

Sarcomeric proteins FATZ-1 and α-actinin-2 associate in a tight fuzzy complex, which forms phase-separated condensates.
Abstract
In sarcomeres, α-actinin cross-links actin filaments and anchors them to the Z-disk. FATZ (filamin-, α-actinin-, and telethonin-binding protein of the Z-disk) proteins interact with α-actinin and other core Z-disk proteins, contributing to myofibril assembly and maintenance. Here, we report the first structure and its cellular validation of α-actinin-2 in complex with a Z-disk partner, FATZ-1, which is best described as a conformational ensemble. We show that FATZ-1 forms a tight fuzzy complex with α-actinin-2 and propose an interaction mechanism via main molecular recognition elements and secondary binding sites. The obtained integrative model reveals a polar architecture of the complex which, in combination with FATZ-1 multivalent scaffold function, might organize interaction partners and stabilize α-actinin-2 preferential orientation in Z-disk. Last, we uncover FATZ-1 ability to phase-separate and form biomolecular condensates with α-actinin-2, raising the question whether FATZ proteins can create an interaction hub for Z-disk proteins through membraneless compartmentalization during myofibrillogenesis.
INTRODUCTION
Voluntary animal movement and involuntary heart beating are produced by the contraction of striated muscles. Sarcomeres, the basic contractile units of striated muscle cells, are composed of arrays of thin (actin) and thick (myosin) filaments that slide past each other during contraction (Fig. 1A). The Z-disk forms the boundary between adjacent sarcomeres, where antiparallel actin filaments (F-actin) are anchored. The mechanical force generated by the interaction between myosin and actin requires a suitably stable anchoring structure. The Z-disk fulfills this role, acting both as a mechanical hub and a signaling platform, allowing transmission of tension during contraction, as well as sensation and transmission of information about biomechanical stress. Accordingly, mutations that disrupt Z-disk architecture and function cause skeletal and cardiac myopathies (1, 2).
Fig. 1. Proteins of the FATZ family display intrinsic disorder.
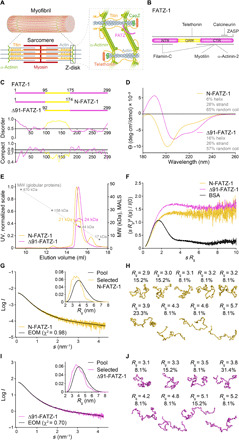
(A) Schematics of the striated muscle sarcomere and close-up view of F-actin/α-actinin/FATZ interactions in Z-disk. (B) Schematics of the FATZ-1 interactome and binding sites reported to date. (C) Schematics of the main FATZ-1 constructs, along with their amino acid boundaries and domain composition. Predicted disordered regions (above 0.5) and compactness (above 0.8) are shown below. (D) Circular dichroism (CD) spectra of N-FATZ-1 and Δ91-FATZ-1, along with calculated secondary structure content. (E) Size exclusion chromatography (SEC)–multiangle light scattering (MALS) analysis of N-FATZ-1 and Δ91-FATZ-1, yielding molecular weights (MWs) of 21 and 24 kDa, respectively. Elution volumes were lower than anticipated relative to globular standards [thyroglobulin (670 kDa), γ-globulin (158 kDa), ovalbumin (44 kDa), and myoglobin (17 kDa)], corresponding to MWs of 50 and 46 kDa for N-FATZ-1 and Δ91-FATZ-1, respectively. UV, ultraviolet. (F) Dimensionless Kratky plots of N-FATZ-1 and Δ91-FATZ-1, as well as of globular bovine serum albumin (BSA) (SASBDB code SASDFQ8). Experimental SEC–small-angle x-ray scattering (SAXS) data of N-FATZ-1 (G) and Δ91-FATZ-1 (I) and corresponding fit to the data of selected ensembles obtained from Ensemble Optimization Method (EOM) (43, 44). Rg distributions of selected ensembles relative to the distribution of a random pool are shown in the insets. Model representatives of the selected EOM ensembles for N-FATZ-1 (H) and Δ91-FATZ-1 (J), along with their Rg (in nanometers) and volume fractions (in percentage). See also figs. S1 to S5 and tables S1 and S2.
The Z-disk is arranged in a remarkable paracrystalline tetragonal lattice of actin filaments through the interplay between α-actinin-2 and titin (3, 4). According to the premyofibril model of myofibrillogenesis, Z-disks originate from Z-bodies, which are protein complexes that grow in size, fuse together, and build mature Z-disks in nascent myofibrils by associating with a plethora of other proteins (5–9). Myofibrils form the sarcomere after assembling with thin and thick filaments in a parallel and repetitive array (Fig. 1A). Among the core proteins identified as being important for Z-body formation are α-actinin-2, FATZ-1 (filamin-, α-actinin-, and telethonin-binding protein of the Z-disk, also known as myozenin-1 or calsarcin-2), ZASP (Z-disk–associated, alternatively spliced, PDZ motif–containing protein), myotilin, filamin-C, and actin (7). However, the molecular details of their interactions and the importance of these interactions for Z-body and thus Z-disk formation are not known.
α-Actinin was originally reported as an F-actin cross-linking protein in muscle Z-disks (10), and four closely related isogenes fulfill similar functions in all cell types (11). α-Actinin-2 is the major Z-disk component, cross-linking antiparallel actin filaments from adjacent sarcomeres and serving as a binding platform for a number of Z-disk proteins such as titin, FATZ-1, ZASP, myotilin, myopalladin, and myopodin (Fig. 1A) (11). Both functions are possible thanks to α-actinin-2’s unique antiparallel dimeric architecture, with each subunit consisting of an N-terminal F-actin binding domain (ABD), connected by a flexible neck region to a central rod domain, comprising four spectrin-like repeats (SRs) and a C-terminal calmodulin-like domain (CAMD) with two pairs of EF-hand motifs (EF1-2 and EF3-4), which do not bind calcium (12). While the ABD interacts with F-actin, the rod and EF3-4 are hotspots for binding to other partners (12). In addition, the rod exhibits a torsional twist of ~90° from one end to the other, which, together with the flexible neck region, is critical for the F-actin cross-linking function of α-actinin-2 (12, 13).
FATZ-1 is a member of the FATZ family, which also includes FATZ-2 (myozenin-2 or calsarcin-1) and FATZ-3 (myozenin-3 or calsarcin-3) (14, 15). The FATZ-1 and FATZ-3 proteins are expressed in skeletal muscle and enriched in fast-twitch fibers, whereas FATZ-2 is highly expressed in cardiac muscle and in slow-twitch skeletal fibers. All three family members interact with filamin-C, α-actinin-2/-3, telethonin, myotilin, and members of the Enigma family of PDZ-LIM proteins, including ZASP (14–19). FATZ proteins also bind to calcineurin (16, 17) and modulate fiber-type composition of skeletal muscle and hypertrophy of cardiac muscle through inhibition of calcineurin (20, 21). FATZ proteins are thus believed to have an important role in myofibrillogenesis not only by serving as an interaction hub for Z-disk proteins but also by localizing calcineurin signaling to the sarcomere (16, 17). Two mutations in the MYOZ2 gene that encodes FATZ-2 have been linked to hypertrophic cardiomyopathy (22, 23), although pathogenic mutations have been rarely found in patients (24–26). MYOZ1 knockout (KO) mice exhibit reduced body weight and fast-twitch muscle mass, resulting from increased calcineurin signaling (20, 21), while MYOZ2 KO mice show accelerated cardiomyopathy in response to pathological biomechanical stress (27, 28). Recently, FATZ-1 and FATZ-3 were shown to be good markers for myofiber maturation during muscle regeneration (29).
FATZ proteins bind to α-actinin-2/-3 SR3-4, filamin-C, and myotilin through their C-terminal region (CTR) (14–18) and to the PDZ domain of Enigma family members via a specific C-terminal recognition motif (Fig. 1B) (19). α-Actinin-2 and filamin-C compete with each other for binding to the CTR of FATZ-1 (15). In addition, a second binding site for filamin-C was reported in the N-terminal region (NTR) of FATZ-1 (18), and the interaction site with another of its binding partners, telethonin, has not been mapped yet. FATZ proteins do not display canonical protein-protein interaction domains, and the molecular mechanism of binding with their many interaction partners remains unknown.
Here, we demonstrate that FATZ proteins contain intrinsically disordered regions (IDRs) and are best described as a “conformational ensemble,” i.e., they are flexible and lack a stable tertiary structure. The results from in vitro binding assays and other biophysical characterization methods, combined with an integrative structural biology approach incorporating x-ray crystallography and small-angle x-ray scattering (SAXS), yield what is best described as a “fuzzy” α-actinin-2/FATZ-1 complex. FATZ-1 might play an organizational role in the Z-disk due to its multivalent scaffolding properties and formation of a tight complex of polar architecture with α-actinin-2. Last, as FATZ-1 is present from the initial stages of myofibrillogenesis (8, 9), both its homotypic interactions revealed by nuclear magnetic resonance (NMR) and the protein’s capacity to phase-separate and form a biomolecular condensate with α-actinin-2 raise the tantalizing question whether FATZ proteins can function as an interaction nucleus for Z-disk partners by promoting membraneless compartmentalization.
RESULTS
Proteins of the FATZ family display intrinsic disorder
Proteins of the FATZ family are found across all vertebrates. Human FATZ-1, FATZ-2, and FATZ-3 share 34 to 40% sequence identity, with FATZ-2 and FATZ-3 being most closely related (fig. S1). FATZ-1 can be divided into three regions based on conservation and residue content [NTR, glycine-rich region (GRR), and CTR; see Fig. 1B, Supplementary Results, figs. S1A and S2, and (14)]. FATZ proteins show predicted intrinsic disorder at N and C termini as well as a central intrinsically disordered low-complexity region (GRR in FATZ-1) (Fig. 1C and figs. S2 and S3).
Because of challenging solubility properties of FATZ-1, we used an Erase-a-Base approach (30) and selected from 14 soluble expressing constructs, N-FATZ-1 and Δ91-FATZ-1, which comprise the NTR and CTR, respectively, and overlap within the GRR (Fig. 1C and Supplementary Results). Circular dichroism (CD) analysis indicated a predominant (ca. 60%) random coil content for both constructs, in line with a high degree of disorder predicted for the GRR, with N-FATZ-1 having a lower α-helical content than Δ91-FATZ-1 (Fig. 1D). The calculation of the compaction index using hydrodynamic radius (Rh) values derived from size exclusion chromatography (SEC) combined with dynamic light scattering (DLS) and viscometry measurements (table S1) (31), together with the analysis of the 222/200 ellipticity ratio, indicated a premolten globule for N-FATZ-1 and an increased content of regular secondary structure for Δ91-FATZ-1. This is in line with limited proteolysis (LP) experiments, revealing a number of defined proteolysis-resistant fragments (see Supplementary Results and fig. S4).
SEC combined with multiangle light scattering (MALS) showed that N-FATZ-1 and Δ91-FATZ-1 are predominantly monomers under the experimental conditions (see Methods). However, these monomers elute with lower retention volumes than would otherwise be expected relative to globular protein standards (see Fig. 1E and table S1), suggesting that the predicted disorder of the proteins affects SEC retention volume. We further characterized N-FATZ-1 and Δ91-FATZ-1 using SEC combined with SAXS. The resulting scattering profiles and dimensionless Kratky plots are characteristic of intrinsically disordered proteins (IDPs), and subsequent modeling indicates that both constructs are best described as a conformational ensemble (Fig. 1, F to J, and Supplementary Results) (32). In addition, we derived the radius of gyration (Rg) of N-FATZ-1 and Δ91-FATZ-1 from analysis of SAXS data (table S2) and obtained Rg/Rh ratios of 1.1 to 1.2 that are, once again, consistent with the presence of IDRs (33). These results, demonstrating the intrinsically disordered/ensemble-state nature of N-FATZ-1 and Δ91-FATZ-1, are further supported by the 1H-15N heteronuclear single-quantum coherence (HSQC) spectra recorded for both constructs, which display a narrow chemical shift range in the proton dimension, as is typical for IDR-containing proteins (fig. S5A).
Proteins of the FATZ family form a tight 2:1 complex with α-actinin-2 dimer
To characterize the binding stoichiometry of FATZ-1, FATZ-2, and FATZ-3 to α-actinin-2, we used SEC-MALS and several α-actinin-2 constructs that included the full-length functional dimer, an engineered α-actinin-2 half-dimer (hd-α-actinin-2), and the isolated dimeric rod domain (rod-α-actinin-2), which lacks the ABD and EF hands (Fig. 2A). SEC-MALS results showed that each of the three FATZ proteins forms a tight complex with α-actinin-2 even in the presence of 250 mM arginine (Fig. 2B and fig. S6A), with a binding stoichiometry of two FATZ molecules per α-actinin-2 dimer, that is, one FATZ molecule per α-actinin-2 subunit or hd-α-actinin-2 (table S1). In relation to the binding of the shorter FATZ-1 constructs, we could not detect complex formation between N-FATZ-1 and rod-α-actinin-2 (Fig. 2C), while Δ91-FATZ-1 formed 2:1, 2:1, and 1:1 complexes with α-actinin-2, rod-α-actinin-2, and hd-α-actinin-2, respectively (Fig. 2D, fig. S6, B and C, and table S1). In SEC-MALS, we also observed a 2:1 complex between Δ91-FATZ-1 and human (nonmuscle) α-actinin-1 (fig. S6D and table S1).
Fig. 2. FATZ-1 forms a tight 2:1 complex with α-actinin-2 dimer via multiple binding sites.
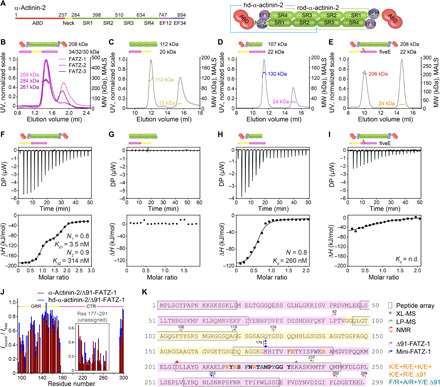
(A) Schematics of the α-actinin-2 constructs, along with their amino acid boundaries and domain composition. SEC-MALS analysis for the interaction of FATZ-1, FATZ-2, and FATZ-3 with α-actinin-2 (B), N-FATZ-1 with rod-α-actinin-2 (C), Δ91-FATZ-1 with hd-α-actinin-2 (D), and fiveE Δ91-FATZ-1 mutant with α-actinin-2 (E). ITC analysis for the interaction of Δ91-FATZ-1 with α-actinin-2 (F), N-FATZ-1 with rod-α-actinin-2 (G), Δ91-FATZ-1 with hd-α-actinin-2 (H), and fiveE Δ91-FATZ-1 mutant with α-actinin-2 (I). n.d., not determined. (J) 1H-15N HSQC signal intensity ratio of 15N Δ91-FATZ-1 bound/free, mapping FATZ-1 primary binding site for α-actinin-2. Unassigned part in FATZ-1 is boxed, and residues are plotted at a random position. (K) Sequence of FATZ-1 showing multiple interaction sites for α-actinin-2 as determined from the peptide array (squared residues), XL-MS (star), LP-MS (residues delimited by arrows), and NMR (arrows). Residues matching the strongest signal peptide in the peptide array are shown in bold. Boundaries for Δ91-FATZ-1 and mini-FATZ-1 are delimited by arrows. Mutations within fiveE Δ91-FATZ-1 and RRE Δ91-FATZ-1 are indicated in orange and dark cyan, respectively. See also figs. S5 to S9 and tables S1, S3, and S7 for statistical analysis and table S8. DP, differential power.
We used isothermal titration calorimetry (ITC) to quantify the interaction affinity. N-FATZ-1 did not interact with rod-α-actinin-2, whereas Δ91-FATZ-1 showed a strong 2:1, 2:1, and 1:1 interaction with α-actinin-2 dimer, dimeric rod-α-actinin-2, and hd-α-actinin-2, respectively, in agreement with SEC-MALS data (Fig. 2, F to H, fig. S6E, and table S3). To fit the isotherm between Δ91-FATZ-1 and α-actinin-2 or rod-α-actinin-2, we used a two-site binding model, which assumes two independent binding events and obtained two Kd (equilibrium dissociation constant) values: Kd1 = 2.6 to 3.5 nM and Kd2 = 294 to 314 nM (Fig. 2F, fig. S6E, and table S3). Using a one-site binding model for the interaction between Δ91-FATZ-1 and hd-α-actinin-2, we obtained a Kd = 260 nM, similar to Kd2 for Δ91-FATZ-1 and α-actinin-2/rod-α-actinin-2 (Fig. 2H and table S3).
Binding of Δ91-FATZ-1 to each of the three α-actinin-2 constructs (α-actinin-2, rod-α-actinin-2, and hd-α-actinin-2) was enthalpy-driven, suggesting predominant polar interactions. Together, SEC-MALS and ITC data revealed that (i) FATZ-1 interacts with SR1-SR2 from one and SR3-SR4 from the adjacent α-actinin-2 subunit via its CTR; (ii) the nanomolar Kd1 for the binding of the first Δ91-FATZ-1 molecule (i.e., for the first binding event) can be explained by the contribution of additional, probably transient interactions between Δ91-FATZ-1 and the adjacent yet nonoccupied binding sites on α-actinin-2, with a corresponding ΔH = −110.8 kJ/mol (fig. S6 and table S3); and (iii) the association of the second Δ91-FATZ-1 molecule by contrast yields a low-micromolar Kd2 because it cannot exploit the same interaction surface as the first one, which is reflected in a halved binding enthalpy and a notably reduced entropic penalty (−TΔS = 62.3 kJ/mol versus 6.2 kJ/mol; fig. S6 and table S3). Accordingly, the binding of Δ91-FATZ-1 to hd-α-actinin-2 is comparable to that of the second Δ91-FATZ-1 molecule to α-actinin-2 (see Discussion for the complete model of the α-actinin-2/FATZ-1 binding mechanism).
FATZ-1 interacts with α-actinin-2 via multiple binding sites
To better map FATZ-1–binding site(s) for α-actinin-2, we used an array of overlapping peptides covering FATZ-1 and detected one region within the CTR, exhibiting the strongest signal, and five additional regions spread over NTR, GRR, and CTR, displaying weaker signals (see fig. S7). We next designed the “fiveE” Δ91-FATZ-1 mutant, based on sequence alignment and metastructure analysis of FATZ proteins, in which five conserved positively charged residues within the CTR were reverted to negatively charged glutamates (K182E+R190E+K217E+K219E+R223E; see Methods). This construct did not form a stable complex with α-actinin-2 as assessed by SEC-MALS (Fig. 2E), in agreement with a very low ITC signal and for which a Kd could not be determined (see Fig. 2I and table S3).
To further narrow down FATZ-1–binding site(s), we used LP and chemical cross-linking coupled with mass spectrometry (LP-MS and XL-MS, respectively) on the α-actinin-2/FATZ-1 complex. LP-MS revealed a protected (i.e., interacting) Δ91-FATZ-1 region spanning residues 119 to 237 (fig. S8). XL-MS, which was performed using a zero-length cross-linker, revealed three specific cross-links, K383/D205, E567/K233, and D893/K176 (see fig. S9; α-actinin-2 residues in italics), supporting the interaction between the CTR of FATZ-1 and the rod of α-actinin-2 (fig. S6E).
We next used two-dimensional (2D) 1H-15N HSQC NMR on 15N-labeled Δ91-FATZ-1 to delineate its binding site(s) for α-actinin-2 at the amino acid level. We could not fully assign Δ91-FATZ-1 due to fast relaxation of cross-peaks in the region comprising residues 177 to 291. We could, however, locate the α-actinin-2–interacting region within the CTR at residues 176 to 292, as the corresponding cross-peaks disappeared or showed a drop in signal intensity (Fig. 2J and fig. S5B). We found a similar signal reduction pattern and thus binding behavior for both α-actinin-2 and hd-α-actinin-2. Further, a decrease in signal intensity upon binding to α-actinin-2 around residues 106 and 128, mapping to the GRR, indicated additional binding site(s) apart from the interacting region found within the CTR, which is in line with ITC and peptide array data (Fig. 2K and figs. S6 and S7).
Last, we designed the trx-Δ91-FATZ-1 and Δ91-FATZ-1-trx constructs by adding thioredoxin (trx) to the N or C terminus of Δ91-FATZ-1 to sterically perturb the secondary binding sites present around residues 110 and 280, detected by NMR, peptide array, and LP-MS (figs. S5B and S7). ITC isotherms for trx-Δ91-FATZ-1 and α-actinin-2 could only be fitted using a one-site binding model, giving a Kd = 1.5 μM and a binding stoichiometry of 1.3 (see fig. S6F and table S3), indicating a major effect of the trx likely on the binding site around residue 110. Although ITC experiments between Δ91-FATZ-1-trx and α-actinin-2 could be fitted using a two-site binding model, Kd values were higher than those of Δ91-FATZ-1 and α-actinin-2/rod-α-actinin-2 (Kd1 = 7.8 nM and Kd2 = 408 nM; fig. S6G and table S3), with two much less pronounced transitions, indicating that the trx also interferes with the C-terminal secondary binding region. Together, our results indicate that the main α-actinin-2 interaction site resides in the CTR of FATZ-1, which, unlike the GRR, has a certain degree of secondary structure (Fig. 1C), and is flanked by additional binding sites (Fig. 2K).
Crystal structures of α-actinin-2/FATZ-1 reveal two linear binding motifs in FATZ-1
We combined the information from the peptide array, fiveE Δ91-FATZ-1 mutant, LP-MS, XL-MS, and NMR and generated a shorter construct, mini-FATZ-1 (residues 176 to 263; Fig. 2K), to aid the crystallization of the α-actinin-2/FATZ-1 complex. Although mini-FATZ-1 was only soluble in the presence of ≥2 M urea, we managed to form a complex with rod-α-actinin-2, obtain crystals that diffracted to 2.7-Å resolution, and solve the structure by molecular replacement (see Methods and table S4). Two distinct fragments of FATZ-1 were visible in the difference electron density. To unambiguously assign their side chains, we used selenomethionine (Se-Met)–labeled mini-FATZ-1 and could identify the position of M192 and M226 by anomalous difference Fourier analysis (Fig. 3A and fig. S10A), which allowed building the two stretches of residues and refining the structure to final Rwork/Rfree values of 20.4%/23.5%. To provide a more complete model, we crystallized the hd-α-actinin-2/Δ91-FATZ-1 complex and collected diffraction data to 3.2-Å resolution. The refinement to the final Rwork/Rfree values of 25.6%/28.4% was bootstrapped using the structure of rod-α-actinin-2/mini-FATZ-1 (Fig. 3B and table S4).
Fig. 3. Crystal structures of α-actinin-2/FATZ-1 reveal two linear binding motifs in FATZ-1.
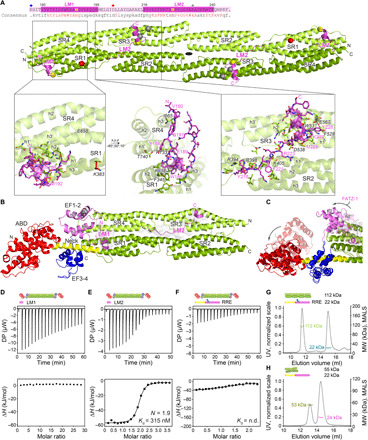
(A) Crystal structure of rod-α-actinin-2/mini-FATZ-1 (in green/magenta), along with the determined FATZ-1 consensus sequence (35 to 80% and 60 to 84% pairwise sequence identity for LM1 and LM2, respectively). Cross-linked residues are indicated by blue, red, and gray stars/balls/sticks on the sequence/structure. Identified Se-Mets are shown in yellow. The rod-α-actinin-2 dimer is assembled through a crystallographic twofold axis between symmetry mates (black circle). Interacting residues (rod-α-actinin-2 in italics), along with helices from SR1/SR2 (h1, h2, and h3) and SR3/SR4 (h1′, h2′, and h3′), are shown in close-up views. (B) Crystal structure of hd-α-actinin-2/Δ91-FATZ-1 (LM1 and LM2 as magenta cartoon and transparent gray surface; hd-α-actinin-2 color-coded as in Fig. 2A). (C) Comparison of unbound [Protein Data Bank (PDB) code 4D1E] and bound (this work) hd-α-actinin-2. ABD and EF1-2 of unbound hd-α-actinin-2 are shown with transparency. ITC analysis for the interaction of LM1 peptide with α-actinin-2 (D), LM2 peptide with α-actinin-2 (E), and RRE Δ91-FATZ-1 mutant with α-actinin-2 (F). SEC-MALS analysis for the interaction of RRE Δ91-FATZ-1 mutant with rod-α-actinin-2 (G) and Δ91-FATZ-1 with E. histolytica rod-α-actinin-2 (H). See also figs. S6 to S11 and S18, tables S1 and S3 to S7, and movie S1.
Comparison of rod-α-actinin-2 in the bound and unbound state revealed that FATZ-1 did not significantly affect the structure of the SRs, as only slight deviations were observed at the edges of each rod subunit [root mean square deviation (RMSD) of 1.17 Å for 844 common Cα atoms of 948 total Cα atoms; see fig. S10B). By contrast, comparison of hd-α-actinin-2 in the bound and unbound state showed larger differences (RMSD of 1.99 Å for 587 common Cα atoms of 856 total Cα atoms), with rotations of 70° and 37° for ABD and EF1-2, respectively (Fig. 3C), as determined with the DynDom server (34).
While movement of the ABD is the result of a built-in flexibility in the neck region required for the protein’s F-actin cross-linking function (12, 35), that of EF1-2 is likely induced by binding to FATZ-1 LM1, which partially occupies the position of EF1-2 in unbound hd-α-actinin-2, decreasing the interface area with the rod from 739 to 464 Å2 (Fig. 3C). The position of ABD and EF1-2 is further stabilized by crystal packing contacts, sharing an interaction interface of 375 Å2. The EF1-2/LM1-interacting region comprises a loop between h3 and h4 that could not be modeled with confidence due to poor electron density in this area of the map, precluding a more detailed analysis (see Methods).
In both α-actinin-2/FATZ-1 structures, one can see two identical stretches of amino acids bound to α-actinin-2, which are conserved among FATZ proteins (62 and 71% average identity for LM1 and LM2, respectively) and conform to the definition of eukaryotic linear motifs (ELMs) (Fig. 3, A and B) (36, 37). Accordingly, they are hereafter called LM1 (residues 180 to 199) and LM2 (residues 216 to 240). LM1 and LM2 are connected by a short 16- and 13-residue linker in rod-α-actinin-2/mini-FATZ-1 and hd-α-actinin-2/Δ91-FATZ-1, respectively, which is not visible in electron density maps and matches the positions of previously identified cross-links (Fig. 3A and fig. S9B). LM1 runs perpendicular to SR4 from one subunit and across the groove formed by SR4 from one and SR1 from the adjacent subunit, being anchored to SR4 h3′. It thereafter forms a five-residue α helix stabilized on SR1 h2 and h3 and returns toward SR4 in a trace that is almost parallel to its N-terminal segment (see Fig. 3, A and B, Supplementary Results, and fig. S10, C and D). LM2 runs in an extended conformation over the groove formed by SR2 h1 and h2. It then crosses over into the groove formed by SR3 h1′ and h2′ and thereafter forms a 12-residue α helix that lies perpendicular to SR3 h2′ and is halved in hd-α-actinin-2/Δ91-FATZ-1 compared to rod-α-actinin-2/mini-FATZ-1.
Interface analysis of rod-α-actinin-2/hd-α-actinin-2 FATZ-1 complexes using the PISA server (38) showed a large interface area of 1578/1887 Å2 contributed by 555/729 Å2 (20/16 residues) and 1023/1158 Å2 (25/22 residues) from LM1 and LM2, respectively (see Supplementary Results and table S5), correlating with the tight nature of the complex. The increased interface area in the hd-α-actinin-2/Δ91-FATZ-1 complex is due to the EF1-2/LM1 interaction and an N-terminally extended LM2-binding site (see Fig. 3, A and B, and Supplementary Results). The P values describing interface compatibility of LM1 and LM2 in rod-α-actinin-2/hd-α-actinin-2 complexes are 0.41/0.29 and 0.65/0.45, respectively. These rather high values indicate low complementarity of hydrophobic residues and are in agreement with a large number of stabilizing polar interactions (table S6) (38), corroborating the enthalpy-driven reaction observed by ITC (fig. S6H). Both LM1 and LM2 show an overall positive electrostatic potential at the binding interface, with a pronounced complementarity at LM2 with respect to the predominantly negatively charged surface of α-actinin-2 rod (fig. S10E).
To dissect the contribution of LM1 and LM2 to the binding affinity, we used ITC with one peptide corresponding to each motif. While we could not detect binding between LM1 peptide and α-actinin-2 (Fig. 3D), LM2 peptide bound to α-actinin-2 with a binding stoichiometry of 1.9 and a Kd of 315 nM (Fig. 3E), which fits that of Δ91-FATZ-1–binding to hd-α-actinin-2 (Kd = 260 nM; see table S3). The Kd of LM1 was predicted to be 882 μM using worm-like chain modeling, which calculates the global Kd value from the individual Kd values of intramolecular binding motifs connected by flexible linkers (39, 40). For this calculation, we used the experimentally determined Kd values of Δ91-FATZ-1 and LM2 for hd-α-actinin-2 and α-actinin-2, respectively (table S3), as well as the length of the LM1-LM2 linker (13 residues) and distance between the two motifs in the hd-α-actinin-2/Δ91-FATZ-1 structure (29.6 Å). The weak and strong affinities of LM1 and LM2 for α-actinin-2 agree with the peptide array data: weak and strong signal for peptides corresponding to LM1 and LM2, respectively (fig. S7), with the strongest signal for the peptide comprising residues 217 to 231. These diverse binding affinities match the structure of rod-α-actinin-2/Δ91-FATZ-1, which was additionally crystallized and displays only a shorter LM2 bound to α-actinin-2 rod [see fig. S10, A (right) and D, and table S4), revealing LM2 as the major binding site. While LM1 and LM2 show a fixed conformation when bound to α-actinin-2, CD spectra of both peptides showed a random coil conformation (fig. S11), suggesting that they are molecular recognition elements (MoREs) [see below and (41, 42)].
To validate our structures, we designed the RRE Δ91-FATZ-1 mutant, which contains three mutations within LM2 residues 217 to 231 (F221R+A225R+Y228E; Fig. 2K). This mutant did not form a complex with α-actinin-2 according to SEC-MALS and the interaction displayed a very low signal in ITC, for which a Kd could not be determined (see Fig. 3, F and G, and tables S1 and S3). Last, Δ91-FATZ-1 did not form a stable complex with the two-SR rod of the distantly related Entamoeba histolytica α-actinin-2 by SEC-MALS (Fig. 3H), showing recognition specificity of FATZ-1.
FATZ-1 forms a fuzzy complex with α-actinin-2 resulting in a polar architecture of the complex
To investigate the conformation and structure of the flexible parts of FATZ-1 in complex with α-actinin-2, we used CD and SAXS. As expected, CD analysis of rod-α-actinin-2 showed that it is mainly α-helical (83%; fig. S11). In addition, analysis of the rod-α-actinin-2/Δ91-FATZ-1 complex revealed a secondary structure content corresponding to that of the stoichiometrically weighted sum of the two individual proteins (fig. S11), indicating no gain in secondary structure for FATZ-1 upon binding to α-actinin-2.
For the SAXS analysis of the α-actinin-2/FATZ-1 complex, we first analyzed rod-α-actinin-2 and hd-α-actinin-2. While rod-α-actinin-2 retains a rigid architecture in solution, the ABD and EF-hand motifs in hd-α-actinin-2 are not spatially fixed relative to the rod but can sample different spatial positions, which was not captured in the crystal structure (see Supplementary Results and figs. S12, A to C and E, and S13, A and B). To confirm that LM1 is a weaker binder for α-actinin-2 than LM2 (Fig. 3), we generated two sets of rod-α-actinin-2/Δ91-FATZ-1 models comprising LM1 fixed or free and subsequently fitted the SAXS data. For both rod-α-actinin-2/Δ91-FATZ-1 and hd-α-actinin-2/Δ91-FATZ-1 complexes, a better fit was obtained with LM1 free (figs. S12, D and F, and S13, C to F), in agreement with ITC and peptide array data. For both complexes, models with LM1 free were selected over those with LM1 fixed, when simultaneously using LM1-fixed and LM1-free pools (Fig. 4, A to D).
Fig. 4. FATZ-1 forms a fuzzy complex with α-actinin-2 resulting in a polar architecture of the complex.
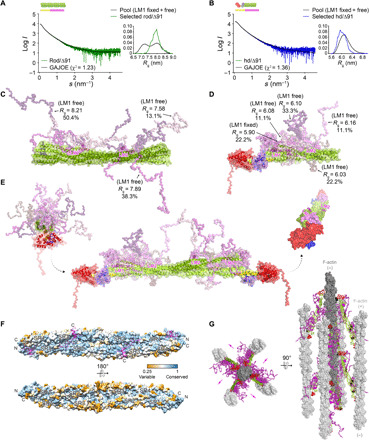
Experimental SAXS data of rod-α-actinin-2/Δ91-FATZ-1 (A) and hd-α-actinin-2/Δ91-FATZ-1 (B), with the corresponding model fits to the data of the selected ensembles. GAJOE, Genetic Algorithm Judging Optimisation of Ensembles. Flexible regions of Δ91-FATZ-1, nonvisible in our determined crystal structures, were generated with EOM (43) keeping LM1 either fixed or free (10,000 models for each). Selected ensemble model representatives for rod-α-actinin-2/Δ91-FATZ-1 (C) and hd-α-actinin-2/Δ91-FATZ-1 (D) (color code as in Fig. 2A), along with their Rg and volume fractions within the ensemble. (E) Integrative model of fuzzy α-actinin-2/Δ91-FATZ-1 built using x-ray crystallography and SAXS models of hd-α-actinin-2/Δ91-FATZ-1. Rotation for LM2 helices of bound FATZ-1 molecules with respect to each other, as well as torsional twist in the rod along the longitudinal α-actinin-2 axis, is shown in the right inset (FATZ-1 flexible parts are omitted for clarity). (F) Surface of the rod-α-actinin-2/FATZ-1 structure showing the sequence conservation of α-actinin interacting residues for FATZ-1 (alignment done using 1505 α-actinins from vertebrates). (G) Model of F-actin/α-actinin-2/FATZ-1 (F-actin in light and dark gray) based on a cryo–electron tomography structure of the Z-disk (47) and our integrative model. See also figs. S11 to S13, tables S1 and S2, and movies S1 to S3.
The notable increase in both Rg and Rg of the cross section of rod-α-actinin-2/Δ91-FATZ-1 compared to that of rod-α-actinin-2, together with an almost unaffected Dmax, suggested that the flexible parts of FATZ-1 do not extend in a parallel but rather perpendicular fashion to the rod of α-actinin-2 (fig. S12B and table S2). In addition, the broad Rg distribution of ensembles selected from the pool generated by Ensemble Optimization Method (EOM) (43, 44), together with the analysis of the generated models and derived structural parameters, revealed that FATZ-1 forms a fuzzy complex with rod-α-actinin-2 and hd-α-actinin-2 (45, 46), as it remains mostly disordered apart from the LM1 and LM2 motifs (Fig. 4, C to E). In our fuzzy complexes, the two FATZ-1 molecules stem radially from both LM2 anchoring points, which are rotated by 155° with respect to each other (Fig. 4E and movie S1), due to the combined effect of the antiparallel dimeric α-actinin-2 architecture and the ~90° torsional twist of its rod (12, 13). Notably, both FATZ-1 molecules bind to the concave side of the α-actinin-2 rod surface, which is significantly more conserved than the convex one (Fig. 4F), resulting in a polar architecture of the complex (Fig. 4E and fig. S13, D and F), as also observed in the α-actinin-2/FATZ-1 complex placed into a recently reported cryo–electron tomography model of the Z-disk (Fig. 4G) (47).
We validated our fuzzy models by comparing calculated and experimentally derived intrinsic viscosity (η), which is a hydrodynamic parameter related to protein conformation (see Supplementary Results and table S1) (48), and additionally refined FATZ-1 conformations in all-atom molecular dynamics (MD) simulations in explicit water. We used the most representative rod-α-actinin-2/Δ91-FATZ-1 model of the EOM ensemble with an LM1-fixed or -free configuration as the starting point, which displayed significant structural fluctuations throughout our relaxation simulations, including compaction and unwinding (movies S2 and S3). In both MD simulations, Δ91-FATZ-1 LM2 remained stably bound to the rod and maintained a conformation close to that in the crystal structure. LM1 behaved similarly in the case of the fixed configuration. In the case of the free configuration, LM1 tended to bind to the rod and form nascent (first Δ91-FATZ-1 molecule) and more prominent (second molecule) intermolecular contacts with α-actinin-2, which, however, were observed at a neighboring position as compared to the crystal structure (see Supplementary Results and movie S3). Apart from a few cursory contacts between periodic images in the case of the LM1-fixed configuration, Δ91-FATZ-1 molecules did not display any specific interaction with each other in either simulation.
FATZ-1 interacts with itself and phase-separates in vitro
As some IDPs participate in multivalent interactions that lead to phase separation and formation of biomolecular condensates (49, 50), we analyzed the FATZ-1 sequence and found blocks of alternating charges, clusters of aromatic residues, and two RGG motifs (see Supplementary Results), which are the typical signatures of proteins undergoing liquid-liquid phase separation (LLPS). Accordingly, bioinformatics analysis using phase separation predictors revealed that FATZ-1 has a high propensity to phase-separate (Fig. 5A and fig. S14, A and B). The highest score was found mostly within the GRR (residues around 100 to 180) using PScore, which is based on the expected number of long-range planar sp2 π-π contacts, and catGRANULES, which was trained on sequence composition weighted by length, disorder, and R/G/F content, among other features (51). This property is conserved not only in FATZ-1 from mammals but also in FATZ-1 from other vertebrates (fig. S14C). FATZ-3 is predicted to display a strong tendency to phase-separate around residues 90 to 115 (central part) and 180 to 225 (C-terminal part), albeit showing a lower global score (fig. S14, A and B). The mostly cardiac paralog FATZ-2 was predicted to exhibit a moderate propensity to phase-separate around residues 80 to 125 and 180 to 230 (fig. S14, A and B).
Fig. 5. FATZ-1 interacts with itself and phase-separates in vitro.
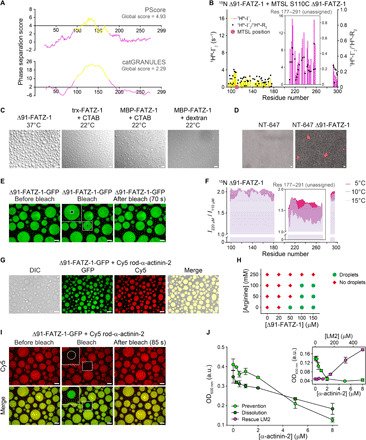
(A) Phase-separation prediction for FATZ-1 using PScore and catGRANULE servers (NTR and CTR in magenta and GRR in yellow). (B) PRE-rate (1HN-Г2) of 15N Δ91-FATZ-1 upon addition of MTSL (14N) S110C Δ91-FATZ-1 (difference of diamagnetic 1HN-R2 and paramagnetic 1HN-R2; bars, left y axis), along with (1HN-Г2)/(1HN-R2 dia) ratio of PRE rate divided by diamagnetic transverse relaxation rate (points, right y axis). (C) Differential interference contrast (DIC) microscopy of FATZ-1 condensates when incubated at 22° or 37°C. CTAB, cetyl-trimethylammonium bromide. (D) Fluorescence image of NT-647–labeled Δ91-FATZ-1 condensates. (E) FRAP of Δ91-FATZ-1-GFP. Photobleached area is circled in the close-up view. (F) 1H-15N HSQC signal intensity ratio of 15N Δ91-FATZ-1 at concentration of 220 μM/110 μM measured at increasing temperatures. (G) DIC/fluorescence images of Δ91-FATZ-1-GFP mixed with Cy5 rod-α-actinin-2. (H) Phase diagram of Δ91-FATZ-1 in function of protein and arginine concentration. (I) FRAP of Cy5 rod-α-actinin-2 mixed with Δ91-FATZ-1-GFP. Photobleached area is circled in the close-up view. Scale bars, 10 μm in all images. (J) Turbidity assay of Δ91-FATZ-1 with increasing concentrations of α-actinin-2. Prevention and dissolution done by triggering phase separation after mixing proteins and by adding α-actinin-2 after forming condensates, respectively. Rescue of phase separation by addition of LM2 peptide is shown in the right inset. Data taken after 14 to 20 min of reaction, represented as means ± SD (n = 3). See also figs. S14 to S16 and table S7 for statistical analysis, table S8, and movies S4 to S8. OD600 nm, optical dentisy at 600 nm; a.u., arbitrary units.
We therefore investigated the potential of FATZ-1 to form biomolecular condensates. First, to experimentally detect whether FATZ-1 interacts with itself, we carried out paramagnetic relaxation enhancement (PRE) NMR experiments (52, 53). We recorded 1H-15N HSQC spectra using a 1:1 molar mixture of 15N Δ91-FATZ-1 plus S-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate (MTSL)–labeled (14N) S110C Δ91-FATZ-1 mutant. A comparison of the relaxation rates between the diamagnetic and paramagnetic forms of the MTSL-label measured at 5°C showed an enhancement of already high relaxation rates in the unassigned FATZ-1 region (Fig. 5B and fig. S15). Accordingly, the relaxation rates in the presence of the paramagnetic label were increased approximately twofold compared to those in its absence, indicating a transient proximity between the MTSL-label in the GRR and the unassigned CTR (Fig. 5B). In addition, residues located in the more compact CTR showed distinct PRE effects, which persisted when normalized by the initial relaxation rates, thus suggesting authentic transient encounters between the GRR and the CTR. Although our data do not provide unambiguous proof that these interactions drive phase separation, bioinformatics predictions show the CTR as being important for FATZ-3 phase separation (fig. S14, A and B), altogether supporting a homotypic interaction for FATZ proteins.
Next, to experimentally assess whether FATZ-1 intermolecular interactions lead to phase separation, we used solutions containing either N-FATZ-1 or Δ91-FATZ-1, hypothesizing that N-FATZ-1 will not phase-separate because it lacks the CTR (Fig. 1C). Only Δ91-FATZ-1 showed spontaneous condensation (i.e., increased turbidity) when the temperature was increased from 4° to 22° or 37°C, whereas N-FATZ-1 remained transparent (Fig. 5C and fig. S16A). Δ91-FATZ-1 formed round-shaped droplets that were constantly fusing over time and reached up to 20 μm in diameter, as indicated by differential interference contrast (DIC) microscopy (see Fig. 5C, fig. S16B, and movie S4). Δ91-FATZ-1 phase separation was reversible when changing the temperature from 4° to 37°C and vice versa (fig. S16A). When the temperature was increased from 4° to 22°C, FATZ-1 precipitated under all experimental conditions due to solubility issues, whereas trx-FATZ-1 phase-separated into condensates that were generally spherical and associated with each other without undergoing fusion (movie S5). Trx-FATZ-1 and maltose-binding protein (MBP)–FATZ-1 formed round-shaped droplets that underwent fusion after addition of 0.5 to 6 mM cationic detergent cetyl-trimethyl-ammonium bromide or 5% of the molecular-crowding agent dextran, which is commonly used in cell-free LLPS studies (Fig. 5C and movies S5 and S6) (54). We further confirmed that the composition of FATZ-1 condensates using NT-647–labeled Δ91-FATZ-1 and Δ91-FATZ-1–green fluorescent protein (GFP). We observed phase separation with both fluorescently labeled variants, indicating that the C-terminally attached GFP does not interfere with phase separation (Fig. 5, D and E). We analyzed the liquid nature of the condensates using fluorescence recovery after photobleaching (FRAP) on Δ91-FATZ-1-GFP. The half-time to recover the fluorescence signal of a photobleached region with a diameter of 2.7 μm was t1/2 = 12.7 s, corresponding to a diffusion coefficient D = 0.096 μm2/s (Fig. 5E, fig. S16C, and movie S7). While this D is two orders of magnitude lower than that of bovine serum albumin (BSA) and catalase (55), t1/2 is in line with typical values observed for liquid condensates (56, 57). Furthermore, 1H-15N HSQC signal intensity measured at 5°C increased linearly with protein concentration for the GRR, whereas a nonlinear increase was observed for the unassigned CTR (Fig. 5F). This effect was more pronounced at higher temperatures, correlating with FATZ-1 homotypic interactions and its in vitro capacity to phase-separate at increased temperature. The CTR showed a faster relaxation behavior due to structural compaction of this segment when free in solution. Therefore, the observation of sizeable intermolecular PRE effects also in the condensate indicates that the reduced conformational flexibility of the CTR prevails after phase separation and facilitates transient encounters between the spin label within the GRR and residues located in the CTR. To conclude, our data indicate that Δ91-FATZ-1 retains structural compaction in the CTR when undergoing LLPS. Last, we generated a liquid-liquid phase diagram, which showed that increasing concentrations of arginine prevent Δ91-FATZ-1 phase separation (Fig. 5H and fig. S16B), suggesting that cation-𝜋 interactions are one of the principal mechanisms behind the formation of FATZ-1 condensates (58).
To investigate whether α-actinin-2 colocalizes with FATZ-1 condensates, we used Cy5-labeled rod-α-actinin-2 and Δ91-FATZ-1-GFP. While we observed a homogeneous distribution of Cy5 rod-α-actinin-2 in the condensates, Cy5 E. histolytica rod, which does not interact with Δ91-FATZ-1 (Fig. 3H), was not enriched (Fig. 5G and fig. S16D). Colocalization between Cy5 rod-α-actinin-2 and Δ91-FATZ-1-GFP was further confirmed by FRAP on Cy5 rod-α-actinin-2. The t1/2 to recover the fluorescence signal of photobleached rod-α-actinin-2 in condensates with a diameter of 4.5 to 9.7 μm was 6.6 to 35.3 s, corresponding to a diffusion coefficient of ~0.5 μm2/s (Fig. 5I, fig. S16E, and movie S8), suggesting that the protein mesh in FATZ-1 condensates is permeable for particles as large as α-actinin-2 and corroborating its selective recruitment.
As the main α-actinin-2–binding sites in FATZ-1 map to the CTR, which is also involved in FATZ-1 homotypic interaction likely driving phase separation, we asked whether increasing amounts of α-actinin-2 could have an impact on the FATZ-1 condensates. We first mixed Δ91-FATZ-1 with increasing concentrations of α-actinin-2 and triggered droplet formation by increasing the temperature to 37°C. As assessed by turbidity assays, which use optical density of a protein solution as a measure of phase separation, α-actinin-2 prevented condensate formation in a concentration-dependent manner (Fig. 5J, fig. S16F, and table S7). We next used preformed Δ91-FATZ-1 droplets and added increasing amounts of α-actinin-2, which led to dissolution of FATZ-1 condensates again in a concentration-dependent manner (Fig. 5J, fig. S16G, and table S7). Last, we added LM2 peptide to the FATZ-1/α-actinin-2 mixture, which “rescued” Δ91-FATZ-1 droplet formation (Fig. 5J, fig. S16, H and I, and table S7), supporting specificity of the interaction inside the droplet. Together, these results demonstrate that increasing the concentration of α-actinin-2 prevents and dissolves FATZ-1 condensates.
α-Actinin-2 stabilizes FATZ proteins at the Z-disk
To determine the contribution of α-actinin-2 to localization of FATZ proteins to the Z-disk, we transfected GFP-tagged FATZ-1 or FATZ-2 into immortalized mouse myoblasts (IMMs) or neonatal rat cardiomyocytes (NRCs) to comply with their skeletal-specific or mostly cardiac-specific expression profile, respectively. Both FATZ-1 and FATZ-2 [wild type (WT) and fiveE mutant] correctly targeted the Z-disk and colocalized with α-actinin-2 and the peripheral Z-disk portion of titin (fig. S17) (59). At the diffraction-limited resolution, no evident alterations in Z-disk morphology were detected. Diffusive GFP signal was specifically detected for fiveE FATZ-1 and FATZ-2 mutants outside of sarcomeric compartments (fig. S17), suggesting their incomplete targeting to the Z-disk most likely due to loss of interaction with α-actinin-2.
To specifically assess the impact of the fiveE mutant on α-actinin-2 binding capabilities of FATZ-1 and FATZ-2, we performed in cellula fluorescence resonance energy transfer (FRET) by acceptor photobleaching. Here, the higher FRET ratio indicates closer physical proximity between the pair of donor-acceptor fluorophores, although it does not provide stoichiometric information about the protein complex (60). Both WT cyan fluorescent protein (CFP)–FATZ proteins and α-actinin-2–yellow fluorescent protein (YFP) colocalized in a nonmuscle COS-1 cell line, which was used to avoid the contribution of endogenous partners, yielding comparable FRET ratios of 0.085 and 0.087 for FATZ-1/α-actinin-2 and FATZ-2/α-actinin-2, respectively (Fig. 6, A and B). By contrast, fiveE FATZ-1 and FATZ-2 mutants exhibited significantly lower FRET ratios than the WT counterparts (0.047 and 0.024; Fig. 6, A and B). Together, the overall reduced proximity of fiveE FATZ-1 and FATZ-2 to α-actinin-2 is reflected by lower Z-disk targeting efficiency compared to the WT, both in IMMs and NRCs, in agreement with their lower affinity for α-actinin-2 (see Fig. 2, E and I). Nevertheless, these mutants still targeted the Z-disk, which can be explained by the contribution of multiple binding partners important for the localization of FATZ-1 in IMMs and FATZ-2 in NRCs.
Fig. 6. α-Actinin-2 stabilizes FATZ proteins at the Z-disk.
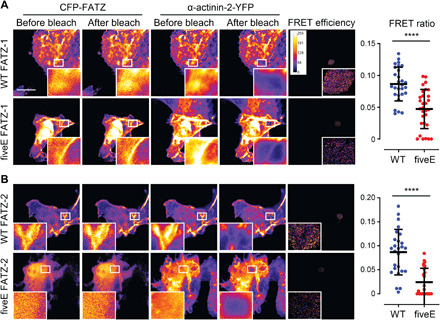
(A) COS-1 cells coexpressing enhanced CFP (ECFP)–tagged FATZ-1 (WT FATZ-1) or corresponding fiveE mutant, in combination with actinin-2-EYFP, as determined in fixed cells by acceptor photobleaching. Representative images are shown before and after bleaching for both proteins. Bleached regions of interest are shown (insets), along with FRET efficiencies used to calculate FRET ratios presented in the adjacent graph [n = 28 (WT) and 30 (fiveE), ****P < 0.0005, Student’s t test). (B) Same cells as in (A) but coexpressing ECFP-tagged FATZ-2 variants in combination with EYFP-α-actinin-2 [n = 26 (WT) and 25 (fiveE), ****P < 0.0005, Student’s t test). Scale bars, 10 μm in all images. See also fig. S17.
DISCUSSION
Proteins of the FATZ family are found in Z-bodies and mature Z-disks and are suggested to have a structural role as an interaction hub by establishing an intricate set of protein-protein interactions linking a diverse set of critical Z-disk components, as well as a role in protein signaling pathways via binding to calcineurin (14, 15, 17). Here, we focused on FATZ-1, which is the most studied family member when it comes to binding to different partners, and its interaction with the major Z-disk protein α-actinin-2 (3, 12).
Previous secondary structure and globularity predictions on FATZ-1 suggested the presence of two α-helical domains displaying a compact globular conformation without significant coiled-coil regions and separated by a GRR (15). Our bioinformatics analyses on FATZ-1, FATZ-2, and FATZ-3 revealed a certain degree of compactness and α-helical content for NTR and CTR, in agreement with the presence of hydrophobic clusters, and an extensive central IDR matching FATZ-1 GRR.
We experimentally confirmed bioinformatics predictions using a battery of biophysical and structural approaches, which revealed a random coil–like conformation for N-FATZ-1 and a premolten globule one for Δ91-FATZ-1, uncovering that both constructs sample an ensemble of states in solution, with no definable tertiary conformation. This ensemble state is likely driven by the IDR encompassing the GRR (as per the NMR result), but the protein retains secondary structure elements (as per the CD result), which is in line with available evidence, suggesting that IDPs populate partially ordered or compact states in the absence of binding partners (31). Together, our data indicate that FATZ-1 is a “modular” IDP with regions of definable secondary structure elements interspersed by structurally heterogeneous regions.
Sequence alignment of vertebrate FATZ-1 showed several stretches of conserved residues at NTR and CTR, two of them mapping to α-actinin-2–binding sites (i.e., LM1 and LM2) (fig. S18). Combining this alignment with reported binding data on FATZ-1, FATZ-2, and FATZ-3, we hypothesize that filamin-C–, telethonin-, and calcineurin-binding sites reside within residues 34 to 83, while the second calcineurin-binding site and the ZASP-binding site reside within residues 265 to 275 and 295 to 299, respectively.
Most of these interactions were identified on a binary level, but it is not clear which higher-order complexes can actually coexist in Z-disks. While titin Z-repeats, myotilin, myopodin, and myopalladin are known to bind to α-actinin-2 EF3-4 via their 1-4-5-8 motif, the ZASP PDZ domain binds to the C terminus of EF3-4 (61–64). The titin Zq motif was shown to bind to the α-actinin-2 rod (SR2-SR3) and modeled as a dimer of α helices running perpendicular to the centre of the rod (59, 65). Our structural analysis of the α-actinin-2/FATZ-1 complex indicates that FATZ-1 and Zq titin should not compete for binding to α-actinin-2. The structure of the hd-α-actinin-2/FATZ-1 complex shows, however, a significant shift in EF1-2 position compared to the equivalent domain in unbound α-actinin-2. Given the reported intramolecular communication between EF-hand lobes in CAMD (35), further experiments are required to test whether FATZ-1 regulates the interaction of different partners to α-actinin-2 EF3-4, ultimately affecting its F-actin bundling function. Our structure also correlates with the reported competition between α-actinin-2 and filamin-C for the CTR of FATZ-1 (15), indicating that the second filamin-C–binding site maps to LM1/LM2 or neighboring regions. Nevertheless, a ternary α-actinin-2/FATZ-1/filamin-C complex is still possible relying on the first filamin-C–binding site in the NTR of FATZ-1 (18). Last, because the predicted second binding site for calcineurin is separated from LM2 by 25 residues, we infer that FATZ-1 could inhibit calcineurin when bound to α-actinin-2, although we cannot exclude competing sterical hindrance effects.
Our crystal structures revealed that the main binding sites in FATZ-1 map to the CTR, in agreement with previous reports (14, 15, 17). Specific interaction with α-actinin-2 is achieved via LM1 and LM2, which classify as ELMs (36, 37), representing two of four islands of highly conserved residues in the CTR (fig. S18) and suggesting evolutionary pressure on the interaction interface. In addition, LM1 and LM2 can be recognized as MoREs, because they undergo disorder-to-order transitions upon binding (41, 42). Together, our data point to LM2, and in particular to residues Tyr218-Tyr228, as the major binding site for α-actinin-2. LM1 binds weakly when alone but, in the context of the full-length protein, is tethered in the polypeptide to the proximity of its binding site by LM2, increasing its local concentration and thus contributing to the interaction.
In addition, both LM1 and LM2 display P-W/F motifs (P187-W188 and P227-Y/F228, respectively). As prolines neighbored by aromatics favor the cis conformation, and the slow switching between cis and trans conformers can influence regulatory networks (66), it is enticing to speculate on the possible function of these elements in the tightly controlled regulation of Z-disk interactions.
Using FRET and colocalization analysis, we tested the in cellula effect of fiveE FATZ-1 and FATZ-2 mutants, which display reduced affinity to α-actinin-2, likely due to loss of hydrogen bonding and electrostatic repulsion arising from K217E + R223E mutations. The obtained FRET ratios for WT FATZes/α-actinin-2 were l significantly lower than the previously reported FRET ratios for other Z-disk complexes (60, 67), which might be explained by the use of full-length protein rather than FRET-optimized truncation variants and also by the intrinsic flexibility of the interacting proteins. FiveE FATZ-1 and FATZ-2 mutants showed lower Z-disk targeting efficiency; however, we detected no evident alterations in Z-disk morphology.
SAXS analysis of the dimeric α-actinin-2 rod uncovered a rigid structure in solution, in contrast to the proposed bending reported from MD simulations (68). CD and SAXS analysis of the complex showed that FATZ-1 retains most of its conformational heterogeneity (apart from LM1 and LM2) when bound to α-actinin-2, featuring as a “random fuzzy” complex, where short binding motifs are interconnected and flanked by disordered regions (45, 46). Peptide array, LP-MS, and NMR revealed additional binding sites spread along the FATZ-1 polypeptide, which contribute to binding affinity, as shown by ITC experiments using Δ91-FATZ-1 constructs, in which secondary binding sites were perturbed by the presence of an N- or C-terminally fused trx.
In summary, the α-actinin-2/FATZ-1 complex is characterized by a central extended and rigid SR core that acts as a stable platform on which the FATZ-1 anchors and goes on to sample multiple spatial positions biased toward orientations perpendicular to the rod domain, while the ABD and EF-hand motifs remain as defined modules peripheral to the rod.
FATZ-1 forms a 2:1 complex with the functional α-actinin-2 dimer, both in solution (as per the SEC-MALS and SAXS data) and in the crystal. This interaction results in two independent binding events as shown by ITC when analyzed using a two-site binding model (Kd1 in the nanomolar range and Kd2 in the low micromolar range), indicating a noncooperative binding. While positive cooperativity can be ruled out as binding isotherms display two transitions, negative cooperativity can be excluded by comparison of binding constants for α-actinin-2 and hd-α-actinin-2, which show that the affinity of the second FATZ-1 molecule for α-actinin-2 is similar to that of the single FATZ-1 molecule binding to hd-α-actinin-2 (Kd1 = 3.5 nM and Kd2 = 314 nM versus Kd = 260 nM). In agreement with this, Δ91-FATZ-1 molecules did not exhibit any specific direct interactions with each other in our relaxation MD simulations.
Our proposed mechanism of binding between α-actinin-2 and FATZ-1 can only be explained in the context of a fuzzy complex, using both main molecular recognition elements and secondary binding sites. In this complex, FATZ-1 displays additional free valences for interactions with multiple partners, and its fuzzy nature is probably less pronounced when fully loaded. The first FATZ-1 molecule binds to α-actinin-2 through both main (LM1 and LM2) and secondary binding sites with nanomolar affinity (Fig. 7A); the binding of the second FATZ-1 to α-actinin-2 is not hindered by the first bound molecule, as it displays a low micromolar affinity similar to that of FATZ-1 for hd-α-actinin-2 but rather does not enjoy the same “bonus” of additional transient interactions as the first one (Fig. 7A). The structure and binding mechanism of the fuzzy α-actinin-2/FATZ-1 complex supports FATZ-1 function as a classical scaffolding protein in Z-disk assembly. The presence of IDRs in fuzzy complexes was shown not only to modulate affinity but also to increase selectivity and improve kinetics by providing nonspecific anchors to binding partners (45).
Fig. 7. Model for the binding mechanism of the fuzzy α-actinin-2/FATZ-1 complex and potential implications in Z-disk ultrastructure and biogenesis.
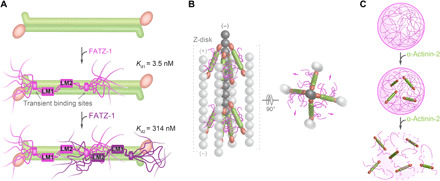
(A) Mechanism of binding between α-actinin-2 (in green) and FATZ-1, showing how the first FATZ-1 molecule (magenta) binds tightly to α-actinin-2 through both main molecular recognition elements (LM1 and LM2) and secondary binding sites, while the second FATZ-1 molecule (dark purple), which does not enjoy the same bonus of additional interactions as the first one, binds with lower affinity. (B) Model of the α-actinin-2/FATZ-1 complex [color code as in (A)] in the Z-disk, showing the polar architecture of the complex due to the binding of both FATZ-1 molecules to the conserved concave side of α-actinin-2. This might provide orientational constraints on α-actinin-2 rod and thus contribute to the paracrystalline tetragonal lattice of actin filaments (light and dark gray; actin dimers shown as a single ball for simplicity). (C) FATZ-1 phase-separates and forms biomolecular condensates with α-actinin-2 [color code as in (A)]. FATZ-1 condensates are prevented and dissolved by increasing concentrations of α-actinin-2, opening a potential novel avenue for sarcomere biogenesis starting from FATZ-1 condensates.
FATZ-2 and FATZ-3 show the same 2:1 binding stoichiometry for α-actinin-2 as found for FATZ-1. This is in line with conserved key interacting residues within LM1 and LM2 binding motifs in the FATZ family. In addition, FATZ-1 displays the same binding stoichiometry with both muscle α-actinin-2 and nonmuscle α-actinin-1, which, together with high sequence conservation of vertebrate α-actinin isogenes, indicates a common binding mechanism between members of FATZ and α-actinin protein families. Nevertheless, differences in binding affinities are expected to occur for different combinations of family members as reported for FATZ-1 and α-actinin-3 (21).
Electron tomography reconstructions of paracrystalline vertebrate Z-disks show a defined orientation for α-actinin-2 rod in cross-linked actin filaments (47, 69). This cannot be explained in the context of the built-in flexibility in the neck region of α-actinin, which allows rotation of the rod along its longitudinal axis (12). Accordingly, orientational constraints are expected to be provided by additional proteins that bind to α-actinin-2 rod and tether it either directly or indirectly to F-actin. Our integrative model of the α-actinin-2/FATZ-1 complex placed into a cryo–electron tomography structure of the Z-disk (47) reveals that both FATZ-1 molecules stem radially from their respective LM2 motifs bound to the concave side of the α-actinin-2 rod surface (Fig. 7B). This polar architecture of the complex is likely to arise from higher conservation of the α-actinin-2 concave side compared to the convex one, which suggests it as a hotspot for protein-protein interactions. In combination with FATZ-1 multivalent scaffolding function (14, 15, 17), the polar architecture of the α-actinin-2/FATZ-1 complex might organize interaction partners within the Z-disk, thus advancing our understanding of the paradigmatic role of scaffold proteins in muscle ultrastructure.
Solubility of FATZ-1 constructs was significantly improved upon addition of arginine. Arginine was shown to reduce FATZ-1 multivalency for biomolecular condensate formation by competing with stabilizing cation-π and π-π interactions, in agreement with the FATZ-1 liquid-liquid phase diagram as a function of protein and arginine concentration. FATZ-1 displays the hallmarks of proteins that undergo LLPS, including IDRs, blocks of alternating charges, clusters of aromatic residues, and two RGG motifs (50). Most of these hallmarks are also found in FATZ-2 and FATZ-3, although both proteins are predicted to have a weaker propensity for phase separation. LLPS depends on external factors such as temperature, pH, and ionic strength. Formation of FATZ-1 biomolecular condensates was triggered by increasing the temperature from 4°C, suggesting that hydrophobic interactions dominate the temperature-dependent behavior, and not charge-charge, cation-π, and π-π interactions, which are rather temperature independent (70, 71). Accordingly, FATZ-1 comprises three P-Xn-G motifs (where n would be typically 4; two in the GRR and another in the CTR), which were reported to drive phase separation in a lower critical solution transition, where condensates form above the threshold temperature (72).
Outlook
Sarcomere assembly starts from Z-bodies, initially termed “punctate concentrates” of α-actinin-2, FATZ, ZASP, myotilin, filamin-C, and actin. During sarcomere biogenesis, Z-bodies grow in size, fuse together, and lastly associate with other proteins to form mature Z-disks in nascent myofibrils (5–9). This process is reminiscent of membraneless organelles with a composition distinct from the surrounding, which originate through a mechanism of phase separation (e.g., Cajal bodies or P bodies) and form discrete puncta, i.e., biomolecular condensates (50, 73). Phase separation is driven by multivalent scaffold molecules that, in turn, create binding valences for the recruitment of client molecules (50, 73–75). Here, we reveal that FATZ-1 displays molecular signatures for LLPS, acting as a scaffold for the formation of biomolecular condensates capable of incorporating the client protein α-actinin-2. Scaffold proteins are essential for condensate formation, while client proteins constitute the majority of components that localize to condensates in a regulated fashion by directly binding to scaffolds, with their concentration being the physical mechanism modulating phase separation (50). Last, we uncover that FATZ-1 condensates are prevented and dissolved by increasing concentrations of α-actinin-2 (Fig. 7C).
This leaves us with the intriguing hypothesis that sarcomere biogenesis starts from biomolecular condensates, whereby FATZ proteins could act as scaffolds that locally concentrate partners essential for initiation of myofibrillogenesis, leading to nucleation of cytoskeletal structures in muscle. In mature sarcomeres, FATZ-1 represents only 1% of the total muscle protein, while the level of α-actinin, which increases during myofibrillogenesis, dominates at 17% (5, 14, 76). Variable cellular protein concentrations could therefore control the phase separation threshold and composition of Z-body condensates. Along these lines, short actin filaments form liquid droplets in the presence of the F-actin cross-linker filamin-A, whose density controls the shape and viscoelastic properties of the condensates (77). These results agree with our bioinformatics predictions on Z-body proteins, which detected an important phase separation propensity also for filamin-C, albeit lower than that of FATZ-1, while the rest of components were not predicted to phase-separate (fig. S14D). A similar principle has recently been described for phase-separated algal organelle the pyrenoid, where the CO2-fixing enzyme Rubisco phase separates with the intrinsically disordered linker protein essential pyrenoid component 1 (78), suggesting a potentially universal molecular organization shared by LLPS condensates. Further experiments are clearly required to characterize the phase diagram of FATZ condensates as a function of concentrations of client Z-body proteins and to assess whether the same principles apply under physiological conditions in living cells.
METHODS
Bioinformatics analysis
Protein sequences and structures were analyzed using a number of servers and bioinformatics tools, namely, ExPASy ProtParam, CIDER, PrDOS, DISOPRED2, IUPred2A, MFDp2, MobiDB, FELLS, JPred, DynaMine, HCA, SMART, PScore, catGRANULE, PLAAC, LARK, DSSP, jsPISA, PIC, DynDom, and Hydropro (for details, see Supplementary Methods). Sequence alignments and phylogenetic trees were prepared using MULTIALIN, ESPript, Clustal Omega, and FigTree.
Protein expression and purification
DNAs encoding human FATZ-1, FATZ-2, and FATZ-3 were cloned into p3NH vector, while those encoding trx-FATZ-1 and MBP-FATZ-1 were cloned into pETM-22 and pET44M vectors, respectively. Soluble FATZ-1 constructs, N-FATZ-1 and Δ91-FATZ-1, were generated using an Erase-a-Base approach as described in Supplementary Methods and further cloned into a modified pET-46 vector. Δ91-FATZ-1–pET-46 was used as a template to generate fiveE, RRE, and S110C Δ91-FATZ-1 mutants, as well as Δ91-FATZ-1-GFP. FATZ-1-p3NH was used as a template to generate mini-FATZ-1. Δ91-FATZ-1 was also cloned into pET-20 vector to generate trx-Δ91-FATZ-1 and Δ91-FATZ-1-trx. Human α-actinin-2 and rod-α-actinin-2 were cloned into pET3d vector, while hd-α-actinin-2 was cloned into a modified pET-8 vector.
Proteins were produced by recombinant expression in Escherichia coli strains BL21 (DE3), Rosetta-2 pLysS (DE3) or Rosetta-2 (DE3) (see Supplementary Methods) using LB medium and induction with 1 mM isopropyl-β-d-thiogalactopyranosid for 5 hours at 37°C. Se-Met–labeled mini-FATZ-1 and 15N-labeled Δ91-FATZ-1 were similarly overexpressed using M9 minimal medium containing Se-Met and 15NH4Cl, respectively. α-Actinin-2 constructs and soluble FATZ constructs were purified by nickel-affinity chromatography on a 5-ml HisTrap FF crude column, anion-exchange chromatography on a 6-ml Resource Q column, and SEC on a HiLoad 26/600 Superdex 200 column. Insoluble FATZ constructs were solubilized in 4 to 6 M urea and further purified as for the soluble constructs (see Supplementary Methods). All proteins were flash-frozen and stored at −80°C. Protein identity and purity were analyzed by SDS–polyacrylamide gel electrophoresis. Proteins were dialysed overnight at 4°C against specific buffers before each experiment and concentrated by ultrafiltration.
Molecular biophysics characterization
Protein conformation was analyzed by CD on a Chirascan spectrometer at 20°C using a 0.5-mm path-length cuvette. Samples were prepared at 1 to 100 μM in 20 mM sodium phosphate (pH 7.5). Protein conformation and binding stoichiometry were assessed by SEC combined with MALS (Wyatt), DLS (Wyatt), and viscometer (Wyatt or Omnisec), using Superdex 200 increase 10/300 or Superose 6 increase (3.2/300 or 10/300) columns, and equilibrated as detailed in Supplementary Methods. Binding affinity was measured by ITC on MicroCal iTC200 or PEAQ-ITC calorimeters, with α-actinin-2 constructs used as titrand at ~20 μM and FATZ-1 constructs or peptides used as ligand at ~200 μM; all prepared in 20 mM sodium phosphate (pH 7.5), 100 mM NaCl, 50 mM arginine, 50 mM glutamic acid, 1 mM β-mercaptoethanol, 1 mM EDTA, and 0.5 mM phenylmethylsulfonyl fluoride. Binding data were processed and fitted using PEAQ-ITC analysis software.
Mapping of interaction sites
Protein interaction sites were mapped using peptide-cellulose conjugates covering the sequence of FATZ-1 and His6-tagged α-actinin-2 (see Supplementary Methods). LP-MS was performed by incubating rod-α-actinin-2/Δ91-FATZ-1 with trypsin or thermolysin at different protease:protein ratios and XL-MS by incubating hd-α-actinin-2/Δ91-FATZ-1 with 2 mM 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide. NMR assignment of N-FATZ-1 and Δ91-FATZ-1 was carried out as described in Supplementary Methods using the experimental parameters provided in table S8. For binding studies, 2D HSQC spectra were recorded at 278 K using a 600-MHz Avance spectrometer. 15N Δ91-FATZ-1 was prepared alone (20 μM) or mixed with either α-actinin-2 (10 μM) or hd-α-actinin-2 (20 μM).
Crystal structure determination
Rod-α-actinin-2/mini-FATZ-1, rod-α-actinin-2/Δ91-FATZ-1, and hd-α-actinin-2/Δ91-FATZ-1 complexes were crystallized by the sitting- and hanging-drop vapor diffusion method as described in Supplementary Methods. X-ray diffraction datasets were collected at the European Synchrotron Radiation Facility (ESRF) and PETRA-III synchrotrons at 100 K (table S4) and processed using programs XDS and XSCALE and reformatted with XDSCONV. The structures were determined by molecular replacement using PHASER and the structure of rod-α-actinin-2 [Protein Data Bank (PDB) code 1HCI] as a search model for the two rod-α-actinin-2 complexes and the structures of ABD, SR1-2, SR3-4, and EF3-4 from α-actinin-2 (PDB code 4D1E) as search models for the hd-α-actinin-2 complex. In all cases, FATZ-1 side chains were built manually with Coot using as a reference the positions of two Se-Met identified by anomalous difference Fourier analysis with PHENIX. Models were finished using iterative rounds of manual building with Coot and refinement with BUSTER and PHENIX.
SAXS analysis and MD simulations
Samples were prepared in 50 mM tris-HCl (pH 7.5) and 150 mM NaCl, and SAXS data were collected at ESRF and PETRA-III synchrotrons at 20°C using both batch and online SEC-SAXS setups (see table S2). For SEC-SAXS, samples were injected onto Superose 6 increase 5/150 or 10/300 columns and data processed with CHROMIXS. SAXS data analysis was performed using programs PRIMUS and GNOM within the ATSAS package. Models of the rod-α-actinin-2/Δ91-FATZ-1 and hd-α-actinin-2/Δ91-FATZ-1 fuzzy complexes were generated using the EOM method and conformational restraints from the crystal structures as described in Supplementary Methods. The obtained models were validated by all-atom MD simulations in explicit water using GROMACS package and Amber99SB-ILDN force field as detailed in Supplementary Methods. Figures and movies were generated with PyMOL and UCSF Chimera.
Characterization of phase-separated condensates
FATZ homotypic interactions were analyzed by PRE NMR using 15N Δ91-FATZ-1 (95 μM) and MTSL-labeled (14N) S110C Δ91-FATZ-1 (95 μM) prepared as detailed in Supplementary Methods. The effect of temperature was assessed by recording 2D HSQC spectra of 15N Δ91-FATZ-1 (110 and 220 μM) at 278, 283, and 288 K. Condensates were formed by concentrating FATZ-1 constructs up to 50 to 200 μM at 4°C and next incubating the protein solution for 5 min at 22° or 37°C. Samples were imaged both in bright field and fluorescence using different microscopes and a homebuilt chamber (see Supplementary Methods). Colocalization experiments were performed by mixing Cy5 human or E. histolytica rod-α-actinin-2 (both at 4.6 μM) with preformed Δ91-FATZ-1-GFP condensates at 37°C or by mixing Cy5 human or E. histolytica rod-α-actinin-2 (both at 4.6 μM) with Δ91-FATZ-1-GFP (at 100 μM) at 4°C and next incubating the protein solution for 5 min at 37°C. FRAP experiments were conducted as detailed in Supplementary Methods. For turbidity experiments, assays were performed by either mixing His6-Δ91-FATZ-1 (at 50 to 65 μM) and α-actinin-2 (at 0.25 to 8 μM) for 5 min at 4°C or by preforming condensates of His6-Δ91-FATZ-1 (at 65 μM) for 5 min at 37°C and then adding α-actinin-2 (at 0.25 to 8 μM). Absorbance was measured at 600 nm and 37°C using a VICTOR Nivo plate reader. Additional experiments were performed by mixing 50 μM His6-Δ91-FATZ-1, 5 μM α-actinin-2, and 20 to 500 μM LM2 peptide.
Cell biophysics characterization
WT and fiveE FATZ-1 and FATZ-2 were cloned into pECFP-C1 and pEGFP-C1 vectors, and α-actinin-2 was cloned into a pEYFP-C1 vector. FATZ-1 and FATZ-2 colocalization with α-actinin-2 in Z-disk was assessed using IMMs and NRCs, respectively. Impact of FATZ-1 and FATZ-2 mutations on α-actinin-2 binding was measured by FRET using COS-1 cells as described in Supplementary Methods.
Acknowledgments
We thank the staff of MX and SAXS beamlines at ESRF and PETRA-III for excellent support, the Mass Spectrometry Facility at Max Perutz Labs for proteomics analyses using the Vienna BioCenter Core Facilities (VBCF) instrument pool, and the VBC Protein Technologies for SEC-MALS analysis. We also thank M. Schosserer [University of Natural Resources and Life Sciences (BOKU)] for usage of the confocal microscope, Wyatt for access to the viscometer and SEC-MALS data analysis, Malvern for ITC data analysis, M. Puchinger for help with CD and protein labeling, M. Somlyay for taking pictures of tubes with protein, S. Żerko for help under optimizing conditions for NMR assignment, G. Wiche (University of Vienna) for providing IMMs, T. Oda (University of Yamanashi) for providing the atomic coordinates of the cryo–electron tomography Z-disk model, N. Frey (University of Kiel) for providing clones for FATZ-2 and FATZ-3, E. Stepinac for providing sortase, A. Sedivy (VBCF) for excellent technical assistance, and Life Science Editors for editing assistance. We are also thankful to J. Lah (University of Ljubljana), S. Keller (TU Kaiserslautern), S. Longhi (Architecture et Fonction des Macromolécules Biologiques), T. Gibson (European Molecular Biology Laboratory, Heidelberg), T. Madl (Medical University of Graz), S. Saha (Institute of Molecular Biotechnology Austria) for insightful discussions on ITC data, MoREs, LMs, and LLPS and B. Sponga for advice and discussions on statistical data analysis. A.S. thanks the tutors of the DLS-CCP4 Data Collection and Structure Solution Workshop for helpful discussions. Last, we thank the anonymous reviewers for careful reading of our manuscript and many insightful comments and suggestions. Funding: K.D.-C.’s research was supported by a Marie Curie Initial Training Network: MUZIC (no. 238423); Austrian Science Fund (FWF) Projects I525, I1593, P22276, P19060, and W1221; Federal Ministry of Economy, Family, and Youth through the initiative “Laura Bassi Centres of Expertise,” funding the Centre of Optimized Structural Studies, no. 253275; the Wellcome Trust Collaborative Award (201543/Z/16); COST action BM1405—Non-globular proteins from sequence to structure, function and application in molecular physiopathology (NGP-NET); WWTF (Vienna Science and Technology Fund) Chemical Biology project LS17-008; Christian Doppler Laboratory for High-Content Structural Biology and Biotechnology; and the Austrian-Slovak Interreg Project B301 StruBioMol, University of Vienna Research Platform Comammox and by the University of Vienna. The work was supported by the Austrian Science Fund FWF grant P30550 to B.Z. M.G. and A.G. were supported by the Wellcome Trust Collaborative Award (201543/Z/16). M.G. holds the British Heart Foundation Chair of Molecular Cardiology. This work was supported by a grant from the Polish National Science Centre to W.K. (MAESTRO, 2015/18/A/ST4/00270). Research in B.W.’s laboratory was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) research FOR 2743, Project ID 403222702/SFB 1381, and Germany’s Excellence Strategy (CIBSS—EXC-2189—Project ID 390939984). D.I.S. and C.M.J. acknowledge the support of the European Union’s Horizon 2020 research and innovation programme “iNEXT Discovery” grant agreement no. 871037. Author contributions: Conceptualization: A.S., J.L.A., T.C.S., C.M.J., J.K., A.G., A.P., G.F., B.Z., M.G., R.K., and K.D.-C. Investigation: A.S., J.L.A., T.C.S., C.M.J., A.R.C., J.K., A.G., F.D., A.P., E.D.A.R., M.P., A.Z.-K., G.M., T.P., P.D., C.S., E.H., B.M., and L.G. Methodology: A.S., J.L.A., J.K., W.K., D.I.S., B.W., B.Z., M.G., R.K., and K.D.-C. Formal analysis: A.S., J.L.A., T.C.S., C.M.J., A.R.C., J.K., A.G., F.D., A.P., E.D.A.R., A.Z.-K., and K.D.-C. Visualization: J.L.A., A.S., T.C.S., J.K., A.G., and A.P. Data curation: J.L.A., T.C.S., C.M.J., J.K., W.K., D.I.S., B.W., B.Z., M.G., R.K., and K.D.-C. Resources: T.P., G.F., W.K., D.I.S., B.W., B.Z., M.G., R.K., and K.D.-C. Supervision: J.L.A., W.K., D.I.S., B.W., B.Z., M.G., R.K., and K.D.-C. Funding acquisition: K.D.-C., R.K., M.G., B.Z., B.W., D.I.S., and W.K. Writing (original draft): J.L.A., A.S., and K.D.-C. Writing (review and editing): All authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The NMR assignments have been deposited in the Biological Magnetic Resonance Data Bank and are available with these links: N-FATZ-1: code 50496, http://deposit.bmrb.wisc.edu/author_view/BMRB/50496_hy_ovvteqaa.str. Δ91-FATZ-1: code 50497, http://deposit.bmrb.wisc.edu/author_view/BMRB/50497_hy_vmbatcvu.str. The SAXS models have been deposited in the Small Angle Scattering Biological Data Bank and are available with these links: sarcomeric IDP FATZ-1 (N-FATZ-1): code SASDJJ6, www.sasbdb.org/data/SASDJJ6/vwgo25jhz8/; sarcomeric IDP FATZ-1 (Δ91-FATZ-1): code SASDJK6, www.sasbdb.org/data/SASDJK6/c1yxymdokh/; sarcomeric F-actin cross-linking protein α-actinin-2 (hd-α-actinin-2): code SASDJL6, www.sasbdb.org/data/SASDJL6/miqtomh44s/; sarcomeric F-actin cross-linking protein α-actinin-2 (hd-α-actinin-2): code SASDJM6, www.sasbdb.org/data/SASDJM6/cj4pgogz43/; sarcomeric fuzzy α-actinin-2/FATZ-1 complex (rod-α-actinin-2/Δ91-FATZ-1): code SASDJN6, www.sasbdb.org/data/SASDJN6/zcgyy2rtq5/; and sarcomeric fuzzy α-actinin-2/FATZ-1 complex (hd-α-actinin-2/Δ91-FATZ-1): code SASDJP6, www.sasbdb.org/data/SASDJP6/2hqhr0dwwc/. The crystal structures have been deposited in the PDB and are available with these links: crystal structure of sarcomeric protein FATZ-1 (mini-FATZ-1) in complex with rod-α-actinin-2: code 7A8T, https://doi.org/10.2210/pdb7A8T/pdb; crystal structure of sarcomeric protein FATZ-1 (δ91-FATZ-1) in complex with rod-α-actinin-2: code 7A8U, https://doi.org/10.2210/pdb7A8U/pdb; and crystal structure of sarcomeric protein FATZ-1 (δ91-FATZ-1) in complex with hd-α-actinin-2: code 7ANK, https://doi.org/10.2210/pdb7ANK/pdb. The 3D models presented in Figs. 3 and 4 are available with these links: Fig. 3A: https://skfb.ly/6YvzT, https://skfb.ly/6YvAN, and https://skfb.ly/6YvB8; Fig. 3B: https://skfb.ly/6YvRV; Fig. 3C: https://skfb.ly/6YvQO; Fig. 4C: https://skfb.ly/6YvTS; Fig. 4D: https://skfb.ly/6YvUE; Fig. 4E: https://skfb.ly/6YvVt; Fig. 4F: https://skfb.ly/6YwQL; and Fig. 4G: https://skfb.ly/6YwZy. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/22/eabg7653/DC1
REFERENCES AND NOTES
- 1.Frank D., Kuhn C., Katus H. A., Frey N., The sarcomeric Z-disc: A nodal point in signalling and disease. J. Mol. Med. 84, 446–468 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Hwang P. M., Sykes B. D., Targeting the sarcomere to correct muscle function. Nat. Rev. Drug Discov. 14, 313–328 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Gautel M., Djinović-Carugo K., The sarcomeric cytoskeleton: From molecules to motion. J. Exp. Biol. 219, 135–145 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Luther P. K., The vertebrate muscle Z-disc: Sarcomere anchor for structure and signalling. J. Muscle Res. Cell Motil. 30, 171–185 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dabiri G. A., Turnacioglu K. K., Sanger J. M., Sanger J. W., Myofibrillogenesis visualized in living embryonic cardiomyocytes. Proc. Natl. Acad. Sci. U.S.A. 94, 9493–9498 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ehler E., Gautel M., The sarcomere and sarcomerogenesis. Adv. Exp. Med. Biol. 642, 1–14 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Sanger J. W., Wang J., Fan Y., White J., Mi-Mi L., Dube D. K., Sanger J. M., Pruyne D., Assembly and maintenance of myofibrils in striated muscle. Handb. Exp. Pharmacol. 235, 39–75 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Stout A. L., Wang J., Sanger J. M., Sanger J. W., Tracking changes in Z-band organization during myofibrillogenesis with FRET imaging. Cell Motil. Cytoskeleton 65, 353–367 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Wang J., Shaner N., Mittal B., Zhou Q., Chen J., Sanger J. M., Sanger J. W., Dynamics of Z-band based proteins in developing skeletal muscle cells. Cell Motil. Cytoskeleton 61, 34–48 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masaki T., Endo M., Ebashi S., Localization of 6S component of α-actinin at Z-band. J. Biochem. 62, 630–632 (1967). [DOI] [PubMed] [Google Scholar]
- 11.Sjöblom B., Salmazo A., Djinović-Carugo K., α-Actinin structure and regulation. Cell. Mol. Life Sci. 65, 2688–2701 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Almeida Ribeiro E. Jr., Pinotsis N., Ghisleni A., Salmazo A., Konarev P. V., Kostan J., Sjöblom B., Schreiner C., Polyansky A. A., Gkougkoulia E. A., Holt M. R., Aachmann F. L., Žagrović B., Bordignon E., Pirker K. F., Svergun D. I., Gautel M., Djinović-Carugo K., The structure and regulation of human muscle α-actinin. Cell 159, 1447–1460 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ylänne J., Scheffzek K., Young P., Saraste M., Crystal structure of the α-actinin rod reveals an extensive torsional twist. Structure 9, 597–604 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Faulkner G., Pallavicini A., Comelli A., Salamon M., Bortoletto G., Ievolella C., Trevisan S., Kojić S., Dalla Vecchia F., Laveder P., Valle G., Lanfranchi G., FATZ, a filamin-, actinin-, and telethonin-binding protein of the Z-disc of skeletal muscle. J. Biol. Chem. 275, 41234–41242 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Takada F., Vander Woude D. L., Tong H. Q., Thompson T. G., Watkins S. C., Kunkel L. M., Beggs A. H., Myozenin: An α-actinin- and γ-filamin-binding protein of skeletal muscle Z lines. Proc. Natl. Acad. Sci. U.S.A. 98, 1595–1600 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frey N., Olson E. N., Calsarcin-3, a novel skeletal muscle-specific member of the calsarcin family, interacts with multiple Z-disc proteins. J. Biol. Chem. 277, 13998–14004 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Frey N., Richardson J. A., Olson E. N., Calsarcins, a novel family of sarcomeric calcineurin-binding proteins. Proc. Natl. Acad. Sci. U.S.A. 97, 14632–14637 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gontier Y., Taivainen A., Fontao L., Sonnenberg A., van der Flier A., Carpen O., Faulkner G., Borradori L., The Z-disc proteins myotilin and FATZ-1 interact with each other and are connected to the sarcolemma via muscle-specific filamins. J. Cell Sci. 118, 3739–3749 (2005). [DOI] [PubMed] [Google Scholar]
- 19.von Nandelstadh P., Ismail M., Gardin C., Suila H., Zara I., Belgrano A., Valle G., Carpen O., Faulkner G., A class III PDZ binding motif in the myotilin and FATZ families binds enigma family proteins: A common link for Z-disc myopathies. Mol. Cell. Biol. 29, 822–834 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frey N., Frank D., Lippl S., Kuhn C., Kögler H., Barrientos T., Rohr C., Will R., Müller O. J., Weiler H., Bassel-Duby R., Katus H. A., Olson E. N., Calsarcin-2 deficiency increases exercise capacity in mice through calcineurin/NFAT activation. J. Clin. Invest. 118, 3598–3608 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seto J. T., Quinlan K. G. R., Lek M., Zheng X. F., Garton F., MacArthur D. G., Hogarth M. W., Houweling P. J., Gregorevic P., Turner N., Cooney G. J., Yang N., North K. N., ACTN3 genotype influences muscle performance through the regulation of calcineurin signaling. J. Clin. Invest. 123, 4255–4263 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Osio A., Tan L., Chen S. N., Lombardi R., Nagueh S. F., Shete S., Roberts R., Willerson J. T., Marian A. J., Myozenin 2 is a novel gene for human hypertrophic cardiomyopathy. Circ. Res. 100, 766–768 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruggiero A., Chen S. N., Lombardi R., Rodriguez G., Marian A. J., Pathogenesis of hypertrophic cardiomyopathy caused by myozenin 2 mutations is independent of calcineurin activity. Cardiovasc. Res. 97, 44–54 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arola A. M., Sanchez X., Murphy R. T., Hasle E., Li H., Elliott P. M., McKenna W. J., Towbin J. A., Bowles N. E., Mutations in PDLIM3 and MYOZ1 encoding myocyte Z line proteins are infrequently found in idiopathic dilated cardiomyopathy. Mol. Genet. Metab. 90, 435–440 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Posch M. G., Perrot A., Dietz R., Ozcelik C., Pankuweit S., Ruppert V., Richter A., Maisch B., Mutations in MYOZ1 as well as MYOZ2 encoding the calsarcins are not associated with idiopathic and familial dilated cardiomyopathy. Mol. Genet. Metab. 91, 207–208 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Posch M. G., Thiemann L., Tomasov P., Veselka J., Cardim N., Garcia-Castro M., Coto E., Perrot A., Geier C., Dietz R., Haverkamp W., Ozcelik C., Sequence analysis of myozenin 2 in 438 European patients with familial hypertrophic cardiomyopathy. Med. Sci. Monit. 14, CR372–CR374 (2008). [PubMed] [Google Scholar]
- 27.Frank D., Kuhn C., van Eickels M., Gehring D., Hanselmann C., Lippl S., Will R., Katus H. A., Frey N., Calsarcin-1 protects against angiotensin-II induced cardiac hypertrophy. Circulation 116, 2587–2596 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Frey N., Barrientos T., Shelton J. M., Frank D., Rütten H., Gehring D., Kuhn C., Lutz M., Rothermel B., Bassel-Duby R., Richardson J. A., Katus H. A., Hill J. A., Olson E. N., Mice lacking calsarcin-1 are sensitized to calcineurin signaling and show accelerated cardiomyopathy in response to pathological biomechanical stress. Nat. Med. 10, 1336–1343 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Yoshimoto Y., Ikemoto-Uezumi M., Hitachi K., Fukada S.-I., Uezumi A., Methods for accurate assessment of myofiber maturity during skeletal muscle regeneration. Front. Cell Dev. Biol. 8, 267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cornvik T., Dahlroth S.-L., Magnusdottir A., Flodin S., Engvall B., Lieu V., Ekberg M., Nordlund P., An efficient and generic strategy for producing soluble human proteins and domains in E. coli by screening construct libraries. Proteins 65, 266–273 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Brocca S., Testa L., Sobott F., Samalikova M., Natalello A., Papaleo E., Lotti M., De Gioia L., Doglia S. M., Alberghina L., Grandori R., Compaction properties of an intrinsically disordered protein: Sic1 and its kinase-inhibitor domain. Biophys. J. 100, 2243–2252 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uversky V. N., Protein intrinsic disorder and structure-function continuum. Prog. Mol. Biol. Transl. Sci. 166, 1–17 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Nygaard M., Kragelund B. B., Papaleo E., Lindorff-Larsen K., An efficient method for estimating the hydrodynamic radius of disordered protein conformations. Biophys. J. 113, 550–557 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qi G., Lee R., Hayward S., A comprehensive and non-redundant database of protein domain movements. Bioinformatics 21, 2832–2838 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Pinotsis N., Zielinska K., Babuta M., Arolas J. L., Kostan J., Khan M. B., Schreiner C., Salmazo A., Ciccarelli L., Puchinger M., Gkougkoulia E. A., de Almeida Ribeiro E. Jr., Marlovits T. C., Bhattacharya A., Djinovic-Carugo K., Calcium modulates the domain flexibility and function of an α-actinin similar to the ancestral α-actinin. Proc. Natl. Acad. Sci. U.S.A. 117, 22101–22112 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davey N. E., Van Roey K., Weatheritt R. J., Toedt G., Uyar B., Altenberg B., Budd A., Diella F., Dinkel H., Gibson T. J., Attributes of short linear motifs. Mol. Biosyst. 8, 268–281 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Kumar M., Gouw M., Michael S., Sámano-Sánchez H., Pancsa R., Glavina J., Diakogianni A., Valverde J. A., Bukirova D., Čalyševa J., Palopoli N., Davey N. E., Chemes L. B., Gibson T. J., ELM-the eukaryotic linear motif resource in 2020. Nucleic Acids Res. 48, D296–D306 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krissinel E., Henrick K., Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Borcherds W., Becker A., Chen L., Chen J., Chemes L. B., Daughdrill G. W., Optimal affinity enhancement by a conserved flexible linker controls p53 mimicry in MdmX. Biophys. J. 112, 2038–2042 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou H.-X., Loops in proteins can be modeled as worm-like chains. J. Phys. Chem. B 105, 6763–6766 (2001). [Google Scholar]
- 41.Oldfield C. J., Cheng Y., Cortese M. S., Romero P., Uversky V. N., Dunker A. K., Coupled folding and binding with α-helix-forming molecular recognition elements. Biochemistry 44, 12454–12470 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Bignon C., Troilo F., Gianni S., Longhi S., Partner-mediated polymorphism of an intrinsically disordered protein. J. Mol. Biol. 430, 2493–2507 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Bernadó P., Mylonas E., Petoukhov M. V., Blackledge M., Svergun D. I., Structural characterization of flexible proteins using small-angle x-ray scattering. J. Am. Chem. Soc. 129, 5656–5664 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Tria G., Mertens H. D. T., Kachala M., Svergun D. I., Advanced ensemble modelling of flexible macromolecules using x-ray solution scattering. IUCrJ 2, 207–217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharma R., Raduly Z., Miskei M., Fuxreiter M., Fuzzy complexes: Specific binding without complete folding. FEBS Lett. 589, 2533–2542 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Tompa P., Fuxreiter M., Fuzzy complexes: Polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 33, 2–8 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Oda T., Yanagisawa H., Cryo-electron tomography of cardiac myofibrils reveals a 3D lattice spring within the Z-discs. Commun. Biol. 3, 585 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gonçalves A. D., Alexander C., Roberts C. J., Spain S. G., Uddin S., Allen S., The effect of protein concentration on the viscosity of a recombinant albumin solution formulation. RSC Adv. 6, 15143–15154 (2016). [Google Scholar]
- 49.Shin Y., Brangwynne C. P., Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Banani S. F., Lee H. O., Hyman A. A., Rosen M. K., Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vernon R. M., Forman-Kay J. D., First-generation predictors of biological protein phase separation. Curr. Opin. Struct. Biol. 58, 88–96 (2019). [DOI] [PubMed] [Google Scholar]
- 52.Gillespie J. R., Shortle D., Characterization of long-range structure in the denatured state of staphylococcal nuclease. I. Paramagnetic relaxation enhancement by nitroxide spin labels. J. Mol. Biol. 268, 158–169 (1997). [DOI] [PubMed] [Google Scholar]
- 53.Xue Y., Podkorytov I. S., Rao D. K., Benjamin N., Sun H., Skrynnikov N. R., Paramagnetic relaxation enhancements in unfolded proteins: Theory and application to drkN SH3 domain. Protein Sci. 18, 1401–1424 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.André A. A. M., Spruijt E., Liquid-liquid phase separation in crowded environments. Int. J. Mol. Sci. 21, 5908 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tyn M. T., Gusek T. W., Prediction of diffusion coefficients of proteins. Biotechnol. Bioeng. 35, 327–338 (1990). [DOI] [PubMed] [Google Scholar]
- 56.Lin Y., Protter D. S. W., Rosen M. K., Parker R., Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol. Cell 60, 208–219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jain S., Wheeler J. R., Walters R. W., Agrawal A., Barsic A., Parker R., ATPase-modulated stress granules contain a diverse proteome and substructure. Cell 164, 487–498 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vernon R. M., Chong P. A., Tsang B., Kim T. H., Bah A., Farber P., Lin H., Forman-Kay J. D., Pi-Pi contacts are an overlooked protein feature relevant to phase separation. eLife 7, e31486 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Young P., Ferguson C., Bañuelos S., Gautel M., Molecular structure of the sarcomeric Z-disk: Two types of titin interactions lead to an asymmetrical sorting of α-actinin. EMBO J. 17, 1614–1624 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hastings R., de Villiers C. P., Hooper C., Ormondroyd L., Pagnamenta A., Lise S., Salatino S., Knight S. J. L., Taylor J. C., Thomson K. L., Arnold L., Chatziefthimiou S. D., Konarev P. V., Wilmanns M., Ehler E., Ghisleni A., Gautel M., Blair E., Watkins H., Gehmlich K., Combination of whole genome sequencing, linkage, and functional studies implicates a missense mutation in titin as a cause of autosomal dominant cardiomyopathy with features of left ventricular noncompaction. Circ. Cardiovasc. Genet. 9, 426–435 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Atkinson R. A., Joseph C., Kelly G., Muskett F. W., Frenkiel T. A., Nietlispach D., Pastore A., Ca2+−independent binding of an EF-hand domain to a novel motif in the α-actinin-titin complex. Nat. Struct. Biol. 8, 853–857 (2001). [DOI] [PubMed] [Google Scholar]
- 62.Au Y., Atkinson R. A., Guerrini R., Kelly G., Joseph C., Martin S. R., Muskett F. W., Pallavicini A., Faulkner G., Pastore A., Solution structure of ZASP PDZ domain: Implications for sarcomere ultrastructure and enigma family redundancy. Structure 12, 611–622 (2004). [DOI] [PubMed] [Google Scholar]
- 63.Beck M. R., Otey C. A., Campbell S. L., Structural characterization of the interactions between palladin and α-actinin. J. Mol. Biol. 413, 712–725 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kostan J., Pavšič M., Puž V., Schwarz T. C., Drepper F., Molt S., Graewert M. A., Schreiner C., Sajko S., van der Ven P. F. M., Svergun D. I., Warscheid B., Konrat R., Fürst D. O., Lenarčič B., Djinović-Carugo K., Molecular basis of F-actin regulation and sarcomere assembly via myotilin. PLOS Biol. 19, e3001148 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chi R. J., Simon A. R., Bienkiewicz E. A., Felix A., Keller T. C. S. III, Smooth muscle titin Zq domain interaction with the smooth muscle α-actinin central rod. J. Biol. Chem. 283, 20959–20967 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gustafson C. L., Parsley N. C., Asimgil H., Lee H.-W., Ahlbach C., Michael A. K., Xu H., Williams O. L., Davis T. L., Liu A. C., Partch C. L., A slow conformational switch in the BMAL1 transactivation domain modulates circadian rhythms. Mol. Cell 66, 447–457.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zou P., Pinotsis N., Lange S., Song Y.-H., Popov A., Mavridis I., Mayans O. M., Gautel M., Wilmanns M., Palindromic assembly of the giant muscle protein titin in the sarcomeric Z-disk. Nature 439, 229–233 (2006). [DOI] [PubMed] [Google Scholar]
- 68.Golji J., Collins R., Mofrad M. R. K., Molecular mechanics of the α-actinin rod domain: Bending, torsional, and extensional behavior. PLOS Comput. Biol. 5, e1000389 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Burgoyne T., Heumann J. M., Morris E. P., Knupp C., Liu J., Reedy M. K., Taylor K. A., Wang K., Luther P. K., Three-dimensional structure of the basketweave Z-band in midshipman fish sonic muscle. Proc. Natl. Acad. Sci. U.S.A. 116, 15534–15539 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dill K. A., Alonso D. O. V., Hutchinson K., Thermal stabilities of globular proteins. Biochemistry 28, 5439–5449 (1989). [DOI] [PubMed] [Google Scholar]
- 71.Lin Y.-H., Forman-Kay J. D., Chan H. S., Theories for sequence-dependent phase behaviors of biomolecular condensates. Biochemistry 57, 2499–2508 (2018). [DOI] [PubMed] [Google Scholar]
- 72.Quiroz F. G., Chilkoti A., Sequence heuristics to encode phase behaviour in intrinsically disordered protein polymers. Nat. Mater. 14, 1164–1171 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Boeynaems S., Alberti S., Fawzi N. L., Mittag T., Polymenidou M., Rousseau F., Schymkowitz J., Shorter J., Wolozin B., Van Den Bosch L., Tompa P., Fuxreiter M., Protein phase separation: A new phase in cell biology. Trends Cell Biol. 28, 420–435 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Forman-Kay J. D., Kriwacki R. W., Seydoux G., Phase separation in biology and disease. J. Mol. Biol. 430, 4603–4606 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li P., Banjade S., Cheng H.-C., Kim S., Chen B., Guo L., Llaguno M., Hollingsworth J. V., King D. S., Banani S. F., Russo P. S., Jiang Q.-X., Nixon B. T., Rosen M. K., Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336–340 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Robson R. M., Goll D. E., Arakawa N., Stromer M. H., Purification and properties of α-actinin from rabbit skeletal muscle. Biochim. Biophys. Acta 200, 296–318 (1970). [DOI] [PubMed] [Google Scholar]
- 77.Weirich K. L., Banerjee S., Dasbiswas K., Witten T. A., Vaikuntanathan S., Gardel M. L., Liquid behavior of cross-linked actin bundles. Proc. Natl. Acad. Sci. U.S.A. 114, 2131–2136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.He S., Chou H.-T., Matthies D., Wunder T., Meyer M. T., Atkinson N., Martinez-Sanchez A., Jeffrey P. D., Port S. A., Patena W., He G., Chen V. K., Hughson F. M., McCormick A. J., Mueller-Cajar O., Engel B. D., Yu Z., Jonikas M. C., The structural basis of Rubisco phase separation in the pyrenoid. Nat. Plants 6, 1480–1490 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mlynek G., Lehner A., Neuhold J., Leeb S., Kostan J., Charnagalov A., Stolt-Bergner P., Djinović-Carugo K., Pinotsis N., The Center for Optimized Structural Studies (COSS) platform for automation in cloning, expression, and purification of single proteins and protein-protein complexes. Amino Acids 46, 1565–1582 (2014). [DOI] [PubMed] [Google Scholar]
- 80.Whitmore L., Wallace B. A., Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers 89, 392–400 (2008). [DOI] [PubMed] [Google Scholar]
- 81.Sreerama N., Woody R. W., Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 287, 252–260 (2000). [DOI] [PubMed] [Google Scholar]
- 82.Cristodero M., Mani J., Oeljeklaus S., Aeberhard L., Hashimi H., Ramrath D. J. F., Lukeš J., Warscheid B., Schneider A., Mitochondrial translation factors of Trypanosoma brucei: Elongation factor-Tu has a unique subdomain that is essential for its function. Mol. Microbiol. 90, 744–755 (2013). [DOI] [PubMed] [Google Scholar]
- 83.Chambers M. C., Maclean B., Burke R., Amodei D., Ruderman D. L., Neumann S., Gatto L., Fischer B., Pratt B., Egertson J., Hoff K., Kessner D., Tasman N., Shulman N., Frewen B., Baker T. A., Brusniak M.-Y., Paulse C., Creasy D., Flashner L., Kani K., Moulding C., Seymour S. L., Nuwaysir L. M., Lefebvre B., Kuhlmann F., Roark J., Rainer P., Detlev S., Hemenway T., Huhmer A., Langridge J., Connolly B., Chadick T., Holly K., Eckels J., Deutsch E. W., Moritz R. L., Katz J. E., Agus D. B., MacCoss M., Tabb D. L., Mallick P., A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Leitner A., Walzthoeni T., Aebersold R., Lysine-specific chemical cross-linking of protein complexes and identification of cross-linking sites using LC-MS/MS and the xQuest/xProphet software pipeline. Nat. Protoc. 9, 120–137 (2014). [DOI] [PubMed] [Google Scholar]
- 85.Combe C. W., Fischer L., Rappsilber J., xiNET: Cross-link network maps with residue resolution. Mol. Cell. Proteomics 14, 1137–1147 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Alberti S., Saha S., Woodruff J. B., Franzmann T. M., Wang J., Hyman A. A., A user’s guide for phase separation assays with purified proteins. J. Mol. Biol. 430, 4806–4820 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schneider C. A., Rasband W. S., Eliceiri K. W., NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Theile C. S., Witte M. D., Blom A. E. M., Kundrat L., Ploegh H. L., Guimaraes C. P., Site-specific N-terminal labeling of proteins using sortase-mediated reactions. Nat. Protoc. 8, 1800–1807 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kazimierczuk K., Zawadzka-Kazimierczuk A., Koźmiński W., Non-uniform frequency domain for optimal exploitation of non-uniform sampling. J. Magn. Reson. 205, 286–292 (2010). [DOI] [PubMed] [Google Scholar]
- 90.Zawadzka-Kazimierczuk A., Koźmiński W., Šanderová H., Krásný L., High dimensional and high resolution pulse sequences for backbone resonance assignment of intrinsically disordered proteins. J. Biomol. NMR 52, 329–337 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Żerko S., Byrski P., Włodarczyk-Pruszyński P., Górka M., Ledolter K., Masliah E., Konrat R., Koźmiński W., Five and four dimensional experiments for robust backbone resonance assignment of large intrinsically disordered proteins: Application to Tau3x protein. J. Biomol. NMR 65, 193–203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kazimierczuk K., Zawadzka A., Koźmiński W., Zhukov I., Random sampling of evolution time space and Fourier transform processing. J. Biomol. NMR 36, 157–168 (2006). [DOI] [PubMed] [Google Scholar]
- 93.Stanek J., Koźmiński W., Iterative algorithm of discrete Fourier transform for processing randomly sampled NMR data sets. J. Biomol. NMR 47, 65–77 (2010). [DOI] [PubMed] [Google Scholar]
- 94.Kazimierczuk K., Zawadzka A., Koźmiński W., Narrow peaks and high dimensionalities: Exploiting the advantages of random sampling. J. Magn. Reson. 197, 219–228 (2009). [DOI] [PubMed] [Google Scholar]
- 95.Kazimierczuk K., Zawadzka A., Koźmiński W., Zhukov I., Lineshapes and artifacts in multidimensional Fourier transform of arbitrary sampled NMR data sets. J. Magn. Reson. 188, 344–356 (2007). [DOI] [PubMed] [Google Scholar]
- 96.Lee W., Tonelli M., Markley J. L., NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zawadzka-Kazimierczuk A., Koźmiński W., Billeter M., TSAR: A program for automatic resonance assignment using 2D cross-sections of high dimensionality, high-resolution spectra. J. Biomol. NMR 54, 81–95 (2012). [DOI] [PubMed] [Google Scholar]
- 98.Schleucher J., Schwendinger M., Sattler M., Schmidt P., Schedletzky O., Glaser S. J., Sørensen O. W., Griesinger C., A general enhancement scheme in heteronuclear multidimensional NMR employing pulsed field gradients. J. Biomol. NMR 4, 301–306 (1994). [DOI] [PubMed] [Google Scholar]
- 99.Iwahara J., Tang C., Marius Clore G., Practical aspects of 1H transverse paramagnetic relaxation enhancement measurements on macromolecules. J. Magn. Reson. 184, 185–195 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A., NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 101.Blanchet C. E., Spilotros A., Schwemmer F., Graewert M. A., Kikhney A., Jeffries C. M., Franke D., Mark D., Zengerle R., Cipriani F., Fiedler S., Roessle M., Svergun D. I., Versatile sample environments and automation for biological solution x-ray scattering experiments at the P12 beamline (PETRA III, DESY). J. Appl. Cryst. 48, 431–443 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pernot P., Round A., Barrett R., De Maria Antolinos A., Gobbo A., Gordon E., Huet J., Kieffer J., Lentini M., Mattenet M., Morawe C., Mueller-Dieckmann C., Ohlsson S., Schmid W., Surr J., Theveneau P., Zerrad L., McSweeney S., Upgraded ESRF BM29 beamline for SAXS on macromolecules in solution. J. Synchrotron Radiat. 20, 660–664 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Panjkovich A., Svergun D. I., CHROMIXS: Automatic and interactive analysis of chromatography-coupled small-angle x-ray scattering data. Bioinformatics 34, 1944–1946 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Franke D., Petoukhov M. V., Konarev P. V., Panjkovich A., Tuukkanen A., Mertens H. D. T., Kikhney A. G., Hajizadeh N. R., Franklin J. M., Jeffries C. M., Svergun D. I., ATSAS 2.8: A comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Cryst. 50, 1212–1225 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Receveur-Brechot V., Durand D., How random are intrinsically disordered proteins? A small angle scattering perspective. Curr. Protein Pept. Sci. 13, 55–75 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Svergun D., Barberato C., Koch M. H. J., CRYSOL - A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 28, 768–773 (1995). [Google Scholar]
- 107.Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D., Konarev P. V., Svergun D. I., New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Cryst. 45, 342–350 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kikhney A. G., Borges C. R., Molodenskiy D. S., Jeffries C. M., Svergun D. I., SASBDB: Towards an automatically curated and validated repository for biological scattering data. Protein Sci. 29, 66–75 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kabsch W., XDS. Acta Crystallogr. D Biol. Crystallogr. 66 ( Pt 2), 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kabsch W., Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liebschner D., Afonine P. V., Baker M. L., Bunkoczi G., Chen V. B., Croll T. I., Hintze B., Hung L. W., Jain S., McCoy A. J., Moriarty N. W., Oeffner R. D., Poon B. K., Prisant M. G., Read R. J., Richardson J. S., Richardson D. C., Sammito M. D., Sobolev O. V., Stockwell D. H., Terwilliger T. C., Urzhumtsev A. G., Videau L. L., Williams C. J., Adams P. D., Macromolecular structure determination using x-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Smart O. S., Womack T. O., Flensburg C., Keller P., Paciorek W., Sharff A., Vonrhein C., Bricogne G., Exploiting structure similarity in refinement: Automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. D Biol. Crystallogr. 68, 368–380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Maupetit J., Gautier R., Tuffery P., SABBAC: Online structural alphabet-based protein backbone reconstruction from alpha-carbon trace. Nucleic Acids Res. 34, W147–W151 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., Lindahl E., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 (2015). [Google Scholar]
- 117.Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J. L., Dror R. O., Shaw D. E., Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Piana S., Donchev A. G., Robustelli P., Shaw D. E., Water dispersion interactions strongly influence simulated structural properties of disordered protein states. J. Phys. Chem. B 119, 5113–5123 (2015). [DOI] [PubMed] [Google Scholar]
- 119.Hoover W. G., Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 (1985). [DOI] [PubMed] [Google Scholar]
- 120.Parrinello M., Rahman A., Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981). [Google Scholar]
- 121.Hess B., Bekker H., Berendsen H. J. C., Fraaije J. G. E. M., LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997). [Google Scholar]
- 122.Corpet F., Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16, 10881–10890 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Robert X., Gouet P., Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Piovesan D., Tabaro F., Paladin L., Necci M., Micetic I., Camilloni C., Davey N., Dosztanyi Z., Meszaros B., Monzon A. M., Parisi G., Schad E., Sormanni P., Tompa P., Vendruscolo M., Vranken W. F., Tosatto S. C. E., MobiDB 3.0: More annotations for intrinsic disorder, conformational diversity and interactions in proteins. Nucleic Acids Res. 46, D471–D476 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mizianty M. J., Peng Z., Kurgan L., MFDp2: Accurate predictor of disorder in proteins by fusion of disorder probabilities, content and profiles. Intrinsically Disord. Proteins 1, e24428 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ward J. J., Sodhi J. S., McGuffin L. J., Buxton B. F., Jones D. T., Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 337, 635–645 (2004). [DOI] [PubMed] [Google Scholar]
- 127.Meszaros B., Erdos G., Dosztanyi Z., IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 46, W329–W337 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Piovesan D., Walsh I., Minervini G., Tosatto S. C. E., FELLS: Fast estimator of latent local structure. Bioinformatics 33, 1889–1891 (2017). [DOI] [PubMed] [Google Scholar]
- 129.Ishida T., Kinoshita K., Prediction of disordered regions in proteins based on the meta approach. Bioinformatics 24, 1344–1348 (2008). [DOI] [PubMed] [Google Scholar]
- 130.Drozdetskiy A., Cole C., Procter J., Barton G. J., JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 43, W389–W394 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cilia E., Pancsa R., Tompa P., Lenaerts T., Vranken W. F., The DynaMine webserver: Predicting protein dynamics from sequence. Nucleic Acids Res. 42, W264–W270 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Konrat R., The protein meta-structure: A novel concept for chemical and molecular biology. Cell. Mol. Life Sci. 66, 3625–3639 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Callebaut I., Labesse G., Durand P., Poupon A., Canard L., Chomilier J., Henrissat B., Mornon J. P., Deciphering protein sequence information through hydrophobic cluster analysis (HCA): Current status and perspectives. Cell. Mol. Life Sci. 53, 621–645 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Letunic I., Bork P., 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 46, D493–D496 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lancaster A. K., Nutter-Upham A., Lindquist S., King O. D., PLAAC: A web and command-line application to identify proteins with prion-like amino acid composition. Bioinformatics 30, 2501–2502 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hughes M. P., Sawaya M. R., Boyer D. R., Goldschmidt L., Rodriguez J. A., Cascio D., Chong L., Gonen T., Eisenberg D. S., Atomic structures of low-complexity protein segments reveal kinked β sheets that assemble networks. Science 359, 698–701 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bolognesi B., Lorenzo Gotor N., Dhar R., Cirillo D., Baldrighi M., Tartaglia G. G., Lehner B., A concentration-dependent liquid phase separation can cause toxicity upon increased protein expression. Cell Rep. 16, 222–231 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kabsch W., Sander C., Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 (1983). [DOI] [PubMed] [Google Scholar]
- 139.Tina K. G., Bhadra R., Srinivasan N., PIC: Protein interactions calculator. Nucleic Acids Res. 35, W473–W476 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ortega A., Amoros D., Garcia de la Torre J., Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 101, 892–898 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 142.Winter L., Staszewska I., Mihailovska E., Fischer I., Goldmann W. H., Schröder R., Wiche G., Chemical chaperone ameliorates pathological protein aggregation in plectin-deficient muscle. J. Clin. Invest. 124, 1144–1157 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Das R. K., Pappu R. V., Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. U.S.A. 110, 13392–13397 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chong P. A., Vernon R. M., Forman-Kay J. D., RGG/RG motif regions in RNA binding and phase separation. J. Mol. Biol. 430, 4650–4665 (2018). [DOI] [PubMed] [Google Scholar]
- 145.Uversky V. N., Santambrogio C., Brocca S., Grandori R., Length-dependent compaction of intrinsically disordered proteins. FEBS Lett. 586, 70–73 (2012). [DOI] [PubMed] [Google Scholar]
- 146.Uversky V. N., Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 11, 739–756 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.A. Kessel, N. Ben-Tal, Introduction to Proteins: Structure, Function, and Motion (Chapman & Hall/CRC Mathematical and Computational Biology Series, CRC Press, Taylor & Francis Group, ed. 2, 2018), pp. lii, 932 pages. [Google Scholar]
- 148.Cooper A., Johnson C. M., Lakey J. H., Nollmann M., Heat does not come in different colours: Entropy-enthalpy compensation, free energy windows, quantum confinement, pressure perturbation calorimetry, solvation and the multiple causes of heat capacity effects in biomolecular interactions. Biophys. Chem. 93, 215–230 (2001). [DOI] [PubMed] [Google Scholar]
- 149.F. W. Putnam, The Plasma Proteins: Structure, Function, and Genetic Control (Academic Press, ed. 2, 1975), pp. v. 1–5. [Google Scholar]
- 150.Axelsson I., Characterization of proteins and other macromolecules by agarose gel chromatography. J. Chromatogr. 152, 21–32 (1978). [Google Scholar]
- 151.Kikhney A. G., Panjkovich A., Sokolova A. V., Svergun D. I., DARA: A web server for rapid search of structural neighbours using solution small angle x-ray scattering data. Bioinformatics 32, 616–618 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Laskowski R. A., Swindells M. B., LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/22/eabg7653/DC1