Yackulic C. B., LeSauter J., Silver R., Organization of suprachiasmatic nucleus projections in Syrian hamsters (Mesocricetus auratus): An anterograde and retrograde analysis. J. Comp. Neurol. 468, 361–379 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varadarajan S., Tajiri M., Jain R., Holt R., Ahmed Q., LeSauter J., Silver R., Connectome of the suprachiasmatic nucleus: New evidence of the core-shell relationship. eNeuro 5, ENEURO.0205–ENEU18.2018 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Evans J. A., Suen T. C., Callif B. L., Mitchell A. S., Castanon-Cervantes O., Baker K. M., Kloehn I., Baba K., Teubner B. J., Ehlen J. C., Paul K. N., Bartness T. J., Tosini G., Leise T., Davidson A. J., Shell neurons of the master circadian clock coordinate the phase of tissue clocks throughout the brain and body. BMC Biol. 13, 43 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Padmanabhan V., Siefert K., Ransom S., Johnson T., Pinkerton J., Anderson L., Tao L., Kannan K., Maternal bisphenol-A levels at delivery: A looming problem? J. Perinatol. 28, 258–263 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuroda N., Kinoshita Y., Sun Y., Wada M., Kishikawa N., Nakashima K., Makino T., Nakazawa H., Measurement of bisphenol A levels in human blood serum and ascitic fluid by HPLC using a fluorescent labeling reagent. J. Pharm. Biomed. Anal. 30, 1743–1749 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Gerona R. R., Woodruff T. J., Dickenson C. A., Pan J., Schwartz J. M., Sen S., Friesen M. W., Fujimoto V. Y., Hunt P. A., Bisphenol-A (BPA), BPA glucuronide, and BPA sulfate in midgestation umbilical cord serum in a northern and central California population. Environ. Sci. Technol. 47, 12477–12485 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kundakovic M., Champagne F. A., Epigenetic perspective on the developmental effects of bisphenol A. Brain Behav. Immun. 25, 1084–1093 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aslanpour S., Han S., Schuurmans C., Kurrasch D. M., Neurog2 Acts as a classical proneural gene in the ventromedial hypothalamus and is required for the early phase of neurogenesis. J. Neurosci. 40, 3549–3563 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mieda M., Okamoto H., Sakurai T., Manipulating the cellular circadian period of arginine vasopressin neurons alters the behavioral circadian period. Curr. Biol. 26, 2535–2542 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Mieda M., Ono D., Hasegawa E., Okamoto H., Honma K., Honma S., Sakurai T., Cellular clocks in AVP neurons of the SCN are critical for interneuronal coupling regulating circadian behavior rhythm. Neuron 85, 1103–1116 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Cheng A. H., Bouchard-Cannon P., Hegazi S., Lowden C., Fung S. W., Chiang C.-K., Ness R. W., Cheng H.-M. M., SOX2-dependent transcription in clock neurons promotes the robustness of the central circadian pacemaker. Cell Rep. 26, 3191–3202.e8 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Nesan D., Thornton H. F., Sewell L. C., Kurrasch D. M., An efficient method for generating murine hypothalamic neurospheres for the study of regional neural progenitor biology. Endocrinology 161, bqaa035 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Androutsellis-Theotokis A., Chrousos G. P., McKay R. D., DeCherney A. H., Kino T., Expression profiles of the nuclear receptors and their transcriptional coregulators during differentiation of neural stem cells. Horm. Metab. Res. 45, 159–168 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brannvall K., Korhonen L., Lindholm D., Estrogen-receptor-dependent regulation of neural stem cell proliferation and differentiation. Mol. Cell. Neurosci. 21, 512–520 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Mieda M., The network mechanism of the central circadian pacemaker of the SCN: Do AVP neurons play a more critical role than expected? Front. Neurosci. 13, 139 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bedont J. L., Rohr K. E., Bathini A., Hattar S., Blackshaw S., Sehgal A., Evans J. A., Asymmetric vasopressin signaling spatially organizes the master circadian clock. J. Comp. Neurol. 526, 2048–2067 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pauls S., Foley N. C., Foley D. K., LeSauter J., Hastings M. H., Maywood E. S., Silver R., Differential contributions of intra-cellular and inter-cellular mechanisms to the spatial and temporal architecture of the suprachiasmatic nucleus circadian circuitry in wild-type, cryptochrome-null and vasoactive intestinal peptide receptor 2-null mutant mice. Eur. J. Neurosci. 40, 2528–2540 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rochester J. R., Bolden A. L., Kwiatkowski C. F., Prenatal exposure to bisphenol A and hyperactivity in children: A systematic review and meta-analysis. Environ. Int. 114, 343–356 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Xu X., Hong X., Xie L., Li T., Yang Y., Zhang Q., Zhang G., Liu X., Gestational and lactational exposure to bisphenol-A affects anxiety- and depression-like behaviors in mice. Horm. Behav. 62, 480–490 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Zhou R., Chen F., Feng X., Zhou L., Li Y., Chen L., Perinatal exposure to low-dose of bisphenol A causes anxiety-like alteration in adrenal axis regulation and behaviors of rat offspring: A potential role for metabotropic glutamate 2/3 receptors. J. Psychiatr. Res. 64, 121–129 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Matsuda S., Matsuzawa D., Ishii D., Tomizawa H., Sutoh C., Nakazawa K., Amano K., Sajiki J., Shimizu E., Effects of perinatal exposure to low dose of bisphenol A on anxiety like behavior and dopamine metabolites in brain. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 39, 273–279 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Xu X., Dong F., Yang Y., Wang Y., Wang R., Shen X., Sex-specific effects of long-term exposure to bisphenol-A on anxiety- and depression-like behaviors in adult mice. Chemosphere 120, 258–266 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Rebuli M. E., Camacho L., Adonay M. E., Reif D. M., Aylor D. L., Patisaul H. B., Impact of low-dose oral exposure to bisphenol A (BPA) on juvenile and adult rat exploratory and anxiety behavior: A CLARITY-BPA consortium study. Toxicol. Sci. 148, 341–354 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosenfeld C. S., Bisphenol A and phthalate endocrine disruption of parental and social behaviors. Front. Neurosci. 9, 57 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wolstenholme J. T., Goldsby J. A., Rissman E. F., Transgenerational effects of prenatal bisphenol A on social recognition. Horm. Behav. 64, 833–839 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heard E., Martienssen R. A., Transgenerational epigenetic inheritance: Myths and mechanisms. Cell 157, 95–109 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miska E. A., Ferguson-Smith A. C., Transgenerational inheritance: Models and mechanisms of non-DNA sequence-based inheritance. Science 354, 59–63 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Nesan D., Vijayan M. M., Maternal cortisol mediates hypothalamus-pituitary-interrenal axis development in zebrafish. Sci. Rep. 6, 22582 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hayward L. S., Wingfield J. C., Maternal corticosterone is transferred to avian yolk and may alter offspring growth and adult phenotype. Gen. Comp. Endocrinol. 135, 365–371 (2004). [DOI] [PubMed] [Google Scholar]
- 58.Bosch O. J., Maternal nurturing is dependent on her innate anxiety: The behavioral roles of brain oxytocin and vasopressin. Horm. Behav. 59, 202–212 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Boudalia S., Berges R., Chabanet C., Folia M., Decocq L., Pasquis B., Abdennebi-Najar L., Canivenc-Lavier M.-C., A multi-generational study on low-dose BPA exposure in Wistar rats: Effects on maternal behavior, flavor intake and development. Neurotoxicol. Teratol. 41, 16–26 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Palanza P. L., Howdeshell K. L., Parmigiani S., vom Saal F. S., Exposure to a low dose of bisphenol A during fetal life or in adulthood alters maternal behavior in mice. Environ. Health Perspect. 110 ( Suppl 3), 415–422 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Coutellier L., Friedrich A. C., Failing K., Wurbel H., Variations in the postnatal maternal environment in mice: Effects on maternal behaviour and behavioural and endocrine responses in the adult offspring. Physiol. Behav. 93, 395–407 (2008). [DOI] [PubMed] [Google Scholar]
- 62.Curley J. P., Champagne F. A., Bateson P., Keverne E. B., Transgenerational effects of impaired maternal care on behaviour of offspring and grandoffspring. Anim. Behav. 75, 1551–1561 (2008). [Google Scholar]
- 63.National Toxicology Program, “NTP research report on the CLARITY-BPA core study: A perinatal and chronic extended-dose-range study of bisphenol A in rats” (Research Report 9, 2018); https://ntp.niehs.nih.gov/ntp/results/pubs/rr/reports/rr09_508.pdf. [PubMed]
- 64.Arambula S. E., Fuchs J., Cao J., Patisaul H. B., Effects of perinatal bisphenol A exposure on the volume of sexually-dimorphic nuclei of juvenile rats: A CLARITY-BPA consortium study. Neurotoxicology 63, 33–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Witchey S. K., Fuchs J., Patisaul H. B., Perinatal bisphenol A (BPA) exposure alters brain oxytocin receptor (OTR) expression in a sex- and region- specific manner: A CLARITY-BPA consortium follow-up study. Neurotoxicology 74, 139–148 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Arambula S. E., Belcher S. M., Planchart A., Turner S. D., Patisaul H. B., Impact of low dose oral exposure to bisphenol A (BPA) on the neonatal rat hypothalamic and hippocampal transcriptome: A CLARITY-BPA consortium study. Endocrinology 157, 3856–3872 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arambula S. E., Jima D., Patisaul H. B., Prenatal bisphenol A (BPA) exposure alters the transcriptome of the neonate rat amygdala in a sex-specific manner: A CLARITY-BPA consortium study. Neurotoxicology 65, 207–220 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vandenberg L. N., Non-monotonic dose responses in studies of endocrine disrupting chemicals: Bisphenol a as a case study. Dose Response 12, 259–276 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Flint S., Markle T., Thompson S., Wallace E., Bisphenol A exposure, effects, and policy: A wildlife perspective. J. Environ. Manag. 104, 19–34 (2012). [DOI] [PubMed] [Google Scholar]
- 70.Kim M. E., Park H. R., Gong E. J., Choi S. Y., Kim H. S., Lee J., Exposure to bisphenol A appears to impair hippocampal neurogenesis and spatial learning and memory. Food Chem. Toxicol. 49, 3383–3389 (2011). [DOI] [PubMed] [Google Scholar]
- 71.Komada M., Asai Y., Morii M., Matsuki M., Sato M., Nagao T., Maternal bisphenol A oral dosing relates to the acceleration of neurogenesis in the developing neocortex of mouse fetuses. Toxicology 295, 31–38 (2012). [DOI] [PubMed] [Google Scholar]
- 72.Desai M., Ferrini M. G., Han G., Jellyman J. K., Ross M. G., In vivo maternal and in vitro BPA exposure effects on hypothalamic neurogenesis and appetite regulators. Environ. Res. 164, 45–52 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Patisaul H. B., Achieving CLARITY on bisphenol A, brain and behaviour. J. Neuroendocrinol. 32, e12730 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vandenberg L. N., Hunt P. A., Gore A. C., Endocrine disruptors and the future of toxicology testing - lessons from CLARITY-BPA. Nat. Rev. Endocrinol. 15, 366–374 (2019). [DOI] [PubMed] [Google Scholar]
- 75.Gerona R., vom Saal F. S., Hunt P. A., BPA: Have flawed analytical techniques compromised risk assessments? Lancet Diabetes Endocrinol. 8, 11–13 (2020). [DOI] [PubMed] [Google Scholar]
- 76.Lehmler H. J., Liu B., Gadogbe M., Bao W., Exposure to Bisphenol A, Bisphenol F, and Bisphenol S in U.S. adults and children: The national health and nutrition examination survey 2013-2014. ACS Omega 3, 6523–6532 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/22/eabd1159/DC1